

Abstract
Microbes entrenched within biofilms can withstand 1000-fold higher concentrations of antibiotics, in part due to the viscous extracellular matrix that sequesters and attenuates antimicrobial activity. Nanoparticle (NP)-based therapeutics can aid in delivering higher local concentrations throughout biofilms as compared to free drugs alone, thereby enhancing efficacy. Canonical design criteria dictate that positively charged nanoparticles can multivalently bind to anionic biofilm components and increase biofilm penetration. However, cationic particles are toxic and rapidly cleared from circulation in vivo, limiting their use. Therefore, we sought to design pH-responsive NPs that change their surface charge from negative to positive in response to the reduced biofilm pH microenvironment. We synthesized a family of pH-dependent, hydrolyzable polymers and employed the layer-by-layer (LbL) electrostatic assembly method to fabricate biocompatible NPs with these polymers as the outermost surface. NP charge conversion rate, dictated by polymer hydrophilicity and side chain structure, ranged from hours to undetectable within the experimental timeframe. LbL NPs with increasingly fast charge conversion rate more effectively penetrated through, and accumulated throughout, wildtype (PAO1) and mutant overexpressing biomass (ΔwspF) P. aeruginosa biofilms. Finally, tobramycin, an antibiotic known to be trapped by anionic biofilm components, was loaded into the final layer of the LbL NP. There was a 3.2-fold reduction in ΔwspF colony forming units for the fastest charge-converting NP as compared to both the slowest charge-converter and free tobramycin. These studies provide a framework for the design of biofilm-penetrating NPs that respond to matrix interactions, ultimately increasing the efficacious delivery of antimicrobials.
Keywords: biofilm, nanoparticle, stimulus-responsive, liposome, drug-delivery
Graphical Abstract
Introduction
Bacterial infections affect 17 million people, cause approximately 550,000 deaths, and cost the United States healthcare system billions of dollars each year.1 Up to 80% of these infections are caused by bacteria that form biofilms.2 Biofilms are micro-aggregates of immobile, sessile bacteria encased within viscous extracellular polymeric substances (EPS),3 thereby protecting them from external stresses such as desiccation, host immune defenses, and exogenous antimicrobials. The EPS can render antibiotics ineffective, ultimately requiring up to 1000-fold higher concentrations of drug in order to eradicate biofilm infections.4-6 During treatment, ensuring a high therapeutic concentration throughout the biofilm matrix is paramount; sublethal drug doses stress microbes and favor the development of antimicrobial resistant strains.7 With the rise of antimicrobial resistance threatening to cause 10 million deaths globally by 2050,8 it is critical to find alternate solutions for effective antibiotic delivery throughout biofilms.
The EPS is a key biological barrier to antimicrobial eradication of biofilms, as it can comprise up to 90% dry weight of a biofilm.3 The EPS is primarily comprised of polysaccharides, proteins, lipids, and extracellular DNA. It establishes various gradients across the biofilm, such as those in metabolic activity, oxygen, and pH. Furthermore, it acts as a physical barrier, attenuating antibiotic penetration due to its mechanical and physicochemical properties.9-14 Pioneering work by Tseng et. al.10 demonstrated how cationic antibiotics, such as tobramycin, are immediately electrostatically sequestered at the biofilm interface by anionic components of the EPS and unable to penetrate further. Designing antimicrobial carriers to overcome the diffusion restrictions caused by the EPS will enhance therapeutic alternatives for biofilm infections.
Nanoparticles (NPs) can help overcome the biofilm barrier15 by efficiently encapsulating antibiotics,16-18 protecting their cargo from degradation,19 and delivering high local concentrations at targeted tissue sites.20 Indeed, in vitro and in vivo biofilm studies demonstrated that NP-encapsulated therapies outperform their free drug counterpart in eliminating biofilm biomass and reducing bacterial viability.21-23 Importantly, NP surface chemistry is a main player in governing the interactions at the biofilm-NP interface and is critical in the design of NPs as drug carriers. A key structure-function relationship has been identified for the migration of NPs within the biofilm.24-26 Cationic, positively charged NP surfaces are superior compared to neutral or anionic NPs at penetrating the local negatively charged microenvironments of the matrix via a ‘catch-and-release’ manner,26-29 in which multivalent charge interactions facilitate reversible binding. However, cationic NPs are toxic30, 31 and are rapidly cleared32-34 when administered systemically, disadvantaging their use in the clinic for the treatment of biofilm infections.
Stimuli-responsive, and more specifically pH-responsive, systems represent a promising strategy for the delivery and release of therapeutics in a site-selective manner.35 While the pH of blood is close to 7.4, that in the biofilm microenvironment is reported as being as low as pH 5.36, 37 Therefore, the inclusion of an anionic pH-responsive surface may mitigate the orthogonal design criteria of biocompatibility and positive surface charge.38 To incorporate a pH-responsive feature onto the surface of NPs, we used the layer-by-layer (LbL) self-assembly technique, a versatile fabrication method used to develop multifunctional drug delivery nanocarriers.39-41 LbL assembly involves the facile adsorption of polyelectrolyte chains onto an oppositely charged colloidal template to form a multilayered structure.42 This modular assembly permits investigation into how precise polymer differences can influence NP surface properties,43 as well as biofilm-NP interactions.
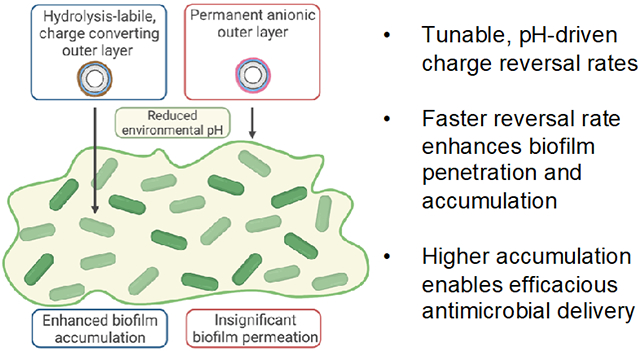
In this work, we generated four distinct polymers characterized by differing backbone hydrophilicity and side-chain flexibility, which led to distinct and predictable charge-reversal rates. We hypothesized that the reversal rate determines biofilm penetration and permeation (Figure 1). Furthermore, by loading NP surfaces with tobramycin, a clinically relevant antibiotic known to be attenuated by the biofilm matrix,10 we hypothesized that the increased NP accumulation due to surface charge reversal rates would lead to more efficacious drug delivery. Overall, this study lays a foundational structure-function model for the future design of pH-responsive nanocarriers to overcome the biofilm matrix and eliminate biofilm-based infections.
Figure 1.

Schematic illustration of charge-reversing nanoparticles (NPs) disseminating throughout biofilms. A reduced pH gradient across the biofilm catalyzes the hydrolysis (dashed red lines) of charge-converting polymers, thereby creating a positively-charged NP surface that can penetrate the biofilm matrix. However, permanently negative NP surfaces have negligible biofilm penetration due to electrostatic repulsion by the biofilm matrix.
Materials and Methods
Materials
All materials were purchased from MilliporeSigma unless otherwise specified. All salt solutions, such as sodium chloride (NaCl, Macron Fine Chemicals) sodium hydroxide (NaOH), calcium chloride dihydrate (CaCl2), magnesium chloride (MgCl2), and sodium bicarbonate (NaHCO3, Mallinckrodt Pharmaceuticals) were dissolved in de-ionized, MilliQ water. Acids and bases, such as sodium hydroxide (NaOH, 1 N, J.T.Baker) and hydrochloric acid (HCl, 6 N, VWR) were used for pH titration unless otherwise stated. Chloroform (TCI America), methanol, and MilliQ water were used to dissolve lipids and cholesterol. All lipids and cholesterol were purchased from Avanti Polar Lipids. 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) was resuspended at 25 mg/mL in chloroform; 1,2-distearoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (sodium salt) (DSPG) was resuspended at 25 mg/mL in a 65:35 chloroform:methanol volumetric ratio; 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DPSE) was dissolved at 5 mg/mL in the same chloroform:methanol mixture; and plant-based cholesterol was dissolved in 50 mg/mL chloroform. Tangential flow filtration (TFF) membranes D02-E100-05-N and C02-E100-05-N were purchased from Spectrum Labs, now incorporated with Repligen. Agar plates were made by dissolving 32g of LB agar into 1 L of MilliQ water, autoclaving, and pouring while still warm.
Charge-converting polymer synthesis
Reaction conditions were taken from literature44 and modified as follows (Figure S1). Poly(allylamine) hydrochloride (PAH, 17.5 kDa) or poly-L-lysine hydrochloride (PLK, 16 kDa, Alamanda Polymers) (100 mg) was dissolved in 3 mL basic MilliQ water, made basic by adding 200 μL of 1 N NaOH while stirring. Citraconic anhydride (CIT, 400 μL) or maleic anhydride (MAL, 400 mg) was added at roughly 2 molar equivalences of cationic polymer primary amines, and 1 N NaOH was added as needed to ensure the reaction was kept at basic conditions (i.e. a pH above 8). For the PAH-CIT-MAL reaction, a 1:1:1 molar equivalence of polymer and each reactant was used. The reaction was kept stirring overnight, and the resulting product was dialyzed (3.5 kD MW cutoff, Spectrum Chemical) against 1000 mL of basic MilliQ water, adjusted with 1 mL of 1N NaOH. The dialysate was replaced after 4, 24, and 48 hours. The resulting purified product was lyophilized and structures were confirmed via 1H nuclear magnetic resonance (NMR) by resuspending in D2O (99.9% pure, Cambridge Isotopes Lab) and running on a 500 MHz Bruker Avance Neo spectrometer.
Nanoparticle preparation
Lipids (DSPC, DSPG, and DSPE for dye conjugation when necessary) and cholesterol were dissolved in their previously described organic solvents and added to a cleaned, oven-dry 50 mL round bottom flask (VWR). Liposomes were always synthesized in a 33:33:34 DSPC:DSPG:cholesterol molar ratio, unless otherwise stated. Thin films were created on the walls of the round bottom flask by evaporating the solvent using a Buchi RotoVap system under heat (> 50 °C) and reduced pressure (< 30 mbar) for at least 1 hour. Dried films were rehydrated in the round bottom flask submerged in a Branson sonicator bath heated to 65 °C by adding a volume of MilliQ water which equaled the total number of milligrams of lipids and cholesterol used, so that the nanoparticle concentration was 1 mg/mL. The rehydrated lipids were sonicated on and off for one minute each and for three total cycles. Afterward, the multilamellar liposomes were extruded through an Avestin LiposoFast LF-50 liposome extruder, complete with a heated sample jacket connected to a Cole-Parmer Polystat Heated Recirculator Bath to maintain a temperate at 65 °C (i.e., above the secondary transition temperature of the lipid mixture). The liposomes were passed through stacked 400 and 200 nm filters three times, then through a 100 nm filter three times, and finally through a 50 nm filter at least once.
Liposomes that were used for downstream fluorescent assays followed the same procedure as described above, except with the following altered lipid composition (31:31:31:7 DSPC:DSPG:cholesterol:DSPE molar ratio), and reacted with NHS ester-linked sulfo-cyanine 5 dye (Cy5, Lumiprobe) in 20 mM sodium bicarbonate with a pH > 8.4 adjusted with 1 N NaOH, as measured with a Hanna electronic pH-meter. The cyanine dye was reacted at > 5-fold molar excess as compared to the primary amine on DSPE. The reaction was stirred overnight in the dark at room temperature and run to completion.
Nanoparticle layering and purification
All nanoparticles were characterized via dynamic light scattering (DLS) using a Malvern ZS90 Particle analyzer, in which 50 μL of sample was added to 700 μL of MilliQ water. Size measurements were run in polystyrene disposable cuvettes (Fisher Scientific) and zeta potential measurements were made using laser Doppler electrophoresis in folded capillary zeta cells (Malvern Panalytical). Data is reported as three independent technical replicates and their standard deviation.
All reported equivalence weight ratios for layering polymers were compared to 1 mg/mL nanoparticle core, where equal volumes of polymer in buffer were added to the nanoparticle core while sonicating for no more than 3-5 seconds. The buffer used for layering polymers onto the nanoparticle core consisted of 50 mM HEPES (1 M, Fisher Bioreagnets) and 40 mM NaCl, so that final the nanoparticle solution was 25 mM HEPES and 20 mM NaCl, as previously reported.45 At least three test weight ratios of polymer and NP were examined for stability before choosing the proper layering ratio. PLK was layered onto anionic NP cores at roughly 0.33 wt. eq., and synthesized polymers, as well as poly(acrylic acid), sodium salt (PAA, 15 kD, 35 wt. % in H2O), were layered at roughly 0.5-1.5 wt. eq, depending on the particle batch.
TFF was used to purify crude layered NPs, whereby the solution was connected to a KrosFlo II (Spectrum Laboratories) system using Teflon-coating tubing. Volumes < 8 mL were filtered through the C02-E100-05-N 100 kD membrane at a 13 mL/min flow rate, whereas larger volumes were filtered through D02-E100-05-N 100kD membranes at an 80 mL/min flow rate. For polycation layering, ionic interactions with the TFF membrane can cause stripping and destabilization, so the membrane was ‘pre-treated’ by keeping a closed loop for 5 minutes while just the polycation solution flowed at a concentration of roughly 0.1 mg/mL. All LbL NPs were washed with at least a 5X volume of MilliQ water, and concentrated down to the desired final concentration. The working concentration of nanoparticles was 1 mg/mL unless otherwise stated.
Purification of dye-conjugated NPs followed the same procedure, except that washing included 10X volume of phosphate-buffered saline (PBS, Lonza Biologics) and then at least 10X volume of water. Wastewater was checked for the removal of the Cy5 fluorophore via fluorescent readings. For subsequent experiments, normalization of nanoparticle mass was carried out based on the purified product and assumed zero nanoparticle loss, with a stock concentration of 1 mg/mL fluorescent nanoparticle.
Bacteria culture
Bacteria were taken from 20% frozen glycerol (VWR) stocks, plated onto pre-poured Luria-Bertani agar (LB, Invitrogen) in standard 100x15 mm plates (VWR), and incubated at 37 °C overnight. CaCl2 and MgCl2 were added to Mueller-Hinton broth (MHB) at 25 and 12.5 μg/mL, respectively, to complete the cation-adjusted Mueller-Hinton broth (CAMHB) recipe. Single colonies were picked, added to 2-3 mL of MHB in Falcon® 14 mL polystyrene round bottom tubes (Corning), and were grown shaking in a shaking incubator (Deville Scientific) at 250 rpm at 37 °C for 16-18 hours. New single colony stocks were picked every 5 days, and new plates were streaked every month. Pseudomonas aeruginosa PAO1 was purchased from ATCC, and the ΔwspF strain was kindly gifted by the Ribbeck Laboratory.
Cell culture and cellular metabolic activity
NIH3T3 mouse fibroblasts were purchased from ATCC and tested monthly for mycoplasma contamination using the Lonza MycoAlert kit (Lonza), which always tested negative. Cells were grown in DMEM medium (Corning) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin (Pen/Strep, Corning Inc.) at 37 °C and 5% CO2. Cells were not used past passage 20.
Cells were grown to confluence, passaged, and 90 μL of cells were seeded onto sterile 96-well plates (Nunc) at a density of 10,000 cells per well. Cells were grown overnight for 24 hours and allowed to settle and adhere. After 24 hours of growth, 10 μL of 10X concentration of treatment was added to the wells, starting at the highest stock concentration of 1 mg/mL for all NPs and 10 mM for citraconic acid. Note that PBS was used as the vehicle control. A positive control set of cells were treated with the lethal proteasome inhibitor MG-132 (Enzo Life Sciences), at a stock concentration of 400 μM and 10% DMSO; all cells had a metabolic activity measurement of <1% using this inhibitor. Cells were incubated with the treatments for 72 hours, and the CellTiter-Blue cell viability assay (Promega) was used according to manufacturer protocols. Briefly, 20 μL of CellTiter-Blue reagent was added to each well and incubated for at least 1 hour before reading the fluorescence (560 ex / 590 em) with a Tecan Infinite M200 Pro plate reader. Blank wells were used to subtract background and metabolic activity was normalized to the PBS controls. Three technical replicates were used for each group.
Nanoparticle charge conversion
Buffers of pH 7.4 and 6.5 were prepared at a concentration of 50 mM of either HEPES or MES, respectively, and titrated with either 6 N HCl or 1 N NaOH. Purified nanoparticles at 1 mg/mL were diluted in equal volumes of buffer in an Eppendorf tube for a final volume of 1 mL and were shaken at 200 rpm either at room temperature or at 37 °C. 50 μL aliquots were removed at the desired timepoints (0, 1, 4, 14, 24, 48, and 72 hours) and the zeta potential was measured in technical triplicate.
Nanoparticle penetration through transwells
Single colonies of either PAO1 or ΔwspF were picked from streaked LB plates and inserted into 2-3 mL of CAMHB grown overnight at 37 °C while shaking at 250 rpm. From this overnight culture, a new 25 mL culture was started by adding 250 μL of overnight culture and allowing bacteria to grow for 4-6 hours shaking at 37 °C, thereby reaching log-phase growth. Bacteria solutions were washed once with fresh CAMHB by spinning for 5 minutes at 6000xg. Bacteria were then resuspended at an OD600 of 1.0, and 50 μL were introduced to the apical side of a 96-well HTS transwell plate (Corning) while 200 μL of CAMHB were added to the basal side. Transwell plates were placed in a humid environment and grown for 48 hours at 37 °C.
After the 48-hour growth, the apical wells were washed twice with 50 μL of fresh CAMHB, and after the final wash, 45 μL of CAMHB was added. A new receiver well was used and filled with 200 μL of CAMHB with 10 mM HEPES (for pH 6.5 and 7.4 solutions) or MES (for pH 6.0), and the apical wells were placed into the receiver. Then, 5 μL of 10-fold concentrated, Cy5 fluorescent nanoparticles were added, so that 5 total micrograms of NP were in each apical well. After the desired incubation time (1, 2, 4, 8, and 24 hours), a new receiver well was filled with 200 μL of CAMHB and replaced the preceding receiver well. 100 μL of the sample was taken out of the old receiver well, mixed with 100 μL of ethanol, transferred into a non-TC coated black 96-well plate (Thermo Fisher), and emission excitation readings at 640 / 670 nm were run. Nanoparticle mass in the receiver wells was calculated based on respective nanoparticle standard curves, and the cumulative mass was recorded at each time point. Each condition had N = 4 technical replicates.
Confocal scanning light microscopy (CSLM) imaging and analysis
Overnight shaking ΔwspF cultures were subcultured into fresh CAMHB and grown for 4-6 hours at 37 °C and 250 rpm to reach log-phase growth. Bacteria were diluted in 10% CAMHB supplemented with 150 mM NaCl, as this reduced nutrient solution encourages the formation of biofilms will still retaining an accurate ionic strength. Bacteria were normalized at an OD600 of 0.01, and 1 mL of solution was added to each well of a glass-bottom, black chimney 24-well plate (Greiner Bio), and allowed to grow statically for 48 hours at 37 °C in a humid environment. Afterward, wells were washed three times with 10% CAMHB and 10 mM HEPES (pH adjusted to either 7.4 or 6.5) supplemented with 150 mM NaCl to remove planktonic, unattached cells. For each wash, 1 mL of medium was added and then removed, ensuring that the biofilm remained hydrated and that volume within the well was never below 1 mL. Washing was performed with a repeater pipette (Integra) at speed 2 to protect the delicate biofilm architecture. On the third wash, 1100 μL of the medium was removed and 100 μL of 100 μg/mL NP solution was added and incubated for either 4 or 24 hours and incubated at 37 °C in a humid environment. After NP incubation, each well was washed three more times with the same procedure as above, and 100 μL of 20 μM SYTO 9 dye was added to stain the biofilm, incubating statically for 30 minutes at room temperature in the dark. Another 3 washes were performed as above before imaging.
Biofilms were imaged using a confocal laser scanning microscope (LSM 800, Zeiss) with a 63x oil immersion magnification lens, and a step size of 0.5 μm. At least three independent wells were used and 3-5 images per well were taken. Images were analyzed using FIJI, in which Z-axis profiles for nanoparticle pixel intensities were plotted as a function of depth throughout the biofilm. Calculated area under the curve (AUC) intensities used the trapezoid rule and normalized x-axis biofilm distance for cumulative amount. For analysis of the middle 50% area under the curve, a similar calculation as stated above was used, except that the pixel intensity in the first and last 25% of the height of the biofilm was removed.
Tobramycin loading and quantification
Concentrations of tobramycin ranging from 0.05 to 1 mg/mL were tested in small batches with a final layering solution of 25 mM HEPES and 20mM NaCl and pH 7.4. The optimal concentration was chosen based on subsequent nanoparticle stability (< 30 nm increase in hydrodynamic diameter and > ∣30∣ mV zeta potential) as measured by DLS. Nanoparticles were then purified via TFF as previously described.
An aliquot of purified, drug-loaded nanoparticles was then placed into a 30 kDa MWCO spin filter and washed 6 times at 4000xg. Elution volume was measured and replaced exactly with 99.9% pure D2O. After 6 washes, the resulting nanoparticle suspension had ≥ 99.5% D2O. To this solution, 25 μg of 3-(trimethylsilyl)propionic-2,2,3,3-d4 (TMSP) acid sodium salt dissolved in D2O was added as a standard for proton quantification via 1H NMR. The 5.75 ppm shift for the tobramycin proton was integrated against the 0.00 ppm shift for the TMSP protons, and the total mass of tobramycin was calculated based on the integration of these peaks. Drug loading was calculated based on an assumption of no loss of colloidal material throughout the entire process.
In vitro reduction of biofilm colony forming units (CFUs)
Biofilms were grown on Calgary Biofilm Devices (CBD, Innovotech) according to manufacturer instructions with slight modifications. Bacteria were grown mid-log phase and diluted to 0.01 OD600. 150 μL was placed in each well and grown for 48 hours shaking at 125 rpm and 37 °C in a humid container. After 48 hours, the CBD lid with pegs was lifted, washed for 10 seconds in a new plate of PBS, and placed in another new plate containing 180 μL of CAMHB medium and 20 μL of various concentrations of NP, free drug, and CAMHB medium used as a no treatment control. Wells were statically incubated, washed, and fresh medium was added either 4 or 8 hours after treatment. The plate was allowed to incubate for a total of 18-24 hours, at which point the pegs were placed in a new microtiter plate with 200 μL of PBS and sonicated for 30 minutes. After 30 minutes, serial dilutions were plated and the bacterial burden was quantified as CFU per mL.
Statistics
All statistics used are described in each of the figure captions, and were analyzed using GraphPad Prism v8. Statistical significance was determined primarily using one- or two-way ANOVA and Tukey’s correction for multiple comparisons.
Results and Discussion
Layered synthetic polymers enable charge-reversible nanoparticle surfaces
In order to leverage the acidic microenvironment of biofilms, a panel of hydrolytically cleavable anionic polymers that could serve as the outer layer of LbL NPs was synthesized (Figure S1). Following a similar strategy from Liu et. al.,44 reacting anhydrides with primary amines on the polymer side chains results in amide bonds with a pendant carboxylic acid. Eventual hydrolysis of the amide results in a terminally charged primary amine. The synthetic polymer panel (Figure 2A) has two orthogonal design elements: the polymer backbone hydrophilicity and the polymer side chain rigidity. First, the two backbones were either the relatively hydrophilic polypeptide backbone, poly-L-lysine (PLK), or the relatively hydrophobic hydrocarbon-based backbone, poly(allylamine) (PAH). We hypothesized that the more hydrophilic backbone would lead to a quicker hydrolysis rate, as hydrolysis depends on the concentration of protons, and therefore, the concentration of water. Second, while the two reactants are five-member anhydride rings, citraconic anhydride (CIT) has an extra methyl group that forces side chain rigidity, unlike maleic anhydride (MAL). The more rigid side chain structure is empirically shown to enhance self-catalyzed amide hydrolysis.44 Following this logic, we hypothesized that polymers functionalized with CIT would hydrolyze faster than those functionalized with MAL. Consequently, the polymer that was conjugated with both CIT and MAL was predicted to hydrolyze at a rate in between the polymer reacted purely with CIT or MAL. Finally, an anionic polymer with no chemical modifications, poly(acrylic) acid (PAA), is used as a negative control. Polymer structures were confirmed with 1H NMR (Figure S2-5) and recorded as subscripts in Figure 2A.
Figure 2.

A synthetic panel of polymers is layered onto nanoparticles and imbues pH-responsive surfaces. A) Extruded, anionic liposomal cores are adsorbed with the polycation poly-L-lysine, and polyanions 1-5 are layered, resulting in 5 distinct LbL NPs. Note copolymer and side chain fractions are calculated from the corresponding 1H NMR spectra (Figure S2-5). B) Hydrodynamic diameters, polydispersity indices, and zeta potentials of the NPs display uniform nanoparticles. C) pH-accelerated charge reversal of NPs with hydrolytically cleavable side chains instills certain NPs with a pH-responsive surface. N = 3 technical replicates for both B and C.
Utilizing the LbL electrostatic assembly method, polymers 1-5 were added as the terminal layer of an LbL NP, defining its surface functionality. The core colloidal template used for all of these experiments was a liposome, primarily because there are FDA-approved antimicrobial formulations that leverage liposome carriers, such as amphotericin B liposome for injection46 and amikacin liposome inhalation suspension.47After layering, these LbL NPs had mean hydrodynamic diameters from 87 to 117 nm, were uniformly distributed with polydispersity indices (PDI) of < 0.20, and had zeta potentials > ∣30 mV∣ as measured by dynamic light scattering (DLS), all of which indicate stable NP formulations (Figure 2B). All LbL NPs layered with polymers 1-5, as well as the citraconic acid byproduct due to hydrolysis, had limited cellular toxicity, especially as compared to liposomes layered with only cationic PLK (Lipo/PLK) at highest functional concentrations (Figures S6-7).
As polymers 1-5 exhibited various aspects of hydrophilicity and rigidity, their use as terminal layers on LbL NPs produced diverse charge reversal rates which were further increased with a reduction of pH (Figure 2C). No LbL NP exhibited charge reversal (i.e. zeta potentials greater than 0 mV) after 72 hours at physiological conditions. However, we chose to also test charge reversal at a modestly reduced pH 6.5, in line with reported acidities for biofilm formed in patients.48 In these conditions, Lipo/PLK/1 became neutrally charged after 4 hours and had a plateaued positive charge after 24 hours. Lipo/PLK/2, which differed in the hydrophilicity of the backbone as compared to Lipo/PLK/1, also showed stimuli-responsive behavior and changed to a slight positive charge around 24 hours of incubation. The introduction of a more flexible side chain to interrupt the locked structure in Lipo/PLK/3 attenuated the charge reversal rate further. Lipo/PLK/4, with a hydrophobic hydrocarbon backbone and the least restricted side chain, showed no stimuli-responsive activity at the reduced pH for the entire 72-hour experiment. Lipo/PLK/4 and Lipo/PLK/5 maintain similar zeta potentials of −37.4 mV versus −42.3 mV after 72 hours, respectively. These data confirmed our initial structure-function hypothesis that both the hydrophilicity and side chain structure of terminal polymers determined charge reversal rates.
Increased charge reversal rate enhances biofilm penetration
With our polymer panel characterized, we wanted to see if there was a relationship between the NP charge, the rate of charge reversal, and infiltration through biofilms. We chose to test our panel of LbL NPs in biofilms produced by the biofilm-producing, opportunistic pathogen Pseudomonas aeruginosa. To specifically evaluate the contribution of biofilm EPS on NPs penetration, we used the well-characterized strain PAO149, 50 and the isogenic wspF deficient mutant. ΔwspF is known to non-specifically overexpress polysaccharides, and thereby creates more robust biofilms.51 Bacterial strains PAO1 or ΔwspF were grown for 48 hours on transwell inserts and NP transport through their produced biofilms was measured for up to an additional 24 hours, as quantified by the fluorescent intensity in the bottom receiver well (Figure 3A). We hypothesized that LbL NPs that charge-converted the fastest would be able to penetrate through biofilms and accumulate more readily in the bottom well, since they would become positive and multivalently bind the matrix, corresponding to increased fluorescent intensity.
Figure 3.

Nanoparticle penetration through biofilms shows pH trend. A) Schematic representation for the transwell assay workflow illustrates how NPs navigate through the biofilm to reach the basal chamber for quantification. B) Cumulative penetration of LbL NPs within the panel for each timepoint collected illustrates how surface charge impacts penetration. More positively charged nanoparticles, including those that alter their surface charge, accumulate to a higher extent than anionic layer-by-layer nanoparticles. C) Significant trends reveal that reduced pH and increased charge reversal rate enhance penetration through biofilms. Error bars represent standard deviation with N = 4 technical replicates, each indicated by filled in circles. Comparisons within and between pH groups were analyzed with two-way ANOVA using Tukey’s multiple comparisons correction, where * means p < 0.05, and ** means p < 0.01.
When incubated at pH 7.4, NP accumulation through the biofilm was lower in the ΔwspF mutant as compared to PAO1 for every formulation and at every timepoint tested (Figure 3B). These results are in line with the notion that higher biofilm biomass (Figure S8) attenuates penetration of exogenous materials more effectively. There was also a general trend for penetration through both biofilms, in that the NPs with the most positive charge, or fastest charge conversion, penetrated more effectively (Figure 3B). These data supported our hypotheses that negatively charged biofilm matrix components electrostatically interact with positive charges on the NP surface and that enhanced interactions can enable more effective penetration.24, 52 The permanently cationic nanoparticle Lipo/PLK showed the greatest penetration through both sets of biofilms at all time points, in agreement with previously reported trends.26, 29 Next, the charge-converting NPs followed in order of its charge conversion rate, for both biofilms at all time points. We note that penetration occurred faster than the hydrolysis rates observed in Figure 2C, most likely due to potential hypoxic heterogeneous microenvironments, as well as reactive oxygen species and enzymes, present within biofilms.53 Furthermore, the penetration of LbL NPs in the biofilm showed a clear correlation with formulations that imply that even small amounts of positive charge can enhance transport.
Moving forward, we chose to focus primarily on the LbL NPs layered with our synthesized polymers (i.e. Lipo/PLK/1-4), as we want to draw conclusions directly from their structures and reversal rates. The greater toxicity of Lipo/PLK suggested a biological incompatibility that would not be translatable, and thus we did not continue using this formulation for subsequent experiments. When investigating biofilm penetration at reduced pHs of 6.5 and 6 (Figure 3C), the same trend, in which enhanced charge reversal rate corresponded to increased penetration through biofilms, held for biofilms produced by both strains. Furthermore, as the exogeneous pH dropped, the same NP formulation penetrated more effectively for a given strain, which we attribute to the accelerated hydrolysis rate. Formulations, such as Lipo/PLK/4, that exhibited a constant anionic charge, saw no penetration benefit when exposed to a reduced pH, further corroborating our hypothesis. While the altered biofilm matrix at reduced pH could be one alternative explanation for the enhanced NP penetration, crystal violet staining of PAO1 and ΔwspF biofilms grown in the same conditions showed no differences in biofilm biomass (Figure S8). Alternative compositional changes to the biofilms as a function of pH may indeed exist, such as the protonation of polysaccharides or other components, though the similar transwell penetration of Lipo/PLK/4 over all pH conditions tested suggest that these structural differences may not play an outstanding role in NP penetration for this assay.
Increased biofilm permeation and accumulation with charge-reversing nanoparticles
To further understand the biofilm-NP interactions during NP passage through the biofilm matrix and assess the spatial distribution of the NPs, we turned to confocal microscopy. Similar to the transwell assay experimental design, we took confocal images of ΔwspF-produced biofilms grown for 48 hours, exogenously adjusted the pH to either 7.4 or 6.5, and incubated with our charge-converting NPs for an additional 24 hours. We saw that the fastest charge-converting NPs interacted more completely than the slowest charge-converting NPs (Figure 4A). Note that the images below portray the biofilm as an average pixel intensity over the entire Z-dimension. To quantify the NP accumulation within the biofilm, we calculated an area under the normalized pixel intensity curve (AUC) as a function of depth throughout the biofilm, thereby quantifying a biofilm accumulation. The highest NP accumulation within biofilms, for both pH 7.4 and 6.5, occurred for Lipo/PLK/1, then Lipo/PLK/2, Lipo/PLK/3, and finally the least accumulation was Lipo/PLK/4 (Figure S9).
Figure 4.

Charge-converting nanoparticles interacted with, and penetrated through, the biofilm matrix more effectively. A) Confocal microscopy images of nanoparticles (red) interacting with the ΔwspF biofilm matrix (green) after 24 hours. Both components are shown as an average pixel intensity throughout the biofilm matrix, collapsed onto a 2D image. The scale bar represents 20 μm. B) Pixel intensity throughout the middle 50% of the biofilm, where the first and last 25% of the biofilm are excluded, was calculated. C) Calculations are shown using an area under the curve formula for both Lipo/PLK/1 and Lipo/PLK/4. Both time and pH were significant sources of variation for Lipo/PLK/1 as analyzed by a two-way ANOVA, whereas only time, and not pH, was significant for Lipo/PLK/4. Error bars represent standard deviation with N = 3 biologically distinct wells with N = 3 technical image replicates per well. Comparisons within and across pH groups were analyzed with two-way ANOVA using Tukey’s multiple comparisons correction, where * means p <0.05, ** means p <0.01, *** means p<0.001, and **** means p<0.0001.
A possible confounding variable in calculating the AUC data in this manner is the effect of gravity. We gently washed the biofilms to maintain their integrity on the glass-bottom well, and perhaps did not completely remove aggregates adsorbed to the bottom of the well. Therefore, we decided to measure the nanoparticle accumulation throughout the middle 50% of the biofilm (Figure 4B) over multiple timepoints. These data follow the same trend, whereby enhanced differences for Lipo/PLK/1 at each time point occurred at a reduced exogenous pH (Figure 4C). From 4 to 24 hours for Lipo/PLK/1 and pH 7.4, there is a 3.95-fold increase in pixel intensity, whereas at pH 6.5 there is an over 8-fold increase. While there are also increases from 4 hours to 24 hours for Lipo/PLK/4 (Figure 4C), this can be explained primarily by the length of the experiment, as a two-way ANOVA analysis determined that only time (p = 0.0008), and not pH (p = 0.8933), significantly accounted for the variation in these data. These results can also explain the previous data (Figure 3B) illustrating increased penetration through the biofilm matrix since anionic NPs will diffuse through the matrix, though much slower. Finally, we were curious if the fastest charge-converting NPs would be able to deliver antimicrobials more efficaciously, as they accumulate more efficiently within the biofilm.
Tobramycin loading onto the NP surface enhances in vitro efficacy
We chose to load tobramycin onto our NPs, a model aminoglycoside regularly prescribed for chronic P. aeruginosa infections.54 Furthermore, the cationic charge on tobramycin causes sequestration by the anionic biofilm matrix10. Contrary to traditional liposomal antibiotic loading methods, whereby therapeutics are loaded into the aqueous interior,18 we adsorbed tobramycin into the NP outermost layer (Figure 5A). Tobramycin was loaded on the exterior surface of the NP since traditional, passive tobramycin encapsulation with the anionic phospholipid DSPG, a requirement for subsequent cationic polymer layering, caused NP aggregation (data not shown). This loading strategy was inspired by works loading peptides onto LbL NPs55 and co-layering cationic antibiotics with anionic polymers.56
Figure 5.

Nanoparticle carriers loaded with tobramycin enhance antibiotic delivery. A) Schematic illustration of tobramycin electrostatically adsorbed into the final layer. B) Tobramycin absorbs onto each nanoparticle with satisfactory loading and encapsulation efficiency. N = 4 independent loading replicates, shown as an average and standard deviation. C) Nanoparticle size and zeta potential comparison before and after loading tobramycin show little difference. N = 3 technical replicates, shown as an average and standard deviation. D) Overnight biofilms treated with dose-matched tobramycin treatments had similar efficacy for PAO1, but a 3-fold reduction in colony-forming units in ΔwspF at the highest concentration. Dark lines in D, sometimes blocked by individual data points, represent the average log(CFU/mL) of biofilms treated with the medium control. N = 4 biological replicates using an independent nanoparticle formulation each with N = 2 technical duplicates. Statistics were calculated using one-way ANOVA with Dunnett’s multiple comparisons correction, comparing means against Lipo/PLK/1.
Layering conditions, such as solution pH and ionic strength,57 are critical for therapeutic loading. Therefore, we tested a series of layering solution pH values, accounting for the range of tobramycin primary amine pKas,58 and saw no meaningful loading differences (Figure S10). We chose to layer at pH 7.4,57 buffered with 25 mM HEPES, and a final concentration of 20 mM sodium chloride. NPs were layered, purified, and dosed all within 24 hours, thereby avoiding potential unintended charge conversion (Figure S11).
Final drug quantifications from three independent formulations were calculated using 1H NMR by integrating the 5.75 ppm shift for the 1" tobramycin proton against a known stock amount of 3-(trimethylsilyl)propionic-2,2,3,3-d4 (TMSP) acid sodium salt, which was also used as a reference compound set at 0.00 ppm. This integration enabled calculation of both the weight loading, which measures the mass of loaded tobramycin per unit mass of nanoparticles, and encapsulation efficiency, which measures the mass of tobramycin retained on the nanoparticles per unit mass of tobramycin used during formulation (i.e. the fraction of drug that got encapsulated from the loading solution). Analyzed results determined that the weight fraction loading and encapsulation efficiencies were 10.9 ± 3.5% and 41.3 ± 5.3% for Lipo/PLK/1, and 15.1 ± 7.1% and 35.2 ± 10.2% for Lipo/PLK/4, respectively (Figure 5B). Importantly, embedding tobramycin into the outermost anionic surface did not result in meaningful differences in NP size and zeta potential (Figure 5C) nor changes in the time required to complete charge conversion for Lipo/PLK/1 (Figure S12). Overall, these data indicated the compatibility of this loading method with our charge-conversation strategy, enabling clinically relevant antibiotics to be loaded onto our NPs for effective delivery.
Each formulation was then tested for its capacity to reduce biofilm CFUs on the Calgary Biofilm Device.59 PAO1 or ΔwspF biofilms were grown for 48 hours, washed, and incubated for 8 hours with either a free tobramycin or a dose-equivalent NP formulation without adjusting the pH of the medium. After 8 hours, the medium was washed and replaced, and biofilms were allowed to grow for another 8 hours before sonicating them off the device and serially diluting them to quantify CFUs (Figure 5D). The 8-hour medium wash timepoint was chosen because intravenous tobramycin dosing occurs every 8 hours during hospitalized infection,60 and Lipo/PLK/1 would have substantially changed its surface charge by this time. While this formulation has not completely charge converted at 8 hours, we had to balance the earlier time with the artificially static nature of this biofilm eradication assay.
All NP treatment groups were equally as effective as free tobramycin in reducing biofilm CFUs for both strains at all given therapeutic concentrations. Furthermore, when ΔwspF biofilms are dosed at 16 μg/mL of tobramycin, Lipo/PLK/1 had a 3.2-fold reduction in ΔwspF biofilm CFU/mL as compared to the free drug. Similar calculations illustrate that Lipo/PLK/4 reduces ΔwspF biofilm CFU/mL with the same efficacy as the free drug at this higher therapeutic concentration. We attribute this increased efficacy to the charge-converting NPs permeating throughout the biofilm, though we recognize that some variation of the in vitro results could have been due to tobramycin being stripped from the nanoparticle surface through electrostatic exchange reactions, releasing prematurely before complete NP penetration. We also note that we only tracked nanoparticle penetration and were not able to attain antibiotic distribution as it penetrates the biofilm matrix. Excitingly, this suggests that the charge-converting NPs aid in the efficacious delivery of tobramycin in the presence of biofilm, potentially enabling a reduced therapeutic dose to elicit a similar benefit. Ultimately, these results provide a framework for engineering tailored, pH-responsive NP surfaces, imbuing those surfaces with antimicrobials, and delivering higher local concentrations of antibiotics more efficaciously.
Conclusions
This work presents surface charge conversion rate as a new design parameter for antibiotic-delivering nanoparticles to maximize biofilm interactions while retaining biocompatibility. Nanoparticles are layered with an outermost, pH-responsive polymer that charge-converts from negative to positive, rendering the polymer surface biocompatible until exposed to the reduced pH of the biofilm environment. Once exposed, the surface becomes positively charged and electrostatically interacts with anionic biofilm matrix components. We synthesized four different polymers, each with varied backbone hydrophilicities and side-chain structures, and determined structure-function properties to tune charge reversal rates from hours to days. The maximum charge reversal rate trended strongly with increased penetration through biofilms grown on transwell inserts and increased permeation throughout biofilms as measured with confocal microscopy pixel intensity areas under the curve. We were then able to load therapeutically-relevant concentrations of tobramycin, a model anti-Pseudomonas antibiotic known to be sequestered by the biofilm matrix, using a new loading strategy that did not alter physiochemical nanoparticle properties nor charge-reversal rates. Finally, the fastest charge-converting nanoparticle reduced bacterial colony-forming units of an overproducing biofilm biomass mutant 3.2-fold compared to free drug, laying the foundation for future efficacious nanoparticle vehicle designs.
Supplementary Material
Acknowledgments
We would like to thank Tamara Dacoba for her thoughtful review and comments on this manuscript, and Walt Massefski for his advice and experimental suggestions regarding tobramycin quantification. Figures 1-5 were made in part with Biorender.com. E.D.-Y. acknowledges the National Research Foundation, Prime Minister’s Office, Singapore for support under its Campus for Research Excellence and Technological Enterprise (CREATE) programme. E.D.-Y. acknowledges the Cystic Fibrosis Foundation (004527H222) for funding part of this work. This research was funded in part by the US Army (W81XWH2110235 to P.T.H.), NIH (F30DK130564 to A.G.B.). A.G.B. acknowledges additional support from NIH grants T32GM007753 and T32GM144273, and the MIT Termeer Fellowship of Medical Engineering and Science.
Footnotes
Supporting Information Statement
Supplemental information includes figures of the polymer synthesis scheme, 1H NMR of synthesized polymers 1-4, cell viability when exposed to layered NPs and hydrolysis byproduct, crystal violet staining of biofilms in buffered pHs, area under the curve for NP permeation, size and zeta measurements for optimized tobramycin loading, NP charge reversal at room temperature, and charge reversal after loading tobramycin.
References
- 1.Wolcott RD; Rhoads DD; Bennett ME; Wolcott BM; Gogokhia L; Costerton JW; Dowd SE, Chronic wounds and the medical biofilm paradigm. J. Wound Care 2010, 19 (2), 45–6, 48-50, 52-3. 10.12968/jowc.2010.19.2.46966. [DOI] [PubMed] [Google Scholar]
- 2.Wolcott R; Dowd S, The Role of Biofilms: Are We Hitting the Right Target? Plast. and Reconstr. Surg 2011, 127, 28S–35S. 10.1097/PRS.0b013e3181fca244. [DOI] [PubMed] [Google Scholar]
- 3.Flemming H-C; Wingender J, The biofilm matrix. Nat. Rev. Microbiol 2010, 8 (9), 623–633. 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 4.Hall CW; Mah T-F, Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev 2017, 41 (3), 276–301. 10.1093/femsre/fux010. [DOI] [PubMed] [Google Scholar]
- 5.Mah T-F, Biofilm-specific antibiotic resistance. Future Microbiol. 2012, 7 (9), 1061–1072. 10.2217/fmb.12.76. [DOI] [PubMed] [Google Scholar]
- 6.Mah T-FC; O'Toole GA, Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9 (1), 34–39. 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 7.Kohanski MA; DePristo MA; Collins JJ, Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell 2010, 37 (3), 311–20. 10.1016/j.molcel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jim O'Neill. Nat. Rev. Drug Discov 2016, 15 (8), 526–526. 10.1038/nrd.2016.160. [DOI] [PubMed] [Google Scholar]
- 9.Sharma D; Misba L; Khan AU, Antibiotics versus biofilm: an emerging battleground in microbial communities. Antimicrob. Resist. & Infect. Control 2019, 8 (1), 76. 10.1186/s13756-019-0533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tseng BS; Zhang W; Harrison JJ; Quach TP; Song JL; Penterman J; Singh PK; Chopp DL; Packman AI; Parsek MR, The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ. Microbiol 2013, 15 (10), 2865–2878. 10.1111/1462-2920.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mah TF; O’Toole GA, Mechanisms of Biofilm Resistance to Antimicrobial Agents. Trends Microbiol. 2001, 9 (1), 34–39. [DOI] [PubMed] [Google Scholar]
- 12.Billings N; Millan M; Caldara M; Rusconi R; Tarasova Y; Stocker R; Ribbeck K, The extracellular matrix Component Psl provides fast-acting antibiotic defense in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2013, 9 (8), e1003526–e1003526. 10.1371/journal.ppat.1003526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nichols WW; Dorrington SM; Slack MP; Walmsley HL, Inhibition of tobramycin diffusion by binding to alginate. Antimicrob. Agents Chemother 1988, 32 (4), 518–23. 10.1128/aac.32.4.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bahamondez-Canas TF; Zhang H; Tewes F; Leal J; Smyth HDC, PEGylation of Tobramycin Improves Mucus Penetration and Antimicrobial Activity against Pseudomonas aeruginosa Biofilms in Vitro. Mol. Pharmaceutics 2018, 15 (4), 1643–1652. 10.1021/acs.molpharmaceut.8b00011. [DOI] [PubMed] [Google Scholar]
- 15.Blanco E; Shen H; Ferrari M, Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol 2015, 33 (9), 941–51. 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez Gomez A; Hosseinidoust Z, Liposomes for Antibiotic Encapsulation and Delivery. ACS Infect. Dis 2020, 6 (5), 896–908. 10.1021/acsinfecdis.9b00357. [DOI] [PubMed] [Google Scholar]
- 17.Barenholz Y., Doxil® — The first FDA-approved nano-drug: Lessons learned. J. Controlled Release 2012, 160 (2), 117–134. 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 18.Gubernator J., Active Methods of Drug Loading into Liposomes: Recent Strategies for Stable Drug Entrapment and Increased in vivo Activity. Expert Opin. Drug Delivery 2011, 8 (5), 565–580. [DOI] [PubMed] [Google Scholar]
- 19.Chenthamara D; Subramaniam S; Ramakrishnan SG; Krishnaswamy S; Essa MM; Lin F-H; Qoronfleh MW, Therapeutic efficacy of nanoparticles and routes of administration. Biomater. Res 2019, 23 (1), 20. 10.1186/s40824-019-0166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yetisgin AA; Cetinel S; Zuvin M; Kosar A; Kutlu O, Therapeutic Nanoparticles and Their Targeted Delivery Applications. Mol. 2020, 25 (9). 10.3390/molecules25092193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cipolla D; Blanchard J; Gonda I, Development of Liposomal Ciprofloxacin to Treat Lung Infection. Pharm. 2016, 8 (1), 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clancy JP; Dupont L; Konstan MW; Billings J; Fustik S; Goss CH; Lymp J; Minic P; Quittner AL; Rubenstein RC; Young KR; Saiman L; Burns JL; Govan JRW; Ramsey B; Gupta R, Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax 2013, 68 (9), 818. 10.1136/thoraxjnl-2012-202230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moura LA; Ribeiro FV; Aiello TB; Duek EADR; Sallum EA; Nociti Junior FH; Casati MZ; Sallum AW, Characterization of the release profile of doxycycline by PLGA microspheres adjunct to non-surgical periodontal therapy. J. Biomater. Sci., Polym. Ed 2015, 26 (10), 573–584. 10.1080/09205063.2015.1045249. [DOI] [PubMed] [Google Scholar]
- 24.Fulaz S; Vitale S; Quinn L; Casey E, Nanoparticle–Biofilm Interactions: The Role of the EPS Matrix. Trends Microbiol. 2019, 27 (11), 915–926. 10.1016/j.tim.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Ikuma K; Decho AW; Lau BLT, When nanoparticles meet biofilms-interactions guiding the environmental fate and accumulation of nanoparticles. Front. in Microbiol 2015, 6, 591–591. 10.3389/fmicb.2015.00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benoit DSW; Sims KR; Fraser D, Nanoparticles for Oral Biofilm Treatments. ACS Nano 2019, 13 (5), 4869–4875. 10.1021/acsnano.9b02816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta A; Das R; Yesilbag Tonga G; Mizuhara T; Rotello VM, Charge-Switchable Nanozymes for Bioorthogonal Imaging of Biofilm-Associated Infections. ACS Nano 2018, 12 (1), 89–94. 10.1021/acsnano.7b07496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta A; Landis R; Rotello V, Nanoparticle-Based Antimicrobials: Surface Functionality is Critical F1000Research 2016, 5 (364). 10.12688/f1000research.7595.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X; Yeh Y-C; Giri K; Mout R; Landis RF; Prakash YS; Rotello VM, Control of nanoparticle penetration into biofilms through surface design. Chem. Commun 2015, 51 (2), 282–285. 10.1039/c4cc07737g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kedmi R; Ben-Arie N; Peer D, The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomater. 2010, 31 (26), 6867–6875. 10.1016/j.biomaterials.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 31.Knudsen KB; Northeved H; Kumar Ek P; Permin A; Gjetting T; Andresen TL; Larsen S; Wegener KM; Lykkesfeldt J; Jantzen K; Loft S; Møller P; Roursgaard M, In vivo toxicity of cationic micelles and liposomes. Nanomed. Nanotechnol. Biol. Med 2015, 11 (2), 467–477. 10.1016/j.nano.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Arvizo RR; Rana S; Miranda OR; Bhattacharya R; Rotello VM; Mukherjee P, Mechanism of anti-angiogenic property of gold nanoparticles: role of nanoparticle size and surface charge. Nanomed. 2011, 7 (5), 580–7. 10.1016/j.nano.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duan X; Li Y, Physicochemical Characteristics of Nanoparticles Affect Circulation, Biodistribution, Cellular Internalization, and Trafficking. Small 2013, 9 (9-10), 1521–1532. 10.1002/smll.201201390. [DOI] [PubMed] [Google Scholar]
- 34.Salvador-Morales C; Zhang L; Langer R; Farokhzad OC, Immunocompatibility properties of lipid-polymer hybrid nanoparticles with heterogeneous surface functional groups. Biomater. 2009, 30 (12), 2231–2240. 10.1016/j.biomaterials.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blum AP; Kammeyer JK; Rush AM; Callmann CE; Hahn ME; Gianneschi NC, Stimuli-Responsive Nanomaterials for Biomedical Applications. J. Am. Chem. Soc 2015, 137 (6), 2140–2154. 10.1021/ja510147n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hidalgo G; Burns A; Herz E; Hay AG; Houston PL; Wiesner U; Lion LW, Functional Tomographic Fluorescence Imaging of pH Microenvironments in Microbial Biofilms by Use of Silica Nanoparticle Sensors. Appl. and Environ. Microbiol 2009, 75 (23), 7426. 10.1128/AEM.01220-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fulaz S; Hiebner D; Barros CHN; Devlin H; Vitale S; Quinn L; Casey E, Ratiometric Imaging of the in Situ pH Distribution of Biofilms by Use of Fluorescent Mesoporous Silica Nanosensors. ACS Appl. Mater. Interfaces 2019, 11 (36), 32679–32688. 10.1021/acsami.9b09978. [DOI] [PubMed] [Google Scholar]
- 38.Gao Y; Wang J; Chai M; Li X; Deng Y; Jin Q; Ji J, Size and Charge Adaptive Clustered Nanoparticles Targeting the Biofilm Microenvironment for Chronic Lung Infection Management. ACS Nano 2020, 14 (5), 5686–5699. 10.1021/acsnano.0c00269. [DOI] [PubMed] [Google Scholar]
- 39.Correa S; Dreaden EC; Gu L; Hammond PT, Engineering nanolayered particles for modular drug delivery. J. Control. Release 2016, 240, 364–386. 10.1016/j.jconrel.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alkekhia D; Hammond PT; Shukla A, Layer-by-Layer Biomaterials for Drug Delivery. Annu. Rev. Biomed. Eng 2020, 22 (1), 1–24. 10.1146/annurev-bioeng-060418-052350. [DOI] [PubMed] [Google Scholar]
- 41.Yan Y; Björnmalm M; Caruso F, Assembly of Layer-by-Layer Particles and Their Interactions with Biological Systems. Chem. Mater 2014, 26 (1), 452–460. 10.1021/cm402126n. [DOI] [Google Scholar]
- 42.Decher G., Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites. Science 1997, 277 (5330), 1232–1237. doi: 10.1126/science.277.5330.1232. [DOI] [Google Scholar]
- 43.Correa S; Boehnke N; Barberio AE; Deiss-Yehiely E; Shi A; Oberlton B; Smith SG; Zervantonakis I; Dreaden EC; Hammond PT, Tuning Nanoparticle Interactions with Ovarian Cancer through Layer-by-Layer Modification of Surface Chemistry. ACS Nano 2020, 14 (2), 2224–2237. 10.1021/acsnano.9b09213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X; Zhang J; Lynn DM, Polyelectrolyte multilayers fabricated from ‘charge-shifting’ anionic polymers: a new approach to controlled film disruption and the release of cationic agents from surfaces. Soft Matter 2008, 4 (8), 1688–1695. 10.1039/B804953J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Correa S; Choi KY; Dreaden EC; Renggli K; Shi A; Gu L; Shopsowitz KE; Quadir MA; Ben-Akiva E; Hammond PT, Highly Scalable, Closed-Loop Synthesis of Drug-Loaded, Layer-by-Layer Nanoparticles. Adv. Funct. Mater 2016, 26, 991–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hann IM; Prentice HG, Lipid-based amphotericin B: a review of the last 10 years of use. Int. J. Antimicrob. Agents 2001, 17 (3), 161–169. 10.1016/S0924-8579(00)00341-1. [DOI] [PubMed] [Google Scholar]
- 47.Shirley M., Amikacin Liposome Inhalation Suspension: A Review in Mycobacterium avium Complex Lung Disease. Drugs 2019, 79 (5), 555–562. 10.1007/s40265-019-01095-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoon SS; Coakley R; Lau GW; Lymar SV; Gaston B; Karabulut AC; Hennigan RF; Hwang S-H; Buettner G; Schurr MJ; Mortensen JE; Burns JL; Speert D; Boucher RC; Hassett DJ, Anaerobic killing of mucoid Pseudomonas aeruginosa by acidified nitrite derivatives under cystic fibrosis airway conditions. J. Clin. Invest 2006, 116 (2), 436–446. 10.1172/JCI24684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holloway BW, Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbiol 1955, 13 (3), 572–81. 10.1099/00221287-13-3-572. [DOI] [PubMed] [Google Scholar]
- 50.Klockgether J; Munder A; Neugebauer J; Davenport CF; Stanke F; Larbig KD; Heeb S; Schöck U; Pohl TM; Wiehlmann L; Tümmler B, Genome diversity of Pseudomonas aeruginosa PAO1 laboratory strains. J. Bacteriol 2010, 192 (4), 1113–21. 10.1128/jb.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hickman JW; Tifrea DF; Harwood CS, A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci 2005, 102 (40), 14422–7. 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Witten J; Ribbeck K, The particle in the spider's web: transport through biological hydrogels. Nanoscale 2017, 9 (24), 8080–8095. 10.1039/c6nr09736g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flemming H-C; Wingender J; Szewzyk U; Steinberg P; Rice SA; Kjellberg S, Biofilms: an Emergent form of Bacterial Life. Nat. Rev. Microbiol 2016, 14, 563–575. [DOI] [PubMed] [Google Scholar]
- 54.Konstan MW; Flume PA; Kappler M; Chiron R; Higgins M; Brockhaus F; Zhang J; Angyalosi G; He E; Geller DE, Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: the EAGER trial. J. Cystic Fibrosis 2011, 10 (1), 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boehnke N; Dolph KJ; Juarez VM; Lanoha JM; Hammond PT, Electrostatic Conjugation of Nanoparticle Surfaces with Functional Peptide Motifs. Bioconjugate Chem. 2020, 31 (9), 2211–2219. 10.1021/acs.bioconjchem.0c00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finbloom JA; Raghavan P; Kwon M; Kharbikar BN; Yu MA; Desai TA, Codelivery of synergistic antimicrobials with polyelectrolyte nanocomplexes to treat bacterial biofilms and lung infections. Sci. Adv 2023, 9 (3), eade8039. doi: 10.1126/sciadv.ade8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Correa S; Boehnke N; Deiss-Yehiely E; Hammond PT, Solution Conditions Tune and Optimize Loading of Therapeutic Polyelectrolytes into Layer-by-Layer Functionalized Liposomes. ACS Nano 2019, 13 (5), 5623–5634. 10.1021/acsnano.9b00792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alkhzem AH; Woodman TJ; Blagbrough IS, Individual pK (a) Values of Tobramycin, Kanamycin B, Amikacin, Sisomicin, and Netilmicin Determined by Multinuclear NMR Spectroscopy. ACS Omega 2020, 5 (33), 21094–21103. 10.1021/acsomega.0c02744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ceri H; Olson ME; Stremick C; Read RR; Morck D; Buret A, The Calgary Biofilm Device: New Technology for Rapid Determination of Antibiotic Susceptibilities of Bacterial Biofilms. J. Clin. Microbiol 1999, 37 (6), 1771. 10.1128/JCM.37.6.1771-1776.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.StatPearls, In Tobramycin, Reyhanoglu G; Reddivari AKR, Eds. Treasure Island Florida, 2022; p https://www.ncbi.nlm.nih.gov/books/NBK551695/. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.