

Abstract
An outstanding question of genome evolution is what stops the invasion of a host genome by transposable elements (TEs). The human genome, harboring the remnants of many extinct TE families, offers an extraordinary opportunity to investigate this problem. ERV9 is an endogenous retrovirus repeatedly mobilized during primate evolution, 15 to 6 million years ago (MYA), which left a trace of over a hundred provirus-like copies and at least 4,000 solitary long terminal repeats (LTRs) in the human genome. Then, its proliferation ceased for unknown reasons, and the family went extinct. We have made a detailed reconstruction of its last active subfamily, ERV9_XII, by examining 115 solitary LTRs from it. These insertions were grouped into 11 sets according to shared nucleotide variants, which could be placed in a sequential order of 10 to 6 MYA. At least 75% of the subfamily was produced 8 to 6 MYA, during a stage of intense proliferation. With new analytical tools, we show that the youngest and most prolific sets may have been produced by effectively instantaneous expansions of corresponding single-sequence variants. The extinction of this family apparently was not a consequence of its slow gradual degeneration, but the outcome of the fixation of specific restrictive alleles in the human-chimpanzee ancestral population. Three species-specific insertions (two in humans and one in chimpanzees) were identified, further supporting that extinction took place when these two species were beginning to diverge. These are the only fixed differences of this kind so far observed between humans and chimpanzees, apart from those belonging to the human endogenous retrovirus K family.
Transposable elements (TEs) constitute a large fraction of the human genome (roughly 45% of the euchromatic component, and an indeterminately much larger amount of the heterochromatin), scattered over all chromosome regions with widely different repeat densities (25). They form an extremely rich community, including many different families pertaining to one or other of four major types: long interspersed repetitive elements (LINEs), short interspersed repetitive elements (SINEs), long terminal repeat (LTR)-containing elements, and DNA transposons. Genomic copy numbers of certain families range in the hundreds of thousands (Alu and LINE1), while in others only a few members can be found (some LTR elements). In general, the number of copies is fairly high, particularly if compared with nonmammalian organisms, such as Drosophila melanogaster, which rarely has more than 100 copies of any family. This is not the only conspicuous difference between mammalian and Drosophila elements at the genome level (19). Individual TE sites are usually fixed in mammalian populations, whereas nearly all sites are occupied at low frequencies in Drosophila wild populations. Thus, all humans share virtually the same array of TE insertions, which date back to the distant past of our evolutionary lineage, so that we also share many TE sites with the other Hominoidea (gibbons, orangutans, gorillas, and chimpanzees). The reasons for these differences are not completely understood, but they probably stem from both the smaller effective population size of mammals than of insects and the involvement of ectopic exchanges in the selective control of TE copy numbers (6). Besides, there has been a marked decline in the overall activity of TEs over the past 35 to 50 million years (MYR) in the lineage leading to humans (25), which also helps to explain the relatively few cases of polymorphism for TE insertions in our populations.
Our genome is plagued with “fossil” remnants of mobilization periods that ceased long ago. And the same happens with the evolutionary history of organisms, some of whose clues can only be found in the fossil record; there are important questions of the evolution of TEs that can only be answered by looking at these genomic fossils. Not the least important of them is what stops the invasion process of a genome by a TE family. The sequenced human genome, harboring thousands of copies from TE families that became “extinct” when they lost their capacity of proliferation, offers an exceptional opportunity to investigate this problem.
Notwithstanding the ultimate beneficial use of particular TE insertions by the host (27, 30) or the importance of TEs as generators of genetic variation in wild populations (23), these sequences generally behave as parasitic, selfish DNAs. Their potential to spread through host genomes and populations relies upon their ability to overreplicate the host DNA in the absence of any selective advantage to their carriers, within an evolutionary context that in many respects recalls an ecological community (7). Several mechanisms that may lead to dynamic equilibria of copy number (11), involving either self-regulation of TEs (40) or the opposing forces of transposition and host fitness effects of increased copy numbers (9), have been proposed and tested against observations. These equilibria may persist at some intermediate value for many generations of the host organism, but finally TEs are expected to be eliminated. Their proliferation may be reduced by their gradual accumulation of degenerating mutations (22) and/or by selection on the host genome to limit the damage caused by TEs (2, 17, 28, 43), leaving behind only the lucky insertions that proceeded to fixation, usually by random drift.
In this paper, we offer some hints of the mechanisms that led to the extinction of an LTR-containing TE family that once thrived in our not-so-distant past. LTR-containing TEs in the human genome are represented mainly by human endogenous retroviruses (HERVs). These fall into three classes, each comprising many families that originated independently from ancient infections of the germ line by different kinds of exogenous retroviruses (25, 41), which have integrated into their chromosomes and then persisted as stable Mendelian factors for multiple generations. Their structure is accordingly quite similar to that of exogenous retroviruses, consisting of an internal sequence with homology to gag, pol, and sometimes env open reading frames, flanked by two LTRs.
HERVs may increase in copy number within the genome either via intracellular retrotransposition (within germ line cells) or through an extracellular infectious phase (reinfection of the germ line). Both pathways are not mutually exclusive. The recent finding of evolutionary constraint in the env gene of several HERV families (1) is consistent with reinfection having been their major means of proliferation. However, apparently this was not the case for the two largest families in terms of copy number (HERV-L and HERV-H). ERV9 is a class I family that was repeatedly mobilized during primate evolution (15, 18), bringing their copy number in the human haploid genome to approximately 120 members distributed on most chromosomes (36), as well as at least 4,000 solitary LTRs (26) produced by recombination between the 5′ and 3′ LTRs of the same insertion. A reconstruction of the evolutionary history of this family through a paleogenomic analysis of their LTRs (15) led to the identification of 14 subfamilies, integrated in a sequential order in one of four main lineages, presumably corresponding to expansion waves from different master copies (5, 16). The age of these subfamilies was estimated so that they could be placed on the phylogenetic tree of primate evolution. The first of them probably appeared after the split of New World and Old World monkeys (≈38 MYA). Then, successive expansions took place, with several subfamilies simultaneously active over long periods of time, particularly in the interval since gibbons began to diverge from higher apes until after the split of gorillas (7 to 15 MYA, according to reference 20). Finally, this high proliferation ceased for unknown reasons, and no new subfamilies have been found since. We have now made a detailed reconstruction of the evolutionary history of the last subfamily of ERV9, named XII, and show that its activity actually went on until the separation of humans and chimpanzees, when it finally ceased, most likely not as a consequence of a more or less slow progressive degeneration of TE sequences but because of a relatively rapid spread of restrictive alleles in the host populations.
MATERIALS AND METHODS
BLASTn, from the BLAST server of the National Center of Biotechnology Information (NCBI [http://www.ncbi.nlm.nih.gov/BLAST]), and the Ensembl Chimpanzee Genome Browser (http://www.ensembl.org/Pan_troglodytes/) were used to search for sequences homologous to the first 676 bp of the consensus LTR for subfamily XII of ERV9 (15) in the annotated human genome database. All hits were carefully examined to eliminate redundant entries. Only sequences that had at least three of the five non-CpG diagnostic differences between subfamily XII and XI (the most similar to XII, according to reference 15) were retained for further analyses. Additional searches were then carried out, using each of the previously identified insertions of subfamily XII as probes. All the sequences obtained in this way (ERV9_XII insertions) were aligned by visual inspection with the aid of BioEdit (available at http://www.mbio.ncsu.edu/BioEdit/bioedit.html). A subfamily consensus was then obtained by choosing the most frequent nucleotide at each position, except when a combination of dinucleotides of the three pairs CpG, CpA, and TpG was present at the same doublet position. In that case, the CpG dinucleotide was chosen as the consensus unless the T or A nucleotides were present in >70% of the sequences. This subfamily consensus is considered the best reconstruction of the founder master element that started the subfamily (15).
Insertions were grouped into different sets according to shared nucleotide variants. A nucleotide position was considered diagnostic of a sequence set whenever >70% of the sequences grouped into it shared the same nucleotide, which differed from that characterizing at least some other similar groups. Groups were made up of at least five sequences, sharing two or more correlated nucleotide variants. At least one of these nucleotide variants had to involve a site not diagnosed as a CpG doublet in the subfamily consensus. Occasionally, two subgroups could be established, each consisting again of at least five sequences but sharing just a single nucleotide variant at a non-CpG doublet. Consensus sequences for each set of ERV9_XII insertions were constructed following the same rules described above for the subfamily.
Phylogenetic reconstructions of the consensus sequences of the different sets of ERV9_XII insertions were carried out both by distance (neighbor joining [NJ] with Kimura's two-parameter model with a transition/transversion ratio of 2) and maximum parsimony (MP) methods implemented in the MEGA2.1 package (available at http://www.megasoftware.net). In MP analyses, we searched for the best trees using the close-neighbor interchange, with default parameter values and random addition of sequences to produce the initial trees.
For the following comparative analyses of the different sets of ERV9_XII insertions, it was necessary first to rid them of CpG dinucleotides, whose very high mutation rate (3) could introduce a significant noise in our analyses. Exclusively for that purpose, a subfamily consensus was constructed with a more stringent condition for the diagnosis of CpG doublets: the CpG dinucleotide was chosen as the consensus unless the T or A nucleotides were present in >90% of the sequences, instead of the 70% threshold routinely applied formerly to derive a consensus. All sites that happened to be CpG under this more stringent condition were removed from the general alignment, as well as all sites corresponding to nonconsensus nucleotide insertions.
To estimate the ages of the different sets of ERV9_XII insertions, we first calculated the average number of nucleotide substitutions from their consensus (K), using Kimura's two-parameter model with a transition/transversion ratio of 2. Assuming 0.16% per MYR as the rate of change of pseudogene sequences in primates (15), the average expansion age of each sequence set was estimated as T = K/0.0016.
The strict master model (SMM) postulates that all the insertions of a given set were instantaneously produced by retrotransposition of the same master element. Assuming that the consensus is the best possible reconstruction of the sequence of that master, expected average pairwise divergence between sequences of the same set was derived by Jurka (21), as in the following equation:
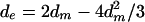 |
(1) |
where dm is the average divergence of the sequences from their consensus, calculated as the relative number of mismatches. To test this null hypothesis, the difference between observed and expected average pairwise divergences (coefficient J, introduced by Jurka) (21) was obtained for each set, and its value was compared with the distribution obtained by generating 1,000 samples with the aid of Seq-Gen (33). Each of these samples simulates the evolution of the corresponding number of DNA sequences along a star phylogeny, with the observed pairwise divergence, assuming the Jukes-Cantor model of nucleotide substitution, and using the corresponding consensus as the ancestral sequence at the root. Although the expected value of J under the SMM is 0, actual values obtained in the simulations can be negative or positive, corresponding to greater or less structuring of the sequence set than predicted by equation 1 (since nucleotide substitutions occur at random and only a sample of sequences is analyzed).
Tentative estimates of the durations of expansion periods were obtained following Tachida (37), assuming a transient master copy model. According to equation 19 in Tachida (37) and replacing terms as in Jurka (21), it can be easily shown that
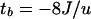 |
(2) |
where tb is the persistence of the expansion period (in units of 2n generations; n is the effective population size), J is Jurka's coefficient, and u is the mutation rate per site per 2n generations, under a Jukes-Cantor mutation scheme. Corresponding estimates in MYR were derived after assuming u = 0.002667 (37), n = 10,000 (38), and a generation time of 20 years.
Phylogenetic reconstruction of the sequences of the whole set of ERV9_XII insertions was carried out by NJ, again using Kimura's two-parameter model of nucleotide substitution with a transition/transversion ratio of 2.
RESULTS
Altogether, 115 different insertions of ERV9_XII were identified, all of them corresponding to solitary LTRs (see the supplemental material). In our previous work (15), only six insertions of this subfamily were found, but their consensus is nearly the same as the one derived from this much larger data set (C instead of T at position 260 and A instead of G at position 419). After aligning all these sequences, we classified them into 10 major groups (Table 1); one of these groups could be split further into two subgroups. In addition, three other sequence sets were considered, each consisting of either three or four sequences. They were assigned to hypothetical categories, denoted by the designation (h) in Table 1, simply to show that although they do not reach the minimum number of elements necessary to merit the consideration of true groups, they fulfilled the other requirements. Finally, nine insertions were excluded from the analyses. Five of them were excluded because they displayed diagnostic features of different groups scattered along their sequences (see the elements listed as unclassified in Table 1). The remaining four (characterized as “others” in Table 1) were excluded either because of they bore insertions or deletions in diagnostic positions (AC084879) or because they formed parts of corresponding pairs of insertions with striking similarities and lie close together in the genome map, all of them associated with telomeric regions. These regions are AL162499 and AL157875 in the long arm of chromosome 13, AC010084 and AC006157 in the short arm of the Y chromosome, and AJ277546 and AC100763 in the short arm of chromosome 11. These three pairs of sequences are most likely to be the result of gene conversions between insertions located nearby and not of extended chromosome rearrangements, because the regions of homology are circumscribed to the LTRs (data not shown). Accordingly, since they could not be considered the products of independent evolution upon insertion into host DNA, the first member of each pair was excluded.
TABLE 1.
Identified insertions of subfamily XII of ERV9 (ERV9_XII)a
| Group | Element |
|---|---|
| A | AL022574, 57085; AC026336, 29532; AL050317, −86922; AL953854, −108369; AL139385, −147125; AL139092, 163973; AC011155, 91661 |
| B | AL157875, 63649; AL138999, 39227; AP002962, −94050; AC097648, −137761; AC000048, 12988; AC003986, −8471; AL158218, 7607; AC138389, 144495 |
| C | AC103975, 173347; AL158081, −42077; AP001836, 57681; AC108103, 38923; AP001132, −98599; AC069259, 148111; AL157815, 93644; AC068319, 211212; AC004972, −18376 |
| D(h) | AC006157, −81510; Z84476, 38665; AC010310, 44647; AL512380, −117517 |
| E | AC006965, 13538; AC024094, 31665; AL122003, −8850; AC068992, −89342; AL354931, −61614; AC020734, 170414 |
| F(h) | AC018494, −72999; AP001531, −133736; AL807740, −18041; Z92547, 57194 |
| G | AL035046, −16198; AL138965, −127529; AL121872, −5721; AP001713, 230094; AC112204, −103372; AL354740, 136182; AL590043, −38830; AL080314, 39315; AL138764, 88744; AL135999, −82737; AL137100HS, − 136754; AL138742, 72894; AC073140, 103540; AL078463, 57229; AL133264, −52570; AL135935, −33118; AC078788, −20327; AL157792, 56910 |
| H | AL590489, −108158; AL034379, 41513; AC092506, 36189; AL157388, 48362; AF222685, −63564; AC011978, −98791; AC009964, 5211; AC008892, 55143; AC093295, −31932 |
| I | AC023050, −32564; AC090156, −36235; AC021755, −93038; AL049780, −167782; AC068599, −99801; AC012516HS, −9016; Pan_4_184812243PS |
| J | AC090440, 16757; AC089985, −111837; AC007106, −71331; AL356307, −61253; AC055717, −13481; AL360155, −96301; AC007793, 5733; AC092581, 71129 |
| K(h) | AC127070, −11247; AC005378, −102045; AC090677, 21801 |
| L | AC023426, 56968; AC113355, 48453; AC097511, 94343; AC010726, 67318; AL354807, 1377; Z84474, −65295; AC108749, −65131; AL133333, 7180; AC104163, −106838; AC121758, 103695; AF338230, 11189; AC103923, 25421 |
| M, subgroup M1 | AC087481, −97398; AC100763, −100041; AC090415, −37530; AL590233, 21417; AC012363, −105041; AC096642, −7471 |
| M, subgroup M2 | AC041005, 5516; AC104641, 87420; AC022821, −72531; AC004668, 79271; AC022203, 28804 |
| Excluded | AC027673, 129631; AC133865, −37661; AC009475, −16004; AC009967, 139531; AC125494, −82562 (unclassified); AL162499, −102147; AC010084, 11514; AJ277546, −109903; AC084879, −34461 (others) |
Each element is identified by its GenBank accession number, followed by the nucleotide position of the 5′ end of the analyzed region. A minus sign indicates sequence orientation opposite to the LTR. Species-specific insertions are indicated by the superscript HS (Homo specific) or PS (Pan specific). The only PS insertion is designated Pan, followed by the chromosome number, orientation, and position of its 5′ end.
By far, the largest part of nucleotide variation in ERV9_XII insertions corresponded to differences within groups. Thus, even after CpG positions are eliminated, the between-group average was 2.73% nucleotide differences, but net divergence (i.e., after average within-group differences were substracted) was only 0.48% (data not shown; within-group distance was approximately twice the distance to the corresponding consensus) (Table 2).
TABLE 2.
Analysis of divergence in the different insertion groups within ERV9_XII
| Group | No. of members | Distance to consensus SEa,b | Age (MYR)c | Observed pairwise divergenced | Je | Pf |
|---|---|---|---|---|---|---|
| A | 7 | 0.017 (0.0022) | 10.6 | 0.0319481 | −0.0011555 | <0.001 |
| B | 8 | 0.014 (0.0017) | 8.8 | 0.0279843 | −0.0000650 | 0.248 |
| C | 9 | 0.013 (0.0017) | 8.1 | 0.0250309 | −0.0006406 | 0.001 |
| E | 6 | 0.013 (0.0021) | 8.1 | 0.0240073 | −0.0009338 | 0.001 |
| G | 18 | 0.010 (0.0009) | 6.3 | 0.0199679 | −0.0000037 | 0.202 |
| H | 9 | 0.013 (0.0016) | 8.1 | 0.0263471 | −0.0000905 | 0.184 |
| I | 7 | 0.013 (0.0017) | 8.1 | 0.0258735 | −0.0001147 | 0.223 |
| J | 8 | 0.011 (0.0015) | 6.9 | 0.0222619 | −0.0001869 | 0.099 |
| L | 12 | 0.010 (0.0012) | 6.2 | 0.0195673 | −0.0000199 | 0.191 |
| M1 | 6 | 0.012 (0.0017) | 7.5 | 0.0224531 | −0.0000231 | 0.315 |
| M2 | 5 | 0.011 (0.0019) | 6.9 | 0.0219264 | −0.0001741 | 0.150 |
| Totalg | 106 | 0.016 (0.0014) | 10.0 | 0.0287413 | −0.0020731 | <0.001 |
Kimura's distance to the corresponding consensus, excluding CpG positions.
Standard error (SE) computed through 500 bootstrap samples.
Estimated age of each set of sequences (Kimura's distance/0.0016).
Average pairwise differences per site between sequences belonging to the same group.
Difference between observed and expected pairwise divergence (Jurka's coefficient).
Relative frequency of occurrences in 1,000 independent simulations of a Jurka's coefficient less than or equal to the observed one.
Including insertions from the three hypothetical groups, designated by (h) in Table 1.
The different groups were not equally well represented, and their abundance ranged from 5% to 17% of the examined sequences (Table 2). The two most abundant groups (G and L) appear to be also the most recent ones, with an estimated age of 6.2 MYR (Table 2), compared with an estimate of 10.0 MYR for the whole subfamily, which is considerably smaller than the value reported in our former paper (13 MYR). The main cause of this difference is our finding of many relatively young sequences of this subfamily since our former BLAST searches of the human genome database, nearly 5 years ago. This new estimate is in very good agreement with the age obtained for the oldest group of the subfamily, namely 10.6 MYR (group A). Therefore, the temporal window of transposition activity of ERV9_XII must have been approximately 4 MYR (6 to 10 MYA). Independent confirmation of the lower limit of this interval was obtained by carrying out a BLAST search of the chimpanzee genome (so far unfinished) with human ERV9_XII sequences. Three insertions were found to be species specific (AL137100 and AC012516 in humans and Pan_4_184812243 in chimpanzee) (Table 1, groups G and I). All the others were shared by both species.
To investigate the evolutionary history of the element during this period, we first made a phylogenetic analysis of the consensus sequences of the different groups, which constitute the best sequence reconstruction of the master elements that gave rise to them. Nucleotide variants of their alignment with the consensus sequences of subfamily ERV9_XI, used as an outgroup (15), subfamily XII, and each of the three hypothetical groups, are shown in Fig. 1. Many of these differences were shared by different groups, suggesting that they can be placed in a sequential order. The phylogenetic tree (Fig. 2) confirmed the presence and ordering of shared variants, showing what seemed to be at first a single, uninterrupted lineage that sequentially gave rise to groups A to D(h) but later split into two lineages, one leading to groups G to H and the other to groups I to M2. The positions of E and F(h) were dubious, since MP analysis places them in the G-H clade (see the supplemental material). The estimated ages of individual groups (Table 2) were not always in good agreement with their sequential order within each lineage. Actually, this finding was not at all unexpected, since each group, except for C and G, showed one or several private differences (Fig. 1 and 2) that indicate that they are not likely to be the direct ancestors of the groups that followed in their lineage. Obviously, there are several transitional stages that left no representatives in the fossil record of the human genome. Each internal node of the phylogenetic tree, except for the dichotomies leading to C and G, bore evidence of the coexistence of at least two master active elements. However, the information on the evolution of the diversity of these masters could not be straightforwardly obtained from the phylogenetic tree, because the evolutionary rates of these active elements may have been rather different. According to parsimony analyses, in the 4 MYR comprising the expansion period of the subfamily, a total of 17 nucleotide changes must have taken place to produce L (see the supplemental material), the master element of the most evolved group, or 0.006 changes per site per MYR (nearly four times higher than the evolutionary rate of pseudogene copies used for dating ERV9 insertions). Of these changes, 10 must have taken place after the splitting of the main lineage into those leading to L and G (the two most recently active groups of the subfamily). However, in this same period, only two changes led to the G master. This may be indicative of heterogeneity in the evolutionary rates of coexisting master elements. On the other hand, our estimates of the ages of the different groups are subject to considerable error (Table 2). This may help to explain some major discrepancies between the age of a group and its position on the phylogenetic tree. Group I, for example, should be relatively old, according to divergence among its representatives, but it occupies an intermediate position in the tree and contains two species-specific insertions. Taking all these factors into consideration, it may be concluded that ERV9_XII sequences experienced a remarkable increase in the diversity of their coexisting active copies in the last two MYR of its history of transpositions.
FIG. 1.

Nucleotide differences among the consensus sequences of the different groups of subfamily XII. The first three rows refer to base positions relative to Fig. 1 in Costas and Naveira (15; see also the supplemental material). Double- and single-underlined positions indicate sites forming part of CpG dinucleotides in the general consensus sequence of subfamily XII, with a 70% or a 90% threshold frequency, respectively (see Materials and Methods). The general consensus sequence of subfamily XI was used as a reference. Dots indicate identity with this reference sequence. Uppercase letters indicate nucleotides present in >70% of the sequences belonging to a group; lowercase letters indicate nucleotides present in 50% to 70% of the sequences in a group.
FIG. 2.

Phylogenetic relationships within ERV9_XII, based on analyses of consensus sequences of the different groups established according to shared nucleotide differences. The displayed tree was obtained by the NJ method, and it is rooted with the general consensus of subfamily XI (ERV9_XI), used as an outgroup. Values indicate the percentages of equally parsimonious trees supporting internal branches (only values of >70% are indicated; three equally most parsimonious trees were obtained). Sequence groups that did not depart significantly from a star phylogeny, after contrasting observed and expected values of Jurka's coefficient, are marked with a star symbol.
A phylogenetic reconstruction of the full set of ERV9_XII sequences (106 insertions) by the NJ method, after excluding CpG positions, is shown in Fig. 3. It can be seen that insertions belonging to the groups previously established according to shared diagnostic differences tended to lie close together in the tree, thus lending further support to our classification. A basal cluster, including insertions from the oldest groups A to C, lying close to the ERV9_XI outgroup, was clearly separated from the other members of the subfamily. The topology of this tree, however, bears several differences with the tree of the consensus sequences depicted in Fig. 2, mainly affecting groups J, M1, and M2, which appear now to be more closely related to the lineage leading to G than to L. This is due to the loss of several phylogenetically informative sites associated to the CpG positions that we removed prior to this analysis (shown by underlining in Fig. 1), precisely to reduce noise in the groupings of insertions and to avoid overestimating the ages of some particular groups. The importance of these sites for correctly inferring the phylogeny of ERV9_XII groups is manifest by the results of our analyses of maximum parsimony. If all nucleotide sites are included in the analysis, only 3 equally most parsimonious trees are obtained (Fig. 2), whereas this number increases to 681 if CpG positions are removed from the alignment (average of 10 MP tree searches; data not shown).
FIG. 3.

Phylogenetic reconstruction of the full set of ERV9_XII sequences (97 solitary LTR insertions) by the NJ method, after CpG positions were excluded. The tree is rooted with the consensus of ERV9_XI. Each sequence is designated by its GenBank accession number, followed by the name of the group it has been assigned to in this work.
At first sight, several groups of insertions in Fig. 3 appear to show star phylogenies, similar to those expected after the instantaneous expansion of a master copy sequence. To address this issue, we compared the observed values of Jurka's coefficient with the values obtained after corresponding sets of 1,000 independent simulations of evolution under a strict master model with instantaneous expansion (see Materials and Methods). The results of these tests are shown in Table 2. The patterns of insertions belonging to groups B, G, H, I, J, L, M1, and M2 (Fig. 2) do not depart significantly from star phylogenies (P > 0.05), so that they may indeed have been produced by expansion of corresponding master elements during relatively short time intervals (“instantaneously”). This is particularly so in the case of groups G and L, the two most prolific ones, which show the lowest absolute values (closest to 0) of Jurka's coefficient. The power of this test (the probability of rejecting the null hypothesis when in fact it is false and the alternative hypothesis is correct) is exemplified by pooling insertions from groups I and D(h), which, according to the results shown in Fig. 3, could superficially appear to fit a star phylogeny. The test correctly rejects the null hypothesis for this pooled set (J = −0.00065807; P < 0.001), whereas it shows a good fit for insertions of group I, thus indicating that insertions of group D(h) originated from a different master copy, in agreement with our former conclusions based on shared nucleotide differences.
Application of Tachida's transient master copy model to the three groups that depart significantly from star phylogenies (namely, A, C, and E) led to point estimates for the persistence of their expansion periods of 1.4, 0.76, and 1.1 MYR, respectively. By contrast, the seven groups that show a good fit to a star phylogeny (i.e., to an “instantaneous” expansion) would have actually expanded for only 0.1 MYR, on average.
DISCUSSION
Depending on the degree of heterogeneity among elements in their probabilities of acting as sources of new copies, two extreme models of expansion of a TE family can be envisioned: SMM, with a single “master” copy as the source of all new insertions, and the random template model, where all copies of the TE family are functionally equivalent (4). The two models make markedly different predictions concerning the topology of the phylogenetic tree, the time of appearance and sequential ordering of shared variants, the divergence between elements within subfamilies, and the distribution of pairwise differences between elements (12). The SMM best fits the existing subfamily data of SINEs and LINEs in the mammalian genome (16), and a master copy is also most likely to be responsible for the high rates of gypsy (a Drosophila LTR retrotransposon) proliferation observed in some laboratory strains (24). On the contrary, it has been experimentally demonstrated that different copia (another Drosophila LTR retrotransposon) variants are capable of multiplication (32), and comparative sequence analyses of several HERV families show evidence of the independent and parallel formation of different subfamilies (8, 13, 14, 15). Therefore, the SMM does not apply to these cases, but neither does the random template model, because the number of simultaneously active lineages always seems to be very small. However, some words of caution are necessary at this point, because the genomic fossil record of TEs is quite imperfect. There may be relatively large gaps, many missing intermediate stages, and many lineages represented only at widely separated time intervals, since only master genes that have been active at a relatively high level over an extended period of time may have a good chance to leave a trace, i.e., a sequence subfamily, in the genomic record (16).
This study was confined to roughly the last four MYR of existence of ERV9 as a TE, when it gave rise to subfamily ERV9_XII, 6 to 10 MYA, in the most recent common ancestor of humans and chimpanzee. Our examination of ERV9_XII sequences reveals that during the first half of this time interval, three major expansion waves of variants of a dominant lineage took place at different times. One of these expansions (group B) may have been instantaneous, meaning that the proliferation rate was probably much higher than the mutation rate per base pair between expansion periods. The other two, groups A and C, should have persisted for 69,000 and 38,000 generations, or 1.4 and 0.76 MYR (assuming a generation time of 20 years), respectively, which is perfectly congruent with our estimations of the ages of the different groups. Then, in the two MYR prior to its extinction, ERV9_XII appears to have been engaged in frenetic activity, which produced at least 75% of the insertions of this subfamily, distributed among eight groups and two lineages. All these groups except E, whose expansion is expected to have persisted for 56,000 generations, may have been produced by “instantaneous” expansion of single-sequence variants. Interestingly, according both to age estimations based on divergence within groups and, above all, to the presence of a few species-specific insertions, several of these groups were most likely simultaneously active just during the first stages of speciation of the genera Homo and Pan. Remarkably, three species-specific insertions have been identified, representing the first reported fixed differences of this kind between humans and chimpanzees, apart from those belonging to the HERV-K family (29). They most probably correspond to insertion polymorphisms in the most recent common ancestor of these two species, which became fixed for alternative alleles after separation of their gene pools.
The human genome harbors nearly half a million copies of roughly 100 HERV families (25). All of these families, except one, are now apparently extinct, i.e., they can spread no further over the genome. The only exception is HERV-K, which has three human-specific subfamilies (8), some of whose insertions are polymorphic in modern human populations and thus may still be capable of movement (42). The ultimate cause of the extinction of a TE family will be the reduction of its proliferation rate below a certain threshold, which depends on the per-nucleotide mutation rate. Thus, in Drosophila, where transpositions are relatively frequent, a TE jumps on average once in 104 to 105 generations, and the mutation rate is 10−9 to 10−8 per bp per generation, so that a copy of a typical element (104 bp) is expected to accumulate at least 1 mutation between jumps (31). This amount may not be considered too serious a risk for losing copy functionality, but if transposition rate is further reduced or the mutation rate is increased, many TEs may certainly die before they have a chance to transpose. But this is not likely to have been the case of ERV9. All the groups that make up subfamily XII appear to have been the result of independent expansions from single sequence variants, each in the elapsed time of the order of 103 to 104 generations, which certainly leaves very few opportunities for the gradual degeneration of the population of sequences.
Another possibility leading to extinction of a TE family is the fixation of restrictive factors in the host population. Host genomes have adopted several defense strategies against TEs and viruses, as part of their intracellular and extracellular conflicts for over a billion years of coevolution. One of the most useful and simple models for analyzing these relationships between TEs and the host genome is offered by Drosophila. In laboratory lines of D. melanogaster, different families of TEs are active in different lines, and transposition rates vary widely among families, with some of them transposing at very high rates and the rest remaining stable. Thus, unstable lines have been found for either gypsy or copia and have been shown to carry permissive alleles, which specifically release the host control on the copy number of the corresponding family; stable lines have been shown to carry alleles that restrict their transposition (see reference 31 and references therein). A repressive state specific for a given family may be established by homology-dependent trans-silencing mechanisms, produced by either transcriptional (inactivation of the promoter) or posttranscriptional (sequence-specific RNA degradation) molecular mechanisms. They were first described with transgenic plants but now appear to have a general role in genome defense against viruses and mobile elements in a broad range of normal organisms (10, 28, 35, 43). However, the best-characterized mechanisms for restricting proviral amplification in both exogenous and endogenous viruses involve different ways of preventing their binding to cell surface receptors, such as the Fv4 gene in mice (39), or hindering preintegration steps of retroviral replication, as in Fv1, Lv1, and Ref1 (2). One of the most remarkable aspects of these different kinds of control mechanisms is that the involved genes are frequently derived from specific TE or provirus copies, not necessarily from the same family that is under its control. Finally, in this succinct list of restriction factors, cytoplasmic RNA/DNA editing enzymes have been added to the intracellular repertoire of defenses in primate genomes, after recent studies of human cell line variation in susceptibility to HIV infection (34). Restrictive and permissive factors are likely to segregate in natural populations of all organisms, and their frequencies are probably the major determinants of the proliferation rates of the different TE families residing in the genome. Sometimes TEs escape from the control of the host and begin to expand in an explosive manner, bringing about a reduction in the relative fitness of the bearers of permissive alleles. Thus, the frequency of restrictive alleles in that population is expected to increase; if they happen to be finally fixed in the species, the corresponding TE family might have been repressed in relatively very few TE generations and so come to a “sudden” extinction just following a period of flourishing activity. This is precisely what seems to have happened with ERV9, which according to our data may have gone extinct in approximately 100,000 years (5,000 generations), after 32 MYR of residence as an active TE in the genome of our ancestors (15), interestingly just before the separation of the human and chimpanzee lineages. It would be very useful to know whether the same pattern applies to the many other families of extinct HERVs harbored by our genome. We are just beginning to understand the genetic basis of trans-silencing mechanisms, and it will probably take a long time to assess the relative strength of the evolutionary forces acting on their variation in natural populations. Until then, we may only guess by examining their putative effects on the populations of TE sequences.
Supplementary Material
Acknowledgments
This work was made possible by a fellowship from the Dirección Xeral de I+D, Xunta de Galicia (Spain), awarded to P.L.-S.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Belshaw, R., V. Pereira, A. Katzourakis, G. Talbot, A. Burt, and M. Tristem. 2004. Long-term reinfection of the human genome by endogenous retroviruses. Proc. Natl. Acad. Sci. USA 101:4894-4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bieniasz, P. D. 2003. Restriction factors: a defense against retroviral infection. Trends Microbiol. 11:286-291. [DOI] [PubMed] [Google Scholar]
- 3.Bird, A. 1980. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 8:1499-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brookfield, J. F. Y. 1986. A model for DNA sequence evolution within transposable element families. Genetics 112:393-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brookfield, J. F. Y. 1993. The generation of sequence similarity in SINEs and LINEs. Trends Genet. 9:38-39. [DOI] [PubMed] [Google Scholar]
- 6.Brookfield, J. F. Y. 1995. Transposable elements as selfish DNA, p. 130-153. In D. J. Sherratt (ed.), Mobile genetic elements. Oxford University Press, New York, N.Y.
- 7.Brookfield, J. F. Y. 2003. Mobile DNAs: The poacher turned gamekeeper. Curr. Biol. 13:R846-R847. [DOI] [PubMed] [Google Scholar]
- 8.Buzdin, A., S. Ustyugova, K. Khodosevich, I. Mamedov, Y. Lebedev, G. Hunsmann, and E. Sverdlov. 2003. Human-specific subfamilies of HERV-K (HML-2) long terminal repeats: three master genes were active simultaneously during branching of hominoid lineages. Genomics 81:149-156. [DOI] [PubMed] [Google Scholar]
- 9.Carr, M., J. R. Soloway, T. E. Robinson, and J. F. Y. Brookfield. 2002. Mechanisms regulating the copy numbers of six LTR retrotransposons in the genome of Drosophila melanogaster. Chromosoma 110:511-518. [DOI] [PubMed] [Google Scholar]
- 10.Casacuberta, J. M., and N. Santiago. 2003. Plant LTR-retrotransposons and MITEs: control of transposition and impact on the evolution of plant genes and genomes. Gene 311:1-11. [DOI] [PubMed] [Google Scholar]
- 11.Charlesworth, B., and D. Charlesworth. 1983. The population dynamics of transposable elements. Genet. Res. 42:1-27. [Google Scholar]
- 12.Clough, J. E., J. A. Foster, M. Barnett, and H. A. Wichman. 1996. Computer simulation of transposable element evolution: random template and strict master models. J. Mol. Evol. 42:52-58. [DOI] [PubMed] [Google Scholar]
- 13.Costas, J. 2002. Characterization of the intragenomic spread of the human endogenous retrovirus family HERV-W. Mol. Biol. Evol. 19:526-533. [DOI] [PubMed] [Google Scholar]
- 14.Costas, J. 2003. Molecular characterization of the recent intragenomic spread of the murine endogenous retrovirus MuERV-L. J. Mol. Evol. 56:181-186. [DOI] [PubMed] [Google Scholar]
- 15.Costas, J., and H. Naveira. 2000. Evolutionary history of the human endogenous retrovirus family ERV9. Mol. Biol. Evol. 17:320-330. [DOI] [PubMed] [Google Scholar]
- 16.Deininger, P. L., M. A. Batzer, C. A. Hutchison III, and M. H. Edgell. 1992. Master genes in mammalian repetitive DNA amplification. Trends Genet. 8:307-311. [DOI] [PubMed] [Google Scholar]
- 17.Desset, S., C. Meignin, B. Dastugue, and C. Vaury. 2003. COM, a heterochromatic locus governing the control of independent endogenous retroviruses from Drosophila melanogaster. Genetics 164:501-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Cristofano, A., M. Strazzullo, T. Parisi, and G. La Mantia. 1995. Mobilization of an ERV9 human endogenous retroviral element during primate evolution. Virology 213:271-275. [DOI] [PubMed] [Google Scholar]
- 19.Eickbush, T. H., and A. V. Furano. 2002. Fruit flies and humans respond differently to retrotransposons. Curr. Opin. Genet. Dev. 12:669-674. [DOI] [PubMed] [Google Scholar]
- 20.Glazko, G. V., and M. Nei. 2003. Estimation of divergence times for major lineages of primate species. Mol. Biol. Evol. 20:424-434. [DOI] [PubMed] [Google Scholar]
- 21.Jurka, J. 1994. Approaches to identification and analysis of interspersed repetitive DNA sequences, p. 294-298. In M. D. Adams, C. Fields, and J. C. Venter (ed.), Automated DNA sequencing and analysis. Academic Press, London, United Kingdom.
- 22.Kaplan, N., T. Darden, and C. H. Langley. 1985. Evolution and extinction of transposable elements in mendelian populations. Genetics 109:459-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kidwell, M. G., and D. Lisch. 1997. Transposable elements as sources of variation in animals and plants. Proc. Natl. Acad. Sci. USA 94:7704-7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim, A. I., N. V. Lyubomirskaya, E. S. Belyaeva, and Y. V. Ilyin. 1994. The introduction of a transpositionally active copy of retrotransposon gypsy into a stable strain of Drosophila melanogaster causes genetic instability. Mol. Gen. Genet. 242:472-477. [DOI] [PubMed] [Google Scholar]
- 25.Lander, E. S., L. M. Linton, B. Birren, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860-921. [DOI] [PubMed] [Google Scholar]
- 26.Lania, L., A. Di Cristofano, M. Strazzullo, G. Pengue, B. Majello, and G. La Mantia. 1992. Structural and functional organization of the human endogenous retroviral ERV9 sequences. Virology 191:464-468. [DOI] [PubMed] [Google Scholar]
- 27.Lorenc, A., and W. Makalowski. 2003. Transposable elements and vertebrate protein diversity. Genetica 118:183-191. [PubMed] [Google Scholar]
- 28.Matzke, M. A., W. Aufsatz, T. Kanno, M F. Mette, and A. J. M. Matzke. 2002. Homology-dependent gene silencing and host defense in plants. Adv. Genet. 46:235-275. [DOI] [PubMed] [Google Scholar]
- 29.Medstrand, P., and D. L. Mager. 1998. Human-specific integrations of the HERV-K endogenous retrovirus family. J. Virol. 72:9782-9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller, W. J., J. F. McDonald, D. Nouaud, and D. Anxolabehere. 1999. Molecular domestication—more than a sporadic episode in evolution. Genetica 107:197-207. [PubMed] [Google Scholar]
- 31.Nuzhdin, S. V. 1999. Sure facts, speculations, and open questions about the evolution of transposable element copy number. Genetica 107:129-137. [PubMed] [Google Scholar]
- 32.Perdue, S., and S. V. Nuzhdin. 2000. Master copy is not responsible for the high rate of copia transposition in Drosophila. Mol. Biol. Evol. 17:984-986. [DOI] [PubMed] [Google Scholar]
- 33.Rambaut, A., and N. C. Grassly. 1997. Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput. Appl. Biosci. 13:235-238. [DOI] [PubMed] [Google Scholar]
- 34.Sawyer, S. L., M. Emerman, and H. S. Malik. 2004. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2:e275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sijen, T., and R. H. Plasterk. 2003. Transposon silencing in the Caenorhabditis elegans germ line by natural RNAi. Nature 426:310-314. [DOI] [PubMed] [Google Scholar]
- 36.Svensson, A. C., T. Raudsepp, C. Larsson, A. Di Cristofano, M. G. Chowdhary, L. Rask, and G. Andersson. 2001. Chromosomal distribution, localization and expression of the human endogenous retrovirus ERV9. Cytogenet. Cell Genet. 92:89-96. [DOI] [PubMed] [Google Scholar]
- 37.Tachida, H. 1996. A population genetic study of the evolution of SINEs. II. Sequence evolution under the master copy model. Genetics 143:1033-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takahata, N. 1993. Allelic genealogy and human evolution. Mol. Biol. Evol. 10:2-22. [DOI] [PubMed] [Google Scholar]
- 39.Taylor, G. M., Y. Gao, and D. A. Sanders. 2001. Fv-4: identification of the defect in Env and the mechanism of resistance to ecotropic murine leukemia virus. J. Virol. 75:11244-11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Townsend, J. P., and D. L. Hartl. 2000. The kinetics of transposable element autoregulation. Genetica 108:229-237. [DOI] [PubMed] [Google Scholar]
- 41.Tristem, M. 2000. Identification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the human genome mapping project database. J. Virol. 74:3715-3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turner, G., M. Barbulescu, M. Su, M. I. Jensen-Seaman, K. K. Kidd, and J. Lenz. 2001. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr. Biol. 11:1531-1535. [DOI] [PubMed] [Google Scholar]
- 43.Vastenhouw, N. L., S. E. Fischer, V. J. Robert, K. L. Thijssen, A. G. Fraser, R. S. Kamath, J. Ahringer, and R. H. Plasterk. 2003. A genome-wide screen identifies 27 genes involved in transposon silencing in C. elegans. Curr. Biol. 13:1311-1316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.