

Abstract
Overgrowth syndromes represent a diverse group of disorders with overlapping features. Interdisciplinary management by a team of experts in vascular anomalies is crucial for establishing the correct diagnosis and optimizing outcomes for these patients. Unique management considerations include increased risk for thrombosis and in some cases, cancer. In recent years, research has demonstrated that these disorders are primarily caused by somatic mutations in growth pathways, particularly the PI3K-mTOR pathway. This improved understanding had led to promising new therapies for this group of patients.
Introduction
Overgrowth syndromes refer to clinically diverse entities defined by excessive proliferation of organs or tissues in association with vascular anomalies. Overgrowth can range from focal to diffuse involvement. Historically, the overgrowth syndromes have been defined by eponymous names (Klippel-Trenaunay Syndrome, Parkes Weber Syndrome). Within the last two decades, our understanding of the molecular mechanisms underlying overgrowth syndromes has progressed at a rapid pace (Table 1). While overgrowth disorders were previously classified on a phenotypic basis according to a pattern of clinical, radiologic, and histologic features, recent discoveries have elucidated that specific genetic changes underlie this group of vascular anomalies. Many of these genetic changes involve cancer pathways and thus confer important implications for treatment.
Table 1.
Clinical summary table of vascular malformations with overgrowth.
| KTS | CLOVES syndrome | FAVA | DCMO | Parkes Weber syndrome | Proteus Syndrome |
PHTS | |
|---|---|---|---|---|---|---|---|
| Capillary malformation* | + | + | − | + | + | + | +/− |
| Venous malformation and/or ectasia | + | + | + | +/− | +/− | + | +/− |
| Arteriovenous malformation | − | +/− | − | − | + | +/− | +/− |
| Lymphatic malformation | +/− | + | − | − | − | + | +/− |
| Overgrowth** | + | + | + | + | + | + | + |
| Digital anomalies | + | + | − | − | − | + | − |
| Thrombosis Risk | + | + | − | − | + | ++ | − |
| Malignant Cancer Risk | − | + | − | − | − | − | +++ |
| Genetic Mutation(s) Identified | PIK3CA | PIK3CA | PIK3CA | GNA11 PIK3CA |
RASA1 EPHB4 |
AKT1 | PTEN |
TS: Klippel-Trenaunay syndrome; CLOVES: congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and skeletal anomalies; FAVA: fibroadipose vascular anomaly; DCMO: diffuse capillary malformation with overgrowth; PHTS: PTEN hamartoma tumor syndrome.
The capillary malformation is often dark red/purple and geographic in KTS and CLOVES. In DCMO, the stain is pink, diffuse, and reticulated.
Overgrowth in KTS is usually out-of-proportion to growth elsewhere, whereas in DCMO the overgrowth keeps pace with growth of the child. Lipomatous overgrowth is a prominent feature of CLOVES syndrome and primarily involves the trunk.
Improvements in the ease and affordability of next generation sequencing has led to significant advances in understanding the overgrowth syndromes. The phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway is involved in several vascular anomalies and accounts for the clinical symptoms seen in many of the overgrowth disorders. Given this understanding, the mTOR inhibitor sirolimus has been used to treat a variety of vascular anomalies, including the overgrowth syndromes.1 Klippel-Trenaunay Syndrome (KTS), Congenital Lipomatous Overgrowth, Vascular Malformations, Epidermal Nevi, Scoliosis/Skeletal and Spinal Syndrome (CLOVES) and fibroadipose vascular anomaly (FAVA) are caused by somatic mutations in the PIK3CA gene and are now collectively referred to as PIK3CA-related overgrowth syndrome (PROS).2 4- PIK3CA mutations can be common in many solid tumors, and targeted therapies including PIK3CA inhibitors have been developed for use in cancer.5 These agents, which target specific somatic mutations, have also shown clinical promise in patients with vascular anomalies.6–8
While many of the overgrowth syndromes are due to changes in PIK3CA, the genetic landscape of vascular anomalies is not limited to mutations in this gene alone. Proteus syndrome results from somatic mutations in AKT1 9. PTEN tumor hamartoma syndrome (PTHS), an overgrowth disorder and a cancer predisposition syndrome, is caused by aberrations in the PTEN gene.10–14 Parkes Weber syndrome, characterized by a high-flow lesion, results from mutations in the RASA1 or EPHB4 genes.15,16 Here, we describe clinical features of the most common overgrowth syndromes, summarize unique management considerations, and then discuss emerging therapies for treatment.
Klippel-Trenaunay syndrome (KTS)
Klippel-Trenaunay syndrome (KTS) is characterized by a capillary lymphatic venous malformation (CLVM) and primarily fatty overgrowth of soft tissue and bone (Fig. 1C). Involvement of the lower extremities is most common. The capillary malformation (or “port wine stain”) usually occurs on the thigh and upper part of the calf (Fig. 1A). Lymphatic vesicles can erupt at this location and cause bleeding and leakage of lymphatic fluid (Fig. 1B). Hyperhidrosis of the capillary malformation is often present, suggesting abnormal innervation to the sweat glands in the affected area.17 Typically, KTS is associated with low-flow vascular lesions. As such, the affected limb does not show signs of hyperdynamic flow and the limb is not warm to touch (as opposed to Parkes Weber syndrome, described later in this article). KTS patients often have chronic debilitating pain and suffer from numerous orthopedic issues that limit movement and impair quality of life. These problems are often managed surgically or with interventional radiology procedures. KTS patients are at risk for thrombotic complications. The marginal venous system within KTS patients demonstrates stagnant flow and competes with a smaller deep venous system, which is often underdeveloped.18,19 There is an increased risk of superficial thrombophlebitis, deep venous thrombosis and pulmonary embolism due to these enlarged marginal veins.19,20 As such, early detection and closure of these large ectatic veins, either by embolization, photocoagulation with endovenous laser, or surgical excision is often pursued.18
Fig. 1.
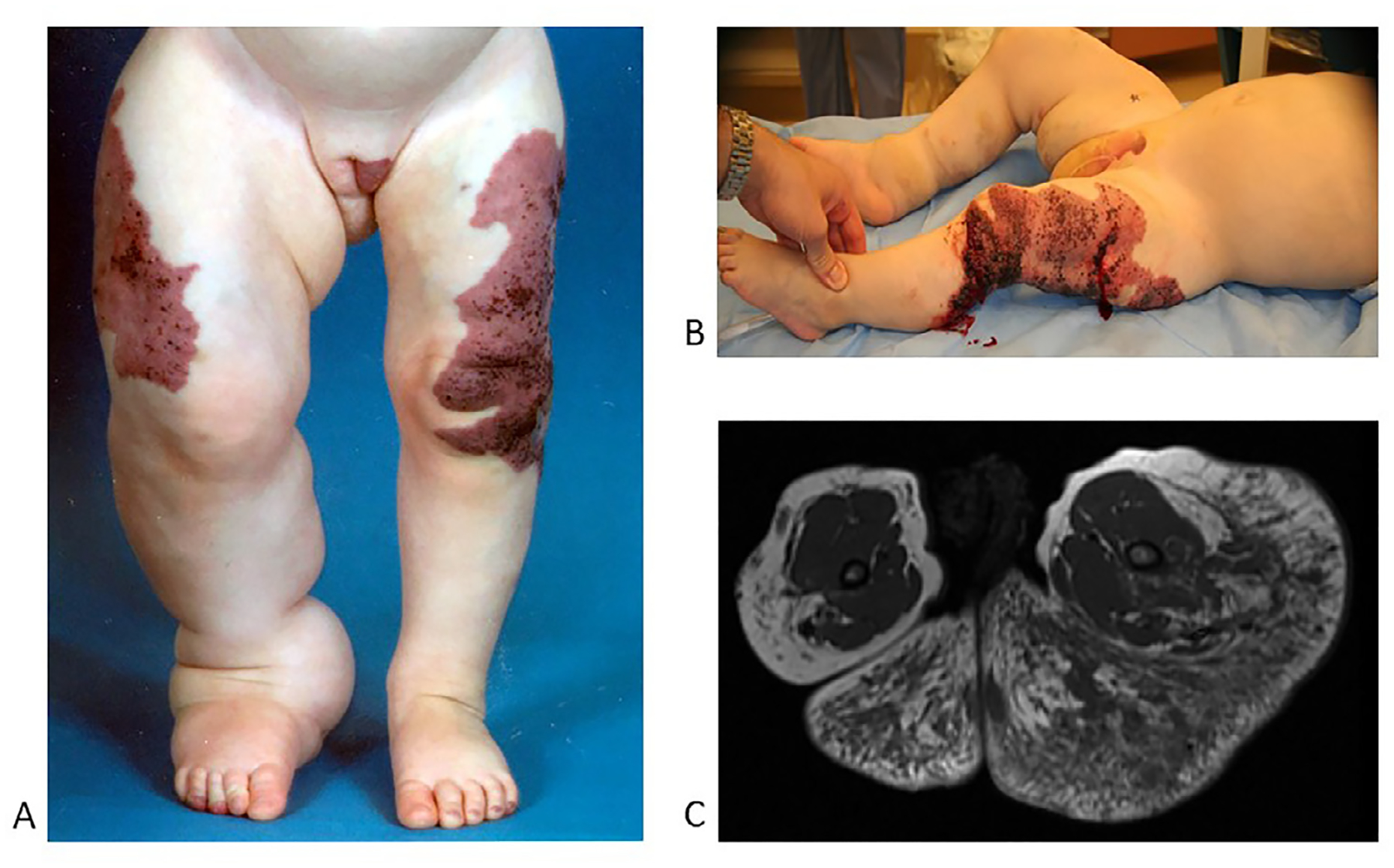
Klippel-Trenaunay syndrome
A. Geographic capillary stain, scattered lymphatic vesicles, enlarged lateral marginal vein, bulky overgrowth of extremity.
B. Bleeding and leakage from blebs on affected extremity.
C. Axial T1 MRI without fat saturation of upper thighs and lower gluteal area. Note that the bulk is composed of excessive extra-fascial fatty overgrowth (hyperintense) with lymphatic malformations (hypointense dark streaks).
In the pelvis, there can be diffuse lymphatic and venous involvement of the anorectal area and surrounding adipose tissue. This can result in chronic rectal bleeding and pain. Hematuria can also result. Recurrent infections are also a feature of this disease, likely due to bacterial proliferation in abnormal lymphatic spaces.21 Some patients with massive and disabling overgrowth undergo surgical debulking to reduce the size of the affected extremity. While this is not curative, it can improve the quality of life for some patients. For KTS patients with chronic rectal bleeding and recurrent need for transfusions, endorectal pull-through can be an option to minimize these symptoms.22 Patients with KTS require close orthopedic monitoring for limb length discrepancy, contractures, muscle atrophy and growth issues. In rare cases, amputation of a limb or overgrown appendage (i.e. a toe) can help with chronic pain and disability.
Patients with KTS require an interdisciplinary approach to care, including the expertise of general surgery, plastic surgery, interventional radiology, orthopedics, and hematology/oncology providers. Compression is a mainstay of therapy and can help to decrease limb swelling and pain. Cutaneous lymphatic vesicles can be treated with sclerotherapy, carbon dioxide laser photovaporation, cauterization, or excision.23 Recently, medical therapies have been helpful in stabilizing disease. Sirolimus has been shown to help some patients and can decrease swelling, pain, and the frequency of infections. However, not all patients respond to this therapy.1 Given that somatic mutations in the PIK3CA gene drive abnormal growth in KTS, the use of oral PIK3CA or AKT inhibitors has been considered for these patients (Fig. 7). Prospective clinical trials for KTS are underway for AKT inhibitors and are in development for PIK3CA inhibitors.
Fig. 7.
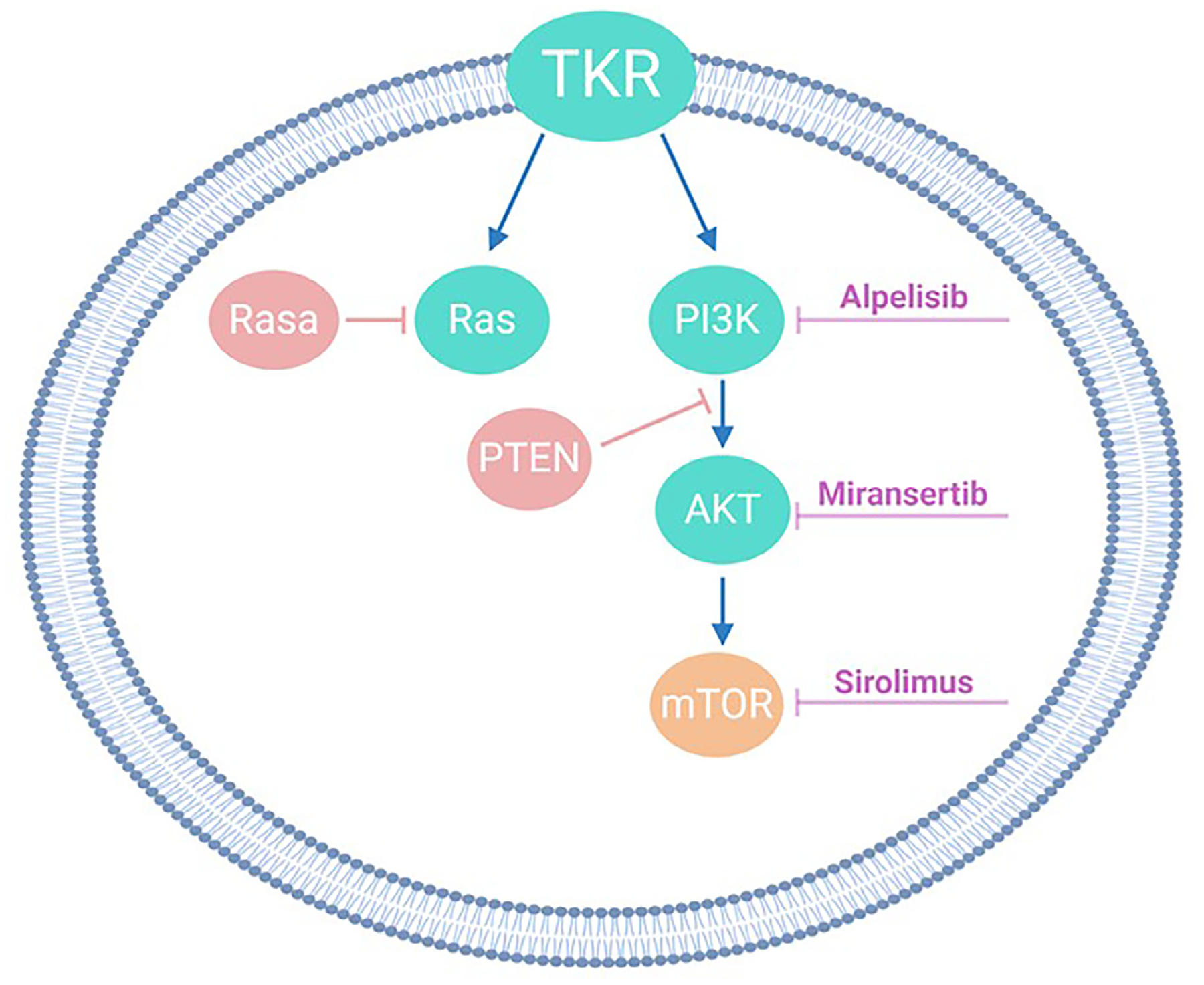
Growth pathways and targeted medical therapies involved in vascular anomalies.
Congenital lipomatous overgrowth, vascular malformations, epidermal nevi, scoliosis/skeletal and spinal syndrome (CLOVES)
CLOVES was first described in 2009 as a unique disorder characterized by lipomatous masses of the trunk, retroperitoneum and pelvis in association with acral deformities.2 It is now considered to be part of the broader umbrella term of PROS given that it is caused by somatic gain-of-function mutations in the PIK3CA gene.3 There is great clinical heterogeneity within this disorder. Truncal lipomatous mass(es) evident at birth are a defining feature of CLOVES (Fig. 2A and C). Fast-flow lesions (AVMs) may be present within the fatty truncal masses, which contributes to the morbidity seen in this disorder.2,24,25 Other clinical features include characteristic triangular shape of extremities with broad- based toes, macrodactyly, and a “sandal-gap” deformity of the first and second toes (Fig. 2B). As with all of the overgrowth disorders, patients with CLOVES often suffer from severe pain and functional impairment. Originally described as CLOVE syndrome, an “S” was added to the definition to capture the occurrence of scoliosis, spinal and skeletal abnormalities that can be found among patients.26 Children with CLOVES are at increased risk of developing seizures and spinal/paraspinal arteriovenous malformations. In the most severe patients, neurologic and renal systems can also be affected. In CLOVES patients, the left kidney is usually larger than the right kidney. There is also an increased incidence of Wilms tumor in patients with CLOVES compared to the general population and thus screening with renal ultrasounds every 3 months for pediatric patients up until 8 years of age should be considered.26 This is similar to the recommendation for patients with Beckwith-Wiedemann syndrome or isolated hemihypertrophy.26 In a single-institution study of 122 patients with CLOVES, the incidence of Wilms tumor was 3.3% (4/122), a significant increase compared to the general population (1/10,000). 26 There was no increased risk of hepatoblastoma. As in Klippel-Trenaunay syndrome, patients with CLOVES are also at high risk for thrombosis and pulmonary embolism, and prophylactic anticoagulation should be considered during periods of immobility or for patients undergoing surgery.20,27
Fig. 2.
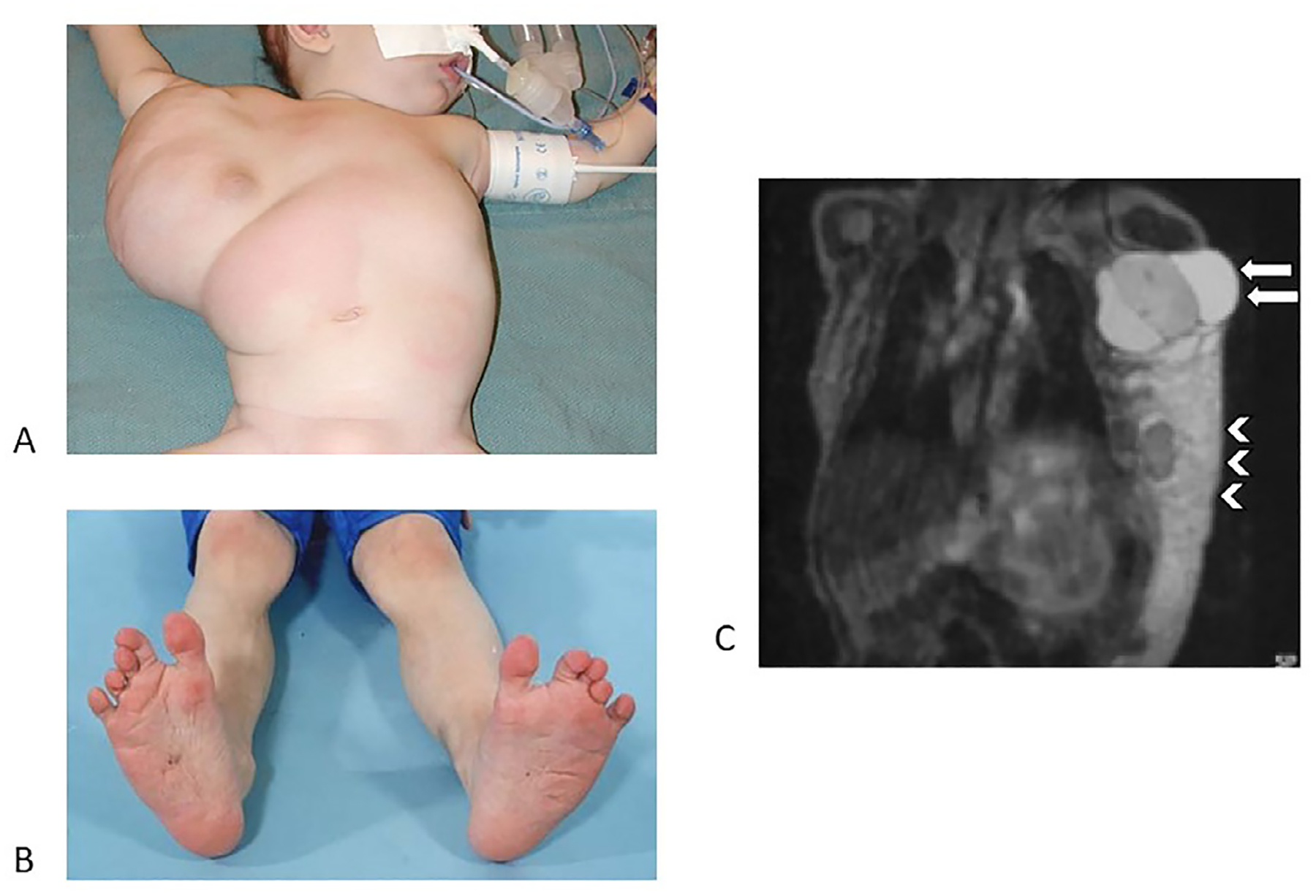
CLOVES
A. Lipomatous overgrowth of the trunk, noted at birth.
B. Classic foot findings of CLOVES, including sandal gap deformity (bilateral here) and triangular shape.
C. Bulky overgrowth is composed of both fatty infiltration and macrocystic lymphatic malformation (arrows denote large, multi-septated areas in the axilla).
Recently, researchers in France created a postnatal mouse model of PROS/CLOVES that partially recapitulated the disease seen in humans.6 They treated these mice with the PIK3CA inhibitor BYL719 (alpelisib). The treated mice had improved survival compared to those who received placebo. Alpelisib was found in their mouse model to be more effective in reducing tumor burden than sirolimus. Based on these results, alpelisib was used to treat a series of 19 patients with PROS, many with life-threatening symptoms. Alpelisib led to symptomatic and radiologic improvement in all patients and had limited reported side effects. While this study showed particular promise, the medication was provided in a compassionate use setting and was not a prospective clinical trial. In the United States, prospective trials evaluating use of alpelisib are emerging. This work will be important to confirm the promising results seen in the compassionate use study, to establish more robust safety data, and to evaluate the durability of response in this patient population.
AKT inhibitors are another promising therapy for use in patients with overgrowth syndromes given the role of AKT in cellular processes such as growth, metabolism, and cell proliferation (Fig. 7).28,29 A Phase I/II study of the AKT inhibitor ARQ-092 is actively recruiting patients (clinicaltrials.gov NCT03094832).
Fibro-adipose vascular anomaly (FAVA)
Fibro-adipose vascular anomaly (FAVA) is a recently recognized disorder defined by fibrofatty replacement of affected muscle (usually the calf) and slow-flow vascular malformations30 (Fig. 3A). Pain is a defining feature, as are joint contractures. FAVA can be often be distinguished from other entities based on the presence of a dominant solid component with phlebectasia. On ultrasound, echogenic (solid) components are present. On MRI, there is moderately hyper-intense T2 signal enhancement (Fig. 3B) and enhancement on post-contrast T1-weighted images (Fig. 3C). On histopathology, dense fibrous tissue, fat, and lymphoplasmacytic aggregates exist within atrophied skeletal muscle. Early involvement of an orthopedic surgeon is critical for patients with this disorder. Unlike many of the other vascular malformations, FAVA usually does not respond to sclerotherapy. Surgery or medical therapy is often favored as a result.31 Cryoablation can also be effective particularly in reducing pain and improving quality of life for these patients and offers a minimally invasive option.32 In some cases, FAVA has also been shown to be due to PIK3CA mutations.
Fig. 3.
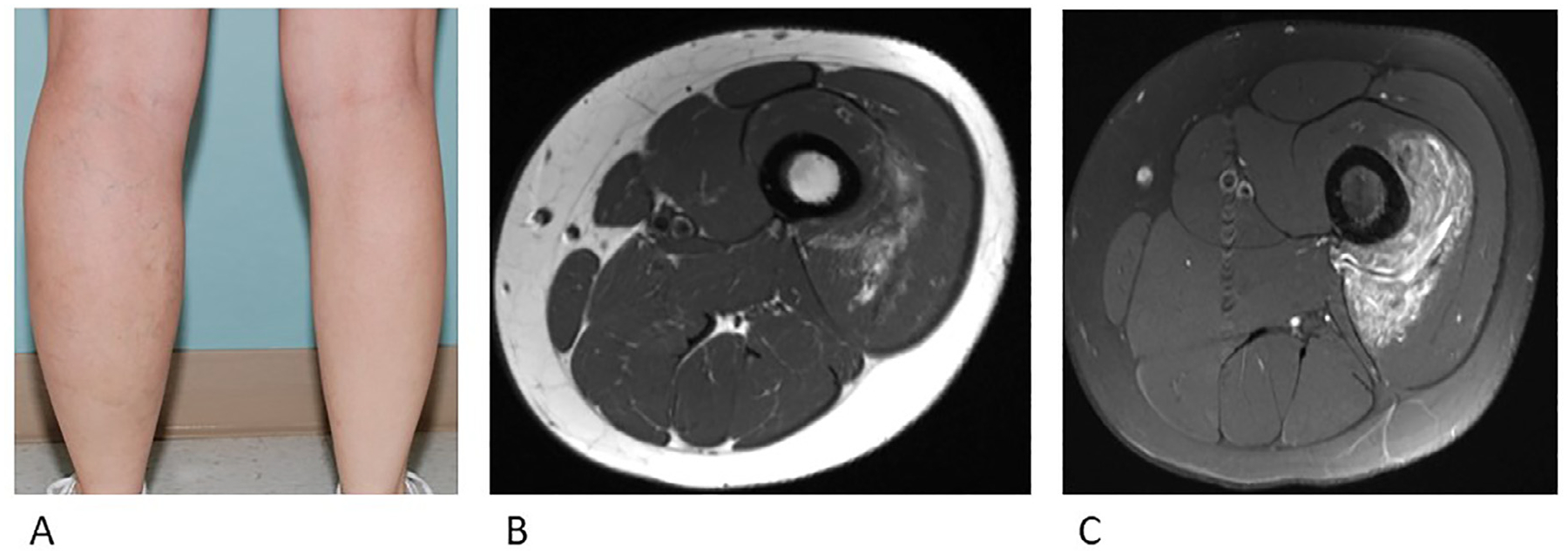
Fibro-adipose vascular anomaly (FAVA)
A. Overgrowth is visible in the affected calf.
B. Axial (a) T1-weighted MRI image demonstrated the muscle replaced by focal heterogeneous FAVA lesion.
c. Axial (b) fat saturated T2-weighted MRI image demonstrates heterogeneous hyperintense lesion.
Diffuse capillary malformation with overgrowth (DCMO)
Diffuse capillary malformation with overgrowth (DCMO) is a clinical entity describing patients with multiple extensive capillary malformations associated with facial asymmetry, limb overgrowth, and foot abnormalities33 (Fig. 4A and B). Patients usually have overgrowth of soft tissue or bone that is out of proportion to the extent of the capillary malformation; this is distinct from the overgrowth seen in Klippel-Trenaunay and CLOVES, in that it does not appear to be lipomatous. Patients with DCMO do not seem to be at increased risk for developing Wilms tumor.34 Recently, somatic mutations in both GNA11 and PIK3CA have been identified in patients with DCMO, suggesting that this clinical diagnosis may be heterogeneous. In other words, patients with GNA11 mutations may lack the features of overgrowth seen in other PROS disorders and may be more likely to have isolated capillary malformations involving an extremity without other features (i.e. hand/foot abnormalities, rapid overgrowth of adipose tissue).35,36
Fig. 4.
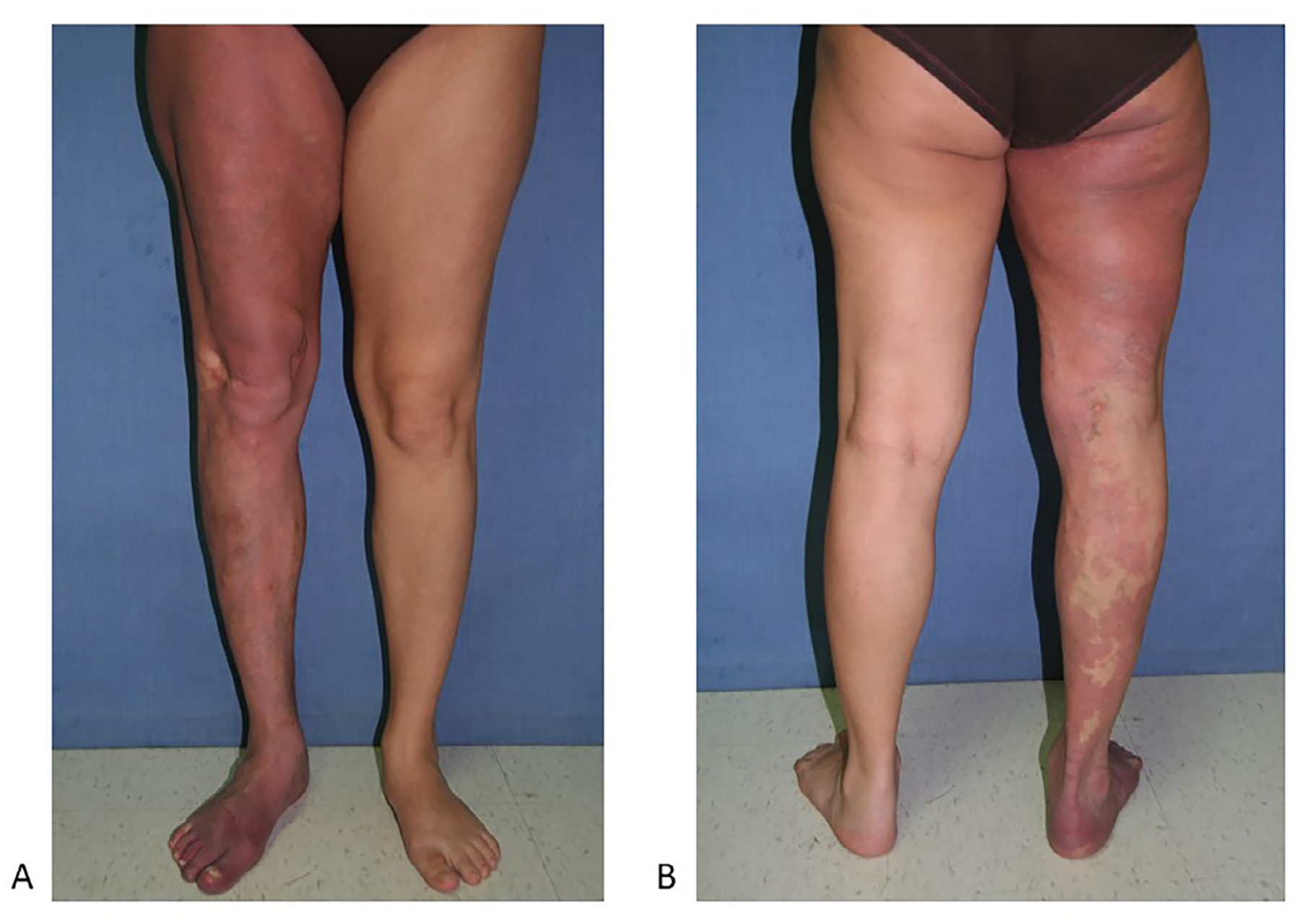
Diffuse capillary malformation with overgrowth (DCMO)
A. Note the diffuse capillary malformation with circumferential overgrowth. This patient also has a leg length discrepancy.
B. Dilated veins are also noted.
Parkes Weber syndrome
Parkes Weber Syndrome, or capillary arteriovenous malformation (CAVM), is defined by a capillary malformation and a fast-flow vascular lesion that is present at birth (Fig. 5A and B)15,37,38. It is less common than KTS. It affects the lower limb most commonly, although involvement of an arm is more common in Parkes Weber than in KTS. The limb is symmetrically enlarged and often leads to a limb length discrepancy. MRI reveals overgrowth in soft tissue and bone, as well as arterial shunting, often diffuse and without a discrete nidus.39 Due to the presence of AV shunting, the extremity is often warm to the touch and a bruit or thrill can often be appreciated. Cardiac overload may be present at birth or develop later in life; patients should be followed regularly for this potential complication.40–43 Some cases of Parkes Weber are caused by germline inactivating mutations in the RASA1 gene as part of capillary malformation-arteriovenous malformation (CM-AVM1) syndrome,38,39,44,45 or mutations in the EPHB4 gene as part of CM-AVM2 syndrome.46,47 In these instances, there is autosomal dominant inheritance. There is high penetrance and wide phenotypic variability, even among members of the same family.46 However, in 25% of patients with clinical symptoms consistent with CM-AVM1 or CM-AVM2, no germline mutation is found.46 Recent studies suggests that the patients with negative germline testing may have mosaic mutations in RASA1.46
Fig. 5.
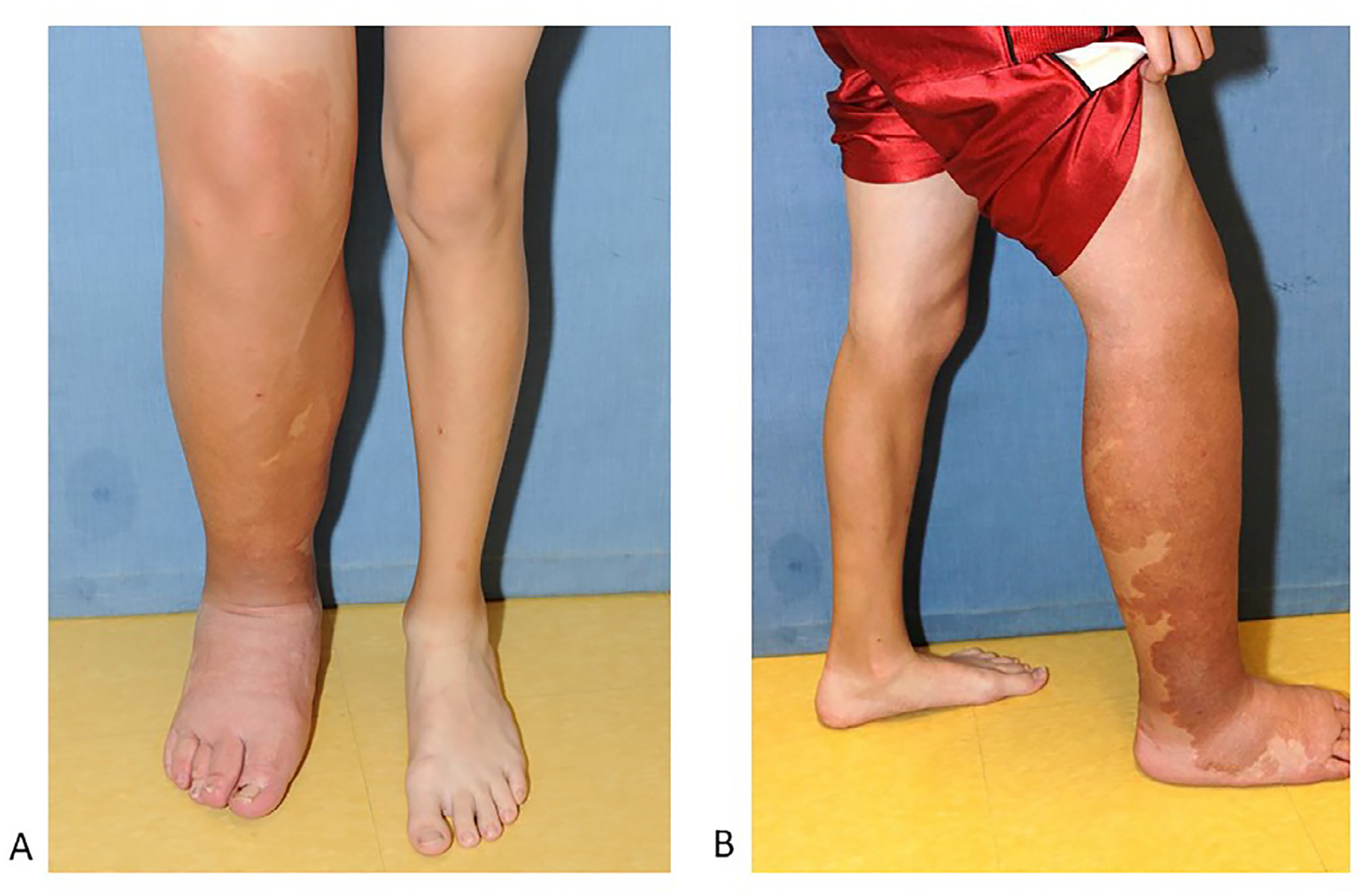
Parkes Weber syndrome.
A Enlarged lower limb and capillary malformation with upward extension. Compression can help with lymphatic swelling.
B. Diffuse stain with overgrowth.
Proteus syndrome
Proteus Syndrome is characterized by asymmetric overgrowth, cerebriform connective tissue nevi, epidermal nevi, and overgrowth of adipose and vascular tissue.9,48,49 Named after the Greek sea god who could transform its shape to evade its captors, Proteus syndrome can be difficult to distinguish from other vascular anomalies. Cerebriform connective tissue nevi are pathognomonic. Hypertrophy is not necessarily congenital in Proteus syndrome. Within the same patients, adipose tissue can be both decreased (lipohypoplasia) or increased (lipohyperplasia) at differing sites. Patients with Proteus syndrome are at increased risk for the development of thrombotic events, and pulmonary embolism is a leading cause of death in these patients. In addition to the stasis associated with vascular malformations and disordered endothelium, Proteus syndrome seems to confer additional thrombotic risk due to the mutations in AK T1. The AK T inhibitor ARQ-092 is currently being evaluated for the treatment of Proteus syndrome in prospective clinical trials50 (clinicaltrials.gov Identifiers NCT03094832 and NCT04316546).
PTEN hamartoma tumor syndrome (PHTS)
The historical eponymic names Bannayan-Riley-Ruvalcaba syndrome and Cowden syndrome have been replaced by the broader term of PTEN Hamartoma Tumor Syndrome (PHTS), given that these disorders are caused by germline mutations in the PTEN gene.14 Unlike the PROS disorders, which represent disorders of somatic mosaicism, PHTS is caused by germline changes inherited in an autosomal dominant manner.10–12 Clinically, the PHTS is characterized by macrocephaly (Fig. 6A1AA, B) A,)BA, B) pigmented penile macules (Fig. 6C), gastrointestinal polyps, thyroid disease, and several benign and malignant neoplasms (Fig. 6A). Macrocephaly is a defining feature and patients’ head circumferences are often at least 4.5 standard deviations above the mean.14 Neurodevelopmental complications are common, and patients may experience delays in speech or motor development. Coordinated care with a neurologist or neurodevelopmental team is essential given the co-occurrence of autism as well as arteriovenous malformations (AVMs). Skeletal abnormalities may include hyper-extensible joints, pectus excavatum, and scoliosis. Hamartomatous growths can be surgically resected if they are painful or bothersome.
Fig. 6.
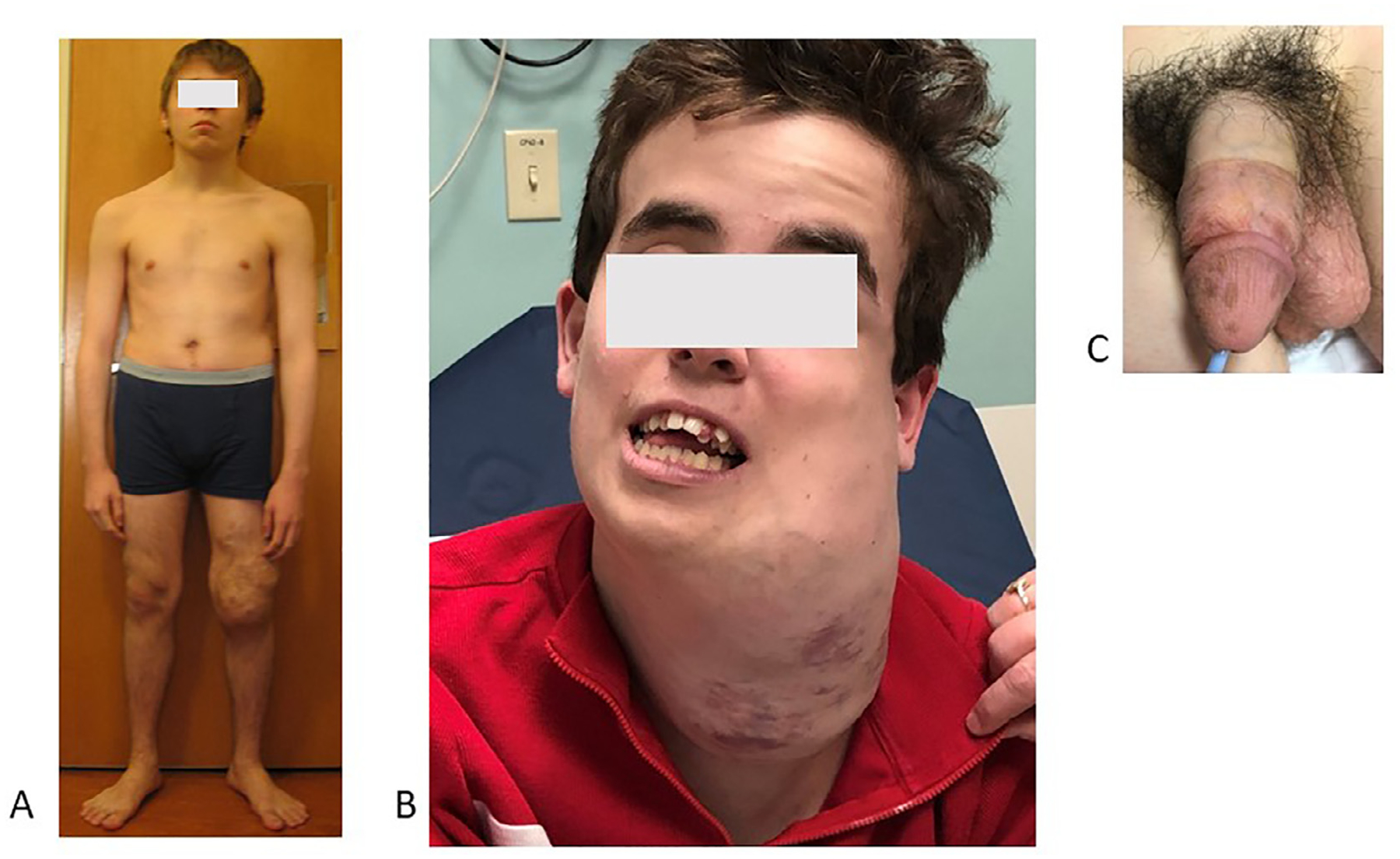
PTEN hamartoma tumor syndrome (PHTS)
A. Patient with soft tissue “hamartomas” of left lower extremity.
B. Overgrowth in this disorder can be profound and severely impact quality-of-life.
C. Penile freckling pathognomonic of patients with PHTS.
Patients with PHTS develop benign and malignant tumors of the breast, thyroid, skin, uterus, and GI tract. In childhood, annual thyroid ultrasounds and close neurodevelopmental follow-up are recommended. Adults with PHTS should be closely followed by an oncology or cancer predisposition team. Clinical breast exams are recommended starting at age 25 years (or 5–10 years before the earliest known breast cancer in the family), breast imaging at 30 years (or earlier if family history revealed breast cancer at earlier age), endometrial biopsy starting at age 30 years, kidney imaging at 30 years, colonoscopy at age 35 years (earlier if symptoms arise or if family members have been diagnosed with colon cancer at a younger age).14 Annual dermatologic exams are important for all patients with PTHS to screen for skin cancer.
Conclusion
Increased use of next generation sequencing and expansion of molecular methods has refined our understanding of many of the overgrowth syndromes. Diverse clinical phenotypes can be caused by changes within a single gene. Improved understanding of the mechanisms involved in the development of overgrowth disorders has led to a genotypic classification of disease and opened new possibilities for targeted therapeutic approaches.
References
- 1.Adams DM, Trenor CC, Hammill AM, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics. 2016;137(2). doi: 10.1542/peds.2015-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alomari AI. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: a descriptive study of 18 cases of CLOVES syndrome. Clin Dysmorphol. 2009;18(1):1–7. doi: 10.1097/MCD.0b013e328317a716. [DOI] [PubMed] [Google Scholar]
- 3.Keppler-Noreuil KM, Rios JJ, Parker VE, et al. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. 2015;167A(2):287–295. doi: 10.1002/ajmg.a.36836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurek KC, Luks VL, Ayturk UM, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90(6):1108–1115. doi: 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adashek JJ, Kato S, Lippman SM, Kurzrock R. The paradox of cancer genes in non-malignant conditions: implications for precision medicine. Genome Med. 2020;12(1):16 02. doi: 10.1186/s13073-020-0714-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venot Q, Blanc T, Rabia SH, et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature. 2018;558(7711):540–546 06. doi: 10.1038/s41586-018-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.López Gutiérrez JC, Lizarraga R, Delgado C, et al. Alpelisib treatment for genital vascular malformation in a patient with congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and spinal/skeletal anomalies and/or scoliosis (CLOVES) syndrome. J Pediatr Adolesc Gynecol. 2019;32(6):648–650. doi: 10.1016/j.jpag.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 8.Kirstein AS, Augustin A, Penke M, et al. The novel phosphatidylinositol-3-kinase (PI3K) inhibitor alpelisib effectively inhibits growth of PTEN-haploinsufficient lipoma cells. Cancers Basel. 2019;11(10). doi: 10.3390/cancers11101586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marsh DJ, Dahia PL, Caron S, et al. Germline PTEN mutations in Cowden syndrome-like families. J Med Genet. 1998;35(11):881–885. doi: 10.1136/jmg.35.11.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marsh DJ, Coulon V, Lunetta KL, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998;7(3):507–515. doi: 10.1093/hmg/7.3.507. [DOI] [PubMed] [Google Scholar]
- 12.Marsh DJ, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8(8):1461–1472. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 13.Eng C, Thiele H, Zhou XP, Gorlin RJ, Hennekam RC, Winter RM. PTEN mutations and proteus syndrome. Lancet. 2001;358(9298):2079–2080. doi: 10.1016/S0140-6736(01)07110-0. [DOI] [PubMed] [Google Scholar]
- 14.Eng C PTEN: one gene, many syndromes. Hum Mutat. 2003;22(3):183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 15.Boon LM, Mulliken JB, Vikkula M. RASA1: variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev. 2005;15(3):265–269. doi: 10.1016/j.gde.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Revencu N, Boon LM, Mulliken JB, et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat. 2008;29(7):959–965. doi: 10.1002/humu.20746. [DOI] [PubMed] [Google Scholar]
- 17.Uller W, Fishman SJ, Alomari AI. Overgrowth syndromes with complex vascular anomalies. Semin Pediatr Surg. 2014;23(4):208–215. doi: 10.1053/j.sempedsurg.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 18.Alomari AI, Burrows PE, Lee EY, Hedequist DJ, Mulliken JB, Fishman SJ. CLOVES syndrome with thoracic and central phlebectasia: increased risk of pulmonary embolism. J Thorac Cardiovasc Surg. 2010;140(2):459–463. doi: 10.1016/j.jtcvs.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 19.Alomari AI. Diversion venography–a modified technique in Klippel-Trenaunay syndrome: initial experience. J Vasc Interv Radiol. 2010;21(5):685–689. doi: 10.1016/j.jvir.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 20.Reis J, Alomari AI, Trenor CC, et al. Pulmonary thromboembolic events in patients with congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and spinal/skeletal abnormalities and Klippel-Trénaunay syndrome. J Vasc Surg Venous Lymphat Disord. 2018;6(4):511–516 07. doi: 10.1016/j.jvsv.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 21.Kulungowski AM, Fishman SJ. Management of combined vascular malformations. Clin Plast Surg. 2011;38(1):107–120. doi: 10.1016/j.cps.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 22.Fishman SJ, Shamberger RC, Fox VL, Burrows PE. Endorectal pull-through abates gastrointestinal hemorrhage from colorectal venous malformations. J Pediatr Surg. 2000;35(6):982–984. doi: 10.1053/jpsu.2000.6947. [DOI] [PubMed] [Google Scholar]
- 23.Dasgupta R, Fishman SJ. Management of visceral vascular anomalies. Semin Pediatr Surg. 2014;23(4):216–220. doi: 10.1053/j.sempedsurg.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Alomari AI, Chaudry G, Rodesch G, et al. Complex spinal-paraspinal fast-flow lesions in CLOVES syndrome: analysis of clinical and imaging findings in 6 patients. Am J Neuroradiol. 2011;32(10):1812–1817 2011 Nov-Dec. doi: 10.3174/ajnr.A2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bloom J, Upton J. CLOVES syndrome. J Hand Surg Am. 2013;38(12):2508–2512. doi: 10.1016/j.jhsa.2013.08.120. [DOI] [PubMed] [Google Scholar]
- 26.Alomari AI. CLOVE(S) syndrome: expanding the acronym. Am J Med Genet A. 2009;149A(2):294 author reply 295. doi: 10.1002/ajmg.a.32632. [DOI] [PubMed] [Google Scholar]
- 27.Keppler-Noreuil KM, Lozier J, Oden N, et al. Thrombosis risk factors in PIK3CA-related overgrowth spectrum and Proteus syndrome. Am J Med Genet C Semin Med Genet. 2019;181(4):571–581 12. doi: 10.1002/ajmg.c.31735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keppler-Noreuil KM, Parker VE, Darling TN, Martinez-Agosto JA. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet C Semin Med Genet. 2016;172(4):402–421 12. doi: 10.1002/ajmg.c.31531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ranieri C, Di Tommaso S, Loconte DC, et al. In vitro efficacy of ARQ 092, an allosteric AKT inhibitor, on primary fibroblast cells derived from patients with PIK3CA-related overgrowth spectrum (PROS). Neurogenetics. 2018;19(2):77–91 05. doi: 10.1007/s10048-018-0540-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alomari AI, Spencer SA, Arnold RW, et al. Fibro-adipose vascular anomaly: clinical-radiologic-pathologic features of a newly delineated disorder of the extremity. J Pediatr Orthop. 2014;34(1):109–117. doi: 10.1097/BPO.0b013e3182a1f0b8. [DOI] [PubMed] [Google Scholar]
- 31.Wang KK, Glenn RL, Adams DM, et al. Surgical management of fibroadipose vascular anomaly of the lower extremities. J Pediatr Orthop. 2020;40(3):e227–e236. doi: 10.1097/BPO.0000000000001406. [DOI] [PubMed] [Google Scholar]
- 32.Shaikh R, Alomari AI, Kerr CL, Miller P, Spencer SA. Cryoablation in fibro-adipose vascular anomaly (FAVA): a minimally invasive treatment option. Pediatr Radiol. 2016;46(8):1179–1186. doi: 10.1007/s00247-016-3576-0. [DOI] [PubMed] [Google Scholar]
- 33.Lee MS, Liang MG, Mulliken JB. Diffuse capillary malformation with overgrowth: a clinical subtype of vascular anomalies with hypertrophy. J Am Acad Dermatol. 2013;69(4):589–594. doi: 10.1016/j.jaad.2013.05.030. [DOI] [PubMed] [Google Scholar]
- 34.Peterman CM, Vadeboncoeur S, Mulliken JB, Fishman SJ, Liang MG. Wilms tumor screening in diffuse capillary malformation with overgrowth and macrocephaly-capillary malformation: a retrospective study. J Am Acad Dermatol. 2017;77(5):874–878. doi: 10.1016/j.jaad.2017.06.014. [DOI] [PubMed] [Google Scholar]
- 35.Couto JA, Ayturk UM, Konczyk DJ, et al. A somatic GNA11 mutation is associated with extremity capillary malformation and overgrowth. Angiogenesis. 2017;20(3):303–306. doi: 10.1007/s10456-016-9538-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goss JA, Konczyk DJ, Smits P, et al. Diffuse capillary malformation with overgrowth contains somatic PIK3CA variants. Clin Genet. 2020;97(5):736–740. doi: 10.1111/cge.13702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hershkovitz D, Bergman R, Sprecher E. A novel mutation in RASA1 causes capillary malformation and limb enlargement. Arch Dermatol Res. 2008;300(7):385–388. doi: 10.1007/s00403-008-0842-5. [DOI] [PubMed] [Google Scholar]
- 38.Wooderchak-Donahue WL, Johnson P, McDonald J, et al. Expanding the clinical and molecular findings in RASA1 capillary malformation-arteriovenous malformation. Eur J Hum Genet. 2018;26(10):1521–1536 10. doi: 10.1038/s41431-018-0196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larralde M, Abad ME, Luna PC, Hoffner MV. Capillary malformation-arteriovenous malformation: a clinical review of 45 patients. Int J Dermatol. 2014;53(4):458–461. doi: 10.1111/ijd.12040. [DOI] [PubMed] [Google Scholar]
- 40.Banzic I, Brankovic M, Maksimović Ž, Davidović L, Marković M, Rančić Z. Parkes Weber syndrome-diagnostic and management paradigms: a systematic review. Phlebology. 2017;32(6):371–383. doi: 10.1177/0268355516664212. [DOI] [PubMed] [Google Scholar]
- 41.Conway AM, Qato K, Nguyen Tran NT, et al. Embolization techniques for arteriovenous malformations in Parkes-Weber syndrome. Ann Vasc Surg. 2020. doi: 10.1016/j.avsg.2020.05.039. [DOI] [PubMed] [Google Scholar]
- 42.Dubrey SW, Hillson R, Dahdal M. High output heart failure caused by extensive arteriovenous malformation: problems and pregnancy. BMJ Case Rep. 2009. 2009doi. doi: 10.1136/bcr.07.2008.0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Epelde F, Guillaumet E, Anarte L, Iglesias-Lepine ML. [High-output cardiac failure due to congenital arteriovenous fistula. Parkes Weber syndrome]. Med Clin Barc. 2013;141(4):187. doi: 10.1016/j.medcli.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 44.Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34(12):1632–1641. doi: 10.1002/humu.22431. [DOI] [PubMed] [Google Scholar]
- 45.Wooderchak-Donahue W, Stevenson DA, McDonald J, Grimmer JF, Gedge F, Bayrak-Toydemir P. RASA1 analysis: clinical and molecular findings in a series of consecutive cases. Eur J Med Genet. 2012;55(2):91–95. doi: 10.1016/j.ejmg.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 46.Revencu N, Fastre E, Ravoet M, et al. mosaic mutations in patients with capillary malformation-arteriovenous malformation. J Med Genet. 2020;57(1):48–52 01. doi: 10.1136/jmedgenet-2019-106024. [DOI] [PubMed] [Google Scholar]
- 47.Wooderchak-Donahue WL, Akay G, Whitehead K, et al. Phenotype of CM-AVM2 caused by variants in EPHB4: how much overlap with hereditary hemorrhagic telangiectasia (HHT). Genet Med. 2019;21(9):2007–2014 09. doi: 10.1038/s41436-019-0443-z. [DOI] [PubMed] [Google Scholar]
- 48.Akgumus G, Chang F, Li MM. Overgrowth syndromes caused by somatic variants in the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin pathway. J Mol Diagn. 2017;19(4):487–497 07. doi: 10.1016/j.jmoldx.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 49.Nathan N, Keppler-Noreuil KM, Biesecker LG, Moss J, Darling TN. Mosaic disorders of the PI3K/PTEN/AKT/TSC/mTORC1 signaling pathway. Dermatol Clin. 2017;35(1):51–60. doi: 10.1016/j.det.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu Y, Fu WZ, He JW, Yue H, Zhang ZL. [A clinical study of Proteus syndrome caused by a mosaic somatic mutation in AKT1 gene]. Zhonghua Nei Ke Za Zhi. 2019;58(7):508–513. doi: 10.3760/cma.j.issn.0578-1426.2019.07.005. [DOI] [PubMed] [Google Scholar]