

Abstract
Cholesteryl ester transfer protein (CETP) is a plasma glycoprotein that facilitates the transfer of cholesteryl esters from the atheroprotective high density lipoprotein (HDL) to the proatherogenic low density lipoprotein cholesterol (LDL) and very low density lipoprotein cholesterol (VLDL) leading to lower levels of HDL but raising the levels of proatherogenic LDL and VLDL. Inhibition of CETP is considered a potential approach to treat dyslipidemia. However, discussions regarding the role of CETP-mediated lipid transfer in the development of atherosclerosis and CETP inhibition as a potential strategy for prevention of atherosclerosis have been controversial. Although many animal studies support the hypothesis that inhibition of CETP activity may be beneficial, negative phase III studies on clinical endpoints with the CETP inhibitor torcetrapib challenged the future perspectives of CETP inhibitors as potential therapeutic agents. The review provides an update on current understanding of the molecular mechanisms involved in CETP activity and its inhibition.
Keywords: Cholesteryl ester transfer protein, High-density lipoprotein, Cholesterol, Inhibition, Dyslipidemia
Introduction
Cardiovascular disease is the most common cause for mortality and morbidity in the developed world, and it is estimated that mortality from cardiovascular diseases will have increased worldwide by 90% by the year 2020 when compared with the situation in 1990 [1]. Guidelines for the prevention and management of cardiovascular disease focus on the reduction of levels of low-density lipoprotein (LDL) cholesterol [2, 3]. Hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (referred to as statins) are commonly used to manage LDL [2, 3]. Despite this existing standard, reduction of coronary events is low, and new and efficacious treatment options are needed [4].
The plasma cholesteryl ester transfer protein (CETP) was first described as a high molecular weight protein stimulating the transfer of cholesteryl ester between lipoproteins in plasma of hypercholesterolemic rabbits [5]. Additional reports on the function of CETP in humans followed a couple of years later [6, 7]. Later it was again demonstrated that CETP is also able to facilitate the transfer of triglyceride and phospholipids [8]. CETP is an important component of the reverse cholesterol transport (RCT) and regulates the concentration of high-density lipoprotein cholesterol (HDL). RCT is chiefly characterized by the transport of cholesterol from peripheral tissues, for instance macrophage foam cells in arterial walls, to the liver for excretion via bile and feces [9].
Various studies suggest that the risk of cardiovascular disease is inversely associated with plasma levels of HDL [10–12], and HDL has been proposed to have potential atheroprotective effects [13]. CETP facilitates the transfer of cholesterol esters from the atheroprotective HDL to the proatherogenic low density lipoprotein cholesterol (LDL) and very low density lipoprotein cholesterol (VLDL), leading to lower levels of HDL but raising the levels of proatherogenic LDL and VLDL. On the other hand, CETP transfers triglycerides (TG) from VLDL or LDL to HDL, leading to TG-enriched HDL, which is more readily hydrolyzed by hepatic lipase resulting in smaller sized HDL particles that more effectively promote reverse cholesterol transport [14]. CETP inhibition might be a powerful tool for increasing HDL cholesterol, decreasing LDL and VLDL cholesterol, and thus reducing the development of atherosclerosis [15].
However, discussions about the role of CETP-mediated lipid transfer and its inhibition for the prevention of cardiovascular diseases have been controversial [16–27], although many animal studies support the hypothesis that inhibition of CETP activity may be beneficial [28–32]. Negative phase III studies on clinical endpoints with the CETP inhibitor torcetrapib challenged the future perspectives of CETP inhibitors as potential therapeutic agents [33–36]. It has been discussed recently whether potential off-target effects of torcetrapib could have contributed to the failure of this CETP inhibitor [17–19], and continuing to study other CETP inhibitors for their potential to improve plasma lipid profiles and reduce cardiovascular risk has been suggested. Indeed, currently several compounds are being investigated in preclinical or clinical studies [17–19, 37, 38].
In this article, we review the current mechanisms involved in CETP activity and CETP inhibition, and their potential implications for treating atherosclerosis.
Structure of cholesteryl ester transfer protein
Information on the structure of CETP is necessary for understanding the interaction of CETP with lipoproteins. CETP belongs to a family of proteins, involved in lipid binding, such as lipopolysaccharide-binding protein (LBP) and bactericidal/permeability-increasing protein (BPI) [39], and lipid transfer [phospholipid transfer protein (PLTP)] [40]. Plasma CETP is a 476 amino-acid residue glycoprotein and has a molecular mass of 74 kDa, of which 28% is attributed to N-glycosylation at residues 88, 240, 341 and 396 [41–43].
At the beginning of CETP research, several laboratories have reported purification of proteins from human plasma active on cholesteryl ester transfer between lipoprotein particles with considerable variability in molecular mass, abundance and specificity. Cloning and sequencing of the CETP-cDNA [42] and the determination of the structure and organization of the human CETP gene [43] have greatly contributed to overcoming this situation. The CETP gene is located at chromosome 16 (16q12–16q21). The gene spans approximately 25 kbp and contains 16 exons (size range 32–250 bp) and 15 introns [43]. The upstream flanking region has regulatory sequences including nuclear factor 1, a sterol regulatory element, hepatocyte nuclear actor 1 and a nuclear receptor binding site that is activated by LXR [44]. A single copy of the CETP gene exists per haploid genome.
Interestingly, the sequence and organization of the CETP gene do not resemble those of other lipid-metabolizing enzymes or apolipoproteins, although there is apparent identity of a pentapeptide sequence (ValLeuThrLeuAla) within the hydrophobic core of the signal sequences of human CETP, apolipoproteins A-IV and A-I, and lipoprotein lipase. This pentapeptide sequence may mediate a specialized function related to lipid metabolism or transport [43].
The availability of structural details of CETP was a prerequisite to replace older and speculative hypotheses regarding the mechanism of CETP-mediated lipid transfer. The crystal structure of CETP has been described at 2.2 Å resolution [45]. Qiu et al. [45] described details regarding the structure of CETP and, on this basis, formulated a structure-based hypothesis regarding the CETP mechanism of action. CETP has a concave surface area that appears to be the part of the molecule that binds lipoproteins [45]. The diameter of the curvature coincides with that of discoidal HDL particles, which suggests that CETP may bind one single HDL particle to this area with only modest structural changes. The surface contains charged and hydrophobic residues that are evenly distributed. This suggests some robustness with respect to activity, which is supported by the absence of detectable activity changes in concave-surface mutants [44]. It was postulated that potential conformational changes may occur to accommodate larger lipoprotein particles [45]. The structure of CETP also revealed a 60-Å-long tunnel structure that demonstrated binding of two neutral lipids and two phospholipids [45]. Two hydrophobic cholesteryl esters fill the tunnel, and the two distinct openings are plugged by phosphatidylcholines at each end [45]. The tunnel openings allow lipid access, which seems to be aided by a flexible helix and by a mobile flap. Point mutations blocking the middle of the tunnel abolish lipid-transfer activities, suggesting that neutral lipids pass through this continuous tunnel [45]. The crystal structure provides evidence for the proposed mechanisms of action of CETP and, in addition, could be a useful basis for future drug design.
Mechanism of action
CETP is secreted mainly from the liver [47]. Significant amounts of CETP mRNA are also expressed in adipose tissues [48, 49]. It was demonstrated that human adipose tissue maintained in organ culture synthesizes and secretes CETP [49].
In the circulation, CETP is frequently bound to HDL [46].
CETP is involved in transport pathways for lipids.
In general, CETP facilitates the transfer of neutral lipids such as cholesteryl ester and triglycerides among lipoprotein particles in two ways, as homoexchange, which describes the bidirectional transfer of the same neutral lipid between lipoproteins or heteroexchange, which describes a net mass transfer of cholesteryl ester and triglycerides between lipoproteins [5–9, 50, 51]. The overall effect of the heteroexchange is the transfer of cholesteryl ester from HDL to triglyceride-rich lipoprotein (TRL like VLDL) and LDL and a transfer of triglycerides from TRL to LDL and HDL [5–9, 50, 51]. CETP may also have an important role in cellular cholesterol homeostasis. Indeed, CETP has a role in selective uptake of HDL cholesterol esters by human adipocytes, suggesting that this pathway may be of quantitative physiological significance in HDL remodeling and adipocyte cholesterol accumulation [52]. Here, we focus on the role of CETP in plasma lipid transport. Figure 1 illustrates the mechanism hypothesis of CETP transfer of cholesteryl ester and triglyceride between HDL and triglyceride-rich lipoproteins. In normolipidemic humans, CETP accounts for redistribution of cholesteryl ester from HDL to LDL particles, particularly of intermediate size and density. Such LDL particles have high affinity for LDL receptor and are therefore rapidly removed from plasma [53].
Fig. 1.

CETP facilitates bi-directional transfer of cholesteryl esters and triglycerides between plasma lipoproteins. The overall effect of the heteroexchange is the transfer of cholesteryl ester from HDL to triglyceride-rich lipoprotein (TRL like VLDL) and LDL and a transfer of triglycerides from TRL to LDL and HDL. CE cholesterylester; VLDL very low density lipoproteins, triglyceride-rich; TG triglycerides; SRB1 scavenger receptor B1; FC free cholesterol; LCAT lecithin:cholesterol acetyltransferase
The key role of CETP in mediating bidirectional transfer of cholesteryl ester and triglyceride among plasma lipoproteins is important to understand the relationship of high plasma triglyceride and low HDL levels and the associated risk to develop cardiovascular diseases.
Basically, CETP is thought to work as a carrier by accepting neutral lipids from a donor particle, transporting them through the aequous phase and delivering them to the acceptor lipoprotein [50]. This transport would involve the following basic steps: binding of CETP to lipoproteins, exchange of lipids at the CETP-lipoprotein interface, the actual transfer of neutral lipids and phospholipids and finally the release of the newly assembled lipoproteins from CETP.
The binding of HDL to CETP is influenced by several factors including particle size, shape of the particle and lipid composition [54]. Binding of lipoproteins occurs most likely at the concave surface of CETP [45]. Biochemical binding studies have shown high binding affinity for nascent disoidal HDL to CETP [54, 55]. According to the structure-based binding model, these particles exactly fit into the radius of the curvature without conformational change [45]. The surface contains charged and hydrophobic residues that are evenly distributed [45]. Polar groups of the CETP-bound phospholipids with amino acids at the HDL surface could interact electrostatically and initiate the transfer of those phospholipids to HDL. CETP-bound triglycerides or cholesteryl ester could fill the hydrophobic pocket temporarily. Upon lipoprotein binding, phospholipids bound to the tunnel openings may merge into the phospholipid monolayer of solubilized neutral lipids and permit access to neutral lipids entering and exiting the tunnel structure. Cholesteryl ester and triglyerides may completely pass through the tunnel, which is considerably wider than the minimal cross-sectional area of cholesteryl esters or triglycerides [45, 56].
The atheroprotective role of HDL and the reverse cholesterol transport
HDL particles are heterogeneous in human plasma with distinct and unique physicochemical properties, intravascular metabolism and biological activities [57]. Types of HDL particles [58] include discoidal and spherical HDL particles. The small discoidal particles (<8 nm diameter) mainly consist of ApoAI embedded in a lipid monolayer consisting of free cholesterol and phospholipids. Spherical HDL particles are larger (>8 nm diameter) and contain a hydrophobic core of cholesterylester and triglycerides. Separation of light HDL2 (9–10 nm diameter; density, d 1.063–1.125 g/ml) and dense HDL particles (8–9 nm diameter; d 1.125–1.21 g/ml) is possible by ultracentrifugation [reviewed in 58].
The imbalance between cholesterol deposition in and removal of cholesterol from the arterial wall is thought to play a role in the development of atherosclerosis [59, 60]. Reverse cholesterol transport (RCT) describes the transport of excess cholesterol from the periphery, for instance, macrophage foam cells in arterial walls to the liver [59]. HDL plays a key role in RCT. The RCT process includes the efflux of free cholesterol from tissue macrophages, the uptake by HDL, its subsequent esterification by lecithin:cholesterol acetyltransferase (LCAT) and transport to the liver for secretion [51, 61, 62]. The efflux of cholesterol from macrophages can occur by several mechanisms including diffusion [63], interaction with the ATP-binding cassette transporters ABCA1 and ABCG1 [64] or scavenger-receptor BI (SR-BI) [65]. The ATP-binding cassette transporters like ABCA1 appear to be the most efficient system accounting for more than 50% of the cholesterol efflux from macrophages to poorly lipidated ApoA-I [62]. After esterification by LCAT, ApoA-I is subsequently converted to HDL. Mature HDL may deliver cholesteryl ester or free cholesterol directly to the liver.
The majority of free cholesterol is transported to the liver by the SR-BI pathway [66, 67]. Cholesteryl ester is mainly transferred to other lipoproteins by CETP in exchange for triglycerides in apolipoprotein B-containing particles like LDL or VLDL. Apo-B-rich particles are taken up by the liver through LDL receptors. The last step in the reverse cholesterol transport pathway is the excretion of free cholesterol or cholesteryl esters [66–70].
HDL has several proposed atheroprotective functions including the improvement of re-endothelization through EPC activation and proliferation [71], the reduction of LDL oxidation and endothelial cell adhesion expression [71]. Administration of exogenous HDL to hypercholesterolemic rabbits led to reduction of free and esterified cholesterol in atherosclerotic plaques and subsequent regression of those plaques [61].
Although particles of all HDL sub-fractions have multiple biological activities including cholesterol efflux capacity, antioxidative, antiinflammatory, antiapoptotic, antithrombotic, vasodilatatory and antiinfective activities, small and protein-rich particles possess particularly potent antiatherogenic properties [20, 73].
There is a substantial body of evidence from basic science, epidemiologic studies and clinical trials showing that raising HDL by therapeutic means may effectively reduce cardiovascular risk [28–32, 74–76]. Caucasian and Asian subjects with exceptional longevity have been described to show elevated HDL and reduced LDL levels with larger particle sizes in both HDL and LDL [74–76].
A therapeutic increase of HDL cholesterol caused by nicotinic acid (niacin) has been described to be effective for the reduction of atherosclerosis progression or clinical cardiovascular events over a broad range of risk levels [20]. Thus, considering the potential impact of increased levels of HDL cholesterol in the reduction of cardiovascular risk, RCT is an important mechanism in its proposed atheroprotective or antiatherogenic role. However, carriers of the apoA-1 Milano mutation [77, 78] have very low HDL cholesterol levels, but no increase in the risk of heart disease. ApoA-I Milano contains an extra cysteine bridge, which enables the protein to form homodimers or heterodimers with apoA-II. This finding led to the speculation that apoA1 Milano has enhanced cardioprotective effects [79–82] on the one hand; on the other hand, some believe that there is functional equivalence between wild-type ApoA-1 and apoA1 Milano [83, 84]. However, in a clinical study infusions of apoA1 Milano-phospholipid complexes led to the regression of atheromas [85, 86]. It is not clear how the Milano mutation impacts the RCT, but a preclincal study suggests that wild-type ApoA-1 and apoA1 Milano are equally efficient at promoting macrophage RCT, which suggests that if there is atheroprotecivity due to the Milano mutation, this is unlikely to be due to enhanced RCT by macrophages [87].
CETP deficiency and genetic variants
Two isoforms of CETP mRNA have been described in humans including a full-length form and a splice variant where exon 9 is deleted [88]. Although the physiological relevance of the splicing for CETP regulation in humans is not fully understood, it is hypothesized that the deletion variant may function as an inhibitor for the secretion of full length CETP [89].
Mutations resulting in CETP deficiencies have been described in Japanese populations but are rare in Western countries [90–93]. Altogether, 13 mutations of the CETP gene have been described including nonsense and missense mutations as well as mutations in the splicing site or promoter [89–93]. These mutations result in net CETP activity loss and are associated with high HDL [44]. An intron 14 splicing defect (Int 14 + 1G → A Int 14 + 1G → A) and an exon 15 missense mutation occur frequently in Japanese people with a heterozygote frequency of 1 and 7%, respectively [44, 92]. Genetic CETP deficiencies account for an estimated 50% of hyperalphalipoproteinemia cases among Japanese people and may lead to up to six-fold higher HDL levels [44, 92, 94]. The relevance of CETP deficiency is still not completely understood, but it is doubted that partial or complete CETP deficiency determines longevity. Indeed, Japanese with the Int 14 + 1G → A mutation have a higher prevalence of ischemic electrocardiogram changes despite HDL levels of more than 1.80 mmol/l [91]. However, it is unclear whether complete CETP deficiency may be proatherogenic [95]. In a small group of people with heterozygote CETP deficiency, carotid intima media thickness has been found to be unaltered [96].
Complete or partial CETP deficiency only occurs infrequently, whereas, in contrast, SNPs including variations in both coding and non-coding regions are quite common [44, 90, 92]. One of the most studied forms is the TaqIB polymorphism in the 277th nucleotide in intron 1 (rs 708272; chromosomal position 55553789 with the B1 allele with a TaqI restriction site being the most common) [44]. It is well established that the B2 allele is a determinant of higher HDL cholesterol, and B1B1 subjects apparently have higher CETP mass and activity compared with B2B2 homozygotes [44, 97]. A pooled analysis concerning the association of cardiovascular disease with the TaqIB polymorphism that was predominantly including Caucasian subjects suggested that that the risk of cardiovascular disease is increased in B2B2 compared with B1B1 carriers, whereas cardiovascular risk appears to be decreased in B2B2 carriers compared to B1B1 carriers from high-risk populations [44]. The authors explain the apparent paradox in part by the selection of subjects towards a lower frequency of B2B2 carriers in the high-risk population. Although many questions are still open, this analysis suggests that in the general population, the B2 allele is associated with a higher cardiovascular risk despite higher HDL. However, the authors conclude that it is unlikely that determination of a single CETP polymorphism or even CETP haplotype analysis would be of clinical benefit in predicting cardiovascular disease in individual subjects. In view of contradictory results concerning the effect of this common CETP variation on the lipid response to (1) lipid-lowering treatment, (2) diet intervention and (3) cardiovascular outcome after statin therapy, the documentation and interpretation of CETP variants for guiding optimal treatment efficacy cannot be recommended [44]. In a recent meta-analysis, three CETP genotypes were found to be associated with moderate inhibition of CETP activity, modestly higher HDL levels and weakly inverse associations with coronary risk [98]. These findings highlight the need for further and probably larger studies to investigate the impact of genetic variants on complex outcomes such as coronary disease.
Rationale for CETP inhibition as a principle of preventative treatment for atherosclerosis
The potential role of CETP in atherogenesis has been discussed for a long period of time, and the controversial debate continues [16–27, 99].
The CETP activity involves two processes. Firstly, CETP activity accounts for a substantial proportion of the cholesterol that is returned to the liver by RCT and subsequently excreted. Secondly, CETP mediates the transport of cholesteryl ester from HDL to apo-B-rich lipoproteins like VLDL or LDL. This process results in a net decrease of atheroprotective HDL, which is thought to be the reason for the potential proatherogenic role of CETP in humans. High CETP activity has been found in patients with metabolic disease including diabetes mellitus type 2 or metabolic syndrome [58]. High CETP activity enriches the triglyceride content of HDL particles. These triglyceride-rich HDL particles are being hydrolyzed by hepatic lipase and destabilized, which involves shedding of apoAI. In consequence, the turnover of this protein is accelerated [72, 91]. Triglyceride-rich small HDL particles do not possess atheroprotective activities [71]. Therefore, CETP inhibition may have a double function; it could first improve the core lipid composition of HDL and, secondly, correct HDL functional defects that are associated with altered composition of HDL in patients with certain metabolic diseases.
The principle of CETP inhibition is supported by numerous animal studies [28–32]. Some species like mice and rats are naturally deficient in CETP. Introduction of the human CETP gene into mice led to a reduction of HDL levels and a net increase of VLDL bidirectional and LDL [100, 101]. A high cholesterol diet led to the development of atherosclerotic lesions that were more severe than in non-transgenic animals [29]. Lesion progression in this model was a function of cholesterol partitioning between the lipoproteins and less a function of plasma cholesterol concentration, which indicates that the CETP-induced alteration in cholesterol distribution might have been the major reason for the more rapid development of arterial lesions in CETP-transgenic mice [29].
Further experimental evidence for the atherogenic potential of CETP and the beneficial effects of inhibiting CETP activity was obtained in rabbits where CETP activity is naturally high. CETP inhibition in cholesterol-fed rabbits by means of antisense oligonucleotides targeted to the liver resulted in higher HDL levels and reduction of atherosclerosis compared to controls [30]. In addition to these findings, administration of the CETP-inhibitor dalcetrapib (JTT-705) to cholesterol-fed rabbits resulted in increased HDL levels and, moreover, reduced atherosclerosis comparable to simvastatin in the same setting [31]. However, different results were obtained in rabbits with severe hypercholesterolemia where administration of dalcetrapib led to high HDL but did not show an effect on the development of atherosclerotic lesions [32]. In this study, the atheromatous area was correlated with triglycerides that were elevated and non-HDL cholesterol levels. This is consistent with the finding that in subjects with familial hypercholesterolemia, increased HDL levels caused by partial CETP deficiency were shown to be insufficient to prevent CHD [102].
In healthy individuals with high triglyceride levels, elevated CETP levels are associated with an increasing risk of future coronary heart disease [103]. Another study in patients with familial hypercholesterolemia confirmed these findings. Higher CETP levels were correlated with decreased HDL, increased LDL, enhanced triglyceride levels and increased progression of atherosclerosis [104]. In addition, high CETP levels were also associated with reduced HDL and LDL particle size. Although statin treatment improved the lipoprotein profile in patients with familial hypercholesterolemia, the effects were less pronounced in patients with high CETP levels. Altogether, the existing evidence supports the hypothesis that pharmacological CETP inhibition may reduce the risk for coronary arterial disease in humans with high triglyceride levels.
CETP inhibitors
Despite the availability of LDL-lowering statins, there remains considerable unmet medical need for novel treatments of atherosclerosis and preventative strategies prompting efforts to develop novel treatment options to fight atherosclerosis. The existing evidence regarding potential atheroprotecive activity of HDL and potential benefits of CETP inhibition have led to numerous efforts to evaluate CETP-inhibiting agents and principles. Several strategies to target CETP inhibition including vaccination, antisense deoxynucleotides and the development of small molecule inhibitors of CETP have been described. CETP vaccines are still under development, although early reports suggested that antibody titers were too low to achieve sufficient CETP inhibition [105]. More recently, the immunogenicity of CETP vaccines could be increased by co-administration of a CpG adjuvant in preclinical models [106]. Administration of antisense deoxynucleotides was studied in small animal models and led to increased HDL levels and reduced atherosclerosis compared to experimental controls [30]. Several small molecule inhibitors of CETP have been developed [17–19, 21, 37, 38]. Results from preclinical and clinical studies conducted with these inhibitors help to understand the complexity of CETP inhibition on the one hand but, on the other hand, lead to important new questions regarding the viability of CETP inhibition in general and in specific populations.
Torcetrapib, a compound that binds to CETP inducing a non-productive complex with HDL [107], has been demonstrated to decrease LDL and increase HDL levels in early clinical studies [108–110].
Monotherapy with 60 mg torcetrapib per day over a period of 8 weeks resulted in a mean +45% increase of HDL, a mean decrease of −8% of LDL and a decrease of −16% of triglycerides versus baseline [109]. Against the background of 20 mg/day atorvastatin therapy, the mean changes versus baseline were +33% for HDL, −16% for LDL and +5% for triglycerides [110]. These impressive results were obtained in double-blinded and placebo-controlled but relatively small studies (n = 162 for monotherapy and n = 174 in the atorvastatin combination at baseline).
HDL increase and LDL decrease have also been observed in large phase III studies [33–36]: the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) study [33], the Investigation of Lipid Level Management Using Coronary Ultrasound (ILLUSTRATE) study [34] and the Rating Atherosclerotic Disease Change by Imaging with New CETP Inhibitor (RADIANCE) studies in patients with familial hypoalphalipoproteinemia (RADIANCE 1) [35] and in patients with mixed hyperlipidemia (RADIANCE 2) [36]. In the ILLUMINATE study, patients treated with torcetrapib for 1 year showed an increase of 72% in HDL and a decrease of 25% in LDL as compared with baseline. However, an increase of 5.4 mmHg in systolic blood pressure, a significant decrease in serum potassium and increases in serum sodium, bicarbonate and aldosterone have also been observed in patients treated with torcetrapib. In addition, there was also a significantly increased risk of cardiovascular events and death in those patients. Post hoc analyses showed an increased risk of death in patients treated with torcetrapib whose reduction in potassium or increase in bicarbonate was greater than the median change [33].
Disappointing results were also obtained in the ILLUSTRATE study, which did not demonstrate a significant decrease in the progression of coronary atherosclerosis in the torcetrapib-atorvastatin group as compared with atorvastatin monotherapy despite a substantial increase in HDL and decrease in LDL [34].
In patients with familial hypercholesterolemia, the use of torcetrapib with atorvastatin, as compared with atorvastatin alone, did not result in further reduction of progression of atherosclerosis, as assessed by a combined measure of carotid arterial wall thickness. In contrast, it was associated with progression of disease in the common carotid segment. Also in this study, these effects occurred despite a large increase in HDL cholesterol levels and a substantial decrease in levels of LDL cholesterol and triglycerides [35].
In vitro, torcetrapib induces aldosterone and cortisol production by an intracellular calcium-mediated mechanism independently of CETP inhibition [111]. The association of torcetrapib treatment with substantial changes in aldosterone levels, changes in electrolytes and hypertension led to discussions of whether off-target effects of torcetrapib have led or at least contributed to the negative results in these large clinical studies [17–26, 112]. However, the reasons for the torcetrapib failure remain elusive, and it cannot be excluded that CETP inhibition may have caused or contributed to the negative results in the torcetrapib studies.
Various efforts to design novel inhibitors of CETP are ongoing [37, 38]. The synthesis of novel tetrahydrochinoline-derived CETP inhibitors has been described [113]. BAY 60-5521 was tested in an early clinical study in humans and proved to be clinically safe and well tolerated in this first human study demonstrating a clear pharmacodynamic effect on CETP inhibition and HDL [114].
The two most advanced CETP inhibitors undergoing late stage clinical trials are dalcetrapib (JTT-705) [115–118] and anacetrapib (MK-0859) [119, 120]. Dalcetrapib increased HDL and demonstrated a favorable safety profile in a phase II study, and no changes in vital signs including blood pressure have been observed [106, 107]. A phase III trial to evaluate the effects of 600 mg dalcetrapib compared with a placebo on mortality and morbidity in approximately 15,600 high-risk patients considered to have stable CHD after recent cardiovascular events is currently in progress (The dal-HEART Program: Dalcetrapip HDL Evaluation, Atherosclerosis and Reverse Cholesterol Transport; ClinicalTrials.gov identifier NCT00658515). The binding mode of dalcetrapib to CETP has been published and is different compared to that of torcetrapib [121–123]. Anacetrapib is an orally active, potent and selective CETP inhibitor. Single and multiple dose studies in healthy volunteers were well tolerated and resulted in favorable changes in the lipid profile without changes in blood pressure [119]. In addition, co-administration with atorvastatin did lead to increased HDL and decreased LDL levels in patients with primary hypercholesterolemia or mixed hyperlipidemia without discernable effects on blood pressure, serum electrolytes or aldosterone levels [120]. Information regarding the structure of the CETP inhibitors described in this article is summarized in Table 1.
Table 1.
CETP inhibitors and their mode of CETP inhibition
| Structure | Compound | Clinical phase | Mode of CETP inhibition |
|---|---|---|---|
|
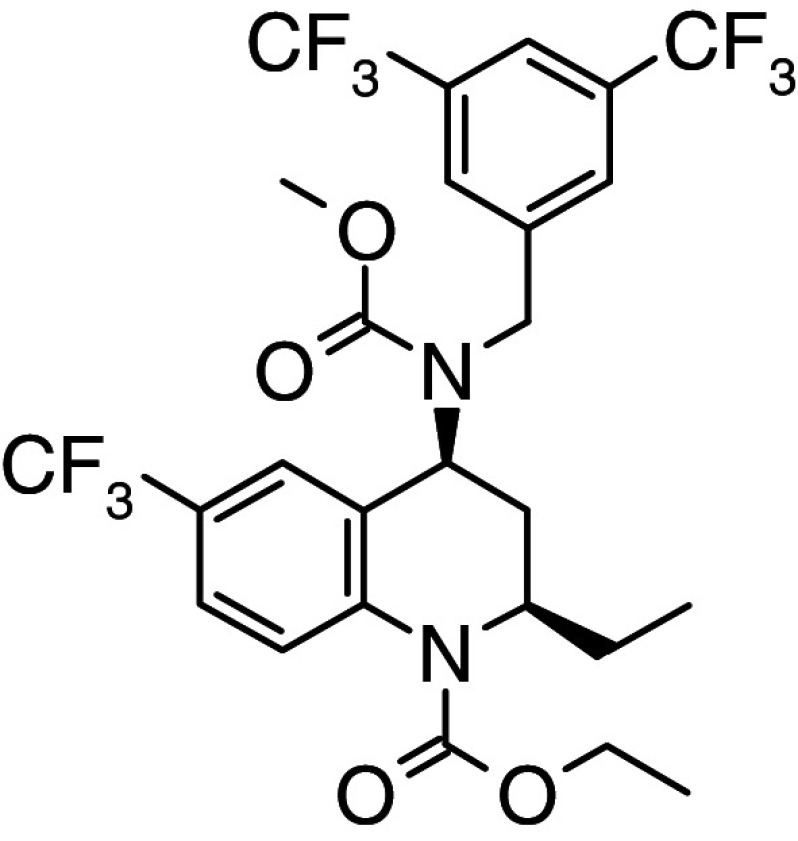
Torcetrapib | Phase III discontinued in 2006 | Reversible binding to CETP resulting in a high affinity nonproductive complex of torcetrapib, HDL and CETP [121, 123] |
|
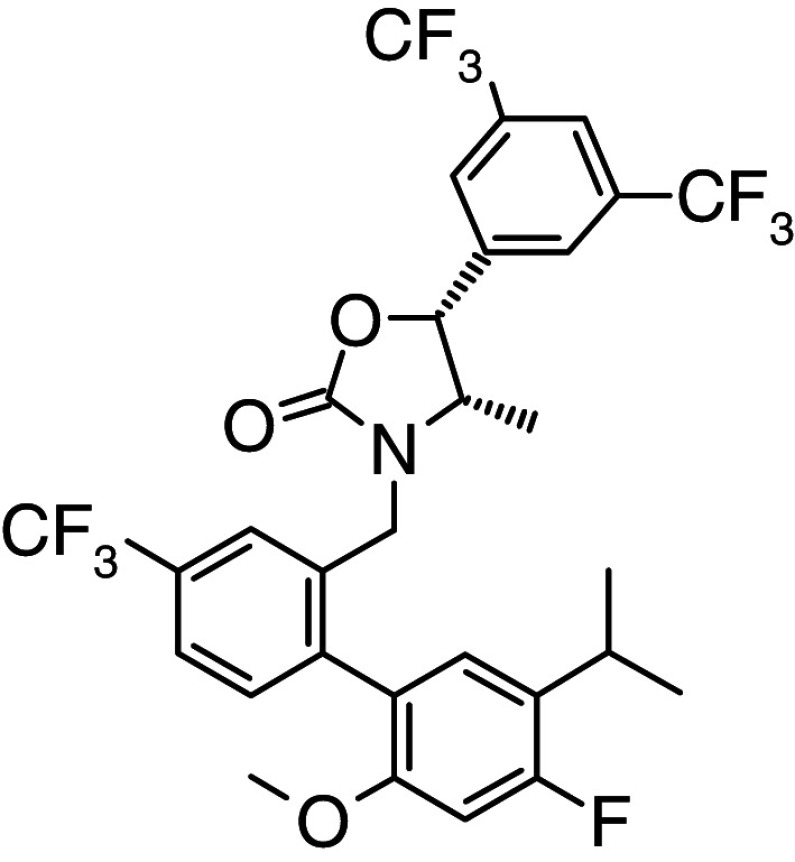
Anacetrapib | Phase III | Binding mode not published |
|
Dalcetrapib | Phase III | Irreversible binding to CETP by disulfide formation of Cys-13 with free SH in active form of dalcetrapib |
| HDL generated is of normal composition [121, 122] | |||
|
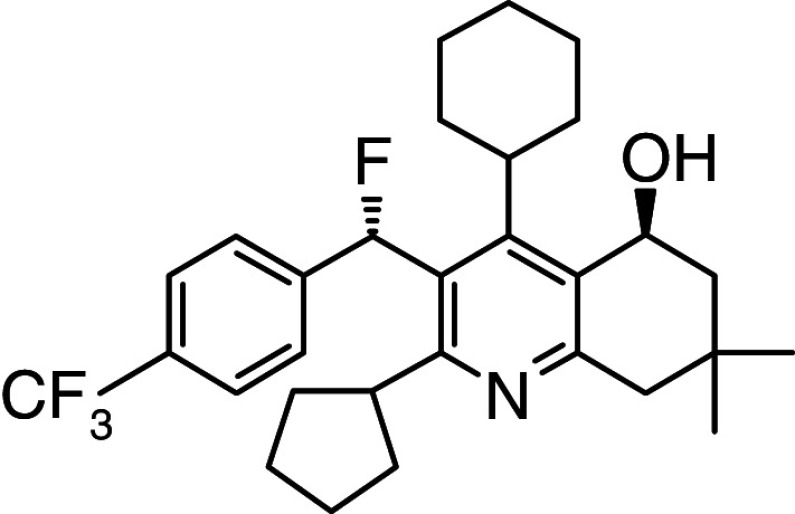
BAY 60-5521 | Phase I | Binding mode not published |

Because of the outcome of the large torcetrapib studies where reasons for the failure remain elusive, smaller studies with these different CETP inhibitors may help to aid future clinical development. A total of 1,623 patients with CHD or CHD equivalents were randomized into a 76-week randomized, double-blind, placebo-controlled phase III study (DEFINE) to assess the tolerability and efficacy of anacetrapib when added to ongoing lipid therapies in patients. The goal of this study is to obtain safety experience with anacetrapib to inform decisions regarding initiation of phase III outcome trials [124].
Overall, the clinical results with anacetrapib and dalcetrapib are promising so far. However, clinical outcome data are still missing with these novel CETP inhibitors.
Only studies with CETP-inhibiting compounds lacking the torcetrapib side effect profile may provide answers to the open question of whether CETP inhibition is indeed a feasible therapeutic approach to minimize CHD risk or treating arteriosclerosis.
References
- 1.Yusuf S, Reddy S, Ounpuu S, Anand S. Global burden of cardiovascular diseases: part I: general considerations, the epidemiologic transition, risk factors, and impact of urbanization. Circulation. 2001;104:2746–2753. doi: 10.1161/hc4601.099487. [DOI] [PubMed] [Google Scholar]
- 2.Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults 1. (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- 3.Grundy SM, Cleeman JI, Merz CN, Brewer HB, Jr, Clark LT, Hunninghake DB, Pasternak RC, Smith SC, Jr, Stone NJ, Coordinating Committee of the National Cholesterol Education Program Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 4.LaRosa JC, He J, Vupputuri S. Effect of statins on risk of coronary disease: a meta-analysis of randomized controlled trials. J Am Med Assoc. 1999;282:2340–2346. doi: 10.1001/jama.282.24.2340. [DOI] [PubMed] [Google Scholar]
- 5.Zilversmit DB, Hughes LB, Balmer J. Stimulation of cholesterol ester exchange by lipoprotein-free rabbit plasma. Biochim Biophys Acta. 1975;409:393–398. doi: 10.1016/0005-2760(75)90036-3. [DOI] [PubMed] [Google Scholar]
- 6.Chajek T, Fielding CJ. Isolation and characterization of a human serum cholesteryl ester transfer protein. Proc Natl Acad Sci USA. 1978;75:3445–3449. doi: 10.1073/pnas.75.7.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deckelbaum RJ, Eisenberg S, Oschry Y, Butbul E, Sharon I, Olivecrona T. Reversible modification of human plasma low density lipoproteins toward triglyceride-rich precursors. A mechanism for losing excess cholesterol esters. J Biol Chem. 1982;257:6509–6517. [PubMed] [Google Scholar]
- 8.Swenson TL, Brocia RW, Tall AR. Plasma cholesteryl ester transfer protein has binding sites for neutral lipids and phospholipids. J Biol Chem. 1988;263:5150–5157. [PubMed] [Google Scholar]
- 9.Glomset JA. The plasma lecithins:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 10.Miller NE, Thelle DS, Forde OH, Mjos OD. The Tromsø heart-study. High-density lipoprotein and coronary heart disease: a prospective case-control study. Lancet. 1977;1(8019):965–968. doi: 10.1016/S0140-6736(77)92274-7. [DOI] [PubMed] [Google Scholar]
- 11.Gordon DJ, Rifkind BM. High-density lipoprotein: the clinical implication of recent studies. N Engl J Med. 1989;321:1311–1316. doi: 10.1056/NEJM198911093211907. [DOI] [PubMed] [Google Scholar]
- 12.Borggreve SE, Hillege HL, Wolffenbuttel BH, de Jong PE, Zuurman MW, van der Steege G, van Tol A, Dullaart RP, PREVEND Study Group An increased coronary risk is paradoxically associated with common cholesteryl ester transfer protein gene variations that relate to higher high-density lipoprotein cholesterol: a population-based study. J Clin Endocrinol Metab. 2006;91:3382–3388. doi: 10.1210/jc.2005-2322. [DOI] [PubMed] [Google Scholar]
- 13.Linsel-Nitschke P, Tall AR. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov. 2005;4:193–205. doi: 10.1038/nrd1658. [DOI] [PubMed] [Google Scholar]
- 14.Sikorski JA. Oral cholesteryl ester transfer protein (CETP) inhibitors: a potential new approach for treating coronary artery disease. J Med Chem. 2006;49(1):1–22. doi: 10.1021/jm058224l. [DOI] [PubMed] [Google Scholar]
- 15.Parini P, Rudel LL. Is there a need for cholesteryl ester transfer protein inhibition? Arterioscler Thromb Vasc Biol. 2003;23:374–375. doi: 10.1161/01.ATV.0000060447.25136.1C. [DOI] [PubMed] [Google Scholar]
- 16.Watts GF. The Yin and Yang of cholesteryl ester transfer protein and atherosclerosis. Clin Sci. 2002;103:595–597. doi: 10.1042/cs1030595. [DOI] [PubMed] [Google Scholar]
- 17.Joy T, Hegele RA. The end of the road for CETP inhibitors after torcetrapib? Curr Opin Cardiol. 2009;24:364–371. doi: 10.1097/HCO.0b013e32832ac166. [DOI] [PubMed] [Google Scholar]
- 18.Neeli H, Rader DJ. Cholesteryl ester transfer protein (CETP) inhibitors: is there life after torcetrapib? Cardiol Clin. 2008;26(4):537–546. doi: 10.1016/j.ccl.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Rennings AJM, Stalenhoef AFH. JTT-705: is there still future for a CETP inhibitor after torcetrapib? Expert Opin Investig Drugs. 2008;17(10):1589–1597. doi: 10.1517/13543784.17.10.1589. [DOI] [PubMed] [Google Scholar]
- 20.Brown BG, Zhao XQ. Nicotinic acid, alone or in combinations, for reduction of cardiovascular risk. Am J Cardiol. 2008;101:58B–62B. doi: 10.1016/j.amjcard.2008.02.039. [DOI] [PubMed] [Google Scholar]
- 21.Hunt JA, Lu Z. Cholesteryl ester transfer protein (CETP) inhibitors. Curr Topics Med Chem. 2009;9:419–427. doi: 10.2174/156802609788340823. [DOI] [PubMed] [Google Scholar]
- 22.Tall AR, Yvan-Charvet L, Wang N. The failure of torcetrapib: was it the molecule or the mechanism? Arterioscler Thromb Vasc Biol. 2007;27:257–260. doi: 10.1161/01.ATV.0000256728.60226.77. [DOI] [PubMed] [Google Scholar]
- 23.Tall AR. CETP inhibitors to increase HDL cholesterol levels. N Engl J Med. 2007;356:1364–1366. doi: 10.1056/NEJMe078029. [DOI] [PubMed] [Google Scholar]
- 24.Gotto AM. Does torcetrapib reduce the progression of atherosclerotic disease? Nat Clin Pract Cardiovasc Med. 2007;4:478–479. doi: 10.1038/ncpcardio0946. [DOI] [PubMed] [Google Scholar]
- 25.Toth PP. Reducing cardiovascular risk by targeting high-density lipoprotein cholesterol. Curr Atheroscler Rep. 2007;9(1):81–88. doi: 10.1007/BF02693933. [DOI] [PubMed] [Google Scholar]
- 26.Howes LG, Kostner K. The withdrawal of torcetrapib from drug development: implications for the future of drugs that alter HDL metabolism. Expert Opin Investig Drugs. 2007;16:1509–1516. doi: 10.1517/13543784.16.10.1509. [DOI] [PubMed] [Google Scholar]
- 27.Potter LK, Sprecher DL, Walker MC, Tobin FL. Mechanism of inhibition defines CETP activity: a mathematical model for CETP in vitro. J Lipid Res. 2009;50(11):2222–2234. doi: 10.1194/jlr.M900015-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barter PJ, Brewer HB, Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:160–167. doi: 10.1161/01.ATV.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 29.Marotti KR, Castle CK, Boyle TP, Lin AH, Murray RW, Melchior GW. Severe atherosclerosis in transgenic mice expressing simian cholesteryl ester transfer protein. Nature. 1993;364:73–75. doi: 10.1038/364073a0. [DOI] [PubMed] [Google Scholar]
- 30.Sugano M, Makino N, Sawada S, Otsuka S, Watanabe M, Okamoto H, Kamada M, Mizushima A. Effect of antisense oligonucleotides against cholesteryl ester transfer protein on the development of atherosclerosis in cholesterol-fed rabbits. J Biol Chem. 1998;273:5033–5036. doi: 10.1074/jbc.273.9.5033. [DOI] [PubMed] [Google Scholar]
- 31.Okamoto H, Yonemori F, Waktiani K, Minowa T, Maeda K, Shinkai H. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature. 2000;406:203–207. doi: 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- 32.Huang Z, Inazu A, Nohara A, Hagashita T, Mabuchi H. Cholesteryl ester transfer protein inhibitor (JTT-705) and the development of atherosclerosis in rabbits with severe hypercholesterolaemia. Clin Sci. 2002;103:587–594. doi: 10.1042/cs1030587. [DOI] [PubMed] [Google Scholar]
- 33.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJP, Komajda M, Lopez-Sendon J, Mosca L, Tardif J, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 34.Nissen SE, Tardif JC, Nicholls SJ, Revkin JH, Shear CL, Duggan WT, Ruzyllo W, Bachinsky WB, Lasala GP. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356:1304–1316. doi: 10.1056/NEJMoa070635. [DOI] [PubMed] [Google Scholar]
- 35.Kastelein JJP, Leuven SI, Burgess L, Evans GW, Kuivenhoven JA, Barter PJ, Revkin JH, Grobbee DE, Riley WA, Shear CL, Duggan WT, Bots ML. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med. 2007;356:1620–1630. doi: 10.1056/NEJMoa071359. [DOI] [PubMed] [Google Scholar]
- 36.Bots ML, Visseren FL, Evans GW, Riley WA, Revkin JH, Tegeler CH, Shear CL, Duggan WT, Vicari RM, Grobbee DE. Torcetrapib and carotid intima-media thickness in mixed dyslipidaemia (RADIANCE 2 study): a randomised, double-blind trial. Lancet. 2007;370:153–160. doi: 10.1016/S0140-6736(07)61088-5. [DOI] [PubMed] [Google Scholar]
- 37.Rano TA, Sieber-McMaster E, Pelton PD, Yang M, Demarest KT, Kuo GH. Design and synthesis of potent inhibitors of cholesteryl ester transfer protein (CETP) exploiting a 1,2,3,4-tetrahydroquinoline platform. Bioorg Med Chem Lett. 2009;19:2456–2460. doi: 10.1016/j.bmcl.2009.03.051. [DOI] [PubMed] [Google Scholar]
- 38.Kuo GH, Rano T, Pelton P, Demarest KT, Gibbs AC, Murray WV, Damiano BP, Connelly MA. Design, synthesis, and biological evaluation of (2R, alphaS)-3,4-dihydro-2-[3-(1,1,2,2-tetrafluoroethoxy)phenyl]-5-[3-(trifluoromethoxy)-phenyl]-alpha-(trifluoromethyl)-1(2H)-quinolineethanol as potent and orally active cholesteryl ester transfer protein inhibitor. J Med Chem. 2009;52:1768–1772. doi: 10.1021/jm801319d. [DOI] [PubMed] [Google Scholar]
- 39.Bruce C, Beamer LJ, Tall AR. The implications of the structure of the bactericidal/permeability increasing protein on the lipid-transfer function of the cholesteryl ester transfer protein. Curr Opin Struct Biol. 1998;8:426–434. doi: 10.1016/S0959-440X(98)80118-8. [DOI] [PubMed] [Google Scholar]
- 40.Kawano K, Qin SC, Lin M, Tall AR, Jiang XC. Cholesteryl ester transfer protein and phospholipid transfer protein have nonoverlapping functions in vivo. J Biol Chem. 2000;275:29477–29481. doi: 10.1074/jbc.M003523200. [DOI] [PubMed] [Google Scholar]
- 41.Tall AR. Plasma cholesteryl ester transfer protein. J Lipid Res. 1993;34:1255–1274. [PubMed] [Google Scholar]
- 42.Drayna D, Jarnagin AS, McLean J, Henzel W, Koehr W, Fielding C, Lawn R. Cloning and sequencing of human cholestryl ester transfer protein cDNA. Nature. 1987;327(61234):632–634. doi: 10.1038/327632a0. [DOI] [PubMed] [Google Scholar]
- 43.Agellon LB, Quinet EM, Gillette TG, Drayna DT, Brown ML, Tall AR. Organization of the human cholesteryl ester transfer protein gene. Biochemistry. 1990;29(6):1372–1376. doi: 10.1021/bi00458a004. [DOI] [PubMed] [Google Scholar]
- 44.Dullaart RPF, Sluiter WJ. Common variation in the CETP gene and the implications for cardiovascular disease and its treatment: an updated analysis. Pharmacogenomics. 2008;9(6):747–763. doi: 10.2217/14622416.9.6.747. [DOI] [PubMed] [Google Scholar]
- 45.Qiu X, Mistry A, Ammirati MJ, Chrunyk BA, Clark RW, Cong Y, Culp JS, Danley DE, Freeman TB, Geoghegan KF, Griffor MC, Hawrylik SJ, Hayward CM, Hensley P, Hoth LR, Karam GA, Lira ME, Lloyd DB, McGrath KM, Stutzman-Engwall KJ, Subashi AK, Subashi TA, Thompson JF, Wang IK, Zhao H, Seddon AP. Crystal structure of cholesteryl ester transfer protein reveals a long tunnel and four bound lipid molecules. Nat Struct Mol Biol. 2007;14:106–113. doi: 10.1038/nsmb1197. [DOI] [PubMed] [Google Scholar]
- 46.Jiang XC, Bruce C, Cocke T, Wang S, Boguski M, Tall AR. Point mutagenesis of positively charged amino acids of cholesteryl ester transfer protein: conserved residues within the lipid transfer/lipopolysaccharide binding protein gene family essential for function. Biochemistry. 1995;34(21):7258–7263. doi: 10.1021/bi00021a042. [DOI] [PubMed] [Google Scholar]
- 47.Pape ME, Ulrich RG, Rea TJ, Marotti KR, Melchior GW. Evidence that the nonparenchymal cells of the liver are the principal source of cholesteryl ester transfer protein in primates. J Biol Chem. 1991;266(20):12829–12831. [PubMed] [Google Scholar]
- 48.Martin LJ, Connelly PW, Nancoo D, Wood N, Zhang ZJ, Maguire G, Quinet E, Tall AR, Marcel YL, McPherson R. Cholesteryl ester transfer protein and high density lipoprotein responses to cholesterol feeding in man: relationship to apoprotein E genotype. J Lipid Res. 1993;34:437–446. [PubMed] [Google Scholar]
- 49.Radeau T, Lau P, Robb M, McDonnell M, Ailhaud G, McPherson R. Cholesteryl ester transfer protein (CETP) mRNA abundance in human adipose tissue: relationship to cell size and membrane cholesterol content. J Lipid Res. 1995;36:2552–2561. [PubMed] [Google Scholar]
- 50.Tall AR. Plasma cholesteryl ester transfer protein. J Lipid Res. 1993;34:1255–1274. [PubMed] [Google Scholar]
- 51.Barter PJ, Brewer HB, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:160–167. doi: 10.1161/01.ATV.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 52.Benoist F, Lau P, McDonnell M, Doelle H, Milne R, McPherson R. Cholesteryl ester transfer protein mediates selective uptake of high density lipoprotein cholesteryl esters by human adipose tissue. J Biol Chem. 1997;272(38):23572–23577. doi: 10.1074/jbc.272.38.23572. [DOI] [PubMed] [Google Scholar]
- 53.Guérin M, Dolphin PJ, Chapman MJ. Preferential cholesteryl ester acceptors among the LDL subspecies of subjects with familial hypercholesterolemia. Arterioscler Thromb. 1994;14:679–685. doi: 10.1161/01.atv.14.5.679. [DOI] [PubMed] [Google Scholar]
- 54.Bruce C, Davidson WS, Kussie P, Lund-Katz S, Phillips MC, Ghosh R, Tall AR. Molecular determinants of plasma cholesteryl ester transfer protein binding to high density lipoprotein. J Biol Chem. 1995;270(19):11532–11542. doi: 10.1074/jbc.270.19.11532. [DOI] [PubMed] [Google Scholar]
- 55.Deckelbaum RJ, Granot E, Oschry Y, Rose L, Eisenberg S. Plasma triglyceride determines structure-composition in low and high density lipoproteins. Arteriosclerosis. 1984;4:225–231. doi: 10.1161/01.atv.4.3.225. [DOI] [PubMed] [Google Scholar]
- 56.Hamilton JA, Deckelbaum RJ. Crystal structure of CETP: new hopes for raising HDL to decrease risk of cardiovascular disease? Nat Struct Mol Biol. 2007;14:95–97. doi: 10.1038/nsmb0207-95. [DOI] [PubMed] [Google Scholar]
- 57.Kontush A, Capman MJ. Functionally defective HDL: a new therapeutic target at the crossroads of dyslipidemia, inflammation and atherosclerosis. Pharmacol Rev. 2006;3:342–374. doi: 10.1124/pr.58.3.1. [DOI] [PubMed] [Google Scholar]
- 58.Kontush A, Guerin M, Chapman MJ. Spotlight on HDL-raising therapies: insights from the torcetrapib trials. Nat Clin Pract Cardiovasc Med. 2008;5(6):329–336. doi: 10.1038/ncpcardio1191. [DOI] [PubMed] [Google Scholar]
- 59.Glomset JA. The plasma lecithin:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 60.Ross R, Glomset JA. Atherosclerosis and the arterial smooth muscle cell: proliferation of smooth muscle is a key event in the genesis of the lesions of atherosclerosis. Science. 1973;180:1332–1339. doi: 10.1126/science.180.4093.1332. [DOI] [PubMed] [Google Scholar]
- 61.Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990;85:1234–1241. doi: 10.1172/JCI114558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moreno PR, Sanz J, Fuster V. Promoting mechanisms of vascular health. J Am Coll Cardiol. 2009;53(25):2315–2325. doi: 10.1016/j.jacc.2009.02.057. [DOI] [PubMed] [Google Scholar]
- 63.Yancey PG, Bortnick AE, Kellner-Weibel G, de-la Llera-Moya M, Phillips MC, Rothblat GH. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:712–719. doi: 10.1161/01.ATV.0000057572.97137.DD. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi Y, Smith JD. Cholesterol efflux to apolipoprotein A1 involves endocytosis and re-secretion in a calcium-dependent pathway. Proc Natl Acad Sci USA. 1999;96:11358–11363. doi: 10.1073/pnas.96.20.11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams DL, Connelly MA, Temel RE, Swarnakar S, Phillips MC, de la Llera-Moya M, Rothblat GH. Scavenger receptor BI and cholesterol trafficking. Curr Opin Lipidol. 1999;10:329–339. doi: 10.1097/00041433-199908000-00007. [DOI] [PubMed] [Google Scholar]
- 66.Brewer HB., Jr High-density lipoproteins: a new potential therapeutic target for the prevention of cardiovascular disease. Arterioscler Thromb Vasc Biol. 2004;24(3):387–391. doi: 10.1161/01.ATV.0000121505.88326.d2. [DOI] [PubMed] [Google Scholar]
- 67.Schwartz CC, Vlahcevic ZR, Berman M, Meadows JG, Nisman RM, Swell L. Central role of high density lipoprotein in plasma free cholesterol metabolism. J Clin Invest. 1982;70(1):105–116. doi: 10.1172/JCI110582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwartz CC, Vlahcevic ZR, Halloran LG, Swell L. An in vivo evaluation in man of the transfer of esterified cholesterol between lipoproteins and into the liver and bile. Biochim Biophys Acta. 1981;663(1):143–162. doi: 10.1016/0005-2760(81)90201-0. [DOI] [PubMed] [Google Scholar]
- 69.Schwartz CC, Berman M, Vlahcevic ZR, Halloran LG, Gregory DH, Swell L. Multicompartmental analysis of cholesterol metabolism in man. Characterization of the hepatic bile acid and biliary cholesterol precursor sites. J Clin Invest. 1978;61(2):408–423. doi: 10.1172/JCI108952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 71.Petoumenos V, Nickenig G, Werner N. High density lipoprotein exerts vasculoprotection via endothelial progenitor cells. J Cell Mol Med. 2008;13(11):4623–4635. doi: 10.1111/j.1582-4934.2008.00472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barter PJ, Baker PW, Rye KA. Effect of high-density lipoproteins on the expression of adhesion molecules in endothelial cells. Curr Opin Lipidol. 2002;13:285–288. doi: 10.1097/00041433-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 73.Kontush A, Chapman MJ. Antiatherogenic small, dense HDL—guardian angel of the arterial wall? Nat Clin Pract Cardiovasc Med. 2006;3:342–374. doi: 10.1038/ncpcardio0500. [DOI] [PubMed] [Google Scholar]
- 74.Atzmon G, Rincon M, Rabizadeh P, Barzilai N. Biological evidence for inheritance of exceptional longevity. Mech Ageing Dev. 2005;126:341–345. doi: 10.1016/j.mad.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 75.Barzilai N, Atzmon G, Schechter C, Schaefer EJ, Cupples AL, Lipton R, Cheng S, Shuldiner AR. Unique lipoprotein phenotype and genotype associated with exceptional longevity. J Am Med Assoc. 2003;290:2030–2040. doi: 10.1001/jama.290.15.2030. [DOI] [PubMed] [Google Scholar]
- 76.Curb JD, Abbott RD, Rodriguez BL, Masaki K, Chen R, Sharp DS, Tall AR. A prospective study of HDL-C and cholesteryl ester transfer protein gene mutations and the risk of coronary heart disease in the elderly. J Lipid Res. 2004;45:948–953. doi: 10.1194/jlr.M300520-JLR200. [DOI] [PubMed] [Google Scholar]
- 77.Franceschini G, Sirtori M, Gianfranceschi G, Sirtori CR. Relation between the HDL apoproteins and AI isoproteins in subjects with the AIMilano abnormality. Metab Clin Exp. 1981;30(5):502–509. doi: 10.1016/0026-0495(81)90188-8. [DOI] [PubMed] [Google Scholar]
- 78.Franceschini G, Calabresi L. Normal vascular function despite low levelsof high-density lipoprotein cholesterol in carriers of the apolipoprotein A-I-Milano mutant. Circulation. 2007;116:2165–2172. doi: 10.1161/CIRCULATIONAHA.107.705657. [DOI] [PubMed] [Google Scholar]
- 79.Franceschini G, Calabresi L, Chiesa G, Parolini C, Sirtori CR, Canavesi M, Bernini F. Increased cholesterol efflux potential of sera from apoAI Milano carriers and transgenic mice. Arterioscler Thromb Vasc Biol. 1999;19:1257–1262. doi: 10.1161/01.atv.19.5.1257. [DOI] [PubMed] [Google Scholar]
- 80.Calabresi L, Canavesi M, Bernini F, Franceschini G. Cell cholesterol efflux to reconstituted high-density lipoproteins containing the apolipoprotein A-IMilano dimer. Biochemistry. 1999;38:16307–16314. doi: 10.1021/bi991246n. [DOI] [PubMed] [Google Scholar]
- 81.Bielicki JK, Oda MN. Apolipoprotein A-IMilano and apolipoprotein A-IParis exhibit an antioxidant activity distinct from that of wild-type apolipoprotein A-I. Biochemistry. 2002;41:2089–2096. doi: 10.1021/bi011716p. [DOI] [PubMed] [Google Scholar]
- 82.Wang L, Sharifi BG, Pan T, Song L, Yukht A, Shah PK. Bone marrow transplantation shows superior atheroprotective effects of gene therapy with apolipoprotein A-I Milano compared with wild-type apolipoprotein A-I in hyperlipidemic mice. J Am Coll Cardiol. 2006;48:1459–1468. doi: 10.1016/j.jacc.2006.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parolini C, Chiesa G, Gong E, Caligari S, Cortese MM, Koga T, Forte TM, Rubin EM. Apolipoprotein A-I and the molecular variant apoAIMilano: evaluation of the antiatherogenic effects in knock-in mouse model. Atherosclerosis. 2005;183:222–229. doi: 10.1016/j.atherosclerosis.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 84.Lebherz C, Sanmiguel J, Wilson J, Rader D. Gene transfer of wild-type apoA-I and apoA-I Milano reduce atherosclerosis to a similar extent. Cardiovasc Diabetol. 2007;6:15. doi: 10.1186/1475-2840-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant apoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 86.Nicholls SJ, Tuzcu EM, Crowe T, Sipahi I, Schoenhagen P, Kapadia S, Hazen SL, Wun CC, Norton M, Ntanios F, Nissen SE. Relationship between cardiovascular risk factors and atherosclerotic disease burden measured by intravascular ultrasound. J Am Coll Cardiol. 2006;47:1967–1975. doi: 10.1016/j.jacc.2005.12.058. [DOI] [PubMed] [Google Scholar]
- 87.Alexander ET, Weibel GL, Joshi MR, Vedhachalam C, de la Llera-Moya M, Rothblat GH, Phillips MC, Rader DJ. Macrophage reverse cholesterol transport in mice expressing apoA-I Milano. Arterioscler Thromb Vasc Biol. 2009;29:1496–1501. doi: 10.1161/ATVBAHA.109.191379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Inazu A, Quinet EM, Wang S, Brown ML, Stevenson S, Barr ML, Moulin P, Tall AR. Alternative splicing of the mRNA encoding human cholesteryl ester transfer protein. Biochemistry. 1992;31:2352–2358. doi: 10.1021/bi00123a021. [DOI] [PubMed] [Google Scholar]
- 89.Quinet E, Yang TP, Marinos C, Tall A. Inhibition of the cellular secretion of cholesteryl ester transfer protein by a variant protein formed by alternative splicing of mRNA. J Biol Chem. 1993;268:16891–16894. [PubMed] [Google Scholar]
- 90.Yamashita S, Hirano KI, Sakai N, Matsuzawa Y. Molecular biology and pathophysiological aspects of plasma cholesteryl ester transfer protein. Biochim Biophys Acta. 2000;1529:257–275. doi: 10.1016/s1388-1981(00)00164-5. [DOI] [PubMed] [Google Scholar]
- 91.Hirano K, Yamashita S, Nakajima N, Arai T, Maruyama T, Yoshida Y, Ishigami M, Sakai N, Kameda-Takemura K, Matsuzawa Y. Genetic cholesteryl ester transfer protein deficiency is extremely frequent in the Omaguri area of Japan. Arterioscler Thromb Vasc Biol. 1997;17:1053–1059. doi: 10.1161/01.atv.17.6.1053. [DOI] [PubMed] [Google Scholar]
- 92.Nagano M, Yamashita S, Hirano K, Takano M, Maruyama T, Ishihara M, Sagehashi Y, Kujiraoka T, Tanaka K, Hattori H, Sakai N, Nakajima N, Egashira T, Matsuzawa Y. Molecular mechanisms of cholesteryl ester transfer protein deficiency in Japanese. J Atheroscler Thromb. 2004;11(3):110–121. doi: 10.5551/jat.11.110. [DOI] [PubMed] [Google Scholar]
- 93.Rhyne J, Ryan MJ, White C, Chimonas T, Miller M. The two novel CETP mutations Gln87X and Gln165X in a compound heterozygous state are associated with marked hyperalphalipoproteinemia and absence of significant coronary artery disease. J Mol Med. 2006;84:647–650. doi: 10.1007/s00109-006-0070-4. [DOI] [PubMed] [Google Scholar]
- 94.Zhong S, Sharp DS, Grove JS, Bruce C, Yano K, Tall AR. Increased coronary heart disease in Japanese American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J Clin Invest. 1996;97:2917–2923. doi: 10.1172/JCI118751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.MacLean PS, Vadlamudi S, Mac Donald KG, Pories WJ, Barakat HA. Suppression of hepatic cholesteryl ester transfer protein expression in obese humans with the development of type 2 diabetes mellitus. J Clin Endocrinol Metabol. 2005;90:2250–2258. doi: 10.1210/jc.2004-1325. [DOI] [PubMed] [Google Scholar]
- 96.Hovingh GK, de Groot E, van der Steeg W, Boekholdt SM, Hutten BA, Kuivenhoven JA, Kastelein JJ. Inherited disorders of HDL metabolism and atherosclerosis. Curr Opin Lipidol. 2005;16:139–145. doi: 10.1097/01.mol.0000162318.47172.ef. [DOI] [PubMed] [Google Scholar]
- 97.Boekholdt SM, Thomson JF. Natural genetic variation as a tool in understanding the role of CETP in lipid levels and disease. J Lipid Res. 2005;44:1080–1093. doi: 10.1194/jlr.R200018-JLR200. [DOI] [PubMed] [Google Scholar]
- 98.Thompson A, DiAngelantonio E, Sarwar N, Erqou S, Salcheen D, Dillaart RPF, Keavney B, Ye Z, Danesh J. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008;299(23):2777–2788. doi: 10.1001/jama.299.23.2777. [DOI] [PubMed] [Google Scholar]
- 99.Sirtori CR, Mombelli G. Viability of developing CETP inhibitors. Cardiovasc Ther. 2008;26:135–146. doi: 10.1111/j.1527-3466.2008.00049.x. [DOI] [PubMed] [Google Scholar]
- 100.Agellon LB, Walsh A, Ayek T, Moulin P, Jiang XC, Shelanski SA, Breslow JL, Tall AR. Reduced high density lipoprotein cholesterol in human cholesteryl ester transfer protein transgenic mice. J Biol Chem. 1991;266:10796–10801. [PubMed] [Google Scholar]
- 101.Jiang XC, Masucci-Magoulas L, Mar J, Lin M, Walsh A, Breslow JL, Tall A. Down-regulation of mRNA for the low density lipoprotein receptor in transgenic mice containing the gene for human cholesteryl ester transfer protein: mechanism to explain accumulation of lipoprotein B particles. J Biol Chem. 1993;268:27406–27412. [PubMed] [Google Scholar]
- 102.Haraki T, Inazu A, Yagi K, Kajinami K, Koizumi J, Mabuchi H. Clinical characteristics of double heterozygotes with familial hypercholesterolemia and cholesteryl ester transfer protein deficiency. Atherosclerosis. 1997;132:229–236. doi: 10.1016/S0021-9150(97)00093-2. [DOI] [PubMed] [Google Scholar]
- 103.Boekholdt SM, Kuivenhoven JA, Wareham NJ, Peters RJG, Jukema JW, Luben R, Bingham SA, Day NE, Kastelein JJP, Khaw KT. Plasma levels of cholesteryl ester transfer protein and the risk of future coronary artery disease in apparently healthy men and women: the prospective EPIC (European Prospective Investigation into Cancer and nutrition)-Norfolk Population Study. Circulation. 2004;110:1418–1423. doi: 10.1161/01.CIR.0000141730.65972.95. [DOI] [PubMed] [Google Scholar]
- 104.De Grooth GJ, Smilde TJ, van Wissen S, Klerkx AHEM, Zwinderman AH, Fruchart JC, Kastelein JJP, Stalenhoef AFH, Kuivenhoven JA. The relationship between cholesteryl ester transfer protein levels and risk factor profile in patients with familial hypercholesterolemia. Atherosclerosis. 2004;173:261–267. doi: 10.1016/j.atherosclerosis.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 105.Davidson MH, Maki K, Umporowicz D, Wheeler A, Rittershaus C, Ryan U. The safety and immunogenicity of a CETP vaccine in healthy adults. Atherosclerosis. 2003;169:113–120. doi: 10.1016/S0021-9150(03)00137-0. [DOI] [PubMed] [Google Scholar]
- 106.Thomas LJ, Hammond RA, Forsberg EM, Geoghegan-Barek KM, Karalius BH, Marsh HC, Jr, Ritterhaus CW. Co-administration of a CpG adjuvant (VaxImmune, CPG 7909) with CETP vaccines increased immunogenicity in rabbits and mice. Hum Vaccin. 2009;5(2):79–84. doi: 10.4161/hv.5.2.6521. [DOI] [PubMed] [Google Scholar]
- 107.Clark RW, Ruggeri RB, Cunningham D, Bamberger MJ. Description of the torcetrapib series of cholesteryl ester transfer protein inhibitors, including mechanism of action. J Lipid Res. 2006;47:537–552. doi: 10.1194/jlr.M500349-JLR200. [DOI] [PubMed] [Google Scholar]
- 108.Clark RW, Sutfin TA, Ruggeri RB, Willauer AT, Sugarman ED, Magnus-Aryitey G, Cosgrove PG, Sand TM, Wester RT, Williams JA, Perlman ME, Bamberger MJ. Raising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein: an initial multidose study of torcetrapib. Arterioscler Thromb Vasc Biol. 2004;24:490–497. doi: 10.1161/01.ATV.0000118278.21719.17. [DOI] [PubMed] [Google Scholar]
- 109.Davidson MH, McKenney JM, Shear CL, Revkin JH. Efficacy and safety of torcetrapib, a novel cholesteryl ester transfer protein inhibitor, in individuals with below-average high-density lipoprotein cholesterol levels. J Am Coll Cardiol. 2006;48:1774–1781. doi: 10.1016/j.jacc.2006.06.067. [DOI] [PubMed] [Google Scholar]
- 110.Mc Kenney JM, Davidson MH, Shear CL, Revkin JH. Efficacy and safety of torcetrapib, a novel cholesteryl ester transfer protein inhibitor, in individuals with below-average high-density lipoprotein cholesterol levels on a background of atorvastatin. J Am Coll Cardiol. 2006;48:1782–1790. doi: 10.1016/j.jacc.2006.06.066. [DOI] [PubMed] [Google Scholar]
- 111.Hu X, Dietz JD, Xia C, Loging Knight DR, WT Smith AH, Yuan H, Perry DA, Keiser J. Torcetrapib induces aldosterone and cortisol production by an intracellular calcium-mediated mechanism independently of cholesteryl ester transfer protein inhibition. Endocrinology. 2009;50(5):2211–2219. doi: 10.1210/en.2008-1512. [DOI] [PubMed] [Google Scholar]
- 112.Forrest MJ, Bloomfield D, Briscoe RJ, Brown PN, Cumiskey AM, Ehrhart J, Hershey JC, Keller WJ, Ma X, McPherson HE, Messina E, Peterson LB, Sharif-Rodriguez W, Siegl PK, Sinclair PJ, Sparrow CP, Stevenson AS, Sun SY, Tsai C, Vargas H, Walker M, 3rd, West SH, White V, Woltmann RF. Torcetrapib-induced blood pressure elevation is independent of CETP inhibition and is accompanied by increased circulating levels of aldosterone. Br J Pharmacol. 2008;154:1465–1473. doi: 10.1038/bjp.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schmeck C, Gielen-Haertwig H, Vakalopoulos A, Bischoff H, Li V, Wirtz G, Weber O. Novel tetrahydrochinoline derived CETP inhibitors. Bioorg Med Chem Lett. 2010;20(5):1740–1743. doi: 10.1016/j.bmcl.2010.01.071. [DOI] [PubMed] [Google Scholar]
- 114.Boettcher M, Heinig R, Schafer A, Wensing G (2008) Safety, tolerability, pharmacodynamics and pharmacokinetics of BAY 60-5521—a new CETP inhibitor. 0th World Conf Clin Pharmacol Ther (July 27–August 1, Quebec), 2008, Abstract T2H023
- 115.Maeda K, Okamoto H, Shinkai H. S-(2-(acylamino)phenyl) 2,2-dimethylpropanethioates as CETP inhibitors. Bioorg Med Chem Lett. 2004;14(10):2589–2591. doi: 10.1016/j.bmcl.2004.02.071. [DOI] [PubMed] [Google Scholar]
- 116.Shinkai H, Maeda K, Yamasaki T, Okamoto H, Uchida I. Bis(2-(Acylamino)phenyl) disulfides, 2-(acylamino)benzenethiols, and S-(2-(acylamino)phenyl) alkanethioates as novel inhibitors of cholesteryl ester transfer protein. J Med Chem. 2000;43(19):3566–3572. doi: 10.1021/jm000224s. [DOI] [PubMed] [Google Scholar]
- 117.De Grooth GJ, Kuivenhoven JA, Stalenhoef AF, de Graaf J, Zwinderman AH, Posma JL, van Tol A, Kastelein JJ. Efficacy and safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in humans: a randomized phase II dose-response study. Circulation. 2002;105(18):2159–2165. doi: 10.1161/01.CIR.0000015857.31889.7B. [DOI] [PubMed] [Google Scholar]
- 118.Stein EA, Stroes ESG, Steiner G, Buckley BM DPhild, Capponi AM, Burgess T, Niesor EJ, Kallend D, Kastelein JJP (2009) Safety and Tolerability of dalcetrapib. Am J Cardiol 104:82–91 [DOI] [PubMed]
- 119.Krishna R, Anderson MS, Bergman AJ, Jin B, Fallon M, Cote J, Rosko K, Chavez-Eng C, Lutz R, Bloomfield DM, Gutierrez M, Doherty J, Bieberdorf F, Chodakewitz J, Gottesdiener KM, Wagner JA. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomized placebo-controlled phase I studies. Lancet. 2007;370(9603):1907–1914. doi: 10.1016/S0140-6736(07)61813-3. [DOI] [PubMed] [Google Scholar]
- 120.Bloomfield D, Carlson GL, Sapre A, Tribble D, McKenney JM, Littlejohn TW, Sisk CM, Mitchel Y, Pasternak RC. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am Heart J. 2009;157(2):352–360. doi: 10.1016/j.ahj.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 121.Cunningham D, Lin W, Hoth LR, Danley DE, Ruggeri RB, Geoghegan KF, Chrunyk BA, Boyd JG. Biophysical and biochemical approach to locating an inhibitor binding site on cholestreyl ester transfer protein. Bioconjug Chem. 2008;19:1604–1613. doi: 10.1021/bc800165n. [DOI] [PubMed] [Google Scholar]
- 122.Okamoto H, Yonemori F, Wakitani K, Minowa T, Maeda K, Shinkai H. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature. 2000;406:203–207. doi: 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- 123.Clark RW, Ruggeri RB, Cunningham D, Bamberger MJ. Description of the torcetrabip series of cholesteryl ester transfer protein inhibitors, including mechanism of action. J Lipid Res. 2006;47:537–552. doi: 10.1194/jlr.M500349-JLR200. [DOI] [PubMed] [Google Scholar]
- 124.Cannon CP, Dansky HM, Davidson M, Gotto AM, Brinton EA, Gould AL, Stepanavage M, Liu SX, Shah S, Rubino J, Gibbons P, Hermanowski-Vosatka A, Binkowitz B, Mitchel Y, Barter P. Determining the efficacy and tolerability of CETP INhibition with AnacEtrapib. Am Heart J. 2009;158(4):513–519. doi: 10.1016/j.ahj.2009.07.028. [DOI] [PubMed] [Google Scholar]