

Abstract

We report a blueprint for the rational design of G protein coupled receptor (GPCR) ligands with a tailored functional response. The present study discloses the structure-based design of cannabinoid receptor type 2 (CB2R) selective inverse agonists (S)-1 and (R)-1, which were derived from privileged agonist HU-308 by introduction of a phenyl group at the gem-dimethylheptyl side chain. Epimer (R)-1 exhibits high affinity for CB2R with Kd = 39.1 nM and serves as a platform for the synthesis of a wide variety of probes. Notably, for the first time these fluorescent probes retain their inverse agonist functionality, high affinity, and selectivity for CB2R independent of linker and fluorophore substitution. Ligands (S)-1, (R)-1, and their derivatives act as inverse agonists in CB2R-mediated cAMP as well as G protein recruitment assays and do not trigger β-arrestin–receptor association. Furthermore, no receptor activation was detected in live cell ERK1/2 phosphorylation and Ca2+-release assays. Confocal fluorescence imaging experiments with (R)-7 (Alexa488) and (R)-9 (Alexa647) probes employing BV-2 microglial cells visualized CB2R expressed at endogenous levels. Finally, molecular dynamics simulations corroborate the initial docking data in which inverse agonists restrict movement of toggle switch Trp2586.48 and thereby stabilize CB2R in its inactive state.
Short abstract
We report a generalizable strategy for structure-based agonist-to-inverse-agonist functional transformation and probe development by ligand modification that modulates the GPCR toggle switch of CB2R.
Introduction
The endocannabinoid system is present in all vertebrates and comprises endogenous ligands, enzymes mediating ligand metabolism, transporters, and the two prominent cannabinoid receptors type 1 and type 2 (CB1R and CB2R).1,2 Exploitation of the therapeutic potential of CB2R has primarily focused on receptor activation with agonists and showed promise to ameliorate a plethora of diseases, such as autoimmune3 and metabolic disorders,4,5 chronic pain,6 and multiple sclerosis.7 By contrast, CB2R antagonists and inverse agonists remain vastly underexplored despite encouraging results in models of arthritis8 and neuroinflammation.9−11 Notably, CB2R antagonist TT-816 is currently being investigated in a phase II clinical trial as an immune checkpoint inhibitor for the treatment of solid tumors.12
Despite the current considerable endeavors to deliver selective CB2R therapeutics,12−19 to date there are no such drugs available on the market. Poor understanding of CB2R localization, expression, and signaling on the molecular level are key factors responsible for this absence.20 Elucidation of CB2R pharmacology has been hampered by the insufficient specificity of monoclonal antibodies21−24 and the scarcity of reliable chemical probes.25 Although some potent, selective, and validated fluorescent probes have been reported,26−28 these function as agonists that disturb cellular homeostasis by triggering downstream signaling and β-arrestin association, followed by agonist-mediated receptor internalization.29,30 These limitations may be addressed by implementation of inverse agonist based fluoroprobes that do not prompt receptor endocytosis. Additionally, inverse agonists tend to possess greater affinity for receptors in the more populous inactive G protein coupled receptor (GPCR) conformation yielding improved specificity and signal-to-noise ratio of probes compared to agonists.31
Historically, development of high-affinity, selective fluorescent CB2R inverse agonists has proven arduous. In the cases reported, fluorophore conjugation completely ablated32 or materially reduced33 affinity. In one example, a study of a series of agonists led to the identification of a specific linker–fluorophore construct endowing inverse agonism in a cAMP assay.34 Development of a potent, selective, and versatile CB2R-targeting inverse agonist scaffold that can be conjugated to a variety of fluorophores and functionalities remains an unmet challenge.
Since its discovery in 1999,35 CB2R-selective agonist HU-308 (Scheme 1) has enjoyed privileged status for the study of CB2R pharmacology.36 HU-308 has been extensively applied to unravel effects of CB2R activation in animal models of pain,37 osteoporosis,38 Parkinson’s disease,39 and amyotrophic lateral sclerosis40 and is currently investigated in phase I clinical trials for mitigation of inflammation.19 The pharmacophore embedded in HU-308 has served in the development of photoswitchable,41 fluorescent,28,42 and ligand-directed covalent probes.26 On the basis of our prior work with HU-308, we focused on this scaffold with the intent of transforming its functional profile from agonist to inverse agonist with minimal structural modification. In this respect, Schapira and Jones have independently discussed the conceptual benefits of working with a set of molecules closely related in structure to enable in-depth understanding of receptor pharmacology.43,44
Scheme 1. Novel Structure-Based Design of HU-308-Derived CB2R-Selective Inverse Agonists That Actuate Trp2586.48 Toggle Switch.

The past two decades witnessed the exponential rise of reported GPCR structures; hence, structure-based ligand design is at present ideally positioned to capitalize on the ongoing revolution.45 Since more than a third of all approved drugs exert their action by GPCR modulation, it is vital to comprehensively investigate and understand receptor pharmacology with functionally orthogonal chemical probes.46 GPCRs of the most populous class A family are distinguished by high homology of the CWxP motif. In particular, the toggle switch of CWxP that modulates receptor activation, Trp2586.48, is conserved within 78% of nonolfactory GPCRs.47 Examination of the X-ray structure of CB2R in its inactive conformation revealed a secondary binding pocket that hosts Trp2586.48. Further investigation by in silico docking suggested that addition of a substituent at C(2′) of HU-308 might constrain Trp2586.48 and hence modulate CB2R activation (Scheme 1).
We report novel inverse agonists that demonstrate avid binding at CB2R with excellent selectivity over the closely related CB1R. The compounds were profiled for their functional response in a comprehensive panel of in vitro (β-arrestin and G protein recruitment) as well as cellular (cAMP, ERK1/2 phosphorylation, Ca2+ signaling) assays. Remarkably, none of the probes activate CB2R-mediated signaling in any of the tested pathways. Fluorescent probes demonstrated excellent specificity and visualized CB2R expressed at endogenous levels in live-cell confocal microscopy experiments. Finally, molecular dynamics simulations investigated structural determinants that prevent receptor activation upon ligand binding and corroborate movement restriction of Trp2586.48. The workflow and key considerations described herein may be used to successfully drive future structure-based switch of functionality involving ligands and proteins beyond HU-308 and CB2R.
Results and Discussion
In Silico Probe Design
We have investigated the recently published active (PDB 8GUS, Figure 1A)48 and inactive (PDB 5ZTY, Figure 1B)49 conformations of CB2R crystallized with agonist HU-308 and antagonist/inverse agonist AM10257, respectively. Close examination of the two receptor conformations revealed that AM10257 reaches into a secondary binding pocket that features a highly conserved CWxP motif in class A GPCRs50 and moreover hosts Trp2586.48, the recently designated single residue toggle switch of CB2R activation.51
Figure 1.

Comparison of active (A, PDB 8GUS, ligand HU-308)48 and inactive (B, PDB 5ZTY, ligand AM10257)49 CB2R conformations. (C) Docking study of HU-308-derived putative inverse agonist (R)-1 in the inactive CB2R conformation (PDB 5ZTY). (R)-1 reaches into the secondary pocket occupied by the toggle switch responsible for CB2R activation, Trp2586.48 (orange), and shares binding interactions virtually identical with those of AM10257.
Comparison of the two receptor conformations combined with in silico docking suggested that a phenyl substituent introduced α to the gem-dimethyl group of HU-308 might occupy the same lipophilic subpocket as the phenyl of AM10257. The phenyl substitution creates a new C(2′) stereocenter at the pendent side chain; accordingly the explicit (S) and (R) designations preceding compound labels denote its absolute configuration. Additionally, structural features were incorporated that proved critical in our prior works to bestow excellent pharmacological profiles, yielding ligands (S)-1 and (R)-1 (Figure 2).26,28,42 Namely, terminal azide was inserted and the allylic alcohol was substituted by an amine to allow facile, stable conjugation to fluorophores and confer improved affinity and selectivity for CB2R. The novel putative HU-308-derived inverse agonist (R)-1 showed binding interactions virtually identical with those of AM10257 in the inactive CB2R conformation (Figure 1C). In particular, the C(2′) phenyl group of (R)-1 oriented toward Trp2586.48 and attained a favorable edge-to-face π-interaction similar to the phenyl of AM10257. The nearly identical interactions are essential as we have hypothesized that the impediment of the upward movement of Trp2586.48 may effectively prevent receptor activation.
Figure 2.

Design of inverse agonists (S)-1 and (R)-1.
Synthesis
Access to (S)-1 and (R)-1 that feature a phenyl group at the homobenzylic position of cannabinoid scaffolds α to a sterically demanding gem-dimethyl group is synthetically challenging and unprecedented. Prior structure–activity relationship studies on cannabinoid ligands focused almost exclusively on the easily accessible benzylic or ω-position of the pendent side chain.52 To the best of our knowledge, there is only a single report of substitution at the homobenzylic position with a methyl group in a structure of Δ8-THC that lacks the sterically congesting gem-dimethyl motif.53
The synthesis of (S)-1/(R)-1 commenced with 10, which was prepared by methylation of 3,5-dimethoxyphenylacetonitrile and subsequent treatment with phenyl lithium (Scheme 2).54,55 Introduction of the alkyl side chain to ketone 10 proved a formidable challenge due to steric hindrance. Initial attempts using established phosphonium ylide routes yielded no reaction even at elevated temperatures.56,57 An extensive screening of Grignard reagents either yielded no reaction or afforded exclusively Grignard reduction product 11. Finally, using a modified procedure for the preparation of alkyl lithiums by Punzalan,58 we employed, for the first time, 5-chloropentyl lithium to forge the tertiary alcohol 12 in 83% yield.59 Subsequent Chugaev elimination of the benzylic alcohol under mild conditions yielded 13 in 95% yield. BBr3-mediated demethylation followed by high-pressure hydrogenation over Pd/C afforded (S)-14/(R)-14 as a racemic mixture in 94% yield over two steps. The synthesis was continued with the racemate to rapidly access material for initial pharmacological evaluation. To this end, Friedel–Crafts allylation with verbenol derivative 15 followed by treatment with (MeO)2SO2 furnished methylated epimeric mixture (S)-16/(R)-16 in 50% yield over the two steps. Subsequent substitution of the primary alkyl chloride with NaN3 and hydrazine-mediated phthalimide deprotection revealed allylic amines (S)-1/(R)-1 in 73% yield. Finally, the synthesis was concluded by functionalization of the diastereomeric mixture (S)-1/(R)-1 with DY-480XL or 4-pentynoic acid to yield mixtures of epimers (S)-2/(R)-2 and (S)-3/(R)-3, respectively.
Scheme 2. Synthesis of Novel CB2R-Selective HU-308-Derived Inverse Agonists.

Reagents and conditions: (a) 1-chloro-5-iodopentane, t-BuLi, n-pentane, Et2O, −78 °C to rt, 83%; (b) KHMDS, CS2, THF, −78 °C to rt and then MeI, 40 °C, 95%; (c) BBr3, CH2Cl2, 0 °C, 97%; (d) Pd/C, H2, EtOAc, rt, 97%; (e) semipreparative SFC, (S)-14, 25%, >99% ee, (R)-14, 20%, 96% ee; (f) 15, pTsOH·H2O, CH2Cl2, rt, 64–71%; (g) (MeO)2SO2, K2CO3, acetone, rt, 78–82%; (h) NaN3, DMF, 50 °C, 88–96%; (i) N2H4·H2O, (E)/(Z)-crotyl alcohol, EtOH, 75 °C, 76–94%; (j) for conjugation conditions and details, see Supporting Information; (k) H2O, microwave irradiation, 150 °C, 99%; (l) 4-nitrobenzoyl chloride, NEt3, DMAP, CH2Cl2, rt, 85%.
We then investigated access to epimers (S)-1 and (R)-1 separately. Following introduction of verbenol fragment 15, screening of conditions to separate the resulting epimers ((S)-16/(R)-16) by silica gel chromatography, HPLC, and supercritical fluid chromatography (SFC) proved unsuccessful. Gratifyingly, we found that enantiomers (S)-14/(R)-14 could be separated by semipreparative SFC using a chiral stationary phase to yield (S)-14 and (R)-14 in >99% ee and 96% ee, respectively. Resorcinols (S)-14 and (R)-14 were then functionalized to yield enantio- and diastereomerically pure (S)-1–(S)-4 and (R)-1–(R)-9. To assign the absolute configuration at the C(2′) stereocenter, (S)-14 was converted to a p-nitrobenzoate (S)-17, whose structure was elucidated by X-ray crystallography.
Pharmacological Profiling
Saturation Binding Assays
We assessed whether the phenyl substitution in (S)-1, (R)-1, and their derivatives (S)-2–(S)-4 and (R)-2–(R)-9 impedes interaction with CB2R. To this end, time-resolved Förster resonance energy transfer (TR-FRET) binding assay was employed to determine the affinities of the new probes at room temperature.28 HEK293 membrane preparations of SNAP-Lumi4-Tb labeled hCB2R were incubated with a fluorescent probe in the presence or absence of a validated inverse agonist, SR-144,528,36 to determine its binding parameters. Gratifyingly, the epimeric mixture (S)-2/(R)-2 demonstrated good affinity for CB2R (Kd = 67.9 nM), suggesting that the C(2′) functionalization was well tolerated and validated the in silico guided design.
Encouraged by the promising result, we studied the impact of configuration at the C(2′) stereocenter on the pharmacological properties by examining each epimer individually. A 12-fold greater binding affinity for CB2R was shown by (R)-1 (Kd = 39.1 nM) in comparison to (S)-1 (Kd = 476 nM). Functionalization of (S)-1 and (R)-1 with 4-pentynoic acid was well tolerated, and the resulting compounds, (S)-3 and (R)-3, retained the stereoisomeric preference with Kd = 2.10 and 0.42 nM, respectively. Conjugation of (S)-1 and (R)-1 with DY-480XL and N-NBD yielded probes (S)-2 and (R)-2 and (S)-4 and (R)-4, respectively. Fluoroprobes (S)-2 and (S)-4 displayed inferior CB2R affinity (Kd = 162 and 158 nM, respectively) compared to the excellent binding potencies of (R)-2 and (R)-4 (Kd = 10.2 and 12.3 nM, respectively). Furthermore, strong agreement was observed between CB2R Kd and Ki values obtained by independent TR-FRET and radioligand binding assays for (R)-2 (Kd = 10.2 nM and Ki = 8.26 nM) and (R)-3 (Kd = 0.42 nM and Ki = 0.66 nM). Collectively, the results further validate the TR-FRET assay and imply that the orthosteric binding pocket of CB2R shows preference for the R-epimer of the parent compound and its derivatives.
We then set out to investigate whether the excellent CB2R affinities of (R)-1–(R)-4 are impacted by linker and fluorophore substitution. To this end, probes (R)-5–(R)-9 were prepared that feature a variety of linker lengths and fluorophores, spanning a wide range of size, lipophilicity, and membrane permeability. When tested by TR-FRET at 37 °C, fluorescein, tetramethylrhodamine (TAMRA), and Alexa488 bearing probes (R)-5, (R)-6, and (R)-7 all emerged as high affinity binders for CB2R with excellent Kd values of 30.3, 2.78, and 24.9 nM, respectively (Table 1). BODIPY 576/589 conjugate (R)-8 showed good binding potency with CB2R, Kd = 44.7 nM. Particularly remarkable was the retention of high affinity displayed by probe (R)-9 (Kd = 25.9 nM) functionalized with Alexa647. These results illustrate substantial improvement over previous work with agonists where functionalization with the highly polar Alexa488 and sterically demanding Alexa647 led to 64-fold and 611-fold drops in affinity, respectively.28 Importantly, fluoroprobes (R)-5, (R)-6, (R)-7, and (R)-9 emit robust fluorescence signals with exquisite specific binding windows when tested at the physiologically relevant temperature, 37 °C (see Figure 3 and Figure S1).
Table 1. TR-FRET-Based Profiling of Binding Affinitya.
|
Kd [nM] |
||||
|---|---|---|---|---|
| probe | dye | CB2R | CB1R | Kd ratio (CB1R/CB2R) |
| (R)-2 | DY-480XL | 18.9 | 1740 | 92 |
| (R)-5 | fluorescein | 30.3 | 1280 | 42 |
| (R)-6 | TAMRA | 2.78 | 396 | 142 |
| (R)-7 | Alexa488 | 24.9 | 3300 | 133 |
| (R)-9 | Alexa647 | 25.9 | 7050 | 272 |
Saturation binding data (Kd) were determined in a TR-FRET assay at 37 °C with membrane preparations from either hCB2R-HEK293 or hCB1R-HEK293 cells. Data shown as a mean, N = 3.
Figure 3.

TR-FRET-based saturation binding profile of (R)-7 (Alexa488) at CB2R determined at 37 °C. Nonspecific binding was determined in the presence of SR-144,528 (10 μM). Data shown as a mean ± SEM, N = 3.
The binding selectivity of fluorescent probes was tested against the closely related CB1R in a saturation binding assay at 37 °C using membrane preparations derived from HEK293 cells expressing hCB1R (Table 1). Fluoroprobes (R)-2, (R)-5, (R)-6, and (R)-7 displayed 42–142-fold selectivity for CB2R over CB1R. Notably, (R)-9 demonstrated an exceptional 272-fold preference for CB2R over CB1R. Collectively, the excellent affinity and selectivity of a range of physicochemically distinct substituents and fluorophores highlight the versatility of novel platform ligand (R)-1.
Kinetic Binding TR-FRET Assay
We were intrigued by the performances of our probes in the saturation binding assay and leveraged TR-FRET to study ligand binding kinetics at a physiologically relevant temperature, 37 °C (Table 2). The results suggest that all compounds, except (S)-1, possess dramatically slower receptor dissociation rates, koff, in comparison to the control inverse agonist, SR-144,528. Therefore, high CB2R affinity of the probes once bound stems from slow receptor dissociation rates. The probes are thus endowed with long receptor residence times, τ, an attribute that has been argued particularly important for GPCRs60 as a better suited determinant, compared to Kd, of ligand–protein interactions in living systems.61,62 Importantly, excellent agreement was found between Kd values obtained in saturation and kinetic binding experiments.
Table 2. TR-FRET-Based Kinetic Profiling at CB2Ra.
| probe | kon [106 M–1 min–1] | koff [10–2 min–1] | τ [min] | kinetic Kd [nM] |
|---|---|---|---|---|
| (S)-1 | 9.39 | 115 | 0.87 | 122 |
| (R)-1 | 12.0 | 13.9 | 7.19 | 11.6 |
| (S)-3 | 44.3 | 4.96 | 20.2 | 1.12 |
| (R)-3 | 88.7 | 2.29 | 43.7 | 0.26 |
| (R)-6 | 3.60 | 1.08 | 92.6 | 3.00 |
| (R)-7 | 1.49 | 2.37 | 42.2 | 15.9 |
| (R)-9 | 1.75 | 1.71 | 58.5 | 9.77 |
| SR-144,528 | 240 | 122 | 0.82 | 5.08 |
Kinetic Kd data were measured at 37 °C in a TR-FRET assay using hCB2R-HEK293 membrane preparations. Data shown as a mean, N = 3.
Functional Profiling: cAMP, G Protein Recruitment, and β-Arrestin
HU-308 is a potent full agonist at CB2R in the [35S]-GTPγS assay, triggers inhibition of cAMP production, promotes recruitment of β-arrestin, stimulates ERK1/2 phosphorylation, and facilitates release of Ca2+ from intracellular stores.36,41,63 Compounding evidence indicates that distinct CB2R agonists favor discrete receptor conformations, leading to preferential activation of one specific signaling pathway over another, a phenomenon known as biased agonism.36,64−66 Accordingly, we have dedicated substantial efforts to comprehensively profile the pharmacological responses elicited by the new probes across known CB2R signaling pathways.
One of the canonical signaling pathways of CB2R involves association with Gαi/o proteins, which elicit reversible inhibition of adenylyl cyclase resulting in a decrease of cellular cAMP levels and suppression of protein kinase A activity.67 Consequently, we have investigated the change in cAMP levels upon probe addition using homogeneous time-resolved fluorescence (HTRF) cAMP assay (Table 3).
Table 3. Functional Characterization in a CB2R cAMP Assaya.
| probe | pEC50 | Emax [%] |
|---|---|---|
| (S)-1 | 5.57 | –44 |
| (R)-1 | 6.95 | –44 |
| (S)-2 | 6.47 | –37 |
| (R)-2 | 7.15 | –31 |
| (S)-3 | 7.39 | –49 |
| (R)-3 | 7.48 | –55 |
| (S)-4b | 4.98 | +44 |
| (R)-4 | 5.91 | –40 |
| ago-3 | 8.47 | +112 |
Potency (pEC50) and Emax data were obtained in a cAMP HTRF assay using hCB2R-CHO cells. Data were normalized to agonist CP-55,940 response (100%) and basal level (0%), unless noted otherwise.
Data were normalized to the response of inverse agonist AM10257 (0%) and basal level (100%). Data shown as a mean, N = 3.
All compounds behaved as inverse agonists, with efficacy (Emax) ranging between −31 and −55%. Both epimers of the parent amine ligand inhibited cAMP production with the (R)-1 stereoisomer demonstrating greater potency (pEC50 = 6.95) than (S)-1 (pEC50 = 5.57). DY-480XL and alkyne functionalized probes, (R)-2 (pEC50 = 7.15) and (R)-3 (pEC50 = 7.48), were favored over (S)-2 (pEC50 = 6.47) and (S)-3 (pEC50 = 7.39) with respect to potency, albeit to a lesser degree. Interestingly, the N-NBD probes (S)-4 and (R)-4 behaved as inverse agonists only at high concentration (pEC50 = 4.98 and 5.91, respectively). As a control, we prepared HU-308-derived agonist probe ago-3 from ago-1 that features the same scaffold as (S)-3 and (R)-3 except that it lacks the C(2′) phenyl substituent (eq 1). In the cAMP assay ago-3 displayed potent receptor activation (pEC50 = 8.47, Emax = 112%). These results provide direct experimental evidence as to the critical role of the phenyl substituent in facilitating the switch in ligand functionality from agonist to inverse agonist.
 |
1 |
To complement the functional response elicited in the cAMP assay, we tested whether the probes trigger association of Gαi protein with CB2R using our recently reported bioluminescence resonance energy transfer (BRET) Gi-CASE assay.68 Membrane preparations harvested from hCB2R-HEK293 T-REx cells that genetically incorporate fluorescent NanoLuciferase donor and Venus acceptor proteins to the Gα and Gγ subunits, respectively, were incubated with a probe, and the change in BRET signal was detected. Agonist binding triggers CB2R activation and dissociation of the Gα and Gβγ subunits resulting in BRET signal reduction. Conversely, inverse agonists elicit increase in BRET intensity by stabilization of inactive CB2R conformation and G protein accumulation beyond the basal level. Compounds (S)-1, (R)-1, (S)-3, and (R)-3 were selected as representatives to circumvent interference among fluorophores in the BRET assay as previously reported.34 The results indicate that all tested compounds behave as potent inverse agonists with respect to G protein recruitment at CB2R (Figure 4 and Table 4).
Figure 4.

BRET-based Gi-CASE membrane assay to characterize G protein recruitment at CB2R. Efficacy, Emax, of the compounds is shown as a mean ± SEM, N = 3–4.
Table 4. Functional Characterization of G Protein Recruitment at CB2R in a BRET Gi-CASE Assaya.
| probe | pEC50 | Emax [%] |
|---|---|---|
| (S)-1 | 6.72 | –21 |
| (R)-1 | 6.80 | –28 |
| (S)-3 | 7.51 | –30 |
| (R)-3 | 7.22 | –25 |
| SR-144,528 | 8.23 | –45 |
Potency (pEC50) and Emax data were obtained in a Gi-CASE BRET-based assay using membrane preparations from hCB2R-HEK293 T-REx cells. Data were normalized to agonist HU-210 response (100%) and basal level (0%). Data are shown as mean, N = 3–4.
Alkyne functionalized probes (S)-3 and (R)-3 (pEC50 = 7.51 and 7.22, respectively) have shown superior potency in comparison to free amines (S)-1 and (R)-1 (pEC50 = 6.72 and 6.80, respectively). Control agonist HU-210 and inverse agonist SR-144,528 demonstrated potency consistent with previously reported [35S]-GTPγS binding assay values, further validating the experimental results (pEC50 = 8.83 and 8.23, respectively).36,69 With respect to efficacy (Emax), probes (S)-1, (R)-1, (S)-3, and (R)-3 induced functional responses between −21 and −30%. Remarkably, comparison of the effects elicited by (S)-1 in the cAMP and Gi-CASE assays (pEC50 = 5.57 and 6.72, respectively) suggests 14-fold increased potency of G protein recruitment over adenylyl cyclase inhibition, a striking bias within a CB2R–Gαi-mediated pathway.
Among the best studied G protein independent signaling pathways of CB2R is the β-arrestin cascade. β-Arrestins bind activated CB2R following receptor phosphorylation, block further G protein mediated signaling, and destine the receptor for internalization.30 Representative compounds were profiled for β-arrestin recruitment in a BRET assay where an increase of the BRET ratio corresponds to recruitment of β-arrestin. Baseline BRET signal was retained by (S)-1, (R)-1, (S)-3, and (R)-3 (Figure 5). In contrast, control agonists HU-308 and HU-210 showed expected recruitment of β-arrestin to CB2R as indicated by increases in BRET intensity (pEC50 = 8.37 and 10.0, respectively). These results imply that (S)-1, (R)-1, (S)-3, and (R)-3 do not activate CB2R toward β-arrestin recruitment.
Figure 5.

BRET-based assay to characterize β-arrestin recruitment at CB2R. L = ligand. Data are a representative of N = 3.
Phosphorylation of ERK
Activation of CB2R is associated with downstream stimulation of mitogen-activated protein kinases, such as ERK1/2, mediated via either Gβγ or β-arrestins.70,71 We have tested representative high-affinity fluorescent probe (R)-2 for CB2R-mediated phosphorylation of endogenous ERK1/2 in a CB2R inducible breast cancer HCC1954 cell line using the AlphaScreen SureFire phospho-ERK assay (Figure 6). Expression of CB2R was optionally induced with doxycycline (DOX), and after 24 h the cells were incubated with a vehicle (0.1% DMSO), CB2R selective agonist JWH13372 (1 μM), or (R)-2 (1 μM) for 30 min. Following cell lysis, lysates were incubated with a mixture containing donor and acceptor beads for 2 h at room temperature and the luminescence emission signal was measured.
Figure 6.

Live cell AlphaScreen SureFire phospho-ERK assay with CB2R inducible breast cancer HCC1954 cell line. Cells were optionally induced with doxycycline (DOX) for 24 h to stimulate expression of CB2R followed by incubation with a vehicle (0.1% DMSO), agonist JWH13372 (1 μM), or (R)-2 (1 μM) for 30 min. Statistical significance was examined by one-way ANOVA followed by Tukey’s post hoc test. ns = nonsignificant, ∗∗, p < 0.01; ∗∗∗, p < 0.001. Data are an average of three independent biological replicates.
In the absence of CB2R expression inducer (DOX), the phosphorylation levels of ERK1/2 remained the same for cells treated with a vehicle, agonist JWH133, and (R)-2. In cells induced to express CB2R with DOX (1 μg/mL), JWH133 effectively stimulated ERK1/2 phosphorylation mediated by CB2R activation, in agreement with previously reported findings.73 Addition of (R)-2 had no effect on the level of phosphorylated ERK1/2, which remained the same as for a vehicle. Data of the phospho-ERK cellular assay imply that (R)-2 does not induce phosphorylation of ERK1/2 by either CB2R-mediated Gβγ or β-arrestin signaling (or by non-CB2R-mediated pathways).
Ca2+ Signaling
Upon activation of CB2R, Ca2+ is often released from intracellular reservoirs.74−76 Our previous work reported that HU-308 and its photoswitchable derivative, azo-HU-308, increase intracellular Ca2+ in the mouse AtT-20 cell line.41 Naturally, we were intrigued to investigate the response elicited by our probes. The epimeric mixture (S)-3/(R)-3 was chosen to avoid fluorophore interference with the Fluo-4AM Ca2+ dye and test both diastereomers simultaneously.
Live AtT-20 cells overexpressing rat CB2R [AtT-20(rCB2R)] were treated with Fluo-4AM Ca2+ dye and imaged by confocal microscopy (Figure 7). Addition of (S)-3/(R)-3 (20 μM) did not elicit increase in Fluo-4AM fluorescence, whereas subsequent addition of agonist HU-308 (20 μM) triggered a robust fluorescence spike. Ionomycin was added at the end of the experiment to saturate Ca2+ levels. This result indicates that neither (S)-3 nor (R)-3 induces Ca2+ release via CB2R activation and suggests that the probes can be displaced by HU-308.
Figure 7.
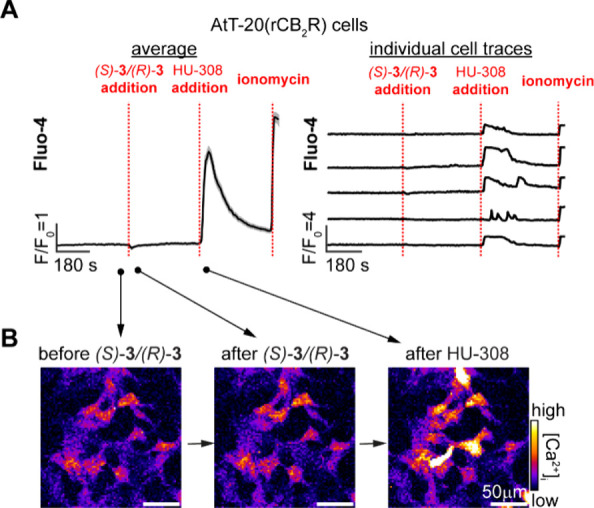
Live cell fluorescent Ca2+ imaging in rat CB2R overexpressing AtT-20 cells [AtT-20(rCB2R)] loaded with Fluo-4AM (2 μM). After initial equilibration, (S)-3/(R)-3 (20 μM) was added, followed by HU-308 (20 μM) and ionomycin (10 μM). Shown are the average responses of 200 cells (A, left), individual traces of five representative cells (A, right), and representative fluorescence images from different time points (B). Averaged data plotted as mean ± SEM, T = 4.
Fluorescence Confocal Microscopy in Live Cells
Having validated that the probes do not trigger CB2R signaling at multiple downstream pathways, we employed (R)-7 and (R)-9 to visualize CB2R by confocal fluorescence microscopy. Probes (R)-7 and (R)-9 were selected due to their bright, photostable, and extensively applied fluorophores Alexa488 and Alexa647. Additionally, the green- and red-shifted fluorescence spectra of (R)-7 and (R)-9 provide flexibility and potential for synergy with additional fluorescent proteins and small molecule dyes for multiplexed imaging studies.
First, AtT-20 cells stably expressing N-terminal SNAP-tagged human CB2R [AtT-20(SNAP-hCB2R)] were coincubated with (R)-7, Janelia Fluor SNAP-549i (JF549i), and Hoechst33342 to label CB2R, SNAP-tags, and nuclei, respectively. Confocal microscopy revealed bright fluorescence of Alexa488 and JF549i delineating the plasma membranes of AtT-20 cells (Figure 8A). Analysis of the corresponding intensity plot showed virtually identical colocalization overlap between the Alexa488 ((R)-7) and JF549i (SNAP-hCB2R) signals (Figure 8A).
Figure 8.

Confocal imaging of (R)-7 in live cell lines. (A) AtT-20(SNAP-hCB2R) cells were labeled for 15 min with (R)-7 (Alexa488, 625 nM, green), SNAP-JF549i (JF549i, 500 nM, red), and Hoechst33342 (Hoechst, 1 μM, blue) to visualize CB2R, SNAP-tags, and nuclei, respectively. Fluorescence intensity profiles across the white line for Alexa488, JF549i, and Hoechst33342 are shown on the right. (B) AtT-20(rCB2R) cells (left) and AtT-20(WT) cells (right) were treated with (R)-7 (625 nM, green) and Hoechst33342 (1 μM, blue) for 15 min and imaged by confocal microscopy. (C) Live BV-2 microglial cells that endogenously express CB2R were incubated with (R)-7 (2.5 μM, green) and Hoechst33342 (1 μM, blue) for 15 min and imaged by confocal microscopy.
Since many cannabinoid ligands tend to accumulate in plasma membranes due to their lipophilic nature, specificity of (R)-7 and (R)-9 for CB2R was evaluated using AtT-20(rCB2R) and AtT-20 wild-type (WT) cells, which do not express CB2R.77 AtT-20(rCB2R) and AtT-20(WT) cells were incubated with (R)-7 and Hoechst33342 and imaged by confocal microscopy. A robust Alexa488 fluorescence signal was detected at the plasma membrane of AtT-20(rCB2R) cells (Figure 8B, left). In stark contrast, AtT-20(WT) cells showed only minimal background fluorescence with no signal stemming from the cellular membrane (Figure 8B, right). These results confirm that (R)-7 specifically labels CB2R at the plasma membrane.
Encouraged by the promising results with (R)-7 in cells overexpressing CB2R, we proceeded to investigate the probes’ ability to detect CB2R at endogenous expression levels. To this end, the murine derived BV-2 microglial cell line was selected due to its extensive use as a high fidelity, primary microglia culture model78 that was applied in the study of neurodegeneration and neuroinflammation.79−81 Importantly, BV-2 cells endogenously express CB2R.82,83 Following incubation of BV-2 cells with (R)-7 and Hoechst33342, an intense Alexa488 signal was observed at the plasma membrane across BV-2 cells (Figure 8C). These results confirm that (R)-7 can visualize CB2R at endogenous expression levels. Importantly, when (R)-9 (Alexa647) was subjected to analogous experiments, it demonstrated equal specificity for CB2R in AtT-20 cells (see Figure S2A,B) combined with strong signal intensity in the BV-2 microglial cell line (see Figure S2C). Finally, these data imply that the performances of (R)-7 and (R)-9 remain uncompromised by interspecies differences and the probes can be employed to investigate both human and murine orthologs of CB2R.
Molecular Dynamics Simulations Unravel Pharmacophore Determinants of Receptor Activation
Molecular dynamics (MD) studies were performed in a membrane environment with inverse agonists (S)-3 and (R)-3 and their agonist counterpart ago-3 to contrast their interactions with CB2R at a molecular level and elucidate their orthogonal functional profiles. To this end, the X-ray structure of CB2R in an inactive state in complex with AM10257 (PDB 5ZTY) was selected as a starting point for 1 μs MD simulations to assess ligand stability and identify rearrangements within the binding site.
All three ligands adopt an L-shape conformation with the pendent alkyl chain hosted in a cleft formed by Phe183ECL2, Tyr1905.39, Trp1945.43, and Thr1143.33 (see Figure 9 and Figures S3 and S4). The resorcinol engages in π–π interactions with Phe183ECL2, while the pinene core is surrounded by aromatic residues (Phe183ECL2, Phe912.61, and Phe942.64). An rmsd plot of ago-3 following a best fit of protein backbone shows an initial rearrangement followed by a periodic “breathinglike” motion of the resorcinol and alkyl chain that oscillates between bent and flat conformations (see Figures S3 and S5). Furthermore, in accord with our earlier work,42 in the CB2R–ago-3 complex the amide group of ago-3 forms a stable hydrogen bond with the carbonyl of Ser902.60.
Figure 9.

Representative frames from molecular dynamics (MD) simulations of CB2R (PDB 5ZTY) in complex with (S)-3 or (R)-3. Superimposition at level of protein backbone of (A) CB2R X-ray structure with AM10257 (light gray) and (R)-3 MD complex (ligand in gold and protein in light yellow) and of (B) the two inverse agonist complexes (S)-3 (ligand in blue and protein light blue) and (R)-3 (ligand in gold and protein in light yellow).
In agreement with the in silico docking (Figure 1), the MD simulations suggest the C(2′) phenyl rings of (S)-3 and (R)-3 mirror that of AM10257 and engage in π–π contacts with Phe1173.36 and Trp2586.48 (see Figure 9A and Figure S6). In contrast to ago-3, no initial rearrangement in the binding poses of (S)-3 and (R)-3 was observed in their rmsd plots (see Figure S7). Ligands (S)-3 and (R)-3 share similar binding modes of the pinene-resorcinol core; however, significant differences are observed in the orientation adopted by gem-dimethyl groups and the C(2′) phenyl rings (see Figure 9B and Figure S8). In particular, the gem-dimethyl group of (S)-3 is rotated clockwise compared to that of (R)-3 and the C(2′) phenyl of (S)-3 is rotated toward Leu2626.44, inducing a minor displacement of helix H6, while that of (R)-3 protrudes deeper toward Trp2586.48. In both cases, the conformation of the Trp2586.48 toggle switch is restricted and CB2R is thus stabilized in its inactive state, in stark contrast to the interactions observed with the agonist complex.
Superimposition of CB2R protein backbone employing the X-ray structure with AM10257 and the representative MD frames in complex with either (S)-3 (see Figure S6) or (R)-3 (Figure 9A) imply that (R)-3 more closely resembles the binding mode of AM10257 in the crystallized complex. In particular, the secondary binding pocket featuring Trp2586.48 and the surrounding residues Phe1173.36 and Leu2626.44 are in excellent agreement between AM10257 and (R)-3.
Finally, the difference in free energy of binding (ΔΔG) was determined for (R)-3 and (S)-3 using molecular mechanics/Poisson–Boltzmann (generalized Born) surface area (MM/PB(GB)SA) calculations (see Table S1). The data imply that binding of (R)-3 is more stable by −0.76 kcal mol–1 (MM/GBSA) and −0.40 kcal mol–1 (MM/PBSA) in comparison to (S)-3. Notably, the calculated ΔΔG values are in agreement with the experimental difference of ΔΔG = −0.9 kcal mol–1 for epimers (S)-3 and (R)-3.
Conclusion
This study describes the in silico guided, structure-based switch of functionality from agonist to inverse agonist of HU-308, a ligand extensively applied to unravel CB2R pharmacology and currently investigated in clinical trials. The novel inverse agonist platform ligands (S)-1 and (R)-1 demonstrated high binding affinity for CB2R and selectivity against CB1R that was retained upon functionalization with a range of chemically distinct substituents and fluorophores. The functional response exerted on CB2R by (S)-1, (R)-1, and their derivatives was evaluated by HTRF and BRET and implied an inverse agonist profile in cAMP as well as G protein recruitment assays, and no induction of β-arrestin–receptor association. Live cell experiments with (R)-2 and (S)-3/(R)-3 demonstrated that the probes do not activate CB2R toward ERK1/2 phosphorylation and Ca2+ signaling pathways, respectively. Fluorescence microscopy experiments with (R)-7 and (R)-9 in AtT-20 cells expressing human and rat CB2R isoforms demonstrated excellent target specificity and species translatability. Treatment of the BV-2 microglial cell line with (R)-7 and (R)-9 allowed imaging of endogenous CB2R in live cells. Finally, MD simulations with (S)-3, (R)-3, and ago-3 corroborate the critical role of the C(2′) phenyl substituent in conferring the functional profile by modulating the CB2R toggle switch Trp2586.48 of the CWxP motif.
More broadly, this work discloses the first ligand platform for CB2R that retains its inverse agonist functional profile, affinity, and selectivity independent of its conjugation to a range of diverse functional groups. The probes introduce a long-sought-after complementarity to an agonist-dominated toolkit to study, elucidate and unlock the full therapeutic potential of CB2R. Moreover, the platform ligands promise broad application and synergy with previously published work which awaited discovery of an inverse agonist.26 The exponential rise in resolved structures of class A GPCRs, many of which are available in inactive, intermediate, and fully active states, has enabled unprecedented insight into the mechanism of receptor activation.50,84,85 Thus, the strategy and experimental framework disclosed herein may aid in the structure-based design of agonists, antagonists, and inverse agonists for GPCRs beyond CB2R.
Acknowledgments
We thank Eric Bald for his help with chiral separations and Isabelle Kaufmann for her assistance with logistics and administration. We are grateful to Raphael Bigler, Paolo Tosatti, Stephan Bachmann, Kurt Püntener, and Manuela Müller for their expertise, insight, and experimental help with high-pressure hydrogenations. We thank René Arnold, Rainer Frankenstein, and Stephan Burkhardt for their help with NMR measurements, Jan Kovacovic for his expertise with high-pressure hydrogenation, and the MoBiAS team for MS analysis. We are grateful to Nils Trapp and Michael Solar for X-ray crystallographic analysis. M.K. and R.C.S. gratefully acknowledge a fellowship by the Scholarship Fund of the Swiss Chemical Industry (SSCI). MD studies have been funded by project code PIR01_00011 “IBISCo”, PON 2014-2020. We would like to thank Jürg Gertsch (University of Bern, Switzerland) for kindly providing the CB2R expressing cells. Research in the laboratory of C.W.G. has been supported by the Austrian Science Fund (FWF) through project P32109. N.T. has been supported by a short-term scientific mission (STSM) grant from the EU COST Action CA 18133 (ERNEST). B.K. gratefully acknowledges the SNF fund 200020_188538. T.H. has been supported by the National Key Research and Development Program of China grant 2022YFA1302903. D.A.S. and D.B.V. gratefully acknowledge funding by Roche postdoctoral fellowship RPF-551.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.3c01461.
Experimental procedures and characterization data for all compounds (PDF)
The authors declare the following competing financial interest(s): M.K., R.C.S., B.K., W.G., U.G., and E.M.C. have filed a patent on CB2R selective modulators and fluorescent probes.
Supplementary Material
References
- Maccarrone M.; Di Marzo V.; Gertsch J.; Grether U.; Howlett A. C.; Hua T.; Makriyannis A.; Piomelli D.; Ueda N.; van der Stelt M. Goods and Bads of the Endocannabinoid System as a Therapeutic Target: Lessons Learned after 30 Years. Pharmacol. Rev. 2023, 75, 885–958. 10.1124/pharmrev.122.000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S.; Thomas K. L.; Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Goncalves E. D.; Dutra R. C. Cannabinoid receptors as therapeutic targets for autoimmune diseases: where do we stand?. Drug Discovery Today 2019, 24, 1845–1853. 10.1016/j.drudis.2019.05.023. [DOI] [PubMed] [Google Scholar]
- Rossi F.; Punzo F.; Umano G. R.; Argenziano M.; Miraglia Del Giudice E. Role of Cannabinoids in Obesity. Int. J. Mol. Sci. 2018, 19, 2690–2700. 10.3390/ijms19092690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumawat V. S.; Kaur G. Therapeutic potential of cannabinoid receptor 2 in the treatment of diabetes mellitus and its complications. Eur. J. Pharmacol. 2019, 862, 172628–172633. 10.1016/j.ejphar.2019.172628. [DOI] [PubMed] [Google Scholar]
- Shang Y.; Tang Y. The central cannabinoid receptor type-2 (CB2) and chronic pain. Int. J. Neurosci. 2017, 127, 812–823. 10.1080/00207454.2016.1257992. [DOI] [PubMed] [Google Scholar]
- Khan H.; Ghori F. K.; Ghani U.; Javed A.; Zahid S. Cannabinoid and endocannabinoid system: a promising therapeutic intervention for multiple sclerosis. Mol. Biol. Rep. 2022, 49, 5117–5131. 10.1007/s11033-022-07223-5. [DOI] [PubMed] [Google Scholar]
- Lunn C. A.; Reich E. P.; Fine J. S.; Lavey B.; Kozlowski J. A.; Hipkin R. W.; Lundell D. J.; Bober L. Biology and therapeutic potential of cannabinoid CB2 receptor inverse agonists. Br. J. Pharmacol. 2008, 153, 226–239. 10.1038/sj.bjp.0707480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A.; Heldt S. A.; Presley C. S.; Guley N. H.; Elberger A. J.; Deng Y.; D’Surney L.; Rogers J. T.; Ferrell J.; Bu W.; Del Mar N.; Honig M. G.; Gurley S. N.; Moore B. M. II Motor, visual and emotional deficits in mice after closed-head mild traumatic brain injury are alleviated by the novel CB2 inverse agonist SMM-189. Int. J. Mol. Sci. 2015, 16, 758–787. 10.3390/ijms16010758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu W.; Ren H.; Deng Y.; Del Mar N.; Guley N. M.; Moore B. M.; Honig M. G.; Reiner A. Mild Traumatic Brain Injury Produces Neuron Loss That Can Be Rescued by Modulating Microglial Activation Using a CB2 Receptor Inverse Agonist. Front. Neurosci. 2016, 10, 449. 10.3389/fnins.2016.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alghamdi S. S.; Mustafa S. M.; Moore B. M. II Synthesis and biological evaluation of a ring analogs of the selective CB2 inverse agonist SMM-189. Bioorg. Med. Chem. Lett. 2021, 33, 116035. 10.1016/j.bmc.2021.116035. [DOI] [PubMed] [Google Scholar]
- TT-816 as Monotherapy or in Combination With a PD-1 Inhibitor in Patients With Advanced Cancers (SEABEAM) (MK3475-E88). ClincialTrials.gov. https://clinicaltrials.gov/study/NCT05525455 (accessed 2023-11-07).
- A Study to Investigate the Efficacy and Safety of RG7774 in Patients With Diabetes Mellitus Type 1 or Type 2 With Treatment-Naive Diabetic Retinopathy (CANBERRA). ClincialTrials.gov. https://www.clinicaltrials.gov/study/NCT04265261 (accessed 2023-09-19).
- NTRX-07. NeuroTherapia. https://www.neurotherapia.com/research (accessed 2024-01-04).
- Safety and Tolerability of NTRX-07 in Healthy Volunteers. ClincialTrials.gov. https://www.clinicaltrials.gov/study/NCT04375436 (accessed 2024-01-04).
- CNTX-6016. Centrexion. https://centrexion.com/science/pipeline/ (accessed 2024-01-04).
- Safety, Tolerability & Pharmacokinetics Study of CNTX-6016 in Healthy Subjects and Subjects With PDN. ClincialTrials.gov. https://clinicaltrials.gov/study/NCT04857957 (accessed 2024-01-04).
- Olorinab - Pfizer. AdisInsight.https://adisinsight.springer.com/drugs/800039670 (accessed 2024-01-04).
- ARDS003. Tetra Bio-Pharma. https://tetrabiopharma.com/pipeline/ (accessed 2023-09-19).
- Whiting Z. M.; Yin J.; de la Harpe S. M.; Vernall A. J.; Grimsey N. L. Developing the Cannabinoid Receptor 2 (CB2) pharmacopoeia: past, present, and future. Trends Pharmacol. Sci. 2022, 43, 754–771. 10.1016/j.tips.2022.06.010. [DOI] [PubMed] [Google Scholar]
- Brownjohn P. W.; Ashton J. C. Spinal cannabinoid CB2 receptors as a target for neuropathic pain: an investigation using chronic constriction injury. Neuroscience 2012, 203, 180–193. 10.1016/j.neuroscience.2011.12.028. [DOI] [PubMed] [Google Scholar]
- Cecyre B.; Thomas S.; Ptito M.; Casanova C.; Bouchard J. F. Evaluation of the specificity of antibodies raised against cannabinoid receptor type 2 in the mouse retina. Naunyn-Schmiedebergs Arch. Pharmacol. 2014, 387, 175–184. 10.1007/s00210-013-0930-8. [DOI] [PubMed] [Google Scholar]
- Marchalant Y.; Brownjohn P. W.; Bonnet A.; Kleffmann T.; Ashton J. C. Validating Antibodies to the Cannabinoid CB2 Receptor: Antibody Sensitivity Is Not Evidence of Antibody Specificity. J. Histochem. Cytochem. 2014, 62, 395–404. 10.1369/0022155414530995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. Y.; Shen H.; Jordan C. J.; Liu Q. R.; Gardner E. L.; Bonci A.; Xi Z. X. CB2 receptor antibody signal specificity: correlations with the use of partial CB2-knockout mice and anti-rat CB2 receptor antibodies. Acta Pharmacol. Sin. 2019, 40, 398–409. 10.1038/s41401-018-0037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. J.; Gao M.; Gao F. F.; Su Q. X.; Wu J. Brain cannabinoid receptor 2: expression, function and modulation. Acta Pharmacol. Sin. 2017, 38, 312–316. 10.1038/aps.2016.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosar M.; Sykes D. A.; Viray A. E. G.; Vitale R. M.; Sarott R. C.; Ganzoni R. L.; Onion D.; Tobias J. M.; Leippe P.; Ullmer C.; Zirwes E. A.; Guba W.; Grether U.; Frank J. A.; Veprintsev D. B.; Carreira E. M. Platform Reagents Enable Synthesis of Ligand-Directed Covalent Probes: Study of Cannabinoid Receptor 2 in Live Cells. J. Am. Chem. Soc. 2023, 145, 15094–15108. 10.1021/jacs.2c13629. [DOI] [PubMed] [Google Scholar]
- Gazzi T.; Brennecke B.; Atz K.; Korn C.; Sykes D.; Forn-Cuni G.; Pfaff P.; Sarott R. C.; Westphal M. V.; Mostinski Y.; Mach L.; Wasinska-Kalwa M.; Weise M.; Hoare B. L.; Miljuš T.; Mexi M.; Roth N.; Koers E. J.; Guba W.; Alker A.; Rufer A. C.; Kusznir E. A.; Huber S.; Raposo C.; Zirwes E. A.; Osterwald A.; Pavlovic A.; Moes S.; Beck J.; Nettekoven M.; Benito-Cuesta I.; Grande T.; Drawnel F.; Widmer G.; Holzer D.; van der Wel T.; Mandhair H.; Honer M.; Fingerle J.; Scheffel J.; Broichhagen J.; Gawrisch K.; Romero J.; Hillard C. J.; Varga Z. V.; van der Stelt M.; Pacher P.; Gertsch J.; Ullmer C.; McCormick P. J.; Oddi S.; Spaink H. P.; Maccarrone M.; Veprintsev D. B.; Carreira E. M.; Grether U.; Nazaré M. Detection of cannabinoid receptor type 2 in native cells and zebrafish with a highly potent, cell-permeable fluorescent probe. Chem. Sci. 2022, 13, 5539–5545. 10.1039/D1SC06659E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarott R. C.; Westphal M. V.; Pfaff P.; Korn C.; Sykes D. A.; Gazzi T.; Brennecke B.; Atz K.; Weise M.; Mostinski Y.; Hompluem P.; Koers E.; Miljus T.; Roth N. J.; Asmelash H.; Vong M. C.; Piovesan J.; Guba W.; Rufer A. C.; Kusznir E. A.; Huber S.; Raposo C.; Zirwes E. A.; Osterwald A.; Pavlovic A.; Moes S.; Beck J.; Benito-Cuesta I.; Grande T.; Ruiz de Martin Esteban S.; Yeliseev A.; Drawnel F.; Widmer G.; Holzer D.; van der Wel T.; Mandhair H.; Yuan C. Y.; Drobyski W. R.; Saroz Y.; Grimsey N.; Honer M.; Fingerle J.; Gawrisch K.; Romero J.; Hillard C. J.; Varga Z. V.; van der Stelt M.; Pacher P.; Gertsch J.; McCormick P. J.; Ullmer C.; Oddi S.; Maccarrone M.; Veprintsev D. B.; Nazare M.; Grether U.; Carreira E. M. Development of High-Specificity Fluorescent Probes to Enable Cannabinoid Type 2 Receptor Studies in Living Cells. J. Am. Chem. Soc. 2020, 142, 16953–16964. 10.1021/jacs.0c05587. [DOI] [PubMed] [Google Scholar]
- Grimsey N. L.; Goodfellow C. E.; Dragunow M.; Glass M. Cannabinoid receptor 2 undergoes Rab5-mediated internalization and recycles via a Rab11-dependent pathway. Biochim. Biophys. Acta 2011, 1813, 1554–1560. 10.1016/j.bbamcr.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zheng C.; Qian J.; Sutton S. W.; Wang Z.; Lv J.; Liu C.; Zhou N. Involvement of beta-arrestin-2 and clathrin in agonist-mediated internalization of the human cannabinoid CB2 receptor. Curr. Mol. Pharmacol. 2015, 7, 67–80. 10.2174/1874467207666140714115824. [DOI] [PubMed] [Google Scholar]
- Weis W. I.; Kobilka B. K. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. 10.1146/annurev-biochem-060614-033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A. G.; MacDonald C.; Glass M.; Hook S.; Tyndall J. D. A.; Vernall A. J. Alkyl indole-based cannabinoid type 2 receptor tools: Exploration of linker and fluorophore attachment. Eur. J. Med. Chem. 2018, 145, 770–789. 10.1016/j.ejmech.2017.11.076. [DOI] [PubMed] [Google Scholar]
- Sexton M.; Woodruff G.; Horne E. A.; Lin Y. H.; Muccioli G. G.; Bai M.; Stern E.; Bornhop D. J.; Stella N. NIR-mbc94, a fluorescent ligand that binds to endogenous CB(2) receptors and is amenable to high-throughput screening. Chem. Biol. 2011, 18, 563–8. 10.1016/j.chembiol.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Oyagawa C. R. M.; Macdonald C.; Grimsey N. L.; Glass M.; Vernall A. J. Chromenopyrazole-based High Affinity, Selective Fluorescent Ligands for Cannabinoid Type 2 Receptor. ACS Med. Chem. Lett. 2019, 10, 209–214. 10.1021/acsmedchemlett.8b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanus L.; Tchilibon S.; Shiloah S.; Goldenberg D.; Horowitz M.; Pertwee R. G.; Ross R. A.; Mechoulam R.; Breuer A.; Fride E. Fride HU-308: A specific agonist for CB2, a peripheral cannabinoid receptor. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 14228–14233. 10.1073/pnas.96.25.14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soethoudt M.; Grether U.; Fingerle J.; Grim T. W.; Fezza F.; de Petrocellis L.; Ullmer C.; Rothenhausler B.; Perret C.; van Gils N.; Finlay D.; MacDonald C.; Chicca A.; Gens M. D.; Stuart J.; de Vries H.; Mastrangelo N.; Xia L.; Alachouzos G.; Baggelaar M. P.; Martella A.; Mock E. D.; Deng H.; Heitman L. H.; Connor M.; Di Marzo V.; Gertsch J.; Lichtman A. H.; Maccarrone M.; Pacher P.; Glass M.; van der Stelt M. Cannabinoid CB2 receptor ligand profiling reveals biased signalling and off-target activity. Nat. Commun. 2017, 8, 13958. 10.1038/ncomms13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBuda C. J.; Koblish M.; Little P. J. Cannabinoid CB2 receptor agonist activity in the hindpaw incision model of postoperative pain. Eur. J. Pharmacol. 2005, 527, 172–174. 10.1016/j.ejphar.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Ossola C. A.; Surkin P. N.; Mohn C. E.; Elverdin J. C.; Fernandez-Solari J. Anti-Inflammatory and Osteoprotective Effects of Cannabinoid-2 Receptor Agonist HU-308 in a Rat Model of Lipopolysaccharide-Induced Periodontitis. J. Periodontol. 2016, 87, 725–734. 10.1902/jop.2016.150612. [DOI] [PubMed] [Google Scholar]
- Rentsch P.; Stayte S.; Egan T.; Clark I.; Vissel B. Targeting the cannabinoid receptor CB2 in a mouse model of l-dopa induced dyskinesia. Neurobiol. Dis. 2020, 134, 104646–104659. 10.1016/j.nbd.2019.104646. [DOI] [PubMed] [Google Scholar]
- Espejo-Porras F.; Garcia-Toscano L.; Rodriguez-Cueto C.; Santos-Garcia I.; de Lago E.; Fernandez-Ruiz J. Targeting glial cannabinoid CB(2) receptors to delay the progression of the pathological phenotype in TDP-43 (A315T) transgenic mice, a model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2019, 176, 1585–1600. 10.1111/bph.14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarott R. C.; Viray A. E. G.; Pfaff P.; Sadybekov A.; Rajic G.; Katritch V.; Carreira E. M.; Frank J. A. Optical Control of Cannabinoid Receptor 2-Mediated Ca(2+) Release Enabled by Synthesis of Photoswitchable Probes. J. Am. Chem. Soc. 2021, 143, 736–743. 10.1021/jacs.0c08926. [DOI] [PubMed] [Google Scholar]
- Westphal M. V.; Sarott R. C.; Zirwes E. A.; Osterwald A.; Guba W.; Ullmer C.; Grether U.; Carreira E. M. Highly Selective, Amine-Derived Cannabinoid Receptor 2 Probes. Chem.—Eur. J. 2020, 26, 1380–1387. 10.1002/chem.201904584. [DOI] [PubMed] [Google Scholar]
- Lee J.; Schapira M. The Promise and Peril of Chemical Probe Negative Controls. ACS Chem. Biol. 2021, 16, 579–585. 10.1021/acschembio.1c00036. [DOI] [PubMed] [Google Scholar]
- Bunnage M. E.; Chekler E. L.; Jones L. H. Target validation using chemical probes. Nat. Chem. Biol. 2013, 9, 195–199. 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- Congreve M.; de Graaf C.; Swain N. A.; Tate C. G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. 10.1016/j.cell.2020.03.003. [DOI] [PubMed] [Google Scholar]
- Hauser A. S.; Attwood M. M.; Rask-Andersen M.; Schioth H. B.; Gloriam D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery 2017, 16, 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivella M.; Caltabiano G.; Cordomi A. The role of Cysteine 6.47 in class A GPCRs. BMC Struct. Biol. 2013, 13, 3. 10.1186/1472-6807-13-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Chang H.; Bouma J.; de Paus L. V.; Mukhopadhyay P.; Paloczi J.; Mustafa M.; van der Horst C.; Kumar S. S.; Wu L.; Yu Y.; van den Berg R.; Janssen A. P. A.; Lichtman A.; Liu Z. J.; Pacher P.; van der Stelt M.; Heitman L. H.; Hua T. Structural basis of selective cannabinoid CB(2) receptor activation. Nat. Commun. 2023, 14, 1447. 10.1038/s41467-023-37112-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Hua T.; Vemuri K.; Ho J. H.; Wu Y.; Wu L.; Popov P.; Benchama O.; Zvonok N.; Locke K.; Qu L.; Han G. W.; Iyer M. R.; Cinar R.; Coffey N. J.; Wang J.; Wu M.; Katritch V.; Zhao S.; Kunos G.; Bohn L. M.; Makriyannis A.; Stevens R. C.; Liu Z. J. Crystal Structure of the Human Cannabinoid Receptor CB2. Cell 2019, 176, 459–467. 10.1016/j.cell.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q.; Yang D.; Wu M.; Guo Y.; Guo W.; Zhong L.; Cai X.; Dai A.; Jang W.; Shakhnovich E. I.; Liu Z. J.; Stevens R. C.; Lambert N. A.; Babu M. M.; Wang M. W.; Zhao S. Common activation mechanism of class A GPCRs. eLife 2019, 8, e50279. 10.7554/eLife.50279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T.; Li X.; Wu L.; Iliopoulos-Tsoutsouvas C.; Wang Y.; Wu M.; Shen L.; Brust C. A.; Nikas S. P.; Song F.; Song X.; Yuan S.; Sun Q.; Wu Y.; Jiang S.; Grim T. W.; Benchama O.; Stahl E. L.; Zvonok N.; Zhao S.; Bohn L. M.; Makriyannis A.; Liu Z. J. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665. 10.1016/j.cell.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bow E. W.; Rimoldi J. M. The Structure-Function Relationships of Classical Cannabinoids: CB1/CB2Modulation. Perspect. Med. Chem. 2016, 8, 17–39. 10.4137/PMC.S32171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman J. W.; Lainton J. A. H.; Banner W. K.; Duncan S. G. Jr.; Jordan R. D.; Yu S.; Martin B. R.; Wiley J. L.; Compton D. R.; Dai D. Side chain methyl analogues of Δ8-THC. Tetrahedron 1997, 53, 1557–1576. 10.1016/S0040-4020(96)01134-9. [DOI] [Google Scholar]
- Sharma R.; Nikas S. P.; Paronis C. A.; Wood J. T.; Halikhedkar A.; Guo J. J.; Thakur G. A.; Kulkarni S.; Benchama O.; Raghav J. G.; Gifford R. S.; Jarbe T. U.; Bergman J.; Makriyannis A. Controlled-deactivation cannabinergic ligands. J. Med. Chem. 2013, 56, 10142–10157. 10.1021/jm4016075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrie J. E. T. Preparation and Properties of Unsymmetrical Benzoins and Related Compounds. Tetrahedron 1998, 54, 5407–5416. 10.1016/S0040-4020(98)00214-2. [DOI] [Google Scholar]
- Zanato C.; Pelagalli A.; Marwick K. F.; Piras M.; Dall’Angelo S.; Spinaci A.; Pertwee R. G.; Wyllie D. J.; Hardingham G. E.; Zanda M. Synthesis, radio-synthesis and in vitro evaluation of terminally fluorinated derivatives of HU-210 and HU-211 as novel candidate PET tracers. Org. Biomol. Chem. 2017, 15, 2086–2096. 10.1039/C6OB02796B. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Iliopoulos-Tsoutsouvas C.; Tong F.; Brust C. A.; Keenan C. M.; Raghav J. G.; Hua T.; Wu S.; Ho J. H.; Wu Y.; Grim T. W.; Zvonok N.; Thakur G. A.; Liu Z. J.; Sharkey K. A.; Bohn L. M.; Nikas S. P.; Makriyannis A. Novel Functionalized Cannabinoid Receptor Probes: Development of Exceptionally Potent Agonists. J. Med. Chem. 2021, 64, 3870–3884. 10.1021/acs.jmedchem.0c02053. [DOI] [PubMed] [Google Scholar]
- Bailey W. F.; Punzalan E. R. Convenient General Method for the Preparation of Primary Alkyllithiums by Lithium-Iodine Exchange. J. Org. Chem. 1990, 55, 5404–5406. 10.1021/jo00306a021. [DOI] [Google Scholar]
- Self-immolation of 5-chloropentyl lithium was averted by preparation at −78 °C prior to electrophile addition and optimization of t-BuLi equivalents. See the Supporting Information for details.
- Hoffmann C.; Castro M.; Rinken A.; Leurs R.; Hill S. J.; Vischer H. F. Ligand Residence Time at G-protein-Coupled Receptors-Why We Should Take Our Time To Study It. Mol. Pharmacol. 2015, 88, 552–560. 10.1124/mol.115.099671. [DOI] [PubMed] [Google Scholar]
- Copeland R. A. The drug-target residence time model: a 10-year retrospective. Nat. Rev. Drug Discovery 2016, 15, 87–95. 10.1038/nrd.2015.18. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discovery 2006, 5, 730–739. 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Sophocleous A.; Landao-Bassonga E.; Van’t Hof R. J.; Idris A. I.; Ralston S. H. The type 2 cannabinoid receptor regulates bone mass and ovariectomy-induced bone loss by affecting osteoblast differentiation and bone formation. Endocrinology 2011, 152, 2141–2149. 10.1210/en.2010-0930. [DOI] [PubMed] [Google Scholar]
- Dhopeshwarkar A.; Mackie K. CB2 Cannabinoid receptors as a therapeutic target-what does the future hold?. Mol. Pharmacol. 2014, 86, 430–437. 10.1124/mol.114.094649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhopeshwarkar A.; Mackie K. Functional Selectivity of CB2 Cannabinoid Receptor Ligands at a Canonical and Noncanonical Pathway. J. Pharmacol. Exp. Ther. 2016, 358, 342–351. 10.1124/jpet.116.232561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood B. K.; Wager-Miller J.; Haskins C.; Straiker A.; Mackie K. Functional selectivity in CB(2) cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB(2) ligands. Mol. Pharmacol. 2012, 81, 250–263. 10.1124/mol.111.074013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New Tools to Interrogate Endocannabinoid Signalling: From Natural Compounds to Synthetic Drugs; Maccarrone M., Ed.; The Royal Society of Chemistry: 2021. [Google Scholar]
- Scott-Dennis M.; Rafani F. A.; Yi Y.; Perera T.; Harwood C. R.; Guba W.; Rufer A. C.; Grether U.; Veprintsev D. B.; Sykes D. A. Development of a membrane-based Gi-CASE biosensor assay for profiling compounds at cannabinoid receptors. Front. Pharmacol. 2023, 14, 1158091. 10.3389/fphar.2023.1158091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manera C.; Malfitano A. M.; Parkkari T.; Lucchesi V.; Carpi S.; Fogli S.; Bertini S.; Laezza C.; Ligresti A.; Saccomanni G.; Savinainen J. R.; Ciaglia E.; Pisanti S.; Gazzerro P.; Di Marzo V.; Nieri P.; Macchia M.; Bifulco M. New quinolone- and 1,8-naphthyridine-3-carboxamides as selective CB2 receptor agonists with anticancer and immuno-modulatory activity. Eur. J. Med. Chem. 2015, 97, 10–18. 10.1016/j.ejmech.2015.04.034. [DOI] [PubMed] [Google Scholar]
- Bouaboula M.; Poinot-Chazel C.; Marchand J.; Canat X.; Bourrie B.; Rinaldi-Carmona M.; Calandra B.; Le Fur G.; Casellas P. Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor. Involvement of both mitogen-activated protein kinase and induction of Krox-24 expression. Eur. J. Biochem. 1996, 237, 704–711. 10.1111/j.1432-1033.1996.0704p.x. [DOI] [PubMed] [Google Scholar]
- Howlett A. C.; Abood M. E. CB1 and CB2 Receptor Pharmacology. Adv. Pharmacol. 2017, 80, 169–206. 10.1016/bs.apha.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman J. W.; Liddle J.; Yu S.; Aung M. M.; Abood M. E.; Wiley J. L.; Martin B. R. 3-(1’,1’-Dimethylbutyl)-1-deoxy-delta8-THC and related compounds: synthesis of selective ligands for the CB2 receptor. Biorg. Med. Chem. 1999, 7, 2905–2914. 10.1016/S0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- Hashiesh H. M.; Sharma C.; Goyal S. N.; Jha N. K.; Ojha S. Pharmacological Properties, Therapeutic Potential and Molecular Mechanisms of JWH133, a CB2 Receptor-Selective Agonist. Front. Pharmacol. 2021, 12, 702675. 10.3389/fphar.2021.702675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti C.; Kipmen-Korgun D.; Osibow K.; Malli R.; Graier W. F. Anandamide initiates Ca(2+) signaling via CB2 receptor linked to phospholipase C in calf pulmonary endothelial cells. Br. J. Pharmacol. 2003, 140, 1351–1362. 10.1038/sj.bjp.0705529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan-Pico P.; Fuentes E.; Bermudez-Silva F. J.; Javier Diaz-Molina F.; Ripoll C.; Rodriguez de Fonseca F.; Nadal A. Cannabinoid receptors regulate Ca(2+) signals and insulin secretion in pancreatic beta-cell. Cell Calcium 2006, 39, 155–162. 10.1016/j.ceca.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Brailoiu G. C.; Deliu E.; Marcu J.; Hoffman N. E.; Console-Bram L.; Zhao P.; Madesh M.; Abood M. E.; Brailoiu E. Differential activation of intracellular versus plasmalemmal CB2 cannabinoid receptors. Biochemistry 2014, 53, 4990–4999. 10.1021/bi500632a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood B. K.; Lopez J.; Wager-Miller J.; Mackie K.; Straiker A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics 2011, 12, 14. 10.1186/1471-2164-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn A.; Lund S.; Hedtjarn M.; Schrattenholz A.; Porzgen P.; Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 2009, 26, 83–94. 10.14573/altex.2009.2.83. [DOI] [PubMed] [Google Scholar]
- Olajide O. A.; Iwuanyanwu V. U.; Adegbola O. D.; Al-Hindawi A. A. SARS-CoV-2 Spike Glycoprotein S1 Induces Neuroinflammation in BV-2 Microglia. Molecular Neurobiology 2022, 59, 445–458. 10.1007/s12035-021-02593-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao K.; Zu H. B. Microglial polarization: novel therapeutic mechanism against Alzheimer’s disease. Inflammopharmacology 2020, 28, 95–110. 10.1007/s10787-019-00613-5. [DOI] [PubMed] [Google Scholar]
- Stansley B.; Post J.; Hensley K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflammation 2012, 9, 115. 10.1186/1742-2094-9-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin A.; Stella N. Arachidonylcyclopropylamide increases microglial cell migration through cannabinoid CB2 and abnormal-cannabidiol-sensitive receptors. Eur. J. Pharmacol. 2003, 474, 195–198. 10.1016/S0014-2999(03)02074-0. [DOI] [PubMed] [Google Scholar]
- Han Q. W.; Shao Q. H.; Wang X. T.; Ma K. L.; Chen N. H.; Yuan Y. H. CB2 receptor activation inhibits the phagocytic function of microglia through activating ERK/AKT-Nurr1 signal pathways. Acta Pharmacol. Sin. 2022, 43, 2253–2266. 10.1038/s41401-021-00853-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A. S.; Kooistra A. J.; Munk C.; Heydenreich F. M.; Veprintsev D. B.; Bouvier M.; Babu M. M.; Gloriam D. E. GPCR activation mechanisms across classes and macro/microscales. Nat. Struct. Mol. Biol. 2021, 28, 879–888. 10.1038/s41594-021-00674-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipek S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 2019, 55, 114–120. 10.1016/j.sbi.2019.03.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.