

Abstract
Apoptosis signal-regulating kinase 1 (ASK1), a member of the MAP kinase kinase kinase, is activated by several death stimuli and is tightly regulated by several mechanisms such as interactions with regulatory proteins and post-translational modifications. Here, we report that dual-specificity phosphatase 13A (DUSP13A) functions as a novel regulator of ASK1. DUSP13A interacts with the N-terminal domain of ASK1 and induces ASK1-mediated apoptosis through the activation of caspase-3. DUSP13A enhances ASK1 kinase activity and thus its downstream factors. Small interfering RNA (siRNA) analyses show that knock-down of DUSP13A in human neuroblastoma SK-N-SH cells reduces ASK1 kinase activity. The phosphatase activity of DUSP13A is not required for the regulation of ASK1. This regulatory action of DSUP13 on ASK1 activity involves competition with Akt1, a negative regulator of ASK1, for binding to ASK1. Taken together, this study provides novel insights into the role of DUSP13A in the precise regulation of ASK1.
Keywords: ASK1, Dual-specificity phosphatase, DUSP13A, MAPK signaling, Apoptosis
Introduction
Apoptosis signal-regulating kinase 1 (ASK1), a member of the serine/threonine kinase family, plays an important role in various cellular processes including cell growth, mitogen-activated protein kinase (MAPK) signaling, and apoptosis. ASK1 activates both the c-Jun N-terminal kinase (JNK) and the p38 signaling pathways by phosphorylating and thereby activating MKK4 (SEK1)/MKK7 and MKK3/MKK6, which are upstream regulators of JNK and p38, respectively [1, 2]. The kinase activity of ASK1 is stimulated by a variety of cellular death signals, including hydrogen peroxide (H2O2 as a reactive oxygen species donor) [3, 4], tumor necrosis factor-α (TNF-α) [5], Fas ligand [6], serum withdrawal [3], and endoplasmic reticulum (ER) stress [7].
ASK1 activity appears to be enhanced by its interaction with several positive cellular partners, such as TNF receptor associated factors (TRAFs) [5, 6, 8], D53L1 [9], G1 to S phase transition protein 1 (GSPT1) [10], and JSAP1/JIP3 [11]. On the other hand, ASK1 activity is suppressed in cells via its interaction with several negative regulatory partners, including thioredoxin [3, 12], glutaredoxin [4], 14-3-3 [13], Hsp90 [14], and Akt1 (protein kinase B) serine/threonine kinase [15].
ASK1 contains three major domains, an N-terminal domain, a central kinase domain, and a C-terminal domain. The N-terminal domain of ASK1 appears to play a negative regulatory role, as the N-terminal deletion mutant of ASK1, ASK1ΔN, shows higher kinase activity and greater pro-apoptotic activity than wild-type ASK1 [16].
ASK1 is regulated by phosphorylation and dephosphorylation. When overexpressed or stimulated, ASK1 undergoes conformational changes in the ASK1 oligomer, which induces trans-autophosphorylation of Thr-845 [17]. Akt1 phosphorylates Ser-83 of ASK1 and negatively regulates the pro-apoptotic activity of ASK1. In addition, protein phosphatase 5 (PP5) dephosphorylates Thr-845 within the kinase domain of ASK1 and thereby inactivates ASK1 [18]. In an effort to identify ASK1-regulating phosphatases, we used the ASK1 N-terminal regulatory domain as bait for immunoprecipitation screening of 46 human protein-tyrosine phosphatases (PTPs). Among several ASK1-interacting phosphatases, dual-specificity phosphatase 13A (DUSP13A) was a novel ASK1-interacting partner. DUSP13A, also known as muscle-restricted dual-specificity phosphatase (MDSP), is encoded by the upstream open reading frame (ORF) of the DUSP13 locus [19]. Two distinct DUSP13 proteins are encoded due to alternative splicing of the ORF of the DUSP13 locus. The second protein is DUSP13B (also known as testis and skeletal muscle-specific dual-specificity phosphatase, TMDP) which is encoded by the downstream ORF. These two proteins share 42% identity in amino acid sequences. DUSP13A is a member of the dual-specificity phosphatase subgroup and is closely related to VH1-related (VHR) protein-tyrosine phosphatase. Expression of DUSP13A is increased during postnatal muscle development. DUSP13A dephosphorylates both phospho-Tyr and phospho-Thr residues but does not target MAP kinases [19].
In the present study, we show that DUSP13A physically interacts with and activates ASK1. Activation of ASK1 by DUSP13A leads to the initiation of the caspase-3 dependent apoptosis pathway. Expression of either wild-type DUSP13A or inactive DUSP13A mutant induces ASK1 autophosphorylation and JNK/p38 activation. Further study shows that DUSP13A induces ASK1-mediated apoptosis by facilitating dissociation of Akt1 from ASK1.
Materials and methods
Cell culture and transfection
Human embryonic kidney (HEK) 293 cells, mouse embryonic fibroblasts (MEF), and human SK-N-SH cells (brain neuroblastoma metastasized to bone marrow), obtained from Korean Cell Line Bank (KCLB) were maintained at 37°C in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and penicillin/streptomycin in the presence of 5% CO2. For transient transfection, 1.4 × 106 cells were plated in each 60-mm cell culture plate, grown overnight, and transfected with DNA using LipofectAMINE (Invitrogen).
Plasmid construction
HA-ASK1 expression plasmid was obtained from Dr. Hidenori Ichijo. The expression constructs of other HA-tagged ASK1 mutants (ASK1 1–460, ASK1 1–666, ASK1 K domain (amino acids 649–940), and ASK1 C-terminal domain (amino acids 941–1,375), were generated by polymerase chain reaction (PCR) and subcloning into pcDNA3 plasmid (Invitrogen). The FLAG- or HA-tagged DUSP13A and DUSP13A DACS mutant (D97A and C128S) were constructed in pcDNA3.1/Zeo plasmid (Invitrogen). HA-tagged Akt1 expression plasmid was subcloned into pcDNA3. His-tagged DUSP13A WT, DUSP13A DACS, and His-MKK6 were constructed in pET28a plasmid (Novagen, Darmstadt, Germany) for protein expression in Escherichia coli. For DUSP13A siRNA expression, the target sequence of DUSP13A used was 5′-aaagGCAGGGAAGTCTTCTTGCA-3′ (sense) and 5′-aaaaTGCAAGAAGACTTCCCT GC-3′ (antisense). The complementary double-stranded siRNA oligonucleotides were inserted into pBabe-Dual vector (Addgene, Cambridge, MA, USA) using BbsI restriction enzyme sites as described [20, 21]. All of the constructs were confirmed by DNA sequencing.
Antibodies and proteins
His-tagged DUSP13A proteins were overexpressed in E. coli and bound to Ni–NTA beads (Qiagen, Hilden, Germany). The bound His-DUSP13A proteins were treated with imidazole to elute DUSP13A from the beads. The eluted DUSP13A proteins were used for in vitro assays. The phosphatase activity of His-DUSP13A and DUSP13A DACS were assayed at 30°C in 100 μl of reaction buffer containing 100 mM Tris–HCl pH 8.2, 40 mM NaCl, 1 mM DTT, 20% glycerol, 500 μM 3-O-Methylfluorescein Phosphate (OMFP; Sigma, St. Louis, MO, USA). The amount of 3-O-methylfluorescein was determined by the absorbance change at 490 nm or fluorescence change of excitation at 485 nm and emission at 525 nm.
Purified His-DUSP13A was used to immunize mice. HA-Akt1 was overexpressed in HEK 293 cells, purified using anti-HA affinity matrix (Roche Diagnostics, Indianapolis, IN, USA), and used in later experiments. Polyclonal anti-Akt1, anti-phospho-Akt1 (Ser-473), anti-phospho-ASK1 (Ser-83), anti-COX IV, anti-cytochrome c, anti-cleaved caspase-3 (Asp-175), and anti-caspase-9 (C9) antibodies were from Cell Signaling Technology (Danvers, MA, USA). Anti-ASK1 agarose bead, anti-ASK1 antibody, and anti-HA antibody were from Santa Cruz Biotechnology. Anti-FLAG M2 affinity agarose beads, anti-FLAG M2 antibody, and anti-tubulin antibody were from Sigma-Aldrich.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNAs were prepared from cells by Trizol (Roche, Basal, Switzerland) and reverse transcription was performed by using M-MLV (Invitrogen). PCR for human DUSP13A was carried out using the following primers: forward 5′-GGCAAGTTCCAGGTGGACAC-3′ and reverse 5′-GTTGTCCAGAACCTGGAGCTG-3′.
Binding assays and immunoblotting analysis
HEK 293 cells were co-transfected with ASK1 and DUSP13A expression plasmids. After 48 h of transfection, cells were washed twice with phosphate buffered saline (PBS) buffer and lysed in PTP lysis buffer (0.5% NP-40, 0.5% Triton X-100, 150 mM NaCl, 20 mM Tris–HCl pH 8.0, 1 mM EDTA, 1% glycerol, 1 mM phenylmethylsulfonyl fluoride and 1 μg/ml aprotinin) for 30 min at 4°C, followed by centrifugation at 13,000 rpm for 30 min. The soluble fractions were incubated with 20 μl of anti-FLAG M2-agarose (Sigma) at 4°C for 4 h with rotation. After binding, the beads were collected by centrifugation at 5,500 rpm for 2 min and washed five times in the PTP lysis buffer. The bound proteins were eluted with the SDS-PAGE sample buffer and then separated by SDS-PAGE, followed by immunoblotting with anti-FLAG antibody (1:10,000 dilution; Sigma) or anti-HA antibody (1:1,000 dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The protein bands were visualized by the ECL detection system (PIERCE, Rockford, IL, USA).
To examine in vivo interaction between the endogenous proteins, cell lysates were prepared from human neuroblastoma SK-N-SH cells as described above and subjected to immunoprecipitation with agarose beads coupled with anti-ASK1 antibody or with DUSP13A-specific mouse IgG followed by incubation with protein A/G agarose for 5 h at 4°C. The bound proteins were analyzed by immunoblotting with anti-ASK1 antibody or mouse anti-DUSP13A IgG.
For in vivo competition assays, HEK 293 cells were transfected with FLAG-ASK1 (1 μg), HA-DUSP13A (0, 0.5, 1, or 2 μg), and HA-Akt1 (1 μg) plasmids. After 48 h of transfection, cells were washed twice with PBS buffer and lysed in PTP lysis buffer for 30 min at 4°C, followed by centrifugation at 13,000 rpm for 30 min. The soluble fractions were incubated with 20 μl of anti-FLAG M2 affinity agarose (Sigma) at 4°C for 4 h with rotation. The bound proteins were eluted with the SDS-PAGE sample buffer and then separated by SDS-PAGE, followed by immunoblotting with anti-FLAG antibody or anti-HA antibody.
ASK1 oligomerization assay
To examine the effect of DUSP13A on the oligomerization of ASK1, HEK 293 cells were transfected with expression plasmids producing FLAG-ASK1 or HA-ASK1, respectively. After 48 h of transfection, both cell lysates were mixed together and incubated in the presence of recombinant DUSP13A WT or mutant. The samples were incubated with 20 μl of anti-FLAG M2 affinity agarose (Sigma) for 5 h at 4°C. The immunoprecipitates were eluted with the SDS-PAGE sample buffer and then separated by SDS-PAGE, followed by immunoblotting with an anti-HA antibody.
In vitro kinase assay
For ASK1 kinase activity assays, transfected HEK 293 cells were harvested and lysed with the lysis buffer containing 0.5% NP-40, 0.5% Triton X-100, 150 mM NaCl/20 mM Tris–HCl pH 8.0, 1 mM EDTA, 1% glycerol, 1 mM phenylmethylsulfonyl fluoride and 1 μg/ml aprotinin. Cell lysates were subjected to centrifugation at 13,000 rpm for 30 min at 4°C. The soluble fraction was then subjected to immunoprecipitation with anti-FLAG M2-agarose by incubation at 4°C for 4 h. Immunoprecipitates were assayed for the ASK1 kinase activities by using His-MKK6 as a substrate. Kinase assays were performed for 30 min at 30°C in the final reaction mixture—1 μCi [γ-32P] ATP, 1 μg of His-MKK6 and His-DUSP13A in kinase reaction buffer (20 mM Tris–HCl, pH 7.5, 20 mM MgCl2). Phosphorylated substrates were visualized and quantified after SDS-PAGE gel electrophoresis and autoradiography. Transfection efficiencies of cells were detected by Western blotting with an anti-FLAG antibody.
Cell viability assay
Cell viability was determined using a CCK-8 cell viability assay kit (DOJINDO Laboratories, Japan). Transfected MEF cells (5 × 103 cells/well) were cultured in DMEM medium supplemented with 5% FBS and incubated for 48 h in a 96-well plate. Then, 10 μl of cell viability assay kit solution was added to each well of the plate. After incubation for 1 h at 37°C in the dark, absorbances were measured at 450 nm using a multiwell plate reader.
Measurement of caspase-3 activity and detection of caspase-3 or -9
Caspase-3 activity was measured as described [9]. MEF cells (2 × 106/60 mm plate) were transfected with both FLAG-ASK1 and FLAG-DUSP13A expression plasmids. After 24 h of transfection, cells were transferred to 100-mm plates and grown to 80% confluence. Then, cells were exposed to serum-free DMEM for 24 h before collection for measurement.
For detection of caspase-3 or -9, SK-N-SH or MEF cells were co-transfected with expression plasmids producing FLAG-ASK1 and FALG-DUSP13A. The cell lysates were electrophoresed on SDS-PAGE and transferred to nitrocellulose membranes followed by immunoblotting with an anti-caspase-3 or anti-caspase-9 antibody.
Detection of cytochrome c release
MEF cells were transfected with FLAG-ASK1 or FLAG-DUSP13A expression plasmid. After 48 h of transfection, cytosolic and mitochondrial fractions were separated with the Qproteome Cell Compartment kit (Qiagen). The presence of cytochrome c in the cytosolic fraction was assessed by immunoblotting with an anti-cytochrome c antibody.
Results
ASK1 interacts with DUSP13A
To identify PTPs that associate with and regulate ASK1, we performed in vitro binding assays with the N-terminal regulatory domain (residues 1–666) of ASK1 and 46 PTPs. Human embryonic kidney (HEK) 293 cells were transiently co-transfected with HA-ASK1 (1–666) and FLAG-PTP expression plasmids. FLAG-PTPs were pulled down by anti-FLAG affinity agarose, followed by immunoblotting with an anti-HA antibody to detect ectopically expressed ASK1 in the pulled-down PTP complexes. We repeated the pull-down experiments three times and found that DUSP13A interacted with ASK1 in all experiments (data not shown).
To confirm the interaction between full-length ASK1 and DUSP13A in mammalian cells, HEK 293 cells were transiently co-transfected with FLAG-ASK1 and HA-DUSP13A expression plasmids. FLAG-ASK1 was pulled down with anti-FLAG affinity agarose, followed by immunoblotting with an anti-HA antibody to detect bound DUSP13A in the pulled-down ASK1 complexes (Fig. 1a). To test if DUSP13A could bring down ASK1 in a reciprocal manner, we performed co-immunoprecipitation assays with the expressed HA-ASK1 and FLAG-DUSP13A proteins and found HA-ASK1 in the immunoprecipitated FLAG-DUSP13A complex (Fig. 1b). DUSP13B, the encoded protein from the downstream ORF of the DUSP13 locus, did not interact with ASK1 (data not shown).
Fig. 1.

Interaction between ASK1 and DUSP13A. a HEK 293 cells were co-transfected using HA-DUSP13A with or without FLAG-ASK1. After 48 h of transfection, cells were lysed and immunoprecipitated with anti-FLAG M2-agarose. Immunoprecipitates were subjected to immunoblot analysis with an anti-HA antibody (top). Middle and bottom panels show expression levels of HA-DUSP13A and FLAG-ASK1 in cell lysates, respectively. b HEK 293 cells were transfected using HA-ASK1 with or without FLAG-DUSP13A. Co-immunoprecipitation was carried out as described above. c Endogenous interaction between ASK1 and DUSP13A is independent of H2O2 and TNF-α. Lysates from SK-N-SH cells treated with H2O2 (1 mM, 1 h) or TNF-α (20 ng/ml, 1 h) were immunoprecipitated with anti-ASK1 (H-300) beads and immunoblotted with an anti-DUSP13A antibody or ASK1-specific antibody. The far right lane (control) shows the immunoblotting of anti-ASK1 (H-300) beads alone used in the immunoprecipitation to confirm no indigenous IgG reactivity. The levels of endogenous proteins were measured by appropriate antibodies. d DUSP13A preferentially interacts with the N-terminal domain of ASK1. HEK 293 cells were co-transfected with FLAG-DUSP13A and either HA-ASK1-WT or its deletion mutants, as indicated. Cell lysates were immunoprecipitated with anti-FLAG M2-agarose. Immunoprecipitates and aliquots of each lysate were subjected to SDS-PAGE followed by immunoblotting with anti-HA and anti-FLAG antibodies, as indicated. ASK1 apoptosis signal-regulating kinase 1, ASK1-FL full-length ASK1, ASK1-N ASK1 N-terminal domain (residues 1–666), ASK1-K ASK1 kinase domain (residues 649–940), ASK1-C ASK1 C-terminal domain (residues 941–1375), DUSP13A dual-specificity phosphatase 13A, IgG immunoglobulin G, SDS-PAGE sodium dodecyl sulfate-polyacrylamide gel electrophoresis, TNF-α tumor necrosis factor-α, HA hemagglutinin
Polyclonal antibody specific for DUSP13A was generated in mice and used to screen for the presence of endogenous DUSP13A in various cell lines. The RT-PCR and immunoblotting analysis data showed that DUSP13A is specifically expressed in human neuroblastoma SK-N-SH cells, whereas it is not detected in HEK 293, Hep3B, and HeLa cells (data not shown). To confirm the observed ASK1–DUSP13A interaction under physiological conditions, we examined the association of endogenous ASK1 and DUSP13A proteins in non-transfected SK-N-SH cells. SK-N-SH cell lysates were immunoprecipitated with an anti-ASK1 polyclonal antibody, and the immunoprecipitates were analyzed by immunoblotting with an anti-DUSP13A antibody (Fig. 1c). The in vivo interaction not change with H2O2 or TNF-α treatment, suggesting that binding of DUSP13A to ASK1 is independent of oxidative stress, at least in part. The N-terminal domain (residues 1–666) of ASK1 (ASK-N) was found to be responsible for the binding to DUSP13A, whereas the kinase domain (residues 649–940, ASK-K) and C-terminal regulatory domain (residues 941–1,375, ASK-C) showed no affinity for DUSP13A (Fig. 1d). Collectively, these data indicate that ASK1 specifically interacts with DUSP13A through the N-terminal domain of ASK1 in an oxidative stress-independent manner.
DUSP13A enhances ASK1-mediated cell death and caspase-3 or -9 cleavage
We next carried out a series of experiments to examine the role of DUSP13A on the regulation of ASK1. Since autophosphorylation of ASK1 is proportional to ASK1 kinase activity, we investigated whether DUSP13A could enhance ASK1 autophosphorylation. HEK 293 cells that do not express endogenous DUSP13A were co-transfected with FLAG-ASK1 and various amounts of FLAG-DUSP13A expression plasmids. With FLAG pull-down immunoprecipitates, the effects of DUSP13A on the ASK1 activity in immunocomplexes were determined by autophosphorylation assays (Fig. 2a). Results show that DUSP13A enhanced ASK1 autophosphorylation in a dose-dependent manner. To further investigate whether ASK1 activity was dependent on the DUSP13A expression level, we performed DUSP13A RNA knock-down experiments using small interfering RNA (siRNA) that specifically targeted DUSP13A mRNA. Immunoblotting analyses and ASK1 kinase assays with cell lysates from SK-N-SH cells transfected with DUSP13A siRNA expression plasmid or control siRNA plasmid revealed that knock-down of DUSP13A expression also reduced ASK1 autophosphorylation and kinase activity (Fig. 2b). We next tested whether DUSP13A enhanced ASK1-induced cell death of HEK 293 cells. ASK1-induced cell death was significantly increased in the presence of DUSP13A while in the absence of DUSP13A ASK1 induced cell death slightly (Fig. 2c), suggesting that DUSP13A acts as an activator in ASK1-mediated cell death. Since ASK1 induces apoptosis by activating caspase-9 and caspase-3 [16], we examined the effect of DUSP13A on ASK1-mediated apoptosis by detection of active caspase-3 or -9. Cells co-transfected with both ASK1 and DUSP13A expression plasmids showed higher caspase-3 or -9 cleavage than cells transfected with the ASK1 expression plasmid alone (Fig. 2d), suggesting that DUSP13A upregulates the ASK1-mediated caspase-3 and -9 activities.
Fig. 2.

DUSP13A activates ASK1 kinase activity. a HEK 293 cells were co-transfected using 1 μg (+) or 2 μg (++) of FLAG-DUSP13A expression plasmid. After 48 h of transfection, cells were lysed and immunoprecipitated with anti-FLAG M2 affinity agarose. ASK1 activities were determined by in vitro phosphorylation assays. b Knock-down of DUSP13A reduces the activation of ASK1. SK-N-SH cells were transfected with plasmid pBabe-Dual-DUSP13A or the empty vector pBabe-Dual. After 48 h of transfection, cell lysates were immunoprecipitated with anti-ASK1 agarose and analyzed for autophosphorylation and in vitro kinase activities of ASK1. In vitro kinase assays were performed using His-MKK6 as a substrate. Kinase activity was normalized to the expression level of ASK1. c MEF cells were transfected with 1 μg of FLAG-ASK1 and 1 μg of FLAG-DUSP13A plasmids as indicated. To minimize the influence of transfection reagents, mock-transfected cells were considered 100% viable. Cell viability was determined by using the CCK-8 kit. Graphs represent the mean of three independent experiments. Error bars indicate ± SEM. d Effect of ASK1 and DUSP13A on cleaved caspase-3 or-9. After transfection, MEF cells were lysed and immunoblotted with anti-caspase-3 and anti-caspase-9 antibodies
DUSP13A interferes with ASK1–Akt1 complex formation in a phosphatase activity-independent manner
Since DUSP13A interacts with the N-terminal domain of ASK1 and activates ASK1, we investigated the mechanism of ASK1 activation by DUSP13A. It has been reported that ASK1 physically associates with Akt1 [15]. In addition, Akt1 phosphorylates ASK1 at Ser-83 and inhibits ASK1-mediated cell death [15]. Therefore, it is possible that DUSP13A may regulate phosphorylation at Ser-83 of ASK1. We first examined whether DUSP13A inhibits phosphorylation of ASK1 at Ser-83. Co-transfection of FLAG-ASK1 and FLAG-DUSP13A expression plasmids decreased the phosphorylation of ASK1 at Ser-83. Further decrease was observed in a dose-dependent manner with up to 2 μg of FLAG-DUSP13A plasmid (Fig. 3a).
Fig. 3.
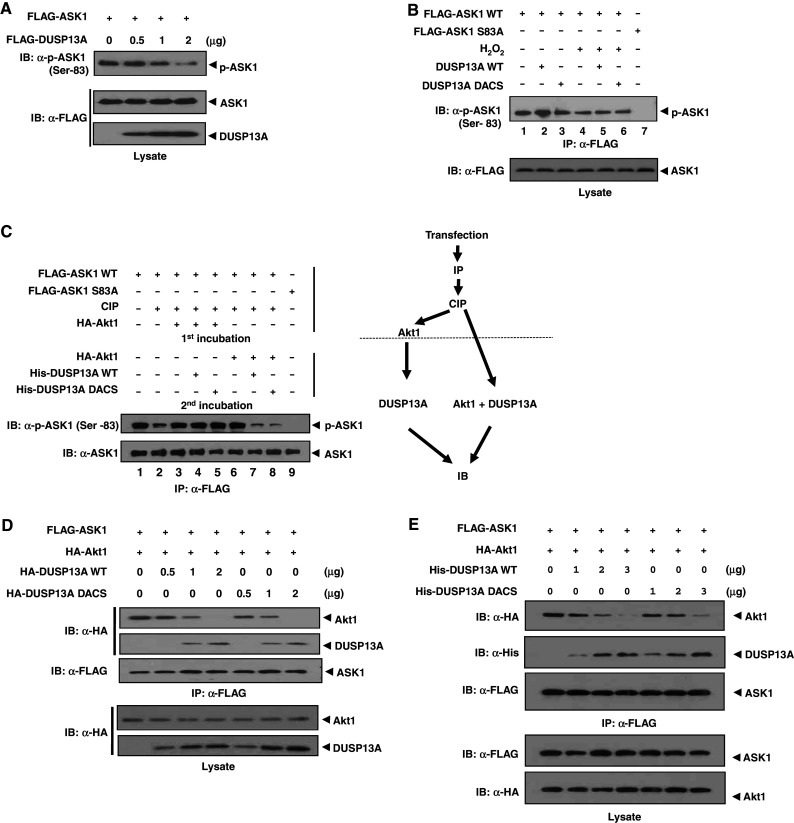
DUSP13A competes with Akt1 for binding to ASK1. a HEK 293 cells were transfected with 1 μg of FLAG-ASK1 and 0.2, 0.5, or 1 μg of FLAG-DUSP13A expression plasmid. After 48 h of transfection, cells were lysed with PTP lysis buffer. Cell lysates were subjected to immunoblotting using anti-FLAG and anti-phospho-ASK1 (Ser-83) antibodies. b HEK 293 cells were transfected with 1 μg of FLAG-ASK1 WT or FLAG-ASK1 S83A plasmid. After 48 h of transfection, cells were treated with or without 1 mM H2O2 for 1 h and ASK1 was immunoprecipitated from the cell extracts using anti-FLAG M2-agarose. Immunoprecipitated ASK1 was incubated with recombinant WT or double mutant His-DUSP13A protein (1 μg) and the phosphorylation status of ASK1 was monitored by immunoblot analysis using anti-phospho-ASK1 (Ser-83) antibody. FLAG-ASK1 S83A was used as a control. c HEK 293 cells were transfected with 1 μg of FLAG-ASK1 plasmid. After 48 h of transfection, cells were lysed and immunoprecipitated with anti-FLAG M2 affinity agarose. The ASK1 immunoprecipitates were treated with calf intestine phosphatase (CIP) in the absence of phosphatase inhibitors. After CIP treatment, ASK1 was incubated with both His-DUSP13A (WT or double mutant) and purified HA-Akt1 at the same time, or with HA-Akt1 first and then with His-DUSP13A (WT or double mutant). Samples were subjected to SDS-PAGE, followed by immunoblotting with anti-FLAG and anti-phospho-ASK1 (Ser-83) antibodies. d Dissociation of Akt1 from ASK1 is DUSP13A dose-dependent in cells. HEK 293 cells were transfected with FLAG-ASK1 (1 μg), HA-DUSP13A WT or inactive mutant (0, 0.5, 1, or 2 μg), and HA-Akt1 (1 μg) plasmids. Associations of DUSP13A and Akt1 with ASK1 were detected by immunoprecipitation with anti-FLAG M2 affinity agarose followed by immunoblotting with an anti-HA antibody. e DUSP13A induces dissociation of Akt1 from ASK1–Akt1 complex in vitro. ASK1–Akt1 complexes were immunoprecipitated with anti-FLAG M2 affinity agarose from HEK 293 cells overexpressing FLAG-ASK1 and HA-Akt1. Recombinant His-DUSP13A (1, 2, or 3 μg) was added and incubated for 2 h at 4°C. The agarose-bound proteins were washed three times with PTP lysis buffer and then separated by SDS-PAGE, followed by immunoblotting analysis
We then examined whether DUSP13A can directly dephosphorylate phospho-Ser-83 in vitro. After transfection of FLAG-ASK1 WT or FLAG-ASK1 S83A plasmid, cells were treated with or without H2O2 and ASK1 was immunoprecipitated from the cell extracts using anti-FLAG M2-agarose. Immunoprecipitated ASK1 was incubated with purified recombinant DUSP13A wild-type (WT) protein. We also used the catalytically inactive double mutant DUSP13A DACS (D97A and C128S) protein as a control. The phosphorylation status of ASK1 was determined by immunoblotting analysis using anti-phospho-ASK1 (Ser-83) antibody (Fig. 3b). Treatment of cells with H2O2 reduced the phosphorylation level of Ser-83 (Fig. 3b, lane 4), suggesting that dephosphorylation of Ser-83 is related to ASK1 activation. However, DUSP13A failed to dephosphorylate pSer-83 of ASK1 in vitro (Fig. 3b, lanes 2 and 5), which suggests that pSer-83 is not a target for dephosphorylation by DUSP13A even though its phosphorylation level is decreased in cells overexpressing DUSP13A (Fig. 3a).
Since DUSP13A does not dephosphorylate pSer-83 of ASK1 but inhibits Akt1 activity toward ASK1, we next examined whether DUSP13A competes with Akt1 for binding to ASK1 in vitro (Fig. 3c). ASK1 was immunoprecipitated and dephosphorylated with calf intestine phosphatase (CIP). When the CIP-treated ASK1 was incubated with Akt1, Akt1 phosphorylated Ser-83 of ASK1. Additional incubation with recombinant DUSP13A did not induce dephosphorylation of pSer-83 (Fig. 3c, lane 4). However, when Akt1 and DUSP13A WT were added at the same time, Akt1 was unable to phosphorylate Ser-83 (Fig. 3c, lane 7). Incubation with DUSP13A DACS mutant also produced the same result (Fig. 3c, lane 8). These results indicate that DUSP13A, regardless of its phosphatase activity, disables Akt1 kinase activity and thus inhibits of Akt1-mediated ASK1 phosphorylation at Ser-83.
We then investigated whether DUSP13A inhibits binding of Akt1 to ASK1 in a dose-dependent manner in cells (Fig. 3d). Associations of DUSP13A and Akt1 with ASK1 were detected by immunoprecipitation with anti-FLAG M2 affinity agarose followed by immunoblotting with an anti-HA antibody. The results showed that the binding of Akt1 to ASK1 gradually decreased, concomitant with an increase of DUSP13A bound to ASK1. The catalytically inactive DUSP13A mutant also bound to ASK1 and inhibited Akt1 binding to ASK1 in a dose-dependent manner. The reduction of complex formation between ASK1 and Akt1 in the presence of DUSP13A suggests that DUSP13A competes with Akt1 for binding to ASK1.
To further confirm that DUSP13A is a direct competitor of Akt1 for binding to ASK1, we performed in vitro binding competition assays (Fig. 3e). Recombinant DUSP13A proteins were incubated with ASK1–Akt1 complexes that were immunoprecipitated from cells overexpressing ASK1 and Akt1. After removal of unbound proteins, levels of DUSP13A and Akt1 bound to ASK1 were detected by immunoblotting analysis. With an increase of DUSP13A WT or mutant, the level of Akt1 bound to ASK1 was reduced in vitro. The in vitro results indicate that DUSP13A acts as a direct competitor of Akt1 for binding to ASK1.
DUSP13A has no effect on Akt1 activity and ASK1 oligomerization
Since DUSP13A interferes with Akt1 action on ASK1, it is also possible that DUSP13A may directly inhibit Akt1 activity by dephosphorylating active Akt1. Since Akt1 is activated when Ser-473 is phosphorylated [22], we investigated the effect of DUSP13A on the phosphorylation status of Ser-473 of endogenous Akt1, as assessed by immunoblotting analysis using an antibody that specifically recognizes phospho-Ser-473 of Akt1 (Fig. 4a). The results indicate that DUSP13A does not dephosphorylate active Akt1.
Fig. 4.

DUSP13A has no effect on Akt1 activity and ASK1 oligomerization. a HEK 293 cells were transfected with 0.5, 1, or 2 μg of FLAG-DUSP13A expression plasmid. After transfection, cells were lysed with PTP lysis buffer. Cell lysates were subjected to immunoblotting using anti-Akt1 and anti-phospho-Akt1 (Ser-473) antibodies. b HEK 293 cells were separately transfected with 1 μg of FLAG-ASK1 or HA-ASK1 plasmid. After 48 h of transfection, cells were lysed in lysis buffer. Both cell lysates were mixed and incubated in the presence of DUSP13A WT or mutant. FLAG-ASK1 was immunoprecipitated with anti-FLAG M2 affinity agarose. Pulled-down ASK1 complexes were immunoblotted with an anti-HA antibody
Since activation of ASK by DUSP13A may result from enhanced ASK1 oligomerization, we tested whether DUSP13A induces ASK1 dimerization to increase ASK1 activity (Fig. 4b). HEK 293 cells expressing HA-ASK1 or FLAG-ASK1 were lysed and then both cell lysates were mixed together and incubated in the presence of recombinant DUSP13A WT or mutant. After further incubation with anti-FLAG affinity agarose, FLAG-ASK1 bound beads were spun down, followed by immunoblotting analysis. DUSP13A did not change the interaction between FLAG-ASK1 and HA-ASK1. These results indicate that DUSP13A does not function as an inducer of ASK1 oligomerization.
DUSP13A enhances ASK1 kinase activity and apoptosis in a manner independent of the phosphatase activity
Given that both DUSP13A DACS mutant and DUSP13A WT compete with Akt1 for binding to ASK1, we further examined whether the catalytically inactive DUSP13A mutant affects ASK1 activity. HEK 293 cells were transfected with FLAG-ASK1 plasmid and ASK1 was immunoprecipitated from the cell extracts using anti-FLAG M2-agarose. Addition of recombinant His-DUSP13A WT to immunoprecipitated ASK1 resulted in activation of both MKK6 phosphorylation and ASK1 autophosphorylation. The DUSP13A mutant was as effective as the WT in its ability to activate ASK1 (Fig. 5a). Thus, the phosphatase activity of DUSP13A was not necessary for ASK1 activity. To confirm the role of DUSP13A in ASK1 signaling in cells, we assessed the endogenous levels of phospho-JNK and phospho-p38, which are ASK1 downstream factors. With co-expression of DUSP13A (WT or DACS) and ASK1, phospho-JNK and phospho-p38 were more apparent than with expression of ASK1 alone (Fig. 5b). We next examined whether the DUSP13A-ASK1 signal was accompanied by apoptotic features such as induction of caspase-3 activity and cleavage of caspase-9 (Fig. 5c, d). Whereas DUSP13A (WT or DACS)-transfected cells showed no significant increase of caspase-3 activity and cleavage of caspase-9, ASK1-expressing cells exhibited apparent increases of caspase-3 activity and cleavage of caspase-9. Cells co-transfected with ASK1 and DUSP13A (WT or DACS) expression plasmids showed higher caspase-3 activity and cleavage of caspase-9 than cells transfected with ASK1 expression plasmid alone (Fig. 5c, d, respectively). It has been reported that activation of ASK1 induces cytochrome c release from mitochondria as well as activation of caspase-9 and caspase-3 [16]. We also confirmed that cytochrome c release from mitochondria into cytosol was significantly enhanced in the presence of both ASK1 and DUSP13A (WT or DACS) (Fig. 5e). Collectively, these data suggest that DUSP13A enhances ASK1 kinase activity and cytochrome c-mediated apoptosis in a manner independent of the phosphatase activity.
Fig. 5.
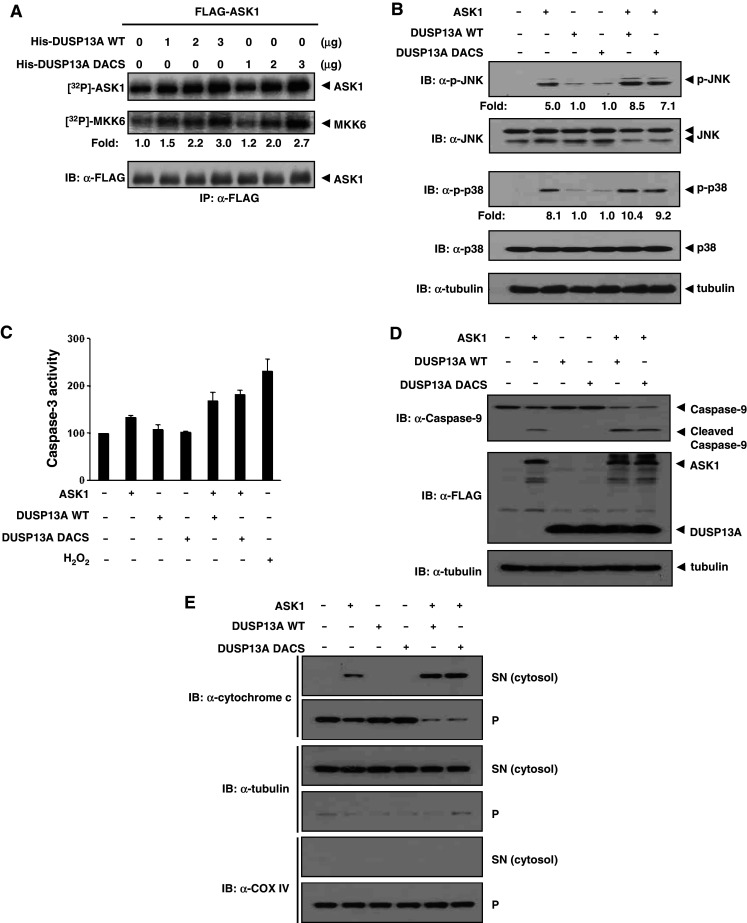
DUSP13A enhances ASK1 kinase activity and apoptosis in a manner independent of the phosphatase activity. a Autophosphorylation activity and in vitro kinase activity relative to the amount of ASK1 are shown as fold increase relative to that of FLAG-ASK1 from transfected HEK 293 cells. Top autophosphorylation assay of ASK1, middle in vitro kinase assay using His-MKK6 as a substrate, bottom immunoblotting of ASK1 with anti-FLAG-M2 antibody. b Overexpression of DUSP13A activates JNK1/p38 activity in the presence of ASK1. After transfection into MEF cells, cells were lysed with lysis buffer and immunoblotted by appropriate antibodies. c Activation of caspase-3 by DUSP13A in MEF cells. After 48 h of transfection, cells were lysed, and the caspase-3 activities in lysates were measured using DEVD-7-amino-4-methylcoumarin as a substrate. Cells treated with 1 mM H2O2 for 16 h were used as a positive control. Graphs represent the mean of three independent experiments. Error bars indicate ± SEM. d Processing of caspase-9 is induced by ASK1 and DUSP13A. After 48 h of transfection, MEF cells were lysed and immunoblotted with anti-caspase-9 antibody. e DUSP13A enhances the ASK1-mediated release of cytochrome c into the cytosol. The release of cytochrome c into the cytosol of transfected MEF was determined by immunoblotting after separation of cytosol (SN) from other organelles (p). The presence of tubulin (cytosol marker) and COX IV (mitochondria marker) in each fraction was revealed by immunoblotting; p pellets, SN supernatants
Discussion
DUSP13A is a member of the DUSP family, which is composed of 61 VH1-like genes in the human genome [23]. Some DUSPs have been shown to inhibit the MAPK pathway while others are known to be activators [24]. DUSP13A was originally identified as a muscle-restricted dual-specificity phosphatase (MDSP), the expression level of which was increased from the third week after birth to adulthood in mouse and which might be involved in postnatal muscle development [19]. However, its physiological substrate has not been identified. Even though DUSP13A is closely related to VH1-related (VHR), it does not target MAPK proteins as substrates [19]. The present study demonstrates that DUSP13A is involved in the positive regulation of the ASK1–MAPK pathway by regulating ASK1 activity. Whereas association of DUSP13A with ASK1 is required for ASK1 activation, treatment with TNF-α or H2O2, two potent ASK1 activators that induce phosphorylation of ASK1, did not modulate the interaction between DUSP13A and ASK1. This suggests that phosphorylation of ASK1 is not necessary for binding to DUSP13A, thus binding of ASK1 to DUSP13A is independent of oxidative stress, at least in part. Moreover, RT-PCR results show that TNF-α and H2O2 have no effect on the transcription of DUSP13A (data not shown). It has been reported that several DUSPs are induced upon stimulation of HEK 293T cells with H2O2 and these dephosphorylate active JNK [25]. Therefore, consistent expression of DUSP13A and stress-independent interaction with ASK1 suggest that DUSP13A may have another role in regulating ASK1 rather than a role as an ASK1 phosphatase. It is possible that DUSP13A functions as an intrinsic activator of ASK1, i.e., it is responsible for basal ASK1 activity in DUSP13A-expressing cells.
Our results show that DUSP13A does not dephosphorylate ASK1 and that DUSP13A phosphatase activity is not necessary for ASK1 activation. The DUSP13A DACS (D97A and C128S) mutant enhanced the activation of ASK1 in the same manner as the wild-type protein. There are similar cases in which the phosphatase activities of DUSPs are not required for the activation of MAPK pathways. Low-molecular-mass dual-specificity phosphatase-3 (LDP-3) enhances activation of JNK and p38, independent of its phosphatase activity [26]. Stress-activated protein kinase (SAPK) pathway-regulating phosphatase 1 (SKRP1) also activates the JNK pathway regardless of its phosphatase activity [27]. There are two possible mechanisms for how DUSP13A enhances activation of the ASK1 pathway. One is that DUSP13A functions as a competitor of negative regulators of ASK1 pathway. The other is that DUSP13A acts as a scaffold protein [26, 27]. In both cases, the phosphatase activity of DUSP13A would not be essential. Based on our results, it appears that DUSP13A enhances ASK1 activity by blocking the access of Akt1, an ASK1 inhibitor, to ASK1. Since the DUSP13A DACS (D97A and C128S) mutant functions as a trapping mutant incapable of catalysis [28, 29], it is likely that the mutant inhibits the access of Akt1 to ASK1 by associating with ASK1. Among several ASK1 regulators, GSPT1 plays a role similar to DUSP13A in regulating ASK1, such that it dissociates 14-3-3, another inhibitor of ASK1, from ASK1 to activate ASK1 [10]. Therefore, the major role of DUSP13A might be to act as a competitor of negative regulators of the ASK1 pathway.
DUSP13A expression is cell type- and developmental stage-specific, which suggests that an additional role for DUSP13A may exist. Since the substrates that are dephosphorylated by DUSP13A are not known, it is important to identify genuine substrates to determine how DUSP13A is involved in biological processes such as development.
Acknowledgments
This work was supported by a grant of the Korea Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (A01-0385-A70604-07M7-00040B) and by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (No. 2009-0072827).
Footnotes
J. E. Park and B. C. Park contributed equally to this work.
References
- 1.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/S0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 2.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 3.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song JJ, Rhee JG, Suntharalingam M, Walsh SA, Spitz DR, Lee YJ. Role of glutaredoxin in metabolic oxidative stress. Glutaredoxin as a sensor of oxidative stress mediated by H2O2 . J Biol Chem. 2002;277:46566–46575. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- 5.Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol. 2000;20:2198–2208. doi: 10.1128/MCB.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science. 1998;281:1860–1863. doi: 10.1126/science.281.5384.1860. [DOI] [PubMed] [Google Scholar]
- 7.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389–395. doi: 10.1016/S1097-2765(00)80283-X. [DOI] [PubMed] [Google Scholar]
- 9.Cho S, Ko HM, Kim JM, Lee JA, Park JE, Jang MS, Park SG, Lee DH, Ryu SE, Park BC. Positive regulation of apoptosis signal-regulating kinase 1 by hD53L1. J Biol Chem. 2004;279:16050–16056. doi: 10.1074/jbc.M305758200. [DOI] [PubMed] [Google Scholar]
- 10.Lee JA, Park JE, Lee DH, Park SG, Myung PK, Park BC, Cho S. G1 to S phase transition protein 1 induces apoptosis signal-regulating kinase 1 activation by dissociating 14-3-3 from ASK1. Oncogene. 2008;27:1297–1305. doi: 10.1038/sj.onc.1210740. [DOI] [PubMed] [Google Scholar]
- 11.Matsuura H, Nishitoh H, Takeda K, Matsuzawa A, Amagasa T, Ito M, Yoshioka K, Ichijo H. Phosphorylation-dependent scaffolding role of JSAP1/JIP3 in the ASK1-JNK signaling pathway. A new mode of regulation of the MAP kinase cascade. J Biol Chem. 2002;277:40703–40709. doi: 10.1074/jbc.M202004200. [DOI] [PubMed] [Google Scholar]
- 12.Gotoh Y, Cooper JA. Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-alpha signal transduction. J Biol Chem. 1998;273:17477–17482. doi: 10.1074/jbc.273.28.17477. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Chen J, Fu H. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc Natl Acad Sci USA. 1999;96:8511–8515. doi: 10.1073/pnas.96.15.8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R, Luo D, Miao R, Bai L, Ge Q, Sessa WC, Min W. Hsp90-Akt phosphorylates ASK1 and inhibits ASK1-mediated apoptosis. Oncogene. 2005;24:3954–3963. doi: 10.1038/sj.onc.1208548. [DOI] [PubMed] [Google Scholar]
- 15.Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatai T, Matsuzawa A, Inoshita S, Mochida Y, Kuroda T, Sakamaki K, Kuida K, Yonehara S, Ichijo H, Takeda K. Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J Biol Chem. 2000;275:26576–26581. doi: 10.1074/jbc.M003412200. [DOI] [PubMed] [Google Scholar]
- 17.Tobiume K, Saitoh M, Ichijo H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J Cell Physiol. 2002;191:95–104. doi: 10.1002/jcp.10080. [DOI] [PubMed] [Google Scholar]
- 18.Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, Ichijo H. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 2001;20:6028–6036. doi: 10.1093/emboj/20.21.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen HH, Luche R, Wei B, Tonks NK. Characterization of two distinct dual specificity phosphatases encoded in alternative open reading frames of a single gene located on human chromosome 10q22.2. J Biol Chem. 2004;279:41404–41413. doi: 10.1074/jbc.M405286200. [DOI] [PubMed] [Google Scholar]
- 20.Kaykas A, Moon RT. A plasmid-based system for expressing small interfering RNA libraries in mammalian cells. BMC Cell Biol. 2004;5:16. doi: 10.1186/1471-2121-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng L, Liu J, Batalov S, Zhou D, Orth A, Ding S, Schultz PG. An approach to genomewide screens of expressed small interfering RNAs in mammalian cells. Proc Natl Acad Sci USA. 2004;101:135–140. doi: 10.1073/pnas.2136685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 23.Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 24.Jeffrey KL, Camps M, Rommel C, Mackay CR. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov. 2007;6:391–403. doi: 10.1038/nrd2289. [DOI] [PubMed] [Google Scholar]
- 25.Teng CH, Huang WN, Meng TC. Several dual specificity phosphatases coordinate to control the magnitude and duration of JNK activation in signaling response to oxidative stress. J Biol Chem. 2007;282:28395–28407. doi: 10.1074/jbc.M705142200. [DOI] [PubMed] [Google Scholar]
- 26.Takagaki K, Satoh T, Tanuma N, Masuda K, Takekawa M, Shima H, Kikuchi K. Characterization of a novel low-molecular-mass dual-specificity phosphatase-3 (LDP-3) that enhances activation of JNK and p38. Biochem J. 2004;383:447–455. doi: 10.1042/BJ20040498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zama T, Aoki R, Kamimoto T, Inoue K, Ikeda Y, Hagiwara M. Scaffold role of a mitogen-activated protein kinase phosphatase, SKRP1, for the JNK signaling pathway. J Biol Chem. 2002;277:23919–23926. doi: 10.1074/jbc.M200838200. [DOI] [PubMed] [Google Scholar]
- 28.Blanchetot C, Chagnon M, Dube N, Halle M, Tremblay ML. Substrate-trapping techniques in the identification of cellular PTP targets. Methods. 2005;35:44–53. doi: 10.1016/j.ymeth.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14:6–16. [PubMed] [Google Scholar]