

Abstract
The UL51 gene of herpes simplex virus type 1 (HSV-1) encodes a phosphoprotein whose homologs are conserved throughout the herpes virus family. Recently, we reported that UL51 protein colocalizes with Golgi marker proteins in transfected cells and that targeting of UL51 protein to the Golgi apparatus depends on palmitoylation of its N-terminal cysteine at position 9 (N. Nozawa, T. Daikoku, T. Koshizuka, Y. Yamauchi, T. Yoshikawa, and Y. Nishiyama, J. Virol. 77:3204-3216, 2003). However, its role in the HSV replication cycle was unknown. Here, we generated UL51-null mutants (FDL51) in HSV-1 to uncover the function of UL51 protein. We show that the mutant plaques were much smaller in size and that maximal titers were reduced nearly 100-fold compared to wild-type virus. Electron microscopy indicated that the formation of nucleocapsids was not affected by the deletion of UL51 but that viral egress from the perinuclear space was severely compromised. In FDL51-infected cells, a large number of enveloped nucleocapsids were observed in the perinuclear space, but enveloped mature virions in the cytoplasm, as well as extracellular mature virions, were rarely detected. These defects were fully rescued by reinsertion of the UL51 gene. These results indicate that UL51 protein is involved in the maturation and egress of HSV-1 virus particles downstream of the initial envelopment step.
Herpes simplex virus (HSV) is a large DNA virus comprised of an icosahedral capsid surrounded by a protein tegument layer and a host-derived membrane or envelope (41). Although the tegument has long been considered an amorphous mass of proteins, recent data have shown that it has an ordered structure and forms an asymmetric cap in mature HSV type 1 (HSV-1) virions (22, 49). Furthermore, several of the 15 or more viral gene products comprising the tegument of mature extracellular HSV-1 virions have been shown to aid in the initiation of the viral replication cycle (34, 41).
UL51 is a component of the tegument (13, 34, 41) that is conserved among herpesvirus family members but is dispensable for HSV-1 replication in cell culture (1, 2, 7, 11, 15, 19, 23, 31, 46). We previously showed that the UL51 gene encodes virion-associated phosphoproteins with molecular masses of 27, 29, and 30 kDa that are expressed during the late phase of infection and, like most tegument proteins (41), localize to the perinuclear region of the cytoplasm in infected cells (13). We also reported that UL51 proteins colocalize with Golgi marker proteins in UL51-transfected cells but colocalize only partially with the Golgi during HSV infection (37). Targeting of UL51 protein to the Golgi apparatus is regulated by posttranslational modification of the protein; specifically, palmitoylation of the N-terminal cysteine at position 9 (Cys-9) of the protein is important for targeting to the Golgi apparatus. UL51 protein is eventually incorporated into virions, localizing primarily to the inside of the viral envelope (37).
HSV egress is a two-stage process whereby nucleocapsid is released into the cytoplasm following envelopment at the nuclear membrane and then undergoes secondary envelopment by Golgi-derived compartments (34, 43). Although this process is still poorly understood, palmitoylated proteins have been shown to play an important role in either virus assembly or egress in a number of viruses, including vaccinia and sindbis virus (21, 25). Given that UL51 gene products are palmitoylated, it is possible that these proteins play a role in virus assembly or egress in infected cells. Indeed, a recombinant virus with a deletion spanning the entire open reading frame (ORF) of UL50 and part of the UL51 ORF (the C-terminal 202 amino acid residues) shows impaired growth compared with wild-type or UL50-deficient virus (6). Although these data imply that UL51 plays a role in viral replication in cell culture, the mechanism by which UL51 gene products act in viral replication remained unclear.
In this study, we generated a recombinant virus in which only the UL51 gene is deleted in order to determine the role of the UL51 gene product during viral replication. Studies with the recombinant virus revealed that the UL51 is involved in the maturation process of virus particles downstream of the initial envelopment.
MATERIALS AND METHODS
Cells and viruses.
Rabbit skin cells and Vero cells used were previously described (27, 28). Wild-type HSV-1(F), a limited-passage isolate, was the prototype strain used in this study (27, 28). YK304 was reconstituted from pYEbac102 containing a full length of infectious clone of HSV-1(F) in which the bacterial artificial chromosome (BAC) sequence was inserted into the intergenic region between UL3 and UL4. YK304 shows the phenotypes identical to those of wild-type HSV-1(F) in vitro and in vivo (45).
Plasmids.
A cosmid containing nucleotides 77933 to 116016 of HSV-1(F) (pBC1007, GenBank accession no. X14112) was kindly provided by Bernard Roizman. pBS1007 was generated as follows: a 3.3-kb BglII-EcoRI fragment of pBC1007 encompassing the entire ORF of UL50 and one-third of the UL52 ORF at the 5′ end (nucleotides 106750 to 110095) was blunt-ended by using T4 DNA polymerase and cloned into blunt-ended EcoRI-HindIII sites in pBluescript II KS(+) (Stratagene).
The Flp recombinase expression plasmid pCP20Zeo was constructed by PCR amplifying the zeocin resistance gene from the pcDNA4/HisMax C cloning vector (Invitrogen) using 5′-CGGATCCTGATCAGCACGTGTTGACAATTAATC-3′ as the forward primer and 5′-GACTGCAGCGCGCTGGAGGATCATCCAGCCGGCG-3′ as the reverse primer. The PCR product was digested with BamHI and PstI and ligated into the BamHI and PstI sites of pCP20 (kindly provided by W. Wackernagel of Universitat Oldenburg, Germany [12]). pGEX-49N and pGEX-49C were constructed by cloning PCR-amplified fragments encoding UL49 amino acids 1 to 167 and 135 to 301, respectively.
Construction of the recombinant viruses.
The mutagenesis procedure was performed as previously described (48), with slight modifications for alteration of the UL51 coding region in pYEbac102. Briefly, linear fragments were prepared by PCR using primer pairs containing 20 to 34 nucleotides for amplification of a kanamycin resistance gene from vector pCR2.1 (Invitrogen), 34 nucleotides of the FLP recognition target (FRT) sequence, and an additional 50 nucleotides homologous to the sequences flanking the HSV-1(F) UL51: 5′-cccctccatcccacaaacacaaaacacacgggttggatgaaaacacgcatGAAGTTCCTATTCTCTAG-AAAGTATAGGAACTTCgacagcaagcgaaccggaat-3′ as the forward primer and 5′-ccctcggaggcggagccgcggctgcaggaggccctggcggtcgttaacgcGAAGTTCCTATACTTTCTA-GAGAATAGGAACTTCcggaaatgttgaatactcatactcttcctttttc-3′ as the reverseprimer (FRT sequences are shown in capital letters). The linear PCR-generated fragments were then electroporated into Escherichia coli DH10Β YEbac201. YEbac201 harbors pYEbac102 (45) and pGETrec, which encodes recombinases E and T under the control of an arabinose-inducible promoter (a kind gift from P. Ioannou, Murdoch Children Research Institute, Australia). Electroporated bacteria were incubated at 37°C for 4 h and plated onto agar plates containing ampicillin (50 μg/ml), chloramphenicol (20 μg/ml), and kanamycin (20 μg/ml) to select for E. coli cells carrying the kanamycin resistance gene inserted into the UL51 locus. HSV-BAC DNA was isolated as previously described (45) and analyzed by PCR with primers that correspond to the UL51 gene of HSV-1, which led to the identification of the mutant HSV-BAC plasmid pFDL51Kan. To remove the kanamycin resistance gene from the mutant HSV-BAC plasmid, the Flp expression plasmid pCP20Zeo was electroporated into E. coli FDL51bacKan, which harbor pFDL51Kan. The bacteria were plated on chloramphenicol-zeocin (20 μg/ml) plates and grown overnight at 30°C. The plates were further incubated at 37°C overnight to remove pCP20Zeo. To confirm the loss of the kanamycin resistance gene, the colonies were restreaked in duplicate on chloramphenicol and choloramphenicol-kanamycin plates and grown overnight at 37°C. Chloramphenicol-resistant and kanamycin-sensitive colonies were selected, and mutated HSV-BAC DNA was extracted and analyzed by PCR to confirm loss of the kanamycin resistance gene. One clone, called pFDL51, was chosen for further analysis.
To generate the UL51 deletion virus FDL51, 1 μg of pFDL51 was transfected into rabbit skin cells in 12-well plates by calcium phosphate precipitation (20) and incubated at 37°C. Viral plaques were observed 6 or 7 days after transfection, and recombinant viruses were plaque purified three times. In the recombinant virus FDL51R, the UL51 sequences deleted from FDL51 were restored by cotransfection of FDL51 viral DNA with the plasmid pBS1007 DNA. Plaques were isolated and purified and analyzed by PCR.
Southern blotting.
Viral DNA was digested with AscI, subjected to electrophoresis on 0.7% agarose gels, and transferred to Hybond-N+ nylon membranes (Amersham Pharmacia). The blots were hybridized with the appropriate DNA probe labeled with horseradish peroxidase using enhanced chemiluminescence direct nucleic acid labeling and detection systems (Amersham Bioscience) according to the manufacturer's instructions.
Real-time quantitative PCR with a fluorogenic probe.
The target sequence of each gene for this assay was selected from conserved regions in alpha-herpesviridae, and primers were designed using the software Primer Express (PE Applied Biosystems, Foster City, Calif.). The forward and reverse primer sequences were 5′-CATCGCCTTCCTTCCAAAACG-3′ and 5′-GACATTGTCGTCCGTCGC-3′ for UL50, 5′-CCCGACGCCTCGTGAAG-3′ and 5′-TGCATGGTGAACCTAGAA-CGAC-3′ for UL51, and 5′-CTCGTTTGCCGCCATCAC-3′ and 5′-AGTACGCCCGCGGG-3′ for UL52. The fluorogenic TaqMan probes with a sequence located between the PCR primers for UL50 (5′-GGAGGATGCCGGTTT-3′), UL51 (5′-CCGCACGTACCACGCC-3′), and UL52 (5′-GGTTCTGTTGCACGAG-3′) were synthesized by PE Applied Biosystems. Reactions were performed in a 50-μl reaction volume with 500 ng of DNase-treated total RNA prepared from each HSV mutant-infected cell line, using the TaqMan Universal master mix (PE Applied Biosystems). Universal thermocycler conditions were used according to the manufacturer's instructions (ABI Prism 7700 sequence detection system; PE Applied Biosystems). Template-negative and reverse transcriptase (RT)-negative reactions served as controls. All samples were run in duplicate, together with reactions to quantify the expression of an internal control gene, 18S rRNA (product no. Hs99999901_s1; PE Applied Biosystems), to normalize for any differences in the amount of total RNA added (44). The copy number of viral RNA transcripts was calculated based on the molecular mass of the plasmid pBS1007, which encoded the target HSV-1 genes, as follows: the cycle threshold (CT) values from samples were plotted on the standard curve, and the copy number was calculated automatically by using Sequence Detector version 1.6 (PE Applied Biosystems), a software package for data analysis. Each sample was tested in duplicate or triplicate, and the mean of the values was taken as the copy number of the sample.
Western blotting and electron microscopy.
Western blotting and electron microscopy were performed as previously described (37).
Antibodies.
Anti-VP22 rabbit polyclonal antibody was prepared as described elsewhere (42). Briefly, two rabbits were immunized with GST-VP22 fusion protein that was expressed and purified from E. coli transformed with pGEX-49N or pGEX49C. Generation of antibodies to UL51, UL46, VP5, and VP16 was previously described (13, 26, 37).
Determination of plaque size.
Confluent monolayers of Vero cells in culture dishes were infected with mutant virus and incubated for 2 h at 37°C for virus adsorption. The medium was replaced with newly prepared medium supplemented with 2% fetal bovine serum and 160 μg/ml of human immunoglobulin (Sigma) (38), and cells were incubated for an additional 48 h at 37°C. For each virus mutant, 20 plaques were photographed and measured using a microscope equipped with a DS camera control unit DS-L1 (Nikon). Photos were processed and analyzed with Adobe Photoshop software.
One-step and multistep growth analysis.
To examine replication kinetics, Vero cells were infected with FDL51, FDL51R, YK304, and HSV-1(F) at a multiplicity of infection (MOI) of 3 or 0.01 and incubated for 2 h at 37°C to allow for virus adsorption. Thereafter, the cell medium was replaced with newly prepared medium containing 2% fetal bovine serum. Cells and supernatants were harvested at the indicated times after infection. Virus progeny were titrated on Vero cells by plaque assays.
RESULTS
Generation of a UL51 deletion mutant by site-specific mutagenesis in E. coli.
To explore the requirement for UL51 during HSV-1 replication in cultured cells, a UL51 deletion mutant virus was generated in the HSV-1(F) strain using the BAC system of recombination in E. coli, in which the recombinases RecE and RecT mediate recombination between linear and circular DNA molecules (48). A linear recombination fragment containing the kanamycin resistance gene flanked by FRT sites and 50-bp regions homologous to HSV-1 sequences was generated by PCR (Fig. 1A) and introduced into the HSV-1 BAC plasmid pYEbac102 by homologous recombination in E. coli (45). Insertion of the kanamycin resistance gene marker resulted in the deletion of the majority of the UL51 ORF (pFDL51Kan) (Fig. 1B). To avoid any disrupting expression of neighboring genes, we deleted only the C-terminal 202 amino acids (of a total of 244 amino acids) of the predicted product of UL51. Proper insertion of the recombination fragment within the HSV-1 genome was confirmed by sequencing (data not shown) and PCR (Fig. 2A).
FIG. 1.

Construction of the HSV-1 UL51 deletion mutant. A schematic map of the HSV-1 genome showing the long (UL) and short (US) unique regions and the internal repeat (IR) and terminal repeat (TR) sequences. (A) Coding regions of proteins in the genomic region relevant for this study are shown. Arrows indicate transcriptional orientations. A recombination fragment containing the kanamycin resistance gene flanked by FRT sites (circled arrows) and UL51 homology regions is substituted for a substantial portion of the UL51 coding region from nucleotide positions 108277 to 108880. (B) Recombination results in the deletion of UL51 by replacement with the kanamycin resistance gene (FDL51Kan). Location of primers used for PCR genotyping is shown (Fig. 2). (C) Using Flp recombinase, the kanamycin resistance gene was excised, leaving behind one FRT sequence (FDL51). (D) Plasmid pBS1007 contains a BglII-EcoRI fragment of the HSV-1 genome used as a Southern blot probe and for the generation of the standard curve for the quantitative real-time PCR assay (Fig. 2).
FIG. 2.

Genotyping of the UL51 deletion mutant. (A) Agarose gel electrophoresis of PCR products containing the UL51 gene region. BAC plasmid DNA or viral genomic DNA was isolated and analyzed by PCR. Numbers below the gel photos indicate the PCR primer pairs used (Fig. 1B and C). (B) Location of the AscI fragment and schematic diagram of the neighboring loci of UL51. Arrows indicate transcriptional orientations. Thick bar represents the region hybridizing with the probe. (C) Southern blot analysis of AscI-digested DNA isolated from mock-, FDL51-, FDL51R-, YK304-, and HSV-1(F)-infected cells using the EcoRI-HindIII fragment of pBS1007 as a probe (Fig. 1D).
An Flp-expressing plasmid, pCP20Zeo, was next transformed into E. coli harboring the mutant HSV-1 BAC pFDL51Kan in order to induce Flp-mediated excision of the FRT-flanked kanamycin resistance cassette. As shown in Fig. 1C and 2A, the 983-bp decrease in size of the mutant HSV-1 BAC plasmid upon excision of the kanamycin cassette was confirmed by PCR analysis (pFDL51). Further verification of excision was performed by PCR using one primer within the kanamycin cassette, which amplifies products from pFDL51Kan but not pFDL51 (Fig. 1B and 2A). pFDL51 was then transfected into rabbit skin cells, and the UL51 deletion mutant strain (FDL51) was harvested and purified. Following construction of the FDL51 strain, a revertant strain in which the UL51 gene was rescued (FDL51R) was generated by cotransfection of the FDL51 viral DNA with the plasmid pBS1007 DNA containing the entire ORFs of UL50 and UL51 and one-third the ORF of UL52.
Viral genomic DNA was purified from the infected cell lysates, and correct recombination was confirmed by PCR (Fig. 2A, right panel). The PCR product amplified from FDL51 DNA using primer pairs flanking the UL51 ORF was smaller than that of the wild-type virus, as expected (Fig. 1C), while that of FDL51R was the same as that of the wild type (Fig. 2A, right panel). The predicted genome structures of mutant viruses were confirmed by Southern blot analysis using probes to UL50, UL51, and UL52 genes (Fig. 2B). Viral DNA purified from infected cell lysates was digested with AscI and analyzed by Southern blotting using the EcoRI-HindIII fragment of pBS1007 as a probe. As expected, the 6.9- and 7.9-kbp AscI fragments detected in the wild-type parent virus YK304, HSV-1(F), and the revertant virus FDL51R were lost in FDL51 and instead, a new 14-kbp fragment was detected (Fig. 2B and C). These results indicate that part of the UL51 sequence was successfully deleted from the viral genome in the FDL51 mutant. Together with the above data, this confirms earlier data that UL51 is not essential for HSV-1 replication in cell culture.
Expression of genes adjacent to UL51 is unaffected in the UL51 deletion mutant.
Although we engineered our mutant to avoid disrupting expression at neighboring loci, the 5′ end of UL52, a primase essential for viral DNA synthesis, has not yet been mapped (10, 18). In addition, the dUTPase UL50 is just 5′ to UL51 (16, 39). As a result, we next examined whether expression from neighboring loci is influenced by deletion of the UL51 sequence, using real-time PCR to quantitate the expression of UL50 and UL52 in our FDL51 mutant. Plasmid DNA standards were used to generate a standard curve, which was used to calculate the copy number of viral transcripts, as previously described (9, 29). Serially diluted pBS1007 was assayed by real-time PCR, using a standard curve for the CT values from the positive control. The CT value for each sample was calculated by determining the point at which the fluorescence exceeded a threshold limit (10 times the standard deviation from the baseline) (24). A wide linear range (from 34 to 108 copies of the control plasmid) was established (Fig. 3B). Samples lacking either template or reverse transcriptase tested negative for each primer and probe pair, confirming the specificity of the primers and the probe (data not shown).
FIG. 3.

Quantitation of gene expression at loci neighboring UL51. (A) Real-time RT-PCR amplification profile of UL50, UL51, and UL52 mRNA in FDL51-infected Vero cells. Data analysis was performed using ABI Prism 7700 sequence detection system software (PE Applied Biosystems), with CT values determined by automated threshold analysis. The fluorescence intensity collected in real time for each sample was plotted against the number of PCR cycles. The black horizontal line represents the threshold setting, set at 10 standard deviations above the baseline. The CT value is defined as the fractional cycle number in which the fluorescence generated within a reaction has crossed the threshold. This value indicates that a sufficient number of amplicons have accumulated to a statistically significant point above the baseline. CT values correlate inversely with target gene expression. To assess relative differences in gene transcript levels, 18S rRNA was used as an RNA control. (B) Standard linear regression for HSV-1 genes. Serially diluted pBS1007 was amplified as described in Materials and Methods. Amplification was detected, and the CT was analyzed by linear regression analysis. The formula and correlation coefficient used are shown in each panel. (C) Log copy numbers of mRNA from UL51-neighboring genes in HSV mutant-infected cells. Shown are average values from two separate experiments in which samples were run in duplicate. Transcript levels from real-time PCR were converted to mRNA copy number.
Next, Vero cells infected with FDL51, FDL51R, YK304, and HSV-1(F) were harvested at 24 h postinfection and total RNA was extracted from the cells and treated with DNase. As shown in Fig. 3A, all viral transcripts except for the UL51 transcript in FDL51-infected cells were detected in the same volume of total RNA that was prepared from each virus-infected cell. The CT value was converted to the copy number based on the standard curve, and the gene expression in infected cells is shown in Fig. 3C.
As expected, expression of UL51 was not detected in FDL51-infected cells but was at wild-type levels in FDL51R-infected cells, indicating that the rescue of the UL51 sequence in FDL51R was successful. Expression of UL50 and UL52 was similar in all cell types, indicating that deletion of the UL51 sequence from the HSV-1 genome has no effect on the expression of neighboring genes.
Furthermore, the expression of UL51 protein was examined by Western blotting. Vero cells infected with FDL51, FDL51R, YK304, and HSV-1(F) were harvested 24 h after infection and were analyzed by using anti-UL51 rabbit polyclonal antibodies. UL46, UL48 (VP16), and UL49 (VP22), all of which are virion-associated tegument proteins synthesized at the late phase of infection, were also examined as controls. As expected, UL51 protein was not detectable in mock- and FDL51-infected cell lysates (Fig. 4), whereas UL46, UL48, and UL49 protein levels were roughly equivalent among all of the cell lysates (Fig. 4), confirming that the effects of the FDL51 mutant are specific.
FIG. 4.
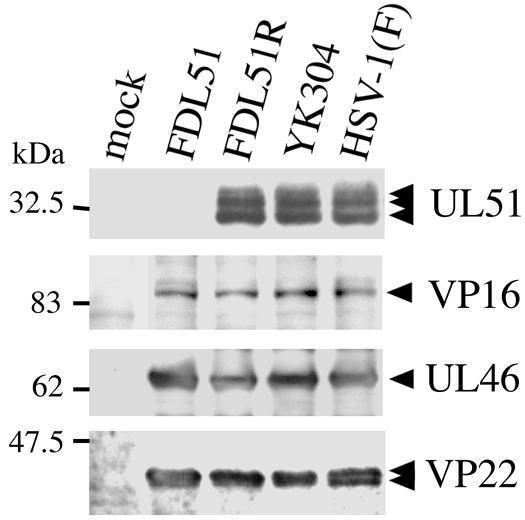
Expression of major tegument proteins. Vero cells were infected with FDL51, FDL51R, YK304, or HSV-1(F) at an MOI of 3. The cells were harvested at 24 h postinfection, and an equivalent amount of protein from each sample was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analyzed by Western blotting with rabbit antiserum against UL51, UL48 (VP16), UL46, or UL49 (VP22) proteins. Molecular mass markers are shown to the left. The arrowheads indicate the position of each protein.
Growth defects of the UL51 deletion mutant.
While UL51 is not essential for HSV-1 replication, previous work showed that a deletion mutant removing both UL50 and UL51 sequences had compromised growth patterns. Thus, we examined the rate of replication of our UL51-null mutant virus by plaque assays and replication kinetics in order to determine whether or not UL51 is involved in viral growth. As shown in Fig. 5a to e, plaques of FDL51 were much smaller than those of wild-type viruses, indicating a growth deficiency in the UL51 deletion mutant. No plaque size reduction was observed in the revertant virus FDL51R, indicating that the growth defect observed in FDL51 mutants was indeed due to the loss of UL51 (Fig. 5b to e). Multistep growth analysis indicated that the viral titer of FDL51 was reduced nearly 100-fold compared to that of HSV-1(F) at 72 h postinfection (MOI = 0.01) (Fig. 5f). Even at an MOI of 3 PFU/cell, the yield of FDL51 was about 50-fold smaller than that of HSV-1(F) (Fig. 5g). In contrast, the growth curve of FDL51R was almost the same as that of the parental virus (Fig. 5f and g). Thus, our results show that UL51 is necessary for the efficient replication of HSV-1 in cultured cells.
FIG. 5.

Growth properties of FDL51 in Vero cells. (a) Plaque formation of FDL51 mutant virus. Vero cells were infected with FDL51, FDL51R, YK304, or HSV-1(F) and were incubated in a medium containing gamma globulin for 48 h. Viral plaques were photographed at 48 h postinfection, and the mean diameters of 20 single plaques per virus mutant were determined as described in Materials and Methods. (b to e) Photos of viral plaques. Vero cells were infected with FDL51 (b), FDL51R (c), YK304 (d), and HSV-1(F) (e). Bars, 100 nm in all panels. (f and g) Growth kinetics of FDL51. Vero cells were infected with FDL51, FDL51R, YK304, or HSV-1(F) at an MOI of 0.01 (f) or 3 (g). At each time point, cells and supernatants were harvested and progeny virus titers were determined by plaque assays, as described in Materials and Methods. The average results of two independent experiments are shown.
UL51 is involved in the egress of virus particles.
We next evaluated the distribution of viral particles in FDL51-infected cells by transmission electron microscopy (TEM). Vero cells infected with FDL51, as well as control cells infected with HSV-1(F), were fixed at 24 h postinfection and processed for TEM. As shown in Fig. 6a, b, and d, extracellular, mature virion was rarely observed on the surface of FDL51-infected cells, whereas a large number of viral particles were observed in HSV-1(F)-infected cells (Fig. 6c and e). In cells infected with FDL51, a number of naked nucleocapsids were readily observed within the cytoplasm although at reduced frequency compared to wild-type virus-infected cells (Fig. 7a and b, arrows). The number of enveloped mature virions budding into cytoplasmic vacuoles in FDL51-infected cells (Fig. 7a, arrowheads) was also reduced in comparison to the control (Fig. 7b, arrowheads). As expected from the results of the immunofluorescence study (data not shown), both FDL51-infected and control cells contained abundant nucleocapsids. However, in FDL51-infected cells, a large number of enveloped nucleocapsids was observed within membranous structures in the perinuclear space (Fig. 7c and d, thick arrows). By contrast, such accumulation of enveloped nucleocapsids in the perinuclear space was rarely observed in control HSV-1(F)-infected cells (data not shown).
FIG. 6.

TEM analysis of extracellular regions of FDL51-infected cells. Vero cells were infected at an MOI of 3 with FDL51 (a, b, and d) or HSV-1(F) (c and e) and analyzed by electron microscopy 24 h postinfection. In FDL51-infected cells, extracellular mature virions were rarely observed in cell-to-cell junctions (b) or on the surfaces of infected cells (a and d), in contrast to wild-type HSV-1(F)-infected cells (c and e). Bars, 2 μm (a to c) or 0.2 μm (d and e).
FIG. 7.

TEM analysis of FDL51-infected Vero cells. Vero cells were infected with FDL51 (a and c to f) or HSV-1(F) (b) and were analyzed by TEM as described in Materials and Methods. In FDL51-infected cells, nucleocapsids in the cytoplasm (a, arrows) and intracytoplasmic vesicles with enveloped matured virions (a, arrowheads) were rarely observed, while they were abundant in wild-type HSV-1(F)-infected cells (b, arrows and arrowheads). Insets show high-magnification views of the squares. No obvious difference was seen in the intranuclear nucleocapsids between cells infected with FDL51 (c and d, arrows) and those with wild-type virus (data not shown). Large numbers of enveloped nucleocapsids accumulated in the perinuclear region of FDL51-infected cells (c and d, thick arrows). A membrane-bound intranuclear structure containing enveloped nucleocapsids (e) and an interwoven membrane-like structure were occasionally observed near the margin of the nucleus in FDL51-infected cells (f). Arrows nucleocapsid; arrowheads, enveloped mature virion in the cytoplasm; thick arrows, enveloped nucleocapsid in the perinuclear region. Bars, 1 μm (a and b) or 0.5 μm (c to f).
Several interesting features were observed in FDL51-infected cells. As shown in Fig. 7e, a membrane-bound intranuclear structure which contained enveloped nucleocapsids was frequently observed. Nuclear vacuoles harboring HSV-1 particles have been reported and are believed to be continuous with the perinuclear cisterna, resulting from the cross-sectioning of nuclear membrane invaginations (14, 35). The enveloped nucleocapsids in intranuclear vacuoles were easily differentiated from the extacellular mature virions since the envelope tightly wrapped around the nucleocapsid appeared as sharp thin lines and lacked the fuzzy appearance of the extacellular mature virions. These features are in common with the enveloped nucleocapsids observed in the perinuclear space, which also supports the idea that the intranuclear vacuoles are continuous with the perinuclear cisterna. Another feature with FDL51 is the presence of distorted, interwoven membrane-like structures located near the margin of the nucleus (Fig. 7f). A similar membrane-like structure has been reported in HSV-infected human amnion FL cells (36), although its significance remains unknown.
Quantitation of the number of virus particles within each compartment of cells infected with FDL51 provides a striking illustration of the effect of UL51 gene deletion (Fig. 8). There was no marked difference in the total number of intracellular virus particles per cells infected with either FDL51 or HSV-1(F), although the number of extracellular mature virions was much smaller in FDL51-infected cells than in HSV-1(F)-infected cells (Fig. 8). In FDL51-infected cells, the number of enveloped nucleocapsids in the perinuclear space was significantly increased. The magnitudes of the decreases in the number of naked nucleocapsids in the cytoplasm and in the number of extracellular mature virions were approximately five- and twentyfold, respectively. Taken together, these findings indicate that the HSV-1 UL51 gene product is important for the egress of virus particles downstream of the initial envelopment step.
FIG. 8.

Distribution of virus particles. Numbers of virus particles present in the different compartments of FDL51-infected Vero cells at 24 h postinfection were determined. Particles were counted in electron micrographs of 20 randomly selected cells for wild-type and FDL51-infected cells. Bars show the average particle numbers in each compartment. (a) Nucleocapsids in the nucleus; (b) enveloped nucleocapsids in the perinuclear space; (c) nucleocapsids in the cytoplasm; (d) enveloped mature virions in the cytoplasm; (e) extracellular mature virions on the outer surface of the plasma membrane.
DISCUSSION
The present study demonstrates that UL51, which encodes an HSV-1 tegument protein, is involved in the egress and/or maturation process of viral replication. Previous work showed that a recombinant virus in which UL50 and a portion of the UL51 sequence were deleted exhibits smaller plaque formation and impaired viral growth in cell culture and that repair of only the UL51 gene was able to rescue these defects (6). Although these results suggest that UL51 plays a role in viral replication, the authors were not able to exclude the possibility that the phenotype observed in the R7107 mutant resulted from decreased expression of the UL52 gene, since deletion of viral genes often affects the expression of neighboring genes (6). Reduced expression of UL52, which encodes the primase of the helicase-primase complex (30) essential for viral DNA replication, leads to poor viral growth in cell culture. Here, we examined the expression levels of UL51-neighboring genes in FDL51-infected cells. Quantitative real-time RT-PCR analysis revealed that the levels of both UL52 and UL50 transcripts in FDL51-infected cells were indistinguishable from the levels observed in cells infected by wild-type and FDL51R revertant virus (Fig. 3). Furthermore, it is known that the deletion of UL50, which encodes dUTPase, induces no replication defects of HSV-1 in cell culture (16, 39, 40), so it is unlikely that mutations of UL50, if any, are responsible for the phenotype of FDL51. Thus, we conclude that the phenotype of FDL51 is due to a lack of UL51, although the possibility that mutations in the neighboring UL52 gene are involved in a part of the phenotype of FDL51 cannot completely be ruled out.
Previous studies from our laboratory have shown that UL51 protein is targeted to the Golgi apparatus (37). The Golgi apparatus plays an important role in assembly and/or maturation of the herpesvirus, particularly in capsid envelopment in the cytoplasm (34, 41). We thus hypothesized that the growth impairment observed in FDL51 mutants (Fig. 5) results from a defect in the assembly of virus particles. In order to test this, we examined viral morphogenesis in FDL51-infected cells by TEM analysis. Although capsid formation in the nucleus of FDL51-infected cells was unaffected, enveloped FDL51 nucleocapsids accumulated in the perinuclear space. Moreover, the number of nucleocapsids in the cytoplasm, as well as the number of extracellular mature virions, was decreased compared to the wild type. These results suggest that UL51 acts downstream of the initial envelopment process, possibly in egress from the perinuclear space or egress beyond the outer nuclear membrane.
A similar accumulation of virus particles in the perinuclear space is observed in cells infected with several other HSV-1 mutants, including UL20, UL48, and UL53 deletion mutants (5, 17, 35). UL20 and UL53 encode a nonglycosylated transmembrane protein and the envelope glycoprotein K, respectively, both of which play important roles in the egress of viral particles as well as in the regulation of membrane fusion (5, 17). UL48 encodes a multifunctional tegument protein known as VP16, which is also required for viral egress beyond the perinuclear space (35). Although these viral proteins have been shown to localize to the nuclear membrane, the nucleus, and the perinuclear cytoplasmic region, the mechanism by which mutations within these genes affect the egress of enveloped perinuclear virions remains unclear. UL51, when expressed in the absence of other viral proteins, localizes precisely to the Golgi apparatus but does so only partially in HSV-1-infected cells. Previous work showed that treatment of pseudorabies virus-infected cells with brefeldin A, an inhibitor of transport between the endoplasmic reticulum and the Golgi apparatus, results in a dramatic accumulation of enveloped virions between inner and outer nuclear membranes (47), suggesting the importance of the Golgi functions in the egress of viral particles from the nucleus. It is possible that the absence of UL51 expression in HSV-1-infected cells may induce the aberrant or uncontrolled trafficking of membrane proteins including glycoprotein K, resulting in the accumulation of enveloped perinuclear virions.
UL51 is similar to UL11 in several respects. Both are incorporated into virions as tegument proteins (13, 33), undergo posttranslational modification of fatty acyl groups such as myristate and palmitate (32, 37), and localize primarily to the Golgi apparatus when expressed in the absence of other viral proteins (8, 36, 37), and palmitoylation of these proteins is required for both Golgi targeting and strong membrane association (32, 37). Furthermore, the deletion of UL11 results in an increase of unenveloped virions within the cytoplasm and a concomitant decrease of extracellular mature virions (4). However, it has also been reported that the deletion of UL11, unlike the case of UL51, does not affect transit across the perinuclear space but affects the envelopment at the inner nuclear membrane (3, 4). Taken together, it is suggested that, although UL51 and UL11 are similar in nature, they play a role at different steps in the process of egress.
Thus, UL51 appears to play multiple roles in viral replication, including egress of virus particles from the perinuclear space and secondary envelopment in the cytoplasm. However, the accumulation of enveloped capsids in the perinuclear space might be due to the result of a secondary effect induced by the uncontrolled trafficking of cytoplasmic vesicles in FDL51-infected cells. The precise mechanism by which UL51 promotes virus assembly remains to be elucidated, and further work to address this question is presently under way in this laboratory.
Acknowledgments
We thank B. Roizman for pBC1007, W. Wackernagel for pCP20, and P. Ioannou for pGETrec. We also thank T. Tsuruguchi, H. Noma, and E. Iwata for technical assistance.
This study was supported by a Grant-in-Aid for Scientific Research and Grants-in-Aid for Priority Areas from the Ministry of Education, Culture, Science, Sports and Technology of Japan and also by a grant from the Japan Society for the Promotion of Science (JSPS-RFTF97L00703).
REFERENCES
- 1.Albrecht, J. C., J. Nicholas, K. R. Cameron, C. Newman, B. Fleckenstein, and R. W. Honess. 1992. Herpesvirus saimiri has a gene specifying a homologue of the cellular membrane glycoprotein CD59. Virology 190:527-530. [DOI] [PubMed] [Google Scholar]
- 2.Baer, R. J., A. T. Bankier, M. D. Biggin, P. L. Deininger, P. J. Farrell, T. J. Gibson, G. Hatfull, G. S. Hudson, S. C. Satchwell, C. Seguin, P. Tuffnell, and B. G. Barrell. 1984. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310:207-211. [DOI] [PubMed] [Google Scholar]
- 3.Baines, J. D., R. J. Jacob, L. Simmerman, and B. Roizman. 1995. The herpes simplex virus 1 UL11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J. Virol. 69:825-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baines, J. D., and B. Roizman. 1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J. Virol. 66:5168-5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baines, J. D., P. L. Ward, G. Campadelli-Fiume, and B. Roizman. 1991. The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress. J. Virol. 65:6414-6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barker, D. E., and B. Roizman. 1990. Identification of three genes nonessential for growth in cell culture near the right terminus of the unique sequences of long component of herpes simplex virus 1. Virology 177:684-691. [DOI] [PubMed] [Google Scholar]
- 7.Baumeister, J., B. G. Klupp, and T. C. Mettenleiter. 1995. Pseudorabies virus and equine herpesvirus 1 share a nonessential gene which is absent in other herpesviruses and located adjacent to a highly conserved gene cluster. J. Virol. 69:5560-5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowzard, J. B., R. J. Visalli, C. B. Wilson, J. S. Loomis, E. M. Callahan, R. J. Courtney, and J. W. Wills. 2000. Membrane targeting properties of a herpesvirus tegument protein-retrovirus Gag chimera. J. Virol. 74:8692-8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broberg, E. K., M. Nygardas, A. A. Salmi, and V. Hukkanen. 2003. Low copy number detection of herpes simplex virus type 1 mRNA and mouse Th1 type cytokine mRNAs by Light Cycler quantitative real-time PCR. J. Virol. Methods 112:53-65. [DOI] [PubMed] [Google Scholar]
- 10.Carrington-Lawrence, S. D., and S. K. Weller. 2003. Recruitment of polymerase to herpes simplex virus type 1 replication foci in cells expressing mutant primase (UL52) proteins. J. Virol. 77:4237-4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chee, M. S., A. T. Bankier, S. Beck, R. Bohni, C. M. Brown, R. Cerny, T. Horsnell, C. A. Hutchison III, T. Kouzarides, J. A. Martignetti, et al. 1990. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 154:125-169. [DOI] [PubMed] [Google Scholar]
- 12.Cherepanov, P. P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9-14. [DOI] [PubMed] [Google Scholar]
- 13.Daikoku, T., K. Ikenoya, H. Yamada, F. Goshima, and Y. Nishiyama. 1998. Identification and characterization of the herpes simplex virus type 1 UL51 gene product. J. Gen. Virol. 79:3027-3031. [DOI] [PubMed] [Google Scholar]
- 14.Darlington, R. W., and L. H. Moss III. 1969. The envelope of Herpesvirus. Prog. Med. Virol. 11:16-45. [PubMed] [Google Scholar]
- 15.Davison, A. J., and J. E. Scott. 1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 67:1759-1816. [DOI] [PubMed] [Google Scholar]
- 16.Fisher, F. B., and V. G. Preston. 1986. Isolation and characterisation of herpes simplex virus type 1 mutants which fail to induce dUTPase activity. Virology 148:190-197. [DOI] [PubMed] [Google Scholar]
- 17.Foster, T. P., and K. G. Kousoulas. 1999. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J. Virol. 73:8457-8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldstein, D. J., and S. K. Weller. 1988. An ICP6::lacZ insertional mutagen is used to demonstrate that the UL52 gene of herpes simplex virus type 1 is required for virus growth and DNA synthesis. J. Virol. 62:2970-2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gompels, U. A., J. Nicholas, G. Lawrence, M. Jones, B. J. Thomson, M. E. Martin, S. Efstathiou, M. Craxton, and H. A. Macaulay. 1995. The DNA sequence of human herpesvirus-6: structure, coding content, and genome evolution. Virology 209:29-51. [DOI] [PubMed] [Google Scholar]
- 20.Graham, F. L., and A. J. van der Eb. 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456-467. [DOI] [PubMed] [Google Scholar]
- 21.Grosenbach, D. W., S. G. Hansen, and D. E. Hruby. 2000. Identification and analysis of vaccinia virus palmitylproteins. Virology 275:193-206. [DOI] [PubMed] [Google Scholar]
- 22.Grunewald, K., P. Desai, D. C. Winkler, J. B. Heymann, D. M. Belnap, W. Baumeister, and A. C. Steven. 2003. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 302:1396-1398. [DOI] [PubMed] [Google Scholar]
- 23.Hamel, F., H. Boucher, and C. Simard. 2002. Transcriptional and translational expression kinetics of the bovine herpesvirus 1 UL51 homologue gene. Virus Res. 84:125-134. [DOI] [PubMed] [Google Scholar]
- 24.Heid, C. A., J. Stevens, K. J. Livak, and P. M. Williams. 1996. Real time quantitative PCR. Genome Res. 6:986-994. [DOI] [PubMed] [Google Scholar]
- 25.Ivanova, L., and M. J. Schlesinger. 1993. Site-directed mutations in the Sindbis virus E2 glycoprotein identify palmitoylation sites and affect virus budding. J. Virol. 67:2546-2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kato, K., T. Daikoku, F. Goshima, H. Kume, K. Yamaki, and Y. Nishiyama. 2000. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 145:2149-2162. [DOI] [PubMed] [Google Scholar]
- 27.Kawaguchi, Y., R. Bruni, and B. Roizman. 1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1delta: ICP0 affects translational machinery. J. Virol. 71:1019-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawaguchi, Y., C. Van Sant, and B. Roizman. 1998. Eukaryotic elongation factor 1δ is hyperphosphorylated by the protein kinase encoded by the UL13 gene of herpes simplex virus 1. J. Virol. 72:1731-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura, H., M. Morita, Y. Yabuta, K. Kuzushima, K. Kato, S. Kojima, T. Matsuyama, and T. Morishima. 1999. Quantitative analysis of Epstein-Barr virus load by using a real-time PCR assay. J. Clin. Microbiol. 37:132-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klinedinst, D. K., and M. D. Challberg. 1994. Helicase-primase complex of herpes simplex virus type 1: a mutation in the UL52 subunit abolishes primase activity. J. Virol. 68:3693-3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lenk, M., N. Visser, and T. C. Mettenleiter. 1997. The pseudorabies virus UL51 gene product is a 30-kilodalton virion component. J. Virol. 71:5635-5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loomis, J. S., J. B. Bowzard, R. J. Courtney, and J. W. Wills. 2001. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 75:12209-12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacLean, C. A., B. Clark, and D. J. McGeoch. 1989. Gene UL11 of herpes simplex virus type 1 encodes a virion protein which is myristylated. J. Gen. Virol. 70:3147-3157. [DOI] [PubMed] [Google Scholar]
- 34.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mossman, K. L., R. Sherburne, C. Lavery, J. Duncan, and J. R. Smiley. 2000. Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J. Virol. 74:6287-6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nii, S., C. Morgan, and H. M. Rose. 1968. Electron microscopy of herpes simplex virus. II. Sequence of development. J. Virol. 2:517-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nozawa, N., T. Daikoku, T. Koshizuka, Y. Yamauchi, T. Yoshikawa, and Y. Nishiyama. 2003. Subcellular localization of herpes simplex virus type 1 UL51 protein and role of palmitoylation in Golgi apparatus targeting. J. Virol. 77:3204-3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pomeranz, L. E., and J. A. Blaho. 2000. Assembly of infectious herpes simplex virus type 1 virions in the absence of full-length VP22. J. Virol. 74:10041-10054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Preston, V. G., and F. B. Fisher. 1984. Identification of the herpes simplex virus type 1 gene encoding the dUTPase. Virology 138:58-68. [DOI] [PubMed] [Google Scholar]
- 40.Pyles, R. B., N. M. Sawtell, and R. L. Thompson. 1992. Herpes simplex virus type 1 dUTPase mutants are attenuated for neurovirulence, neuroinvasiveness, and reactivation from latency. J. Virol. 66:6706-6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, 4th ed. Lippincott-Williams & Wilkins, Philadelphia, Pa.
- 42.Shiba, C., T. Daikoku, F. Goshima, H. Takakuwa, Y. Yamauchi, O. Koiwai, and Y. Nishiyama. 2000. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J. Gen. Virol. 81:2397-2405. [DOI] [PubMed] [Google Scholar]
- 43.Skepper, J. N., A. Whiteley, H. Browne, and A. Minson. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment → deenvelopment → reenvelopment pathway. J. Virol. 75:5697-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taddeo, B., A. Esclatine, and B. Roizman. 2002. The patterns of accumulation of cellular RNAs in cells infected with a wild-type and a mutant herpes simplex virus 1 lacking the virion host shutoff gene. Proc. Natl. Acad. Sci. USA 99:17031-17036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka, M., H. Kagawa, Y. Yamanashi, T. Sata, and Y. Kawaguchi. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Telford, E. A., M. S. Watson, K. McBride, and A. J. Davison. 1992. The DNA sequence of equine herpesvirus-1. Virology 189:304-316. [DOI] [PubMed] [Google Scholar]
- 47.Whealy, M. E., J. P. Card, R. P. Meade, A. K. Robbins, and L. W. Enquist. 1991. Effect of brefeldin A on alphaherpesvirus membrane protein glycosylation and virus egress. J. Virol. 65:1066-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang, Y., F. Buchholz, J. P. Muyrers, and A. F. Stewart. 1998. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 20:123-128. [DOI] [PubMed] [Google Scholar]
- 49.Zhou, Z. H., D. H. Chen, J. Jakana, F. J. Rixon, and W. Chiu. 1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J. Virol. 73:3210-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]