

Abstract
RAS mutations occur in approximately 30% of tumors worldwide and have a poor prognosis due to limited therapies. Covalent targeting of KRAS G12C has achieved significant success in recent years, but there is still a lack of efficient therapeutic approaches for tumors with non-G12C KRAS mutations. A highly promising approach is to target the MAPK pathway downstream of RAS, with a particular focus on RAF kinases. First-generation RAF inhibitors have been authorized to treat BRAF mutant tumors for over a decade. However, their use in RAS-mutated tumors is not recommended due to the paradoxical ERK activation mainly caused by RAF dimerization. To address the issue of RAF dimerization, type II RAF inhibitors have emerged as leading candidates. Recent clinical studies have shown the initial effectiveness of these agents against RAS mutant tumors. Promisingly, type II RAF inhibitors in combination with MEK or ERK inhibitors have demonstrated impressive efficacy in RAS mutant tumors. This review aims to clarify the importance of RAF dimerization in cellular signaling and resistance to treatment in tumors with RAS mutations, as well as recent progress in therapeutic approaches to address the problem of RAF dimerization in RAS mutant tumors.
Key words: RAS mutations, RAF dimerization, RAF inhibitors, Cancer therapy, Drug resistance
Graphical abstract
Homo- and heterodimers of RAF kinases play a pivotal role in the signaling transduction and drug resistance in RAS mutant tumors. The red pentagrams represent mutated RAS proteins.
1. Introduction
Approximately 30% of human tumors harbor RAS mutations, among the most common oncogenic changes1. Individuals diagnosed with tumors that carry RAS mutations face a higher likelihood of metastasis and typically experience an unfavorable prognosis2. Nonetheless, the development of precise treatments for tumors with RAS mutations is urgently needed. Among the RAS GTPase family members, KRAS has considerably greater oncogenic potential than NRAS and HRAS3. Therefore, particular focus should be placed on managing tumors with KRAS mutations. KRAS mutations are the most common and are associated with approximately 90% of pancreatic cancers, 50% of colorectal cancers (CRCs), and 25% of lung cancers4. However, KRAS mutations exhibit significantly reduced sensitivity to numerous targeted inhibitors compared to patients with NRAS and HRAS mutations5. Reduced sensitivity, along with a high occurrence of drug resistance, leads to low rates of response and short durations of treatment effectiveness in individuals with KRAS mutant tumors. Nevertheless, the KRAS proteins’ distinctive molecular structure and extensive biological impact make it difficult to be targeted directly. Solid tumors commonly exhibit KRAS mutations, with G12D, G12V, and G13D being the most prevalent. Less frequent mutations, such as G12C, G12A, and G12R6, make up less than 10% of cases. Several inhibitors are being developed that directly target mutant KRAS proteins4, but various oncogenic mutant KRAS proteins function differently7. Only KRAS G12C inhibitors (KRAS G12Ci) are therapeutically beneficial and have received clinical approval for use in treating individuals with advanced non-small cell lung cancer (NSCLC)8,9, while effective medications are still lacking for non-G12C KRAS mutant cancers. Hence, it is crucial and challenging to investigate specific treatment approaches for RAS mutant tumors, specifically those with non-G12C KRAS mutations.
KRAS mutations regulate the growth and survival of cancer cells mainly by triggering the RAF-MEK-ERK (MAPK) signaling pathway, which has been heavily studied to develop drugs10,11. Multiple membrane receptors utilize the MAPK pathway as a central hub for transmitting growth signals12. Following external stimulation from growth factors, receptor tyrosine kinases (RTKs) initiate the activation of RAS-GTPases, subsequently attracting RAF kinases to the cellular membrane. This event stimulates the formation of homo or heterodimers of RAF, resulting in autophosphorylation of RAF, followed by the activation of MEK and ERK via a phosphorylation cascade13. ERK then relocates to the cell nucleus and regulates the activation of numerous transcription factors. Sustained activation of MAPK signaling, facilitated by RAS mutations, drives tumorigenesis. Most MAPK pathway inhibitors could cause the reactivation of ERK signaling when resistance develops10,14. Thus, effective treatment of RAS mutant tumors relies on achieving robust and enduring suppression of ERK signaling15,16.
Investigations into essential kinases downstream of KRAS have attracted significant attention in academic research. In particular, there is an increasing fascination with the advancement of medications targeting distinct RAF isoforms or genetic variations. Targeting RAF upstream of the RAF-MEK-ERK cascade is a promising strategy, as the signal is amplified stepwise from RAF to ERK. Nevertheless, the simultaneous elimination of MEK1/2 or ERK1/2 leads to fatality in adult mice, whereas the concurrent elimination of BRAF and CRAF is well tolerated17. Additionally, clinical evidence suggests that targeting BRAF V600 has greater efficacy than targeting MEK in patients with BRAF V600 mutant melanoma18. Notably, these RAF monomer inhibitors can induce ERK activation in RAS mutant tumor cells19, making them inappropriate for treating RAS mutant tumors. ERK signaling reactivation relies heavily on the homo and heterodimerization of RAF20,21. Preclinical and clinical studies of next-generation RAF dimer inhibitors are ongoing and have obtained promising results, although some drugs still exhibit minimal paradoxical ERK activation in certain RAS mutant cell lines22. This review highlights the significance of RAF dimerization in MAPK signaling and the regulatory mechanisms of RAF dimers. This paper also summarizes the progress in developing RAF dimer inhibitors for RAS mutant tumors, explicitly focusing on resistance mechanisms and therapeutic strategies to combat resistance.
2. Two main strategies for targeting oncogenic RAS
2.1. Targeting oncogenic RAS proteins directly
Two main strategies for targeting oncogenic RAS signaling have been identified: direct and indirect (Fig. 1). Since KRAS mutations account for the most significant proportion of RAS, current direct targeting strategies focus on KRAS mutations. There are many obstacles to directly targeting oncogenic KRAS proteins because of their strong affinity for GTP and the lack of small molecule drug binding pockets23. KRAS was considered an “undruggable” target until KRAS G12Ci made breakthroughs in recent years. The KRAS G12C covalent inhibitors, such as sotorasib and adagrasib, can bind to the switch II pocket of KRAS G12C-GDP and effectively lock it in the inactive state24. Recent clinical success in targeting KRAS G12C has inspired scientists to search for novel strategies targeting alternative KRAS mutants. MRTX1133, a selective and non-covalent KRAS G12D inhibitor, potently inhibits both the activated and inactivated states of KRAS G12D, exhibiting more than 1000-fold more excellent antitumor activity against mutant KRAS G12D tumor cells than against wild-type (WT) KRAS tumor cells25. However, its bioavailability is currently limited and requires further optimization26. In addition to small molecule inhibitors, several novel therapeutics targeting specific KRAS mutations, such as the KRAS G12D degrader (ASP3082)27, KRAS G12D siRNAs (siG12D-LODER28 and iExosomes29) and KRAS G12D/V-specific T cell receptor (TCR)-engineered T cell therapy30, have also been developed and entered clinical trials (Table 1). These mutant-specific KRAS therapies exhibit reduced toxicity toward normal cells but require individualized development of each KRAS mutant and may be more susceptible to drug resistance.
Figure 1.

Strategies for targeting oncogenic RAS signaling. (A) Direct targeting strategies are categorized into targeting mutant-specific KRAS and targeting pan-KRAS or pan-RAS, including small molecule inhibitors (KRAS G12C–sotorasib complex, PDB: 6OIM; KRAS G12D–MRTX1133 complex, PDB: 7T47; KRAS G12V–BI-2865 complex, PDB: 8AZZ; KRAS G13D–BI-2865 complex, PDB: 8B00.)25,53, PROTAC27,54, gene editing therapy (siRNA29, CRISPR/Cas955,56), TCR T-cell therapy57,58, CAR T-cell therapy59,60, cancer vaccines33, mutant KRAS-specific dimeric IgA61, and cyclic peptides62,63. (B) Indirect targeting strategies include targeting upstream of RAS (such as RTKs and SHP2/SOS1) and targeting downstream of RAS (such as RAF-MEK-ERK and PI3K-AKT-mTOR). PROTAC, proteolysis targeting chimeras; TCR, T-cell receptor; CAR, chimeric antigen receptor; RISC, RNA-induced silencing complex; NRP-1, receptor neuropilin-1.
Table 1.
Therapies directly targeting oncogenic KRAS signaling in clinical trials.
| Therapy | Target | Mechanism | Phase | Trial number | Ref. |
|---|---|---|---|---|---|
| KRAS G12C | |||||
| Sotorasib (AMG510) | KRASG12C | Covalent inhibitor | Approved | NCT04303780, NCT03600883, NCT04185883, NCT04380753 | 8 |
| Adagrasib (MRTX849) | KRASG12C | Covalent inhibitor | Approved | NCT04330664, NCT03785249, NCT04613596, NCT04793958 | 9 |
| Divarasib (GDC-6036) | KRASG12C | Covalent inhibitor | III | NCT04449874, NCT04302025, NCT03178552, NCT04589845, NCT04929223 | 64 |
| JDQ443 | KRASG12C | Covalent inhibitor | III | NCT05999357, NCT05329623, NCT05358249, NCT04699188, NCT05714891, NCT05445843, NCT05132075 | 41 |
| LY3537982 | KRASG12C | Covalent inhibitor | III | NCT04956640, NCT06119581 | 65 |
| D-1553 | KRASG12C | Covalent inhibitor | II | NCT05383898, NCT05492045, NCT04585035, NCT05379946 | 66 |
| JAB-21822 | KRASG12C | Covalent inhibitor | II | NCT05009329, NCT05002270, NCT05194995, NCT05276726, NCT06008288, NCT05288205 | 67 |
| GFH925 | KRASG12C | Covalent inhibitor | II | NCT05756153, NCT05005234, NCT05497336, NCT05504278 | 68 |
| BI 1823911 | KRASG12C | Covalent inhibitor | I | NCT04973163 | 69 |
| JNJ-74699157 | KRASG12C | Covalent inhibitor | I | NCT04006301 | 70 |
| MK-1084 | KRASG12C | Covalent inhibitor | I | NCT05067283 | 71 |
| BPI-421286 | KRASG12C | Covalent inhibitor | I | NCT05315180 | 72 |
| GH35 | KRASG12C | Covalent inhibitor | I | NCT05010694 | 73 |
| RMC-6291 | KRASG12C | Form a tri-complex with cyclophilin A and RAS | I | NCT06128551, NCT05462717 | 74 |
| KRAS G12D | |||||
| RMC-9805 | KRASG12D | Form a tri-complex with cyclophilin A and RAS | I | NCT06040541 | 75 |
| HRS-4642 | KRASG12D | Small molecule inhibitor | I | NCT05533463 | 76 |
| MRTX1133 | KRASG12D | Non-covalent inhibitor | I/II | NCT05737706 | 25 |
| ASP3082 | KRASG12D | Degraders | I | NCT05382559 | 27 |
| siG12D-LODER | KRASG12D | siRNA | II | NCT01188785, NCT01676259 | 28 |
| iExosomes | KRASG12D | Exosomes with siRNA | I | NCT03608631 | 29 |
| LUNA18 | KRASG12D | Cyclic peptide inhibitor | I | NCT05012618 | 63 |
| Anti-KRAS G12D mTCR PBL | KRASG12D | TCR-engineered T cell therapy | I/II | NCT03745326 | 30 |
| KRAS G12V | |||||
| Anti-KRAS G12V mTCR PBL | KRASG12V | TCR-engineered T cell therapy | I/II | NCT03190941 | 30 |
| KRAS G12V-specific TCR transduced autologous T cells | KRASG12V | TCR-engineered T cell therapy | I/II | NCT04146298 | – |
| YK0901 immunotherapy | KRASG12V | TCR-engineered T cell therapy | I | NCT05933668 | – |
| FH-A11KRASG12V-TCR | KRASG12V | TCR-engineered T cell therapy | I | NCT06043713 | – |
| Pan-(K)RAS | |||||
| RSC-1255 | KRASG12V, KRASG13D | Inhibiting vacuolar-ATPase | I | NCT04678648 | 32 |
| mRNA-5671/V941 | KRASG12C/D/V, KRASG13D |
Cancer vaccines | I | NCT03948763 | 34 |
| ELI-002 2P | KRASG12D/R | Cancer vaccines | I/II | NCT05726864, NCT04853017 | 33 |
| RMC-6236 | KRASG12D/V, KRASG13D, KRASQ61K, RASWT |
Form a tri-complex with cyclophilin A and RAS | I/II | NCT06128551, NCT06162221, NCT05379985 | 36,37 |
PBL, peripheral blood lymphocyte; TCR, T-cell receptor.
Conversely, pan-KRAS therapies aim to encompass a broader patient population, including those who display resistance to mutant-specific KRAS therapies. The development of pan-KRASi is underway. For instance, the recently reported pan-KRASi BI-2865 can bind to KRAS-GDP and block GTP-GDP nucleotide exchange, thereby inhibiting WT KRAS and various KRAS mutants such as G12 A/C/D/F/V/S and G13C/D, and sparing NRAS and HRAS31. Several pan-KRAS therapies have already been tested in clinical trials32,33. RSC-1255 inhibits oncogenic KRAS signaling by inhibiting vacuolar ATPase activity, showing heightened potency against the KRAS G13D and G12V mutants32. A phase I trial of RSC-1255 in advanced solid tumor malignancies is ongoing (NCT04678648). Cancer vaccines have emerged as another promising approach for targeting pan-KRAS mutant tumors. One such mRNA cancer vaccine, mRNA-5671/V94134, has been developed to specifically target the KRAS G12C/D/V and G13D mutants, exhibiting potential immunostimulatory and antitumor effects (NCT03948763). Another cancer vaccine, ELI-00233, comprises amphiphile-modified mutant KRAS peptides and amphiphile-modified CpG oligonucleotides. This vaccine activates the autoimmune system by delivering antigenic peptides into the lymphatic system, eliminating tumor cells. Its clinical trial program includes ELI-002 2P (2 peptides, including KRAS G12D/R, NCT04853017) and ELI-002 7P (7 peptides, including KRAS G12 A/C/D/R/S/V and G13D, NCT05726864). Pan-RAS therapies have also been proposed to inhibit all three RAS isoforms (KRAS, HRAS, and NRAS). However, the tolerability of pan-RAS therapies may be a novel clinical issue, as adult mice die from ablation of all three RAS isoforms35. RMC-6236 can inhibit RAS activation by inducing the formation of a tri-complex of cyclophilin A and RAS36. Thus, RMC-6236 can result in significant and sustained regressions in tumors with WT and mutant RAS variants, particularly those harboring mutations at the KRAS G12 locus. RMC-6236 is currently being evaluated in patients with RAS mutant solid tumors (NCT05379985), and the preliminary results indicated that the objective response rate (ORR) was 36% in KRAS G12X mutant patients with pancreatic cancer and NSCLC37. However, further studies are needed to fully investigate the potential of pan-KRAS and pan-RAS therapies to overcome drug resistance, and attention must be given to the increased toxicity, especially in combination therapies.
2.2. Targeting oncogenic RAS signaling indirectly
Targeting oncogenic RAS signaling indirectly through inhibiting key related targets in both the upstream and downstream of mutant RAS represents a significant strategy4. SOS1 and SHP2 are crucial targets upstream of RAS38,39. Inhibition of these two targets has demonstrated antitumor activity against pan-KRAS mutants. Hence, they are also referred to as indirect pan-KRASi35. With the potential to inhibit reactivation of the RAS/MAPK pathway, multiple combination regimens are being evaluated in clinical trials, such as BI 170196340, TNO15541, and RMC-463042. Additionally, extensive efforts have been made to develop inhibitors that block major kinases within the downstream pathway of RAS, such as RAF-MEK-ERK and PI3K-AKT-mTOR. However, the intricate crosstalk between signaling pathways poses challenges for indirect targeting strategies. Single-agent multitarget inhibitors may hold promise. The novel dual RAF/MEK inhibitor VS-6766 has emerged as a potential therapeutic option for various RAS-mutated tumors43. Combinatorial therapies based on these inhibitors, including vertical and horizontal combinations, have shown remarkable efficacy in clinical trials and could overcome resistance. Vertical combinations can induce more effective and durable inhibition of MAPK signaling by targeting multiple targets of the RAS/MAPK pathway44, 45, 46. Horizontal combinations that simultaneously target RAS signaling and other RAS-related pathways may result in increased antitumor activity, for example, by simultaneously inhibiting the PI3K pathway or the CDK4/6 pathway47,48. These combinations have demonstrated a synergistic impact in preclinical models, albeit with the need to strike a delicate equilibrium between efficacy and tolerability in clinical trials. As a critical target in the MAPK pathway, RAF kinase inhibitors have undergone several generations of iterations. Based on in-depth studies of the RAS-RAF activation process, RAF dimer inhibitors have been developed. Notably, several clinical trials have demonstrated their potential as mono- and combination therapies in treating patients with RAS mutant tumors49, 50, 51, 52.
3. Importance of RAF dimerization in RAS/MAPK signaling
3.1. Structural and functional features of RAF kinases
3.1.1. Structure of RAF kinases
RAF kinases are composed of an N-terminal regulatory region (NTR) and a C-terminal kinase structural domain (KD) and consist of three conserved regions (CRs). The NTR includes a RAS-binding structural domain (RBD), a cysteine-rich structural domain (CRD), and a serine/threonine-rich region (Fig. 2A)77. The RAF protein exists as a monomer in the cytoplasm in an inactive conformation, which is inhibited by intramolecular interactions between the NTR and KD. Upon activation of RTK, RAS-GTP recruits and attaches to the RAF-RBD, leading to phosphorylation of specific residues78. This enables the anchoring of the RAF protein to the cell membrane and causes lateral dimerization and activation of RAF kinases79.
Figure 2.

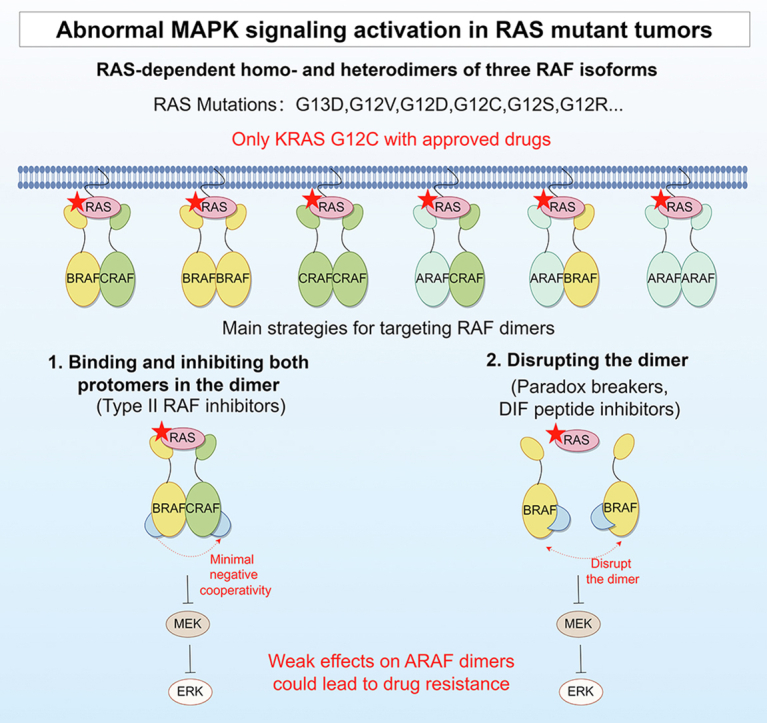
Structural and functional features of RAF dimers. (A) Domain structures of the three RAF isoforms. There are three conserved regions (CRs) of RAF kinases. CR1 contains a RAS-binding structural domain (RBD) and a cysteine-rich structural domain (CRD). CR2 is a serine/threonine-rich region, and CR3 contains a kinase structural domain. (B) The structure of the BRAF dimer bound to 14-3-3 (PDBID: 6U2H)84. Dimeric 14-3-3 promotes BRAF dimer formation (side-to-side), and the image was generated with the NGL Viewer85. (C) The role of RAF dimers in MAPK signaling in normal and RAS/RAF mutant cells. All three RAF isoforms can form RAF homo or heterodimers (six in total). BRAF and CRAF dimers play important roles in MAPK signaling and exhibit more excellent kinase activity than ARAF dimers, which were recently found to be involved in drug resistance. Both normal cells and RAS mutant tumor cells activate downstream ERK signaling through RAS-dependent RAF dimers, and RAS mutations result in sustained and significant activation of ERK signaling. BRAF class I mutations function as RAS-independent BRAF mutant monomers with the highest kinase activity. BRAF class II mutations are characterized by RAS-independent BRAF mutant dimers with moderate BRAF kinase activity. BRAF class III mutations tend to form heterodimers with wild-type CRAF, activated by RAS-dependent RAF dimers with the lowest kinase activity. The red pentagrams represent mutated RAS/RAF proteins.
The RAF protein has a typical kinase domain structure that consists primarily of the N lobe, the C lobe, and the activation loop. The activation loop connects the two lobes and is located near the ATP binding site78. Typically, this activation loop encompasses 20–30 amino acid residues, starting with a DFG sequence (Asp-Phe-Gly) and ending with an APE sequence (Ala-Pro-Glu)80. The DFG-Asp side chain is near the ATP binding site in active RAF kinases, resulting in the DFG-in conformation. Conversely, the DFG-Asp side chain is distant from the ATP binding site in inactive RAF kinases, leading to the denoted DFG-out conformation81. The conformations can also be defined based on the DFG-Phe residues82. Moreover, the N lobe contains five β-folds and an αC-helix78, wherein a lysine in the β3 chain (β3-Lys) can create a “salt bridge” with a glutamic acid in the αC-helix (αC-Glu). This interaction is characteristic of the αC-helix-in conformation in active RAF kinase83. The RAF kinase adopts the αC-helix-out conformation without this interaction, which is inactive. The αC-helix-in conformation transition is essential for the activation of RAF kinase and impacts the function of RAFi.
3.1.2. The three RAF isoforms play different roles in RAS signaling
Despite sharing common activators and substrates, the three isoforms of RAF (ARAF, BRAF, and CRAF/RAF1) exhibit distinct characteristics in kinase activity, regulatory mechanisms, and functional roles86. RAF kinases play an essential intermediate role in the MAPK signaling cascade and undergo several critical events for activation. These processes include recruitment to the cell membrane and activation via RAS activation, RAF dimerization, phosphorylation at distinct structural domains, attachment to scaffold protein complexes, and regulation of activity by chaperone proteins87. Research on RAF knockout mice indicated that the three RAF isoforms have distinct physiological functions during development88. Nonetheless, the specific mechanisms through which these isoforms contribute to the progression of RAS mutant tumors in different tumors are still poorly understood. BRAF exhibits the highest kinase activity in activating downstream ERK signaling, whereas ARAF and CRAF exhibit comparatively lower kinase activity. Notably, CRAF could play a crucial role in the progression of RAS mutant tumors88,89. CRAF, rather than BRAF, is responsible for triggering ERK reactivation90. Nevertheless, selective CRAF inhibition promotes paradoxical activation due to RAF dimerization91, whereas suppressing CRAF has been found to enhance the efficacy of MEKi in KRAS mutant tumors90. Limited studies have been conducted on the function of ARAF, but recent investigations have revealed its capacity to regulate RAS activity92. Specifically, ARAF competitively hinders the binding of the GAP protein NF1 to RAS, thereby impeding the negative regulatory impact of the GAP protein on RAS. Importantly, this regulatory function of ARAF is independent of its kinase activity. Cells exhibiting elevated ARAF expression show an augmented baseline level of RAS-GTP, leading to drug resistance. There is a need to gain a broader understanding of the functions of RAF isoforms in different types of tumors, which may provide valuable perspectives on the distinct reactions of RAS mutant tumors to particular treatment approaches.
3.2. RAF dimerization in normal and oncogenic RAS/RAF signaling
MAPK signaling in both normal cells (wild-type RAS/RAF) and RAS mutant tumor cells depends on RAF dimers. RAF dimerization plays a vital role in MAPK signaling reactivation and drug resistance. However, only a few RAF mutants act through RAF dimers, and monomeric BRAF V600 locus mutations account for most of these mutations. Breakthroughs were made in the field of BRAF V600E inhibitors a decade ago, which have been approved for clinical use93. Therapeutic strategies for other RAF dimer-driven RAF mutant tumors and RAS mutant tumors are still under investigation.
3.2.1. Structure and detection of RAF dimers
In addition to RAF dimers, dimerization was also observed in RAS, MEK, and ERK kinases, highlighting the importance of dimerization in the MAPK signaling pathway94. However, the specific mechanisms and functions of these dimers remain unclear. The crystal structures of RAF dimers have been recently revealed, providing insights into the structural foundation of kinase activation and drug development (Fig. 2B)13,84,95,96. Two RAF protomers combine to form a RAF dimer in a lateral arrangement (side-to-side). The dimer interface (DIF) contains a specific cluster of residues essential for dimerization and presents potential therapeutic targets for RAF dimer inhibition79,97. The conformation of DIF can be altered by key residues that, for example, inhibit RAF dimerization (BRAF R509H and CRAF R401H) or promote RAF dimerization (BRAF E586K and CRAF E478K)98. Recent studies on RAF dimerization have focused on BRAF and CRAF, as ARAF is the least inclined to dimerize because of its unique structural characteristics and lack of stability in DIF98,99.
Current methods for detecting and studying RAF dimers are still under development. The coimmunoprecipitation (co-IP) method is widely used as the classical experimental approach for investigating the interaction between RAF dimers by tagging RAF proteins differently. Co-IP can only probe RAF protein interactions solely in cell-free lysates, which requires the level of RAF expression and stability of the RAF dimers. Alternatively, proximity ligation assays can be employed to detect RAF dimerization in fixed cells98,100. Additionally, Lavoie et al. proposed a biosensor system utilizing bioluminescence resonance energy transfer to detect RAF dimers in living cells101. The split luciferase system has emerged as a novel method for investigating RAF dimers in living cells. This system involves the fusion of RAF proteins with various luciferases, such as firefly (Photinus) luciferase, click beetle (Pyrearinus) luciferase, and Nanoluc luciferase102, 103, 104. When the target fusion protein interacts and brings two luciferase fragments close together, the split luciferase protein undergoes reorganization, activating catalytically active luciferase. The split luciferase system allows researchers to study the real-time dynamics of RAF dimers within living cells.
3.2.2. RAF dimerization in normal cell signaling
All three RAF isoforms can form homo and heterodimers, which are crucial for kinase activity under normal physiological conditions79. The prevailing belief is that when RAS-GTP is activated, and scaffold complexes such as SHOC2-PP1C are present, they recruit autoinhibited RAF monomers to the cell membrane. This process promotes intracellular colocalization and ultimately facilitates RAF dimerization105,106. The RAF dimer exhibits functional asymmetry, where one protomer can function as an activator to enhance the activity of the other protomer, termed trans-activation107. Despite variations in their capacity to induce ERK signaling, full activation of all RAF isoforms depends on RAF dimerization21. Linda et al. observed endogenous BRAF/CRAF heterodimers under physiological conditions, which are the predominant RAF dimers in normal cells108. Additionally, the BRAF/CRAF heterodimers were found to be negatively regulated by ERK signaling (by inducing phosphorylation of the BRAF T753 site). Remarkably, even the kinase-inactive form of BRAF can activate ERK signaling by forming a dimer with CRAF108, underscoring the importance of RAF dimerization in RAF activation.
3.2.3. RAF dimerization in oncogenic RAS signaling
In RAS mutant tumors, RAF dimerization is critical for MEK-ERK signaling and contributes to drug resistance to MAPK pathway inhibitors. Although a complete understanding of the formation preferences of RAF dimers and their specific mechanisms remains elusive, it is believed that different RAF dimers may uniquely contribute to the progression of different types of tumors. In 2001, Weber et al. discovered that active RAS G12V could stimulate BRAF/CRAF heterodimer formation109. The BRAF/CRAF heterodimer plays a dominant role in RAS signaling due to its high kinase activity compared to other RAF dimers98. It is closely associated with treatment response and has significant clinical implications110. Quantitative proteomic studies have further revealed that ARAF/CRAF dimers are more abundant than BRAF/CRAF dimers in KRAS mutant tumors88. Notably, the tumorigenesis process depends on the dimerization of CRAF rather than on its activity88. In KRAS mutant tumors, MEKi promotes BRAF/CRAF dimer formation and induces a rebound in MEK-ERK signaling, leading to drug resistance20,111. Furthermore, recent studies on the mechanisms of RAF dimerization have focused on the regulation of RAF dimers by scaffolding proteins. Scaffolding proteins are essential for RAF dimer formation and activation, such as SHOC220,112. Recent studies have drawn attention to the synthetic lethal interaction between SHOC2 and MEKi in RAS mutant tumors113. In these tumors, the absence of SHOC2 hinders MEKi-induced BRAF/CRAF dimers, resulting in more potent and sustained inhibition of ERK signaling20. Notably, SHOC2 may play an essential role in the resistance mechanisms of RAS/MAPK pathway inhibitors, such as KRAS G12Ci114. Scaffolding proteins provide temporal and spatial control over ERK signaling activation and may be potential targets in RAS mutant tumors.
The first-generation RAF inhibitors, which bind to one of the protomers and induce RAF dimerization, result in allosterically significant activation of the other protomer and subsequent ERK activation77,115. Consequently, these inhibitors are ineffective and may even be contraindicated for RAS mutant tumors. New-generation RAF inhibitors employ different strategies to target and inhibit RAF dimers, with RAF dimer inhibitors showing particular promise. Nonetheless, the existing RAF dimer inhibitors cannot completely inhibit the activity of all RAF dimers, especially homo and heterodimers of ARAF, and induce ERK reactivation by ARAF dimerization18,116. Therefore, to effectively inhibit ERK signaling and treat RAS mutant tumors, it is imperative to focus on targeting RAF dimers.
3.2.4. Only a few RAF mutants act through RAF dimers
Approximately 8% of human tumors carry BRAF mutations81. Mutations in the BRAF gene are predominant within the V600 motif and constitute 98.4% of the total mutations, whereas mutations within the non-V600 motif account for a mere 1.6% of the total mutations78. Three types of BRAF mutations are classified according to the characteristics of the mutant kinase activity and its dependence on RAS and RAF dimerization (Fig. 2C)12,115. BRAF class I mutations include various mutations at the BRAF V600 locus (such as BRAF V600 E/K/D/R). Unlike wild-type BRAF, which requires RAS-GTP and RAF dimerization for catalytic activation, BRAF class I mutations exhibit the highest kinase activity and function as RAS-independent BRAF mutant monomers. Monomeric BRAF V600E inhibitors, such as vemurafenib, are effective against BRAF class I mutations and have been approved for clinical use. BRAF class II mutations (such as BRAF K601, L597, G469, and G464), characterized by RAS-independent BRAF mutant dimers, exhibit moderate BRAF kinase activity. BRAF class II mutations are sensitive to RAF dimer inhibitors115. BRAF class III mutations (such as BRAF G466, N581, D594, and G596) with impaired kinase activity tend to form dimers with wild-type CRAF and bind tightly to RAS-GTP117. Thus, BRAF class III mutations are activated by RAS-dependent RAF dimers and exhibit the lowest kinase activity. These mutations are often accompanied by mutations in upstream oncogenes such as RAS and NF1, further complicating matters. Combination therapies targeting both upstream (RTKi or SHP2i) and downstream signaling (MEKi or ERKi) may be potential therapeutics for BRAF class III mutations118.
In addition to point mutations, fusion proteins arising from translocations and in-frame deletions can also trigger continuous activation of the BRAF kinase81,119. These BRAF fusion proteins are predominantly observed in melanoma and gliomas120. They typically contain structural domains of protein kinase but lack the NTR, which is responsible for the self-inhibition of BRAF. Consequently, this absence promotes continuous RAF activation through a RAS-independent dimer121, making the fusion proteins susceptible to inhibition by RAF dimer inhibitors115. In addition, BRAF in-frame deletions are most frequently observed in pancreatic and lung tumors100. These deletions cause a reduction in the length of the β3/αC-helix loop of the BRAF kinase, leading to the stabilization of the αC-helix-in conformation and promoting dimer formation119. While these deletions are not sensitive to the RAF monomer inhibitor vemurafenib, they exhibit sensitivity to RAF dimer inhibitors100. Chen et al. proposed that this discrepancy arises from promoting BRAF homodimer formation100. Nonetheless, some researchers have suggested that resistance to vemurafenib is independent of RAF dimerization but attributed to the spatial blocking effect and conformational changes associated with the deletions119,122.
In contrast, mutations in the ARAF and CRAF genes occur less frequently than BRAF mutations. The mutational hotspots in the ARAF and CRAF genes differ significantly from those observed in BRAF mutations12. A complete understanding of these mutations’ activation mechanisms and consequences on MAPK signaling has yet to be achieved. Wenjing et al. discovered that ARAF S214 mutations could enhance MEK phosphorylation by forming a persistent activation dimer92. This process is independent of RAS activity and resembles BRAF class II mutations. CRAF fusions also act as RAF dimers and are thus resistant to RAF monomer inhibitors but sensitive to RAF dimer inhibitors123. Although there are specific variations, RAF dimerization plays a significant role in these RAF mutants.
4. Therapeutic strategies to target RAF dimers in RAS mutant tumors
Various approaches have been employed to target RAF dimers in tumors harboring RAS mutations (Table 2). Most presently developed RAFi bind directly to the ATP-binding site of the RAF protein, thereby impeding the activation of downstream MEK/ERK, so-called ATP competition inhibitors. Based on the conformation of the bound RAF kinase, the currently developed RAF inhibitors can be classified into three groups: type I, type I1/2, and type II RAFi (Fig. 3A‒D)22,124. Another distinct class of RAF inhibitors are paradox breakers, which disrupt RAF dimers125. Furthermore, allosteric inhibitors directly targeting DIF are called DIF peptide inhibitors (type IV RAFi)126. Notably, type II RAFi and DIF peptide inhibitors have demonstrated preliminary antitumor efficacy against RAS mutant tumors (Fig. 4). It is imperative to devote particular attention to treating tumors with KRAS mutations. Compared to NRAS mutant tumors, KRAS mutant tumors exhibit reduced sensitivity to RAFi5. Among these RAFi, only type II RAFi have demonstrated activity against KRAS mutant tumors in both preclinical and clinical settings, although to a lesser degree than BRAF V600E mutations.
Table 2.
Characteristics of inhibitors targeting RAF dimers.
| Inhibitors | Conformation binding to RAF | Cell-free assay (IC50: nmol/L) |
Effects on RAF dimers (confirmed experimentally) | Preclinical effects | Phase | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| ARAF | BRAF | CRAF | BRAFV600E | ||||||
| Type II RAF inhibitors | |||||||||
| Tovorafenib (MLN2480) | DFG-out/αC-helix-in | 2300 | 10.1 | 0.7 | 7.1 | BRAF/CRAF, BRAF/BRAF, CRAF/CRAF | Effective in BRAF and NRAS mutants in melanoma | Phase III (NCT05566795) | 103,104,130,131 |
| Naporafenib (LXH254) | DFG-out/αC-helix-in | 6.4 | 0.21 | 0.072 | 28 | BRAF/CRAF, BRAF/BRAF, CRAF/CRAF | Effective in BRAF and NRAS mutants, moderately effective in KRAS mutants | Phase II (NCT04417621) | 18,131, 132, 133 |
| Belvarafenib (HM95573) | DFG-out/αC-helix-in | 152 | 116 | 23 | 9 | BRAF/CRAF, BRAF/BRAF, CRAF/CRAF, ARAF/BRAF, ARAF/CRAF | Effective in BRAF and NRAS mutants in melanoma | Phase II (NCT04589845) | 116,132,133 |
| Lifirafenib (BGB283) | DFG-out/αC-helix-in | 5.6 | 32 | 6.7 | 23 | BRAF/CRAF | Effective in BRAF mutants in CRC | Phase I (NCT03905148) | 104,134,135 |
| LY3009120 | DFG-out/αC-helix-in | NA | 9.1 | 15 | 5.8 | BRAF/CRAF, BRAF/BRAF, CRAF/CRAF | Effective in BRAF, NRAS, and KRAS mutants in melanoma, CRC, lung, and pancreatic cancer | Phase I (NCT02014116) | 102,103 |
| CCT3833 | DFG-out/αC-helix-in | NA | 420 | 33 | 34 | NA | Effective in KRAS mutants in CRC, lung, and pancreatic cancer | Phase I (NCT02437227) | 81,136 |
| BGB3245 | NA | NA | NA | NA | NA | NA | Effective in BRAF and NRAS mutants in melanoma | Phase I (NCT05580770) |
49,137, 138 |
| TAK632 | DFG-out/αC-helix-in | NA | 8.3 | 1.4 | 2.4 | BRAF/CRAF, BRAF/BRAF, CRAF/CRAF | Effective in BRAF, NRAS, and KRAS mutants in melanoma, CRC, lung, and pancreatic cancer | Preclinical | 102,103,129 |
| AZ628 | DFG-out/αC-helix-in | NA | 105 | 29 | 34 | BRAF/CRAF, CRAF/CRAF | Effective in BRAF and NRAS mutants in melanomas, CRC, and thyroid cancer | Preclinical |
81,101, 102,139 |
| RAF709 | DFG-out/αC-helix-in | NA | 1.5 | 0.4 | 1 | BRAF/CRAF | Effective in BRAF, NRAS, and KRAS mutants in melanomas, CRC, and lung cancer | Preclinical | 132,140 |
| BGB659 | DFG-out/αC-helix-in | NA | NA | NA | NA | BRAF/CRAF, BRAF/BRAF | Effective in BRAF mutants in melanoma | Preclinical | 115,141 |
| DCC3084 | DFG-out/αC-helix-in | 903 | 71 | 34 | 2 | BRAF/CRAF | Effective in BRAF and KRAS mutants in melanoma, CRC, lung, and pancreatic cancer | Preclinical | 131 |
| IHMT-RAF-128 | DFG-out/αC-helix-in | NA | 5.4 | 3.6 | 5.9 | NA | Effective in BRAF and RAS mutants in solid tumors | Preclinical | 142 |
| REDX05358 | NA | NA | NA | NA | NA | NA | Effective in BRAF, NRAS, and KRAS mutants in melanoma, lung cancer, and CRC | Preclinical | 143 |
| BDTX4933 | NA | NA | NA | NA | NA | BRAF/CRAF, BRAF/BRAF, CRAF/CRAF | Effective in BRAF and NRAS mutants in melanoma | Preclinical | 133 |
| Paradox breakers | |||||||||
| PLX8394 | DFG-in/αC-helix-out/R506-out | NA | 14 | 23 | 3.8 | BRAF/CRAF, BRAF/BRAF | Effective in BRAF mutants in melanoma and CRC | Phase I (NCT02428712) | 125,141,144 |
| PLX7904 | DFG-in/αC-helix-out/R506-out | NA | 140 | 91 | 4.2 | BRAF/CRAF | Effective in BRAF mutants in melanoma and CRC | Preclinical |
22,103, 144,145 |
| Compound Ia | NA | NA | <5E-10 | NA | 1.77E-09 | BRAF/BRAF | Effective in BRAF mutants in melanoma | Preclinical | 146,147 |
| Hybrid 6 | DFG-out/αC-helix-in/R506-out | NA | 56 | NA | 48 | BRAF/BRAF | Effective in BRAF mutants in melanoma | Preclinical | 148 |
| DIF peptide inhibitors | |||||||||
| DI1 | NA | NA | NA | NA | NA | BRAF/CRAF, BRAF/BRAF | Effective in KRAS mutants in lung cancer | Preclinical | 98 |
| Pep17 | NA | NA | NA | NA | NA | BRAF/BRAF | Effective in NRAS mutants in melanoma | Preclinical | 126 |
| Braftide | NA | NA | 364 | NA | NA | BRAF/CRAF, BRAF/BRAF | Effective in KRAS mutants in CRC | Preclinical | 149 |
DIF, dimer interface; IC50, half maximal inhibitory concentration; CRC, colorectal cancer; NA, not available.
Figure 3.

The conformations of RAF kinases bound to type I, I1/2, and II RAFi. The three main conformations of the RAF kinase domain are shown in (A–D). The crystal structure of each conformation is shown above. Below the crystal structure is a schematic representation showing the conformations of the DFG motif (green) and the αC-helix (red). (A) SB-590885 (type I RAFi) interacts with BRAF kinase in the DFG-in/αC-helix-in conformation (PDB: 2FB8). (B) Dabrafenib (type I1/2 RAFi) binds to BRAF kinase in the DFG-in/αC-helix-out conformation (PDB:5CSW). (C) Naporafenib (type II RAFi) binds to both BRAF protomers in the DFG-out/αC-helix-in conformation (PDB: 8F7P)150. (D) Comparison of the conformations of type I1/2 RAFi (teal for dabrafenib, PDB: 5CSW) and type II RAFi (green for naporafenib, PDB: 8F7P) bound to BRAF kinase. The DFG-Phe residues point toward the αC-helix in the DFG-in conformation, and the DFG-Phe residues point away from the αC-helix in the DFG-out conformation. The image was created with SWISS-MODEL151. (E–F) Structures of two typical type II RAFi currently under clinical investigation (tovorafenib and naporafenib), as well as their specific conformations for binding to the WT BRAF kinase (PDB: 8F7O and 8F7P)150.
Figure 4.

Role of RAF inhibitors on RAF dimers. Only type II RAFi and DIF peptide inhibitors are effective against RAS mutant tumors. These compounds effectively inhibit the kinase activity of BRAF and CRAF dimers but show weak effects on ARAF dimers, which may lead to drug resistance. The red pentagrams represent mutated RAS/RAF proteins. DIF, dimer interface.
4.1. Inability of type I and type I1/2 RAFi to inhibit RAF dimers
Type I RAFi interact with RAF kinases via the DFG-in and αC-helix-in conformation. Type I RAFi (such as GDC0879 and SB590885) can adopt a stable, closed, and active conformation22,127,128. Nevertheless, these inhibitors are only effective against tumor cells that are activated by RAF monomer-driven ERK signaling (BRAF class I mutations). Type I RAFi stabilize both protomers in RAF dimers in the αC-helix-in conformation, thus significantly promoting RAF dimerization, which leads to paradoxical ERK activation104. RAS mutant tumor cells often show reduced sensitivity or even resistance to type I RAFi compared to cells with BRAF V600E mutations128.
Type I1/2 RAFi, such as vemurafenib, dabrafenib, and encorafenib, can attach to RAF kinases in the DFG-in and αC-helix-out conformation. These inhibitors, usually called first-generation RAFi, show potent inhibitory effects on BRAF class I mutant tumor cells. When applied to mutant RAS and WT RAS/RAF cells, which function as RAF dimers, type I1/2 RAFi paradoxically activate ERK signaling77. The critical factors responsible for this could be the compounds binding to RAF kinase, alleviating RAF autoinhibition, inducing RAF dimerization, and enhancing RAS‒RAF interactions77. Type I1/2 RAFi promote RAF dimerization, but the effect is weaker than type I and II RAFi103,104. Paradoxical activation of ERK occurs because of the partial disruption of the DIF caused by the drug-binding RAF protomer. This process prevents the inhibitors from binding to the other drug-free RAF protomer, called negative allostery, and even significantly activates the drug-free RAF protomer81,115. Increasing the concentration of type I1/2 RAFi can occupy both protomers of the RAF dimer, effectively inhibiting ERK signaling129. However, this is difficult to achieve in patients, resulting in poor efficacy of these drugs in RAF dimer-driven tumors.
The US Food and Drug Administration (FDA) has approved the use of vemurafenib and dabrafenib in treating metastatic melanoma in patients harboring BRAF V600 E/K mutations. While most melanoma patients experience tumor regression and prolonged survival152, these medications seldom eradicate the tumor, and a notable percentage of patients still encounter both inherent and acquired resistance153. The clinical effectiveness of these medications is relatively low in nonmelanoma cancers that feature activating BRAF V600 mutations154,155. Paradoxical activation of ERK is a crucial reason for the poor outcome and limited efficacy of these agents in patients with RAS mutant tumors. They even trigger adverse reactions such as keratoacanthomas and cutaneous squamous cell carcinomas (SCCs) that lead to drug discontinuation, limiting their use in RAS mutant tumors156. The risk of progression can be reduced by inhibiting two nodes of the MAPK pathway to prevent ERK reactivation. The standard treatment for BRAF V600 E/K metastatic melanoma now involves combining type I1/2 RAFi with MEKi (such as vemurafenib and cobimetinib, dabrafenib and trametinib)157,158. Encorafenib has entered phase III clinical trials and has also shown therapeutic benefits when combined with MEKi159.
4.2. Type II RAFi show efficacy in RAS mutant tumors
Type II RAFi are classified as compounds that adopt the DFG-out, αC-helix-in conformation. Usually, type II RAFi are also referred to as RAF dimer inhibitors. Tovorafenib (MLN2480) and naporafenib (LXH254) are typical type II RAFi. Their chemical structures and a detailed view of the conformations in complex with wild-type BRAF kinases are shown in Fig. 3E‒F)150. The central thiazole ring of tovorafenib is positioned between Thr529 and Lys483. The bisubstituted pyrimidine ring of tovorafenib establishes hydrogen bonds with the BRAF kinase hinge (Cys532). The carbonyl oxygen forms a hydrogen bond with DFG-Asp594, and the neighboring amide nitrogen establishes a hydrogen bond with Glu501 in the αC-helix. The trifluoromethyl-substituted pyridine ring of tovorafenib can bind to the hydrophobic pocket formed by rotation of the DFG motif (DFG-out). Like in the case of tovorafenib, in the BRAF-naporafenib complex, the central substituted phenyl ring binds within the spatial region between Thr529 and Lys483. The trifluoromethyl pyridyl moiety of naporafenib occupies the hydrophobic pocket created by flipping the DFG motif (DFG-out). Naporafenib also forms hydrogen bonds with Cys532 in the kinase hinge, Glu501 in the αC-helix, and Asp594 in the DFG motif. These hydrogen bonds are postulated to contribute to the potent medicinal properties of these compounds160. These two inhibitors induce a DFG-out conformation, leading to a slight alteration in the relative orientation of the N-lobe and C-lobe, potentially leading to cooperative effects in both protomers in the dimer150.
The interactions between type II RAFi and RAF kinase stabilize the αC-helix-in conformation in both RAF protomers, facilitating the formation of RAF dimers161,162. Indeed, despite differences in the degree and preference for inducing RAF dimerization, almost all ATP competitive inhibitors facilitate the formation of RAF dimers in a RAS-dependent manner101. However, due to the low negative allostery, type II RAFi potently bind to both RAF protomers and repress the kinase function of these RAF dimers102,116. RAF dimerization was observed after 10 min of exposure to most inhibitors, with peak formation occurring after 4–6 h103. Nevertheless, the suppressive impact of type II RAFi on kinase activity does not always correlate with the promoting effect of RAF dimers103.
To overcome the paradoxical ERK activation in RAS mutant tumors, inhibiting the kinase activity of all RAF isoforms, including RAF monomers and dimers, could be an essential strategy. Type II RAFi are generally considered pan-RAF inhibitors because they theoretically bind to RAF monomers and dimers with similar potency and inhibit their kinase activity102,134. Therefore, these compounds are not expected to induce paradoxical activation of ERK signaling. These inhibitors have demonstrated a mild pro-phosphorylation effect on MEK and ERK only at very low concentrations22, while higher concentrations of the compound occupying both RAF protomers can inhibit their activation potently102,116. Recent studies have shown that some type II RAFi potently inhibit BRAF and CRAF monomers and dimers, while their activity against ARAF is relatively weak18,116,150, which could be the critical mechanism for ERK signaling reactivation and drug resistance. In addition, type II RAFi can have toxic effects on normal cells due to their extensive inhibition, although they inhibit mutant RAF monomers and dimers more potently than the wild-types22. Consequently, additional research and clinical investigations are necessary to assess their therapeutic index fully.
Some type II RAFi, such as TAK632129, AZ628139, RAF709140, and DCC3084131, are currently under investigation in preclinical research. These inhibitors have shown promising antitumor effects on tumor cells with KRAS and NRAS mutations. Other type II RAFi, such as tovorafenib50, naporafenib163, belvarafenib (HM95573)116, lifirafenib (BGB283)164 and LY3009120165, have undergone clinical trials. These agents have demonstrated initial clinical benefits as monotherapy or when combined with MEKi or ERKi in individuals with KRAS and NRAS-mutated tumors. Furthermore, several multikinase drugs, such as sorafenib and regorafenib, can be classified as type II RAFi132. Screening for inhibitors that inhibit RAF dimers from multitarget compounds is also a new direction101, although the extent of their effectiveness and toxicity in RAS mutant tumors still requires further substantiation. In addition, ponatinib, a tyrosine kinase inhibitor targeting BCR-ABL, adopts a unique conformation known as αC-helix-CENTRE166. Building upon the insights gained from ponatinib, Xiomaris et al. developed a novel BRAF dimer inhibitor called ponatinib hybrid inhibitor 1 (PHI1)166. This inhibitor demonstrates second-site positive cooperativity within the BRAF dimer, resulting in increased suppression of the second protomer when the first protomer is already bound. However, PHI1 shows limited efficacy in RAS mutant tumors. Furthermore, although the effects of some pan-RAF inhibitors on RAF dimerization have not been determined, such as exarafenib167 and XL281168, these agents have progressed to clinical trials and exhibited preliminary antitumor activity in NRAS and KRAS mutant tumors.
4.3. Paradox breakers offer new opportunities
RAF dimer disruption is a novel strategy for overcoming paradoxical ERK signaling. These RAFi are called “paradox breakers”, exemplified by PLX7904 and PLX8394, which have undergone structural modifications derived from vemurafenib144. Paradox breakers occupy one active site of RAF dimers, causing a conformational change with DFG-in, αC-helix-out, and R506-out22. These agents disrupt the DIF of RAF kinases, particularly BRAF/BRAF and BRAF/CRAF dimers22,144. This disruption prevents RAF dimerization and subsequently inhibits paradoxical ERK activation125,144. Notably, PLX8394 demonstrates a higher affinity for mutant dimeric BRAF (including BRAF fusions and splice variants) than for wild-type BRAF in normal cells141. Thus, its therapeutic index could be higher than that of type II RAFi. However, these compounds show weak effects on CRAF homodimers and ARAF dimers and even induce their activation, potentially resulting in drug resistance141. Since they are αC-helix-out inhibitors with negative allostery, their potency can be limited in RAS mutant tumors driven by homo and heterodimers of three RAF isoforms22. As predicted, both preclinical and clinical evidence suggests that paradox breakers exhibit greater efficacy in treating BRAF class I and II mutant tumors, while their effectiveness in treating BRAF class III and RAS mutant tumors is comparatively limited141,169, 170, 171. The same applies to recently reported paradox breakers: compound Ia, which offers good brain penetration146,147, and hybrid 6, which adopts a DFG-out and αC-helix-in conformation148, but neither shows potent activity against RAS mutant tumors.
4.4. DIF peptide inhibitors offer a promising approach
DIF peptide inhibitors, which directly target the DIF of RAF proteins, are considered promising therapeutic approaches for inhibiting RAF dimers. Most residues engaged in DIF interactions are in a contiguous sequence, enabling the design of short peptides specifically targeting DIF. Given that BRAF has the highest level of dimerization among the RAF isoforms, current efforts in developing peptide inhibitors have primarily concentrated on BRAF. In this context, Freeman et al. created a peptide inhibitor named DI1 that specifically targeted the DIF of BRAF and consists of 19 amino acids (residues 503–521)98. DI1 successfully dissociated the BRAF/CRAF dimer. It demonstrated remarkable efficacy in inhibiting the proliferation of NSCLC cells with KRAS and BRAF G466V mutations, both driven by RAS-dependent RAF dimers. DI1 could also inhibit pMEK signaling activated by the BRAF G464V and L597V mutations, driven by RAS-independent BRAF dimers. However, it was ineffective against tumor cells with BRAF V600E mutations, which function as BRAF monomers. In contrast, Amber et al. developed a 10-mer peptide inhibitor called braftide149. Braftide also targeted the DIF of BRAF and could dissociate BRAF/BRAF and BRAF/CRAF dimers. Notably, braftide selectively degraded both the BRAF and MEK kinases. Researchers posited that this dual-action mechanism may potentially delay ERK reactivation. Braftide did show activity in CRC cell lines harboring KRAS mutations, and its combination with dabrafenib effectively suppressed paradoxical ERK activation. Thus, simultaneous targeting of DIF may be synergistic with ATP competition inhibitors. Another avenue being explored involves the utilization of cyclic peptides. Compared with linear peptides, cyclic peptides exhibited enhanced binding affinity to the BRAF kinase domain, suggesting they may possess greater potency in inhibiting RAF dimers126.
Collectively, these studies support the prospective utilization and development of novel DIF peptide inhibitors, promising for preventing the negative cooperativity and paradoxical ERK activation. The activation of kinases in all three RAF isoforms necessitates RAF dimerization, and DIF is extensively conserved across these isoforms, with only marginal disparities. Thus, inhibitors that target DIF can potentially function as pan-RAF inhibitors107,149. However, the impact of these developed DIF peptide inhibitors on ARAF and CRAF dimers has yet to be comprehensively elucidated. Further investigations and validations are needed to thoroughly ascertain these agents' efficacy in treating RAS mutant tumors.
5. Clinical outcomes of type II RAFi monotherapy in RAS mutant tumors
Among the research strategies for targeting RAF dimers, only type II RAFi are being evaluated in clinical trials for RAS mutant tumors (Table 3). When used as monotherapies, these inhibitors have shown initial antitumor activity in individuals with RAS-mutated tumors (Table 4). However, further research is still needed to exploit these agents’ potential for clinical use thoroughly.
Table 3.
Clinical trials of inhibitors targeting RAF dimers in RAS/RAF mutant tumors.
| Intervention | Indication | Cancer | Phase | Trial number | Start year | Status | Published results | Ref. |
|---|---|---|---|---|---|---|---|---|
| Tovorafenib (MLN2480) | ||||||||
|
BRAF/NRAS mutants | Solid tumors | I | NCT01425008 | 2011 | Completed | YES | 50 |
|
MAPK alterations | LGG | I | NCT03429803 | 2018 | Active, not recruiting | YES (Abstract) | 172,173 |
|
BRAF mutants | LGG | II | NCT04775485 | 2021 | Recruiting | YES (Abstract) | 175 |
|
RAF mutants | LGG | III | NCT05566795 | 2023 | Recruiting | NA | |
|
MAPK alterations | Solid tumors | I/II | NCT04985604 | 2021 | Recruiting | NA | |
|
BRAF/KRAS mutants | NSCLC | I | NCT02327169 | 2015 | Completed | YES | clinicaltrials.gov |
|
BRAF/NRAS mutants | Melanoma | I | NCT02723006 | 2016 | Terminated | YES | clinicaltrials.gov |
| Naporafenib (LXH254) | ||||||||
|
MAPK alterations | Solid tumors | I | NCT02607813 | 2016 | Terminated | YES | 163,176 |
|
BRAF/NRAS/KRAS mutants | NSCLC Melanoma |
Ib | NCT02974725 | 2017 | Active, not recruiting | YES | 177 |
|
BRAF/NRAS mutants | Melanoma | II | NCT04417621 | 2020 | Active, not recruiting | YES (Abstract) | 52 |
|
BRAF mutants | CRC | Ib | NCT04294160 | 2020 | Recruiting | NA | |
| Belvarafenib (HM95573) | ||||||||
|
Not mentioned | Solid tumors | I | NCT02405065 | 2015 | Completed | YES | 116 |
|
BRAF/NRAS/KRAS mutants | Solid tumors | I | NCT03118817 | 2017 | Completed | YES | 116 |
|
BRAF mutants | Solid tumors | II | NCT04589845 | 2021 | Recruiting | NA | |
|
RAS/RAF mutants | Solid tumors | Ib | NCT03284502 | 2017 | Recruiting | YES (Abstract) | 178,179 |
|
NRAS mutants | Melanoma | I | NCT04835805 | 2021 | Recruiting | NA | |
| LY3009120 | ||||||||
|
BRAF/NRAS/KRAS mutants | Solid tumors | I | NCT02014116 | 2013 | Terminated | YES | 165 |
| Lifirafenib (BGB283) | ||||||||
|
BRAF/NRAS/KRAS mutants | Solid tumors | I | NCT02610361 | 2013 | Completed | YES | 164 |
|
BRAF/NRAS/KRAS mutants | Solid tumors | I | NCT03641586 | 2015 | Completed | NA | |
|
KRAS mutants | Solid tumors | Ib | NCT03905148 | 2019 | Recruiting | YES (Abstract) | 51 |
| CCT3833 | ||||||||
|
BRAF/RAS mutants | Solid tumors | I | NCT02437227 | 2015 | Completed | YES | 136 |
| BGB3245 | ||||||||
|
BRAF mutants | Solid tumors | I | NCT04249843 | 2020 | Recruiting | YES | 49 |
|
BRAF/NRAS/KRAS mutants | Solid tumors | I/IIa | NCT05580770 | 2023 | Recruiting | NA | |
| PLX8394 | ||||||||
|
BRAF mutants | Solid tumors | I/IIa | NCT02428712 | 2015 | Active, not recruiting | YES (Abstract) | 169 |
|
BRAF mutants | Solid tumors | I/IIa | NCT02012231 | 2014 | Terminated | NA | |
MEKi, MEK inhibitor (same with other inhibitors); LGG, low-grade glioma; NSCLC, non-small cell lung cancer; CRC, colorectal cancer; NA, not available.
Table 4.
Clinical outcomes of type II RAFi monotherapies in RAS mutant tumors.
| Phase | Trial number | Pts | Administration | Efficacy in RAS mutant tumors | Safety | Ref. |
|---|---|---|---|---|---|---|
| I | NCT01425008 | 149 | Single-agent tovorafenib: RP2D: 200 mg Q2D or 600 mg QW. MTD: 200 mg Q2D or 600 mg QW, over 28-day cycles. |
1. NRAS-mutant melanoma in Q2D expansion phase (n = 14): DCR: 36% (5/14); PR: 7% (1/14); SD: 29% (4/14); PD: 64% (9/14). |
|
50 |
| I | NCT02607813 | 87 | Single-agent naporafenib: 100–1200 mg QD or 200–800 mg BID. |
1. KRAS-mutant and BRAF-mutant cancers (n = 87): DCR: 37% (32/87); PR: 2% (2/87); SD: 34% (30/87); PD: 41% (36/87). |
|
163,176 |
| I |
NCT02405065 NCT03118817 |
135 | Single-agent belvarafenib: 450 mg BID. |
|
|
116 |
| I | NCT02014116 | 51 | Single-agent LY3009120: RP2D: 300 mg BID. |
|
|
165 |
| I | NCT02610361 | 131 | Single-agent lifirafenib: RP2D: 30 mg daily. MTD: 40 mg daily. |
1. KRAS or NRAS-mutant tumors (n = 66): DCR: 53% (35/66); PR: 3% (2/66); SD: 50% (33/66); PD: 24% (16/66). 1 pt with KRAS-mutant endometrial cancer and 1 pt with KRAS-mutant NSCLC showed PR. But KRAS or NRAS-mutant CRC (n = 20) showed no responses. |
|
164 |
| I | NCT04249843 (Abstract) | 42 | Single-agent BGB3245: MTD: 40 mg QD. |
1. Objective responders included 2 pts with NRAS-mutant melanoma and 1 pt with KRAS G12D appendiceal cancer. |
|
49 |
RP2D, recommended phase II dose; MTD, maximum tolerated dose; Q2D, once every other day; QW, once weekly; QD, once daily; BID, twice a day; DCR, disease control rate; PR, partial response; SD, stable disease; PD, progressive disease; CRC, colorectal cancer; NSCLC, non-small cell lung cancer; DLT, dose-limiting toxicity; TEAEs, treatment-emergent adverse events; SCC, squamous cell carcinoma; pts, patients.
5.1. Tovorafenib
Tovorafenib potently blocks the kinase function of BRAF and CRAF monomers, as well as their homo and heterodimers104,150. Crystal structure analysis revealed that this drug binds to both protomers of the BRAF dimer, although it has a comparatively weaker impact on the ARAF monomers and dimers150. The efficacy of tovorafenib in treating BRAF and NRAS mutant melanoma has been shown in preclinical and clinical research50. Furthermore, tovorafenib possesses an excellent capability to enter the central nervous system (CNS)130. It has shown promising antitumor efficacy in clinical trials involving pediatric low-grade glioma (LGG) patients with RAF mutations172, 173, 174. The FDA granted it the “breakthrough therapy designation” in 2021, highlighting its potential in treating intracranial tumors. The drug exhibited modest efficacy in individuals with NRAS-mutated melanoma, as evidenced by a disease control rate (DCR) of 36% (5/14) and a partial response (PR) rate of 7% (1/14) (NCT01425008)50. The overall safety profile of tovorafenib was deemed acceptable in a cohort of 149 treated patients. A total of 68% (67/99) of the patients experienced grade ≥3 treatment-emergent adverse events (TEAEs), with anemia accounting for 14% (14/99) and maculopapular rash accounting for 8% (8/99). In addition, the incidence of cutaneous SCC was less than 1% (1/149), significantly lower than that of type I1/2 RAFi. Notably, tovorafenib is the only type II RAFi that has advanced to a phase III clinical trial, determining its efficacy in RAF mutant LGG (NCT05566795). However, the effectiveness of tovorafenib in RAS mutant tumors remains to be investigated.
5.2. Naporafenib
Naporafenib is a potent type II RAFi that effectively inhibits both monomers and dimers of BRAF and CRAF while exhibiting relatively lower activity against ARAF (approximately 30- to 50-fold less activity)18. Minimal paradoxical ERK activation occurs due to the occupancy of naporafenib in both protomers of the RAF dimer160. The efficacy of naporafenib in models with BRAF V600E and NRAS mutations has been shown in vitro and in vivo, although its activity in KRAS mutants is only moderate18,160,180. Furthermore, wild-type cell lines lacking RAS/RAF mutations exhibit predominant insensitivity to naporafenib, which may be attributed to their lack of dependence on the MAPK pathway. Consistent with the findings of preclinical investigations, a phase I dose-scaling trial revealed that most individuals with KRAS mutant tumors experienced disease progression following naporafenib monotherapy (NCT02607813)176. Only a minority of patients had stable disease (SD, 30/87), and two achieved a confirmed PR (2/87)176. The adverse events of naporafenib were manageable, with grade ≥3 TEAEs occurring in 24% of patients (21/87). While naporafenib monotherapy has limited efficacy in RAS mutant tumors, combination regimens involving napafenib have achieved significant efficacy in clinical trials.
5.3. Belvarafenib
Belvarafenib has been shown to selectively inhibit BRAF V600E and wild-type CRAF more potently than wild-type ARAF and BRAF. CRISPR-Cas9 analysis demonstrated that belvarafenib was more effective against cells expressing only BRAF or CRAF than against cells expressing only ARAF116. Immunoprecipitation experiments have further confirmed that this drug facilitates the dimerization of all RAF isoforms, including ARAF, and impedes kinase activity by binding to both RAF dimer protomers116. Moreover, it displays potent inhibitory effects on the growth of melanoma cells harboring BRAF V600E and NRAS mutations, although it induces minimal paradoxical ERK activation (compared to vemurafenib)116. In clinical trials involving belvarafenib monotherapy116, a PR of 20% (2/10) was observed in individuals with NRAS-mutated melanoma, while no PR (0/10) was shown in individuals with KRAS-mutated CRC. The complicating drug resistance mechanisms in KRAS mutant CRC may account for the variable response rates181. However, PR was observed in two patients with sarcoma harboring the KRAS G12V mutation and bladder cancer carrying the KRAS G12D mutation, indicating its potential for treating specific KRAS mutant malignancies. The drug demonstrated good tolerability in the study involving 135 participants, and no SCC cases were observed. Furthermore, belvarafenib displayed effective blood–brain barrier penetration and exhibited potent antitumor activity against melanoma brain metastasis with BRAF/NRAS mutations in mice182, possibly superior to that of tovorafenib131. Although additional clinical validation is warranted, this compound presents a potential novel therapeutic option for intracranial tumors.
5.4. Lifirafenib
Lifirafenib is a potent inhibitor of both pan-RAF and EGFR and can effectively inhibit the kinase activity of the three RAF isoforms with a slow off-rate134. The drug exhibits a marked capacity to stimulate BRAF/CRAF dimerization, although further investigation is needed to determine its impact on other types of RAF dimers104,135. Preclinical investigations have shown that this drug is effective in CRC cells harboring BRAF V600E or EGFR mutations but has a decreased response in cells with RAS mutations134. In a clinical trial involving 66 patients with KRAS- or NRAS-mutated tumors164, lifirafenib monotherapy demonstrated a PR in a patient diagnosed with KRAS-mutated endometrial cancer and another patient diagnosed with KRAS-mutated NSCLC. Conversely, no response was observed in KRAS or NRAS mutant CRC patients (n = 20). The drug exhibited manageable side effects, good tolerability, and no occurrence of SCC. Additionally, the activation of EGFR feedback is a significant mechanism for the development of resistance to RAFi 183,184. Lifirafenib has exhibited the capacity to hinder EGFR feedback activation and achieve sustained suppression of pERK134. Although the efficacy of EGFR inhibition may contribute to its effectiveness, the optimal therapeutic window achievable through combined RAF/EGFR inhibition has yet to be determined.
5.5. LY3009120
As a pan-RAF inhibitor, LY3009120 exhibits comparable affinities for ARAF, BRAF, and CRAF100,102. This compound stimulates homo and heterodimerization of BRAF and CRAF and subsequently binds to both protomers of the RAF dimer and prevents its kinase activity102. LY3009120 demonstrates reduced paradoxical ERK activation in RAS mutant tumor cells compared to RAF monomer inhibitors102. LY3009120-induced paradoxical ERK activation does not rely on classical RAS proteins (H/N/KRAS) but does require the MRAS/SHOC2 complex185. These findings indicate that upstream scaffolding protein complexes play a significant role in MAPK pathway signaling. In preclinical research, LY3009120 demonstrated remarkable effectiveness against tumors harboring BRAF, KRAS, or NRAS mutations102,180. Unexpectedly, the phase I clinical trial for LY3009120 was prematurely halted because of its inadequate clinical effectiveness (NCT02014116)165. The trial enrolled a total of 51 individuals diagnosed with BRAF- or RAS-mutated tumors. Surprisingly, no one in the study exhibited a complete or partial response, and only eight individuals (15.7%) displayed stable disease. The researchers suspected poor patient selection might have contributed to these disappointing outcomes. In addition, the short half-life and limited tumor-targeting ability of LY3009120 resulted in ineffective inhibition of pERK within the tumor microenvironment. The clinical side effects of LY3009120 must also be considered. Off-target effects may restrict the dosage of LY3009120 since it selectively affects a wide range of protein kinases, such as p38, ephrin receptors, and JNK102.
6. Mechanisms of resistance and combination therapies
The understanding of resistance mechanisms to type II RAFi is relatively limited compared to that of KRAS G12Ci. Insights from the resistance mechanisms of KRAS G12Ci can help explain from multiple perspectives how RAS mutant tumor cells escape RAS/MAPK signaling pathway inhibition. Reactivation of the MAPK pathway and activation of other alternative pathways are common mechanisms leading to resistance to MAPK pathway inhibitors. These findings have implications for other inhibitors targeting the MAPK pathway, prompting researchers to explore comparable adaptive strategies.
6.1. Insights from the KRAS G12Ci
6.1.1. Mechanisms of resistance to KRAS G12Ci
While there are variations in the specific resistance mechanisms across different tumor types, reactivation of the RAS/MAPK pathway is a prevalent cause of resistance to KRAS G12Ci186. Jenny et al. reported that shortly after KRAS G12C inhibition, certain quiescent cells can generate new KRAS G12C. These mutant proteins can maintain the active, drug-insensitive state by activating EGFR and aurora kinase A signaling187. This rapid adaptive mechanism leads to predominantly partial responses to KRAS G12Ci in patients with NSCLC. A study of 38 patients (primarily NSCLC and CRC) receiving adagrasib showed that 45% (17/38) of patients demonstrated resistance188. The main resistance mechanisms can be classified into four groups: i) KRAS alterations, such as KRAS G12D/R/V/W, G13D, and Q61H, which reactivate KRAS; KRAS R68S, H95D/Q/R, and Y96C, which occur in the switch II pocket and hinder drug binding; and KRAS G12C amplification. ii) RAS/MAPK/PI3K alterations, including the activation of NRAS and PI3K; the activation of the downstream effector kinases BRAF, CRAF, and MEK1; and loss-of-function mutations in PTEN and NF1. iii) RTK alterations, including the activation of MET, ALK, RET, and FGFR3. iv) Tumor cells undergo histopathological changes, transitioning from lung adenocarcinoma to squamous lung cancer. Another study of 43 patients treated with sotorasib reported similar resistance mechanisms114. The sensitivity of different secondary genetic alterations to different KRAS G12Ci may vary, with certain secondary mutations conferring resistance to both drugs (e.g., Y96D and Y96S are resistant to either sotorasib or adagrasib), while others are resistant to only one drug188,189. The complexity of the problem is further exacerbated by the emergence of multiple resistance mechanisms in some patients and the presence of subclonal heterogeneity of resistance mechanisms within the tumor114.
6.1.2. Approaches to address KRAS G12Ci resistance
To address the resistance mechanisms to KRAS G12Ci, various therapeutic strategies can be employed: i) KRAS G12Ci with novel mechanisms of action. An example is the Y96D mutation, which hinders adagrasib binding and consequently leads to resistance. However, the tri-complex active-state KRAS G12Ci RM-018 can still bind and inhibit KRAS G12C/Y96D mutant and overcome resistance186. ii) Pan-KRASi with broader targets31. Pan-KRASi can potentially reduce the KRAS-GTP burden by various KRAS mutants and may be effective against secondary KRAS mutations. iii) Targeting drug-modified targets. Zhang et al. reported an immune-based therapy based on neoepitopes generated by KRAS G12Ci (ARS1620) that may prove effective against drug resistance190,191. iv) Combination regimens with other drugs. Vertical combination strategies with other RAS/MAPK pathway inhibitors or horizontal combination strategies with other signaling pathway inhibitors can prolong resistance and achieve sustained inhibition. Preclinical evidence suggests that the resistance of KRAS G12Ci can be overcome by combining with EGFRi, SOS1i, SHP2i, type II RAFi, and MEKi114,192,193. A phase III clinical trial involving 160 patients with chemorefractory CRC reported that the combination of sotorasib and panitumumab (EGFRi) significantly extended progression-free survival (PFS) compared to the standard treatment regimen194. However, the overall response (OS) did not significantly improve with the combination regimen, possibly due to the limited duration of follow-up (median follow-up of 7.8 months). Another phase I clinical trial reported an ORR of 62.5% for the combination of divarasib plus cetuximab (EGFRi) in patients with metastatic CRC who had not previously received KRAS G12Ci (n = 24)195, and the ORR of divarasib alone is 35.9%64. The median PFS for patients receiving the combination regimen was 8.1 months (95% CI: 5.5–12.3). Among the other five patients who had previously received KRAS G12Ci treatment, three achieved PR, and two had SD with the combination regimen. In addition to EGFRi, clinical trials combining SOS1i, SHP2i, mTORi, CDK4/6i, and anti-PD-1/PD-L1 immunotherapy with KRAS G12Ci are currently underway (NCT04330664, NCT04185883, and NCT03785249).
6.2. Mechanisms of resistance to type II RAFi
Extensive research has been conducted on the mechanisms of resistance to RAF monomer inhibitors, yet limited knowledge exists regarding resistance mechanisms specific to type II RAFi. ARAF mutations can cause compensatory activation of ERK signaling when BRAF and CRAF are inhibited, potentially acting as a shared resistance mechanism for type II RAFi116. Kelli et al. reported that ARAF played a significant role in conferring resistance to naporafenib18. Decreased ARAF expression may increase the susceptibility of RAS mutant cells to this medication. Resistance was linked to the kinase activity and dimerization properties of ARAF, resulting in paradoxical ERK activation in cells expressing only ARAF. Similarly, Ivana et al. discovered that acquired ARAF mutations (such as ARAF G387) decreased the flexibility of the hinge region and hindered the attachment of belvarafenib to RAF proteins. This resulted in resistance against belvarafenib in an ARAF kinase- and dimer-dependent manner in BRAF- and NRAS-mutated melanoma116. Despite resistance to other type II RAFi (naporafenib and AZ628), these ARAF mutations remained vulnerable to downstream MEKi and ERKi. The combination of belvarafenib with MEKi (cobimetinib) could overcome the resistance driven by ARAF mutations.
6.3. Combination therapy based on type II RAFi in RAS mutant tumors
The MAPK pathway relies predominantly on linear cascade phosphorylation for signal transduction. Still, its regulation is remarkably complex, involving multiple feedback loops at different cascade levels and interactions with other signaling pathways196. Therefore, combination approaches are considered a more promising strategy for treating RAS mutant tumors (Fig. 5). Both preclinical (Table 5) and clinical (Table 6) evidence have demonstrated that combination therapies based on type II RAFi show significant efficacy in treating RAS mutant tumors. In addition to vertical and horizontal combination strategies, synergistic effects of type II RAFi with several chemotherapeutic agents and anti-PD-1/PD-L1 immunotherapy have also been reported.
Figure 5.

Combination regimens based on type II RAFi in RAS mutant tumors. Type II RAF inhibitors demonstrate synergistic antitumor activity when combined with other targeted agents, chemotherapeutic agents, or immunotherapies in RAS mutant tumors.
Table 5.
Preclinical evidence of type II RAFi combination regimens in RAS mutant tumors.
| Combination regimens | Synergistic effect | Ref. |
|---|---|---|
| Combined with MAPK pathway inhibitors | ||
| Tovorafenib+TAK733 (MEKi) | RAS mutants in melanoma and CRC | 197 |
| Naporafenib+Trametinib (MEKi) | NRAS and HRAS mutants in bladder cancer | 198 |
| Belvarafenib+Cobimetinib (MEKi) | NRAS mutants in melanoma | 116,182 |
| Lifirafenib+Mirdametinib/Selumetinib/Trametinib (MEKi) | KRAS and NRAS mutants in melanoma, CRC, and lung cancer |
14,135, 137 |
| LY3009120+Cobimetinib (MEKi) | NRAS mutants in melanoma; KRAS mutants in CRC and lung cancer | 5,199 |
| LY3009120+Trametinib (MEKi)/LY3214996 (ERKi) | NRAS and HRAS mutants in rhabdomyosarcoma | 200 |
| BGB3245+Mirdametinib (MEKi) | NRAS mutants in melanoma | 137 |
| TAK632+TAK733 (MEKi) | NRAS mutants in melanoma | 129 |
| AZ628+Cobimetinib/Selumetinib (MEKi) | KRAS mutants in CRC and lung cancer | 5,111 |
| RAF709+Trametinib (MEKi) | KRAS mutants in CRC, lung, and pancreatic cancer |
14,140, 201 |
| DCC3084+Cobimetinib/Binimetinib (MEKi) | KRAS mutants in lung and pancreatic cancer | 131 |
| Naporafenib+Sotorasib (KRAS G12Ci) | Lung cancer cells that are resistant to KRAS G12Ci | 114 |
| Naporafenib+SHP099 (KRAS SHP2i) | KRAS mutants in gastroesophageal adenocarcinomas | 202 |
| Combined with other signaling pathway inhibitors | ||
| LY3009120+Abemaciclib (CDK4/6i) | NRAS mutants in melanoma; KRAS mutants in CRC and lung cancer | 203 |
| Naporafenib+Volasertib (PLK1i) | NRAS mutants in lung cancer | 180 |
| TAK632+Saracatinib/Bosutinib (SRCi) | KRAS mutants in CRC, lung, and pancreatic cancer | 136 |
| AZ628+Dasatinib (SRCi) | KRAS mutants in breast cancer | 204 |
| AZ628+BP-1-102 (STAT3i) | KRAS mutants in lung cancer | 205 |
| AZ628+Dactolisib (PI3Ki/mTORi) | KRAS mutants in CRC | 206 |
| LY3009120+Verteporfin (YAPi) | KRAS mutants in pancreatic cancer | 207 |
| Tovorafenib+Selinexor (CRM1i) | KRAS mutants in multiple myeloma | 208 |
| Tovorafenib+Bortezomib (proteasome inhibitor) | KRAS mutants in multiple myeloma | 209 |
| TAK632+Bortezomib/Carfilzomib (proteasome inhibitor) | KRAS and NRAS mutants in multiple myeloma | 210 |
| Combined with chemotherapeutic agents | ||
| LY3009120+Cytarabine | KRAS and NRAS mutants in AML | 211,212 |
| AZ628+5-FU+Irinotecan | KRAS mutants in CRC | 213 |
| Combined with immunotherapy | ||
| Belvarafenib+Atezolizumab (anti-PD-L1) | NRAS mutants in melanoma | 182 |
AML, acute myeloid leukemia; CRC, colorectal cancer.
Table 6.
Clinical outcomes of type II RAFi combination regimens in RAS mutant tumors.
| Phase | Trial number | Pts | Administration | Efficacy in RAS mutant tumors | Safety | Ref. |
|---|---|---|---|---|---|---|
| Ib | NCT02974725 | 66 | Naporafenib+Trametinib (MEKi):
|
|
|
177 |
| II | NCT04417621 (Abstract) | 134 | Naporafenib+LTT462 (ERKi): Naporafenib 400 mg BID +LTT462 200 mg QD. |
NRAS-mutant, immuno-resistant melanoma (n = 29): DCR: 62% (18/29); PR: 21% (6/29); SD: 41% (12/29); PD: 31% (9/29). |
|
52 |
| Naporafenib+Trametinib (MEKi): Naporafenib 200 mg BID +trametinib 1 mg QD. |
NRAS-mutant, immuno-resistant melanoma (n = 24): DCR: 67% (16/24); PR: 4% (1/24); SD: 63% (15/24); PD: 17% (4/24). |
|||||
| Naporafenib+Ribociclib (CDK4/6i): Naporafenib 400 mg BID +ribociclib 400 mg QD. |
NRAS-mutant, immuno-resistant melanoma (n = 15): DCR: 33% (5/15); PR: 7% (1/15); SD: 27% (4/15); PD: 40% (6/15). |
|||||
| Ib | NCT03284502 (Abstract) | 118 | Belvarafenib + Cobimetinib (MEKi):
|
|
|
178, 179 |
| Ib | NCT03905148 (Abstract) | 56 | Lifirafenib + Mirdametinib (MEKi): Lifirafenib 15–20 mg QD +mirdametinib 2–4 mg QD or BID. |
|
|
51 |
| I | NCT02607813 | 43 | Naporafenib + PDR001 (anti-PD-1): Naporafenib 400 mg BID + PDR001 400 mg Q4W. |
|
|
176 |
MEKi, MEK inhibitor (same with other inhibitors); QD, once daily; BID, twice a day; QOD, three times a week; Q4W, every four weeks; DCR, disease control rate; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; NSCLC, non-small cell lung cancer; CRC, colorectal cancer; LGSOC, low-grade serous ovarian carcinoma; DLT, dose-limiting toxicity; TEAEs, treatment-emergent adverse events; pts, patients.
6.3.1. Combined with other MAPK pathway inhibitors
Multiple preclinical studies have substantiated the synergistic antitumor efficacy of type II RAFi when combined with MEKi (especially cobimetinib116, trametinib14, and mirdametinib135,137) in tumors harboring KRAS and NRAS mutations. Notably, type II RAFi exhibit more pronounced synergistic effects with MEKi than type I1/2 RAFi5. However, not all MEKi exhibit the same degree of synergy, which may be attributed to their impact on RAF‒MEK complexes5. Combination therapy with type II RAFi and MEKi induces the BRAF/CRAF complex and prevents MEK from dissociating from the RAF complex. This reduces MEK dimerization and MEK-ERK interactions, leading to sustained inhibition of ERK activation14.
Type II RAFi, in combination with MEKi or ERKi, has demonstrated significant clinical efficacy in treating RAS mutant tumors. In a phase Ib clinical trial, naporafenib plus trametinib (MEKi) displayed preliminary antitumor efficacy in melanoma patients with NRAS mutations, resulting in a DCR of 73% (22/30) and a PR rate of 30% (9/30) (NCT02974725)177. However, this combination therapy did not yield satisfactory results in KRAS mutant NSCLC, with only one PR observed out of 25 patients. Another phase II trial demonstrated the superiority of naporafenib plus trametinib in NRAS-mutated melanoma individuals who had experienced advancement after prior immunotherapy (NCT04417621). The DCR was 67% (16/24), and the PR rate was 4% (1/24) in this cohort52. For these patients, naporafenib plus LTT462 (ERKi) also exhibited strong efficacy (DCR of 62%). Furthermore, belvarafenib plus cobimetinib (MEKi) showed impressive antitumor activity against NRAS-mutated, immuno-resistant melanoma, resulting in a PR rate of 45% (5/11) (NCT03284502)178,179. In addition to its efficacy in melanoma, the combination of type II RAFi and MEKi also shows promise for treating other solid tumors. This is exemplified by the efficacy of lifirafenib plus mirdametinib in patients with RAS-mutated gynecologic tumors (NCT03905148)51. Clinical trials are ongoing to evaluate other combination regimens of type II RAFi plus MEKi to treat tumors with RAS mutations, including tovorafenib plus pimasertib (NCT04985604), belvarafenib plus cobimetinib (NCT04835805), and BGB3245 plus mirdametinib (NCT05580770).
The remarkable efficacy of type II RAFi, when combined with MEKi or ERKi, highlights the importance of achieving continuous and effective suppression of MAPK signaling to treat RAS-mutated tumors135. Hence, combining type II RAFi with other MAPK pathway inhibitors may also yield significant synergistic effects. An example of this is the frequent reactivation of RAS-GTP caused by negative feedback from ERK5, and the combination of naporafenib plus either sotorasib (KRAS G12Ci) or SHP099 (SHP2i) showed synergistic effects on KRAS-mutated lung cancer and gastric cancer separately114,202. Based on these findings, it can be inferred that type II RAFi may also have synergistic antitumor effects when combined with drugs such as SOS1i or EGFRi.
6.3.2. Combined with inhibitors targeting other signaling pathways
The complex interaction between the MAPK pathway and other pathways greatly influences the emergence of innate or acquired resistance in RAS-mutated tumors. Some RAS mutant cells exhibit reduced sensitivity to type II RAFi, in which the RAS mutation does not serve as the primary oncogenic driver gene214. Consequently, combining type II RAFi with inhibitors targeting the crosstalk pathways is an attractive approach.
Specifically, the combination of type II RAFi with inhibitors of the cell cycle pathway (CDK4/6i and PLK1i) exhibits synergistic effects. The combination of LY3009120 plus abemaciclib (CDK4/6i) resulted in increased G0/G1 arrest in tumor cells with KRAS and NRAS mutations, mainly due to increased suppression of phospho-Rb and cyclin D1203. A phase II trial showed the synergistic effect of combining naporafenib and ribociclib (CDK4/6i) in immune-resistant melanoma patients with NRAS mutations (NCT04417621)52. The study showed moderate efficacy, with a DCR of 33% (5/15) and a PR of 7% (1/15). However, this combination's efficacy was weaker than that of naporafenib in combination with MEKi or ERKi therapies52. PLK1 is another prominent cell cycle regulator and interacts with CRAF, promoting ERK activation and tumor progression215. Both in vitro and in vivo studies have demonstrated that coadministration of PLK1i has synergistic effects on KRAS-mutated tumors216. Notably, PLK1i exhibit heightened efficacy in treating KRAS mutant CRC217,218, and ongoing clinical studies are investigating the potential therapeutic benefits (NCT03829410). In NSCLC cells harboring NRAS mutations, the combination of naporafenib and volasertib (PLK1i) was found to be lethal, leading to the arrest of the cell cycle at the G2/M phase180.
Previous studies have shown that simultaneous targeting of the SRC/STAT3 (SRCi, STAT3i) pathway in combination with type II RAFi has synergistic antitumor effects. Specifically, the combination of AZ628 and dasatinib (SRCi) successfully inhibited the growth of KRAS-mutated breast cancer cells204. The synergistic effects of AZ628 and BP-1-102 (STAT3i) were observed in lung cancer cells with KRAS mutations by abrogating the MAPK pathway205. Moreover, CCT3833 targets both pan-RAF and SRC. Although the exact mechanism of action of CCT3833 on RAF dimers has not been determined, docking studies have proposed that CCT3833 acts as a type II RAFi136. Preclinical evidence has shown that CCT3833 exhibits significant activity in KRAS-mutated CRC, pancreatic cancer, and lung cancer136. CCT3833 showed a favorable effect on one patient diagnosed with spindle cell sarcoma, who had the KRAS G12V mutation and did not respond to other inhibitors136. The activation of the SRC/STAT3 pathway has a complex interaction with RAS-MAPK signaling that needs to be further elucidated219. These results offer encouraging preclinical evidence for combining RAF and SRC inhibition to treat tumors with RAS mutations.
Moreover, blocking the MAPK pathway has been discovered to significantly trigger the PI3K/AKT/mTOR pathway220. Consequently, AZ628 plus dactolisib (PI3Ki/mTORi) demonstrated synergistic effects on CRC cells with KRAS mutations206. In KRAS mutant pancreatic cancer, verteporfin (YAPi) could enhance LY3009120 by inhibiting AKT compensatory activation207. Additionally, proteasome inhibitors, such as bortezomib and carfilzomib, are vital clinical significance in treating multiple myeloma221. The activation of RAS or RAF mutations increases proteasomal activity, leading to resistance against proteasome inhibitors210. Notably, the combination of type II RAFi with proteasome inhibitors exhibited a synergistic inhibitory effect in multiple myeloma with RAS mutations, such as tovorafenib plus bortezomib and TAK632 plus carfilzomib209,210.
6.3.3. Combined with chemotherapeutic agents
Type II RAFi could enhance the efficacy of several DNA-damaging chemotherapeutic agents and synergistically inhibit tumor growth. Carlota et al. reported that IKKα (p45) was required for efficient DNA repair. DNA damage rapidly activates IKKα (p45), dependent on MAPK signaling213. Based on this, researchers proposed that RAF inhibition could synergistically enhance the efficacy of DNA-damaging chemotherapeutic agents. In patient-derived KRAS mutant CRC tumoroids, AZ628 showed improved therapeutic potential when combined with 5-FU and irinotecan, particularly in tumors resistant to chemotherapy213. Similarly, in RAS mutant acute myeloid leukemia cells, LY3009120 showed increased potency in combination with cytarabine, which inhibited cell proliferation mainly by disrupting DNA synthesis211. However, a clinical trial evaluating the regimen of tovorafenib plus paclitaxel showed insufficient efficacy and disease progression in patients with KRAS- or BRAF-mutated NSCLC (NCT02327169). The distinct mechanism of action of paclitaxel might cause this lack of synergistic effects. Paclitaxel can bind to microtubule proteins to inhibit cell division rather than directly inducing DNA damage222.
6.3.4. Combined with immunotherapy
Multiple inhibitors targeting RAS signaling could modulate the tumor immune microenvironment4. Type II RAFi have exhibited the potential to improve the efficacy of anti-PD-1/L1 immunotherapy. Yu et al. discovered that belvarafenib plus atezolizumab (anti-PD-L1) showed more significant inhibitory activity against NRAS mutant melanoma in mice than single drug treatment182. The combined treatment significantly boosted the infiltration of CD8+ T cells, and the improved inhibition of tumor growth was strongly associated with the increase in CD8+ T cell levels within the tumor microenvironment. A clinical trial involving 43 patients has provided additional evidence supporting the potential synergistic effects of type II RAFi in combination with anti-PD-1 immunotherapy (NCT02607813)176. Among patients with NRAS-mutated melanoma, the combination of naporafenib and PDR001 (anti-PD-1) demonstrated a DCR of 52% (11/21), with one patient achieving a complete response (CR). In patients with KRAS-mutated NSCLC, the regimen resulted in a DCR of 46% (10/22), with three patients achieving PR. Unfortunately, another clinical trial investigating tovorafenib in combination with nivolumab was prematurely terminated (NCT02723006). Moreover, the efficacy of combining type II RAFi with MEKi in RAS mutant tumors was found to rely on the presence of CD8+ T cells14. The presence of T cells within the tumor was reduced by MEKi (trametinib), but this effect was reversed when combined with type II RAFi (BGB283). This combination increased the proportion of CD8+ T cells and caused long-lasting immune infiltration. CD8+ T cells were required for tumor continued inhibition, while anti-PD-L1 therapy activated these cells and showed increased efficacy14. Consequently, this investigation offers a rationale for conducting triple therapy trials involving type II RAFi, MEKi, and anti-PD-1/L1 therapy. Currently, an ongoing phase I clinical trial is evaluating the efficacy of belvarafenib plus cobimetinib (MEKi) plus nivolumab (anti-PD-1) in advanced melanoma patients with NRAS mutations who previously received anti-PD-1/L1 therapy (NCT04835805). Additional research is necessary to gain a complete understanding of the exact underlying mechanisms involved. However, the utilization of type II RAFi plus anti-PD-1/L1 in a dual regimen, or with the addition of MEKi in a triple regimen, can show potential in treating RAS-mutated tumors.
7. Summary and prospects
Although significant breakthroughs have been made in targeting KRAS G12C, directly targeting RAS remains challenging given the structural specificity and diverse biological functions, particularly in non-G12C RAS mutant tumors. Therefore, a promising research strategy is inhibiting crucial downstream kinases of RAS, particularly RAF. However, there is currently limited knowledge about the role of different RAF homo and heterodimers in various tumors with different RAS mutations. Nonetheless, the notion that RAF dimerization is vital for the emergence of drug resistance via ERK reactivation is now well accepted. To address the issue of RAF dimerization, new-generation RAFi employ strategies such as inhibiting RAF dimer activity, disrupting RAF dimer formation, and targeting DIF.
Currently available inhibitors targeting RAF dimers exhibit various effects on different types of RAF monomers and dimers, some of which may result in drug insensitivity or resistance. Complete suppression of MAPK signaling is necessary to achieve significant antitumor effects in RAS mutant tumors6. Additionally, the crosstalk between MAPK signaling pathway and other signaling pathways is complex. Thus, targeting specific RAS mutations and the RAF dimer responsible for driving ERK signaling is imperative7. It is crucial to use drugs with well-defined actions and mechanisms to ensure their effectiveness, as failure to do so may result in paradoxical ERK activation.
Previous studies on RAF dimers have focused mainly on BRAF and CRAF dimers due to their high kinase activity and dimerization capacity. However, emerging studies indicate the significance of ARAF dimers in drug resistance mechanisms, although ARAF exhibits a decreased dimer formation ability. The impact of most inhibitors targeting RAF dimers on ARAF monomers and dimers is unknown. Based on the existing data, it is plausible that most type II RAFi may exhibit inadequate inhibition of ARAF150, and further investigations are imperative to validate this hypothesis. The underlying structural and mechanistic rationale behind the ineffective inhibition of ARAF requires further exploration. Therapeutically, the lack of ARAF inhibition can have positive and negative implications18. On the one hand, potential outcomes may include decreased effectiveness and the emergence of drug resistance. On the other hand, sparing ARAF could potentially widen the therapeutic window since patients may be intolerant to completely inhibiting the MAPK pathway150.
Current inhibitors targeting RAF dimers have shown varying efficacy in different tumor types. Paradox breakers have limited efficacy in treating RAS mutant tumors, while type II RAFi have demonstrated significant therapeutic benefits in patients with NRAS- and KRAS-mutated tumors, particularly in melanoma. Notably, type II RAFi, such as belvarafenib, tovorafenib, and the recently published DCC3084 and BDTX4933, exhibit promising potential for penetrating the blood–brain barrier131,133. This heightened permeability of type II RAFi may offer clinical advantages for patients with brain metastases and CNS tumors compared to first-generation RAFi. However, the effectiveness of type II RAFi monotherapy is limited in individuals with CRC and NSCLC. The reasons for variations in drug sensitivity across different tumor types remain to be thoroughly investigated. The efficacy of type II RAFi monotherapy in RAS mutant tumors should be viewed cautiously due to the sparing of ARAF, and single-target inhibitors typically reactivate MAPK signaling.
The mechanisms underlying resistance to type II RAFi are poorly understood, although recent evidence suggests a potential association with reactivation of ARAF dimer-induced ERK signaling. It is crucial to investigate the effectiveness and resistance mechanisms of type II RAFi in RAS mutant tumors, particularly the combination therapies targeting the identified resistance mechanisms. Ongoing clinical trials have shown exciting results in combining type II RAFi with MEKi or ERKi for managing NRAS and KRAS mutant tumors. This approach has shown promising efficacy in patients with melanomas who have received prior immunotherapy. Due to the critical role of RAF dimerization in ERK signaling reactivation, which often leads to drug resistance, type II RAFi can potentially address the issue of drug resistance in other MAPK pathway inhibitors. These clinical findings suggest that type II RAFi may be a promising approach for clinical application against RAS mutant tumors. In addition to combination with MAPK pathway inhibitors, other combination regimens involving type II RAFi have also shown promise in RAS mutant tumor cells. These include combinations with inhibitors targeting other pathways, DNA-damaging chemotherapeutic agents, and anti-PD-1/L1 immunotherapy, although further validation through clinical trials is needed to confirm their effectiveness. Thus, targeting RAF dimers is expected to be further studied and developed for treating RAS mutant tumors.
Nevertheless, combination therapies often result in increased toxicity, potentially limiting their clinical utility. While achieving significant efficacy is important, another crucial goal is minimizing combination therapies' effects on normal cells to improve patient tolerability. In addition to developing and optimizing combination regimens that target specific RAS mutant tumors, improving drug targeting strategies represents a viable approach. One prevalent issue is the inadequate localization of small-molecule kinase inhibitors to tumors due to oral administration. Interactions with diverse food substances can affect the plasma concentration of drugs223. Thus, the different pharmacokinetics and distributions within the body hinder the optimal synergistic effect of coadministration. To address this issue, exploring novel strategies, such as utilizing nanomaterials and other tumor-targeted co-delivery systems, may offer promising prospects224,225. These systems could enable the precise administration and controlled release of drugs within the body, thereby ensuring continuous suppression of tumor cell growth, achieving the best therapeutic effect at the smallest dose, and reducing the adverse effects of combined treatments.
Declaration of Generative AI and AI-assisted technologies in the writing process
While preparing this paper, the authors used Home for Researchers (www.home-for-researchers.com) to improve readability and language. After using this tool, the authors reviewed and edited the content as needed and took full responsibility for the publication's content.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (82073059, 82073314), the Sichuan Science and Technology Program (2023NSFSC1838, China), and the Sichuan University Postdoctoral Interdisciplinary Innovation Fund (JCXK2212, China).
Author contributions
Hongwei Xia and Feng Bi conceived and supervised the work. Huanhuan Yin summarized the literature, drafted the manuscript, and plotted the tables and figures. Qiulin Tang proofread the tables and figures. Hongwei Xia and Feng Bi revised and edited the manuscript. All authors approved the final manuscript.
Conflicts of interest
All authors declare no conflicts of interest.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Hongwei Xia, Email: hw.xia@scu.edu.cn.
Feng Bi, Email: bifeng@scu.edu.cn.
References
- 1.Murugan A.K., Grieco M., Tsuchida N. RAS mutations in human cancers: roles in precision medicine. Semin Cancer Biol. 2019;59:23–35. doi: 10.1016/j.semcancer.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Yaeger R., Cowell E., Chou J.F., Gewirtz A.N., Borsu L., Vakiani E., et al. RAS mutations affect pattern of metastatic spread and increase propensity for brain metastasis in colorectal cancer. Cancer. 2015;121:1195–1203. doi: 10.1002/cncr.29196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nussinov R., Tsai C.J., Jang H. Oncogenic ras isoforms signaling specificity at the membrane. Cancer Res. 2018;78:593–602. doi: 10.1158/0008-5472.CAN-17-2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Punekar S.R., Velcheti V., Neel B.G., Wong K.K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat Rev Clin Oncol. 2022;19:637–655. doi: 10.1038/s41571-022-00671-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yen I., Shanahan F., Merchant M., Orr C., Hunsaker T., Durk M., et al. Pharmacological induction of RAS-GTP confers RAF inhibitor sensitivity in KRAS mutant tumors. Cancer Cell. 2018;34:611–625.e7. doi: 10.1016/j.ccell.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Moore A.R., Rosenberg S.C., McCormick F., Malek S. RAS-targeted therapies: is the undruggable drugged?. Nat Rev Drug Discov. 2020;19:533–552. doi: 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Timar J., Kashofer K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer metastasis rev. 2020;39:1029–1038. doi: 10.1007/s10555-020-09915-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blair H.A. Sotorasib: first approval. Drugs. 2021;81:1573–1579. doi: 10.1007/s40265-021-01574-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhillon S. Adagrasib: first approval. Drugs. 2023;83:275–285. doi: 10.1007/s40265-023-01839-y. [DOI] [PubMed] [Google Scholar]
- 10.Kun E., Tsang Y.T.M., Ng C.W., Gershenson D.M., Wong K.K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev. 2021;92 doi: 10.1016/j.ctrv.2020.102137. [DOI] [PubMed] [Google Scholar]
- 11.Pan X., Pei J., Wang A., Shuai W., Feng L., Bu F., et al. Development of small molecule extracellular signal-regulated kinases (ERKs) inhibitors for cancer therapy. Acta Pharm Sin B. 2022;12:2171–2192. doi: 10.1016/j.apsb.2021.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ullah R., Yin Q., Snell A.H., Wan L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin Cancer Biol. 2022;85:123–154. doi: 10.1016/j.semcancer.2021.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Park E., Rawson S., Li K., Kim B.W., Ficarro S.B., Pino G.G., et al. Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature. 2019;575:545–550. doi: 10.1038/s41586-019-1660-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong A., Piva M., Liu S., Hugo W., Lomeli S.H., Zoete V., et al. Durable suppression of acquired MEK inhibitor resistance in cancer by sequestering MEK from ERK and promoting antitumor T-cell immunity. Cancer Discov. 2021;11:714–735. doi: 10.1158/2159-8290.CD-20-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stalnecker C.A., Der C.J. RAS, wanted dead or alive: advances in targeting RAS mutant cancers. Sci Signal. 2020;13 doi: 10.1126/scisignal.aay6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samatar A.A., Poulikakos P.I. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 17.Blasco R.B., Francoz S., Santamaría D., Cañamero M., Dubus P., Charron J., et al. C-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell. 2011;19:652–663. doi: 10.1016/j.ccr.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monaco K.-A., Delach S., Yuan J., Mishina Y., Fordjour P., Labrot E., et al. LXH254, a potent and selective ARAF-sparing inhibitor of BRAF and CRAF for the treatment of MAPK-driven tumors. Clin Cancer Res. 2021;27:2061–2073. doi: 10.1158/1078-0432.CCR-20-2563. [DOI] [PubMed] [Google Scholar]
- 19.Poulikakos P.I., Zhang C., Bollag G., Shokat K.M., Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones G.G., Del Río I.B., Sari S., Sekerim A., Young L.C., Hartig N., et al. SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat Commun. 2019;10:2532. doi: 10.1038/s41467-019-10367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang M., Maloney R., Jang H., Nussinov R. The mechanism of Raf activation through dimerization. Chem Sci. 2021;12:15609–15619. doi: 10.1039/d1sc03444h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karoulia Z., Wu Y., Ahmed T.A., Xin Q., Bollard J., Krepler C., et al. An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell. 2016;30:485–498. doi: 10.1016/j.ccell.2016.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Escher T.E., Satchell K.J.F. RAS degraders: the new frontier for RAS-driven cancers. Mol Ther. 2023;31:1904–1919. doi: 10.1016/j.ymthe.2023.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boike L., Henning N.J., Nomura D.K. Advances in covalent drug discovery. Nat Rev Drug Discov. 2022;21:881–898. doi: 10.1038/s41573-022-00542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hallin J., Bowcut V., Calinisan A., Briere D.M., Hargis L., Engstrom L.D., et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat Med. 2022;28:2171–2182. doi: 10.1038/s41591-022-02007-7. [DOI] [PubMed] [Google Scholar]
- 26.Ji X., Li Y., Kong X., Chen D., Lu J. Discovery of prodrug of MRTX1133 as an oral therapy for cancers with KRAS(G12D) mutation. ACS Omega. 2023;8:7211–7221. doi: 10.1021/acsomega.3c00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tolcher A.W., Park W., Wang J.S., Spira A.I., Janne P.A., Lee H.J., et al. Trial in progress: a phase 1, first-in-human, open-label, multicenter, dose-escalation and dose-expansion study of ASP3082 in patients with previously treated advanced solid tumors and KRAS G12D mutations. J Clin Oncol. 2023;41:TPS764. [Google Scholar]
- 28.Varghese A.M., Ang C., Dimaio C.J., Javle M.M., Gutierrez M., Yarom N., et al. A phase II study of siG12D-LODER in combination with chemotherapy in patients with locally advanced pancreatic cancer (PROTACT) J Clin Oncol. 2020;38:TPS4672. [Google Scholar]
- 29.Surana R., LeBleu V.S., Lee J.J., Smaglo B.G., Zhao D., Lee M.S., et al. Phase I study of mesenchymal stem cell (MSC)-derived exosomes with KRASG12D siRNA in patients with metastatic pancreatic cancer harboring a KRASG12D mutation. J Clin Oncol. 2022;40:TPS633. [Google Scholar]
- 30.Wang Q.J., Yu Z., Griffith K., Hanada K., Restifo N.P., Yang J.C. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol Res. 2016;4:204–214. doi: 10.1158/2326-6066.CIR-15-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim D., Herdeis L., Rudolph D., Zhao Y., Böttcher J., Vides A., et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature. 2023;619:160–166. doi: 10.1038/s41586-023-06123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tolani B., Celli A., Yao Y., Tan Y.Z., Fetter R., Liem C.R., et al. Ras-mutant cancers are sensitive to small molecule inhibition of V-type ATPases in mice. Nat Biotechnol. 2022;40:1834–1844. doi: 10.1038/s41587-022-01386-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Reilly E.M., Wainberg Z.A., Weekes C.D., Furqan M., Kasi P.M., Devoe C.E., et al. AMPLIFY-201, a first-in-human safety and efficacy trial of adjuvant ELI-002 2P immunotherapy for patients with high-relapse risk with KRAS G12D- or G12R-mutated pancreatic and colorectal cancer. J Clin Oncol. 2023;41:2528. [Google Scholar]
- 34.Barbier A.J., Jiang A.Y., Zhang P., Wooster R., Anderson D.G. The clinical progress of mRNA vaccines and immunotherapies. Nat Biotechnol. 2022;40:840–854. doi: 10.1038/s41587-022-01294-2. [DOI] [PubMed] [Google Scholar]
- 35.Hofmann M.H., Gerlach D., Misale S., Petronczki M., Kraut N. Expanding the reach of precision oncology by drugging all KRAS mutants. Cancer Discov. 2022;12:924–937. doi: 10.1158/2159-8290.CD-21-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spira A.I., Starodub A.N., Arbour K.C., Sommerhalder D., Wolpin B.M., Barve M., et al. Abstract PR010: preliminary safety and pharmacokinetic profiles of RMC-6236, a first-in-class, RAS-selective, tri-complex RASMULTI(ON) inhibitor in patients with KRAS mutant solid tumors on the phase 1 trial RMC-6236-001. Mol Cancer Therapeut. 2023;22:PR010. [Google Scholar]
- 37.Arbour K.C., Punekar S., Garrido-Laguna I., Hong D.S., Wolpin B., Pelster M.S., et al. 652O Preliminary clinical activity of RMC-6236, a first-in-class, RAS-selective, tri-complex RAS-MULTI(ON) inhibitor in patients with KRAS mutant pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC) Ann Oncol. 2023;34:S458. [Google Scholar]
- 38.Luo G., Wang B., Hou Q., Wu X. Development of son of sevenless homologue 1 (SOS1) modulators to treat cancers by regulating RAS signaling. J Med Chem. 2023;66:4324–4341. doi: 10.1021/acs.jmedchem.2c01729. [DOI] [PubMed] [Google Scholar]
- 39.Song Z., Wang M., Ge Y., Chen X.P., Xu Z., Sun Y., et al. Tyrosine phosphatase SHP2 inhibitors in tumor-targeted therapies. Acta Pharm Sin B. 2021;11:13–29. doi: 10.1016/j.apsb.2020.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waizenegger I.C., Lu H., Thamer C., Savarese F., Gerlach D., Rudolph D., et al. Abstract 2667: trial in progress: phase 1 study of BI 1823911, an irreversible KRASG12C inhibitor targeting KRAS in its GDP-loaded state, as monotherapy and in combination with the pan-KRAS SOS1 inhibitor BI 1701963 in solid tumors expressing KRASG12C mutation. Cancer Res. 2022;82:2667. [Google Scholar]
- 41.Weiss A., Lorthiois E., Barys L., Beyer K.S., Bomio-Confaglia C., Burks H., et al. Discovery, preclinical characterization, and early clinical activity of JDQ443, a structurally novel, potent, and selective covalent oral inhibitor of KRASG12C. Cancer Discov. 2022;12:1500–1517. doi: 10.1158/2159-8290.CD-22-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falchook G., Li B.T., Marrone K.A., Bestvina C.M., Langer C.J., Krauss J.C., et al. OA03.03 Sotorasib in combination with RMC-4630, a SHP2 inhibitor, in KRAS p.G12C-mutated NSCLC and other solid tumors. J Thorac Oncol. 2022;17:S8. [Google Scholar]
- 43.Guo C., Chénard-Poirier M., Roda D., de Miguel M., Harris S.J., Candilejo I.M., et al. Intermittent schedules of the oral RAF-MEK inhibitor CH5126766/VS-6766 in patients with RAS/RAF-mutant solid tumours and multiple myeloma: a single-centre, open-label, phase 1 dose-escalation and basket dose-expansion study. Lancet Oncol. 2020;21:1478–1488. doi: 10.1016/S1470-2045(20)30464-2. [DOI] [PubMed] [Google Scholar]
- 44.Hindley C., Biondo A., Brothwood J., Dao K.H., Kandola N., Lyons J., et al. A combination vertical inhibition approach with inhibitors of SHP2 and ERK provides improved activity in KRAS-mutant pancreatic and colorectal cancer models. Eur J Cancer. 2022;174:S57. [Google Scholar]
- 45.Coma S., Chowdhury S., Pachter J.A. Abstract 1263: dual RAF/MEK inhibitor VS-6766 enhances antitumor efficacy of KRAS-G12C inhibitors through a vertical pathway inhibition strategy. Cancer Res. 2021;81:1263. [Google Scholar]
- 46.Reissig T.M., Ladigan-Badura S., Steinberg A., Maghnouj A., Li T., Verdoodt B., et al. Lasting response by vertical inhibition with cetuximab and trametinib in KRAS-mutated colorectal cancer patient-derived xenografts. Mol Oncol. 2023;17:2396–2414. doi: 10.1002/1878-0261.13510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Portelinha A., Thompson S., Smith R.A., Da Silva Ferreira M., Asgari Z., Knezevic A., et al. ASN007 is a selective ERK1/2 inhibitor with preferential activity against RAS-and RAF-mutant tumors. Cell Rep Med. 2021;2 doi: 10.1016/j.xcrm.2021.100350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodwin C.M., Waters A.M., Klomp J.E., Javaid S., Bryant K.L., Stalnecker C.A., et al. Combination therapies with CDK4/6 Inhibitors to treat KRAS-mutant pancreatic cancer. Cancer Res. 2023;83:141–157. doi: 10.1158/0008-5472.CAN-22-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schram A.M., Subbiah V., Sullivan R., Cosman R., Liu J., Sbar E.I., et al. Abstract CT031: a first-in-human, phase 1a/1b, open-label, dose-escalation and expansion study to investigate the safety, pharmacokinetics, and antitumor activity of the RAF dimer inhibitor BGB-3245 in patients with advanced or refractory tumors. Cancer Res. 2023;83:CT031. [Google Scholar]
- 50.Rasco D.W., Medina T., Corrie P., Pavlick A.C., Middleton M.R., Lorigan P., et al. Phase 1 study of the pan-RAF inhibitor tovorafenib in patients with advanced solid tumors followed by dose expansion in patients with metastatic melanoma. Cancer Chemother Pharmacol. 2023;92:15–28. doi: 10.1007/s00280-023-04544-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Solomon B., Gao B., Subbiah V., Millward M., Rosen L., Desai J., et al. Abstract CT033: safety, pharmacokinetics, and antitumor activity findings from a phase 1b, open-label, dose-escalation and expansion study investigating RAF dimer inhibitor lifirafenib in combination with MEK inhibitor mirdametinib in patients with advanced or refractory solid tumors. Cancer Res. 2023;83:CT033. [Google Scholar]
- 52.Lebbe C., Long G.V., Robert C., Hamid O., Atkinson V.G., Shoushtari A.N., et al. LBA40 Phase II study of multiple LXH254 drug combinations in patients (pts) with unresectable/metastatic, BRAF V600- or NRAS-mutant melanoma. Ann Oncol. 2022;33:S1407. [Google Scholar]
- 53.Canon J., Rex K., Saiki A.Y., Mohr C., Cooke K., Bagal D., et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 54.Bond M.J., Chu L., Nalawansha D.A., Li K., Crews C.M. Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Cent Sci. 2020;6:1367–1375. doi: 10.1021/acscentsci.0c00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao Q., Ouyang W., Kang B., Han X., Xiong Y., Ding R., et al. Selective targeting of the oncogenic KRAS G12S mutant allele by CRISPR/Cas9 induces efficient tumor regression. Theranostics. 2020;10:5137–5153. doi: 10.7150/thno.42325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee W., Lee J.H., Jun S., Lee J.H., Bang D. Selective targeting of KRAS oncogenic alleles by CRISPR/Cas9 inhibits proliferation of cancer cells. Sci Rep. 2018;8 doi: 10.1038/s41598-018-30205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leidner R., Sanjuan Silva N., Huang H., Sprott D., Zheng C., Shih Y.P., et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N Engl J Med. 2022;386:2112–2119. doi: 10.1056/NEJMoa2119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu D., Chen Y., Jiang M., Wang J., Li Y., Ma K., et al. KRAS G12V neoantigen specific T cell receptor for adoptive T cell therapy against tumors. Nat Commun. 2023;14:6389. doi: 10.1038/s41467-023-42010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minehart J., Eguchi T., Morello A., Adusumilli P. OA14.03 Clinical rationale and preclinical evidence for chimeric antigen receptor (CAR) T cell therapy clinical trial in KRAS-mutant lung cancer. J Thorac Oncol. 2019;14:S244. [Google Scholar]
- 60.Asimgil H., Ertetik U., Çevik N.C., Ekizce M., Doğruöz A., Gökalp M., et al. Targeting the undruggable oncogenic KRAS: the dawn of hope. JCI Insight. 2022;7 doi: 10.1172/jci.insight.153688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Biswas S., Mandal G., Anadon C.M., Chaurio R.A., Lopez-Bailon L.U., Nagy M.Z., et al. Targeting intracellular oncoproteins with dimeric IgA promotes expulsion from the cytoplasm and immune-mediated control of epithelial cancers. Immunity. 2023;56:2570–2583.e6. doi: 10.1016/j.immuni.2023.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou Y., Zou Y., Yang M., Mei S., Liu X., Han H., et al. Highly potent, selective, biostable, and cell-permeable cyclic d-peptide for dual-targeting therapy of lung cancer. J Am Chem Soc. 2022;144:7117–7128. doi: 10.1021/jacs.1c12075. [DOI] [PubMed] [Google Scholar]
- 63.Tanada M., Tamiya M., Matsuo A., Chiyoda A., Takano K., Ito T., et al. Development of orally bioavailable peptides targeting an intracellular protein: from a hit to a clinical KRAS inhibitor. J Am Chem Soc. 2023;145:16610–16620. doi: 10.1021/jacs.3c03886. [DOI] [PubMed] [Google Scholar]
- 64.Sacher A., LoRusso P., Patel M.R., Miller W.H., Jr., Garralda E., Forster M.D., et al. Single-agent divarasib (GDC-6036) in solid tumors with a KRAS G12C Mutation. N Engl J Med. 2023;389:710–721. doi: 10.1056/NEJMoa2303810. [DOI] [PubMed] [Google Scholar]
- 65.Murciano-Goroff Y.R., Heist R.S., Kuboki Y., Koyama T., Ammakkanavar N.R., Hollebecque A., et al. Abstract CT028: a first-in-human phase 1 study of LY3537982, a highly selective and potent KRAS G12C inhibitor in patients with KRAS G12C-mutant advanced solid tumors. Cancer Res. 2023;83:CT028. [Google Scholar]
- 66.Li Z., Song Z., Zhao Y., Wang P., Jiang L., Gong Y., et al. D-1553 (garsorasib), a potent and selective inhibitor of KRAS(G12C) in patients with NSCLC: phase 1 study results. J Thorac Oncol. 2023;18:940–951. doi: 10.1016/j.jtho.2023.03.015. [DOI] [PubMed] [Google Scholar]
- 67.Li J., Zhao J., Cao B., Fang J., Li X., Wang M., et al. A phase I/II study of first-in-human trial of JAB-21822 (KRAS G12C inhibitor) in advanced solid tumors. J Clin Oncol. 2022;40:3089. [Google Scholar]
- 68.Zhou Q., Yang N., Zhao J., Zhao M., Yu Y., Yuan Y., et al. Abstract CT030: phase I study of IBI351 (GFH925) monotherapy in patients with advanced solid tumors: updated results of the phase I study. Cancer Res. 2023;83:CT030. [Google Scholar]
- 69.Heymach J., Kotecki N., Prenen H., Alonso G., Lindsay C., Barve M., et al. 665P first-in-human, phase Ia/b, dose-escalation/expansion study of KRAS G12C inhibitor BI 1823911, as monotherapy and combined with anticancer therapies, in patients (pts) with advanced or metastatic solid tumours harbouring a KRAS G12C mutation. Ann Oncol. 2023;34:S468. [Google Scholar]
- 70.Wang J., Martin-Romano P., Cassier P., Johnson M., Haura E., Lenox L., et al. Phase I study of JNJ-74699157 in patients with advanced solid tumors harboring the KRAS G12C mutation. Oncol. 2022;27:536-e53. doi: 10.1093/oncolo/oyab080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rojas C., Lugowska I., Juergens R., Sacher A., Weindler S., Sendur M., et al. 663P Safety and preliminary efficacy of the KRAS G12C inhibitor MK-1084 in solid tumors and in combination with pembrolizumab in NSCLC. Ann Oncol. 2023;34:S466–S467. [Google Scholar]
- 72.Zhu X., Lu Y., Guo J., Yan D., Li B., Chen X., et al. Abstract 5443: BPI-421286: a highly potent small molecule inhibitor targeting KRASG12C mutation. Cancer Res. 2022;82:5443. [Google Scholar]
- 73.Wu X., Song W., Cheng C., Liu Z., Li X., Cui Y., et al. Small molecular inhibitors for KRAS-mutant cancers. Front Immunol. 2023;14 doi: 10.3389/fimmu.2023.1223433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jänne P.A., Bigot F., Papadopoulos K., Eberst L., Sommerhalder D., Lebellec L., et al. Abstract PR014: preliminary safety and anti-tumor activity of RMC-6291, a first-in-class, tri-complex KRASG12C (ON) inhibitor, in patients with or without prior KRASG12C (OFF) inhibitor treatment. Mol Cancer Therapeut. 2023;22:PR014. [Google Scholar]
- 75.Menard M.J., Chow C., Chen K., Blaj C., Shifrin N.T., Courtney H., et al. Abstract 3475: RMC-9805, a first-in-class, mutant-selective, covalent and orally bioavailable KRASG12D(ON) inhibitor, promotes cancer-associated neoantigen recognition and synergizes with immunotherapy in preclinical models. Cancer Res. 2023;83:3475. [Google Scholar]
- 76.Zhou C., Li W., Song Z., Zhang Y., Zhang Y., Huang D., et al. LBA33 A first-in-human phase I study of a novel KRAS G12D inhibitor HRS-4642 in patients with advanced solid tumors harboring KRAS G12D mutation. Ann Oncol. 2023;34:S1273. [Google Scholar]
- 77.Jin T., Lavoie H., Sahmi M., David M., Hilt C., Hammell A., et al. RAF inhibitors promote RAS-RAF interaction by allosterically disrupting RAF autoinhibition. Nat Commun. 2017;8:1211. doi: 10.1038/s41467-017-01274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lavoie H., Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16:281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 79.Rajakulendran T., Sahmi M., Lefrançois M., Sicheri F., Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461:542–545. doi: 10.1038/nature08314. [DOI] [PubMed] [Google Scholar]
- 80.Modi V., Dunbrack R.L., Jr. Defining a new nomenclature for the structures of active and inactive kinases. Proc Natl Acad Sci U S A. 2019;116:6818–6827. doi: 10.1073/pnas.1814279116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karoulia Z., Gavathiotis E., Poulikakos P.I. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer. 2017;17:676–691. doi: 10.1038/nrc.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lakkaniga N.R., Balasubramaniam M., Zhang S., Frett B., Li H.Y. Structural characterization of the aurora kinase B "DFG-flip" using metadynamics. AAPS J. 2019;22:14. doi: 10.1208/s12248-019-0399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gagic Z., Ruzic D., Djokovic N., Djikic T., Nikolic K. In silico methods for design of kinase inhibitors as anticancer drugs. Front Chem. 2019;7:873. doi: 10.3389/fchem.2019.00873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liau N.P.D., Wendorff T.J., Quinn J.G., Steffek M., Phung W., Liu P., et al. Negative regulation of RAF kinase activity by ATP is overcome by 14-3-3-induced dimerization. Nat Struct Mol Biol. 2020;27:134–141. doi: 10.1038/s41594-019-0365-0. [DOI] [PubMed] [Google Scholar]
- 85.Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlic A., Rose P.W. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018;34:3755–3758. doi: 10.1093/bioinformatics/bty419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cook F.A., Cook S.J. Inhibition of RAF dimers: it takes two to tango. Biochem Soc Trans. 2021;49:237–251. doi: 10.1042/BST20200485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Simanshu D.K., Nissley D.V., McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Venkatanarayan A., Liang J., Yen I., Shanahan F., Haley B., Phu L., et al. CRAF dimerization with ARAF regulates KRAS-driven tumor growth. Cell Rep. 2022;38 doi: 10.1016/j.celrep.2022.110351. [DOI] [PubMed] [Google Scholar]
- 89.McCormick F. C-Raf in KRAS mutant cancers: a moving target. Cancer Cell. 2018;33:158–159. doi: 10.1016/j.ccell.2018.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lito P., Saborowski A., Yue J., Solomon M., Joseph E., Gadal S., et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697–710. doi: 10.1016/j.ccr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morgan C.W., Dale I.L., Thomas A.P., Hunt J., Chin J.W. Selective CRAF inhibition elicits transactivation. J Am Chem Soc. 2021;143:4600–4606. doi: 10.1021/jacs.0c11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Su W., Mukherjee R., Yaeger R., Son J., Xu J., Na N., et al. ARAF protein kinase activates RAS by antagonizing its binding to RASGAP NF1. Mol Cell. 2022;82:2443–2457.e7. doi: 10.1016/j.molcel.2022.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patel H., Yacoub N., Mishra R., White A., Long Y., Alanazi S., et al. Current advances in the treatment of BRAF-mutant melanoma. Cancers. 2020;12:482. doi: 10.3390/cancers12020482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Simanshu D.K., Philips M.R., Hancock J.F. Consensus on the RAS dimerization hypothesis: strong evidence for lipid-mediated clustering but not for G-domain-mediated interactions. Mol Cell. 2023;83:1210–1215. doi: 10.1016/j.molcel.2023.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kondo Y., Ognjenović J., Banerjee S., Karandur D., Merk A., Kulhanek K., et al. Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science. 2019;366:109–115. doi: 10.1126/science.aay0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jambrina P.G., Rauch N., Pilkington R., Rybakova K., Nguyen L.K., Kholodenko B.N., et al. Phosphorylation of RAF kinase dimers drives conformational changes that facilitate transactivation. Angew Chem Int Ed Engl. 2016;55:983–986. doi: 10.1002/anie.201509272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Röring M., Herr R., Fiala G.J., Heilmann K., Braun S., Eisenhardt A.E., et al. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J. 2012;31:2629–2647. doi: 10.1038/emboj.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Freeman A.K., Ritt D.A., Morrison D.K. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell. 2013;49:751–758. doi: 10.1016/j.molcel.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yuan J., Ng W.H., Lam P.Y.P., Wang Y., Xia H., Yap J., et al. The dimer-dependent catalytic activity of RAF family kinases is revealed through characterizing their oncogenic mutants. Oncogene. 2018;37:5719–5734. doi: 10.1038/s41388-018-0365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen S.H., Zhang Y., Van Horn R.D., Yin T., Buchanan S., Yadav V., et al. Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov. 2016;6:300–315. doi: 10.1158/2159-8290.CD-15-0896. [DOI] [PubMed] [Google Scholar]
- 101.Lavoie H., Thevakumaran N., Gavory G., Li J.J., Padeganeh A., Guiral S., et al. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat Chem Biol. 2013;9:428–436. doi: 10.1038/nchembio.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peng S.B., Henry J.R., Kaufman M.D., Lu W.P., Smith B.D., Vogeti S., et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell. 2015;28:384–398. doi: 10.1016/j.ccell.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 103.Miyamoto K., Sawa M. Development of highly sensitive biosensors of RAF dimerization in cells. Sci Rep. 2019;9:636. doi: 10.1038/s41598-018-37213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rohrer L., Spohr C., Beha C., Griffin R., Braun S., Halbach S., et al. Analysis of RAS and drug induced homo- and heterodimerization of RAF and KSR1 proteins in living cells using split Nanoluc luciferase. Cell Commun Signal. 2023;21:136. doi: 10.1186/s12964-023-01146-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kwon J.J., Hajian B., Bian Y., Young L.C., Amor A.J., Fuller J.R., et al. Structure-function analysis of the SHOC2-MRAS-PP1C holophosphatase complex. Nature. 2022;609:408–415. doi: 10.1038/s41586-022-04928-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liau N.P.D., Johnson M.C., Izadi S., Gerosa L., Hammel M., Bruning J.M., et al. Structural basis for SHOC2 modulation of RAS signalling. Nature. 2022;609:400–407. doi: 10.1038/s41586-022-04838-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hu J., Stites E.C., Yu H., Germino E.A., Meharena H.S., Stork P.J.S., et al. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154:1036–1046. doi: 10.1016/j.cell.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rushworth L.K., Hindley A.D., O'Neill E., Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol. 2006;26:2262–2272. doi: 10.1128/MCB.26.6.2262-2272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Weber C.K., Slupsky J.R., Kalmes H.A., Rapp U.R. Active RAS induces heterodimerization of CRAF and BRAF. Cancer Res. 2001;61:3595–3598. [PubMed] [Google Scholar]
- 110.Boussemart L., Girault I., Malka-Mahieu H., Mateus C., Routier E., Rubington M., et al. Secondary tumors arising in patients undergoing BRAF inhibitor therapy exhibit increased BRAF-CRAF heterodimerization. Cancer Res. 2016;76:1476–1484. doi: 10.1158/0008-5472.CAN-15-2900-T. [DOI] [PubMed] [Google Scholar]
- 111.Lamba S., Russo M., Sun C., Lazzari L., Cancelliere C., Grernrum W., et al. RAF suppression synergizes with MEK inhibition in KRAS mutant cancer cells. Cell Rep. 2014;8:1475–1483. doi: 10.1016/j.celrep.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 112.Boned Del Río I., Young L.C., Sari S., Jones G.G., Ringham-Terry B., Hartig N., et al. SHOC2 complex-driven RAF dimerization selectively contributes to ERK pathway dynamics. Proc Natl Acad Sci U S A. 2019;116:13330–13339. doi: 10.1073/pnas.1902658116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sulahian R., Kwon J.J., Walsh K.H., Pailler E., Bosse T.L., Thaker M., et al. Synthetic lethal interaction of SHOC2 depletion with MEK inhibition in RAS-driven cancers. Cell Rep. 2019;29:118–134.e8. doi: 10.1016/j.celrep.2019.08.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhao Y., Murciano-Goroff Y.R., Xue J.Y., Ang A., Lucas J., Mai T.T., et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. 2021;599:679–683. doi: 10.1038/s41586-021-04065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yao Z., Torres N.M., Tao A., Gao Y., Luo L., Li Q., et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28:370–383. doi: 10.1016/j.ccell.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yen I., Shanahan F., Lee J., Hong Y.S., Shin S.J., Moore A.R., et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature. 2021;594:418–423. doi: 10.1038/s41586-021-03515-1. [DOI] [PubMed] [Google Scholar]
- 117.Yao Z., Yaeger R., Rodrik-Outmezguine V.S., Tao A., Torres N.M., Chang M.T., et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548:234–238. doi: 10.1038/nature23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yaeger R., Corcoran R.B. Targeting alterations in the RAF-MEK pathway. Cancer Discov. 2019;9:329–341. doi: 10.1158/2159-8290.CD-18-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Foster S.A., Whalen D.M., Özen A., Wongchenko M.J., Yin J., Yen I., et al. Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell. 2016;29:477–493. doi: 10.1016/j.ccell.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 120.Ross J.S., Wang K., Chmielecki J., Gay L., Johnson A., Chudnovsky J., et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer. 2016;138:881–890. doi: 10.1002/ijc.29825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sievert A.J., Lang S.S., Boucher K.L., Madsen P.J., Slaunwhite E., Choudhari N., et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U S A. 2013;110:5957–5962. doi: 10.1073/pnas.1219232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Niu Y., Zhang Y., Yao X. Resistance mechanism of the oncogenic β3-αC deletion mutation in BRAF kinase to dabrafenib and vemurafenib revealed by molecular dynamics simulations and binding free energy calculations. Chem Biol Drug Des. 2019;93:177–187. doi: 10.1111/cbdd.13399. [DOI] [PubMed] [Google Scholar]
- 123.Jain P., Fierst T.M., Han H.J., Smith T.E., Vakil A., Storm P.B., et al. CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene. 2017;36:6348–6358. doi: 10.1038/onc.2017.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brummer T., McInnes C. RAF kinase dimerization: implications for drug discovery and clinical outcomes. Oncogene. 2020;39:4155–4169. doi: 10.1038/s41388-020-1263-y. [DOI] [PubMed] [Google Scholar]
- 125.Tutuka C.S.A., Andrews M.C., Mariadason J.M., Ioannidis P., Hudson C., Cebon J., et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol Cancer. 2017;16:112. doi: 10.1186/s12943-017-0684-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Beneker C.M., Rovoli M., Kontopidis G., Röring M., Galda S., Braun S., et al. Design and synthesis of type-IV inhibitors of BRAF kinase that block dimerization and overcome paradoxical MEK/ERK activation. J Med Chem. 2019;62:3886–3897. doi: 10.1021/acs.jmedchem.8b01288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.King A.J., Patrick D.R., Batorsky R.S., Ho M.L., Do H.T., Zhang S.Y., et al. Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res. 2006;66:11100–11105. doi: 10.1158/0008-5472.CAN-06-2554. [DOI] [PubMed] [Google Scholar]
- 128.Hoeflich K.P., Herter S., Tien J., Wong L., Berry L., Chan J., et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009;69:3042–3051. doi: 10.1158/0008-5472.CAN-08-3563. [DOI] [PubMed] [Google Scholar]
- 129.Nakamura A., Arita T., Tsuchiya S., Donelan J., Chouitar J., Carideo E., et al. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013;73:7043–7055. doi: 10.1158/0008-5472.CAN-13-1825. [DOI] [PubMed] [Google Scholar]
- 130.Sun Y., Alberta J.A., Pilarz C., Calligaris D., Chadwick E.J., Ramkissoon S.H., et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro Oncol. 2017;19:774–785. doi: 10.1093/neuonc/now261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bulfer S.L., Le Bourdonnec B., Zwicker J.D., Ahn Y.M., Al-Ani G., Al-Hashimi H., et al. DCC-3084, a RAF dimer inhibitor, broadly inhibits BRAF class I, II, III, BRAF fusions, and RAS-driven solid tumors leading to tumor regression in preclinical models. Cancer Res. 2023;83:4045. [Google Scholar]
- 132.Adamopoulos C., Ahmed T.A., Tucker M.R., Ung P.M.U., Xiao M., Karoulia Z., et al. Exploiting allosteric properties of RAF and MEK inhibitors to target therapy-resistant tumors driven by oncogenic BRAF signaling. Cancer Discov. 2021;11:1716–1735. doi: 10.1158/2159-8290.CD-20-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Han Y.C., Ng P.Y., Ogawa L.S., Yang S.N., Chen M., Ishiyama N., et al. Preclinical characterization of a brain penetrant RAF inhibitor, BDTX-4933, targeting oncogenic BRAF Class I/II/III and RAS mutations. Cancer Res. 2023;83:3415. [Google Scholar]
- 134.Tang Z., Yuan X., Du R., Cheung S.H., Zhang G., Wei J., et al. BGB-283, a novel RAF kinase and EGFR inhibitor, displays potent antitumor activity in BRAF-mutated colorectal cancers. Mol Cancer Therapeut. 2015;14:2187–2197. doi: 10.1158/1535-7163.MCT-15-0262. [DOI] [PubMed] [Google Scholar]
- 135.Yuan X., Tang Z., Du R., Yao Z., Cheung S.H., Zhang X., et al. RAF dimer inhibition enhances the antitumor activity of MEK inhibitors in K-RAS mutant tumors. Mol Oncol. 2020;14:1833–1849. doi: 10.1002/1878-0261.12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Saturno G., Lopes F., Niculescu-Duvaz I., Niculescu-Duvaz D., Zambon A., Davies L., et al. The paradox-breaking panRAF plus SRC family kinase inhibitor, CCT3833, is effective in mutant KRAS-driven cancers. Ann Oncol. 2021;32:269–278. doi: 10.1016/j.annonc.2020.10.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Briker L., Johnson M., Kamal A., Dummer R., Levesque M., Eichhoff O. Vertical inhibition of the MAPK pathway as potential treatment strategy in NRAS-mutant melanoma. Eur J Cancer. 2022;174:S52. [Google Scholar]
- 138.Chen Y., Sun H., Deng Y., Ma Y., Huang H., Liu Y., et al. The clinical and genomic distinctions of Class1/2/3 BRAF-mutant colorectal cancer and differential prognoses. Biomark Res. 2023;11:11. doi: 10.1186/s40364-022-00443-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McDermott U., Sharma S.V., Dowell L., Greninger P., Montagut C., Lamb J., et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci U S A. 2007;104:19936–19941. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shao W., Mishina Y.M., Feng Y., Caponigro G., Cooke V.G., Rivera S., et al. Antitumor properties of RAF709, a highly selective and potent inhibitor of RAF kinase dimers, in tumors driven by mutant RAS or BRAF. Cancer Res. 2018;78:1537–1548. doi: 10.1158/0008-5472.CAN-17-2033. [DOI] [PubMed] [Google Scholar]
- 141.Yao Z., Gao Y., Su W., Yaeger R., Tao J., Na N., et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med. 2019;25:284–291. doi: 10.1038/s41591-018-0274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang A., Liu J., Li X., Zou F., Qi Z., Qi S., et al. Discovery of a highly potent pan-RAF inhibitor IHMT-RAF-128 for cancer treatment. Eur J Pharmacol. 2023;952 doi: 10.1016/j.ejphar.2023.175752. [DOI] [PubMed] [Google Scholar]
- 143.Mason H., Scrace S., Testar R., Rainard J., Talab F., Poonawala R., et al. Abstract 5160: development of REDX05358, a novel highly selective and potent pan RAF inhibitor and a potential therapeutic for BRAF and RAS mutant tumors. Cancer Res. 2017;77:5160. [Google Scholar]
- 144.Zhang C., Spevak W., Zhang Y., Burton E.A., Ma Y., Habets G., et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–586. doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
- 145.Le K., Blomain E.S., Rodeck U., Aplin A.E. Selective RAF inhibitor impairs ERK1/2 phosphorylation and growth in mutant NRAS, vemurafenib-resistant melanoma cells. Pigment Cell Melanoma Res. 2013;26:509–517. doi: 10.1111/pcmr.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bonfill-Teixidor E., Iurlaro R., Handl C., Wichmann J., Arias A., Cuartas I., et al. Activity and resistance of a brain-permeable paradox breaker BRAF inhibitor in melanoma brain metastasis. Cancer Res. 2022;82:2552–2564. doi: 10.1158/0008-5472.CAN-21-4152. [DOI] [PubMed] [Google Scholar]
- 147.Wichmann J., Rynn C., Friess T., Petrig-Schaffland J., Kornacker M., Handl C., et al. Preclinical characterization of a next-generation brain permeable, paradox breaker BRAF inhibitor. Clin Cancer Res. 2022;28:770–780. doi: 10.1158/1078-0432.CCR-21-2761. [DOI] [PubMed] [Google Scholar]
- 148.Arora R., Linders J.T.M., Aci-Sèche S., Verheyen T., Van Heerde E., Brehmer D., et al. Design, synthesis and characterisation of a novel type II B-RAF paradox breaker inhibitor. Eur J Med Chem. 2023;250 doi: 10.1016/j.ejmech.2023.115231. [DOI] [PubMed] [Google Scholar]
- 149.Gunderwala A.Y., Nimbvikar A.A., Cope N.J., Li Z., Wang Z. Development of allosteric BRAF peptide inhibitors targeting the dimer interface of BRAF. ACS Chem Biol. 2019;14:1471–1480. doi: 10.1021/acschembio.9b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tkacik E., Li K., Gonzalez-Del Pino G., Ha B.H., Vinals J., Park E., et al. Structure and RAF family kinase isoform selectivity of type II RAF inhibitors tovorafenib and naporafenib. J Biol Chem. 2023;299 doi: 10.1016/j.jbc.2023.104634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chapman P.B., Hauschild A., Robert C., Haanen J.B., Ascierto P., Larkin J., et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sun C., Wang L., Huang S., Heynen G.J., Prahallad A., Robert C., et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118–122. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 154.Kopetz S., Desai J., Chan E., Hecht J.R., O'Dwyer P.J., Maru D., et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33:4032–4038. doi: 10.1200/JCO.2015.63.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hyman D.M., Puzanov I., Subbiah V., Faris J.E., Chau I., Blay J.Y., et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373:726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Su F., Viros A., Milagre C., Trunzer K., Bollag G., Spleiss O., et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Larkin J., Ascierto P.A., Dréno B., Atkinson V., Liszkay G., Maio M., et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 158.Long G.V., Stroyakovskiy D., Gogas H., Levchenko E., de Braud F., Larkin J., et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–1888. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 159.Dummer R., Flaherty K.T., Robert C., Arance A., de Groot J.W.B., Garbe C., et al. COLUMBUS 5-Year update: a randomized, open-label, phase III trial of encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. J Clin Oncol. 2022;40:4178–4188. doi: 10.1200/JCO.21.02659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ramurthy S., Taft B.R., Aversa R.J., Barsanti P.A., Burger M.T., Lou Y., et al. Design and discovery of N-(3-(2-(2-hydroxyethoxy)-6-morpholinopyridin-4-yl)-4-methylphenyl)-2-(trifluoromethyl)isonicotinamide, a selective, efficacious, and well-tolerated RAF inhibitor targeting RAS mutant cancers: the path to the clinic. J Med Chem. 2020;63:2013–2027. doi: 10.1021/acs.jmedchem.9b00161. [DOI] [PubMed] [Google Scholar]
- 161.Hatzivassiliou G., Song K., Yen I., Brandhuber B.J., Anderson D.J., Alvarado R., et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 162.Wang X., Kim J. Conformation-specific effects of Raf kinase inhibitors. J Med Chem. 2012;55:7332–7341. doi: 10.1021/jm300613w. [DOI] [PubMed] [Google Scholar]
- 163.Janku F., Iyer G., Spreafico A., Yamamoto N., Bang Y.J., Elez E., et al. A phase I study of LXH254 in patients (pts) with advanced solid tumors harboring MAPK pathway alterations. J Clin Oncol. 2018;36:2586. [Google Scholar]
- 164.Desai J., Gan H., Barrow C., Jameson M., Atkinson V., Haydon A., et al. Phase I, open-label, dose-escalation/dose-expansion study of lifirafenib (BGB-283), an RAF family kinase inhibitor, in patients with solid tumors. J Clin Oncol. 2020;38:2140–2150. doi: 10.1200/JCO.19.02654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sullivan R.J., Hollebecque A., Flaherty K.T., Shapiro G.I., Rodon Ahnert J., Millward M.J., et al. A phase I study of LY3009120, a pan-RAF inhibitor, in patients with advanced or metastatic cancer. Mol Cancer Therapeut. 2020;19:460–467. doi: 10.1158/1535-7163.MCT-19-0681. [DOI] [PubMed] [Google Scholar]
- 166.Cotto-Rios X.M., Agianian B., Gitego N., Zacharioudakis E., Giricz O., Wu Y., et al. Inhibitors of BRAF dimers using an allosteric site. Nat Commun. 2020;11:4370. doi: 10.1038/s41467-020-18123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang T.S., Lee C., Severson P., Pelham R.J., Williams R., Miller N.L.G. Exarafenib (KIN-2787) is a potent, selective pan-RAF inhibitor with activity in preclinical models of BRAF class II/III mutant and NRAS mutant melanoma. Cancer Res. 2023;83:4927. [Google Scholar]
- 168.Dickson M.A., Gordon M.S., Edelman G., Bendell J.C., Kudchadkar R.R., LoRusso P.M., et al. Phase I study of XL281 (BMS-908662), a potent oral RAF kinase inhibitor, in patients with advanced solid tumors. Invest N Drugs. 2015;33:349–356. doi: 10.1007/s10637-014-0191-5. [DOI] [PubMed] [Google Scholar]
- 169.Ahnert J.R., Yaeger R., Tsai F.Y.C., Janku F., Butowski N.A., Allen C.E., et al. Safety and efficacy of the novel BRAF inhibitor FORE8394 in patients with advanced solid and CNS tumors: results from a phase 1/2a study. J Clin Oncol. 2023;41:3006. [Google Scholar]
- 170.Okimoto R.A., Lin L., Olivas V., Chan E., Markegard E., Rymar A., et al. Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer. Proc Natl Acad Sci U S A. 2016;113:13456–13461. doi: 10.1073/pnas.1610456113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Koumaki K., Kontogianni G., Kosmidou V., Pahitsa F., Kritsi E., Zervou M., et al. BRAF paradox breakers PLX8394, PLX7904 are more effective against BRAFV600Ε CRC cells compared with the BRAF inhibitor PLX4720 and shown by detailed pathway analysis. Biochim Biophys Acta, Mol Basis Dis. 2021;1867 doi: 10.1016/j.bbadis.2020.166061. [DOI] [PubMed] [Google Scholar]
- 172.Wright K., Krzykwa E., Greenspan L., Chi S., Yeo K.K., Prados M., et al. CTNI-19. Phase I trial of DAY101 in pediatric patients with radiographically recurrent or progressive low-grade glioma (LGG) Neuro Oncol. 2020;22:ii46. [Google Scholar]
- 173.Wright K., Kline C., Abdelbaki M., Ebb D., Sayour E., Elster J., et al. PNOC014: phase IB study results of DAY101 (tovorafenib) for children with low-grade gliomas (LGGs) and other RAS/RAF/MEK/ERK pathway-activated tumors. Neuro Oncol. 2022;24:vii84. [Google Scholar]
- 174.Kilburn L., Landi D., Leary S., Ziegler D., Baxter P., Franson A., et al. FIREFLY-1 (PNOC026): phase 2 study of pan-RAF inhibitor tovorafenib in pediatric and young adult patients with RAF-altered recurrent or progressive low-grade glioma or advanced solid tumors. Neuro Oncol. 2022;24:vii89. [Google Scholar]
- 175.Khuong-Quang D.-A., Nysom K., Landi D.B., Ziegler D.S., Driever P.H., Leary S., et al. Clinical activity of pan-RAF inhibitor tovorafenib in the registrational pediatric low-grade glioma arm of the phase 2 FIREFLY-1 (PNOC026) study. J Clin Oncol. 2023;41 [Google Scholar]
- 176.Janku F., Kim T.M., Iyer G., Spreafico A., Elez E., de Jonge M., et al. First-in-human study of naporafenib (LXH254) with or without spartalizumab in adult patients with advanced solid tumors harboring MAPK signaling pathway alterations. Eur J Cancer. 2024;196 doi: 10.1016/j.ejca.2023.113458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.de Braud F., Dooms C., Heist R.S., Lebbe C., Wermke M., Gazzah A., et al. Initial evidence for the efficacy of naporafenib in combination with trametinib in NRAS-mutant melanoma: results from the expansion arm of a phase Ib, open-label study. J Clin Oncol. 2023;41:2651–2660. doi: 10.1200/JCO.22.02018. [DOI] [PubMed] [Google Scholar]
- 178.Shin S.J., Lee J., Kim T.M., Kim J.S., Kim Y.J., Hong Y.S., et al. A phase Ib trial of belvarafenib in combination with cobimetinib in patients with advanced solid tumors: interim results of dose-escalation and patients with NRAS-mutant melanoma of dose-expansion. J Clin Oncol. 2021;39:3007. [Google Scholar]
- 179.Kim T.W., Lee J., Kim T.M., Shin S.J., Han S.W., Kim J.S., et al. 529P A phase Ib trial of belvarafenib in combination with cobimetinib in patients (pts) with RAS- or RAF- mutated (m) solid tumors: updated safety data and indication-specific efficacy results. Ann Oncol. 2021;32:S595. [Google Scholar]
- 180.Park S., Kim T.M., Cho S.Y., Kim S., Oh Y., Kim M., et al. Combined blockade of polo-like kinase and pan-RAF is effective against NRAS-mutant non-small cell lung cancer cells. Cancer Lett. 2020;495:135–144. doi: 10.1016/j.canlet.2020.09.018. [DOI] [PubMed] [Google Scholar]
- 181.Knickelbein K., Zhang L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015;2:4–12. doi: 10.1016/j.gendis.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kim Y.Y., Park H., Song T., Choi K., Dolton M., Mao J., et al. Belvarafenib penetrates the BBB and shows potent antitumor activity in a murine melanoma brain metastasis model. Clin Exp Metastasis. 2023;40:137–148. doi: 10.1007/s10585-023-10198-7. [DOI] [PubMed] [Google Scholar]
- 183.Prahallad A., Sun C., Huang S., Di Nicolantonio F., Salazar R., Zecchin D., et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 184.Corcoran R.B., André T., Atreya C.E., Schellens J.H.M., Yoshino T., Bendell J.C., et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAF(V600E)-mutant colorectal cancer. Cancer Discov. 2018;8:428–443. doi: 10.1158/2159-8290.CD-17-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lai L.P., Fer N., Burgan W., Wall V.E., Xu B., Soppet D., et al. Classical RAS proteins are not essential for paradoxical ERK activation induced by RAF inhibitors. Proc Natl Acad Sci U S A. 2022;119 doi: 10.1073/pnas.2113491119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tanaka N., Lin J.J., Li C., Ryan M.B., Zhang J., Kiedrowski L.A., et al. Clinical acquired resistance to KRAS(G12C) inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021;11:1913–1922. doi: 10.1158/2159-8290.CD-21-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Xue J.Y., Zhao Y., Aronowitz J., Mai T.T., Vides A., Qeriqi B., et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. 2020;577:421–425. doi: 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Awad M.M., Liu S., Rybkin, Arbour K.C., Dilly J., Zhu V.W., et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N Engl J Med. 2021;384:2382–2393. doi: 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Koga T., Suda K., Fujino T., Ohara S., Hamada A., Nishino M., et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies: insights from in vitro experiments. J Thorac Oncol. 2021;16:1321–1332. doi: 10.1016/j.jtho.2021.04.015. [DOI] [PubMed] [Google Scholar]
- 190.Freed-Pastor W.A., Aguirre A.J. Getting a handle on KRAS inhibitor resistance with hapten-mediated anti-tumor immunity. Cancer Cell. 2022;40:908–910. doi: 10.1016/j.ccell.2022.08.018. [DOI] [PubMed] [Google Scholar]
- 191.Zhang Z., Rohweder P.J., Ongpipattanakul C., Basu K., Bohn M.F., Dugan E.J., et al. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell. 2022;40:1060–1069.e7. doi: 10.1016/j.ccell.2022.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ryan M.B., Fece de la Cruz F., Phat S., Myers D.T., Wong E., Shahzade H.A., et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRAS(G12C) inhibition. Clin Cancer Res. 2020;26:1633–1643. doi: 10.1158/1078-0432.CCR-19-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Amodio V., Yaeger R., Arcella P., Cancelliere C., Lamba S., Lorenzato A., et al. EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov. 2020;10:1129–1139. doi: 10.1158/2159-8290.CD-20-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fakih M.G., Salvatore L., Esaki T., Modest D.P., Lopez-Bravo D.P., Taieb J., et al. Sotorasib plus panitumumab in refractory colorectal cancer with mutated KRAS G12C. N Engl J Med. 2023;389:2125–2139. doi: 10.1056/NEJMoa2308795. [DOI] [PubMed] [Google Scholar]
- 195.Desai J., Alonso G., Kim S.H., Cervantes A., Karasic T., Medina L., et al. Divarasib plus cetuximab in KRAS G12C-positive colorectal cancer: a phase 1b trial. Nat Med. 2023;30:271–278. doi: 10.1038/s41591-023-02696-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Drosten M., Barbacid M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell. 2020;37:543–550. doi: 10.1016/j.ccell.2020.03.013. [DOI] [PubMed] [Google Scholar]
- 197.Cunniff E.G.C., Zhang J., Chouitar J., Mettetal J., Nakamura K., Arita T., et al. Use of combination treatment with the investigational RAF kinase inhibitor MLN2480 and the investigational MEK kinase inhibitor TAK-733 on the growth of BRAF-mutant and RAS-mutant preclinical models of melanoma and CRC. J Clin Oncol. 2013;31 [Google Scholar]
- 198.Bekele R.T., Samant A.S., Nassar A.H., So J., Garcia E.P., Curran C.R., et al. RAF1 amplification drives a subset of bladder tumors and confers sensitivity to MAPK-directed therapeutics. J Clin Invest. 2021;131 doi: 10.1172/JCI147849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kohtamäki L., Arjama M., Mäkelä S., Ianevski P., Välimäki K., Juteau S., et al. High-throughput ex vivo drug testing identifies potential drugs and drug combinations for NRAS-positive malignant melanoma. Transl Oncol. 2022;15 doi: 10.1016/j.tranon.2021.101290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Garcia N., Del Pozo V., Yohe M.E., Goodwin C.M., Shackleford T.J., Wang L., et al. Vertical inhibition of the RAF-MEK-ERK cascade induces myogenic differentiation, apoptosis, and tumor regression in H/NRAS(Q61X) mutant rhabdomyosarcoma. Mol Cancer Therapeut. 2022;21:170–183. doi: 10.1158/1535-7163.MCT-21-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhou Z.W., Ambrogio C., Bera A.K., Li Q., Li X.X., Li L., et al. KRAS(Q61H) preferentially signals through MAPK in a RAF dimer-dependent manner in non-small cell lung cancer. Cancer Res. 2020;80:3719–3731. doi: 10.1158/0008-5472.CAN-20-0448. [DOI] [PubMed] [Google Scholar]
- 202.Li T., Kikuchi O., Zhou J., Wang Y., Pokharel B., Bastl K., et al. Developing SHP2-based combination therapy for KRAS-amplified cancer. JCI Insight. 2023;8 doi: 10.1172/jci.insight.152714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chen S.H., Gong X., Zhang Y., Van Horn R.D., Yin T., Huber L., et al. RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene. 2018;37:821–832. doi: 10.1038/onc.2017.384. [DOI] [PubMed] [Google Scholar]
- 204.Park S.M., Hwang C.Y., Choi J., Joung C.Y., Cho K.H. Feedback analysis identifies a combination target for overcoming adaptive resistance to targeted cancer therapy. Oncogene. 2020;39:3803–3820. doi: 10.1038/s41388-020-1255-y. [DOI] [PubMed] [Google Scholar]
- 205.Wang Z., Yin M., Chu P., Lou M. STAT3 inhibitor sensitized KRAS-mutant lung cancers to RAF inhibitor by activating MEK/ERK signaling pathway. Aging (Albany NY) 2019;11:7187–7196. doi: 10.18632/aging.102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Thakuri P.S., Gupta M., Joshi R., Singh S., Tavana H. Synergistic inhibition of kinase pathways overcomes resistance of colorectal cancer spheroids to cyclic targeted therapies. ACS Pharmacol Transl Sci. 2019;2:275–284. doi: 10.1021/acsptsci.9b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhao X., Wang X., Fang L., Lan C., Zheng X., Wang Y., et al. A combinatorial strategy using YAP and pan-RAF inhibitors for treating KRAS-mutant pancreatic cancer. Cancer Lett. 2017;402:61–70. doi: 10.1016/j.canlet.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 208.Suzuki R., Kitamura Y., Ogiya D., Ogawa Y., Kawada H., Ando K. Anti-tumor activity of the pan-RAF inhibitor TAK-580 in combination with KPT-330 (selinexor) in multiple myeloma. Int J Hematol. 2022;115:233–243. doi: 10.1007/s12185-021-03244-1. [DOI] [PubMed] [Google Scholar]
- 209.Suzuki R., Kitamura Y., Nakamura Y., Akashi H., Ogawa Y., Kawada H., et al. Anti-tumor activities of the new oral pan-RAF inhibitor, TAK-580, used as monotherapy or in combination with novel agents in multiple myeloma. Oncotarget. 2020;11:3984–3997. doi: 10.18632/oncotarget.27775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shirazi F., Jones R.J., Singh R.K., Zou J., Kuiatse I., Berkova Z., et al. Activating KRAS, NRAS, and BRAF mutants enhance proteasome capacity and reduce endoplasmic reticulum stress in multiple myeloma. Proc Natl Acad Sci U S A. 2020;117:20004–20014. doi: 10.1073/pnas.2005052117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Khoury J.D., Tashakori M., Yang H., Loghavi S., Wang Y., Wang J., et al. Pan-RAF inhibition shows anti-leukemic activity in RAS-mutant acute myeloid leukemia cells and potentiates the effect of sorafenib in cells with FLT3 mutation. Cancers. 2020;12:3511. doi: 10.3390/cancers12123511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Park J., Park H., Byun J.M., Hong J., Shin D.Y., Koh Y., et al. Pan-RAF inhibitor LY3009120 is highly synergistic with low-dose cytarabine, but not azacitidine, in acute myeloid leukemia with RAS mutations. Oncol Lett. 2021;22:745. doi: 10.3892/ol.2021.13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Colomer C., Margalef P., Villanueva A., Vert A., Pecharroman I., Solé L., et al. IKKα kinase regulates the DNA damage response and drives chemo-resistance in cancer. Mol Cell. 2019;75:669–682.e5. doi: 10.1016/j.molcel.2019.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Temko D., Tomlinson I.P.M., Severini S., Schuster-Böckler B., Graham T.A. The effects of mutational processes and selection on driver mutations across cancer types. Nat Commun. 2018;9:1857. doi: 10.1038/s41467-018-04208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wu J., Ivanov A.I., Fisher P.B., Fu Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. Elife. 2016;5 doi: 10.7554/eLife.10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yang Z., Liang S.Q., Saliakoura M., Yang H., Vassella E., Konstantinidou G., et al. Synergistic effects of FGFR1 and PLK1 inhibitors target a metabolic liability in KRAS-mutant cancer. EMBO Mol Med. 2021;13 doi: 10.15252/emmm.202013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Luo J., Emanuele M.J., Li D., Creighton C.J., Schlabach M.R., Westbrook T.F., et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Yu C., Luo D., Yu J., Zhang M., Zheng X., Xu G., et al. Genome-wide CRISPR-cas9 knockout screening identifies GRB7 as a driver for MEK inhibitor resistance in KRAS mutant colon cancer. Oncogene. 2022;41:191–203. doi: 10.1038/s41388-021-02077-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ram P.T., Iyengar R. G protein coupled receptor signaling through the src and stat3 pathway: role in proliferation and transformation. Oncogene. 2001;20:1601–1606. doi: 10.1038/sj.onc.1204186. [DOI] [PubMed] [Google Scholar]
- 220.Wee S., Jagani Z., Xiang K.X., Loo A., Dorsch M., Yao Y.M., et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–4293. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 221.Kumar S.K., Jacobus S.J., Cohen A.D., Weiss M., Callander N., Singh A.K., et al. Carfilzomib or bortezomib in combination with lenalidomide and dexamethasone for patients with newly diagnosed multiple myeloma without intention for immediate autologous stem-cell transplantation (ENDURANCE): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2020;21:1317–1330. doi: 10.1016/S1470-2045(20)30452-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Mosca L., Ilari A., Fazi F., Assaraf Y.G., Colotti G. Taxanes in cancer treatment: activity, chemoresistance and its overcoming. Drug Resist Updates. 2021;54 doi: 10.1016/j.drup.2020.100742. [DOI] [PubMed] [Google Scholar]
- 223.Veerman G.D.M., Hussaarts K., Jansman F.G.A., Koolen S.W.L., van Leeuwen R.W.F., Mathijssen R.H.J. Clinical implications of food–drug interactions with small-molecule kinase inhibitors. Lancet Oncol. 2020;21:e265–e279. doi: 10.1016/S1470-2045(20)30069-3. [DOI] [PubMed] [Google Scholar]
- 224.Sriraman S.K., Geraldo V., Luther E., Degterev A., Torchilin V. Cytotoxicity of PEGylated liposomes co-loaded with novel pro-apoptotic drug NCL-240 and the MEK inhibitor cobimetinib against colon carcinoma in vitro. J Control Release. 2015;220:160–168. doi: 10.1016/j.jconrel.2015.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.He Z.D., Zhang M., Wang Y.H., He Y., Wang H.R., Chen B.F., et al. Anti-PD-L1 mediating tumor-targeted codelivery of liposomal irinotecan/JQ1 for chemo-immunotherapy. Acta Pharmacol Sin. 2021;42:1516–1523. doi: 10.1038/s41401-020-00570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]