

Abstract
目的
探讨不同程度肾损伤的Alport综合征(alport syndrome, AS)患者远期听力变化与特点。
方法
收集2007年1月至2022年9月确诊AS并完善听力学检查患者的肾脏病理、基因检测、听力检查等临床资料, 进行听力及肾功能的远期随访。
结果
研究纳入AS患者70例, 随访到33例, 失访率52.9%, 随访时间1.1~15.8年, 16例患者随访时间>10年。随访患者男25例, 女8例, 随访年龄3.4~27.8岁。10例确诊AS时听力异常者随访期间均出现进行性听力下降, 随访新增听力异常者3例, 于病程5~6年时出现, 均为感音神经性耳聋, 随访13例听力下降患者中仅3例接受助听器治疗。7例发展为终末期肾病(end-stage renal disease, ESRD), 男性多见(6/7), ESRD组与非ESRD组远期听力损失率比较差异有统计学意义(P=0.013), 肾病进展变化与远期听力水平无相关(P>0.05)。28例完善肾脏活检, 均见基底膜厚薄不均及不同程度足细胞病变, 足细胞病变严重程度与远期听力损失率相关(P=0.048), 与听力损失严重程度无关(P>0.05)。11例进行基因检测, COL4A5 突变多见(8/11), 突变类型与听力表型无明显相关(P>0.05)。
结论
AS患者听力可进行性下降, 远期听力进展异质性较高, 在病程5~6年时出现听力下降的概率高; 听力异常与肾病状态、肾组织病理改变及突变基因密切相关, 需要重视远期听力随访并尽早听力干预。
Keywords: Alport综合征, 儿童, 远期随访, 听力变化
Abstract
Objective
To investigate long-term auditory changes and characteristics of Alport syndrome (AS) patients with different degrees of renal injury.
Methods
Retrospectively analyzing clinical data of patients diagnosed AS from January 2007 to September 2022, including renal pathology, genetic detection and hearing examination.A long-term follow-up focusing on hearing and renal function was conducted.
Results
This study included 70 AS patients, of which 33(25 males, 8 females, aged 3.4-27.8 years) were followed up, resulting in a loss rate of 52.9%.The follow-up period ranged from 1.1 to 15.8 years, with 16 patients followed-up for over 10 years.During the follow-up, 10 patients presenting with hearing abnormalities at the time of diagnosis of AS had progressive hearing loss, and 3 patients with new hearing abnormalities were followed up, which appeared at 5-6 years of disease course.All of which were sensorineural deafness.While only 3 patients with hearing abnormalities among 13 patients received hearing aid intervention.Of these patients, 7 developed end-stage renal disease (ESRD), predominantly males (6/7).The rate of long-term hearing loss was significantly different between ESRD group and non-ESRD group (P=0.013).There was no correlation between the progression of renal disease and long-term hearing level (P>0.05).kidney biopsies from 28 patients revealed varying degrees of podocyte lesion and uneven thickness of basement membrane.The severity of podocyte lesion was correlated with the rate of long-term hearing loss (P=0.048), and there was no correlation with the severity of hearing loss (P>0.05).Among 11 cases, the COL4A5 mutationwas most common (8 out of 11), but there was no significant correlation between the mutation type and hearing phenotype (P>0.05).
Conclusion
AS patients exhibit progressive hearing loss with significant heterogeneity over the long-term..THearing loss is more likely to occur 5-6 years into the disease course.Hearing abnormalities are closely related to renal disease status, kidney tissue pathology, and gene mutations, emphasizing the need for vigilant long-term hearing follow-up and early intervention.
Keywords: alport syndrome, children, long-term monitoring, hearing changes
遗传性感音神经性聋人群中,Alport综合征(alport syndrome,AS)是其较为常见的病因。2018年AS列入我国《第一批罕见病目录》[1],是一种以血尿、蛋白尿、进行性肾功能减退为特征,部分患者合并感音神经性耳聋、眼部病变等肾外表现的综合征,发病率约为1 /5 000[2]。AS由编码Ⅳ型胶原α3α4α5链的 COL4A3/4/5 基因突变引起,X连锁遗传最为常见(85%),常染色体显性遗传和常染色体隐性遗传少见(15%)。听力损害是AS常见的肾外症状[3],既往研究发现听力受损可能是肾衰竭的危险因素。但儿童期确诊AS的患者远期听力情况及其与病理及肾病进展的关系少有文献报道,远期听力损失的变化程度也常因基因和表观遗传等因素影响表现出较大的表型异质性。本研究纳入儿童时期确诊的AS患者,进行远期听力随访监测,并基于不同程度肾损伤分析AS患者的听力预后。
1. 资料与方法
1.1. 研究对象
检索2007年1月至2022年9月就诊于我院,确诊AS并具有完整听力学资料的患者70例,所有患者均于2023年9月统一进行随访。远期随访定义为随访时间在确诊AS后1年以上,本研究随访的患者均为病程>1年,共随访到患者33例。收集33例随访患者就诊时的完整临床资料及随访结果,包括姓名、性别、就诊年龄、明确诊断年龄、听力损失的年龄、听力学检查、耳鼻喉专科体检、肾功能检查、肾脏活检、有无视力损害、基因检测结果;随访听力检查、听力变化、肾功能变化等。
AS的诊断标准[4]:临床主要表现为持续性肾小球源性血尿或血尿伴蛋白尿的患者,具有以下任一项即可诊断为AS:①肾组织电镜示肾小球基底膜致密层撕裂、分层、薄厚不均或篮网状改变;②肾脏组织基底膜Ⅳ型胶原α3α4α5链免疫荧光染色异常或皮肤基底膜Ⅳ型胶原α5链免疫荧光染色异常;③基因检测示 COL4A3 、 COL4A4 或 COL4A5 基因具有致病性变异。本研究已通过首都医科大学附属北京儿童医院医学伦理委员会审批,已获得入组患者家长/监护人知情同意。
1.2. 听力检查
所有纳入研究患者均在住院时已接受常规耳鼻喉专科查体,以排除外中耳疾病;并进行纯音测听检查及声导抗检查,了解听力损失程度。
听力学诊断标准:根据世卫组织(2021)听力损失程度分级标准[5],取500、1 000、2 000、4 000 Hz四个频率的平均听阈将听力损失分为轻度(20~<35 dB)、中度(35~<50 dB)、中重度(50~<65 dB)、重度(65~<80 dB)、极重度(80~<95 dB)、全聋(≥95 dB)。本研究依据患者听力损伤程度,将听力检查结果分为轻度、中度、中重度及以上组。
1.3. 肾脏活检
全麻下经皮肾脏穿刺活检,取材、固定、脱水、浸透、包埋、切片、染色等步骤处理标本,电镜下观察肾小管、肾小球的结构、基底膜厚度纤维丝及微管、肾小管上皮细胞内特殊包涵体、肾小管基底膜的电子致密物沉积等,了解肾脏的病理改变和病理类型。
足突融合是足细胞损伤的显著特征之一,目前无具体量化标准判定电镜下足细胞的损伤程度,本研究以单个肾小球足细胞足突融合情况进行分组描述足细胞的损伤程度以量化肾脏损伤情况:无融合、小部分融合、大部分融合、广泛融合组[6-8]。
1.4. 基因检测
采集患者及其直系亲属外周血,利用高通量测序方法检测目标基因中是否存在基因突变(全外显子测序/Panel),对患者直系亲属进行Sanger验证。
1.5. 随访方案
横断面随访我院病历系统中检索到的具有完整病历资料及听力学检查数据的70例AS患者,随访患者均为病程>1年。随访内容包括患者听力下降时间、近3个月听力程度及近3个月肾功能变化情况。
听力随访:随访过程中采取两种方案:①于耳鼻喉科复诊时完成纯音测听检查,随访医生收集并分析患者听力检查结果;②由患者及家属共同完成听力随访问卷(问卷参考2021世界听力报告,多数患者在安静和噪声环境下的听力体验),由随访医生评估患者听力损失程度。
肾功能随访:本研究依据随访时患者的肾功能情况分为两组:终末期肾病(end stage renal disease,ESRD)组和非终末期肾病组(非ESRD)组。ESRD的定义[9]为肾小球滤过率(GFR) < 1.50/1.73 m2或接受肾脏替代治疗(血液透析、腹膜透析、肾移植)。
1.6. 统计学处理
采用SPSS 25.0统计软件进行数据分析。分类变量采用频数、百分比进行统计描述,分类变量比较采用χ2检验,超过25%单元格的论理频数 < 5时,采用Fisher精确检验;P < 0.05为差异有统计学意义。
2. 结果
2.1. 一般资料
本研究纳入AS患者70例,共随访33例,失访率为52.9%。随访患者中男25例,女8例;确诊年龄2.3~15.6岁,平均8.8岁;伴随眼部病变10例,初始听力异常者10例(30.3%)。所有患者住院治疗时均未发生终末期肾病28例,完成肾脏活检,11例完善基因检测。随访间隔时间为1.1~15.8年,16例患者随访时间>10年。截至随访日期,患者年龄为3.4~27.8岁,平均17.1岁;随访听力异常者新增3例;7例发展为终末期肾病,其中6例接受替代治疗,1例因肾衰竭死亡。
2.2. 听力随访结果
随访确诊AS合并听力异常患者13例,均为感音神经性听力下降。13例患者听力受损程度见图 1。其中,5例随访时间 < 5年,2例随访时间为5~10年,6例随访时间>10年。10例确诊AS时伴有听力异常者出现进行性听力下降,3例确诊AS时听力正常,患者随访时间>10年,均于病程5~6年出现不同程度双侧对称性听力下降。仅3例患者接受双侧助听器辅助治疗,助听下可正常交流。
图 1.
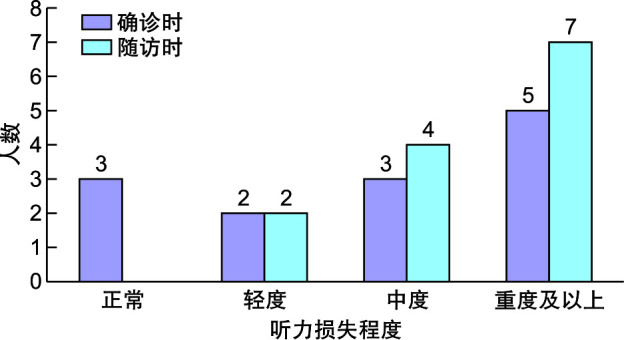
Alport综合征患者确诊肾病及随访时听力损失程度
2.3. 听力变化与ESRD
随访患者中有7例发展为ESRD,男6例,女1例。6例患者接受透析或肾移植治疗,1例因肾衰竭去世。确诊AS时即出现听力异常者6例,远期随访(3~11年)听力均出现进行性听力下降(随访8年者听力曲线,见图 2),仅1例患者接受了助听器辅助治疗;病情发展为ESRD但听力于确诊AS时及远期随诊时均正常者1例。ESRD组与非ESRD组远期听力损失率比较差异有统计学意义(P=0.013),但两组间听力损失程度差异无统计学意义(P>0.05),见表 1。
图 2.
接受肾移植患者(随访时间8年)听力前后比较
确诊AS时听力曲线(蓝色);随访时听力曲线(红色)。
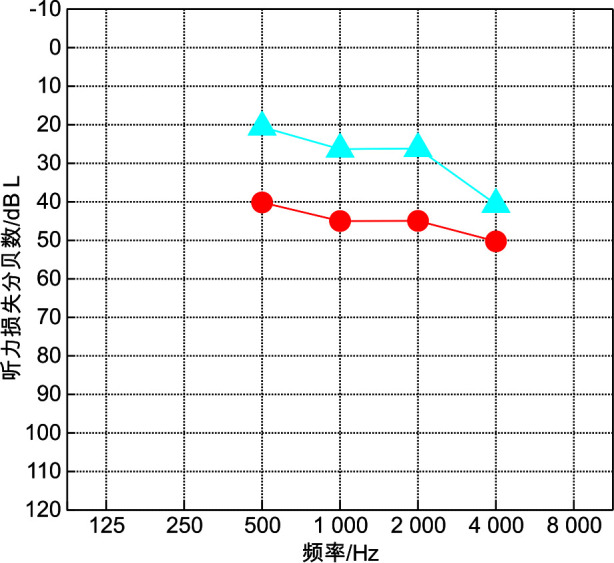
表 1.
ESRD组和非ESRD组听力损失情况比较 例
| 听力程度 | ESRD组 | 非ESRD组 |
| 听力正常者 | 1 | 20 |
| 轻度 | 0 | 2 |
| 中度 | 2 | 3 |
| 中重度及以上 | 4 | 3 |
2.4. 听力变化与肾脏病理
28例患者就诊时完善肾脏病理活检并进行电镜检查。所有患者均表现为基底膜厚薄不均、密度不均、断裂分层或篮网状改变;均有不同程度足细胞病变,见图 3。随访患者中,足细胞病变越严重,远期听力损失患者的数量越多,差异有统计学意义(P=0.048);足细胞病变严重程度与听力损失程度间未发现相关性(P>0.05),见表 2。
图 3.
电镜下肾小球基底膜及足细胞改变
基底膜厚薄不均(绿色箭头);足细胞不同程度融合(红色箭头)。
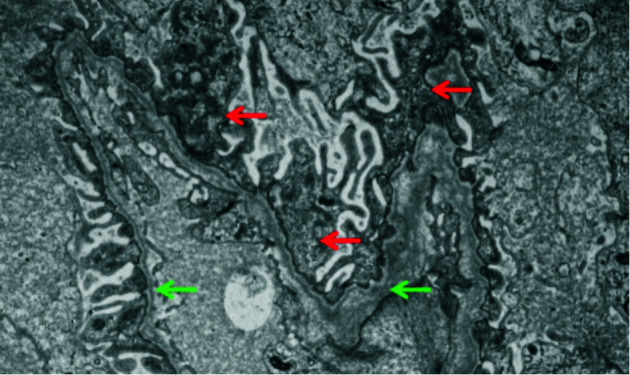
表 2.
不同程度足突病变听力损失情况比较 例
| 听力程度 | 电镜足突病变 | |||
| 无融合 | 小部分融合 | 大部分融合 | 广泛融合 | |
| 听力正常 | 0 | 7 | 7 | 3 |
| 轻度 | 0 | 1 | 0 | 1 |
| 中度 | 0 | 0 | 1 | 3 |
| 中重度及以上 | 0 | 1 | 1 | 3 |
2.5. 听力变化与基因结果
随访患者中11例患者进行了基因检测,检出两种Alport综合征的致病基因: COL4A3 、 COL4A5 ,未发现 COL4A4 突变基因的患者。获得4例患者及家属Sanger验证结果,3例为X连锁遗传(1例为全外显子测序;2例为Panel),1例为常染色体隐性遗传(Panel),见图 4。不同突变基因间未发现听力表型及听力严重程度的差异(P>0.05),见表 3。其中有2例患者为 COL4A5 致病基因突变,突变位点及遗传方式相同,但表现为不同听力表型,见表 4。
图 4.
Alport综合征家系图
a:X连锁遗传;b:常染色体隐性遗传。
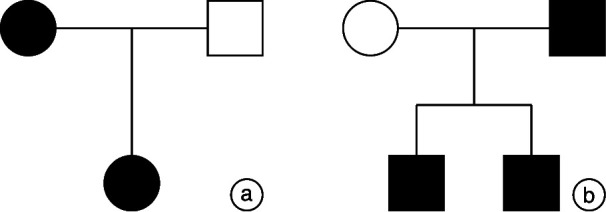
表 3.
不同突变基因类型听力损失情况及严重程度比较 例
| 听力程度 | 突变基因类型 | ||
| COL4A3 | COL4A4 | COL4A5 | |
| 正常 | 2 | 0 | 4 |
| 轻度 | 0 | 0 | 0 |
| 中度 | 1 | 0 | 3 |
| 中重度及以上 | 0 | 0 | 1 |
表 4.
两个COL4A5基因突变患者听力比较
| 病例 | 突变基因 | 突变位点 | 遗传方式 | 初始听力 | 随访听力 |
| 患者1 | COL4A5 | c.2060G>A p.Gly687Glu | X连锁遗传 | 右45/左35 | 中度 |
| 患者2 | COL4A5 | c.2060G>A p.Gly687Glu | X连锁遗传 | 右15/左15 | 正常 |
3. 讨论
肾脏、耳蜗和眼底基底膜结构的重要组成成分是IV型胶原蛋白,由α链组装成3种不同的异源性三聚体(αlαlα2,α3α4α5和α5α6)构成,其中α3α4α5三聚体是组织特异性的关键部分[10]。编码α3α4α5链的 COL4A3 、 COL4A4 和 COL4A5 基因突变引起结构扭曲、链条终止降解等导致AS。因此,患有AS的患者会基于组织特异性的胶原蛋白结构出现除肾脏损伤外的组织受累,在常见的肾外表现中以耳部病变最为多见[11]。
AS导致的听力损失在既往研究中多被描述为后天性,患者新生儿听力筛查通过,通常在儿童晚期或青少年早期通过听力检测首次发现,表现为双侧中高频听力进行性下降[3]。本研究随访患者中出现听力损失均于肾脏病变时或之后发现,通过对患者进行远期随访,且16例随访时间超过10年,听力损失的发生率从最初确诊AS的30.3%发展到随访时的39.4%,表现为进行性下降的特点。一项将听力损失作为次要研究终点的临床试验,在长达6年的时间跨度里随访了51例AS患儿,同样发现听力损失的患儿人数从研究开始时的10%增加到研究结束时的18%[12]。两个研究均证实AS听力进行性发展的特点,但听力损失的发生率差异性较大。AS听力损失的发生率10%~83%不等,取决于肾脏病变、基因突变类型及遗传方式等[9],因此,需要综合评估以判断远期听力预后情况。
Fátima等[3]回顾AS的临床表现,认为听力变化是肾病进展预后的关键因素;Boeckhaus等[12]在药物临床试验中发现听力损失儿童的尿蛋白含量较高,即听力损失与肾功能更快丧失有关。本研究随访患者中,发生ESRD者中85.7%(6/7)为听力异常患者,ESRD组听力损失发生率较非ESRD组更高,同样提示肾脏病变和听力下降的相关性。听力和肾病的同步进展,除Ⅳ型胶原蛋白结构受基因控制外,Moon等[9]认为可能与长期肾脏疾病发展出现后遗症有关,长期使用肾病药物产生一定耳毒性,肾功能改变使得离子代谢异常内耳功能障碍,免疫力减弱病毒感染等。所有发生ESRD患者中,85.7%(6/7)为男性,中重度听力损失多见,仅1例女性,远期听力表现为中度听力下降。本研究发生ESRD患者进行基因检测3例,均为X连锁遗传型,既往文献报道,大部分AS患者为X连锁遗传型,女性存在X染色体的随机失活及2条X染色体的互补作用,X连锁遗传型女性患者表型普遍轻于男性患者,进展至ESRD的风险低于男性[13]。
本研究也发现不同肾脏病理改变,均在疾病进展过程中出现不同程度的听力异常,足细胞病变越重,听力损失患者比例越高。其中,3例患者在最初就诊时听力正常,随访时发现均在病程的第5~6年出现听力下降,提示延长时间跨度进行听力随访的必要性。但肾脏足细胞病变严重程度,与远期听力下降程度间并未出现明显相关性。考虑可能原因为随访过程中患者失访率较高,远期听力随访方式欠精准,需进一步扩大样本量,加强对AS患者的长期随访,获得完整数据集[12]。随访困难同样也反映出患者及家属对耳部病变的重视程度较低,本研究随访的33例患者中,听力损失的13例患者均存在不同程度的交流困难,但仅3例进行了听力干预。其干预覆盖非常有限,而进行干预则可有效提高患者的生活质量。有研究认为获得明确的基因诊断及了解听力损失对生活质量的影响可能会增加父母对听力干预的接受程度,并影响干预决策[8]。笔者提倡在治疗肾脏病变的同时,需要加强肾病科医师及患者对AS听力受损的关注度,定期随访听力并及时进行听力干预[14]。
本研究共获得11例患者的基因数据,8例为 COL4A5 基因突变,3例为 COL4A3 基因突变,与以往数据一致,Morinière等[15]发现 COL4A5 基因突变占所有基因突变的80%~85%。但两种基因型的远期听力随访异常患者的比例并未发现明显统计学差异。考虑与样本量较小且随访依从性低有关,可扩大样本量后进行验证。本研究中有2例患者为 COL4A5 基因相同位点的突变,1例为中度听力损失,1例听力正常。既往研究也发现即使相同基因突变,耳部临床表现差异仍较大,需与突变位点、突变方式、突变类型、遗传方式等结合进行综合评价。据报道,AS的临床表型也受表观遗传学控制,即在DNA序列没有改变的情况下,通过DNA甲基化、基因印迹、X染色体失活等多种方式在疾病进程中控制了表观遗传的沉默,使得基因功能发生遗传学改变[16-17]。曾有动物实验数据表明,噪声暴露可能是解释人类听觉表型可变性的因素[18],提示环境因素也是造成临床表型差异的因素之一。
AS患者耳部病变表现为进行性听力下降,且多在病程中后期进展,病程5~6年时出现听力下降的概率高。因此要更加重视远期听力随访的必要性。听力远期变化程度与肾病变化、病理改变及突变基因密切相关,需进一步纳入大量病例研究及动物实验明确听力临床表型的远期特点及可能影响因素。
Funding Statement
国家呼吸系统疾病临床医学研究中心呼吸专项(No:HXZX-20210201);首都医科大学教育教学改革研究课题(No:2022JYY268);首都医科大学教育教学改革研究课题(No:2022JYZ026);首都医科大学国自然培育基金(No:PYZ21141)
Footnotes
利益冲突 所有作者均声明不存在利益冲突
References
- 1.伍 琳琳, 刘 栋, 董 宁宁. Alport综合征的药物研发进展. 肾脏病与透析肾移植杂志. 2023;32(3):270–275. [Google Scholar]
- 2.Nozu K, Nakanishi K, Abe Y, et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin Exp Nephrol. 2019;23(2):158–168. doi: 10.1007/s10157-018-1629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alves FR, de A Quintanilha Ribeiro F. Revision about hearing loss in the Alport's syndrome, analyzing the clinical, genetic and bio-molecular aspects. Braz J Otorhinolaryngol. 2005;71(6):813–819. doi: 10.1016/S1808-8694(15)31253-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alport综合征协作组, 国家肾脏疾病临床医学研究中心, 北京医学会罕见病分会 Alport综合征诊治专家共识(2023版) 中华医学杂志. 2023;103(20):1507–1525. [Google Scholar]
- 5.Chadha S, Kamenov K, Cieza A. The world report on hearing, 2021. Bulletin of the World Health Organization. 2021;99(4):242–242A. doi: 10.2471/BLT.21.285643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.任 献国, 张 沛, 杜 丽芳, et al. 儿童紫癜性肾炎足突融合与肾脏病理分级相关性研究. 中国实用儿科杂志. 2013;28(7):512–515. [Google Scholar]
- 7.倪 军, 殷 佳珍, 朱 斌, et al. Alport综合征听力异常与肾损伤及其预后相关性分析. 中国实用内科杂志. 2014;34(3):285–288. [Google Scholar]
- 8.张 娇, 关 静, 王 秋菊. 儿童听力损失的遗传学研究进展. 临床耳鼻咽喉头颈外科杂志. 2023;37(3):181–185. doi: 10.13201/j.issn.2096-7993.2023.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon IS, Bang MY, Shim DB, et al. Severe to profound hearing loss in patients with progressed Alport's syndrome. Acta Otolaryngol. 2009;129(9):982–987. doi: 10.1080/00016480802545588. [DOI] [PubMed] [Google Scholar]
- 10.Kalluri R, Shield CF, Todd P, et al. Isoform switching of type Ⅳ collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest. 1997;99(10):2470–2478. doi: 10.1172/JCI119431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.郑 琦敏, 顾 向晨, 谢 静远. 奥尔波特综合征致病机制及新治疗方法的研究进展. 中华肾脏病杂志. 2023;9(9):716–721. [Google Scholar]
- 12.Boeckhaus J, Strenzke N, Storz C, et al. Characterization of Sensorineural Hearing Loss in Children with Alport Syndrome. Life(Basel) 2020;10(12):360. doi: 10.3390/life10120360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamamura T, Nozu K, Fu XJ, et al. Natural History and Genotype-Phenotype Correlation in Female X-Linked Alport Syndrome. Kidney Int Rep. 2017;2(5):850–855. doi: 10.1016/j.ekir.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kashtan CE, Gross O. Correction to: Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. 2021;36(3):731. doi: 10.1007/s00467-020-04892-x. [DOI] [PubMed] [Google Scholar]
- 15.Morinière V, Dahan K, Hilbert P, et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J Am Soc Nephrol. 2014;25(12):2740–2751. doi: 10.1681/ASN.2013080912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.张 宏文. X连锁Alport综合征女性患者临床表型差异的可能机制. 临床儿科杂志. 2011;29(2):189–191. [Google Scholar]
- 17.Egger G, Liang G, Aparicio A, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 18.Meehan DT, Delimont D, Dufek B, et al. Endothelin-1 mediated induction of extracellular matrix genes in strial marginal cells underlies strial pathology in Alport mice. Hear Res. 2016;341:100–108. doi: 10.1016/j.heares.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]