

Abstract
Somatostatin and cortistatin exert multiple biological actions through five receptors (sst1-5); however, not all their effects can be explained by activation of sst1-5. Indeed, we recently identified novel truncated but functional human sst5-variants, present in normal and tumoral tissues. In this study, we identified and characterized three novel truncated sst5 variants in mice and one in rats displaying different numbers of transmembrane-domains [TMD; sst5TMD4, sst5TMD2, sst5TMD1 (mouse-variants) and sst5TMD1 (rat-variant)]. These sst5 variants: (1) are functional to mediate ligand-selective-induced variations in [Ca2+]i and cAMP despite being truncated; (2) display preferential intracellular distribution; (3) mostly share full-length sst5 tissue distribution, but exhibit unique differences; (4) are differentially regulated by changes in hormonal/metabolic environment in a tissue- (e.g., central vs. systemic) and ligand-dependent manner. Altogether, our results demonstrate the existence of new truncated sst5-variants with unique ligand-selective signaling properties, which could contribute to further understanding the complex, distinct pathophysiological roles of somatostatin and cortistatin.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-009-0240-y) contains supplementary material, which is available to authorized users.
Keywords: Somatostatin receptor, Truncated receptor, Cortistatin, Mouse models, Metabolism
Introduction
Somatostatin (SST) is a well-known pleiotropic polypeptide, which is widely distributed throughout the organism and exerts a number of (patho)-physiological functions, including inhibition of endocrine and exocrine secretions and of tumoral cell growth, modulation of neurotransmission, regulation of metabolic, digestive and immune functions. To exert its multiple functions, SST can act through a family of five G-protein couple receptors (GPCR) with seven transmembrane domains (TMD) termed sst1–5 [1–5]. The ssts also seem to mediate most actions of cortistatin (CST), a peptide with strong homology to SST that shares the majority, but not all of the biological actions of SST [1, 6–10]. In spite of the wealth of knowledge gathered in recent years on the patho-physiology of SST/CST and their receptors, there are still a number of unsolved questions. This has led several authors to propose the existence of additional SST or CST receptors and/or signaling mechanisms, either related or not to the sst family, to explain their findings, which indicated that SST and CST can cause distinct effects at both central and peripheral levels [7, 10–14]. However, the existence of such postulated additional SST/CST receptors, or that of specific receptors for SST or CST has not been clearly demonstrated so far, although there is some evidence suggesting that MAS related gene receptor (MgrX2) as well as the GH secretagogue receptor (GHS-R1a) may play such a role by selectively binding CST, and not SST [13, 15–17].
After their discovery, initial studies on seven TMD domain GPCRs were mainly focused on the region of the receptor with the most obvious implications in ligand binding (i.e., the N-terminus), whereas the C-terminal region received somewhat less attention. However, the GPCR C-terminus is now recognized as a critical domain for the regulation of GPCR functions, and over the last years, it has been widely demonstrated that many splice variants of a given GPCR show sequence variations within the C-terminal domain, as is the case for the receptors for growth hormone-releasing hormone (GHRH-R) and gonadotropin-releasing hormone (GnRH-R) [18–22]. In fact, such a diversity of functions of the GPCR C-terminus and its related splicing processing mechanism has led to term this region the ‘magic tail’ [18]. Furthermore, there is increasing evidence that the splice mechanism of GPCR at the C-tail can also result in truncated receptors with fewer than the typical seven TMDs, as has been shown for the receptors for prostaglandin (EP1-4), corticotrophin-releasing hormone, GHRH and gonadotropin-releasing hormone [20–25]. In this context, our group has demonstrated recently the existence of two new truncated but functional splice variants of the human sst5, of five and four TMDs, accordingly termed sst5TMD5 and sst5TMD4. These receptors show a unique expression pattern in normal tissues as well as in different pituitary tumor types and display distinct responses to SST and CST [26].
Laboratory rodents are widely used as animal models to study the consequences that a patho-physiologic state (i.e., fasting, obesity) can cause on specific tissue and cell functions because of their easy experimental manipulation. Likewise, rodents, especially mice, are ideally suited to study the pathophysiological importance of gene products because of the feasibility to generate genetically modified mice over- or under-expressing the product of interest (i.e., SST knockout). Consequently, the current study focused on the identification of potential new sst5 variants in different rodent models, and their characterization under normal or pathophysiological states. The results obtained provide the first evidence for the existence of three truncated but functional sst5 variants in mouse (named sst5TMD4, sst5TMD2 and sst5TMD1) and one in rat (named sst5TMD1). These receptors, which show high inter-specific sequence identity, unique tissue expression patterns and distinct ligand-selective signaling properties, may help to better understand the complex pathophysiology of the SST/CST/sst system.
Materials and methods
Reagents
Unless indicated otherwise, all chemical products and tissue culture reagents were purchased from Sigma–Aldrich. D-MEM was obtained from Cambrex (Milan, Italy), fetal bovine serum (FBS) was purchased from PAA (Pasching, Austria), tissue culture plasticware was obtained from TPP (Trasadingen, Switzerland), mouse/rat somatostatin-14 (SST) and cortistatin-14 (CST) were purchased from Phoenix Pharmaceuticals (Karlsruhe, Germany).
Animal models
All experimental procedures were approved by the Animal Care and Use Committees of the University of Cordoba, University of Illinois at Chicago and the Jesse Brown VA Medical Center. Male and female C57Bl/6 J, Swiss and FVB mice and Wistar rats were purchased from “Centro de Instrumentación Científica” (University of Granada, Spain) or from Jackson Laboratories (Bar Harbor, ME). ob/ob mice and their lean littermate controls were purchased from Jackson Laboratories. SST-KO mice were provided by Dr. Ute Hochgeschwender [27]. MT-GHRH mice were provided by Dr. Robert E. Hammer [28]. All animals were housed under standard conditions of light (12-h light, 12-h dark cycle; lights on at 0700 h) and temperature (22–24°C), with free access to tap water and food (standard rodent chow; LabDiet, Framingham, MA). All animals were handled daily at least 1 week prior to euthanasia to acclimate to personnel and handling procedures. Animals were killed by decapitation, without anesthesia, under fed conditions unless otherwise specified. Trunk blood was collected for hormone and metabolite determinations, and fresh tissues were collected for primary tissue cell culture or were frozen in liquid nitrogen and stored at −80°C until their further analysis of mRNA levels by conventional or qrtRT–PCR (see below). Descriptions of the experiments conducted follows:
Distribution of sst5 variants in mouse tissues
Expression of sst5 variants was screened by qrtRT-PCR in a variety of mouse tissues using specific primers for each transcript (Table 1; primer sets 7–10).
Table 1.
Specific primers used for amplification of the different mouse and rat sst5 transcripts (msst5 and rsst5, respectively) in conventional RT-PCR (RT-PCR I and II) and quantitative real-time RT-PCR (RT-PCR III)
| Set no. | Name | Primer sequence | Nucleotide position | Product size (bp) | Tm (ºC) |
|---|---|---|---|---|---|
| RT-PCR I primers | |||||
| 1 | msst5-5′UTR | Sn: GGCTCCTCTAACATTGTCT | 260 | 1,844 | 58 |
| msst5-3′UTR | As: GTCCATTCTCTTTCCAGTCTT | 2,104 | |||
| 2 | rsst5-5′UTR | Sn: GCACCCTGTCCTGCACAGAGACACG | 97 | 1,268 | 64 |
| rsst5-3′UTR | As: ACACTGCCACCGAGAGGAGTCCAAC | 1,364 | |||
| RT-PCR II primers | |||||
| 3 | msst5-nested5′a | Sn: GGTCATCTATGTGGTGTTGCGGT | 491 | 1,617 | 62 |
| msst5-nested3′a | As: GTGTGTCCATTCTCTTTCCAGTCTT | 2,108 | |||
| 4 | msst5-nested5′b | Sn: CTATGTGGTGTTGCGGTACG | 497 | 1,398 | 60 |
| msst5-nested3′b | As: GGGCTGAACACACATGGATACCTTT | 1,895 | |||
| 5 | msst5-nested5′c | Sn: CGGTATTAGTGCCTGTGCTCTACTT | 429 | 1,045 | 62 |
| msst5-nested3′c | As: CTCTCATATCCCAGAAGACAACACC | 1,474 | |||
| 6 | rsst5-nested5′ | Sn: CGGCTGCTTCCAGTGGTAACCATA | 197 | 1,164 | 64 |
| rsst5-nested3′ | As: TGCCACCGAGAGGAGTCCAACTTGT | 1,360 | |||
| RT-PCR III (qrtRT-PCR) primers | |||||
| 7 | Qrt-msst5 | Sn: ACCCCCTGCTCTATGGCTTT | 1,215 | 105 | 61 |
| As: GCTCTATGGCATCTGCATCCT | 1,319 | ||||
| 8 | Qrt-msst5TMD4 | Sn: GTCCACCCTCTCCGCTCA | 415 | 131 | 61 |
| As: GCAGGTTCGCAGAGGACATC | 545 | ||||
| 9 | Qrt-msst5TMD2 | Sn: CAGTTCACCCGTACTGTGGCAT | 358 | 132 | 61 |
| As: CACAGCTTCAGGGTGGGTAA | 489 | ||||
| 10 | Qrt-msst5TMD1 | Sn: AACGTGTATATCCAGACAAGAGTGG | 217 | 152 | 61 |
| As: TCCCAGAAGACAACACCACA | 368 | ||||
| 11 | Qrt-rsst5 | Sn: GCTCATGTCTCTGCCGCTCTT | 648 | 281 | 61 |
| As: AGCCCACAAACACCAGCACCACGAC | 928 | ||||
| 12 | Qrt-rsst5TMD1 | Sn:ACGCCAAGATGAAGACAAGAGT | 194 | 154 | 61 |
| As:CAGAACCCAGAAGACAACACC | 347 | ||||
| Set no. | Name | Primer Sequence | Products size (bp) | Tm (ºC) | |
|---|---|---|---|---|---|
| Primers to clone msst5 variants in pcDNA3.1 | |||||
| 13 | Se_Start codon | Sn: ATGGAGCCCCTCTCTTTGAC | 1,716 (msst5); 668 (msst5TMD4) | 61 | |
| As_3′UTR | As: CCTTGCTTTTCCTTGTGCTG | 713 (msst5TMD2); 938 (msst5TMD1) | |||
| 14 | Sense_EcoRI_Kozak | Sn: TAATGAATTCCCACCATGGAGCCCCTCTC | 1,187 (msst5) | 61 | |
| Antisense_XhoI_msst5/TMD1 | As: TAATCTCGAGTCAGCAAACTCTCAT | 406 (msst5TMD1) | |||
| 15 | Sense_EcoRI_Kozak | Sn: TAATGAATTCCCACCATGGAGCCCCTCTC | 601 (msst5TMD4) | 61 | |
| Antisense_XhoI_ msst5TMD4/2 | As: TAATCTCGAGTCAGCAAACTCTCAT | 646 (msst5TMD2) | |||
| Primers to clone 3xHA-tagged-msst5 variants in pcDNA3.1 | |||||
| 16 | Sense_HindIII_Kozak | Sn: ACTTAAGCTTGGTACCACCATGTAC | 117 | 61 | |
| Antisense_EcoRI | As: TAATGAATTCCATCAGCGTAATCTG | ||||
| 17 | Sense_EcoRI | Sn:TAATGAATTCTTATGGAGCCCCTCTC | 1,184 (msst5) | 61 | |
| Antisense_XhoI_ msst5/TMD1 | As: TAATCTCGAGTCAGCAAACTCTCAT | 403 (msst5TMD1) | |||
| 18 | Sense_EcoRI | Sn:TAATGAATTCTTATGGAGCCCCTCTC | 598 (msst5TMD4) | 61 | |
| Antisense_XhoI_ msst5TMD4/2 | As: TAATCTCGAGTCAGCAAACTCTCAT | 643 (msst5TMD2) | |||
Effect of fasting on the hypothalamus (HPT) and pituitary (PIT) expression of mouse sst5 variants
To evaluate the effect of fasting on sst5 variants expression, male mice (10 weeks of age) were assigned to one of four groups (n = 5–8): 12, 24, 48 h fasting or control feeding ad libitum and HPT and PIT were collected for further analysis as previously reported [29].
Effect of obesity on the HPT and PIT expression of mouse sst5 variants
To study the effect of the obese state on sst5 variant expression, tissue samples were obtained from two models of obesity as previously reported [30, 31]: (1) ob/ob mice and their littermate controls (n = 6; 10 weeks of age); (2) diet-induced obesity (DIO) mice, fed a low-fat diet (LFD) or a high-fat diet (HFD) diet. Briefly, DIO groups of male mice (n = 6–7) were placed at 4 weeks of age on either a LFD (10% kcal from fat, used as control; Research Diets, Brunswick, NJ) or HFD (60% kcal from fat) and killed at 20 weeks of age.
Effect of leptin on the HPT and PIT expression of mouse sst5 variants
To evaluate the impact of leptin replacement on sst5 variants expression in the ob/ob mouse, HPT and PIT from ob/ob male mice (10 weeks of age) used in a recent study [30] were further evaluated. Briefly, groups of ob/ob mice (n = 5) were implanted with osmotic mini-pumps (sc) containing either recombinant mouse leptin or vehicle for 7 days. This study included a group of ob/ob mice pair-fed to match the food intake of leptin-treated mice to differentiate between direct effects of leptin and those mediated indirectly by leptin-induced reduction in food intake and weight loss.
Expression of sst5 variants on the HPT and PIT of SST-KO mice
To explore the potential role of endogenous SST in the regulation of sst5 variant expression, tissues samples from male SST-knockout (SST−/−) and SST-intact (SST+/+) mice were used as previously reported [32, 33]. Briefly, SST+/+ and SST−/− mice (n = 5–8 per group; 9–11 weeks old) were fed ad libitum or subjected to food deprivation for 48 h. After this period, mice were killed by decapitation and tissues collected for analysis.
Expression of mouse sst5 variants on PIT tumors
qrtRT-PCR was used to determine whether sst5 variants were present in hyperplastic and adenomatous PIT samples from male mice expressing the MT-hGHRH transgene and normal PIT samples of age-matched wild-type controls previously generated by our laboratory [34].
RNA isolation and reverse transcription (RT)
Tissues and cell cultures were processed for recovery of total RNA using the Absolutely RNA RT–PCR Miniprep Kit (Stratagene, La Jolla, CA) or Trizol reagent (Invitrogen, Barcelona, Spain) with deoxyribonuclease treatment (Stratagene; Promega, Madison, WI) as previously described [31, 32, 35]. The amount of RNA recovered was determined using the Ribogreen RNA quantification kit (Molecular Probes, Eugene, OR). Total RNA (1–2 μg for whole tissue extract and 0.25 μg for primary cell cultures) was reversed transcribed with enzyme and buffers supplied in the cDNA First Strand Synthesis kit (MRI Fermentas, Hanover, MD). cDNA was treated with ribonuclease H (1U; MRI Fermentas).
Isolation and sequence of the sst5 variants by standard RT-PCR
Initial amplification and sequencing of truncated sst5 variant transcripts were performed by standard RT-PCR, named type I, using tissue extracts (HPT and PIT) from adult C57Bl/6 J, Swiss and FVB mice and Wistar rats. Primers used for standard PCR type I (Table 1; Primer sets 1–2) were located upstream of the start codon (sense primer: msst5-5′UTR and rsst5-5′UTR) and downstream of the stop codon (antisense primer: msst5-3′UTR and rsst5-3′UTR) of the mouse and rat sst5 gene (Genbank accession no. NM_011425.1 and NM_012882, respectively). PCR reactions (type I) were performed using the iCycler IQTM system (BioRad, Madrid, Spain), 1.25U EcoTaq polymerase (Ecogen, Barcelona, Spain), 200 μM dNTPs, 2 mM MgCl2 (Ecogen), 0.2 μM of primers, 1 M betaine and 100 ng of cDNA as template in a 25 μl-volume reaction with a program consisting of the following steps: (1) 95°C for 10 min, (2) 38 cycles of denaturation (95°C for 30 s), annealing (58–64°C for 30 s) and extension (72°C for 30–120 s), and (3) final extension of 10 min at 72°C. All PCR products were run on agarose gel, stained with ethidium bromide and column-purified using QuiaQuick gel extraction kit (Qiagen, GmBH, Germany). All PCR products were sequenced to confirm target specificity at the Genomic Unity of the University of Cordoba using an ABI Prism 3130XL Genetic analyzer (Applied Biosystems, Madrid, Spain). Bioinformatics analyses of the isolated fragments were performed to blast the sequences and deeply analyze the novel spliced sst5 transcripts. Then, a second nested PCR (named type II) was carried out to re-amplify single PCR products with nested primer pairs (Table 1; primer sets 3–6) located more internally in the mouse and rat sst5 gene than the primers used in PCR type I.
Verification and quantification of the truncated sst5 variant expression using qrtRT–PCR
Primers used for standard and qrtRT–PCR (Table 1; primers sets 7–12) were selected using Oligo 6.0 (Molecular Biology Insights, Inc., Cascade, CO) and Primer3 (http://frodo.wi.mit.edu/primer3/input.htm) software [36]. Details regarding the development, validation and application of a qrtRT-PCR to measure expression levels of mouse transcripts, including full-length sst5, have been reported previously [29, 31, 32]. Similarly, sets of primers were designed and validated to specifically amplify and quantify the truncated sst5TMD4/2/1 variants in different RT-PCR samples. Specifically, to determine the starting copy number of cDNA, RT samples were PCR amplified, and the signal was compared with that of a standard curve run on the same plate. Specific standard curves consisted of 1, 101, 102, 103, 104, 105 and 106 copies of synthetic cDNA template for each of the transcripts of interest (full-length and truncated sst5 variants) were constructed. In addition, total RNA samples that were not reversed transcribed and a no DNA control were run on each plate to control for genomic DNA contamination and to monitor potential exogenous contamination, respectively. Also, to control for variations in the amount of RNA used in the RT reaction and the efficiency of the RT reaction, the mRNA copy number of the transcript of interest was adjusted by the mRNA copy number of cyclophilin A (used as housekeeping gene), where cyclophilin A mRNA levels did not significantly vary between experimental groups within tissue type (data not shown).
Cloning, epitope-tagging and transfection of mouse sst5 variants
cDNA sequences encoding the whole open reading frame (ORF) of all mouse sst5 variants (full-length and truncated sst5) were generated by a RT-PCR strategy using mouse RNA (pituitary and fat) as a template, the iMAX-II polymerase kit (iNtRON Biotechnology, Seongnam, Korea) and 0.2 μM of primers (Table 1; primer sets 13–18). The PCR products were amplified and purified from the gel and then fragments were sequenced at least three times to verify the correct ORF. These fragments were subsequently used for two sets of studies:
Functional studies
PCR fragments of the sst5 variants were introduced into the pCDNA3.1 vector using specific primers containing a Kozak sequence and a EcoRI site (sense primer) and a XhoI site (antisense primer) (Table1; primer sets 14–15) and then stable transfected into CHO-K1 cells as previously described [26, 37]. Cells stable transfected with each sst5 variant were used to evaluate the modulation of free cytosolic [Ca2+]i in single cells by microfluorimetry and the measurement of cAMP levels in response SST-14 and CST-14 (see below).
Cellular localization studies
PCR fragments of the sst5 variants were introduced into pCDNA3.1 vector using specific primers containing EcoRI and XhoI sites (Table1; primer sets 17–18), and a 3xHA tag was synthesized (Table 1; primer set 16), self-annealed and inserted (in phase upstream of the sst5 variant fragments) into the HindIII and EcoRI sites acquiring the different mouse 3xHA-tag-sst5 expressing vectors. The triple ligation of all sst5 variants was validated by PCR sequence. These 3xHA-tag-sst5 vectors were transiently transfected into CHO-K1 cells as previously described [37], and then cells were used for confocal localization assays (see below).
In both sets of studies, the cloning and transfection of sst5 variants were performed employing the reagents and methods reported previously [26, 37]. Expression analysis validations in CHO-K1 cells transfected with each sst5 variant by qrtRT-PCR showed an equally high transfection efficiency level (data not shown).
Cell culture models
CHO-K1 cells
Transient and stable cells transfected with sst5 variants were cultured as previously described [37].
Primary PIT cell and HPT N6 cell cultures
PIT of adult male C57Bl/6 J mice (10 weeks of age) were dispersed into single cells and cultured in serum containing α-medium, as previously described [30, 31]. N6 cells, a mouse HPT cell line originally developed by Belsham et al. [38], were cultured in monolayer in serum containing α-medium, as previously described [39, 40]. After 24 h of culture (200,000 cells/well, 24-well plates), media was removed, and wells were washed in serum free media and subsequently treated with SST-14 and CST-14 (100 nM) for 24 h, and then total cellular RNA was extracted for determination of sst5 variant expression by qrtRT–PCR.
Measurement of free cytosolic calcium concentration ([Ca2+]i) in single cells
CHO-K1 cells stably transfected with each of the sst5 variants were grown onto 25 mm-Ø glass coverslips for 24 h (50,000 cells/coverslip, 35-mm plates) and incubated for 30 min at 37°C with 2.5 μM of the Ca2+ indicator dye Fura-2 AM (Molecular Probes) in phenol red-free DMEM containing 20 mM NaHCO3 (pH 7.4). Coverslips were washed with phenol red-free DMEM, mounted on the stage of a Nikon Eclipse TE200-E microscope (Nikon, Tokyo, Japan) with an attached back thinned-CCD cooled digital camera (ORCAII BT; Hamamatsu Photonics, Hamamatsu, Japan). Cells were examined under a ×40 oil immersion objective during exposure to alternating 340- and 380-nm light beams, and the intensity of light emission at 505 nm was measured every 5 s as previously reported [35, 37]. Changes in [Ca2+]i after SST-14 and CST-14 (100 nM) administration were recorded as background substrate ratios of the corresponding excitation wavelength (F340/F380) using MetaFluor Software (Imaging Corp., West Chester, PA).
cAMP measurements
To investigate the responsiveness of CHO-K1 cells stably transfected with each of the sst5 variants or transfected with the empty vector (used as a control), cells were plated (10,000 cells/well, 96-well plates) and cultured in serum containing F12 media. After 24 h of culture, media was removed and cells washed in serum-free media for 40 min, and then preincubated with F12 containing 1 nM of 3-isobutyl-1-methylxanthine (IBMX) to prevent enzymatic degradation of cAMP. After 30 min of preincubation with IBMX, cells were incubated for an additional 30-min period with control (media), SST-14 or CST-14 (100 nM) in the presence of IBMX and 100 nM of forskolin to evaluate the ability of SST and CST to inhibit forskolin-stimulated cAMP production. The experiment was terminated by adding lysis reagent to each well, and lysates were recovered and stored at −80°C for analysis of intracellular cAMP accumulation, as assessed by cAMP Biotrack EIA system following Protocol 3 of the manufacturer’s instructions (GE Healthcare, Barcelona, Spain).
Subcellular localization and confocal microscopy
Transiently transfected CHO-K1 cells with each sst5 variant plasmids containing a 3xHA epitope were cultured on round 18-mm Ø glass coverslips (50,000 cells/well, 12-well plates) in serum containing media, as previously described [37]. After 24 h of culture at 37°C, media was removed and cells washed in serum free media and fixed in 4% paraformaldehyde for 10 min at room temperature. After fixation, cells were rinsed sequentially with PBS (three times, 5 min each), permeabilized with 50% metanol-PBS (3 min at −20°C) and 100% metanol (3 min at −20°C), blocked with PBS-0.5% BSA (2 h at 4°C) and then incubated overnight (4°C) with primary antibody for 3xHA epitope (1:1,000; self-made) [26]. Finally, cells were incubated with secondary antibody (Alexa Fluor 488 goat anti-rabbit IgG; 1:500 in PBS; 2 h at 4°C; Invitrogen, Eugene, OR), washed in PBS and mounted with glycerol PBS. The signal was visualized using a Leica Espectral TCS-SP2-AOBS confocal scanning microscope (Leica Corp., Heidelberg, Germany) and then analyzed with ImageJ software (National Institutes of Health, Bethesda, MD). Controls consisted of transiently transfected CHO-K1 cell preparations processed as described above with the exception of excluding primary antibody to 3xHA epitope.
Statistical analysis
Samples from all groups within an experiment were processed at the same time; therefore, the in vivo effects of genotype/fasting/obesity and the in vitro effects of SST/CST were assessed by one- or two-way ANOVA followed by a Newman-Keuls test for multiple comparisons or by Student’s t test, as appropriate. P < 0.05 was considered significant. All data are expressed as means ± SEM. The in vivo effects of genotype/fasting/obesity were obtained from a minimum of five animals per group. Results from in vitro studies were obtained from at least three separate, independent experiments carried out on different days and with different cell preparations. For single cell analysis, a minimum of 20 cells were analyzed per experiment. All statistical analyses were performed using the GB-STAT software package (Dynamic Microsystems, Inc. Silver Spring, MD).
GenBank submission
The new sequence of the mouse and rat sst5 variants were submitted to Genbank (accession nos. GQ359775, GQ359776, GQ359777 and GQ359778 for the mouse sst5TMD4, sst5TMD2, sst5TMD1 and rat sst5TMD1 variants, respectively).
Results
Identification of mouse and rat sst5 variants using standard RT–PCR
Three specific bands in mouse tissues and one band in rat tissues corresponding to novel variants of the sst5 gene were obtained by standard RT-PCR (type I and II, see “Materials and methods” section) using primers specified in Table 1. Each sst5 variant was separately amplified by RT-PCR (type III) using selective-sst5 variant primers (see below). All PCR products did not appear using total RNA samples that were not reversed transcribed or in PCR that did not contain samples, indicating that they were not the result of genomic contamination or potential exogenous contamination, respectively. cDNA sequencing and bioinformatic analysis of the isolated fragments revealed that these products have a complete identity with discontinuous segments of the mouse and rat sst5 gene with full identity against two different and distant areas of the sst5 genomic sequence [GeneID nos. 20609 (mouse) and 25354 (rat); Fig. 1a and supplemental Figs. 1a, 2a]. The new truncated sst5 variants described herein are not generated by a classical alternative splicing mechanism since the sst5 gene lacks canonical introns within its coding sequence (CDS), which is fully encoded within the exon-3 (mouse) or the exon-2 (rat) of the sst5 genomic sequences published to date in the NCBI website (GeneID nos. 20609 and 25354, respectively). Thus, the generation of new truncated sst5 variants should involve the presence of cryptic introns with non-canonical donor and acceptor splice sites within the CDS and the 3′UTR region, resulting in new receptors that maintain the same N-terminal region (CDS-1; Fig. 1b–d, left panel) as full-length sst5 isoform (Fig. 1a, left panel), but have different, shorter C-terminal tails (CDS-2; Fig. 1b–d, left panel) with 4 (mouse sst5TMD4), 2 (mouse sst5TMD2) and 1 (mouse and rat sst5TMD1) TMD (Fig. 1b–d, middle and right panels and supplemental Fig. 1a, 2a, b). Therefore, assuming that the start of the transcription is unaltered in the novel sst5 variants, the complete CDS of 576, 621 and 384 nucleotides (for the mouse sst5TMD4, sst5TMD2 and sst5TMD1 variants, respectively), and of 348 nucleotides (for the rat sst5TMD1 variant) would encode proteins of 191, 206, 127 and 115 amino acids, respectively (Supplemental Fig. 1a, 2). It is interesting to note that the mouse and rat sst5TMD1 variants showed high interspecific nucleotide (87%) and amino acid (86%) sequence identity (Supplemental Fig. 2c–d).
Fig. 1.
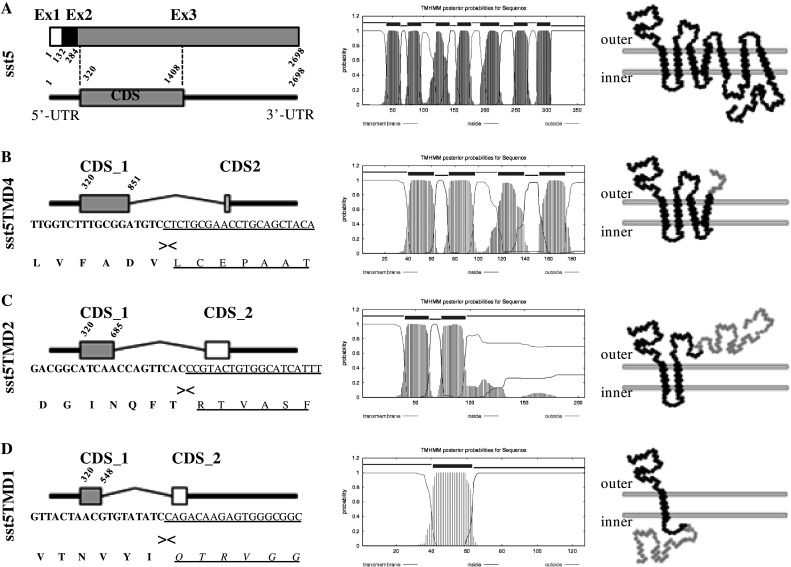
Molecular identification and characterization of novel truncated mouse sst5 variants. a Left panel schematic representation of the full-length sst5 mRNA constituted by three exons: exon 1 (Ex1; white box; nucleotide nos. 1–132), exon 2 (Ex2; black box: nucleotide nos. 133–284) and exon 3 (Ex3; grey box; nucleotide nos. 285–2698). Coding region (CDS) is located and codified within the Ex3 (nucleotide nos. 320–1408). b, c Left panel, schematic representation of the truncated sst5 variant mRNA constituted by the same 5′UTR sequence (straight black line on the left) as full-length sst5 (including Ex1, Ex2 and first 34 bp of Ex3), two coding regions (CDS_1 and CDS_2) and a 3′UTR sequence (straight black line on the right). The N-terminal regions of the CDS_1 (grey boxes) are similar in all sst5 variants (full-length and truncated) but have different length (nucleotide nos. 320–857 in sst5TMD4, 320–685 in sst5TMD2 and 320–548 in sst5TMD1), while CDS_2 of each truncated sst5 variant (white boxes) is constituted by unique sequences originating by splicing of a portion of the CDS and/or 3′-UTR of the full-length sst5. The nucleotide and amino-acid sequences of the junction between the CDS_1 and CDS_2 of the different truncated sst5 variants are shown below the corresponding schematic representation of each sst5 variant. a–d Middle panel hydrophobicity profiles of full-length and truncated sst5 variants obtained from the program TMHMM Server v.2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0) (CBS, Denmark) and right panel predicted transmembrane domain structure of full-length and truncated sst5 variants. In grey are shown the different C-terminal tails of each truncated sst5 variant compared with full-length sst5 variant
Verification and quantification of the mouse sst5 variant expression in different tissues using qrtRT PCR
Our aim was to accurately compare the level of expression of the new truncated sst5 variants with that of full-length sst5 in various tissues. Therefore, sets of primers were designed and validated to amplify specifically the truncated mouse sst5TMD4, 2 and 1 variants by qrtRT–PCR. To this end, one of the primers was designed to span the two partial coding regions (CDS-1/CDS-2) of the different truncated sst5 variants [on top of the joining site (open arrows in Fig. 2a)]. Using these new sets of primers and the one previously validated to amplify sst5 [29], a single PCR product was consistently obtained, which was found to be of the expected size (105 bp for full-length sst5, 131 bp for sst5TMD4, 132 bp for sst5TMD2 and 152 bp for sst5TMD1 by separation in agarose gel; Fig. 2b) in all tissues analyzed. The temperature of dissociation of these PCR products differed (Fig. 2c), indicating that distinct products were generated as confirmed by the sequencing analysis. Analysis of the expression pattern of the sst5 variants in normal tissues shows that full-length and truncated sst5 variants are widely and differentially expressed in the mouse tissues tested (Fig. 2d and supplemental Fig. 3). Specifically, we found that sst5 was predominantly expressed in the pituitary (PIT) and hypothalamus (HPT) (21,163 ± 4,343 and 4,638 ± 473 copies, respectively; Fig. 2d). Similarly, sst5TMD2 and sst5TMD1 were highly expressed at the PIT and HPT level compared with other tissues (PIT: 15,185 ± 3,437 and 1,044 ± 207 copies; HPT: 2,950 ± 490 and 197 ± 35 copies, respectively). In contrast, the expression level of sst5TMD4 variant was very low in those tissues (PIT: 15 ± 2 and HPT: 5 ± 2). Analysis of the other tissues revealed a heterogeneous pattern of expression depending on the sst5 variant and the tissue screened. Thus, sst5 and sst5TMD2 are the most represented variants at the systemic level, whereas sst5TMD4 is scarcely expressed, being only detectable in the gut, ovary, uterus, fat and mammary gland, and sst5TMD1 was preferentially found in heart and at low levels in other tissues where sst5 and sst5TMD2 are expressed (Fig. 2d and supplemental Fig. 3).
Fig. 2.

Verification and quantification of the full-length and truncated mouse sst5 variants in mouse tissues. a Schematic representation of the full-length sst5 and novel truncated sst5 variant transcripts. CDS are shown by boxes (grey boxes CDS of sst5 or CDS_1 of each truncated sst5 variant; white boxes CDS_2 of each truncated sst5 variant). Arrow heads are used to indicate the relative positions of primers sets used in quantitative real-time RT-PCR (qrtRT-PCR; closed arrows indicated primers within a CDS, while open arrows indicate primers spanning CDS_1 and CDS_2). b Representative agarose gel showing PCR products amplified by qrtRT-PCR using mouse pituitary as template. All products (sst5 variants and cyclophilin used as a housekeeping gene) were size separated on an agarose gel containing EtBr, column-purified and sequenced to confirm target specificity. PCR product of the sst5TMD4 variant could not be amplified if pituitary cDNA was used as a template, indicating that this variant was not present in this tissue; however, the sst5TMD4 variant transcript was detected in other tissues (i.e., fat; Supplemental Figure 3). c Melting curves of qrtRT-PCR products with different Tm of dissociation (80.9°C for full-length sst5, 79.4°C for sst5TMD2 and 86.8°C for sst5TMD1) generated from the same mouse pituitary sample. d Absolute cDNA copy number/0.1 μg total RNA of full-length and truncated sst5 variant transcripts in the pituitary, hypothalamus, cortex and cerebellum of male C57Bl/6 mice, as determined by qrtRT-PCR. Values represent means ± SEM (n = 5 mice)
Subcellular localization of mouse sst5 variants in Chinese hamster ovary (CHO)-K1 cells
Analysis of the subcellular localization of each of the sst5 variants (3xHA-tagged receptors; Fig. 3) in CHO-K1 cells by confocal microscopy revealed that whereas the full-length sst5 variant is preferentially located at the plasma membrane (Fig. 3a), the truncated sst5 variants are mainly located at intracellular compartments (Fig. 3b–d). Nonetheless, a proportion of all of the truncated sst5 variants was also located at the plasma membrane (Fig. 3b–d).
Fig. 3.

Subcellular localization of full-length and truncated mouse sst5 variants. Top panel Schematic representation of the recombinant protein of full-length (a) and truncated sst5 variants [sst5TMD4 (b), sst5TMD2 (c) and sst5TMD1 (d)] containing the 3x hemagglutinin (3xHA) epitope in the N-terminal domain. Black boxes represent 3xHA epitope, and grey and white boxes represent CDS_1 and CDS_2, respectively. Bottom panel Representative images of the subcellular localization of the full-length (a) and truncated sst5 variants [sst5TMD4 (b), sst5TMD2 (c) and sst5TMD1 (d)] transfected in CHO-K1 cells using confocal microscopy. White arrows indicate regions of plasma membrane where truncated sst5 variants are visualized. Scale bar 10 μm
Measurement of free cytosolic Ca2+ concentration ([Ca2+]i)
The ability of each sst5 variant to modify the [Ca2+]i kinetics in response to their natural ligands, SST and CST, was evaluated in CHO-K1 cells stably transfected with full-length sst5, sst5TMD4, sst5TMD2 or sst5TMD1 (Fig. 4a, b). This approach demonstrated that cells transfected with sst5 responded to SST and CST in a similar manner in terms of the proportion of responsive cells (47 and 40% respectively) and the profile of the response evoked, which showed comparable temporal and quantitative features (Fig. 4a). Similar to sst5, cells transfected with sst5TMD1 showed a comparable, albeit lower proportion of cells responsive to SST and CST (20 and 11%, respectively). In striking contrast, cells transfected with sst5TMD4 responded almost exclusively (48%) to SST, since only 2% of those cells responded to CST. Interestingly, an opposite situation was observed for sst5TMD2, in that only 13% of the cells responded to SST by showing delayed responses (28 s), whereas CST evoked rapid and clear [Ca2+]i increases in 51% of the cells transfected with this receptor. SST or CST treatment did not evoke any changes in [Ca2+]i in non-transfected CHO-K1 cells or in cells transfected with the empty vector (used as controls; data not shown).
Fig. 4.
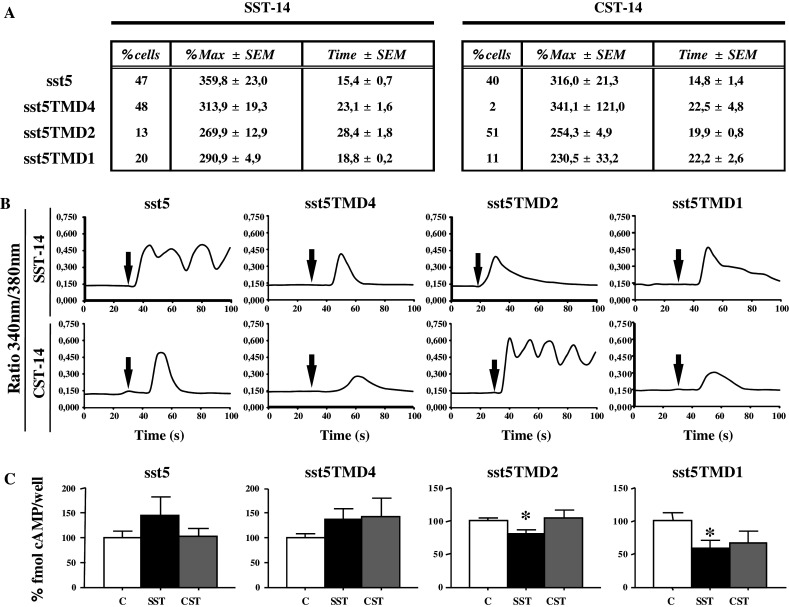
Functional characterization of full-length sst5 and truncated variants in terms of activation of signal transduction ([Ca2+]i and cAMP) in response to somatostatin (SST) and cortistatin (CST). CHO-K1 cells were stable-transfected with full-length or truncated sst5 variants, and the effect of SST and CST challenge (100 nM) was evaluated. a Percentage of transfected cells showing changes in [Ca2+]i in response to SST and CST. Percentage of maximum response (%Max) and time of response to SST and CST administration are also indicated. b Representative profiles of changes in [Ca2+]i in transfected cells in response to SST and CST administration (arrow). c Effect of SST and CST treatment on the forskolin-induced cAMP production in transfected cells (C control, n = 4 experiments). Asterisk (*P < 0.05) indicates values that differ from control-treated cells
cAMP measurement
To further analyze the functionality of the truncated sst5 variants, we examined the ability of SST and CST to modify forskolin-stimulated cAMP production in transfected CHO-K1 cells. As shown in Fig. 4c, SST significantly diminished cAMP production in cells expressing sst5TMD2 or sst5TMD1 (20% and 40% respectively compared with control). In marked contrast, we observed a trend toward an opposite, stimulatory effect of SST in cells transfected with full-length sst5 or with truncated sst5TMD4 (45% and 36%, respectively, compared with control), although these differences did not reach statistical significance. On the other hand, CST did not cause any significant effect in cells transfected with full-length or truncated sst5 variants (Fig. 4c).
Metabolic regulation of the HPT and PIT expression of mouse sst5 variants
Given the fact that the PIT and HPT full-length sst5 mRNA levels can be regulated under different states of energy balance and by different metabolic signals [2, 29], we sought to study the PIT and HPT transcriptional regulation of the new truncated sst5 variants in a series of mice models under different metabolic/pathologic conditions previously characterized in our laboratory, which include: fasting, diet-induced obesity (DIO), obese mice due to leptin-deficiency (ob/ob), mice expressing the metallothionein promoter-human GHRH transgene (MT-hGHRH) and somatostatin knockout mice (SST-KO) [29–33] (Fig. 5a and Supplemental Figure 4). In addition, changes in the expression levels of all sst5 variants were evaluated in primary PIT cell cultures as well as in the murine hypothalamic cell line N6 [38] in response to treatments with endogenous ligands for these receptors (SST and CST; Fig. 5b, c, respectively). It should be noted that, as mentioned above, expression levels of sst5TMD4 were not detected or were extremely low in the normal mouse PIT and HPT; therefore, the analysis of this variant was not included in the corresponding figures.
Fig. 5.
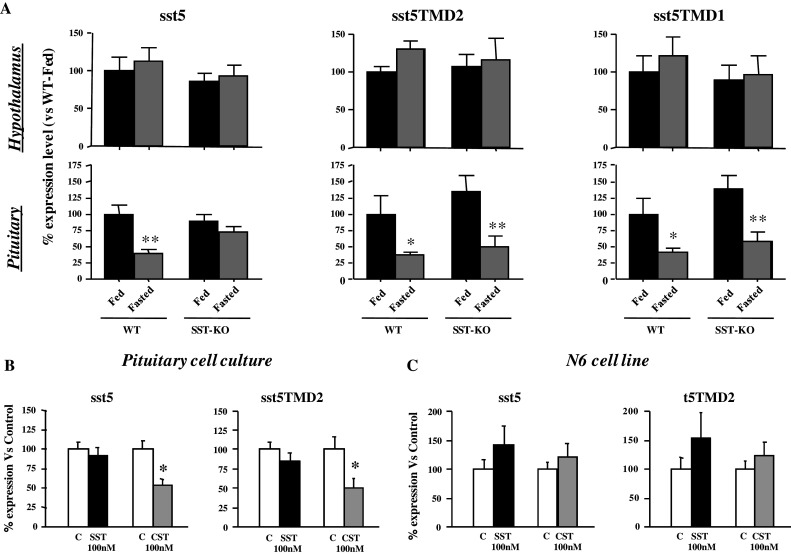
Regulation of hypothalamic and pituitary mRNA levels of full-length and truncated mouse sst5 variants by their endogenous ligands, somatostatin (SST) and cortistatin (CST). a sst5, sst5TMD2 and sst5TMD1 mRNA levels in the hypothalamus (top panel) and pituitary (bottom panel) of fed and fasted (48 h) SST+/+ (WT) and SST−/− (SST-KO) male mice (n = 5–8 mice/group). b, c Effects of 24-h treatment of SST or CST (100 nM) on sst5 and sst5TMD2 mRNA levels in primary pituitary cell cultures from male mice (b) and in the murine hypothalamic cell line N6 (c). The mRNA copy number was determined by qrtRT–PCR, and the values were adjusted by cyclophilin A copy number as an internal control. Values are expressed as % of fed WT mice (a set at 100%) or as % of control-treated cells (b, c set at 100% within experiment). Values represent the mean ± SEM of n = 5–7 mice/group (a) or three independent experiments (b, c 3–4 replicates/treatment/experiment). Asterisks (*P < 0.05; **P < 0.01) indicate values that differ within genotype in response to fasting (a) or from control-treated cells (b, c)
Effect of negative energy balance conditions (fasting time-course)
Fasting evoked a significant marked decrease in the expression levels of all sst5 variants in the PIT, but did not significantly alter HPT mRNA levels (Supplemental Fig. 4a)
Effect of conditions of positive energy balance (obesity)
No significant differences were observed in expression levels of sst5 variants in PIT or HPT between obese mice fed on a high-fat diet (HFD) as compared with mice fed on a low-fat diet (LFD) used as control (Supplemental Fig. 4b). Likewise, no differences were found between ob/ob mice and lean control animals (Supplemental Fig. 4c). Moreover, when the effect of leptin replacement on the PIT and HPT expression of sst5 variants was analyzed in ob/ob mice, we found that leptin infusion did not significantly alter the expression levels of PIT and HPT sst5 variants (vehicle control ob/ob compared with leptin-treated ob/ob and pair-fed ob/ob mice; Supplemental Fig. 4d).
Presence of sst5 variants in PIT tumors (MT-hGHRH transgenic mice)
We have previously observed that the full-length sst5 variant is highly expressed in PIT tumors of MT-GHRH mice and that the expression levels of this receptor are upregulated in the hyperplastic PIT (4 month-old) compared to age-matched controls, while adenoma formation (mice >10 months of age) is associated with a decline in sst5 expression compared to WT levels (Supplemental Fig. 4e, left panel) [34]. Therefore, we sought to determine if the novel truncated sst5 variants were also present in mouse PIT tumors and whether the expression levels of these variants were regulated in the hyperplastic and adenomatous state. Our results demonstrate that, similar to that found in normal pituitaries, sst5TMD2 and sst5TMD1 are present in both hyperplastic and adenomatous PIT tumors of MT-GHRH mice. In striking contrast to that found for full-length sst5, we found that expression levels of sst5TMD2 or sst5TMD1 were not altered in hyperplastic or adenomatous pituitaries compared with their respective controls (Supplemental Fig. 4e, middle and right panels).
Effect of lack of SST (SST-KO mice)
Given the essential role of SST in regulating the expression levels of its own receptors [2], we also sought to determine the potential function of SST as a modulator of the expression levels of the new truncated sst5 variants. To this end, PIT and HPT of SST-KO mice were analyzed. We observed that lack of endogenous SST did not modify PIT or HPT mRNA levels of any sst5 variant under normal-fed conditions (Fig. 5a). Similarly, food deprivation for 48 h did not alter HPT expression levels of sst5 variants in SST-KO as compared with wild-type control mice. Of note, however, whereas 48 h-fasting decreased PIT mRNA levels of all sst5 variants (full-length and truncated) in wild-type (control) animals, in SST-KO mice full-length sst5 expression was not reduced, while the mRNA levels for the two truncated variants did decrease in fasted SST-KO (Fig. 5a).
Analysis of sst5 variant expression in PIT and HPT mouse cell cultures
Finally, to address the potential direct regulatory actions of SST and CST, we examined their effects on the expression levels of sst5 variants in primary PIT cell cultures (Fig. 5b) and in the mouse HPT cell line N6 (Fig. 5c). Use of qrtRT–PCR indicated that sst5 and sst5TMD2 mRNAs were expressed in both PIT and HPT cell cultures (Fig. 5b, c, respectively), whereas mRNA for sst5TMD4 and sst5TMD1 were not detected in cultured cells, in line with the low levels of expression for these variants observed in whole PIT and HPT extract compared to those of sst5 and sst5TMD2 (Fig. 2d). Interestingly, results from these treatments indicated a differential regulatory ability for SST and CST. Indeed, SST treatments did not significantly modify PIT or HPT mRNA levels of sst5 and sst5TMD2 variants in cultured cells, a situation similar to that found in whole PIT and HPT of wild-type controls compared with SST-KO mice (Fig. 5b, c). In contrast, CST treatments reduced by half the PIT mRNA levels of both sst5 and sst5TMD2 variants (Fig. 5b), while having no effect on HPT sst5 variants (Fig. 5c).
Discussion
The GPCRs superfamily of receptors, which includes the sst1-5 family, is associated with a number of important physio-pathological functions [41–43]. This explains the increasing interest in this research area, which is also supported by the fact that roughly 30% of marketed drugs target GPCRs, as is the case for synthetic SST analogs used to control cell growth and hormonal hypersecretion in pituitary adenomas and other neuroendocrine tumors [44, 45]. However, despite their biological importance and extensive study, there are still functions associated with these receptors that cannot be explained by the GPCRs known to date. Accordingly, over the last years several laboratories have been searching for novel, additional GPCR variants, and this has led in some cases to the identification of new, functional variants of GPCR bearing fewer than the typical seven TMDs that constitute the hallmark for this receptor superfamily [21, 22, 46, 47]. In particular, our laboratory has recently identified two new spliced variants of the human sst5 with unique properties that could contribute to mediate SST and CST signaling in normal and tumoral cells [26]. Given that rodents are widely used to study the physiological importance of gene products, in the present study we followed the lead of our previous work on human sst5 and focused our efforts on the identification and characterization of new variants of sst5 in rats and mice. As a result, we have isolated, for the first time, novel variants of rodent sst5 with different size and sequence as compared to the full-length sst5s that are generated by splicing of cryptic introns at the sst5 mRNA. Specifically, we have identified three truncated sst5 variants in mouse (named sst5TMD4, sst5TMD2 and sst5TMD1) and one in rat (named sst5TMD1), displaying unique properties and the latter, mouse and rat sst5TMD1 showing high inter-specific nucleotide and amino acid sequence identity.
GPCRs are commonly encoded by genes with an exon-intron gene structure, wherein alternative splicing, especially within the C- and N-terminal tails, originates proteins with different lengths and sequences, although generally conserving the typical seven TMDs [48, 49]. In striking contrast, the new truncated sst5 variants described herein are not originated by classical alternative splicing, since the sst5 gene lacks canonical introns (starting with GT and ending with AG, the so-called GT-AG rule) within its CDS [50]. Thus, the generation of new truncated sst5 variants involves the presence of cryptic introns with non-canonical donor and acceptor splice sites resulting in new receptors that maintain the same N-terminal region as full-length sst5s, but bear different, shorter C-terminal tails with 4, 2 or 1 TMD (mouse sst5TMD4, sst5TMD2 or mouse and rat sst5TMD1, respectively) arising from a splicing of a portion of the canonical CDS and a portion of the 3′-UTR. These findings were not completely unexpected based on the fact that previous results from other laboratories studying different systems had suggested the existence of additional sst5 variants [12, 51]. Also, generation of gene products involving non-canonical cryptic introns is not rare, and over the last years several gene products derived from this type of splicing are being identified [52, 53], including the new truncated human sst5 variants reported by our group [26]. Interestingly, generation of this type of spliced proteins has been shown to involve new signals and mechanisms, which are still poorly understood and require further study [54, 55]. These include the presence of GC-rich motifs in the spliced genes, which could enhance the recognition of non-canonical introns resulting in new gene products [53, 56]. In fact, it has been reported that the relative gene density increases more than tenfold as GC content increases from 30 to 50% [56], as is the case for the sst5 gene in several species [66.3%, 64.8%, 64.4%, 58.16% and 55.07% for human, baboon, pig, rat and mouse, respectively (Genbank accession nos. NM_001053, EF639293, NM_001038229, NM_012882 and NM_011425.1, respectively)]. Indeed, while the overall GC content of the human and mouse genome is ~40%, it is noteworthy that the GC content in the full-length and truncated sst5 variants spans from 55 to 60%, thus supporting the idea that the sst5 gene is a suitable target for the splicing machinery of non-canonical events.
An initial analysis of the expression pattern of the full-length and truncated sst5 variants in normal and tumoral tissues showed a wide but differential distribution of these receptors, which was tissue- and variant-dependent. Specifically, our results using qrtRT-PCR show that the overall predominant sst5 variants at the central and peripheral levels are full-length sst5 and sst5TMD2, which are highly expressed in endocrine/metabolic tissues as HPT, PIT, digestive tract, etc., whereas the other truncated receptors were less frequently and abundantly expressed. In fact, at the central level (HPT, cortex and cerebellum), we found that the expression of sst5 variants were also differentially distributed, with sst5 being the most abundant receptor in HPT and cerebellum, followed by sst5TMD2 and sst5TMD1, while mRNA levels of sst5TMD4 were not detected in those tissues. Interestingly, and similar to the results shown previously by others [57], sst5 mRNA levels were not detected in mouse cortex; however, quite interestingly, all truncated sst5 variants were present at different levels in this tissue (sst5TMD2 ≫> sst5TMD4 > sst5TMD1), suggesting that truncated sst5 variants may play a physiologically important role in this tissue. Therefore, future studies need to be performed to elucidate the possible function of the truncated variants in these tissues.
Examination of the subcellular distribution of 3xHA-tagged receptors expressed in CHO-K1 cells by confocal microscopy indicated that all truncated sst5 variants display a predominant cytoplasmic localization, whereas full-length sst5 is mainly located at the plasma membrane, a situation that closely mimics that found recently for human sst5 variants [26]. Nonetheless, a proportion of all of the truncated sst5 variants were also located at the plasma membrane in an amount that seems sufficient to convey a potent ligand-induced response that is comparable to that exerted through the full-length receptor (see below). Although intracellular localization of GPCR is a common transient event due to the typical process of ligand-induced endocytosis or heterotypic interaction of receptors [58, 59], the question arises, why do truncated sst5 variants have a different subcellular localization than full-length sst5? A plausible explanation could be related with recent data showing that the signaling properties and cellular distribution of GPCR can be influenced by GPCR interacting proteins (GIP) that assemble with the intracellular regions of the receptors, most notably with the C-terminal tail [18, 19, 60–62]. Although the C-terminal tails of GPCRs were promptly recognized as being important for the fine-tuning of G protein activation, their roles as ‘magic’ hooks to fish for GPCR-associated proteins have only recently been established. In this scenario, it seems reasonable to link this property with the observation that many GPCR splice variants [46, 63–65], including the human [26], mouse and rat sst5 variants (this study), differ in their C-terminal tail, and therefore, it could be speculated that the C-termini of these truncated sst5 variants could associate with distinct intracellular scaffolding/signaling molecules, which would selectively define their specific subcellular localization and signal transduction properties. Although additional studies are obviously required to validate this idea, our hypothesis is supported by recent functional and immunolocalization studies using AtT20 and HEK293 cells expressing the full-length sst5 or truncated C-terminal mutants of the receptor, indicating that the C-terminal tail motifs are crucial in the intracellular localization and trafficking regulation of the sst5 [61, 66] and also by a recent study showing that different regions of the sst5 are associated with the activation of different signaling pathways [67].
In spite of the fact that all novel sst5 variants possess less than seven TMDs and display different C-terminal tails than the full-length sst5, these truncated receptors maintain an intact N-terminus that has also been found to contribute to the ligand-induced activation mechanism. In fact, previous results of our laboratory have shown that human sst5 truncated variants exhibit a similar, but reduced ability to be activated by SST and CST compared with the full-length sst5 [26]. Therefore, to investigate whether the rodent sst5 variants were functional, we analyzed the cellular response ([Ca2+]i and cAMP measurement) of CHO-K1 cells transfected with each of the sst5 variants after treatment with the endogenous ligands, SST and CST. Application of a Ca2+ imaging approach demonstrated that, despite their truncated nature, sst5TMD4/2/1 variants were functional receptors able to mediate increases in [Ca2+]i. Specifically, [Ca2+]i levels were increased with SST and CST treatment in ~40–45% of the cells transfected with full-length sst5, which is consistent with previous reports showing a similar [Ca2+]i increase in response to SST or a specific sst5 agonist in AtT-20 cells expressing sst5, as well as in cell lines transfected with human sst5 [68, 69]. Interestingly, only a minor percentage of cells transfected with sst5TMD1 (~11–20%) responded to SST and CST showing similar profiles. In clear contrast, whereas sst5TMD4 was almost exclusively activated by SST and not by CST (48% vs. 2%), cells transfected with sst5TMD2 responded quite selectively to CST and not to SST (51% vs. 13%). These results closely resemble those found for human truncated sst5TMD5 and sst5TMD4, which, despite not being orthologues for the mouse receptors, displayed a strong functional analogy in that they were selectively activated by SST and CST, respectively [26]. This suggests that shortening of sst5 unveils molecular features that enable the selectivity of the responses to these strikingly similar ligands, although this seems to include species-specific features, which will require additional detailed analysis.
To further investigate the functionality of the truncated sst5 variants, we studied the levels of sst5-mediated cAMP accumulation after SST and CST challenge. Our results showed that SST but not CST significantly inhibited forskolin-induced cAMP production in cells transfected with sst5TMD2 and 1. In clear contrast, SST tended to potentiate forskolin-induced cAMP accumulation in cells transfected with full-length sst5 and sst5TMD4. Although this latter observation did not reach statistical significance, this trend is in line with previous data showing that mouse and human sst5 could cause a stimulatory effect on cAMP accumulation in response to SST or specific sst5 agonists in some [68, 70, 71], but not all [70] cellular systems. All together, our results clearly demonstrate that truncated sst5 variants are functional, as shown by their ability to mediate selective, ligand-induced regulation in [Ca2+]i and cAMP levels in transfected cells, and that the different structure/sequence of each variant could result in the distinct functional properties shown by each individual receptor. Nevertheless, although these functional results are not conclusive, they provide primary, convincing evidence for the relevant ability of the new truncated sst5 receptors with 4, 2 and 1 TMD to mediate selective activation of intracellular signaling pathways in response to specific ligands (SST vs. CST). In fact, these results are consistent with previous findings showing that comparable splice variants with less than seven TMDs, such as the human truncated sst5 variants sst5TMD5 and sst5TMD4 [26], conserve the ability to convey selective ligand-induced responses. Furthermore, even receptor variants containing only the extracellular region of TMD receptors [i.e., corticotrophin-releasing factor receptor type 2 α (CRFR2α)] [23] have been reported to be capable of binding and modulating ligand activity.
SST and CST mediate a variety of biological effects, but the most well known are those occurring at the HPT and PIT level (i.e., inhibition of GH, TSH and ACTH release) [2, 5, 43]. Our results using qrtRT-PCR show, for the first time in mice, that the expression levels (copy number/0.1 μg total RNA) of the full-length sst5 are the highest in the PIT and HPT compared with other tissues. A similar situation was found for the truncated sst5TMD2 and sst5TMD1 receptors, whereas the expression levels of the sst5TMD4 variant were extremely low at the HPT and PIT level. Subsequently, use of different mice models under distinct metabolic conditions revealed that truncated sst5 variants are differentially regulated in the PIT and HPT depending of the metabolic insult considered (i.e., fasting and obesity). Specifically, we observed that the expression level of both sst5TMD2 and 1 variants were downregulated in the PIT after a 12 h-fasting period, whereas HPT mRNA levels were unresponsive to this metabolic insult similar to that previously observed with the full-length sst5 variant [29]. However, we cannot exclude the possibility that our measurement of whole hypothalamic extracts may have masked nuclei-specific changes (arcuate, paraventricular, ventromedial) in sst5 variant expression in the different mouse models analyzed in the present study. Of note, other studies have also reported similar changes in pituitary sst expression in the fasted male rat [72], suggesting that fasting-mediated changes in pituitary sst5 responsiveness (plausibly including truncated receptor variants) could be common across species and thus that this phenomenon is likely to be of functional relevance. Actually, in contrast to the parallel regulation of full-length and truncated sst5 variants in the PIT in response to fasting, obesity (DIO and ob/ob mice) did not alter expression levels of sst5 variants in the HPT and PIT, indicating that, depending on the metabolic insult (fasting vs. obesity), sst5 variants are also differentially regulated in a tissue-specific manner.
These results raised the question of which factors may contribute to the regulation of PIT and HPT sst5 variants expression in response to metabolic stress. One factor may be endogenous SST, because previous studies have indicated that fasting and obesity could be associated with changes in SST tone [2, 32, 73–75], and SST can directly regulate the expression of its own receptors [4, 76, 77]. Therefore, we sought to determine if loss of endogenous SST (SST-KO compared with SST-intact control mice) could modulate the expression of truncated sst5 variants in PIT and HPT. We found that, similar to that seen with full-length sst5, mRNA levels of truncated sst5TMD2 and 1 variants were not altered in HPT and PIT of SST-KO mice under fed conditions compared with SST controls. These observations are also consistent with the in vitro results showing that SST did not alter mRNA levels of full-length sst5 or truncated sst5TMD2 in mouse primary PIT cell cultures and HPT N6 cells. In contrast, CST significantly decreased both full-length sst5 and truncated sst5TMD2 in PIT cell cultures, demonstrating, for the first time, that the modulation of full-length and truncated sst5 variants synthesis is ligand (SST vs. CST)- and tissue (PIT vs. HPT)-dependent in mouse. On the other hand, 48 h-fasting resulted in a significant decrease of truncated sst5TMD2 and 1 mRNA levels in the PIT of both SST-intact and SST-KO mice, whereas fasting inhibited full-length sst5 mRNA levels only in the PIT of SST-intact but not in SST-KO mice [29]. Therefore, our results demonstrate that the fasting-induced suppression of sst5 variants is variant-specific and that, at the PIT level, endogenous SST tone is a critically required regulator for the fasting-induced fall observed in the full-length sst5 synthesis, but not for the truncated sst5 variants.
In summary, we report for the first time the existence of truncated variants of the rodent sst5, one in rat and three in mouse, which may be of pathophysiological relevance because they: (1) are widely and distinctly expressed across tissues and they also display differential subcellular distribution compared to the full-length sst5; (2) are functional as shown by their ability to mediate ligand-selective changes in [Ca2+]i and cAMP production; (3) their expression is regulated in a tissue-specific manner (central vs. systemic) by changes in hormonal and metabolic environment (i.e., fasting and lack of endogenous SST); (4) their expression is also regulated in a ligand-dependent manner, since CST but not SST was able to induce a downregulation on the sst5 variant mRNA levels in primary PIT cell cultures. The increasing multiplicity of protein networks associated with GPCRs generates more complexity in the understanding of the physiology, pharmacology and pathology related to this receptor family. However, the diversity of new functional sst may also shed some light into this complexity and could pave the way to discover novel drugs and tools to treat some endocrine pathologies associated with the SST- and CST-axis such as pituitary and other neuroendocrine tumors, cancer, immune disorders, etc.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknownledgements
This work is supported by grants from Research Grants BFU2007-60180/BFI and BFU2008-01136/BFI (Ministerio de Ciencia e Innovación), BIO-139 and CTS-1705 (Junta de Andalucía), Ayudas predoctorales de formación en investigación en salud del Fondo de Investigación Sanitaria (FIS, ISCIII: FI06/00804; to JCC), Programa Nacional de becas de FPU (FPU-AP20052473, to MDG) and Programa Ramón y Cajal del Ministerio de Educación y Ciencia (RYC-2007-00186, to RML), Spain, NIDDK 30677 and VA Merit (to RDK) and IPSEN Pharmaceuticals (to J.P.C). CIBER is an initiative of Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación, Spain. The authors would like to thank Dr. Ute Hochgeschwender (Oklahoma Medical Research Foundation, Oklahoma City, OK), who generously provided us with the original SST-KO mice, Dr. Ralph L. Brinster (University of Pennsylvania, Philadelphia PA) for the original MT-hGHRH transgenic mice and Dr. Belsham (University of Toronto, Canada) for kind provision of hypothalamic N6 cells.
Abbreviations
- SST
Somatostatin
- CST
Cortistatin
- sst
Somatostatin receptor
- TMD
Transmembrane domain
- HPT
Hypothalamus
- PIT
Pituitary
- DIO
Diet-induced obesity
- MT-hGHRH
Metallothionein promoter-human growth hormone releasing hormone
- SST-KO
Somatostatin knock-out
- 3xHA
3x hemagglutinin
- qrtRT-PCR
Quantitative real-time retrotranscriptase-polymerase chain reaction
References
- 1.Gahete MD, Duran-Prado M, Luque RM, Martinez-Fuentes AJ, Vazquez-Martinez R, Malagon MM, Castaño JP. Are somatostatin and cortistatin two siblings in regulating endocrine secretions? In vitro work ahead. Mol Cell Endocrinol. 2008;286:128–134. doi: 10.1016/j.mce.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 2.Luque RM, Park S, Kineman RD. Role of endogenous somatostatin in regulating GH output under basal conditions and in response to metabolic extremes. Mol Cell Endocrinol. 2008;286:155–168. doi: 10.1016/j.mce.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Moller LN, Stidsen CE, Hartmann B, Holst JJ. Somatostatin receptors. Biochim Biophys Acta. 2003;1616:1–84. doi: 10.1016/S0005-2736(03)00235-9. [DOI] [PubMed] [Google Scholar]
- 4.Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999;20:157–198. doi: 10.1006/frne.1999.0183. [DOI] [PubMed] [Google Scholar]
- 5.Viollet C, Lepousez G, Loudes C, Videau C, Simon A, Epelbaum J. Somatostatinergic systems in brain: networks and functions. Mol Cell Endocrinol. 2008;286:75–87. doi: 10.1016/j.mce.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 6.Broglio F, Grottoli S, Arvat E, Ghigo E. Endocrine actions of cortistatin: in vivo studies. Mol Cell Endocrinol. 2008;286:123–127. doi: 10.1016/j.mce.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 7.de Lecea L. Cortistatin: functions in the central nervous system. Mol Cell Endocrinol. 2008;286:88–95. doi: 10.1016/j.mce.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 8.de Lecea L, Castaño JP. Cortistatin: not just another somatostatin analog. Nat Clin Pract Endocrinol Metab. 2006;2:356–357. doi: 10.1038/ncpendmet0219. [DOI] [PubMed] [Google Scholar]
- 9.de Lecea L, Criado JR, Prospero-Garcia O, Gautvik KM, Schweitzer P, Danielson PE, Dunlop CL, Siggins GR, Henriksen SJ, Sutcliffe JG. A cortical neuropeptide with neuronal depressant and sleep-modulating properties. Nature. 1996;381:242–245. doi: 10.1038/381242a0. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Rey E, Delgado M. Emergence of cortistatin as a new immunomodulatory factor with therapeutic potential in immune disorders. Mol Cell Endocrinol. 2008;286:135–140. doi: 10.1016/j.mce.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Castaño JP, Delgado-Niebla E, Duran-Prado M, Luque RM, Sanchez-Hormigo A, Gracia-Navarro F, Garcia-Navarro S, Kineman RD, Malagon MM. New insights in the mechanism by which SRIF influences GH secretion. J Endocrinol Invest. 2005;28:10–13. [PubMed] [Google Scholar]
- 12.Patel YC, Panetta R, Escher E, Greenwood M, Srikant CB. Expression of multiple somatostatin receptor genes in AtT-20 cells: evidence for a novel somatostatin-28 selective receptor subtype. J Biol Chem. 1994;269:1506–1509. [PubMed] [Google Scholar]
- 13.Siehler S, Nunn C, Hannon J, Feuerbach D, Hoyer D. Pharmacological profile of somatostatin and cortistatin receptors. Mol Cell Endocrinol. 2008;286:26–34. doi: 10.1016/j.mce.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Tallent M, Liapakis G, O’Carroll AM, Lolait SJ, Dichter M, Reisine T. Somatostatin receptor subtypes SSTR2 and SSTR5 couple negatively to an L-type Ca2+ current in the pituitary cell line AtT-20. Neuroscience. 1996;71:1073–1081. doi: 10.1016/0306-4522(95)00510-2. [DOI] [PubMed] [Google Scholar]
- 15.Deghenghi R, Papotti M, Ghigo E, Muccioli G. Cortistatin, but not somatostatin, binds to growth hormone secretagogue (GHS) receptors of human pituitary gland. J Endocrinol Invest. 2001;24:RC1–RC3. doi: 10.1007/BF03343800. [DOI] [PubMed] [Google Scholar]
- 16.Robas N, Mead E, Fidock M. MrgX2 is a high potency cortistatin receptor expressed in dorsal root ganglion. J Biol Chem. 2003;278:44400–44404. doi: 10.1074/jbc.M302456200. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Zhou YB, Zhang GG, Cai Y, Duan XH, Teng X, Song JQ, Shi Y, Tang CS, Yin XH, Qi YF. Cortistatin attenuates vascular calcification in rats. Regul Pept. 2010;159:35–43. doi: 10.1016/j.regpep.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Bockaert J, Marin P, Dumuis A, Fagni L. The ‘magic tail’ of G protein-coupled receptors: an anchorage for functional protein networks. FEBS Lett. 2003;546:65–72. doi: 10.1016/S0014-5793(03)00453-8. [DOI] [PubMed] [Google Scholar]
- 19.Bockaert J, Roussignol G, Becamel C, Gavarini S, Joubert L, Dumuis A, Fagni L, Marin P. GPCR-interacting proteins (GIPs): nature and functions. Biochem Soc Trans. 2004;32:851–855. doi: 10.1042/BST0320851. [DOI] [PubMed] [Google Scholar]
- 20.Havt A, Schally AV, Halmos G, Varga JL, Toller GL, Horvath JE, Szepeshazi K, Koster F, Kovitz K, Groot K, Zarandi M, Kanashiro CA. The expression of the pituitary growth hormone-releasing hormone receptor and its splice variants in normal and neoplastic human tissues. Proc Natl Acad Sci USA. 2005;102:17424–17429. doi: 10.1073/pnas.0506844102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neill JD, Musgrove LC, Duck LW. Newly recognized GnRH receptors: function and relative role. Trends Endocrinol Metab. 2004;15:383–392. doi: 10.1016/j.tem.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Rekasi Z, Czompoly T, Schally AV, Halmos G. Isolation and sequencing of cDNAs for splice variants of growth hormone-releasing hormone receptors from human cancers. Proc Natl Acad Sci USA. 2000;97:10561–10566. doi: 10.1073/pnas.180313297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen AM, Perrin MH, Digruccio MR, Vaughan JM, Brar BK, Arias CM, Lewis KA, Rivier JE, Sawchenko PE, Vale WW. A soluble mouse brain splice variant of type 2alpha corticotropin-releasing factor (CRF) receptor binds ligands and modulates their activity. Proc Natl Acad Sci USA. 2005;102:2620–2625. doi: 10.1073/pnas.0409583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasegawa H, Negishi M, Ichikawa A. Two isoforms of the prostaglandin E receptor EP3 subtype different in agonist-independent constitutive activity. J Biol Chem. 1996;271:1857–1860. doi: 10.1074/jbc.271.4.1857. [DOI] [PubMed] [Google Scholar]
- 25.Miyata I, Shiota C, Ikeda Y, Oshida Y, Chaki S, Okuyama S, Inagami T. Cloning and characterization of a short variant of the corticotropin-releasing factor receptor subtype from rat amygdala. Biochem Biophys Res Commun. 1999;256:692–696. doi: 10.1006/bbrc.1999.0392. [DOI] [PubMed] [Google Scholar]
- 26.Duran-Prado M, Gahete MD, Martinez-Fuentes AJ, Luque RM, Quintero A, Webb SM, Benito-Lopez P, Leal A, Schulz S, Gracia-Navarro F, Malagon MM, Castaño JP. Identification and characterization of two novel truncated but functional isoforms of the somatostatin receptor subtype 5 differentially present in pituitary tumors. J Clin Endocrinol Metab. 2009;94:2634–2643. doi: 10.1210/jc.2008-2564. [DOI] [PubMed] [Google Scholar]
- 27.Zeyda T, Diehl N, Paylor R, Brennan MB, Hochgeschwender U. Impairment in motor learning of somatostatin null mutant mice. Brain Res. 2001;906:107–114. doi: 10.1016/S0006-8993(01)02563-X. [DOI] [PubMed] [Google Scholar]
- 28.Hammer RE, Brinster RL, Rosenfeld MG, Evans RM, Mayo KE. Expression of human growth hormone-releasing factor in transgenic mice results in increased somatic growth. Nature. 1985;315:413–416. doi: 10.1038/315413a0. [DOI] [PubMed] [Google Scholar]
- 29.Luque RM, Park S, Kineman RD. Severity of the catabolic condition differentially modulates hypothalamic expression of growth hormone-releasing hormone in the fasted mouse: potential role of neuropeptide Y and corticotropin-releasing hormone. Endocrinology. 2007;148:300–309. doi: 10.1210/en.2006-0592. [DOI] [PubMed] [Google Scholar]
- 30.Luque RM, Huang ZH, Shah B, Mazzone T, Kineman RD. Effects of leptin replacement on hypothalamic-pituitary growth hormone axis function and circulating ghrelin levels in ob/ob mice. Am J Physiol Endocrinol Metab. 2007;292:E891–E899. doi: 10.1152/ajpendo.00258.2006. [DOI] [PubMed] [Google Scholar]
- 31.Luque RM, Kineman RD. Impact of obesity on the growth hormone axis: evidence for a direct inhibitory effect of hyperinsulinemia on pituitary function. Endocrinology. 2006;147:2754–2763. doi: 10.1210/en.2005-1549. [DOI] [PubMed] [Google Scholar]
- 32.Luque RM, Gahete MD, Hochgeschwender U, Kineman RD. Evidence that endogenous SST inhibits ACTH and ghrelin expression by independent pathways. Am J Physiol Endocrinol Metab. 2006;291:E395–E403. doi: 10.1152/ajpendo.00038.2006. [DOI] [PubMed] [Google Scholar]
- 33.Luque RM, Kineman RD. Gender-dependent role of endogenous somatostatin in regulating growth hormone-axis function in mice. Endocrinology. 2007;148:5998–6006. doi: 10.1210/en.2007-0946. [DOI] [PubMed] [Google Scholar]
- 34.Luque RM, Soares BS, Peng XD, Krishnan S, Cordoba-Chacon J, Frohman LA, Kineman RD. Use of the metallothionein promoter-human growth hormone-releasing hormone (GHRH) mouse to identify regulatory pathways that suppress pituitary somatotrope hyperplasia and adenoma formation due to GHRH-receptor hyperactivation. Endocrinology. 2009;150:3177–3185. doi: 10.1210/en.2008-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez-Fuentes AJ, Moreno-Fernandez J, Vazquez-Martinez R, Duran-Prado M, de la Riva A, Tena-Sempere M, Dieguez C, Jimenez-Reina L, Webb SM, Pumar A, Leal-Cerro A, Benito-Lopez P, Malagon MM, Castaño JP. Ghrelin is produced by and directly activates corticotrope cells from adrenocorticotropin-secreting adenomas. J Clin Endocrinol Metab. 2006;91:2225–2231. doi: 10.1210/jc.2006-0235. [DOI] [PubMed] [Google Scholar]
- 36.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 37.Duran-Prado M, Bucharles C, Gonzalez BJ, Vazquez-Martinez R, Martinez-Fuentes AJ, Garcia-Navarro S, Rhodes SJ, Vaudry H, Malagon MM, Castaño JP. Porcine somatostatin receptor 2 displays typical pharmacological sst2 features but unique dynamics of homodimerization and internalization. Endocrinology. 2007;148:411–421. doi: 10.1210/en.2006-0920. [DOI] [PubMed] [Google Scholar]
- 38.Belsham DD, Cai F, Cui H, Smukler SR, Salapatek AM, Shkreta L. Generation of a phenotypic array of hypothalamic neuronal cell models to study complex neuroendocrine disorders. Endocrinology. 2004;145:393–400. doi: 10.1210/en.2003-0946. [DOI] [PubMed] [Google Scholar]
- 39.Kineman RD, Gahete MD, Luque RM. Identification of a mouse ghrelin gene transcript that contains intron 2 and is regulated in the pituitary and hypothalamus in response to metabolic stress. J Mol Endocrinol. 2007;38:511–521. doi: 10.1677/JME-06-0026. [DOI] [PubMed] [Google Scholar]
- 40.Luque RM, Kineman RD, Tena-Sempere M. Regulation of hypothalamic expression of KiSS-1 and GPR54 genes by metabolic factors: analyses using mouse models and a cell line. Endocrinology. 2007;148:4601–4611. doi: 10.1210/en.2007-0500. [DOI] [PubMed] [Google Scholar]
- 41.Dalm VA, Hofland LJ, Lamberts SW. Future clinical prospects in somatostatin/cortistatin/somatostatin receptor field. Mol Cell Endocrinol. 2008;286:262–277. doi: 10.1016/j.mce.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 42.Hofland LJ. Somatostatin and somatostatin receptors in Cushing’s disease. Mol Cell Endocrinol. 2008;286:199–205. doi: 10.1016/j.mce.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 43.Lania A, Mantovani G, Spada A. Genetic abnormalities of somatostatin receptors in pituitary tumors. Mol Cell Endocrinol. 2008;286:180–186. doi: 10.1016/j.mce.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 44.Schonbrunn A. Selective agonism in somatostatin receptor signaling and regulation. Mol Cell Endocrinol. 2008;286:35–39. doi: 10.1016/j.mce.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siehler S. Cell-based assays in GPCR drug discovery. Biotechnol J. 2008;3:471–483. doi: 10.1002/biot.200800001. [DOI] [PubMed] [Google Scholar]
- 46.Kilpatrick GJ, Dautzenberg FM, Martin GR, Eglen RM. 7TM receptors: the splicing on the cake. Trends Pharmacol Sci. 1999;20:294–301. doi: 10.1016/S0165-6147(99)01355-3. [DOI] [PubMed] [Google Scholar]
- 47.Leung PK, Chow KB, Lau PN, Chu KM, Chan CB, Cheng CH, Wise H. The truncated ghrelin receptor polypeptide (GHS-R1b) acts as a dominant-negative mutant of the ghrelin receptor. Cell Signal. 2007;19:1011–1022. doi: 10.1016/j.cellsig.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 48.Hawrylyshyn KA, Michelotti GA, Coge F, Guenin SP, Schwinn DA. Update on human alpha1-adrenoceptor subtype signaling and genomic organization. Trends Pharmacol Sci. 2004;25:449–455. doi: 10.1016/j.tips.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 49.McWilliams DF, Watson SA, Crosbee DM, Michaeli D, Seth R. Coexpression of gastrin and gastrin receptors (CCK-B and delta CCK-B) in gastrointestinal tumour cell lines. Gut. 1998;42:795–798. doi: 10.1136/gut.42.6.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rogers J, Wall R. A mechanism for RNA splicing. Proc Natl Acad Sci USA. 1980;77:1877–1879. doi: 10.1073/pnas.77.4.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tallent M, Dichter MA, Reisine T. Evidence that a novel somatostatin receptor couples to an inward rectifier potassium current in AtT-20 cells. Neuroscience. 1996;73:855–864. doi: 10.1016/0306-4522(96)00079-6. [DOI] [PubMed] [Google Scholar]
- 52.Burset M, Seledtsov IA, Solovyev VV. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–4375. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murray JI, Voelker RB, Henscheid KL, Warf MB, Berglund JA. Identification of motifs that function in the splicing of non-canonical introns. Genome Biol. 2008;9:R97. doi: 10.1186/gb-2008-9-6-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 55.Smith DJ, Query CC, Konarska MM. “Nought may endure but mutability”: spliceosome dynamics and the regulation of splicing. Mol Cell. 2008;30:657–666. doi: 10.1016/j.molcel.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clark F, Thanaraj TA. Categorization and characterization of transcript-confirmed constitutively and alternatively spliced introns and exons from human. Hum Mol Genet. 2002;11:451–464. doi: 10.1093/hmg/11.4.451. [DOI] [PubMed] [Google Scholar]
- 57.Fehlmann D, Langenegger D, Schuepbach E, Siehler S, Feuerbach D, Hoyer D. Distribution and characterisation of somatostatin receptor mRNA and binding sites in the brain and periphery. J Physiol Paris. 2000;94:265–281. doi: 10.1016/S0928-4257(00)00208-4. [DOI] [PubMed] [Google Scholar]
- 58.Reubi JC, Waser B, Liu Q, Laissue JA, Schonbrunn A. Subcellular distribution of somatostatin sst2A receptors in human tumors of the nervous and neuroendocrine systems: membranous versus intracellular location. J Clin Endocrinol Metab. 2000;85:3882–3891. doi: 10.1210/jc.85.10.3882. [DOI] [PubMed] [Google Scholar]
- 59.Stroh T, Sarret P, Tannenbaum GS, Beaudet A. Immunohistochemical distribution and subcellular localization of the somatostatin receptor subtype 1 (sst1) in the rat hypothalamus. Neurochem Res. 2006;31:247–257. doi: 10.1007/s11064-005-9013-7. [DOI] [PubMed] [Google Scholar]
- 60.Hukovic N, Panetta R, Kumar U, Rocheville M, Patel YC. The cytoplasmic tail of the human somatostatin receptor type 5 is crucial for interaction with adenylyl cyclase and in mediating desensitization and internalization. J Biol Chem. 1998;273:21416–21422. doi: 10.1074/jbc.273.33.21416. [DOI] [PubMed] [Google Scholar]
- 61.Jacobs S, Schulz S. Intracellular trafficking of somatostatin receptors. Mol Cell Endocrinol. 2008;286:58–62. doi: 10.1016/j.mce.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 62.Tulipano G, Schulz S. Novel insights in somatostatin receptor physiology. Eur J Endocrinol. 2007;156(Suppl 1):S3–S11. doi: 10.1530/eje.1.02354. [DOI] [PubMed] [Google Scholar]
- 63.Claeysen S, Sebben M, Becamel C, Bockaert J, Dumuis A. Novel brain-specific 5-HT4 receptor splice variants show marked constitutive activity: role of the C-terminal intracellular domain. Mol Pharmacol. 1999;55:910–920. [PubMed] [Google Scholar]
- 64.Fagni L, Chavis P, Ango F, Bockaert J. Complex interactions between mGluRs, intracellular Ca2+ stores and ion channels in neurons. Trends Neurosci. 2000;23:80–88. doi: 10.1016/S0166-2236(99)01492-7. [DOI] [PubMed] [Google Scholar]
- 65.Namba T, Sugimoto Y, Negishi M, Irie A, Ushikubi F, Kakizuka A, Ito S, Ichikawa A, Narumiya S. Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature. 1993;365:166–170. doi: 10.1038/365166a0. [DOI] [PubMed] [Google Scholar]
- 66.Wente W, Stroh T, Beaudet A, Richter D, Kreienkamp HJ. Interactions with PDZ domain proteins PIST/GOPC and PDZK1 regulate intracellular sorting of the somatostatin receptor subtype 5. J Biol Chem. 2005;280:32419–32425. doi: 10.1074/jbc.M507198200. [DOI] [PubMed] [Google Scholar]
- 67.Peverelli E, Lania AG, Mantovani G, Beck-Peccoz P, Spada A. Characterization of intracellular signaling mediated by human somatostatin receptor 5: role of the DRY motif and the third intracellular loop. Endocrinology. 2009;150:3169–3176. doi: 10.1210/en.2008-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Akbar M, Okajima F, Tomura H, Majid MA, Yamada Y, Seino S, Kondo Y. Phospholipase C activation and Ca2+ mobilization by cloned human somatostatin receptor subtypes 1–5, in transfected COS-7 cells. FEBS Lett. 1994;348:192–196. doi: 10.1016/0014-5793(94)00603-2. [DOI] [PubMed] [Google Scholar]
- 69.Ben-Shlomo A, Wawrowsky KA, Proekt I, Wolkenfeld NM, Ren SG, Taylor J, Culler MD, Melmed S. Somatostatin receptor type 5 modulates somatostatin receptor type 2 regulation of adrenocorticotropin secretion. J Biol Chem. 2005;280:24011–24021. doi: 10.1074/jbc.M501998200. [DOI] [PubMed] [Google Scholar]
- 70.Carruthers AM, Warner AJ, Michel AD, Feniuk W, Humphrey PP. Activation of adenylate cyclase by human recombinant sst5 receptors expressed in CHO-K1 cells and involvement of Galphas proteins. Br J Pharmacol. 1999;126:1221–1229. doi: 10.1038/sj.bjp.0702401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cervia D, Zizzari P, Pavan B, Schuepbach E, Langenegger D, Hoyer D, Biondi C, Epelbaum J, Bagnoli P. Biological activity of somatostatin receptors in GC rat tumour somatotrophs: evidence with sst1-sst5 receptor-selective nonpeptidyl agonists. Neuropharmacology. 2003;44:672–685. doi: 10.1016/S0028-3908(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 72.Park S, Sohn S, Kineman RD. Fasting-induced changes in the hypothalamic-pituitary-GH axis in the absence of GH expression: lessons from the spontaneous dwarf rat. J Endocrinol. 2004;180:369–378. doi: 10.1677/joe.0.1800369. [DOI] [PubMed] [Google Scholar]
- 73.Ishikawa M, Mizobuchi M, Takahashi H, Bando H, Saito S. Somatostatin release as measured by in vivo microdialysis: circadian variation and effect of prolonged food deprivation. Brain Res. 1997;749:226–231. doi: 10.1016/S0006-8993(96)01163-8. [DOI] [PubMed] [Google Scholar]
- 74.Tannenbaum GS, Epelbaum J, Colle E, Brazeau P, Martin JB. Antiserum to somatostatin reverses starvation-induced inhibition of growth hormone but not insulin secretion. Endocrinology. 1978;102:1909–1914. doi: 10.1210/endo-102-6-1909. [DOI] [PubMed] [Google Scholar]
- 75.Zhou X, De Schepper J, Vergeylen A, Luis O, Delhase M, Hooghe-Peters EL. Cafeteria diet-induced obese rats have an increased somatostatin protein content and gene expression in the periventricular nucleus. J Endocrinol Invest. 1997;20:264–269. doi: 10.1007/BF03350298. [DOI] [PubMed] [Google Scholar]
- 76.Luque RM, Park S, Peng XD, Delgado E, Gracia-Navarro F, Kineman RD, Malagon MM, Castaño JP. Homologous and heterologous in vitro regulation of pig pituitary somatostatin receptor subtypes, sst1, sst2 and sst5 mRNA. J Mol Endocrinol. 2004;32:437–448. doi: 10.1677/jme.0.0320437. [DOI] [PubMed] [Google Scholar]
- 77.Ramirez JL, Mouchantaf R, Kumar U, Otero Corchon V, Rubinstein M, Low MJ, Patel YC. Brain somatostatin receptors are up-regulated in somatostatin-deficient mice. Mol Endocrinol. 2002;16:1951–1963. doi: 10.1210/me.2002-0068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.