

Abstract
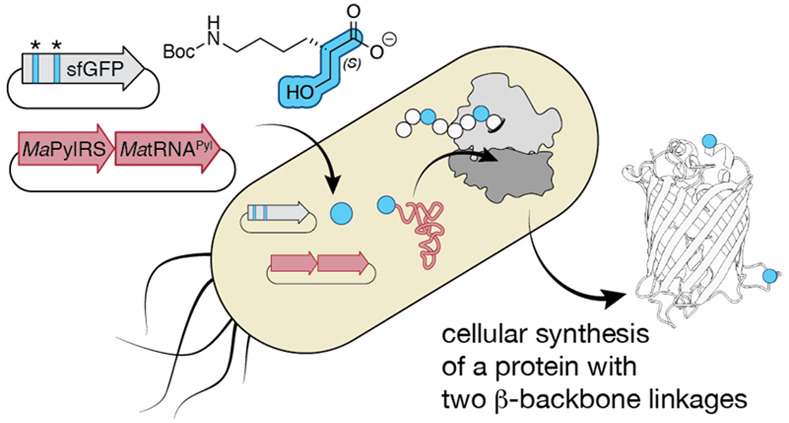
The programmed synthesis of sequence-defined biomaterials whose monomer backbones diverge from those of canonical α-amino acids represents the next frontier in protein and biomaterial evolution. Such next-generation molecules provide otherwise nonexistent opportunities to develop improved biologic therapies, bioremediation tools, and biodegradable plastic-like materials. One monomer family of particular interest for biomaterials includes β-hydroxy acids. Many natural products contain isolated β-hydroxy acid monomers, and polymers of β-hydroxy acids (β-esters) are found in polyhydroxyalkanoate (PHA) polyesters under development as bioplastics and drug encapsulation/delivery systems. Here we report that β2-hydroxy acids possessing both (R) and (S) absolute configuration are substrates for pyrrolysyl-tRNA synthetase (PylRS) enzymes in vitro and that (S)-β2-hydroxy acids are substrates in cellulo. Using the orthogonal MaPylRS/MatRNAPyl synthetase/tRNA pair, in conjunction with wild-type E. coli ribosomes and EF-Tu, we report the cellular synthesis of model proteins containing two (S)-β2-hydroxy acid residues at internal positions. Metadynamics simulations provide a rationale for the observed preference for the (S)-β2-hydroxy acid and provide mechanistic insights that inform future engineering efforts. As far as we know, this finding represents the first example of an orthogonal synthetase that acylates tRNA with a β2-hydroxy acid substrate and the first example of a protein hetero-oligomer containing multiple expanded-backbone monomers produced in cellulo.
Short abstract
The aminoacyl-tRNA synthetase PylRS from M. alvus acylates tRNAPyl with β2-hydroxy acid substrates and supports their incorporation at multiple sites into a protein in vivo.
Introduction
There is great interest in the synthesis and study of sequence-defined biomaterials whose monomer backbones diverge from those of canonical α-amino acids. Such hetero-oligomers can adopt new and known secondary1 and tertiary structures2−4 that confer novel functions including enhanced proteolytic resistance5−7 and membrane permeability.8,9 As a class, sequence-defined biomaterials provide otherwise nonexistent opportunities to expand and evolve protein and biomaterial structure and function and develop improved biologic therapies.10,11
One monomer family of particular interest for biomaterials consists of β-hydroxy acids (β-HAs) (Figure 1A). β-HAs embody both an expanded backbone and a hydroxy versus amino nucleophile and assemble into polymeric biomaterials known as β-esters. Isolated β-hydroxy acid esters are found in therapeutically relevant natural products (enterobactin),12 biosurfactants with environmental applications (surfactin),13 and FDA-approved therapeutics (romidepsin) (Figure 1B).14 Polymeric β-esters are found naturally in polyhydroxyalkanoate (PHA) polyesters currently in development as bioplastics.15
Figure 1.
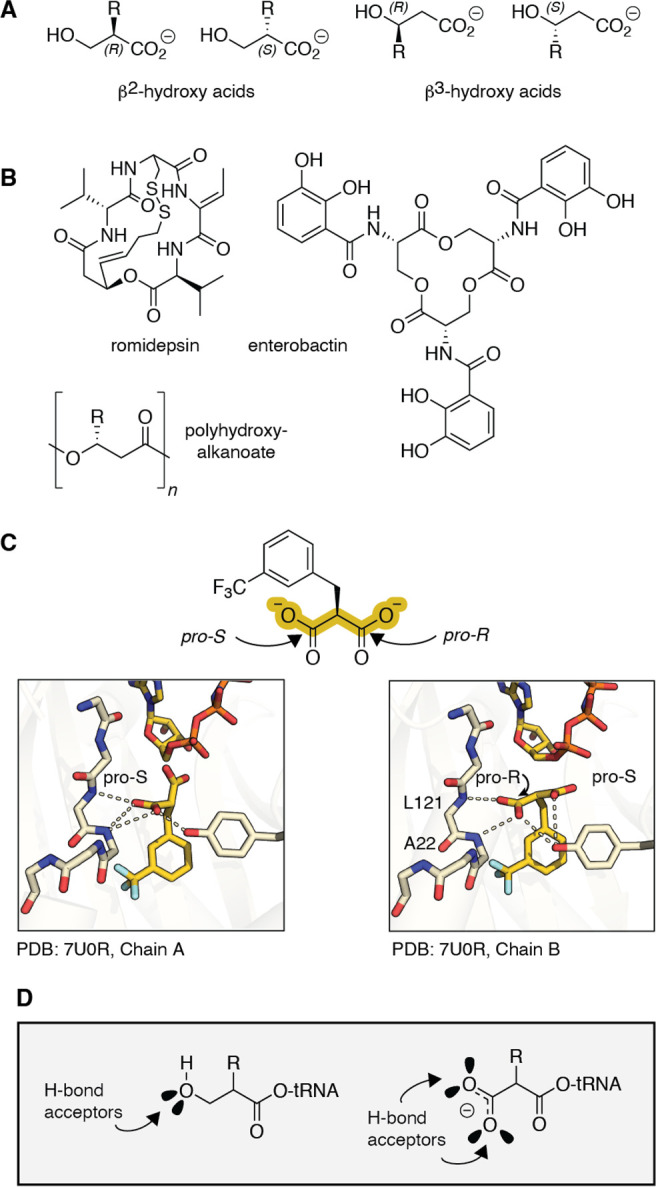
β-Hydroxy acids in natural products and as substrates for pyrrolysyl-tRNA synthetase (PylRS) variants. (A) Structures of β2- and β3-hydroxy acids and (B) natural products and biomaterials that contain β2- or β3-hydroxy acid esters. (C) Structure of MaFRSA bound to m-CF3-2-benzylmalonate (PDB 7U0R) illustrates the absence of a bound active site water and direct H-bonds from L121 and A122 (Ma numbering). (D) This work tests the hypothesis that β2-hydroxy acids will also act as substrates for PylRS enzymes by virtue of their ability to accept one or two backbone H-bonds.
Despite enormous interest in sequence-defined biomaterials containing non-α-amino acid monomers, progress towards their in vivo biosynthesis has been exceptionally slow.16−21 There exists one report in which a single β2-ester has been introduced into a protein using the wild-type E. coli ribosome in vitro,17 but there are no examples in which any β2-hydroxy acid has been introduced into a ribosomal product in vivo. One challenge limiting the in vivo ribosomal synthesis of sequence-defined β-ester biomaterials has been the absence of an orthogonal aminoacyl-tRNA synthetase (aaRS)/tRNA pair that acylates tRNA with a β-hydroxy acid substrate at a level that supports protein biosynthesis.
Previous work has shown that the widely employed and orthogonal pyrrolysyl-tRNA synthetase from Methanomethylophilus alvus (MaPylRS),22,23 as well as an engineered derivative with two active site Ala substitutions (MaFRSA),24 acylate tRNA with a number of non-α-amino acid substrates, including those in which the α-NH2 group is replaced with α-H, α-OH, α-SH, α-N-methyl, α-N-formyl, and α-carboxy substituents.25−28 Examination of the structure of MaFRSA bound to one such α-carboxy substrate, m-CF3-2-benzylmalonate (PDB: 7U0R), provided two insights into how PylRS enzymes might engage expanded backbone monomers such as a β2-HA. First, although the structure of PylRS bound to pyrrolysine shows the substrate α-amine coordinated to the homodimeric enzyme in a single conformation via a well-defined active site water molecule,29 that water is absent in the structure of MaFRSA bound to m-CF3-2-benzylmalonate (Figure 1C), presumably to accommodate the expanded size of the α-substituent. In the absence of this bound water, new amide backbone H-bonds from residues L121 and A122 engage the α-carboxy group. Second, in the structure of FRSA bound to m-CF3-2-benzylmalonate, the two subunits of the dimeric enzyme bind the prochiral m-CF3-2-benzylmalonate substrate in stereochemically distinct configurations. In one active site, the pro-S carboxylate of m-CF3-2-benzylmalonate engages the backbone amides of L121 and A122; in the other, the substrate rotates, and the pro-R carboxylate is coordinated instead (Figure 1C). These results imply that the backbone H-bonds from residues L121 and A122 are highly stabilizing, and PylRS can accommodate substrates retaining the same absolute configuration as natural l-α-amino acids as well as their enantiomers. Some evidence that PylRS accepts certain d-α-amino acid substrates has been reported.26
Like the α-carboxy group of a malonic acid, a β2-hydroxyl group of tRNA acylated with a β2-HA can also accept at least one, and perhaps two, H-bonds from the synthetase amide backbone (Figure 1D). Here we report that β2-hydroxy acids possessing both (R) and (S) absolute configurations are substrates for PylRS enzymes in vitro. Further, we report the unexpected finding that only (S)-β2-hydroxy acids—whose absolute configuration maps onto a d-α-amino acid—are substrates in cellulo. Using the orthogonal MaPylRS/MatRNAPyl synthetase/tRNA pair and classic E. coli expression strains (BL21 and C321.ΔA.exp), we report the cellular synthesis of model proteins containing up to two (S)-β2-HA residues at internal positions. Metadynamics simulations provide a clear rationale for the observed enantioselective preference for incorporation of an (S)-β2-hydroxy acid and provide mechanistic insights useful for future translational engineering efforts. As far as we know, this finding represents the first example of an orthogonal synthetase that acylates tRNA with a β2-hydroxy acid substrate and the first cellular biosynthesis of a protein hetero-oligomer containing multiple expanded backbone monomers.
Results
MaPylRS Acylates tRNAPyl with a β2-Hydroxy Acid Substrate in Vitro
To assess if PylRS-like enzymes would acylate tRNA with a β2-hydroxy acid substrate, we performed in vitro tRNA acylation reactions using purified enzymes and directly analyzed the products using intact tRNA LC-HRMS (Figure 2A). We began with MaPylRS and compared the yields of acylated tRNAPyl from reactions containing the known substrates (S)-α-NH2–Nε-Boc-Lysine ((S)-α-ΝΗ21) and (S)-α-OH-Nε-Boc-Lysine ((S)-α-OH 2) as well as the enantiopure β2-hydroxy acid (β2-OH) analogs 3 and 4. Reactions performed using 12.5 μM MaPylRS, 25 μΜ MatRNAPyl, and 10 mM 1 or 2 and incubated for 2 h at 37 °C generated the expected monoacylated tRNA products 1-acyl-tRNAPyl (23188.4 Da) and 2-acyl-tRNAPyl (23189.6 Da) (Figure 2B, Supporting Information Figure 1) in yields of 56% and 59%, respectively (Figure 2C). As observed previously,27MaPylRS-promoted acylation of tRNAPyl with (S)-α-OH 2 also generated a diacylated tRNAPyl product (Figure 2C). Analogous reactions supplemented with (S)-β2-OH 3 or (R)-β2-OH 4 also generated the expected diastereomeric monoacylated tRNA products 3-acyl-tRNAPyl and 4-acyl-tRNAPyl (23203.6 Da) in yields of 27% and 79%, respectively (Figure 2B). Again, under these conditions both substrates also generated detectable levels of diacylated tRNAPyl: 68% for (S)-β2-OH 3 and 11% for (R)-β2-OH 4 (Figure 2C). No acylated tRNAPyl was observed in the absence of enzyme, substrate, or tRNA (Supporting Information Figure 1 and 3). These data imply that β2-OH acids 3 and 4 are both substrates for MaPylRS, as anticipated by the FRSA-bound structure of the pro-chiral substrate m-CF3-2-benzylmalonate (Figure 1C and D).
Figure 2.
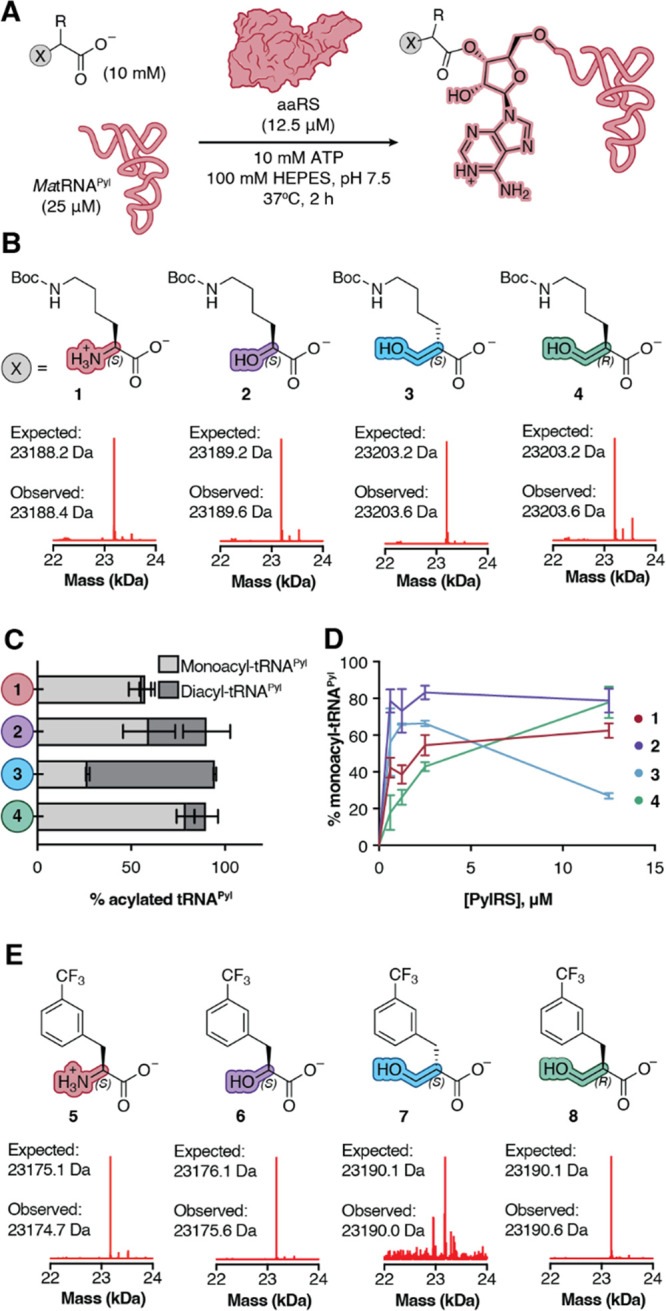
MaPylRS andMaFRSA acylate tRNAPyl with β2-hydroxy acid substrates in vitro. (A) Workflow for and (B) deconvoluted mass spectra of in vitro tRNAPyl acylation reactions containing 12.5 μM MaFRSA and supplemented with monomers 1–4. Signal is normalized to the highest signal in the respective traces. Expected masses shown correspond to monoacylated products. (C) Plot illustrating the relative yields of mono- and diacylated tRNAPyl generated during in vitro acylation reactions containing 12.5 μM PylRS and supplemented with 10 mM of monomer 1, 2, 3, or 4. (D) Plot illustrating yield of monoacylated tRNAPyl as a function of [MaPylRS]. The analogous plot showing the yield of mono- + diacylated tRNAPyl as a function of [MaPylRS] is shown in Supporting Information Figure 2. (E) Deconvoluted mass spectra of in vitro tRNAPyl acylation reactions containing 12.5 μM MaFRSA and supplemented with monomers 5–8. Signal is normalized to the highest signal in respective traces. Expected masses shown correspond to monoacylated products. Control tRNAPyl acylation reactions in which tRNAPyl, enzyme, or substrate is omitted are shown in Supporting Information Figures 1 and 3.
It has been estimated that the concentration of a single aaRS enzyme expressed from an endogenous promoter in E. coli falls in the low μM range.30 Thus, these initial reactions, performed at high enzyme concentration (12.5 μM, 50 mol % of tRNAPyl), could mask reactivity differences that are relevant under conditions that better mimic the cellular environment. To more carefully characterize the relative reactivity of monomers 1–4, we evaluated the yield of both mono- and diacylated tRNAPyl as the concentration of MaPylRS was reduced stepwise from 12.5 μM (50 mol %) to 625 nΜ (2.5 mol %) (Figure 2D). At low PylRS concentrations, the yield of tRNAPyl monoacylated with (S)-β2-OH 3 was comparable to that of (S)-α-OH 2 and higher than that of (S)-α-NH21 (Figure 2D); at higher enzyme concentrations, the diacylated product predominates. Yields of tRNAPyl monoacylated with (R)-β2-OH 4 at low enzyme concentration were slightly lower than yields of tRNAPyl monoacylated with (S)-α-ΝΗ21.
MaFRSA Acylates tRNAPyl with a β2-Hydroxy Acid Substrate in Vitro
The PylRS derivative FRSA contains two active site mutations (N166A and V168A, M. alvus numbering) that favor substrates with substituted Phe side chains.24 One of the best substrates for FRSA is the α-amino acid m-CF3-Phe 5 (Figure 2E). To determine if MaFRSA would also acylate tRNA with a β2-HA, we performed in vitro tRNA acylation reactions supplemented with m-CF3–Phe 5 alongside analogous reactions containing (S)-α-OH 6 and the enantiopure β2-OH analogs 7 and 8. Reactions performed with 12.5 μM MaFRSA, 25 μΜ MatRNAPyl, and 10 mM 5 or 6 cleanly generated the expected monoacylated tRNA products 5-acyl-tRNAPyl (23174.7 Da) and 6-acyl-tRNAPyl (23175.6 Da) (Figure 2E). Quantification of the monoacyl-tRNA and unacylated tRNA pools indicates yields of 45% and 29% for 5-acyl-tRNAPyl and 6-acyl-tRNAPyl, respectively (Supporting Information Figure 3). Aminoacylation reactions supplemented with 10 mM (S)-β2-HA monomer 7 yielded only a single low signal peak in the TIC corresponding to the molecular weight of monoacyl-tRNA (23190.0 Da) with a yield of <1% 7-acyl-tRNAPyl (Supporting Information Figure 3). By contrast, supplementation of an analogous aminoacylation reaction with (R)-β2-HA 8 yielded two peaks in the TIC corresponding to monoacylated (23190.6 Da) and diacylated 8-acyl-tRNAPyl (23420.1 Da) (Figure 2E) in 28% and 11% yield, respectively. Interestingly, while MaPylRS processes (S)-β2-OH 3 more efficiently than (R)-β2-OH 4, MaFRSA shows the opposite preference, processing (R)-β2-HA 8 more efficiently than (S)-β2-HA 7.
MaPylRS Supports in Vivo Synthesis of a Protein Containing a Single β2-HA
We next asked whether the in vitro tRNAPyl acylation efficiencies observed with β2-OH substrates would support the incorporation of these monomers into proteins biosynthesized in E. coli. Experiments were performed using the recoded E. coli strain C321.ΔΑ.exp, which lacks all endogenous TAG codons and release factor 1 (RF1).31 Cells were cotransformed with a pMega plasmid32 encoding the MaPylRS/MatRNAPyl pair (pMega-MaPylRS) as well as a pET22b plasmid encoding sfGFP with an in-frame TAG codon at position 3 (sfGFP-3TAG) (Figure 3A). MatRNAPyl naturally decodes TAG codons,33 making successful translation of full-length sfGFP dependent on the concentration and activity of acyl-tRNAPyl. Test expressions were performed in the presence of 0.5 to 2 mM 1–4, and both OD600 and 528 nm emission (F528) were monitored as a function of time (Supporting Information Figure 4). Although the rate of increase in F528/OD600 was greater for monomers with a natural α-backbone ((S)-α-NH21 and (S)-α-OH 2), by 24 h all growth curves had reached saturation, and hence this time point was used for comparisons of F528/OD600.
Figure 3.
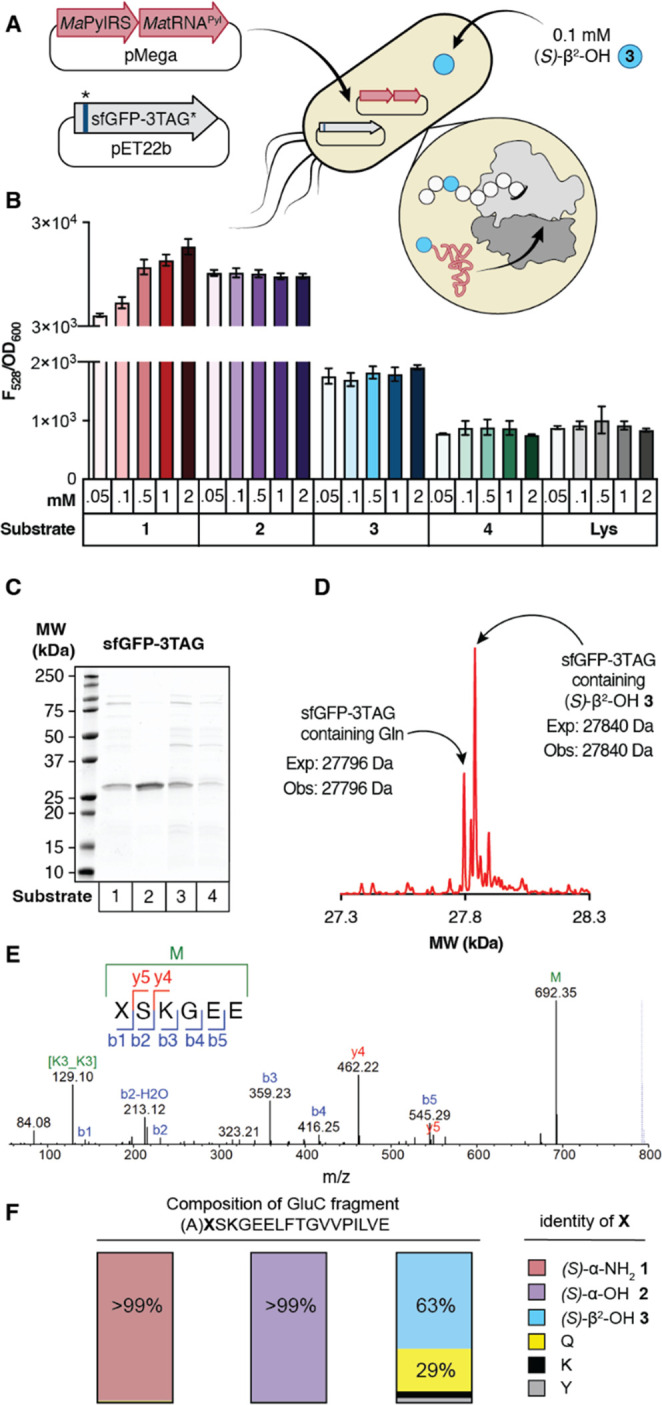
MaPylRS supports in vivo synthesis of a protein containing a single β2-HA. (A) Workflow for protein expression in C321.ΔΑ.exp E. coli transformed with pMega-MaPylRS and pET22b-sfGFP-3TAG. (B) Plot of F528/OD600 values measured 24 h after induction with 1 mM IPTG as a function of substrate identity and concentration. n = 2 biological replicates, 3 technical replicates per biological replicate. (C) SDS-PAGE of sfGFP-3TAG expressed in the presence of 1–4. (D) Deconvoluted mass spectrum of sfGFP-3TAG expressed in the presence of (S)-β2-OH 3. (E) MS/MS profile of peptide fragment XSKGEE (where X is (S)-β2-OH 3) observed after GluC digestion of sfGFP-3TAG expressed in the presence of (S)-β2-OH 3. During collision dissociation, the Boc group of 3 is lost, and subsequent fragmentation generates the commonly observed b and y ions. We note that the analogous peptide fragment corresponding to incorporation of Gln at position 3 (AQSKGEE) is too hydrophilic to be retained on the column and cannot be quantified via LC-MS/MS. (F) Evaluation of the fidelity of incorporation of (S)-α-NH21, (S)-α-OH 2, or (S)-β2-OH 3 at position 3 of sfGFP-3TAG using a larger GluC digestion product that could be quantified. Here, X denotes position 3, and the preceding A is included when growths included (S)-α-NH21, Gln, Lys, or Tyr (as the products contain only amide bonds) and not when growths included (S)-α-OH 2 or (S)-β2-OH 3 (as the single ester bond undergoes hydrolysis). Analysis of this longer fragment reveals that more than 99% of the sfGFP-3TAG produced from growths supplemented with (S)-α-NH21 or (S)-α-OH 2 contains the requisite monomer at position 3. When the growths are supplemented with (S)-β2-OH 3, 63% of the sfGFP-TAG produced contains this monomer at position 3. The remaining material contains Gln (28.6%), Lys (4.4%), or Tyr (2.9%).
Comparison of F528/OD600 values after 24 h revealed a clear concentration-dependent increase when cultures were supplemented with (S)-α-NH21 relative to those in which substrate was withheld (ΔAA) or supplemented with Lys, which is not a substrate for MaPylRS (Figure 3B).34 Although no concentration dependence of the F528/OD600 value was observed when cultures were supplemented with (S)-α-OH BocK 2, the F528/OD600 values observed after 24 h were comparable to those observed in the presence of 0.5 mM (S)-α-NH21. Cultures supplemented with (S)-β2-OH 3 also showed an increase in F528/OD600 relative to those in which the substrate was withheld (ΔAA) or supplemented with Lys. Interestingly, like growths supplemented with (S)-α-OH 2, the F528/OD600 values observed after 24 h, although low, were independent of the concentration of (S)-β2-OH 3 over an 80-fold range of concentration. No increases in F528/OD600 relative to background were observed when cultures were supplemented with (R)-β2-OH 4. Identical protein expression assays with C321.ΔΑ.exp cells harboring MaFRSA and supplemented with either enantiomer of β2-ΟΗ-m-CF3-Phe yielded no significant sfGFP expression over a ΔAA control (Supporting Information Figure 5).
Two experiments were performed in an attempt to increase the F528/OD600 values of cells expressing sfGFP in the presence of (S)-β2-OH 3. The growth experiments described above were repeated using either Top10 or BL21 (DE3) E. coli in place of C321.ΔΑ.exp. We also examined whether the F528/OD600 values of growths supplemented with (S)-β2-OH 3 could be improved via the mutation of MatRNAPyl. Previous work has emphasized the effect of tRNA identity on the efficiency of noncanonical α-amino acid incorporation into proteins.35 A rationally evolved, orthologous tRNAPyl from the organism Methanosarcina barkeri (MbtRNAPyl-opt) bearing mutations at the base of the acceptor and T-stems improved the incorporation of certain noncanonical α-amino acid substrates into proteins expressed in BL21 (DE3) and Top10 E. coli,36 and mutations at the identical sites in an evolved EctRNASec increased the incorporation of selenocysteine at TAG codons when grown in a derivative strain of C321.ΔA.37 Changing neither the expression strain nor the tRNA body improved the observed F528/OD600 values of growths supplemented with (S)-β2-OH 3 (Supporting Information Figure 6).
To confirm that (S)-β2-OH 3 was introduced into sfGFP-3TAG, we isolated protein from a preparative growth of C321.ΔΑ.exp cells transformed with pMega-MaPylRS and pET22b-sfGFP-3TAG and supplemented with 0.1 mM (S)-β2-OH 3 and characterized its identity using SDS-PAGE (Figure 3C) and LC-HRMS (Figure 3D). SDS-PAGE of cultures supplemented with (S)-β2-OH 3 produced a major protein product of ∼28 kDa whose mobility was comparable to proteins expressed in the presence of (S)-α-NH21 and (S)-α-OH 2 (Figure 3C). The deconvoluted mass spectrum of purified sfGFP-3TAG expressed in the presence of (S)-β2-OH 3 included a major peak at 27840 Da, corresponding to the expected molecular mass of sfGFP with (S)-β2-OH 3 at position 3 but lacking residues 1–2 due to ester hydrolysis. A second, smaller peak at 27796 Da was also observed, corresponding to the mass of sfGFP with Gln at position 3; in this case, residue 2 is retained. Thus, although (S)-β2-OH 3 and (R)-β2-OH 4 are substrates for PylRS in vitro, with activities approaching or exceeding that of (S)-α-NH21, only (S)-β2-OH 3 is incorporated into proteins in cells, and with approximately one-tenth the efficiency anticipated on the basis of aaRS activity in vitro. Approximately 20–30 mg/L of sfGFP-3TAG could be isolated from C321.ΔΑ.exp cells supplemented with (S)-α-NH21 or (S)-α-OH 2, whereas only 2 mg/L could be isolated from cells supplemented with (S)-β2-OH 3. GluC digestion and peptide mapping of sfGFP-3TAG from (S)-β2-OH 3 supplemented growths provided unambiguous evidence for the presence of the expected β2-hydroxy acid at position 3 of sfGFP (Figure 3E) and with reasonable fidelity: roughly 63% of the sfGFP-3TAG produced contains (S)-β2-OH 3 at position 3 with some contamination with Gln (28.6%), Lys (4.4%), or Tyr (2.9%) (Figure 3F).
To evaluate whether an intact protein containing an intact β2-hydroxy acid ester could be isolated, we designed three additional sfGFP expression plasmids. In one, residue E213 was replaced by an amber codon; in a second, residue K214 was replaced by an amber codon; and in the third an amber codon was inserted between E213 and K214. E213 and K214 can function as the N- and C-termini of a split sfGFP variant that assembles from two independent polypeptides.38 We reasoned that these sites would be well-suited to accommodate an internal β2-hydroxy acid monomer without disrupting either the sfGFP fold or chromophore maturation. C321.ΔΑ.exp cells were cotransformed with pMega-MaPylRS and a pET22b plasmid encoding sfGFP with an in-frame TAG codon at position 213 (pET22b-sfGFP-213TAG), position 214 (pET22b-214TAG), or between positions 213 and 214 (pET22b-213TAG214). Growths were supplemented with 0.1 mM 2, 3, or 4 as described previously, and both F528 and OD600 were monitored as a function of time (Supporting Information Figure 7). Again, although the rate of increase in F528 was greater for growths containing (S)-α-OH 2 than those containing (S)-β2-OH 3 or (R)-β2-OH 4, by 24 h all growth curves had reached saturation, and this time point was used for comparisons of F528/OD600. We observed a robust increase in the F528/OD600 signal of growths expressing sfGFP-213TAG, sfGFP-214TAG, or sfGFP-213TAG214 in the presence of (S)-α-OH 2 and a modest increase in F528/OD600 when cultures expressing sfGFP-213TAG214 were supplemented with (S)-β2-OH 3 (Figure 4A).
Figure 4.
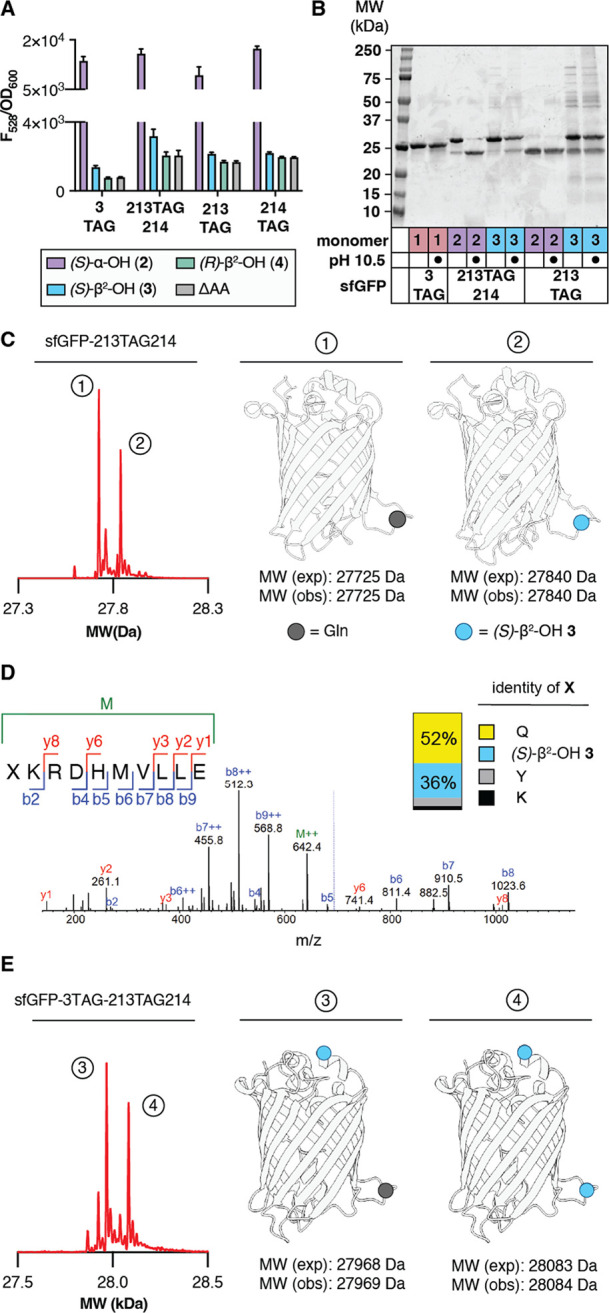
MaPylRS supports the in vivo synthesis of sfGFP containing one or two β2-hydroxy acid monomers. (A) Plot of F528/OD600 values of C321.ΔΑ.exp Ε. coli transformed with pMega-MaPylRS and the indicated sfGFP expression plasmid measured 24 h after induction with 1 mM IPTG as a function of monomer identity. (B) SDS-PAGE of the indicated isolated sfGFP variants after treatment with CAPS buffer (pH 10.5) or Milli-Q for 2 h at 37 °C. (C) Deconvoluted mass spectrum of sfGFP-213TAG214 expressed in the presence of (S)-β2-OH 3 reveals two products. One contains a single residue of 3, and the other contains a single residue of Gln. (D) MS/MS profile for peptide XKRDHMVLLE (where X is (S)-β2-OH 3) observed after GluC digestion of sfGFP-213TAG214 expressed in the presence of 0.1 mM (S)-β2-OH 3. Inset shows the fidelity of incorporation of (S)-β2-OH 3 relative to Gln, Tyr, or Lys. (E) Deconvoluted mass spectrum of sfGFP-3TAG-213TAG214 grown in defined media lacking Gln in the presence of 0.1 mM (S)-β2-OH 3. Two products are observed, whose masses correspond to (1) sfGFP with the addition of two copies of (S)-β2-OH 3 and (2) sfGFP with the addition of one copy of (S)-β2-OH 3 and a single Gln residue. GluC mapping data are found in Supporting Information Figure 9.
sfGFP variants were isolated from preparative growths of C321.ΔΑ.exp cells transformed with pMega-MaPylRS and either sfGFP-3TAG, sfGFP-213TAG, or sfGFP-213TAG214 and supplemented with (S)-α-NH21, (S)-α-OH 2, or (S)-β2-OH 3. The products were characterized by SDS-PAGE, with and without base treatment (pH 10.5), to confirm the presence of an ester bond (Figure 4B), as well as by LC-HRMS (Figure 4C). SDS-PAGE analysis of sfGFP-213TAG214 isolated from C321.ΔΑ.exp cultures supplemented with (S)-α-ΟΗ 2 and without base treatment shows two bands: one that migrates with the MW expected for intact sfGFP (∼28 kDa) and one that migrates with the MW expected for the ester hydrolysis product (∼24 kDa). SDS-PAGE analysis of an analogous sample after base treatment led to virtually complete loss of the 28 kDa band and an increase in the intensity of the 24 kDa band, consistent with the presence of a base-labile ester bond. SDS-PAGE analysis of sfGFP-213TAG214 grown in the presence of (S)-β2-OH 3 also yielded two bands at ∼24 and 28 kDa prior to base treatment. Base treatment led to a partial loss of the 28 kDa band and an increase in intensity of the 24 kDa band, suggesting that a fraction of the sample contained a base-labile ester linkage. Only a truncated product was isolated from growths programmed with sfGFP-213TAG and supplemented with (S)-α-OH 2; in the case of an analogous growth supplemented with (S)-β2-OH 3, both full length and truncated protein was observed, and the ratio was unaffected by base treatment. This observation implies that an ester linkage within sfGFP-213TAG is more hydrolytically labile than an ester linkage within sfGFP-213TAG214.
The successful internal incorporation of (S)-β2-OH 3 into sfGFP-213TAG214 was confirmed by LC-HRMS. sfGFP isolated from growths programmed with sfGFP-213TAG214 and supplemented with 0.1 mM (S)-β2-OH 3 contained two sfGFP variants with a yield of approximately 16 mg/L. One variant contained a single residue of 3, and the other contained a single residue of Gln (Figure 4C). GluC digest followed by peptide mapping to quantitatively assess fidelity indicates that 36% of the sfGFP-213TAG214 produced contains (S)-β2-OH 3 at position 3. The remaining material contains Gln (52%), Lys (3.6%), or Tyr (8.4%; Figure 4D).
As predicted by gel analysis, sfGFP isolated from growths programmed with sfGFP-213TAG and supplemented with (S)-β2-OH 3 revealed the presence of Gln at position 213 of sfGFP as well as a pair of protein fragments whose approximate molecular weights (∼4 and ∼23.6 kDa) correspond to those expected if sfGFP was fragmented at position 213. However, the exact mass of the large (23625 Da) fragment was 18 Da less than that predicted on the basis of sequence alone (Supporting Information Figure 8A). It is possible that ester hydrolysis in this case is promoted by the Asn residue at position 212, which could induce ester cleavage via the pathway used by asparagine lyase self-cleaving enzymes, the product of which undergoes dehydration (Supporting Information Figure 8B).39
MaPylRS Supports the in Vivo Synthesis of sfGFP Containing Two β2-HA Monomers
Having identified that (S)-β2-OH 3 could be introduced into sfGFP at position 3 as well as between positions 213 and 214, we next asked whether this monomer could be introduced at both positions simultaneously. C321.ΔA.exp cells were transformed with pMega-MaPylRS as well as a plasmid encoding sfGFP with TAG codons at position 3 as well as between E213 and K214 (pET22b-sfGFP-3TAG-213TAG214) and grown in the presence of 0.1 mM (S)-β2-OH 3 or 0.1 mM (S)-α-OH 2. LC-HRMS analysis of the isolated sfGFP generated in the presence of (S)-β2-OH 3 confirmed incorporation at both positions, albeit with Gln contamination (Supporting Information Figure 9). However, expression of sfGFP-3TAG-213TAG214 with 0.1 mM (S)-β2-OH 3 in defined media lacking Gln27,40 led to only two products. One contained (S)-β2-OH 3 at position 3 and glutamine at position 213-214 (27969 Da); the other contained (S)-β2-OH 3 at both positions (28084 Da) (Figure 4E). GluC digest of sfGFP-3TAG-213TAG214 grown in the presence of 0.1 mM (S)-β2-OH 3 followed by GluC mapping again unambiguously confirmed the incorporation of (S)-β2-OH 3 at both positions (Supporting Information Figure 9B).
Metadynamics Simulation Probe Enantioselectivity of the PTC with Respect to β2-OH-Monomers
For over 30 years, the level of noncanonical α-amino acid incorporation at a stop codon has been used as a proxy for aaRS activity in vivo.41 This proxy fails for the β2-backbone monomers studied here. Although MaPylRS acylates tRNAPyl with both (S)-β2-OH 3 and (R)-β2-OH 4 at levels comparable to (S)-α-ΝΗ21 and (S)-α-ΟH 2in vitro, only (S)-β2-OH 3 is introduced into protein in cells. We turned to metadynamics to learn more about how (S)-α-NH21, (S)-α-OH 2, (S)-β2-OH 3, and (R)-β2-OH 4 are accommodated within the ribosomal A-site when loaded on tRNAPyl, as previously reported simulations emphasize the important relationship between nucleophile positioning within the PTC and bond formation.42 These metadynamics simulations made use of a reduced ribosome model (RRM) containing fMet-tRNAfMet in the P site and 1-acyl-tRNAfMet, 2-acyl-tRNAfMet, 3-acyl-tRNAfMet, or 4-acyl-tRNAfMet in the A site. Two 100 ns metadynamics simulations were initiated using two distinct monomer poses (Supporting Information Figure 10), and the results were averaged. One pose aligned the A-site nucleophile with the nucleophilic atom of the A-site Met in the 2.1 Å cryo-EM model used to build the RRM;42 this pose placed the α-NH2, α-OH, or β2-OH nucleophile as close as possible to the P-site carbonyl. The second pose was generated by rotating the psi (ψ) angle of the A-site nucleophile monomer by 180°; this pose placed the nucleophile farther from the P-site carbonyl.
Previous results suggest that reactivity within the PTC is related to two distinct parameters: the Nα–Csp2 distance between the A-site nucleophile (Nα) and the P-site carbonyl electrophile (Csp2), and the Bürgi–Dunitz attack angle (αBD). Monomers that react readily within the PTC populate a conformational space characterized by a Nα–Csp2 distance of <4 Å and a αBD value between 76 and 115°.42 Examination of plots showing αBD as a function of the Nα–Csp2 distance reveals global minima that differentiate highly reactive ((S)-α-NH21, (S)-α-OH 2), moderately reactive ((S)-β2-OH 3), and nonreactive ((R)-β2-OH 4) monomers (Figure 5A). The free energy surface for an RRM containing 1-acyl-tRNAfMet in the A-site is defined by an Nα–Csp2 distance of 3.9 ± 0.2 Å and an αBD of 92.3° ± 13.5° averaged for all final poses within 1 kcal/mol of the global energy minimum (Figure 5B, darkest blue). Both of these values are comparable to those reported for an RRM with Met-tRNAfMet in the A-site (Nα–Csp2 = 3.7 Å and αBD = 76°).42 Simulations with 2-acyl-tRNAfMet in the A-site yielded comparable values (Nα–Csp2 = 3.9 ± 0.1 Å and αBD = 69.8 ± 3.6°), which is fully consistent with the high reactivity of (S)-α-OH 2in vivo.
Figure 5.

Metadynamics simulations of tRNA acylated with α- and β2-hydroxy and amino acids prior to bond formation in a reduced ribosome model (RRM). (A) Free energy surface (FES) plots of 30 Å RRM containing a P-site tRNAMet acylated with fMet and A-site tRNAMet acylated with (S)-α-NH21, (S)-α-OH 2, (S)-β2-OH 3, or (R)-β2-OH 4, plotted along the collective variables of Bürgi–Dunitz angle αBD and Nα–Csp2 distance. Each FES shown is the average of two metadynamics runs starting from orientations of the A-site monomers that differ by a 180° rotation about the ψ angle (Supporting Information Figure 10). The color scale represents the free energy in kilocalories per mole (kcal/mol), where the global minima are set at 0 and therefore the various heights of the energy scales are based on the energetics of the fluctuations of the A-site monomers. Average Nα–Csp2 distances and αBD angles for the 0–1 kcal/mol energy minima of each plot are displayed below (gray boxes). (B) Representative conformation and relative geometry of the P-site (gray) and A-site (colored) monomers from the frames at the free energy minimum of each FES plot. Poses were chosen to highlight the Nα–Csp2 distance (black dotted line) and the Bürgi–Dunitz angle αBD at the free energy minimum. The tRNAs to which each monomer is attached as well as the RRM surrounding the two monomers have been omitted for clarity. (C) Pairwise interaction difference analysis (PIA) plots displaying the pairwise-distance differences between the C1′ carbons of nucleotides within 5 Å of each A-site monomer ((S)-α-NH21, (S)-α-OH 2, (S)-β2-OH 3, or (R)-β2-OH 4) along the metadynamics trajectories. Heatmaps were constructed by subtracting the C1′ distances within the RRM containing tRNA charged with the indicated monomer from the C1′ distances within the analogous RRM containing (S)-α-NH21. Darker red indicates greater distances between nucleotides for RRMs containing the indicated monomer ((S)-α-OH 2, (S)-β2-OH 3, or (R)-β2-OH 4) relative to that containing (S)-α-NH21, whereas darker blue indicates smaller distances.
The free energy surfaces for an RRM containing 3-acyl-tRNAfMet or 4-acyl-tRNAfMet are defined by different global minima values. For the RRM containing 3-acyl-tRNAfMet in the A-site, we observe low energy poses characterized by Nα–Csp2 distances of 4.5 ± 0.3 Å and αBD values of 72.1° ± 9.8°. The Nα–Csp2 distance for 3-acyl-tRNAfMet falls outside the Nα–Csp2 distance range for monomers predicted to be highly reactive in the ribosome and suggests that the relatively low incorporation of (S)-β2-OH 3 is due in part to poor sampling of conformations within the PTC that supports rapid bond formation. The free energy surface of an RRM containing 4-acyl-tRNAfMet is defined by a similar averaged Nα–Csp2 distance and αBD but with much smaller standard deviations (Nα–Csp2 = 4.8 ± 0.1 Å and αBD = 88.8° ± 1.7°). These differences suggest that 3-acyl-tRNAfMet can achieve Nα–Csp2 distances that permit modest incorporation of (S)-β2-OH 3 into a ribosomal product, whereas 4-acyl-tRNAfMet cannot. Indeed, the energy minima of metadynamics trajectories for monomer 4 produces a minimized structure in which the β-OH of 4-acyl-tRNAfMet is turned away from the P-site carbonyl electrophile (Figure 5B). This analysis provides one plausible explanation for the observed in vivo differences in the reactivity of A-site tRNAs esterified with (S)-β2-OH 3 and (R)-β2-OH 4.
We next examined the overall structure of the PTC during each trajectory to learn more about conformational changes in the ribosome that might facilitate the productive incorporation of (S)-β2-OH 3 and not (R)-β2-OH 4. We first calculated the average distance between the C1′ atoms of all rRNA bases within 5 Å of either acyl-tRNAfMet in the P- or A-site of RRMs containing 1-acyl-tRNAfMet, 2-acyl-tRNAfMet, 3-acyl-tRNAfMet, or 4-acyl-tRNAfMet (Figure 5C). The matrix of pairwise interactions in each of the four minimized RRMs provides an effective map of how the PTC responds to structurally and stereochemically distinct α-NH2, α-OH, and β2-OH monomers. To visualize differences between the maps, the pairwise interaction distances calculated from RRMs containing 2-acyl-tRNAfMet, 3-acyl-tRNAfMet, or 4-acyl-tRNAfMet in the A-site were subtracted from the pairwise interaction distances calculated from the RRM containing 1-acyl-tRNAfMet. The resulting pairwise interaction difference analysis (PIA) plots provide a comprehensive view of how the internal architecture of the PTC varies in a monomer-dependent fashion (Figure 5C).43,44 In a PIA plot, distances between rRNA bases that are greater in the presence of 2, 3-, or 4-acyl-tRNAfMet relative to 1-acyl-tRNAfMet are represented in red, whereas distances that are shorter are represented in blue.
Examination of the PIA plot comparing RRMs containing 1-acyl-tRNAfMet or 2-acyl-tRNAfMet shows significant differences at only two rRNA bases (U2504, located within H89, and C2063, part of the highly conserved A2450-C2063 non-Watson–Crick base pair). In both cases, the differences reflect an expansion of the PTC when bound to 2-acyl-tRNAfMet. The PIA plot comparing RRMs containing 1-acyl-tRNAfMet or 3-acyl-tRNAfMet is highly similar to the plot comparing the RRMs for 1-acyl-tRNAfMet and 2-acyl-tRNAfMet with the largest differences again involving lengthened interactions with A2504 and C2063.
In contrast, the PIA plot comparing the RRMs for 1-acyl-tRNAfMet and 4-acyl-tRNAfMet is different. First, it highlights many pairwise interactions that are markedly shorter when 4-acyl-tRNAfMet occupies the A-site, including those involving U2506, U2585, and A2062. Changes involving U2506 and U2585 are especially notable, as both have been implicated as critical for induced conformational changes required for efficient bond formation.45 U2585 is believed to shield the P-site peptidyl-tRNA from hydrolysis in the uninduced state and rotate away in the induced state to expose the ester bond for nucleophilic attack by the A-site monomer.45 Indeed, recent cryo-EM structures of E. coli ribosomes with aminobenzoic acid monomers in the A-site show U2585 locked in the uninduced conformation, prohibiting access to the P-site peptidyl-tRNA.46 Overall, the decreased pairwise distances within the RRM containing 4-acyl-tRNAfMet may indicate an inability to support the dynamic movements necessary for efficient bond formation. These differences in RRM structure provide a second explanation for the observed in vivo enantioselective preference for (S)-β2-OH 3 over (R)-β2-OH 4. More broadly, they emphasize that the complex mechanism of translation, including (but not limited to) EF-Tu- and tRNA-induced conformational changes and essential bound water molecules46 must be considered in future ribosome or monomer engineering efforts.
Discussion
The programmed synthesis of sequence-defined biomaterials whose monomer backbones diverge from canonical α-amino acids is of enormous current interest. Such next-generation molecules provide strategies for improved biologic therapies, tools for bioremediation, and plastic-like materials that biodegrade. But progress toward sequence-defined non-protein materials l has been exceptionally slow.17−21 Although most elements of the translational machinery tolerate even wildly divergent α-amino acid side chains,41 altered backbones are tolerated predominantly in vitro,47 at small scale, under nonphysiological conditions, and with efficiencies and fidelities that have not been rigorously evaluated.
Here we report that β2-hydroxy acids possessing both (R) and (S) absolute configuration are excellent substrates for pyrrolysyl-tRNA synthetase (PylRS) enzymes in vitro and that certain β2-hydroxy acids are also substrates in cellulo. One unexpected finding is that the (S)-β2-hydroxy acid incorporated successfully into protein by the ribosome in vivo possesses an absolute configuration that maps onto a d-α-amino acid, not an l-α-amino acid. Although the structure of the PylRS variant FRSA bound to 2-benzylmalonate27 provides an explanation for the absence of enantio-preference at the aaRS level, the preference for d-like (S)-β2-OH 3 in the PTC was a surprise. Metadynamics simulations and recent cryo-EM structures46 provide evidence that the preference for tRNA charged with (S)-β2-OH 3 has less to do with inherent stereochemistry than with differences in the ability to induce conformational changes within the PTC necessary for favorable bond formation. A better understanding of the interactions necessary to facilitate P-site electrophile deshielding could be used to identify d-α-amino acids that are successfully elongated in vivo, another long-sought goal.48
Overall, the combined biochemical and computational approach reported here provides the first example of orthogonal cellular translation through a β2-backbone and the first example of a protein hetero-oligomer containing two β-backbone monomers at predetermined positions. Although further work is necessary to improve yield, understand sequence context, and showcase the benefits embodied by a sequence-defined non-α-peptide backbone, these results represent an important stepping stone toward cell-based synthesis of β2-HA/α-AA hybrids with new-to-nature functionalities. They also deepen our understanding of how the WT E. coli ribosome accommodates substrates with non-native stereochemistry and backbone configurations (or not) and suggest that translation factor49,52 and/or ribosome engineering,17,50,51 as well as alternative chemical and biochemical approaches,19,20 may be needed to achieve robust levels of expanded backbone linkages within proteins produced in cells.
Acknowledgments
We are grateful to members of the Schepartz, Chatterjee, and Cate laboratories for insightful comments and suggestions. We also thank Dr. Chandrima Majumdar for productive discussions about induced state ribosomal conformations. This work was supported by the NSF Center for Genetically Encoded Materials (C-GEM; CHE 2002182). A.S. is a Chan-Zuckerberg Biohub-San Francisco Investigator and an ARC Innovation Investigator.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.3c01366.
Supplementary figures, tables, data, experimental methods, computational methods, and supplementary references (PDF)
Author Contributions
Study conception and design: N.X.H., A.S.; preparation of materials: N.X.H.; data collection: N.X.H., A.M.A., B.S.; analysis and interpretation of results: N.X.H., Z.Z., A.M.A., A.S.; and manuscript preparation: N.X.H., A.S.
The authors declare the following competing financial interest(s): N.X.H. and A.S. are coinventors on an international patent application that incorporates methods outlined in this manuscript.
Supplementary Material
References
- Cheng R. P.; Gellman S. H.; DeGrado W. F. β-Peptides: From Structure to Function. Chem. Rev. 2001, 101, 3219–3232. 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- Qiu J. X.; Petersson E. J.; Matthews E. E.; Schepartz A. Toward β-Amino Acid Proteins: A Cooperatively Folded β-Peptide Quaternary Structure. J. Am. Chem. Soc. 2006, 128, 11338–11339. 10.1021/ja063164+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels D. S.; Petersson E. J.; Qiu J. X.; Schepartz A. High-Resolution Structure of a β-Peptide Bundle. J. Am. Chem. Soc. 2007, 129, 1532–1533. 10.1021/ja068678n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon S.; Shin H. S.; Gong J.; Eom J.-H.; Jeon A.; Yoo S. H.; Chung I. S.; Cho S. J.; Lee H.-S. Self-Assembled Peptide Architecture with a Tooth Shape: Folding into Shape. J. Am. Chem. Soc. 2011, 133, 17618–17621. 10.1021/ja2082476. [DOI] [PubMed] [Google Scholar]
- Frackenpohl J.; Arvidsson P. I.; Schreiber J. V.; Seebach D. The Outstanding Biological Stability of β- and γ-Peptides toward Proteolytic Enzymes: An In Vitro Investigation with Fifteen Peptidases. ChemBioChem. 2001, 2, 445–455. 10.1002/1439-7633(20010601)2:6<445::AID-CBIC445>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Hook D. F.; Bindschädler P.; Mahajan Y. R.; Šebesta R.; Kast P.; Seebach D. The Proteolytic Stability of ‘Designed’ β-Peptides Containing α-Peptide-Bond Mimics and of Mixed α,β-Peptides: Application to the Construction of MHC-Binding Peptides. Chem. Biodivers. 2005, 2, 591–632. 10.1002/cbdv.200590039. [DOI] [PubMed] [Google Scholar]
- Cheloha R. W.; Maeda A.; Dean T.; Gardella T. J.; Gellman S. H. Backbone Modification of a Polypeptide Drug Alters Duration of Action in Vivo. Nat. Biotechnol. 2014, 32, 653–655. 10.1038/nbt.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwochert J.; Turner R.; Thang M.; Berkeley R. F.; Ponkey A. R.; Rodriguez K. M.; Leung S. S. F.; Khunte B.; Goetz G.; Limberakis C.; et al. Peptide to Peptoid Substitutions Increase Cell Permeability in Cyclic Hexapeptides. Org. Lett. 2015, 17, 2928–2931. 10.1021/acs.orglett.5b01162. [DOI] [PubMed] [Google Scholar]
- Hosono Y.; Uchida S.; Shinkai M.; Townsend C. E.; Kelly C. N.; Naylor M. R.; Lee H.-W.; Kanamitsu K.; Ishii M.; Ueki R. Amide-to-Ester Substitution as a Stable Alternative to N-Methylation for Increasing Membrane Permeability in Cyclic Peptides. Nat. Commun. 2023, 14, 1416. 10.1038/s41467-023-36978-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeur E.; Guéret S. M.; Adihou H.; Gopalakrishnan R.; Lemurell M.; Waldmann H.; Grossmann T. N.; Plowright A. T. New Modalities for Challenging Targets in Drug Discovery. Angewandte Chemie International Edition 2017, 56, 10294–10323. 10.1002/anie.201611914. [DOI] [PubMed] [Google Scholar]
- Varanko A.; Saha S.; Chilkoti A. Recent Trends in Protein and Peptide-Based Biomaterials for Advanced Drug Delivery. Adv. Drug Delivery Rev. 2020, 156, 133–187. 10.1016/j.addr.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.; Nolan E. M. Heavy-Metal Trojan Horse: Enterobactin-Directed Delivery of Platinum(IV) Prodrugs to Escherichia Coli. J. Am. Chem. Soc. 2022, 144, 12756–12768. 10.1021/jacs.2c03324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan C. N. Environmental Applications for Biosurfactants. Environ. Pollut. 2005, 133, 183–198. 10.1016/j.envpol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- West A. C.; Johnstone R. W. New and Emerging HDAC Inhibitors for Cancer Treatment. J. Clin. Invest. 2014, 124, 30–39. 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Yang J.; Loh X. J. Polyhydroxyalkanoates: Opening Doors for a Sustainable Future. NPG Asia Mater. 2016, 8, e265–e265. 10.1038/am.2016.48. [DOI] [Google Scholar]
- Niquille D. L.; Hansen D. A.; Mori T.; Fercher D.; Kries H.; Hilvert D. Nonribosomal Biosynthesis of Backbone-Modified Peptides. Nature Chem. 2018, 10, 282–287. 10.1038/nchem.2891. [DOI] [PubMed] [Google Scholar]
- Czekster C. M.; Robertson W. E.; Walker A. S.; Söll D.; Schepartz A. In Vivo Biosynthesis of a β-Amino Acid-Containing Protein. J. Am. Chem. Soc. 2016, 138, 5194–5197. 10.1021/jacs.6b01023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Ji X.; Gao M.; Dedkova L. M.; Hecht S. M. In Cellulo Synthesis of Proteins Containing a Fluorescent Oxazole Amino Acid. J. Am. Chem. Soc. 2019, 141, 5597–5601. 10.1021/jacs.8b12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakis E.; Magyari S.; Piel J. In-Vivo Production of Diverse β-Amino Acid-Containing Proteins. Angew. Chem. Int. Ed. 2022, 10.1002/anie.202202695. [DOI] [PubMed] [Google Scholar]
- Roe L.; Schissel C. K.; Dover T. L.; Shah B.; Hamlish N. X.; Zheng S.; Dilworth D. A.; Wong N.; Zhang Z.; Chatterjee A. Backbone Extension Acyl Rearrangements Enable Cellular Synthesis of Proteins with Internal Β2-Peptide Linkages. bioRxiv 2023, 10.1101/2023.10.03.560714. [DOI] [Google Scholar]
- Dunkelmann D. L.; Piedrafita C.; Dickson A.; Liu K. C.; Elliott T. S.; Fiedler M.; Bellini D.; Zhou A.; Cervettini D.; Chin J. W. Adding α,α-Disubstituted and β-Linked Monomers to the Genetic Code of an Organism. Nature 2024, 625, 603–610. 10.1038/s41586-023-06897-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan G.; James C.; Krzycki J. Pyrrolysine Encoded by UAG in Archaea: Charging of a UAG-Decoding Specialized tRNA. SCIENCE 2002, 296, 1459–1462. 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]
- Willis J. C. W.; Chin J. W. Mutually Orthogonal Pyrrolysyl-tRNA Synthetase/tRNA Pairs. Nat. Chem. 2018, 10, 831–837. 10.1038/s41557-018-0052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-S.; Fang X.; Wallace A. L.; Wu B.; Liu W. R. A Rationally Designed Pyrrolysyl-tRNA Synthetase Mutant with a Broad Substrate Spectrum. J. Am. Chem. Soc. 2012, 134, 2950–2953. 10.1021/ja211972x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindman N. A.; Bobeica S. C.; Liu W. R.; van der Donk W. A. Facile Removal of Leader Peptides from Lanthipeptides by Incorporation of a Hydroxy Acid. J. Am. Chem. Soc. 2015, 137, 6975–6978. 10.1021/jacs.5b04681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T.; Yanagisawa T.; Sakamoto K.; Yokoyama S. Recognition of Non-Alpha-Amino Substrates by Pyrrolysyl-tRNA Synthetase. J. Mol. Biol. 2009, 385, 1352–1360. 10.1016/j.jmb.2008.11.059. [DOI] [PubMed] [Google Scholar]
- Fricke R.; Swenson C. V.; Roe L. T.; Hamlish N. X.; Shah B.; Zhang Z.; Ficaretta E.; Ad O.; Smaga S.; Gee C. L.; et al. Expanding the Substrate Scope of Pyrrolysyl-Transfer RNA Synthetase Enzymes to Include Non-α-Amino Acids in Vitro and in Vivo. Nat. Chem. 2023, 15, 960–971. 10.1038/s41557-023-01224-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinck M.; Piedrafita C.; Robertson W. E.; Elliott T. S.; Cervettini D.; de la Torre D.; Chin J. W. Genetically Programmed Cell-Based Synthesis of Non-Natural Peptide and Depsipeptide Macrocycles. Nat. Chem. 2023, 15, 61–69. 10.1038/s41557-022-01082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa T.; Ishii R.; Fukunaga R.; Kobayashi T.; Sakamoto K.; Yokoyama S. Crystallographic Studies on Multiple Conformational States of Active-Site Loops in Pyrrolysyl-tRNA Synthetase. J. Mol. Biol. 2008, 378, 634–652. 10.1016/j.jmb.2008.02.045. [DOI] [PubMed] [Google Scholar]
- Jakubowski H.; Goldman E. Quantities of Individual Aminoacyl-tRNA Families and Their Turnover in Escherichia Coli. J. Bacteriol. 1984, 158, 769–776. 10.1128/jb.158.3.769-776.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. B. F.; Xu J.; Shen Z.; Takimoto J. K.; Schultz M. D.; Schmitz R. J.; Xiang Z.; Ecker J. R.; Briggs S. P.; Wang L. RF1 Knockout Allows Ribosomal Incorporation of Unnatural Amino Acids at Multiple Sites. Nat. Chem. Biol. 2011, 7, 779–786. 10.1038/nchembio.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie M. J.; Rovner A. J.; Goodman D. B.; Aerni H.-R.; Haimovich A. D.; Kuznetsov G.; Mercer J. A.; Wang H. H.; Carr P. A.; Mosberg J. A.; et al. Genomically Recoded Organisms Expand Biological Functions. Science 2013, 342, 357–360. 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B.; Gong W.; Ferguson T.; James C.; Krzycki J.; Chan M. A New UAG-Encoded Residue in the Structure of a Methanogen Methyltransferase. SCIENCE 2002, 296, 1462–1466. 10.1126/science.1069556. [DOI] [PubMed] [Google Scholar]
- Blight S. K.; Larue R. C.; Mahapatra A.; Longstaff D. G.; Chang E.; Zhao G.; Kang P. T.; Green-Church K. B.; Chan M. K.; Krzycki J. A. Direct Charging of tRNACUA with Pyrrolysine in Vitro and in Vivo. Nature 2004, 431, 333–335. 10.1038/nature02895. [DOI] [PubMed] [Google Scholar]
- Guo J.; Melançon III C. E.; Lee H. S.; Groff D.; Schultz P. G. Evolution of Amber Suppressor tRNAs for Efficient Bacterial Production of Proteins Containing Nonnatural Amino Acids. Angew. Chem. 2009, 121, 9312–9315. 10.1002/ange.200904035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C.; Xiong H.; Reynolds N. M.; Söll D. Rationally Evolving tRNAPyl for Efficient Incorporation of Noncanonical Amino Acids. Nucleic Acids Res. 2015, 43, e156–e156. 10.1093/nar/gkv800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyer R.; Robotham S. A.; Brodbelt J. S.; Ellington A. D. Evolving tRNA(Sec) for Efficient Canonical Incorporation of Selenocysteine. J. Am. Chem. Soc. 2015, 137, 46–49. 10.1021/ja510695g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pédelacq J.-D.; Cabantous S.; Tran T.; Terwilliger T. C.; Waldo G. S. Engineering and Characterization of a Superfolder Green Fluorescent Protein. Nat. Biotechnol. 2006, 24, 79–88. 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- Rawlings N. D.; Barrett A. J.; Bateman A. Asparagine Peptide Lyases: A Seventh Catalytic Type of Proteolytic Enzymes*⧫. J. Biol. Chem. 2011, 286, 38321–38328. 10.1074/jbc.M111.260026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharp J. M.; Ad O.; Amikura K.; Ward F. R.; Garcia E. M.; Cate J. H. D.; Schepartz A.; Söll D. Initiation of Protein Synthesis with Non Canonical Amino Acids In Vivo. Angew. Chem., Int. Ed. 2020, 59, 3122–3126. 10.1002/anie.201914671. [DOI] [PubMed] [Google Scholar]
- Liu C.; Schultz P. Adding New Chemistries to the Genetic Code. Annual Review of Biochemistry 2010, 79, 413–444. 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- Watson Z.; Knudson I.; Ward F. R.; Miller S. J.; Cate J. H.; Schepartz A.; Abramyan A. M. Atomistic Simulations of the E. Coli Ribosome Provide Selection Criteria for Translationally Active Substrates. Nat. Chem. 2023, 15, 913. 10.1038/s41557-023-01226-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramyan A. M.; Stolzenberg S.; Li Z.; Loland C. J.; Noé F.; Shi L. The Isomeric Preference of an Atypical Dopamine Transporter Inhibitor Contributes to Its Selection of the Transporter Conformation. ACS Chem. Neurosci. 2017, 8, 1735–1746. 10.1021/acschemneuro.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michino M.; Boateng C. A.; Donthamsetti P.; Yano H.; Bakare O. M.; Bonifazi A.; Ellenberger M. P.; Keck T. M.; Kumar V.; Zhu C.; et al. Toward Understanding the Structural Basis of Partial Agonism at the Dopamine D3 Receptor. J. Med. Chem. 2017, 60, 580–593. 10.1021/acs.jmedchem.6b01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin Schmeing T.; Huang K. S.; Strobel S. A.; Steitz T. A. An Induced-Fit Mechanism to Promote Peptide Bond Formation and Exclude Hydrolysis of Peptidyl-tRNA. Nature 2005, 438, 520–524. 10.1038/nature04152. [DOI] [PubMed] [Google Scholar]
- Majumdar C.; Walker J. A.; Francis M. B.; Schepartz A.; Cate J. H. D. Aminobenzoic Acid Derivatives Obstruct Induced Fit in the Catalytic Center of the Ribosome. ACS Cent. Sci. 2023, 9, 1160. 10.1021/acscentsci.3c00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y.; Suga H. The RaPID Platform for the Discovery of Pseudo-Natural Macrocyclic Peptides. Acc. Chem. Res. 2021, 54, 3604–3617. 10.1021/acs.accounts.1c00391. [DOI] [PubMed] [Google Scholar]
- Englander M. T.; Avins J. L.; Fleisher R. C.; Liu B.; Effraim P. R.; Wang J.; Schulten K.; Leyh T. S.; Gonzalez R. L.; Cornish V. W. The Ribosome Can Discriminate the Chirality of Amino Acids within Its Peptidyl-Transferase Center. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6038–6043. 10.1073/pnas.1424712112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruna K.; Alkazemi M. H.; Liu Y.; Söll D.; Englert M. Engineering the Elongation Factor Tu for Efficient Selenoprotein Synthesis. Nucleic Acids Res. 2014, 42, 9976–9983. 10.1093/nar/gku691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete F. A. C.; Griffin W. C.; Chan Y.-C.; Martin M. I.; Alejo J. L.; Natchiar S. K.; Knudson I.; Altman R. B.; Schepartz A.; Miller S. J.; et al. β-Amino Acids Reduce Ternary Complex Stability and Alter the Translation Elongation Mechanism. bioRxiv 2024, 2024.02.24.581891. 10.1101/2024.02.24.581891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmied W. H.; Tnimov Z.; Uttamapinant C.; Rae C. D.; Fried S. D.; Chin J. W. Controlling Orthogonal Ribosome Subunit Interactions Enables Evolution of New Function. Nature 2018, 564, 444–448. 10.1038/s41586-018-0773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht S. M. Expansion of the Genetic Code Through the Use of Modified Bacterial Ribosomes. J. Mol. Biol. 2022, 434, 167211. 10.1016/j.jmb.2021.167211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.