

Abstract
We developed an in vitro translation extract from Krebs-2 cells that translates the entire open reading frame of the hepatitis C virus (HCV) strain H77 and properly processes the viral protein precursors when supplemented with canine microsomal membranes (CMMs). Translation of the C-terminal portion of the viral polyprotein in this system is documented by the synthesis of NS5B. Evidence for posttranslational modification of the viral proteins, the N-terminal glycosylation of E1 and the E2 precursor (E2-p7), and phosphorylation of NS5A is presented. With the exception of NS3, efficient generation of all virus-specific proteins is CMM dependent. A time course of the appearance of HCV products indicates that the viral polyprotein is cleaved cotranslationally. A competitive inhibitor of the NS3 protease inhibited accumulation of NS3, NS4B, NS5A, and NS5B, but not that of NS2 or structural proteins. CMMs also stabilized HCV mRNA during translation. Finally, the formyl-[35S]methionyl moiety of the initiator tRNAMet was incorporated exclusively into the core protein portion of the polyprotein, demonstrating that translation initiation in this system occurs with high fidelity.
Hepatitis C virus (HCV) is an enveloped virus that belongs to the genus Hepacivirus in the family Flaviviridae (which also includes flaviviruses and pestiviruses) and is the leading cause of chronic hepatitis and liver cirrhosis in humans in the developed world (77). The genome of HCV is a ∼9.6-kb-long positive-strand RNA that is translated into a polyprotein of approximately 3,010 amino acids (58). For some HCV strains, evidence for an alternative open reading frame that overlaps the core protein gene has also been reported (83, 88). Translation of the HCV RNA is accomplished by binding of ribosomes to an internal ribosome entry site (IRES) (80). A salient feature of the HCV IRES is its ability to recruit ribosomes with the aid of only two canonical initiation factors (eIF3 and eIF2) (29, 55), in contrast to most other IRESs, e.g., IRESs from picornaviruses, which also require the eIF4 initiation factors (52, 54, 73).
HCV does not replicate in cultured cells. However, viral protein detection and mapping were achieved by using transient expression systems (1, 18, 20) and, more recently, the replicon system (38, 57). These studies have suggested that the viral polyprotein is cleaved co- and posttranslationally at specific sites into at least 10 polypeptides ordered from the N terminus as follows: C-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B. Cleavages within the structural region and at the p7/NS2 junction are thought to be mediated by host cell signal peptidase(s), which is located in the lumen of the endoplasmic reticulum (ER) and cleaves behind stretches of hydrophobic amino acids. The first cleavage product (C; core) is highly basic and constitutes the major component of the nucleocapsid. Envelope proteins E1 and E2 are type I transmembrane glycoproteins. Processing of the NS region is mediated by two overlapping virus-specific proteases. The NS2-NS3 zinc-dependent autoproteinase is essential for cleavage at the NS2/3 site. The NS3 serine protease utilizes NS4A as a cofactor to efficiently cleave the polyprotein at all sites downstream of the NS3 carboxy terminus, i.e., at the NS3/4A, NS4A/4B, NS4B/5A, and NS5A/5B sites (2). The NS3 protein also functions as an RNA helicase, which together with NS5B (an RNA-dependent RNA polymerase), other NS proteins, and host factors forms a membrane-associated RNA replication complex (14, 16, 17, 48).
Our understanding of HCV gene expression would greatly benefit from the development of a cell-free system, akin to those developed for picornaviruses, that allows coupled translation-replication of the viral genome (3, 45, 74). Systems developed so far translate HCV RNA with low fidelity, yielding only complete polypeptides of the structural proteins and a number of aberrant products (23, 24). No NS5B could be detected, making these systems of little use for the studies of coupled translation-replication. Synthesis of predominantly structural proteins was also observed in translation systems for other members of the family Flaviviridae (40, 46, 76, 85). Here, we describe an extract from Krebs-2 cells that in the presence of canine microsomal membranes (CMMs) translates HCV RNA completely and accurately. CMMs are known to mediate processing, such as signal peptide cleavage, membrane insertion, translocation, and core glycosylation of the proteins (84). CMM requirements for processing of HCV and flavivirus proteins in vitro have also been demonstrated (20, 23, 36, 72). However, we show for the first time that CMMs are required to support not only signal peptidase activity but also most NS3 protease-mediated cleavages of the HCV polyprotein. In addition, our data suggest that CMMs stabilize HCV mRNA during translation.
MATERIALS AND METHODS
Materials.
Plasmid pCV-H77C (a kind gift of Jens Bukh and Robert Purcell, National Institutes of Health, Bethesda, MD) contains the full-length genotype 1a HCV cDNA under the transcriptional control of the T7 promoter (89). Micrococcal-nuclease-treated rabbit reticulocyte lysate (RRL) and CMMs were purchased from Promega. Mouse monoclonal antibody against NS5B (5B-3B1) was kindly provided by Darius Moradpour (University Hospital Freiburg, Freiburg, Germany) (47). Subgenomic HCV replicon, pFK I389/NS3-3′/wt, and rabbit antisera against NS3 and NS5A were a kind gift from Ralf Bartenschlager (Johannes Gutenberg University Mainz, Mainz, Germany) (38). A rabbit antiserum against the core protein (RR8) was kindly provided by Michinori Kohara (Tokyo Metropolitan Institute of Medical Science, Bunkyo-ku, Tokyo, Japan) (26). E2-specific rat monoclonal antibody 6/1a was kindly provided by Jane McKeating (The Rockefeller University, New York, NY) (15). E1-specific mouse monoclonal antibody 3D5-C3 was kindly provided by Michael Houghton (Chiron Corporation, Emeryville, CA) (65). Compound A was kindly provided by Daniel Lamarre and Michael G. Cordingley [Boehringer Ingelheim (Canada) Ltd., Laval, Quebec, Canada] (51, 79).
Preparation of HCV RNAs and f[35S]Met-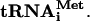
pCV-H77C was linearized with XbaI and treated with mung bean nuclease (New England Biolabs) to remove the unpaired vector-derived nucleotides. DNA templates for subgenomic RNA synthesis were generated by PCR amplification of pCV-H77C with the forward primer 5′-GCATGCTAATACGACTCACTATAGCC-3′. The reverse primers used to amplify the regions of pCV-H77C encoding proteins C, C-E1, C-E2, C-p7, and C-NS2 were 5′-GCGCACTTGTTAGGCTGAAGCGGGC-3′, 5′-GACGTGGGTTTACGCGTCGACGCC-3′, 5′-GAGGTTCTCCAATTACGCCTCCGCTTGGG-3′, 5′-GGCCACCTCCGTGTCCTATGCGTATGC-3′, and 5′-GTACGCCGTGATGGGCTACAGCAACC-3′, respectively (each primer contained a stop codon, which is shown in boldface). pT7HCV-Luc-3′UTR encoding luciferase under the control of the HCV IRES and 3′ untranslated region (3′ UTR) was prepared as follows: pFK I389/NS3-3′/wt (38) was used as a template for PCR. A KpnI/BamHI fragment containing the HCV 3′UTR (231 nucleotides) was amplified using primers 5′-GCGCGGTACCACGGGGAGCTAAACACTCCA-3′ (forward) and 5′-GCGCGGATCCTCTAGACATGATCTGCAGAGAGGCC-3′ (reverse) (the underlined nucleotides correspond to the restriction sites). After digestion, the fragment was ligated together with pT7HCVluc (71) linearized with KpnI and BamHI. pT7HCV-Luc-3′UTR was linearized with BamHI. The linearized plasmids and subgenomic DNA templates were transcribed by T7 RNA polymerase. Synthesis of uncapped RNA transcripts was performed at 37°C for 2.5 h with the RiboMAX system (Promega) as recommended by the manufacturer. Full-size and subgenomic HCV RNAs (referred to as HCV RNA-C, C-E1, C-E2, C-p7, and C-NS2, as appropriate) and HCV-Luc-3′ UTR mRNA were further purified by treatment with RNase-free DNase (Roche), phenol-chloroform extraction, 2 M LiCl precipitation, and CHROMA SPIN-100 column (Clontech) chromatography. RNA integrity was verified by electrophoresis on a formaldehyde-denaturing agarose gel. To prepare f[35S]Met-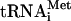 , total tRNA isolated from calf liver (100 μg; Novogen) was aminoacylated with [35S]methionine, translation grade (1 mCi/ml final concentration; 1,200 Ci/mmol), and formylated using leucovorin and a preparation of aminoacyl-tRNA synthetases from Escherichia coli (0.9-mg/ml final concentration; Sigma). The reaction was carried out in a total volume of 0.4 ml as specified previously (76), with the exception that deacylation of the unformylated tRNA was omitted and f[35S]Met-
, total tRNA isolated from calf liver (100 μg; Novogen) was aminoacylated with [35S]methionine, translation grade (1 mCi/ml final concentration; 1,200 Ci/mmol), and formylated using leucovorin and a preparation of aminoacyl-tRNA synthetases from Escherichia coli (0.9-mg/ml final concentration; Sigma). The reaction was carried out in a total volume of 0.4 ml as specified previously (76), with the exception that deacylation of the unformylated tRNA was omitted and f[35S]Met-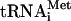 was purified by acidic phenol extraction rather than by chromatography on DEAE-cellulose (68). A specific activity of 2 × 105 cpm/μg RNA was obtained.
was purified by acidic phenol extraction rather than by chromatography on DEAE-cellulose (68). A specific activity of 2 × 105 cpm/μg RNA was obtained.
Preparation of cell extracts.
Krebs-2 ascites cells were propagated in mice, and translation extract was prepared essentially as described previously (75). To deplete the endogenous methionine pool, washed cells were suspended in methionine-free Dulbecco's modified minimal essential medium (DMEM) (ICN Biochemicals Inc.) that was supplemented with 4 mM l-glutamine, 2% dialyzed fetal bovine serum (FBS) (Invitrogen Corp.), 0.03% sodium bicarbonate, 20 U/ml penicillin, and 20 μg/ml streptomycin. The cells were maintained in suspension (5 × 106 cells/ml) at 37°C for 2 h. This procedure was found to significantly enhance protein labeling in vitro. Following incubation, the cells were collected by centrifugation and washed (three times) with an isotonic buffer (3) (buffer A [75]). After the final wash, the compact cell pellet was resuspended in 2 volumes of a buffer containing 25 mM HEPES-KOH, pH 7.3, 50 mM KCl, 1.5 mM MgCl2, 1 mM dithiothreitol (DTT) (buffer B [75]). The cells were allowed to swell on ice for 20 min and then were broken with 20 to 30 strokes of a tight-fitting Dounce homogenizer. One-ninth volume of the buffer containing 25 mM HEPES-KOH, pH 7.3, 1 M K(OAc), 30 mM MgCl2, 30 mM DTT (buffer C [75]) was added to the homogenate, and the homogenate was centrifuged at 18,000 × g for 20 min at 4°C. The supernatant was flash frozen and stored at −70°C. Suspension cultures of HeLa S3 cells were maintained as described previously (3). Extracts from HeLa cells were prepared using the protocol described above. Huh7 cells were grown in monolayers in DMEM (Invitrogen Corp.) containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin with passages at a dilution of 1:3 every 3 days. The cells were maintained in 175-cm2 flasks, except for the last passage, when 15-cm petri dishes were used. At least 40 dishes with cells at 60 to 90% confluency are required to obtain a sizable amount (3 ml) of the extract. The cells were dislodged by brief (1-min) washing with trypsin-EDTA (Invitrogen Corp.) and further incubation with a fresh portion of trypsin-EDTA (2.5 ml) at room temperature for 3 to 4 min. Trypsinization was stopped by adding 5 ml 20% FBS in DMEM. Cell aggregates were disrupted by pipetting, and cell suspension was transferred to a conical centrifuge tube containing cold 20% FBS in DMEM. Cells were collected by centrifugation, washed (three times) with buffer A, and processed for the preparation of the extract in the same way as described for Krebs-2 cells.
Cell-free synthesis and analysis of HCV proteins.
Conditions for HCV protein synthesis in Krebs-2, Huh-7, and HeLa S10 cells were similar to those described previously (74, 75), with the exception that translation reaction mixtures contained higher concentrations of potassium and magnesium salts as specified below. The extracts were made mRNA dependent by treatment with micrococcal nuclease and were not subjected to either dialysis or Sephadex G-25 chromatography (75). Translation reaction mixtures (20 μl) contained 9 μl of S10, 1 μl of CMMs (or CMM buffer [50 mM triethanolamine, pH 7.3, 2 mM DTT, and 250 mM sucrose] where appropriate), 2 μl of a master mix (74), 2 μl of a salt solution (1 M KCl, 5 mM MgCl2, and 2.5 mM spermidine, unless otherwise specified), 1 μl of [35S]methionine (10 mCi/ml; 1,200 Ci/mmol), and 0.4 μg HCV RNA (unless otherwise specified). Translation in RRL was performed as recommended by the manufacturer (Promega), with the exception that KCl and MgCl2 were included in the reaction cocktail (at 100 mM and 0.75 mM final concentrations, respectively). The reactions were carried out at 32°C for 3 h (unless otherwise specified). These conditions were found to be optimal for the expression of HCV proteins. For deglycosylation of HCV glycoproteins, samples (20 μl) were supplemented with an equal volume of buffer 1 (5 mM EDTA, 2% β-mercaptoethanol, and 2% sodium dodecyl sulfate [SDS]), and proteins were denatured by heating them at 95°C for 5 min. The protein buffer solution was then exchanged for buffer 2 (50 mM sodium phosphate, pH 7.5, 5 mM EDTA, and 0.1% SDS) by centrifugation of the samples through CHROMA SPIN-10 columns (Clontech) at room temperature. After the addition of an equal volume of buffer 3 (50 mM sodium phosphate, pH 7.5, 5 mM EDTA, 1% β-mercaptoethanol, and 2% NP-40) containing Complete protease inhibitor cocktail (Roche), samples were divided into two aliquots. One aliquot was incubated at 37°C overnight in the absence of peptide N-glycosidase F (PNGase F) (Roche), while another was incubated at 37°C overnight in the presence of 4 U of PNGase F. Proteins were concentrated by trichloroacetic acid (TCA) precipitation, washed with 70% acetone (twice), and dissolved in SDS sample buffer (by incubation at 95°C for 10 min) for polyacrylamide gel electrophoresis (PAGE) analysis. For N-formyl-methionine labeling in Krebs-2 S10, 2 × 105 cpm of f[35S]Met-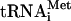 and 0.1 mM unlabeled methionine were added to a 20-μl reaction mixture. Proteins were analyzed by SDS-15% PAGE (acrylamide/bis-acrylamide ratio, 99:1) and fluorography. Incorporation of [35S]methionine into TCA-insoluble material was measured as described previously (75). Formulations for preparing Western blots and detecting protein bands using a Western Lightning chemiluminescence kit were as recommended by the manufacturer (Perkin-Elmer Life Sciences, Inc.). For detection of NS3, NS5B, and NS5A, the antibodies were used diluted 1:2,500 in phosphate-buffered saline-5% nonfat dry milk. Anti-core, anti-E1, and anti-E2 antibodies were used diluted 1:100, 1:500, and 1:50, respectively, in phosphate-buffered saline-0.5% nonfat dry milk.
and 0.1 mM unlabeled methionine were added to a 20-μl reaction mixture. Proteins were analyzed by SDS-15% PAGE (acrylamide/bis-acrylamide ratio, 99:1) and fluorography. Incorporation of [35S]methionine into TCA-insoluble material was measured as described previously (75). Formulations for preparing Western blots and detecting protein bands using a Western Lightning chemiluminescence kit were as recommended by the manufacturer (Perkin-Elmer Life Sciences, Inc.). For detection of NS3, NS5B, and NS5A, the antibodies were used diluted 1:2,500 in phosphate-buffered saline-5% nonfat dry milk. Anti-core, anti-E1, and anti-E2 antibodies were used diluted 1:100, 1:500, and 1:50, respectively, in phosphate-buffered saline-0.5% nonfat dry milk.
Tryptic peptide mapping.
Tryptic digestion of 35S-labeled polypeptides immobilized on the polyvinylidene difluoride membrane was performed essentially as described previously (39). Separation of the digestion products in two dimensions was done according to method B (76) using plastic-coated thin-layer cellulose plates (10 by 10 cm; Kodak).
Assay of mRNA degradation.
For Northern blot analysis, 200-μl reaction mixtures were programmed with the indicated mRNAs in the presence of unlabeled l-methionine (20 μM) at 32°C. At the times indicated, 20 μl was withdrawn from the reaction mixtures. Translation was stopped by the addition of 180 μl of 0.4-mg/ml proteinase K (made up in the buffer containing 100 mM NaCl, 20 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% SDS, and 50 μg/ml E. coli tRNA). After incubation at 37°C for 15 min, total RNA was phenol-chloroform extracted, separated on a formaldehyde-agarose gel, and transferred onto a nylon membrane (Hybond-N; Amersham Biosciences). To confirm that equal amounts of total RNA were loaded in each lane, blots were stained with Blot Stain Blue (Sigma), and the intensities of the bands of rRNA were compared. The blots were then hybridized with a fragment of randomly primed 32P-labeled HCV or luciferase cDNA using ExpressHyb hybridization solution (BD Biosciences Clontech) according to the company's protocol. Quantifications were carried out using the BAS-2000 phosphorimaging system (FUJI Medical Systems USA).
RESULTS
Full-size HCV RNA translation in different cell-free systems.
We sought to establish conditions that would allow translation of the entire HCV genome in vitro. In a pilot study, the RNA transcribed from the infectious H77C HCV cDNA clone (89) was used to program nuclease-treated translation extracts (hereafter referred as S10) from different sources, such as human hepatoma Huh7, Krebs-2, and HeLa S3 cells, and a commercially available RRL. Translation assays were performed either in the absence or in the presence of CMMs. SDS-PAGE analyses of [35S]methionine-labeled proteins synthesized in these translation systems are shown in Fig. 1. CMM-supplemented Huh7 and Krebs-2 S10 extracts synthesized proteins with similar patterns (Fig. 1A). The ∼74-kDa protein (p74; putative NS3) was readily identified among the translation products (p74 migrated slightly more slowly than NS3 encoded by a subgenomic type 1b HCV replicon RNA [38], presumably due to strain-specific differences in the mobilities of these proteins [data not shown]). Other major translation products were p68, p58, p32, p29, p24, p21, p20, and p18. (For unknown reasons, p32 was synthesized only in this set of experiments.) In the absence of CMMs, translation yielded little (lane 3, Huh7 S10 preparation 2, and lane 5, Krebs-2 S10) or none (lane 1, Huh7 S10 preparation 1) of the processed products. Programming HCV RNA into CMM-supplemented HeLa S10 or RRL that was optimized for potassium and magnesium salt concentrations yielded a set of polypeptides similar to those produced in Krebs-2 S10 (Fig. 1B). However, HeLa S10 and RRL also accumulated a significant amount of unprocessed heterogeneous polypeptides, rendering the bands of high-molecular-weight products barely discernible above background.
FIG. 1.

Products of full-size HCV RNA translation in different cell-free systems. (A) Translation in two preparations of Huh7 S10 (preparation 1, lanes 1 and 2; preparation 2, lanes 3 and 4) and in Krebs-2 S10 (5 and 6). +, present; −, absent. (B) Translation in HeLa S10 (lanes 1 to 3) and in RRL (lanes 4 to 6). The reactions were programmed with HCV RNA in the presence of [35S]methionine at 32°C for 4 h under standard conditions (see Materials and Methods). CMMs (1 μl) were included in the reaction mixtures where indicated. Translation products were separated by SDS-15% PAGE and detected by autoradiography. Analyses of reactions that did not contain mRNA are shown in lanes 1 and 4 of panel B. The positions of the prestained molecular weight protein markers (MBI; Fermentas) are shown at the left. Arrows at the right indicate the major HCV RNA translation products.
Identification of HCV proteins in vitro.
Since Krebs-2 S10 efficiently translates HCV RNA and could be prepared in large quantities, this system was chosen for further use. To confirm the authenticity of the in vitro-synthesized proteins, they were resolved by electrophoresis, transferred to a nitrocellulose membrane, and detected first by autoradiography (Fig. 2A, left) and then by Western blotting using polyclonal antibodies against the core protein, NS3, and NS5B (right). CMMs stimulated HCV polyprotein processing (Fig. 2A, left; compare lane 4 to 2). Western analysis performed on the same blot allowed the unambiguous identification of p20 as the core protein, p74 as NS3, and p68 as NS5B (Fig. 2A, right). Importantly, all immunoreactive proteins were revealed in HCV RNA-programmed reactions, but not in mock-programmed reactions (Fig. 2A, right, compare lane 2 to 1 and 4 to 3). This excludes the possibility that some cellular proteins that cross-react with the antibodies and comigrate with the translation products are the sources of the signals on the immunoblots. In a separate Western blot analysis using an anti-NS5A rabbit polyclonal antibody, p58, which was formed almost exclusively in the presence of CMMs, was identified as NS5A (Fig. 2B). We also suggest that p24 corresponds to NS4B. This assignment is strongly supported by the fact that the appearance of p24 is sensitive to an NS3 protease inhibitor while that of other low-molecular-weight products is resistant (see Fig. 6B). It is noteworthy that anti-NS3 also detected an ∼100-kDa band. We reasoned that since cleavage at the NS3/4A site is a fast intramolecular reaction (1), p100 is likely to be the uncleaved NS2-NS3 (hereafter designated NS2-3). To corroborate this conclusion, we performed tryptic fingerprinting of p100, putative NS2 (p21), and NS3 (p74) after resolving these proteins by electrophoresis and blotting them onto a polyvinylidene difluoride membrane. These analyses revealed that indeed p100 shares the peptides characteristic of p21 and p74 (Fig. 3).
FIG. 2.

Detection of HCV antigens in HCV RNA-programmed Krebs-2 S10. (A) The assays were performed in the absence (−; lanes 1 and 3) or presence (+; lanes 2 and 4) of HCV RNA and in the absence (lanes 1 and 2) or presence (lanes 3 and 4) of CMMs as described in the legend to Fig. 1. Translation products were separated by SDS-15% PAGE and transferred onto a nitrocellulose membrane. The autoradiograph of the membrane (left) and Western blots of NS3, NS5B, and the core protein (C) (right) are shown. For Western blotting, the membrane was first probed with anti-NS3 and then sequentially stripped and reprobed with anti-NS5B and anti-Core. (B) Detection of NS5A. HCV RNA translation and separation of HCV-specific proteins by PAGE were as described for panel A. After transfer to nitrocellulose and autoradiography, the blots were reacted with an HCV NS5A-specific rabbit antiserum. The autoradiograph of the middle portion of the membrane and Western blot are shown on the left and right, respectively. An arrowhead indicates a nonspecific band. The positions of the prestained molecular weight protein markers are shown at the left.
FIG. 6.

Translation and processing of HCV polyprotein in Krebs-2 S10 as effected by the indicated concentrations of compound A, a hexapeptide-like competitive inhibitor of HCV NS3 protease (51). (A) Compound A, when used in a 0.25 to 1,000 nM concentration range, does not inhibit HCV RNA translation. Reaction mixtures containing the indicated concentrations of compound A were programmed with HCV RNA under standard conditions (serial dilutions of compound A were made up in 0.1% dimethyl sulfoxide, and 1 μl of each dilution was added to a 20-μl reaction mixture). Portions (1 μl) of each sample were assayed for TCA-insoluble radioactivity. (B) Compound A affects NS3 protease-mediated, but not signal peptidase-mediated, processing of the HCV polyprotein. Portions of the reaction mixtures in panel A were subjected to SDS-15% PAGE analysis and autoradiography. To improve detection of the high-molecular-weight products, the exposure to the gel of the film used for scanning the upper part of the figure was reduced three times compared to that used for printing the lower part of the figure. HCV-specific proteins whose appearance is sensitive and resistant to compound A are indicated at the left and right, respectively.
FIG. 3.

Comparison of tryptic peptide maps of p100, p21 (NS2), and p74 (NS3). The polypeptides were separated by SDS-PAGE, transferred to a polyvinylidene difluoride membrane, and digested with trypsin as described in Materials and Methods. Two-dimensional fingerprints were obtained after high-voltage electrophoresis and thin-layer chromatography (TLC) of the digestion products. On the map of p100, arrows and arrowheads indicate the major p21 (NS2)- and p74 (NS3)-specific peptides, respectively.
To further characterize the HCV RNA translation products and, specifically, to identify HCV glycoproteins, the full-size HCV RNA was translated in parallel with 3′-truncated subgenomic HCV RNAs encoding C, C-E1, C-E2, C-p7, and C-NS2 portions of the viral polyprotein (Fig. 4A). As expected, HCV RNA C was translated to yield the core protein exclusively (Fig. 4A, lane 2). HCV RNA C-E1 translation gave rise, in addition to the core protein, to p29 (putative fully glycosylated E1; gp31) and several proteins that do not have counterparts among the products of the full-size HCV RNA translation (Fig. 4A, compare lane 3 to 7). The translation products of HCV RNAs C-E2, C-p7, and C-NS2 were similar; notably, they included p18 in addition to the products synthesized from HCV RNA C-E1. To determine which of these products were glycosylated, they were treated with PNGase F, which removes both high-mannose and complex glycans (43). (It should be noted that prior to deglycosylation, the proteins were denatured by being heated in the presence of SDS and were passed through a spin column [see Materials and Methods]. The latter procedure, which was initially intended to remove excess SDS, which inhibits PNGase, was also found to remove the core protein and some unprocessed polypeptides that interfere with the analysis of the mature HCV glycoproteins.) As is best seen from the analysis of HCV RNA C-E1 translation products (Fig. 4A, lanes 4 and 5), PNGase caused p29 to disappear due to its conversion to a faster-migrating form (an ∼18.5-kDa polypeptide). Both p29 and p18.5 reacted with an E1-specific monoclonal antibody in Western blotting, in accordance with their assignments as fully glycosylated and deglycosylated E1, respectively (Fig. 4C, bottom). Importantly in addition to p29, the anti-E1 antibody detected one major (p18) and two minor (p25 and p22) products of the full-size HCV RNA translation (Fig. 4C, bottom, lane 2). It is likely that the sharp band p18 corresponds to E1 that lacks carbohydrate, while the smeared band p18.5 results from incomplete deglycosylation of E1 by PNGase. Therefore, for simplicity, we will designate p18 and p18.5 together E1*** (to distinguish them from E1, which bears carbohydrate) (Fig. 4B and C). Furthermore, given that p25 and p22 are sensitive to PNGase digestion, they are most likely the hypoglycosylated subsets of E1 (hereafter named E1* and E1**, respectively). Surprisingly, only fully glycosylated E1 (p29) was formed upon translation of HCV RNA C-E1, while the translation of longer mRNAs also yielded a substantial amount of deglycosylated and hypoglycosylated E1 forms (p18, p22, and p25) (Fig. 4A, compare lane 3 to lanes 4 to 7, and B and C, compare lane 4 to lanes 2, 6, 8, and 10). It is not immediately clear why this is so. With regard to HCV E2 (gp70) formation, no distinct band corresponding to the glycoprotein was evident upon analysis of the HCV RNA translation products by Western blotting. Instead, the anti-E2 antibody recognized a set of poorly resolved diffuse bands, which correspond to polypeptides ranging from 44 to 74 kDa and whose presence is not due to the cross-reactivity of the cellular extract proteins with the antibody (Fig. 4C, top, compare lanes 1 and 2). PNGase digestion of the products translated from HCV RNAs (fullsize; C-p7 and C-NS2) dramatically reduced the intensities of anti-E2-reactive heterogeneous bands; moreover, it generated a polypeptide whose molecular mass was similar to that of the E2-p7 protein backbone (p44) (Fig. 4B and C). Importantly, a smaller polypeptide (p39), presumably lacking the p7 moiety, was formed when the products of translation of HCV RNA C-E2 were deglycosylated (Fig. 4B and C, lanes 7). Taken together, these results imply that, at least in vitro, E2 undergoes glycosylation while being a part of its immediate precursor, E2-p7.
FIG. 4.

Products of subgenomic HCV RNA translation and identification of HCV glycoproteins. pCV-H77C was used as a template for DNA amplifications and subsequent synthesis of the subgenomic HCV RNAs as described in Materials and Methods. (A) Subgenomic HCV RNAs harboring the terminator codons after the core (C), E1 (C-E1), E2 (C-E2), p7 (C-p7), and NS2 (C-NS2) coding regions and full-size HCV RNA were translated in Krebs-2 S10 under standard conditions. Translation products were analyzed by SDS-15% PAGE and autoradiography. The positions of HCV-specific proteins are indicated on the right. (B) Autoradiograph of the full-size and subgenomic HCV RNA translation products that were incubated in either the absence (−) or presence (+) of PNGase F, as indicated. Nonstructural HCV proteinsare indicated on the left and HCV glycoproteins on the right of the autoradiogram. Asterisks and arrowheads indicate PNGase-deglycosylated E2-p7/E2 and E1, respectively. E1* and E1** are putative hypoglycosylated forms and E1*** is an unglycosylated form of E1 produced in the course of HCV RNA translation. (C) Products of translation of HCV RNAs shown in panel B were blotted onto a nitrocellulose membrane and probed with the antibodies against HCV E2 and E1 glycoproteins, as indicated. In panels A to C in lanes 1, no mRNA was added to the reaction samples.
Optimization of the conditions of HCV RNA translation in vitro.
To obtain higher yields of HCV-specific proteins in vitro, HCV RNA-programmed reactions were optimized with respect to CMM amounts and potassium and magnesium ion concentrations. Increasing the amount of CMMs from 0 to 5% of the reaction volume increased the proportion of processed polypeptides (Fig. 5A). Concentrations above 5% of the CMM fraction brought about no further difference in the polypeptide synthesis pattern. Therefore, CMMs were used at a concentration of 5% in all subsequent experiments. The yield of mature HCV proteins was maximal at a relatively high potassium ion concentration (160 mM, including 60 mM contributed by S10) both in the absence and presence of CMMs (Fig. 5B). The optimal magnesium chloride concentration was 2.5 mM (including 2 mM concentration contributed by S10) (data not shown). Under optimal salt conditions, there was almost linear HCV RNA dose dependence of incorporation of [35S]methionine into proteins (data not shown). Importantly, the proteins encoded in the 3′ portion of the viral genome (i.e., NS5A and NS5B) were most abundant at 20 to 30 μg/ml HCV RNA (Fig. 5C, lanes 4 and 5).
FIG. 5.

CMMs (A), potassium ion (B), and HCV RNA (C) concentration dependences of HCV protein synthesis in Krebs-2 S10. (A) HCV RNA was translated in the presence of the indicated amounts of CMMs (given as percent [vol/vol] of the total reaction volume) as described in the legend to Fig. 1. (B) Potassium ion concentration optimum of translation. Mg2+ was used at the optimal (2.5 mM) concentration. CMMs were present (+) in the reactions at 5% of the reaction volume where indicated. In lane 9, no HCV RNA or CMMs were added to the reaction sample containing 160 mM potassium ion concentration. (C) The indicated concentrations of HCV RNA were translated in the presence of CMMs (at 5% reaction volume) under optimal ionic conditions (160 mM K+ and 2.5 mM Mg2+). Other conditions were as specified above. The samples were analyzed by SDS-15% (A and C) or 10% (B) PAGE and autoradiography. The positions of HCV-specific proteins are indicated on the right.
Analysis of HCV polyprotein processing using an NS3 serine protease inhibitor.
The NS3 protease is competitively inhibited by specific penta- or hexapeptides derived from the amino-terminal NS3 cleavage products (69, 79). One potent and highly specific inhibitor of the NS3 protease activity (compound A) was shown to reduce replication of a subgenomic serotype 1b HCV RNA to undetectable levels in Huh7 cells (51).
To confirm the specificity of HCV polyprotein cleavage reactions in vitro, HCV RNA was programmed into CMM-supplemented Krebs-2 S10 in the presence of increasing concentrations of compound A. Compound A did not inhibit the incorporation of [35S]methionine into protein when tested in the 0.25-to-1,000 nM range of concentrations (Fig. 6A). However, consistent with its described function, compound A caused a dramatic dose-dependent decrease in the accumulation of mature viral proteins derived from the NS3-NS5B portion of the polyprotein (Fig. 6B). At 50 nM compound A, the NS4B and NS5A bands disappeared and the intensities of the NS3, NS2-3, and NS5B bands were significantly reduced. The reduction in the amounts of these proteins was accompanied by the accumulation of the high-molecular-weight precursor polypeptides (p160 and p230, putative NS3-5A, and NS3-5B, respectively). Preferential accumulation of unidentified p130 in the presence of the inhibitor was also evident. Interestingly, compound A inhibited the appearance of NS3 and NS5B only partially (by ∼70% and 80%, respectively) (Fig. 6B). In contrast, inhibition of NS4B and NS5A production reached 100%. The 50% inhibitory concentrations for compound A were 45 nM for NS3, 25 nM for NS5B, and 15 nM for NS4B and NS5A (data not shown). Some possible reasons for this differential inhibition of processing events are discussed below. In contrast to its potent inhibition of NS3-mediated cleavages, compound A failed to inhibit the cleavages believed to be carried out by a cellular signal peptidase(s), i.e., appearances of proteins C, E1 (E1* and E1***), and NS2. This is further proof of the high specificity of the inhibition. Earlier studies demonstrated that compound A inhibits proteolytic activity of NS3 in genotype 1b HCV (51). Our data show that compound A is also efficient in inhibiting genotype 1a NS3. Thus, our system should be useful for testing the specificities and potencies of other NS3 inhibitors.
Kinetics of appearance of HCV proteins.
The time course of HCV protein appearance was examined using mock- and membrane-supplemented Krebs-2 S10 extract (Fig. 7A). In the presence of CMMs, the first discrete band appeared after 20 min of incubation and corresponded to fully glycosylated E1. The core protein was barely detectable at 20 min, presumably because it possesses fewer methionine residues (two residues) than E1 (seven residues). No conclusion could be drawn about the timing of the appearance of E2-p7, because of its heterogeneous electrophoretic profile. NS3 appeared at 40 min, concomitantly with the bulk of C, E1***, and NS2. NS4B, NS5A, NS5B, and NS2-3 appeared between 60 and 80 min. Further incubation led to an increase in the accumulation of NS4B (indicating that it is formed by cleavage of a precursor polypeptide), but not other proteins. Importantly, NS3 appeared at the same time independently of the presence or absence of CMMs (lanes 2 and 9). Thus, CMMs are not critical for cleavage at NS2-NS3 and NS3-NS4A junctions. In addition to NS3, an ∼160-kDa polypeptide (putative NS3-5A) was synthesized in the absence of CMMs, along with a set of other poorly resolved high-molecular-weight products (Fig. 7A, lane 4). Most of these products degraded upon prolonged incubation without being converted into stable polypeptides. A pertinent question is whether the precursors for the nonstructural proteins are cleaved co- or posttranslationally. Our results favor the first possibility. Indeed, in the presence of CMMs, no distinct polypeptides larger than NS2-3 could be revealed, and even NS2-3 does not seem to serve as a precursor for NS2 and NS3, considering its stability. To further explore the possibility of a precursor-product relationship, duplicate samples were withdrawn from the membrane-supplemented translation reactions at different time points. One sample was supplemented with SDS sample buffer and analyzed directly, while translation products in another sample were chased in the presence of cycloheximide. With the possible exception of NS4B, no proteins appeared or became more abundant during the chase period (Fig. 7B). Also, no reduction in the intensity of the NS2-3 band as a result of the chase was evident. Cumulatively, these data indicate that secondary processing plays a minor or no role in the formation of the HCV proteins. Interestingly, band NS5A shifted slightly upward upon chase (Fig. 7B) or prolonged incubation (Fig. 7A), indicating posttranslational modification, most likely phosphorylation (33, 49). Indeed, NS5A undergoes phosphorylation in vitro, since an increase in the electrophoretic mobility of NS5A, but not other HCV proteins, was observed as a result of the phosphatase treatment (Fig. 7C).
FIG. 7.

Time course of synthesis of HCV polypeptides in Krebs-2 S10. (A) HCV RNA (20 μg/ml) was translated in a 100-μl total reaction volume in either the absence (−) or presence (+) of CMMs, as indicated. At the time points indicated, aliquots (10 μl) of the translation samples were withdrawn, supplemented with the SDS sample buffer, and analyzed by SDS-15% PAGE and autoradiography. (B) HCV RNA was programmed into a CMM-supplemented reaction (100 μl) as described above. At the time points indicated, two aliquots (10 μl each) were withdrawn. One aliquot (p, pulse) was immediately supplemented with the SDS sample buffer, while another (ch, chase) was additionally incubated in the presence of cycloheximide (0.6 mM final concentration). The total incubation time for all chased samples was 180 min. HCV-specific proteins and the positions of the prestained protein markers are indicated. (C) Alkaline calf intestinal phosphatase (CIP) treatment of HCV RNA translation products. HCV RNA wastranslated in a 40-μl total volume reaction in the presence of CMMs for 3 h as described for panel A. After the adjustment of the MgCl2 concentration to 10 mM, the reaction mixture was divided into two portions. One portion was incubated with CIP (40 U; New England BioLabs) for 30 min at 37°C, while another was incubated with the control buffer. The samples were analyzed by SDS-PAGE as described above. The position of dephosphorylated NS5A [NS5A (−P)] is indicated by an arrow.
N-Formyl-[35S]methionine-labeled products.
RRL was reported to initiate translation at multiple spurious sites when programmed with poliovirus or HCV RNA (10, 23). It was therefore important to determine whether initiation on HCV RNA in Krebs-2 S10 extract occurred with fidelity. To this end, f[35S]Met-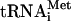 (which is used exclusively for initiation of translation and donates a labeled formyl-methionyl residue to the N-terminal position of the polyprotein [67]) was used. Control experiments demonstrated that addition of f[35S]Met-
(which is used exclusively for initiation of translation and donates a labeled formyl-methionyl residue to the N-terminal position of the polyprotein [67]) was used. Control experiments demonstrated that addition of f[35S]Met-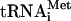 to an encephalomyocarditis virus (EMCV) RNA-programmed reaction resulted in labeling of only the N-terminal leader peptide (L) and its precursor (L-P1-2A) (Fig. 8A, lane 4), in agreement with published data (6, 30, 50). When f[35S]Met-
to an encephalomyocarditis virus (EMCV) RNA-programmed reaction resulted in labeling of only the N-terminal leader peptide (L) and its precursor (L-P1-2A) (Fig. 8A, lane 4), in agreement with published data (6, 30, 50). When f[35S]Met-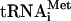 was used as a substrate in reactions programmed with HCV RNA, the core protein was the only mature protein that incorporated the label (Fig. 8B). The proteolytic release of the f[35S]Met-labeled core protein from the nascent chain was CMM dependent, as expected (compare lanes 6 to 8 to lanes 10 to 12). Thus, only a single initiation site that corresponds to the start of the polyprotein functions in our translation system. Surprisingly, N-terminal labeling of the core protein with formyl-methionine was already optimal after a 30-min incubation and decreased slightly with time, while labeling with [35S]methionine reached maximum only after a 60-min incubation. Conceivably, this would be the expected result if the formyl-[35S]methionyl residue in the initiator tRNAMet was rapidly chased by unlabeled methionine, which is present in excess in the reaction mixture, thereby permitting only the first round of initiation to occur with the labeled substrate. In contrast, the steady-state level of charging of the elongator tRNAMet with [35S]methionine should ensure continuous protein labeling. Early studies of initiation of protein synthesis in vitro also revealed that the incorporation of N-formyl-[35S]methionine into nascent polypeptides is complete within 5 min, while utilization of the elongator tRNAMet lasts significantly longer (50).
was used as a substrate in reactions programmed with HCV RNA, the core protein was the only mature protein that incorporated the label (Fig. 8B). The proteolytic release of the f[35S]Met-labeled core protein from the nascent chain was CMM dependent, as expected (compare lanes 6 to 8 to lanes 10 to 12). Thus, only a single initiation site that corresponds to the start of the polyprotein functions in our translation system. Surprisingly, N-terminal labeling of the core protein with formyl-methionine was already optimal after a 30-min incubation and decreased slightly with time, while labeling with [35S]methionine reached maximum only after a 60-min incubation. Conceivably, this would be the expected result if the formyl-[35S]methionyl residue in the initiator tRNAMet was rapidly chased by unlabeled methionine, which is present in excess in the reaction mixture, thereby permitting only the first round of initiation to occur with the labeled substrate. In contrast, the steady-state level of charging of the elongator tRNAMet with [35S]methionine should ensure continuous protein labeling. Early studies of initiation of protein synthesis in vitro also revealed that the incorporation of N-formyl-[35S]methionine into nascent polypeptides is complete within 5 min, while utilization of the elongator tRNAMet lasts significantly longer (50).
FIG. 8.

Comparison of [35S]methionine- and formyl-[35S]methionine-labeled products of translation of HCV RNA in Krebs-2 S10. (A) Control translation of EMCV RNA showing that the f[35S]Met-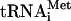 preparation used specifically labels only the N-terminal protein moieties in the viral polyprotein. The reaction mixtures containing either [35S]methionine (lane 2) or f[35S]Met-
preparation used specifically labels only the N-terminal protein moieties in the viral polyprotein. The reaction mixtures containing either [35S]methionine (lane 2) or f[35S]Met-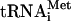 (lane 4) were incubated with EMCV RNA (20 μg/ml) at 34°C for 90 min, as described previously (74). Analyses of the samples that did not receive the exogenous mRNA and were incubated with either [35S]methionine or f[35S]Met-
(lane 4) were incubated with EMCV RNA (20 μg/ml) at 34°C for 90 min, as described previously (74). Analyses of the samples that did not receive the exogenous mRNA and were incubated with either [35S]methionine or f[35S]Met-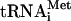 are shown in lanes 1 and 3, respectively. (B) HCV RNA (20 μg/ml) was translated for the times indicated in the absence (−; lanes 6 to 8) or presence (+; lanes 10 to 12) of CMMs, using f[35S]Met-
are shown in lanes 1 and 3, respectively. (B) HCV RNA (20 μg/ml) was translated for the times indicated in the absence (−; lanes 6 to 8) or presence (+; lanes 10 to 12) of CMMs, using f[35S]Met-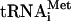 as a substrate. [35S]methionine-labeled HCV RNA translation products synthesized after the indicated time intervals are shown in lanes 2 to 4 for comparison. In lanes 1, 5, and 9, no HCV RNA was added and the reaction mixtures were incubated for 180 min. The samples were analyzed by SDS-15% PAGE and autoradiography. The positions of virus-specific proteins are indicated on the right. Arrows indicate formyl-[35S]methionine-labeled products.
as a substrate. [35S]methionine-labeled HCV RNA translation products synthesized after the indicated time intervals are shown in lanes 2 to 4 for comparison. In lanes 1, 5, and 9, no HCV RNA was added and the reaction mixtures were incubated for 180 min. The samples were analyzed by SDS-15% PAGE and autoradiography. The positions of virus-specific proteins are indicated on the right. Arrows indicate formyl-[35S]methionine-labeled products.
CMMs enhance HCV protein expression via several mechanisms.
The results presented above clearly demonstrate the requirement for the membranous structures in maturation of HCV proteins. Also, in Krebs-2 S10 extract, [35S]methionine incorporation into the total translation product is substantially higher in the presence of CMMs, and this stimulation is CMM dose dependent (Fig. 1A and 9A). One possible explanation for this effect is that unprocessed polypeptides formed in the absence of CMMs are less stable than the mature viral proteins. This is indeed the case, since during the first 40 min of incubation, incorporation increases linearly with time and is similar in CMM- and mock-supplemented reactions (Fig. 9B). However, after a 60-min incubation, the curves diverge, and by 4 h almost half of the amount of protein synthesized in the absence of CMMs is converted to TCA-soluble material. Thus, membrane association and proper processing of HCV precursor polypeptides prevent their nonspecific proteolysis.
FIG. 9.

CMMs prevent nonspecific proteolysis of HCV RNA translation products. (A) HCV RNA was translated in the presence of the indicated amounts of CMMs (given as percent [vol/vol] of the total reaction volume) for 4 h as described in the legend to Fig. 5A. Aliquots (1 μl) of the translation samples were assayed for TCA-insoluble radioactivity. (B) HCV RNA was translated in the absence (−) or presence (+) of CMMs, as described in the legend to Fig. 7A. At the time points indicated, aliquots (1 μl) of the translation samples were withdrawn and assayed for TCA-insoluble radioactivity. Results are averages for three assays with standard deviations from the mean.
We next considered the possibility that CMMs also enhance HCV protein expression by stabilizing HCV RNA. Figure 10A shows a time course experiment in which the decay of HCV RNA has been assessed by Northern blotting. In the absence of CMMs, the half-life of HCV RNA was about 100 min, with ∼25% of the intact RNA remaining after a 180-min incubation (Fig. 10B). However, in the presence of CMMs, the half-life of HCV RNA was more than 180 min (in fact, more than 67% of HCV RNA remained intact by 180 min). HCV RNA stabilization by CMMs was also observed in HeLa S10 and RRL (data not shown). Interestingly, in Krebs-2 S10, luciferase mRNA flanked by the HCV 5′ and 3′ UTRs (Luc-HCV) was also stabilized by CMMs (Fig. 10C and D). However, CMMs only marginally stabilized a polyadenylated luciferase mRNA harboring the EMCV IRES (Luc-EMC) (Fig. 10E). Notably, the half-life of Luc-EMCV mRNA (∼40 min) appeared to be much shorter than that of Luc-HCV mRNA. In contrast, capped and polyadenylated Luc mRNA exhibited remarkable stability, with almost 90% of the mRNA recoverable after 3 h of incubation (Fig. 10F). Taken together, these data indicate that CMMs stabilize HCV RNA and that this process is HCV 5′ and/or 3′ UTR sequence specific. Augmented HCV RNA stability could contribute to the stimulation of [35S]methionine incorporation by CMMs (Fig. 9). It is also clear that the cessation of protein synthesis after 60 min, in both the absence and presence of CMMs (Fig. 9B), is not due to mRNA degradation, since the majority of HCV RNA remains intact at that time.
FIG. 10.

CMMs increase stability of HCV RNA in vitro. HCV RNA (A) or HCV-Luc-3′ UTR mRNA (Luc-HCV) (C) was used at equimolar concentrations (20 μg/ml and 5 μg/ml, respectively) to program Krebs-2 S10 translation reactions in the absence (−) or presence (+) of CMMs, as indicated. Total RNA was isolated at the indicated times from the aliquots of the reaction mixtures. mRNA integrity was analyzed by formaldehyde-0.8% (for HCV RNA) or 1% (for Luc-HCV mRNA) agarose gel electrophoresis and Northern blotting as described in Materials and Methods. Arrows indicate the positions of 28S and 18S rRNAs visualized by staining. Quantifications of the signals from panels A and C are shown in panels B and D, respectively. The rates of decay of polyadenylated luciferase mRNAs (5 μg/ml), containing either the EMCV IRES (Luc-EMCV) (E) or cap structure (Luc) (F) (71) in Krebs-2 S10 in the absence or presence of CMMs, are shown for comparison.
DISCUSSION
Previous attempts to translate the entire open reading frame of the HCV RNA, using either RRL or HeLa cell extracts, were by and large unsuccessful (8, 22, 23). The failure to synthesize all the viral proteins could be attributed to low fidelity of translation initiation, premature termination of translation, HCV RNA degradation, or a combination of these factors. Furthermore, there are no reports showing complete translation of flavivirus or pestivirus genomes. In this study, we investigated the potential utility of different translation extracts for the expression of HCV proteins. Ribosomes of Krebs-2 S10 could complete translation of the HCV open reading frame to produce NS5B in amounts detectable not only by radiolabeling, but also by Western blotting (Fig. 2). The fidelity of translation is demonstrated by the absence of products arising from initiation at spurious internal sites (Fig. 8B). Evidence for posttranslational modification of viral proteins, glycosylation of E1 and E2-p7, and phosphorylation of NS5A is presented.
The most important variables affecting HCV protein expression in our system are the CMMs and potassium salt concentrations. Although Krebs-2 S10 extract contains some amount of membranes, exogenously added CMMs drastically enhanced HCV protein expression in this system. Our results implicate several mechanisms in this stimulation. First, CMMs stabilize HCV RNA in a process that appears to involve HCV 5′ and/or 3′ UTRs (Fig. 10). Second, CMMs mediate proper HCV polyprotein processing, and the mature viral proteins appear to be more stable than their unprocessed precursors (Fig. 7A and 9B). Maximal protein synthesis was achieved by employing a relatively high K+ concentration (160 mM) (Fig. 5B). This is not surprising, since high K+ concentrations ensure a high rate of polypeptide elongation (44, 82) while allowing efficient initiation on the HCV IRES. It is noteworthy that HCV proteins were expressed to higher levels when potassium chloride, rather than acetate, was used to supplement the reaction mixture (data not shown). In the translation systems of other viral RNAs, the ratio of authentic products to incorrect products also increased as a result of partial or complete substitution of KCl for K(OAc) (28, 76).
Even under optimal conditions, there was a cessation of translation after ∼60 min of incubation (Fig. 9B). This inhibition cannot be fully explained by the decay of HCV mRNA, which is mostly intact at this time (Fig. 10B). It remains to be studied whether ribosome recycling on HCV RNA ceased due to inactivation of some labile component(s) of the extract or other factors. The pattern of HCV proteins in vitro was variable, depending on the particular extract and CMM preparations. For instance, it is unclear why Huh7 preparation 2 yielded more processed products in the absence of added CMMs than preparation 1 (Fig. 1A). Some inadequately controlled cell culture conditions or differences in cell homogenization and/or subcellular fractionation could account for these variations. Also for unknown reasons, with most batches of CMMs, there was very low or no production of the unidentified p32 polypeptide, which appears as a major band in Fig. 1. Finally, some CMM preparations yielded little E1 compared to E1***, apparently due to their low glycosylation activities (Fig. 5C and 6B).
Initiation of translation.
HCV RNA translation is unique because it requires fewer initiation factors than picornavirus or cellular mRNAs (55). While picornavirus IRESes take advantage of eIF4G and eIF4A to recruit the 40S ribosomal subunit, the HCV IRES binds to the 40S subunit directly. The binding occurs even when high-salt-washed 40S subunits, essentially free of all initiation factors or Met-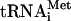 , are used (31, 35). Although in cells most 40S subunits contain eIF3 and other associated factors (19), the direct HCV IRES-40S interaction has been implicated in the formation of the physiological 48S preinitiation complex (comprising the HCV IRES RNA, the 40S subunit, eIF3, a ternary eIF2 · GTP · Met-tRNAi complex, and possibly other factors) (41). There is also a critical question, which was raised recently, of whether the HCV IRES, akin to the cricket paralysis virus IRES (53, 86), can initiate translation without assistance from the eIF2 · GTP · Met-
, are used (31, 35). Although in cells most 40S subunits contain eIF3 and other associated factors (19), the direct HCV IRES-40S interaction has been implicated in the formation of the physiological 48S preinitiation complex (comprising the HCV IRES RNA, the 40S subunit, eIF3, a ternary eIF2 · GTP · Met-tRNAi complex, and possibly other factors) (41). There is also a critical question, which was raised recently, of whether the HCV IRES, akin to the cricket paralysis virus IRES (53, 86), can initiate translation without assistance from the eIF2 · GTP · Met-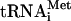 complex (cf. references 34 and 60). The dispensability of Met-
complex (cf. references 34 and 60). The dispensability of Met-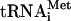 for HCV IRES activity according to this notion would explain why this activity is only marginally compromised upon mutating the authentic initiation codon to a non-AUG codon (59). Our results clearly show that the formyl-[35S]methionyl residue of f[35S]Met-
for HCV IRES activity according to this notion would explain why this activity is only marginally compromised upon mutating the authentic initiation codon to a non-AUG codon (59). Our results clearly show that the formyl-[35S]methionyl residue of f[35S]Met-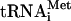 is incorporated into the core protein, which corresponds to the N-terminal portion of the polyprotein (Fig. 8B). Thus, the initiator tRNAMet, and by inference eIF2 and GTP, are used to initiate translation on the HCV IRES, at least in vitro. However, the simultaneous operation of two mechanisms of translation initiation, with and without participation of the ternary complex, cannot be rigorously ruled out. It is noteworthy that we could not detect an alternative form of the HCV core protein (p17), which would be expected to incorporate N-formyl-[35S]methionine. This form (termed F, for frameshift protein) was suggested to result from initiation at the initiator codon of the core protein sequence and a subsequent −2/+1 frameshift between codons 8 and 14 by a fraction of elongating ribosomes (83, 88). However, recent studies also demonstrated that ribosomal slipping occurs only in HCV isolates that possess an A-rich region (10 A residues) between nucleotides 363 and 374 of their genomes (8, 61, 81). Since this stretch is absent from strain H77 (89), p17 synthesis is not anticipated in our system.
is incorporated into the core protein, which corresponds to the N-terminal portion of the polyprotein (Fig. 8B). Thus, the initiator tRNAMet, and by inference eIF2 and GTP, are used to initiate translation on the HCV IRES, at least in vitro. However, the simultaneous operation of two mechanisms of translation initiation, with and without participation of the ternary complex, cannot be rigorously ruled out. It is noteworthy that we could not detect an alternative form of the HCV core protein (p17), which would be expected to incorporate N-formyl-[35S]methionine. This form (termed F, for frameshift protein) was suggested to result from initiation at the initiator codon of the core protein sequence and a subsequent −2/+1 frameshift between codons 8 and 14 by a fraction of elongating ribosomes (83, 88). However, recent studies also demonstrated that ribosomal slipping occurs only in HCV isolates that possess an A-rich region (10 A residues) between nucleotides 363 and 374 of their genomes (8, 61, 81). Since this stretch is absent from strain H77 (89), p17 synthesis is not anticipated in our system.
Polyprotein processing.
In agreement with processing of the C-NS2 region by host signal peptidases, the appearance of the core protein, E1 forms, and NS2 was resistant to compound A (Fig. 6B). Given that E1 is the earliest protein to appear (Fig. 7A), nascent C-E1 and E1-E2 cleavages should be realized first. Cleavage at C-E1 either precedes cleavage at E1-E2 or occurs simultaneously with it, since there was no C-E1 precursor detectable. Presumably, both these cleavages require ongoing translation, since only very small amounts of C and E1 were released after chase in the presence of cycloheximide (Fig. 7B). We did not reveal any cotranslational cleavage at the E2-p7 junction, as only a single deglycosylated polypeptide, E2-p7, was detected by the anti-E2 antibody (Fig. 4C, top, lane 3). However, the release of E2 from the N terminus of p7 is also inefficient in vivo (12, 36, 66). Cleavage at the p7-NS2 junction is fast and complete, since no E2-NS2 precursor polypeptide was apparent. In contrast, a rather stable E2-NS2 precursor accumulated in the cells infected with vaccinia virus recombinants or harboring stably replicating full-length HCV genomes (7, 57). The explanation for these differences in processing efficiency between in vitro and in vivo systems is not clear.
Cleavage between NS2 and NS3 is accomplished by NS2-3 autoprotease. For several HCV strains, efficient cleavage at this site in vitro was reported to require synthesis of the protein precursor in the presence of CMMs (63). In the case of HCV strain H77, cleavages at the NS2-3 and NS3-4A junctions and the ultimate NS3 release occur efficiently in the absence of added CMMs in both Krebs-2 S10 and RRL (Fig. 1). Given the remarkably high stability of NS2-3 in vitro (Fig. 7A and B), the bulk of mature NS2 and NS3 should arise from processing of the nascent chain. Previous studies also noted that the posttranslational NS2-3 autocleavage is inefficient, unless special conditions (such as the presence of detergents) are met (56, 78).
Our results conform to the accepted pathway of HCV polyprotein processing by NS3 protease (42). For most proteins, there is no clear precursor-product relationship (Fig. 7C), in agreement with the results obtained in vivo with the use of the recombinant vaccinia virus system (1). Interestingly, compound A failed to completely inhibit the release of NS3 and NS5B from the nascent precursor polypeptides (Fig. 6B). Previous studies indicated that there is a temporal hierarchy in NS3-mediated polyprotein processing events (1, 21). The NS3-NS4A cleavage is believed to be the first event in the processing cascade, and in contrast to other cleavages, is an intramolecular reaction (1). This, as well as the fact that processing at the NS2-NS3 junction is carried out autocatalytically by a distinct protease (NS2-3) (9), could account for the partial resistance of NS3 appearance to compound A. Cleavage at the NS5A-NS5B junction has been reported to be more efficient than at other sites (1, 70). This peculiarity may be responsible for the inability of compound A to completely block NS5B appearance. Our data are not compatible with the notion that NS5A and NS5B mature in the cytosol and target membranes posttranslationally (cf. references 5 and 64). As shown here (Fig. 5A), only cleavages at NS2-NS3 and NS3-NS4A junctions are CMM independent, whereas little NS4B, NS5A, or NS5B is formed upon HCV RNA translation in the absence of CMMs. NS4B, NS5A, and NS5B are integral membrane proteins associated with the ER and Golgi apparatus (5, 13, 25, 27, 48, 64). We presume that membrane association of the corresponding precursor polypeptides is a prerequisite for maturation of these proteins. In this regard, membrane targeting of NS3 via interaction with NS4A (87) could be responsible for the fast cleavage at the NS4A-4B, NS4B-5A, and NS5A-5B junctions due to the proximity of NS3 and the substrate even after the excision of NS3 from the nascent chain (cf. reference 2). Figure 7A reveals, in fact, that there is a time interval of only 20 min between the release of NS3 and the appearance of the downstream products. It should be noted that NS3-dependent processing, in contrast to that catalyzed by signal peptidases, should occur at the cytosolic rather than the lumen side of the ER membrane. In addition, NS protein precursors are likely to target membranes directly via the amphipathic helixes in NS4B and NS5A or the hydrophobic insertion sequence in NS5B (5, 13, 14, 48, 64). In contrast, structural protein precursors exploit the signal recognition particle-dependent pathway for membrane association and processing (62). Deeper understanding of the mechanism by which membranes facilitate maturation of the HCV NS proteins is needed, as it could guide the design of novel inhibitors of NS3.
Glycosylation of HCV E1 and E2.
As evidenced by a dramatic increase in electrophoretic mobility upon PNGase treatment, both E1 and E2-p7 in vitro are heavily modified by N-linked glycosylation (Fig. 4B and C). Western blot analysis of the products of full-size HCV RNA translation revealed four products differing in the extent of glycosylation of E1 antigens, p29 (E1), p25 (E1*), p22 (E1**), and p18 (E1***). Notably, four similar glycosylation forms of E1 are synthesized in PK-15 cells transiently expressing HCV glycoproteins (Fig. 2B in reference 12). Given that band E1 is already prominent after a 20-min translation, all E1 maturation steps, including processing, translocation into the microsomal vesicles, and glycosylation with high-mannose oligosaccharides, proceed very fast. E1*** appeared later than E1 (after 40 min) and was stable upon subsequent incubation; moreover, no redistribution of E1 forms was seen upon chase (Fig. 7A and B). The reasons why a sizable fraction of E1 glycosylation is not coupled with processing are not known. Alternatively, E1*, E1**, and E1*** may result from partial or complete deglycosylation of E1 within microsomal vesicles. We also noticed that only fully glycosylated E1 appeared upon translation of a subgenomic C-E1 HCV RNA. Hence, E2 synthesis, or at least ribosomal traversing of the HCV RNA sequence downstream of the E1 gene, may be required for the production of deglycosylated and hypoglycosylated E1 forms. In contrast, the glycosylation of E1 in vivo was reported to improve upon expression of the downstream amino acid sequence (11). The reason for these differences is not clear. E2 in vitro appeared as a heavily glycosylated fusion protein, E2-p7 (Fig. 4C, top). Its high heterogeneity is consistent with the presence of the multiple glycosylation forms. However, little E2-p7 lacking carbohydrate was evident, in contrast to the situation with E1. For comparison, E2-p7 that is expressed in vivo migrates as a broad but well-defined ∼72-kDa band (36). Thus, our in vitro system is deficient in providing uniform and complete glycosylation of E2, as well as its release from the E2-p7 precursor.
In summary, we have described a robust in vitro system that allows us to translate HCV RNA completely and accurately and to follow polyprotein processing. Our studies demonstrate the utility of this system for assaying the efficiency and specificity of NS3 protease inhibitors. They also underscore the importance of membranes for sequence-specific cleavage of the C-terminal half of the HCV polyprotein. Despite efficient expression of NS5B and other components of the HCV RNA replication complex in Krebs-2 S10, our attempts to detect [32P]CTP incorporation into minus- and plus-strand HCV RNAs in this system were not successful (data not shown). It is possible that Krebs-2 cells lack some host factors that are required for HCV RNA replication. Given that Huh7 cells efficiently support replication of both subgenomic and full-length HCV replicons (4, 37, 38, 57), they appear to be superior to Krebs-2 cells as a potential source of HCV RNA replication-competent extracts. It would also be interesting to study whether these systems can support HCV RNA encapsidation, which has been recently recapitulated in vitro (32).
Acknowledgments
We thank Jens Bukh and Robert Purcell for the pCV-H77C clone; Ralf Bartenschlager, Michinori Kohara, Darius Moradpour, Jane A. McKeating, and Michael Houghton for the antibodies against HCV proteins and other reagents; Daniel Lamarre and Michael G. Cordingley for compound A and helpful discussions; and Colin Lister for excellent technical assistance.
This work was supported by grants from the Canadian Institute of Health Research (CIHR) to A.P. and N.S. M.L.-L. was the recipient of a CIHR postdoctoral fellowship. N.S. is a CIHR Distinguished Scientist and a Howard Hughes Medical Institute International Scholar.
REFERENCES
- 1.Bartenschlager, R., L. Ahlborn-Laake, J. Mous, and H. Jacobsen. 1994. Kinetic and structural analyses of hepatitis C virus polyprotein processing. J. Virol. 68:5045-5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartenschlager, R., and V. Lohmann. 2000. Replication of hepatitis C virus. J. Gen. Virol. 81:1631-1648. [DOI] [PubMed] [Google Scholar]
- 3.Barton, D. J., B. J. Morasco, and J. B. Flanegan. 1996. Assays for poliovirus polymerase, 3DPol, and authentic RNA replication in HeLa S10 extracts. Methods Enzymol. 275:35-57. [DOI] [PubMed] [Google Scholar]
- 4.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972-1974. [DOI] [PubMed] [Google Scholar]
- 5.Brass, V., E. Bieck, R. Montserret, B. Wolk, J. A. Hellings, H. E. Blum, F. Penin, and D. Moradpour. 2002. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 277:8130-8139. [DOI] [PubMed] [Google Scholar]
- 6.Campbell, E. A., and R. J. Jackson. 1983. Processing of the encephalomyocarditis virus capsid precursor protein studied in rabbit reticulocyte lysates incubated with N-formyl-[35S]methionine-tRNAfMet. J. Virol. 45:439-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrere-Kremer, S., C. Montpellier, L. Lorenzo, B. Brulin, L. Cocquerel, S. Belouzard, F. Penin, and J. Dubuisson. 2004. Regulation of hepatitis C virus polyprotein processing by signal peptidase involves structural determinants at the p7 sequence junctions. J. Biol. Chem. 279:41384-41392. [DOI] [PubMed] [Google Scholar]
- 8.Choi, J., Z. Xu, and J. H. Ou. 2003. Triple decoding of hepatitis C virus RNA by programmed translational frameshifting. Mol. Cell. Biol. 23:1489-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Francesco, R., P. Neddermann, L. Tomei, C. Steinkuhler, P. Gallinari, and A. Folgori. 2000. Biochemical and immunologic properties of the nonstructural proteins of the hepatitis C virus: implications for development of antiviral agents and vaccines. Semin. Liver Dis. 20:69-83. [DOI] [PubMed] [Google Scholar]
- 10.Dorner, A. J., B. L. Semler, R. J. Jackson, R. Hanecak, E. Duprey, and E. Wimmer. 1984. In vitro translation of poliovirus RNA: utilization of internal initiation sites in reticulocyte lysate. J. Virol. 50:507-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubuisson, J., S. Duvet, J. C. Meunier, A. Op De Beeck, R. Cacan, C. Wychowski, and L. Cocquerel. 2000. Glycosylation of the hepatitis C virus envelope protein E1 is dependent on the presence of a downstream sequence on the viral polyprotein. J. Biol. Chem. 275:30605-30609. [DOI] [PubMed] [Google Scholar]
- 12.Dubuisson, J., H. H. Hsu, R. C. Cheung, H. B. Greenberg, D. G. Russell, and C. M. Rice. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68:6147-6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elazar, M., K. H. Cheong, P. Liu, H. B. Greenberg, C. M. Rice, and J. S. Glenn. 2003. Amphipathic helix-dependent localization of NS5A mediates hepatitis C virus RNA replication. J. Virol. 77:6055-6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elazar, M., P. Liu, C. M. Rice, and J. S. Glenn. 2004. An N-terminal amphipathic helix in hepatitis C virus (HCV) NS4B mediates membrane association, correct localization of replication complex proteins, and HCV RNA replication. J. Virol. 78:11393-11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flint, M., C. Maidens, L. D. Loomis-Price, C. Shotton, J. Dubuisson, P. Monk, A. Higginbottom, S. Levy, and J. A. McKeating. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73:6235-6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao, L., H. Aizaki, J. W. He, and M. M. Lai. 2004. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 78:3480-3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gosert, R., D. Egger, V. Lohmann, R. Bartenschlager, H. E. Blum, K. Bienz, and D. Moradpour. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 77:5487-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grakoui, A., C. Wychowski, C. Lin, S. M. Feinstone, and C. M. Rice. 1993. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67:1385-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hershey, J. W. B., and W. C. Merrick. 2000. Pathway and mechanism of initiation of protein synthesis, p. 33-88. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 20.Hijikata, M., N. Kato, Y. Ootsuyama, M. Nakagawa, and K. Shimotohno. 1991. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. USA 88:5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hijikata, M., H. Mizushima, Y. Tanji, Y. Komoda, Y. Hirowatari, T. Akagi, N. Kato, K. Kimura, and K. Shimotohno. 1993. Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc. Natl. Acad. Sci. USA 90:10773-10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Honda, M., E. A. Brown, and S. M. Lemon. 1996. Stability of a stem-loop involving the initiator AUG controls the efficiency of internal initiation of translation on hepatitis C virus RNA. RNA 2:955-968. [PMC free article] [PubMed] [Google Scholar]
- 23.Honda, M., L. H. Ping, R. C. Rijnbrand, E. Amphlett, B. Clarke, D. Rowlands, and S. M. Lemon. 1996. Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology 222:31-42. [DOI] [PubMed] [Google Scholar]
- 24.Honda, M., R. Rijnbrand, G. Abell, D. Kim, and S. M. Lemon. 1999. Natural variation in translational activities of the 5′ nontranslated RNAs of hepatitis C virus genotypes 1a and 1b: evidence for a long-range RNA-RNA interaction outside of the internal ribosomal entry site. J. Virol. 73:4941-4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hugle, T., F. Fehrmann, E. Bieck, M. Kohara, H. G. Krausslich, C. M. Rice, H. E. Blum, and D. Moradpour. 2001. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology 284:70-81. [DOI] [PubMed] [Google Scholar]
- 26.Ishida, S., M. Kaito, M. Kohara, K. Tsukiyama-Kohora, N. Fujita, J. Ikoma, Y. Adachi, and S. Watanabe. 2001. Hepatitis C virus core particle detected by immunoelectron microscopy and optical rotation technique. Hepatol. Res. 20:335-347. [DOI] [PubMed] [Google Scholar]
- 27.Ivashkina, N., B. Wolk, V. Lohmann, R. Bartenschlager, H. E. Blum, F. Penin, and D. Moradpour. 2002. The hepatitis C virus RNA-dependent RNA polymerase membrane insertion sequence is a transmembrane segment. J. Virol. 76:13088-13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson, R. J. 1991. Potassium salts influence the fidelity of mRNA translation initiation in rabbit reticulocyte lysates: unique features of encephalomyocarditis virus RNA translation. Biochim. Biophys. Acta 1088:345-358. [DOI] [PubMed] [Google Scholar]
- 29.Ji, H., C. S. Fraser, Y. Yu, J. Leary, and J. A. Doudna. 2004. Coordinated assembly of human translation initiation complexes by the hepatitis C virus internal ribosome entry site RNA. Proc. Natl. Acad. Sci. USA 101:16990-16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kazachkov, Y. A., T. V. Chernovskaya, E. Siyanova, Y. V. Svitkin Yu, T. Ugarova, and Y. V. I. Agol. 1982. Leader polypeptides encoded in the 5′-region of the encephalomyocarditis virus genome. FEBS Lett. 141:153-156. [DOI] [PubMed] [Google Scholar]
- 31.Kieft, J. S., K. Zhou, R. Jubin, and J. A. Doudna. 2001. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA 7:194-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein, K. C., S. J. Polyak, and J. R. Lingappa. 2004. Unique features of hepatitis C virus capsid formation revealed by de novo cell-free assembly. J. Virol. 78:9257-9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koch, J. O., and R. Bartenschlager. 1999. Modulation of hepatitis C virus NS5A hyperphosphorylation by nonstructural proteins NS3, NS4A, and NS4B. J. Virol. 73:7138-7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koev, G., R. F. Duncan, and M. M. Lai. 2002. Hepatitis C virus IRES-dependent translation is insensitive to an eIF2α-independent mechanism of inhibition by interferon in hepatocyte cell lines. Virology 297:195-202. [DOI] [PubMed] [Google Scholar]
- 35.Kolupaeva, V. G., T. V. Pestova, and C. U. Hellen. 2000. An enzymatic footprinting analysis of the interaction of 40S ribosomal subunits with the internal ribosomal entry site of hepatitis C virus. J. Virol. 74:6242-6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin, C., B. D. Lindenbach, B. M. Pragai, D. W. McCourt, and C. M. Rice. 1994. Processing in the hepatitis C virus E2-NS2 region: identification of p7 and two distinct E2-specific products with different C termini. J. Virol. 68:5063-5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lohmann, V., S. Hoffmann, U. Herian, F. Penin, and R. Bartenschlager. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007-3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lohmann, V., F. Körner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110-113. [DOI] [PubMed] [Google Scholar]
- 39.Luo, K. X., T. R. Hurley, and B. M. Sefton. 1991. Cyanogen bromide cleavage and proteolytic peptide mapping of proteins immobilized to membranes. Methods Enzymol. 201:149-152. [DOI] [PubMed] [Google Scholar]
- 40.Lyapustin, V. N., Y. V. Svitkin, T. Y. Ugarova, V. A. Lashkevich, and V. I. Agol. 1986. A tentative model of formation of structural proteins of tick-borne encephalitis virus (flavivirus). FEBS Lett. 200:314-316. [DOI] [PubMed] [Google Scholar]
- 41.Lytle, J. R., L. Wu, and H. D. Robertson. 2002. Domains on the hepatitis C virus internal ribosome entry site for 40S subunit binding. RNA 8:1045-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Major, M. E., and S. M. Feinstone. 1997. The molecular virology of hepatitis C. Hepatology 25:1527-1538. [DOI] [PubMed] [Google Scholar]
- 43.Maley, F., R. B. Trimble, A. L. Tarentino, and T. H. Plummer, Jr. 1989. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 180:195-204. [DOI] [PubMed] [Google Scholar]
- 44.Mathews, M. B., and M. Osborn. 1974. The rate of polypeptide chain elongation in a cell-free system from Krebs II ascites cells. Biochim. Biophys. Acta 340:147-152. [DOI] [PubMed] [Google Scholar]
- 45.Molla, A., A. V. Paul, and E. Wimmer. 1991. Cell-free, de novo synthesis of poliovirus. Science 254:1647-1651. [DOI] [PubMed] [Google Scholar]
- 46.Monckton, R. P., and E. G. Westaway. 1982. Restricted translation of the genome of the flavivirus Kunjin in vitro. J. Gen. Virol. 63:227-232. [DOI] [PubMed] [Google Scholar]
- 47.Moradpour, D., E. Bieck, T. Hugle, W. Wels, J. Z. Wu, Z. Hong, H. E. Blum, and R. Bartenschlager. 2002. Functional properties of a monoclonal antibody inhibiting the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 277:593-601. [DOI] [PubMed] [Google Scholar]
- 48.Moradpour, D., V. Brass, E. Bieck, P. Friebe, R. Gosert, H. E. Blum, R. Bartenschlager, F. Penin, and V. Lohmann. 2004. Membrane association of the RNA-dependent RNA polymerase is essential for hepatitis C virus RNA replication. J. Virol. 78:13278-13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neddermann, P., A. Clementi, and R. De Francesco. 1999. Hyperphosphorylation of the hepatitis C virus NS5A protein requires an active NS3 protease, NS4A, NS4B, and NS5A encoded on the same polyprotein. J. Virol. 73:9984-9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oberg, B. F., and A. J. Shatkin. 1972. Initiation of picornavirus protein synthesis in ascites cell extracts. Proc. Natl. Acad. Sci. USA 69:3589-3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pause, A., G. Kukolj, M. Bailey, M. Brault, F. Do, T. Halmos, L. Lagace, R. Maurice, M. Marquis, G. McKercher, C. Pellerin, L. Pilote, D. Thibeault, and D. Lamarre. 2003. An NS3 serine protease inhibitor abrogates replication of subgenomic hepatitis C virus RNA. J. Biol. Chem. 278:20374-20380. [DOI] [PubMed] [Google Scholar]
- 52.Pause, A., N. Méthot, Y. Svitkin, W. C. Merrick, and N. Sonenberg. 1994. Dominant negative mutants of mammalian translation initiation factor eIF-4A define a critical role for eIF-4F in cap-dependent and cap-independent initiation of translation. EMBO J. 13:1205-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pestova, T. V., and C. U. Hellen. 2003. Translation elongation after assembly of ribosomes on the Cricket paralysis virus internal ribosomal entry site without initiation factors or initiator tRNA. Genes Dev. 17:181-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pestova, T. V., C. U. Hellen, and I. N. Shatsky. 1996. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol. Cell. Biol. 16:6859-6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pestova, T. V., I. N. Shatsky, S. P. Fletcher, R. J. Jackson, and C. U. Hellen. 1998. A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev. 12:67-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pieroni, L., E. Santolini, C. Fipaldini, L. Pacini, G. Migliaccio, and N. La Monica. 1997. In vitro study of the NS2-3 protease of hepatitis C virus. J. Virol. 71:6373-6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pietschmann, T., V. Lohmann, A. Kaul, N. Krieger, G. Rinck, G. Rutter, D. Strand, and R. Bartenschlager. 2002. Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol. 76:4008-4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reed, K. E., and C. M. Rice. 2000. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr. Top. Microbiol. Immunol. 242:55-84. [DOI] [PubMed] [Google Scholar]
- 59.Reynolds, J. E., A. Kaminski, H. J. Kettinen, K. Grace, B. E. Clarke, A. R. Carroll, D. J. Rowlands, and R. J. Jackson. 1995. Unique features of internal initiation of hepatitis C virus RNA translation. EMBO J. 14:6010-6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rivas-Estilla, A. M., Y. Svitkin, M. Lopez Lastra, M. Hatzoglou, A. Sherker, and A. E. Koromilas. 2002. PKR-dependent mechanisms of gene expression from a subgenomic hepatitis C virus clone. J. Virol. 76:10637-10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roussel, J., A. Pillez, C. Montpellier, G. Duverlie, A. Cahour, J. Dubuisson, and C. Wychowski. 2003. Characterization of the expression of the hepatitis C virus F protein. J. Gen. Virol. 84:1751-1759. [DOI] [PubMed] [Google Scholar]
- 62.Santolini, E., G. Migliaccio, and N. La Monica. 1994. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J. Virol. 68:3631-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santolini, E., L. Pacini, C. Fipaldini, G. Migliaccio, and N. Monica. 1995. The NS2 protein of hepatitis C virus is a transmembrane polypeptide. J. Virol. 69:7461-7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmidt-Mende, J., E. Bieck, T. Hugle, F. Penin, C. M. Rice, H. E. Blum, and D. Moradpour. 2001. Determinants for membrane association of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 276:44052-44063. [DOI] [PubMed] [Google Scholar]
- 65.Selby, M., A. Erickson, C. Dong, S. Cooper, P. Parham, M. Houghton, and C. M. Walker. 1999. Hepatitis C virus envelope glycoprotein E1 originates in the endoplasmic reticulum and requires cytoplasmic processing for presentation by class I MHC molecules. J. Immunol. 162:669-676. [PubMed] [Google Scholar]
- 66.Selby, M. J., E. Glazer, F. Masiarz, and M. Houghton. 1994. Complex processing and protein:protein interactions in the E2:NS2 region of HCV. Virology 204:114-122. [DOI] [PubMed] [Google Scholar]
- 67.Smith, A. E. 1973. The initiation of protein synthesis directed by the RNA from encephalomyocarditis virus. Eur. J. Biochem. 33:301-313. [DOI] [PubMed] [Google Scholar]
- 68.Stanley, W. M., Jr. 1974. Specific aminoacylation of the methionine-specific tRNA's of eukaryotes. Methods Enzymol. 29:530-547. [DOI] [PubMed] [Google Scholar]
- 69.Steinkuhler, C., G. Biasiol, M. Brunetti, A. Urbani, U. Koch, R. Cortese, A. Pessi, and R. De Francesco. 1998. Product inhibition of the hepatitis C virus NS3 protease. Biochemistry 37:8899-8905. [DOI] [PubMed] [Google Scholar]
- 70.Steinkuhler, C., A. Urbani, L. Tomei, G. Biasiol, M. Sardana, E. Bianchi, A. Pessi, and R. De Francesco. 1996. Activity of purified hepatitis C virus protease NS3 on peptide substrates. J. Virol. 70:6694-6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Svitkin, Y. V., H. Imataka, K. Khaleghpour, A. Kahvejian, H. D. Liebig, and N. Sonenberg. 2001. Poly(A)-binding protein interaction with eIF4G stimulates picornavirus IRES-dependent translation. RNA 7:1743-1752. [PMC free article] [PubMed] [Google Scholar]
- 72.Svitkin, Y. V., V. N. Lyapustin, V. A. Lashkevich, and V. I. Agol. 1984. Differences between translation products of tick-borne encephalitis virus RNA in cell-free systems from Krebs-2 cells and rabbit reticulocytes: involvement of membranes in the processing of nascent precursors of flavivirus structural proteins. Virology 135:536-541. [DOI] [PubMed] [Google Scholar]
- 73.Svitkin, Y. V., A. Pause, A. Haghighat, S. Pyronnet, G. Witherell, G. J. Belsham, and N. Sonenberg. 2001. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 7:382-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Svitkin, Y. V., and N. Sonenberg. 2003. Cell-free synthesis of encephalomyocarditis virus. J. Virol. 77:6551-6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Svitkin, Y. V., and N. Sonenberg. 2004. An efficient system for cap- and poly(A)-dependent translation in vitro. Methods Mol. Biol. 257:155-170. [DOI] [PubMed] [Google Scholar]
- 76.Svitkin, Y. V., T. Y. Ugarova, T. V. Chernovskaya, V. N. Lyapustin, V. A. Lashkevich, and V. I. Agol. 1981. Translation of tick-borne encephalitis virus (flavivirus) genome in vitro: synthesis of two structural polypeptides. Virology 110:26-34. [DOI] [PubMed] [Google Scholar]
- 77.Tan, S. L., A. Pause, Y. Shi, and N. Sonenberg. 2002. Hepatitis C therapeutics: current status and emerging strategies. Nat. Rev. Drug Discov. 1:867-881. [DOI] [PubMed] [Google Scholar]
- 78.Thibeault, D., R. Maurice, L. Pilote, D. Lamarre, and A. Pause. 2001. In vitro characterization of a purified NS2/3 protease variant of hepatitis C virus. J. Biol. Chem. 276:46678-46684. [DOI] [PubMed] [Google Scholar]
- 79.Tsantrizos, Y. S., G. Bolger, P. Bonneau, D. R. Cameron, N. Goudreau, G. Kukolj, S. R. LaPlante, M. Llinas-Brunet, H. Nar, and D. Lamarre. 2003. Macrocyclic inhibitors of the NS3 protease as potential therapeutic agents of hepatitis C virus infection. Angew. Chem. Int. 42:1356-1360. [DOI] [PubMed] [Google Scholar]
- 80.Tsukiyama-Kohara, K., N. Iizuka, M. Kohara, and A. Nomoto. 1992. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 66:1476-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Varaklioti, A., N. Vassilaki, U. Georgopoulou, and P. Mavromara. 2002. Alternate translation occurs within the core coding region of the hepatitis C viral genome. J. Biol. Chem. 277:17713-17721. [DOI] [PubMed] [Google Scholar]
- 82.Villa-Komaroff, L., N. Guttman, D. Baltimore, and H. F. Lodish. 1975. Complete translation of poliovirus RNA in a eukaryotic cell-free system. Proc. Natl. Acad. Sci. USA 72:4157-4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Walewski, J. L., T. R. Keller, D. D. Stump, and A. D. Branch. 2001. Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA 7:710-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Walter, P., and G. Blobel. 1983. Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol. 96:84-93. [DOI] [PubMed] [Google Scholar]
- 85.Wengler, G., and M. Beato. 1979. In vitro translation of 42 S virus-specific RNA from cells infected with the flavivirus West Nile virus. Virology 96:516-529. [DOI] [PubMed] [Google Scholar]
- 86.Wilson, J. E., T. V. Pestova, C. U. Hellen, and P. Sarnow. 2000. Initiation of protein synthesis from the A site of the ribosome. Cell 102:511-520. [DOI] [PubMed] [Google Scholar]
- 87.Wolk, B., D. Sansonno, H. G. Krausslich, F. Dammacco, C. M. Rice, H. E. Blum, and D. Moradpour. 2000. Subcellular localization, stability, and trans-cleavage competence of the hepatitis C virus NS3-NS4A complex expressed in tetracycline-regulated cell lines. J. Virol. 74:2293-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu, Z., J. Choi, T. S. Yen, W. Lu, A. Strohecker, S. Govindarajan, D. Chien, M. J. Selby, and J. Ou. 2001. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J. 20:3840-3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yanagi, M., R. H. Purcell, S. U. Emerson, and J. Bukh. 1997. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl. Acad. Sci. USA 94:8738-8743. [DOI] [PMC free article] [PubMed] [Google Scholar]