

Abstract
Background
Disaccharide Intolerance Type I (Mendelian Interance in Man database: *222900) is a rare inborn error of metabolism resulting from mutation in sucrase-isomaltase (Enzyme Catalyzed 3.2.1.48). Usually, infants with SI deficiency come to attention because of chronic diarrhea and nutritional evidence of malabsorption.
Case Presentation
We describe an atypical presentation of this disorder in a 10-month-old infant. In addition to chronic diarrhea, the child displayed severe and chronic hypercalcemia, the evaluation of which was negative. An apparently coincidental right orbital hemangioma was detected. Following identification of the SI deficiency, an appropriately sucrose-restricted, but normal calcium diet regimen was instituted which led to cessation of diarrhea, substantial weight gain, and resolution of hypercalcemia.
Conclusions
This case illustrates that, similar to congenital lactase deficiency (Mendelian Interance in Man database: *223000, Alactasia, Hereditary Disaccharide Intolerance Type II), hypercalcemia may complicate neonatal Sucrase-Isomaltase deficiency. Hypercalcemia in the presence of chronic diarrhea should suggest disaccharide intolerance in young infants.
Background
Sucrase-isomaltase (SI, EC 3.2.1.48) is an enterocyte-specific small intestine brush-border membrane disaccharidase. It is required for hydrolysis of dietary sucrose and some starches. Because absorption cannot take place until monosaccharides are produced, SI deficiency leads to increased luminal disaccharides and consequent osmotic diarrhea. Although generally thought to have a later presentation [1] than other primary intestinal malabsorption syndromes, recent reports of infants with SI deficiency indicate the existence of an early onset form (presentation of diarrhea at 2–16 weeks) that is complicated by severe malnutrition [2]. Starnes and Walsh [3] observed renal calculi in two adults with sucrase deficiency, but neither patient had evidence of hypercalcemia. In contrast, hypercalcemia frequently complicates congenital lactase deficiency [4]. The present case illustrates the occurrence of hypercalcemia and nephrocalcinosis as a clinically significant complication of SI deficiency in an infant and suggests that common mechanisms may lead to hypercalcemia in both SI and congenital lactase deficiency.
Case Presentation
The patient was a 10 month old caucasian female infant presenting with failure to thrive (FTT), metabolic acidosis, and hypercalcemia. She was the product of a term uncomplicated pregnancy in a 31-year-old and delivered without complications. She had been growing well and thriving until weaned from breast milk to formula at age 4 months when she began to have frequent stools and poor weight gain. Formula and diet changes were attempted without improvement. She was seen by a pediatric gastroenterologist and underwent colonoscopy, a biopsy from which showed eosinophilic inflammation. Upper GI tract endoscopy was interrupted because of sedation problems. She was treated with glucocorticoids and her formula was changed to Neocate (49 mg Ca/100 mL) with some initial improvement. The glucocorticoids were discontinued at age 9 months but she still gained weight poorly; routine chemistry analyses showed evolving hypercalcemia (outpatient calcium peak 13.7 mg/dL, Figure 1) and persistent metabolic acidosis. She was admitted to the hospital for evaluation and treatment of hypercalcemia, FTT, and metabolic acidosis.
Figure 1.
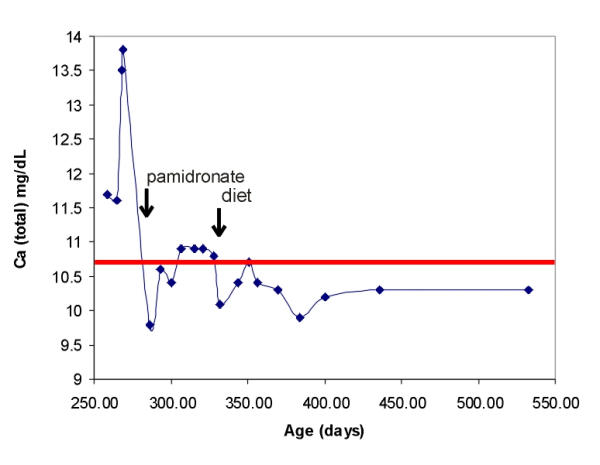
Course of hypercalcemia and response to dietary intervention
Other than skin changes consistent with wasting, her examination was benign. There was no abnormal flatus or bloating, but there had been intermittent blood in the stool with some mucous. Laboratory studies are shown in Table 1. The intact parathyroid hormone (PTH) was appropriately suppressed to 6 pg/mL with concomitant serum calcium of 13.8 mg/dL and ionized calcium (Ca2+) 1.98 mmol/L (normal 1.20–1.38); other pertinent laboratory values included: phosphorus 5.7 mg/dL; magnesium 2.6 mg/dL; alkaline phosphatase 79 IU/L; PTH-related peptide (PTH-rp) appropriately undetectable at <0.2 pmol/L (normal < 2.8); 25-hydroxy Vitamin D 16 ng/mL (reference lab normal range for age 3–17 years = 13–67); and 1,25-dihydroxy Vitamin D level 32 pg/mL (reference lab normal range for age 3–17 years = 27–71). The urine calcium was 11.9 mg/dL with urine creatinine < 10 mg/dL. Renal ultrasound demonstrated nephrocalcinosis. The hemi-skeletal bone age was interpreted to be delayed to that of a 3–6 month old. Stools were positive for leukocytes and trace reducing substances. Normal karyotype and normal fluorescent in-situ hybridization (FISH) for the elastin gene locus on chromosome 7 (46, XX, negative ELN 7q11.23) along with a negative echocardiogram helped exclude Williams syndrome. She initially was treated with intravenous normal saline followed by furosemide with only transient improvement. Recurrent hypercalcemia prompted a 3-day course of intravenous pamidronate (0.5 mg/kg per dose) leading to sustained high-normal serum calcium (Figure 1). She was discharged home. She was empirically treated with Polycitra™ for the persistent metabolic acidosis, but on-going FTT prompted reevaluation.
Table 1.
Initial admission and follow up studies of case report.
| Metabolite | Admission #1 Age 9 mos | Admission #2 Age 10 mos | Age 17 mos |
| Na (mmol/L) | 144 | 140 | 139 |
| K (mmol/L) | 3.9 | 5.7 | 4.1 |
| Cl (mmol/L) | 103 | 101 | 103 |
| CO2 (mmol/L) | 22 | 19 | 24 |
| BUN (mg/dL) | 18 | 32 | 14 |
| Creatinine (mg/dL) | 0.4 | 0.2 | 0.3 |
| Glucose (mg/dL) | 96 | 69 | 92 |
| AST (U/L) | 33 | 73 | |
| ALT (U/L) | 9 | 24 | |
| Alkaline Phosphatase (U/L) | 79 | 152 | |
| LDH (U/L) | 928 | 748 | |
| Pre-albumin (g/dL) | 20.0 | 18.1 | |
| Total Protein (g/dL) | 8.0 | 7.9 | |
| Albumin (g/dL) | 4.5 | 5.2 | |
| Calcium (mg/dL) | 13.8 | 10.9 | 10.3 |
| Ca 2+ (mmol/L) | 1.98 | 1.44 | |
| Phosphorus (mg/dL) | 5.7 | 7.7 | 5.1 |
| PTH (pg/mL) | 6 | 10 | |
| PTH-rp (pmol/L) | <0.2 | ||
| VIP (ng/mL) | < 5 | ||
| Urine Ca/Cr | 11.9/<10 | 2.8 | 0.36 |
| 25-hydroxy Vitamin D (ng/mL) | 16 | 16 | |
| 1,25-dihydroxy Vitamin D (pg/mL) | 32 | 47 | |
| Mg (mg/dL) | 2.6 | 2.3 | |
| T4 (ug/dL) | 8.6 | ||
| TSH (uIU/mL) | 2.63 | ||
| Calcitonin (pg/mL) | 9.9 | ||
| Vitamin A (μg/dL) | 33 | ||
| Gastrin (pg/mL) | 158 |
At time of second admission the infant was taking Neocate supplemented with 5 jars of baby food fruits per day plus Polycitra™. Gross motor development was delayed but other developmental axes were intact. Family history was significant for Hashimoto's thyroiditis in the mother, maternal aunt, and maternal grandfather. The parents were both of European ethnic origin and there was no consanguinity. Physical examination showed an alert, nondysmorphic infant with severe reduction in muscle mass and subcutaneous fat. Frequent loose watery stools were noted. A skeletal survey showed striking linear bone density increase in the epiphyses in an area adjacent to the growth plate (Figure 2A). Follow up admission laboratory studies are shown in Table 1. Specifically, the calcitonin was 9.9 pg/mL (normal < 12) and the Vitamin A concentration was 33 μg/dL (20–43). The urine calcium was 11.1 mg/dL and calcium/creatinine ratio of 2.8. EKG showed a normal QTc. Renal ultrasound confirmed nephrocalcinosis (Figure 2B). Upper endoscopy with biopsy revealed areas of mild focal chronic inflammatory cells in the stomach. Colonoscopy showed no bowel wall inflammation but copious watery stool. Stool sodium was 17 mmol/L.
Figure 2.
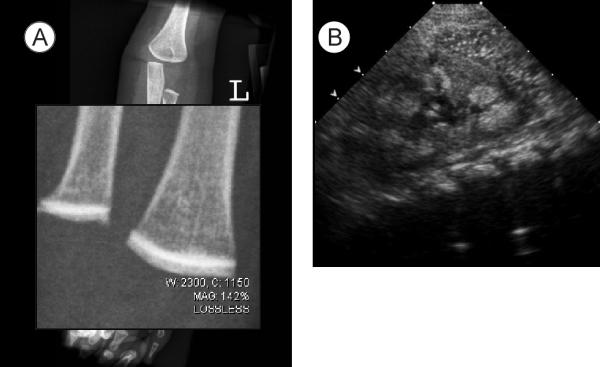
Diagnostic imaging studies. A. Increased bone density in area adjacent to distal femoral growth plate; B. renal ultrasound demonstrating left medullary nephocalcinosis
The persistence of hypercalcemia with diarrhea prompted a nuclear medicine scan with 1.5 mCi of 111indium pentetreotide to pursue a possible paraneoplastic process [5] which showed uptake in the right orbital region. A head CT and MRI demonstrated an infiltrating lesion in the right orbit along the inferior and lateral compartment of the orbit involving the intra- and extra-coronal space. There was no obvious lytic change or periostosis; however, there were flow voids consistent with a vascular malformation. No abnormalities in extraocular movements, globe, or retina were observed. These findings were interpreted as consistent with a benign orbital hemangioma. Serial follow up studies confirm that the lesion is not growing; indeed there is evidence of fatty involution.
Small bowel biopsies taken at the time of endoscopy revealed: lactase 34.6 μM/min/g (24.5 +/- 18, abnormal less than 15), sucrase 0 μM/min/g (54.4 +/- 25.4, abnormal less than 25), maltase 22.7 μM/min/g (160.8 +/- 52.8, abnormal less than 100) leading to the diagnosis of sucrase-isomaltase deficiency. Sucrose free diet with Sucraid™ enzyme replacement was prescribed followed by changing feeds to 3232A base formula with supplemental anhydrous dextrose (Ca 94 mg/100 mL) resulting in a prompt cessation of diarrhea. Urine calcium/creatinine ratio decreased to 0.58. Her weight has steadily improved (Figure 3). Residual muscle weakness in the lower extremities is being treated with physical therapy.
Figure 3.
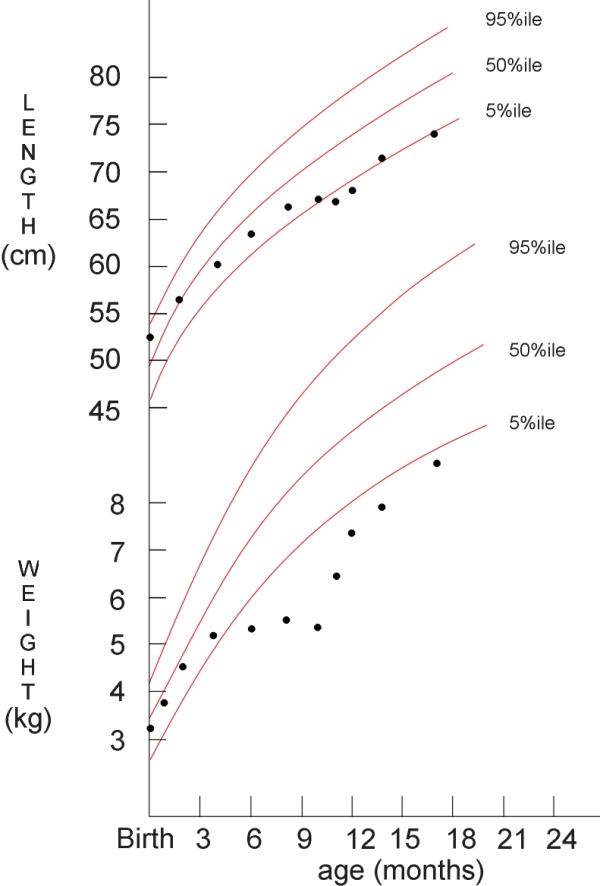
Growth response to dietary intervention
Causes of hypercalcemia in infancy
An extensive evaluation of our patient failed to identify any abnormality in calcium regulation. It should be emphasized that our patient demonstrated nephrocalcinosis prior to alkalinization with Polycitra™ to combat her metabolic acidosis. Urine alkalinization is a common strategy to treat urate and cysteine renal stones. Animal and preliminary human studies suggest acid-phosphate replacement (vs. neutral-phosphate therapy) is of equal benefit and does not perturb calcium/phosphorus metabolism in children with phosphate-wasting rickets [6]. Infants with hypercalcemia may have no obvious symptoms, but constitutional symptoms such as poor appetite, irritability, vomiting, FTT, or occasionally seizures may occur [7]. Classically, hypercalcemia leads to increased urine output and diminished stool frequency. Physical signs may include hypotonia, dehydration, and bradycardia. Laboratory abnormalities can include short QTc and elevated creatinine. Increased urinary calcium is an expected compensatory response with associated increased risk of renal calculi. The differential diagnosis of hypercalcemia in infants includes hyperparathyroidism, familial hypocalciuric hypercalcemia, hypervitaminosis, paraneoplastic processes, and Williams syndrome.
Hyperparathyroidism may be either sporadic or familial arising from either adenoma or parathyroid hyperplasia. Familial hyperparathyroidism may be confined to parathyroid adenoma (HRPT1 #145000) or may involve the broader endocrine disturbances of multiple endocrine neoplasia Types I (#131100) and IIA (#171400). MENI results from mutations in menin, a regulator of TGFβ signaling in a variety of tissues [8]. MENIIA results form mutations in RET a cell surface growth factor receptor with intrinsic tyrosine kinase activity [9]. Familial hypocalciuric hypercalcemia (#145980), generally autosomal dominant and caused by mutations in the renal calcium sensing receptor [10,11], is excluded rather easily based on urinary calcium levels of the patient and parents.
Both hypervitaminoses D and A are potential causes of hypercalcemia. Prolonged feeding with over fortified milk or vitamin preparations has been reported as sources of excess intake of vitamin D. Abnormal vitamin D metabolism resulting from inflammatory diseases such as sarcoidosis or tuberculosis or as seen in neonatal fat necrosis are additional potential causes, albeit highly unlikely in young infants [12]. The mechanism is thought to be due to increased 1á-hydroxylase activity leading to increased production of 1,25-dihydroxy Vitamin D.
While neoplasia with osseous metastasis is a potential cause of hypercalcemia, tumors may also secrete materials such as PTH-rp or other osteoclast-activating factors, cytokines, or other growth factors that modulate bone remodeling leading to calcium adsorption from bone. Chronic immobilization can also lead to enhanced bone resorption.
Williams syndrome is a well described birth defect complex characterized by FTT, branch pulmonary artery stenosis, supravalvular aortic stenosis, dysmorphic facies, mental retardation, and infantile hypercalcemia. Williams syndrome results from a constitutional deletion in chromosome 7q11.23 [13]. The cause of hypercalcemia associated with Williams syndrome is not well understood but has been postulated to be associated with enhanced intestinal absorption of calcium. The presence or absence of significant hypercalcemia does not correlate with the deletion size [14].
Sucrase-isomaltase deficiency
Sucrase-isomaltase deficiency is a rare autosomal recessive disorder in which there is complete absence of sucrase and most of the maltase digestive activity. The usual presentation generally is at weaning from breast milk when infants are first exposed to the offending carbohydrates [15-17]. Upon ingestion of disaccharides and oligosaccharides, the failure to breakdown sucrose into fructose and glucose results in an osmotic-fermentative diarrhea. Occasional patients may come to attention in late childhood or adulthood but careful history often indicates that symptoms have appeared earlier [18,19]. The mainstay of treatment is dietary restriction of sucrose-containing foods. Effective enzyme replacement with whole yeast cells has been reported [20]. Purified yeast enzyme, sacrosidase, is a highly effective adjunct to dietary restriction [21].
The enzyme itself is composed of 1827 amino acids encoded by a 3364 bp mRNA. The gene locus has 30 exons spanning 106.6 kB. Pro-SI is anchored in the membrane by a 20 amino acid membrane-spanning segment. This is followed by a 22 amino acid stalk on which sit the 2 globular catalytic domains. The catalytic domains are similar to one another (41% amino acid identity) indicating that they have arisen by duplication and divergence [22]. One estimate indicated a 2% mutant heterozygote frequency in European Americans [23] but, given the rarity of diagnosed SI deficiency, this seems high. Sucrase deficiency may be especially common in indigenous Greenlanders (estimated 5%) and is accompanied most often by lactase deficiency [24,25]. Initial studies demonstrated that most SI mutations result in lack of enzyme protein (i.e. null mutation) [26]. Mutation may alter the post-translational processing of the enzyme only for the asparagines-linked high-mannose groups but not the mature oligosaccharides. The resulting enzyme is both mislocalized and unstable [27]. Another mutation, Q1098P, was found to cause retention of SI in the transport pathway between the endoplasmic reticulum and Golgi apparatus [28]. An unusual mutation, Q117R, that alters the structure close to the O-glycosylated stalk domain, leads to random distribution of the enzyme to both the apical and basolateral cell membranes [29]. An unusual human mutation that causes SI to be secreted rather than retained in the plasma membrane demonstrates that the enzyme is required to be at the enterocyte cell surface [30].
Disaccharidase deficiency and hypercalcemia
A single case report potentially relates SI deficiency with disturbance of calcium metabolism. In that study, two adults presented with renal calculi and nephrocalcinosis [3]. Lactase deficiency may give rise to metabolic bone disease [31]. More pertinently, Saarela et al described 7 of 10 infants with congenital lactase deficiency who had hypercalcemia at the time of initial presentation [4]. Five of these 7 infants had medullary nephrocalcinosis. The mechanism by which congenital lactase deficiency induces hypercalcemia is uncertain. There may be roles for chronic metabolic acidosis and dehydration in promoting both increased bone mobilization of calcium and thus increasing the risk of renal deposition. Non-hydrolyzed lactose may also exert a direct enhancing effect on calcium absorption in the ileum. In other conditions with chronic diarrhea, such as inflammatory bowel disease, renal stones have been composed of calcium phosphate or oxalate [32]. Alternatively, it is possible that both SI and lactase are connected mechanistically to intestinal calcium homeostasis through molecular interactions with calcium transporters. In this regard, SI, lactase, and the receptor for 1,25 OH-VitD3 (VDR) share transcriptional regulation through the homeodomain transcription factor CDX2 [33,34]. The possibility that SI or lactase deficiency could cause secondary up-regulation of VDR awaits exploration in cell culture or animal models.
Conclusions
We conclude that SI deficiency should be added to the list of disorders that can cause infantile hypercalcemia and particularly may be considered when accompanied by FTT and watery diarrhea. In addition, infants with SI deficiency should have careful monitoring of serum calcium. Prolonged periods of diet noncompliance may increase the risk of chronic renal disease secondary to nephrocalcinosis. Infants presenting with persistent, watery diarrhea should be evaluated for SI.
Competing interests
None declared
Authors contributions
JWB – provided care during second admission, composed the manuscript
BR – provided care during second admission, performed endoscopy, and made the primary diagnosis
WT – provided long term primary care including longitudinal followup
SSB – performed and interpreted the specialized diagnostic enzyme assays
WHM – performed and interpreted the nuclear medicine imaging studies
MCM – performed and interpreted the neuroimaging studies
SMB – provided dietary management during and after second hospitalization
NLG – provided anaesthesia care and coordinated multiple procedures during second hospitalization
IDS – evaluated/managed the patient primarily and contributed/edited much of the manuscript
All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
John W Belmont, Email: jbelmont@bcm.tmc.edu.
Barbara Reid, Email: jbelmont@bcm.tmc.edu.
William Taylor, Email: jbelmont@bcm.tmc.edu.
Susan S Baker, Email: sbaker@upa.chob.edu.
Warren H Moore, Email: wmoore@bcm.tmc.edu.
Michael C Morriss, Email: cmorriss@bcm.tmc.edu.
Susan M Podrebarac, Email: smpodreb@TexasChildrensHospital.org.
Nancy Glass, Email: nglass@bcm.tmc.edu.
I David Schwartz, Email: dschwartz@richmed.medpark.sc.edu.
References
- Peterson ML, Herber R. Intestinal sucrase deficiency. Trans Assoc Am Physicians. 1967;80:275–283. [PubMed] [Google Scholar]
- Newton T, Murphy MS, Booth IW. Glucose polymer as a cause of protracted diarrhea in infants with unsuspected congenital sucrase-isomaltase deficiency. J Pediatr. 1996;128:753–756. doi: 10.1016/s0022-3476(96)70325-6. [DOI] [PubMed] [Google Scholar]
- Starnes CW, Welsh JD. Intestinal sucrase-isomaltase deficiency and renal calculi. N Engl J Med. 1970;282:1023–1024. doi: 10.1056/NEJM197004302821809. [DOI] [PubMed] [Google Scholar]
- Saarela T, Simila S, Koivisto M. Hypercalcemia and nephrocalcinosis in patients with congenital lactase deficiency. J Pediatr. 1995;127:920–923. doi: 10.1016/s0022-3476(95)70028-5. [DOI] [PubMed] [Google Scholar]
- Sandhu FA, Martuza RL. Craniofacial hemangiopericytoma associated with oncogenic osteomalacia: case report. J Neurooncol. 2000;46:241–247. doi: 10.1023/A:1006352106762. [DOI] [PubMed] [Google Scholar]
- Alon U, Moore W, Schwartz ID. Effect of acid-phosphate vs neutral phosphate on urine acidity, acid-base balance and mineral homeostasis in children with familial hypophosphatemic rickets (XLH). Pediatric Research. 1999;45:328A. [Google Scholar]
- Rodd C, Goodyer P. Hypercalcemia of the newborn: etiology, evaluation, and management. Pediatr Nephrol. 1999;13:542–547. doi: 10.1007/s004670050654. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, Pasini B, Hoppener JW, van Amstel HK, Romeo G, et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature. 1994;367:375–376. doi: 10.1038/367375a0. [DOI] [PubMed] [Google Scholar]
- Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, Levi T, Seidman CE, Seidman JG. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 1993;75:1297–1303. doi: 10.1016/0092-8674(93)90617-y. [DOI] [PubMed] [Google Scholar]
- Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81:239–297. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- Schwartz ID, Root AW. Hypercalcemia associated with normal 1,25-dihydroxyvitamin D concentrations in a neonate with factor IX deficiency. J Pediatr. 1989;114:509–510. doi: 10.1016/s0022-3476(89)80593-1. [DOI] [PubMed] [Google Scholar]
- Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993;5:11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- Wang MS, Schinzel A, Kotzot D, Balmer D, Casey R, Chodirker BN, Gyftodimou J, Petersen MB, Lopez-Rangel E, Robinson WP. Molecular and clinical correlation study of Williams-Beuren syndrome: No evidence of molecular factors in the deletion region or imprinting affecting clinical outcome. Am J Med Genet. 1999;86:34–43. doi: 10.1002/(SICI)1096-8628(19990903)86:1<34::AID-AJMG7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Baudon JJ, Veinberg F, Thioulouse E, Morgant G, Aymard P, Charritat JL. Sucrase-isomaltase deficiency: changing pattern over two decades. J Pediatr Gastroenterol Nutr. 1996;22:284–288. doi: 10.1097/00005176-199604000-00010. [DOI] [PubMed] [Google Scholar]
- Treem WR. Congenital sucrase-isomaltase deficiency. J Pediatr Gastroenterol Nutr. 1995;21:1–14. doi: 10.1097/00005176-199507000-00001. [DOI] [PubMed] [Google Scholar]
- Treem WR. Clinical heterogeneity in congenital sucrase-isomaltase deficiency. J Pediatr. 1996;128:727–729. doi: 10.1016/s0022-3476(96)70320-7. [DOI] [PubMed] [Google Scholar]
- Ringrose RE, Preiser H, Welsh JD. Sucrase-isomaltase (palatinase) deficiency diagnosed during adulthood. Dig Dis Sci. 1980;25:384–387. doi: 10.1007/BF01308064. [DOI] [PubMed] [Google Scholar]
- Mainguet P, Vanderhoeden R, Loeb H, Eggermont E. Congenital maltase-sucrase and maltase-isomaltase deficiency in an adult. Digestion. 1973;8:353–359. doi: 10.1159/000197335. [DOI] [PubMed] [Google Scholar]
- Harms HK, Bertele-Harms RM, Bruer-Kleis D. Enzyme-substitution therapy with the yeast Saccharomyces cerevisiae in congenital sucrase-isomaltase deficiency. N Engl J Med. 1987;316:1306–1309. doi: 10.1056/NEJM198705213162104. [DOI] [PubMed] [Google Scholar]
- Treem WR, McAdams L, Stanford L, Kastoff G, Justinich C, Hyams J. Sacrosidase therapy for congenital sucrase-isomaltase deficiency. J Pediatr Gastroenterol Nutr. 1999;28:137–142. doi: 10.1097/00005176-199902000-00008. [DOI] [PubMed] [Google Scholar]
- Hunziker W, Spiess M, Semenza G, Lodish HF. The sucrase-isomaltase complex: primary structure, membrane-orientation, and evolution of a stalked, intrinsic brush border protein. Cell. 1986;46:227–234. doi: 10.1016/0092-8674(86)90739-7. [DOI] [PubMed] [Google Scholar]
- Welsh JD, Poley JR, Bhatia M, Stevenson DE. Intestinal disaccharidase activities in relation to age, race, and mucosal damage. Gastroenterology. 1978;75:847–855. [PubMed] [Google Scholar]
- Gudmand-Hoyer E, Fenger HJ, Kern-Hansen P, Madsen PR. Sucrase deficiency in Greenland. Incidence and genetic aspects. Scand J Gastroenterol. 1987;22:24–28. doi: 10.3109/00365528708991851. [DOI] [PubMed] [Google Scholar]
- Asp NG, Berg NO, Dahlqvist A, Gudmand-Hoyer E, Jarnum S, McNair A. Intestinal disaccharidases in Greenland Eskimos. Scand J Gastroenterol. 1975;10:513–519. [PubMed] [Google Scholar]
- Gray GM, Conklin KA, Townley RR. Sucrase-isomaltase deficiency. Absence of an inactive enzyme variant. N Engl J Med. 1976;294:750–753. doi: 10.1056/NEJM197604012941403. [DOI] [PubMed] [Google Scholar]
- Lloyd ML, Olsen WA. A study of the molecular pathology of sucrase-isomaltase deficiency. A defect in the intracellular processing of the enzyme. N Engl J Med. 1987;316:438–442. doi: 10.1056/NEJM198702193160804. [DOI] [PubMed] [Google Scholar]
- Ouwendijk J, Moolenaar CE, Peters WJ, Hollenberg CP, Ginsel LA, Fransen JA, Naim HY. Congenital sucrase-isomaltase deficiency. Identification of a glutamine to proline substitution that leads to a transport block of sucrase-isomaltase in a pre-Golgi compartment. J Clin Invest. 1996;97:633–641. doi: 10.1172/JCI118459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spodsberg N, Jacob R, Alfalah M, Zimmer KP, Naim HY. Molecular basis of aberrant apical protein transport in an intestinal enzyme disorder. J Biol Chem. 2001;276:23506–23510. doi: 10.1074/jbc.C100219200. [DOI] [PubMed] [Google Scholar]
- Jacob R, Zimmer KP, Schmitz J, Naim HY. Congenital sucrase-isomaltase deficiency arising from cleavage and secretion of a mutant form of the enzyme. J Clin Invest. 2000;106:281–287. doi: 10.1172/JCI9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metabolic bone disease as a result of lactase deficiency. Nutr Rev. 1979;37:72–73. doi: 10.1111/j.1753-4887.1979.tb02210.x. [DOI] [PubMed] [Google Scholar]
- Clark JH, Fitzgerald JF, Bergstein JM. Nephrolithiasis in childhood inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1985;4:829–834. doi: 10.1097/00005176-198510000-00026. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Miyamoto K, Li B, Taketani Y, Kitano M, Inoue Y, Morita K, Pike JW, Takeda E. The caudal-related homeodomain protein Cdx-2 regulates vitamin D receptor gene expression in the small intestine. J Bone Miner Res. 1999;14:240–247. doi: 10.1359/jbmr.1999.14.2.240. [DOI] [PubMed] [Google Scholar]
- Goda T. Regulation of the expression of carbohydrate digestion/absorption-related genes. Br J Nutr. 2000;84:S245–248. doi: 10.1079/096582197388626. [DOI] [PubMed] [Google Scholar]