

Abstract
Mammalian cytochrome P450 drug-metabolizing enzymes rarely cleave carbon–carbon (C-C) bonds and the mechanisms of such cleavages are largely unknown. We identified two unusual cleavages of non-polar, unstrained C(sp2)-C(sp3) bonds in the FDA-approved tyrosine kinase inhibitor pexidartinib that are mediated by CYP3A4/5, the major human phase I drug metabolizing enzymes. Using a synthetic ketone, we rule out the Baeyer-Villiger oxidation mechanism that is commonly invoked to address P450-mediated C-C bond cleavages. Our studies in 18O2 and H218O enriched systems reveal two unusual distinct mechanisms of C-C bond cleavage: one bond is cleaved by CYP3A-mediated ipso-addition of oxygen to a C(sp2) site of N-protected pyridin-2-amines, and the other occurs by a pseudo-retro-aldol reaction after hydroxylation of a C(sp3) site. This is the first report of CYP3A-mediated C-C bond cleavage in drug metabolism via ipso-addition of oxygen mediated mechanism. CYP3A-mediated ipso-addition is also implicated in the regioselective C-C cleavages of several pexidartinib analogs. The regiospecificity of CYP3A-catalyzed oxygen ipso-addition under environmentally friendly conditions may be attractive and inspire biomimetic or P450-engineering methods to address the challenging task of C-C bond cleavages.
Keywords: CYP3A, carbon-carbon bond cleavage, ipso addition, retro-aldol, pexidartinib
Graphical Abstract
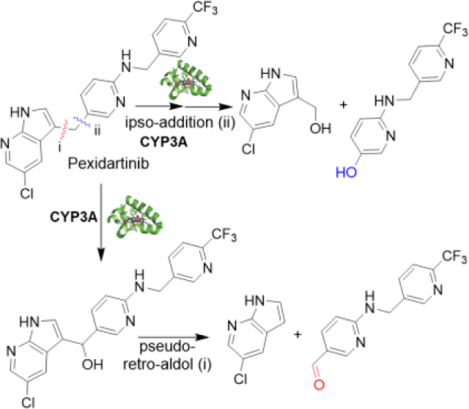
Mammalian cytochrome P450 drug metabolizing enzymes rarely cleave C-C bonds. Here we report the mechanisms of two unusual CYP3A-mediated cleavages of non-polar, unstrained C(sp2)-C(sp3) bonds in the metabolism of tyrosine kinase inhibitor pexidartinib. One bond is cleaved by CYP3A-mediated ipso-addition of activated oxygen, and the other occurs by a pseudo-retro-aldol reaction after hydroxylation of a C(sp3) site.
Introduction
Selective oxidative cleavage of carbon–carbon (C-C) single bonds is a challenge in both chemistry and enviroscience[1] that offers the potential to realize free molecular editing[2] and to address the degradation of plastic polymer waste. The cleavage or activation of a C–C bond may also produce synthetically complex compounds that would be difficult to achieve in other ways.[3] Current methods for cleaving C-C bonds rely primarily on transition-metal catalysts, and substrates that possess a high degree of ring strain or feature polar functional groups such as carbonyls, nitriles, of imines are preferred. The cleavage of unstrained and nonpolar C–C bonds is challenging,[4] and reports of catalytic C(aryl)-C(alkyl) bond activation have been rare.[5] Rhodium-catalyzed hydrogenolysis of 2,2’-diphenols to monophenols has been achieved with the aid of directing groups,[6] and selective cleavage and borylation of C(aryl)-CH3 bonds has been achieved without directing groups or transition metals.[4] Jiao et al. developed oxidative reactions to scissor C(aryl)-C(alkyl) bonds using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as the oxidant to yield anilines, demonstrating the feasibility of selective cleavage of non-polar unstrained C–C bonds despite DDQ toxicity, high cost, and difficulty in removing DDQH2 waste.[7] Novel processes for selective cleavage of unactivated C(sp2)-C(sp3) bonds under mild reaction conditions are thus of great interest.[4a]
Cytochrome P450s (P450s) are thiolate-ligated heme enzymes with remarkable catalytic versatility.[8] P450s catalyze an extraordinary breadth of physiologically important oxidations for specialized biosynthetic pathways[2] and xenobiotic detoxification including hydroxylation, dealkylation, deamination, dehalogenation, cyclization,[9] carbon–nitrogen (C–N) bond formation,[10] and C–C bond cleavage.[11] In a typical catalytic cycle, a P450 activates molecular oxygen, inserts one oxygen atom into a substrate, and reduces the second oxygen to water using electrons provided by β-Nicotinamide adenine dinucleotide 2′-phosphate (NADPH) via a P450 reductase.[12] P450 mediated C–C bond cleavages in sterol metabolism are achieved by CYP51,[13] CYP11A,[14] CYP17A,[15] CYP19A,[16] and CYP125.[17] Bacterial P450s OleT (CYP152L1), BSβ (CYP152A1), and P450BioI (CYP107H1) catalyze C–C bond cleavage of fatty acids.[18]
Drug metabolizing P450s such as CYP3A and CYP1A2 are responsible for the phase I metabolism of xenobiotics and of 70–80% of pharmaceuticals in humans.[19] Mammalian drug metabolizing P450s have been reported to cleave C–C bonds,[11a] but the mechanism(s) of these cleavages are not fully understood, and evidence for direct cleavage of C(sp2)-C(sp3) single bonds is very limited.[20] For the most abundant Phase I human drug metabolizing enzymes, CYP3A4/5, the only previous reports of C-C bond cleavages in drugs have been for C(sp3)–C(sp3) bonds in tipranavir, noscapine, and aplidine[11a] and for one C(sp2)–C(sp3) bond in nefazodone.[21]
Pexidartinib (PEX, TURALIO™), an FDA-approved small-molecule CSF-1R kinase inhibitor drug, is indicated for treatment of adults with tenosynovial giant cell tumor not amenable to improvement with surgery.[22] Because life-threatening hepatotoxicity was reported in 3.3% of patients treated with PEX,[23] we studied PEX metabolism to elucidate the mechanism of its toxicity. Using liquid chromatography mass spectrometry (LC-MS)-based metabolomic approaches, we identified unusual PEX metabolites whose formation in human liver microsomes (HLM) results from CYP3A-dependent cleavage of either of the C(sp2)-C(sp3) bonds of a bridging methylene. Given the rarity of C-C bond cleavages in drug metabolism, and the limited data about their mechanisms, we explored the basis for these reactions. We determine that one C(sp2)-C(sp3) bond cleavage results from CYP3A-mediated ipso-addition of oxygen to the 5 position of the 5-alkylated N-protected pyridin-2-amine, and the other cleavage occurs by a retro-aldol mechanism after hydroxylation of the bridging methylene. These efforts provide new insights into the mechanism of CYP3A-mediated uncommon C(sp2)-C(sp3) bond cleavages.
Results and Discussion
Metabolomic Profiling Reveals Two PEX C(sp2)–C(sp3) Bond Cleavages in HLM.
Our previous studies have shown LC-MS-based metabolomics to be a powerful tool for identifying unpredicted pathways or novel reactions in drug metabolism.[9b, 24] Principal component analysis of the ions generated from LC-Q Exactive MS analysis of PEX incubation in HLM with and without the co-factor NADPH revealed two clusters corresponding to the PEX and control groups that are clearly separated in the score plot (Fig. 1a, inset). The S-plot generated from orthogonal projection to latent structures-discriminant analysis (OPLS-DA) displays the top-ranking PEX metabolite ions contributing to group separation (labeled in Fig. 1a). The exact masses, mass errors, predicted formulas and proposed structures of PEX metabolites in HLM are presented in Table S1 and Fig. S1, and the relative abundances of ions for metabolites M1-M31 from HLM are presented in Fig. S2a. The analysis resolves eleven metabolites that contain all parent compound atoms, including five mono-oxygenations, three di-oxygenations, and a ketone. We resolve five aniline-derived fragments (M12-M16) and two benzyl-derived fragments (M22-M23) inferred to result from N-dealkylation (Fig. 1b) and additional modifications. We also identify five metabolites apparently derived from C–C bond cleavage of PEX: M24-M26 and M27-M28 (Fig. 1c) are inferred to result from cleavage at positions i and ii, respectively, of Fig. 1b. Manual re-analysis of ions with low counts revealed two additional fragments consistent with C–C bond cleavage, M30 and M31, as well as a metabolite that contains all parent non-hydrogen atoms, M29 [PEX+2O-2H]. Formation of all these metabolites is NADPH-dependent, as confirmed by the trend plots of M24, M26, M27, and M28 shown in Fig. 1d. Only trace amounts of four metabolites are detected on incubating PEX with H2O2 in PBS (Fig. S2b). Adding catalase slightly decreases formation of only a few metabolites in CYP3A4 or HLM (blue and red bars in Figs. S2c and S2d), establishing that all of these metabolites are generated largely by enzyme catalysis. Some metabolites can be generated in CYP3A4 or HLM with H2O2 (green bars in Figs. S2c and S2d). Metabolite production in HLM is time-dependent as shown in Fig. S3a. Representative chromatograms of time-courses for M28 and M27 are displayed in Fig. 1e and Fig. S3b, respectively. Structures of seven of the metabolites (M24–M28, M30 and M31) inferred based on their exact masses, MS/MS fragments and predicted formulas (Figs. S4–S8) correspond to scission products that implicate enzyme-mediated C(sp2)-C(sp3) bond cleavages at either site i or ii (Fig. 1b) during PEX metabolism in HLM. We confirmed the inferred structures of M24 through M30 by comparative LC-MS/MS analysis of standards obtained by chemical synthesis (Schemes S1–S5) or commercial purchase.
Figure 1.
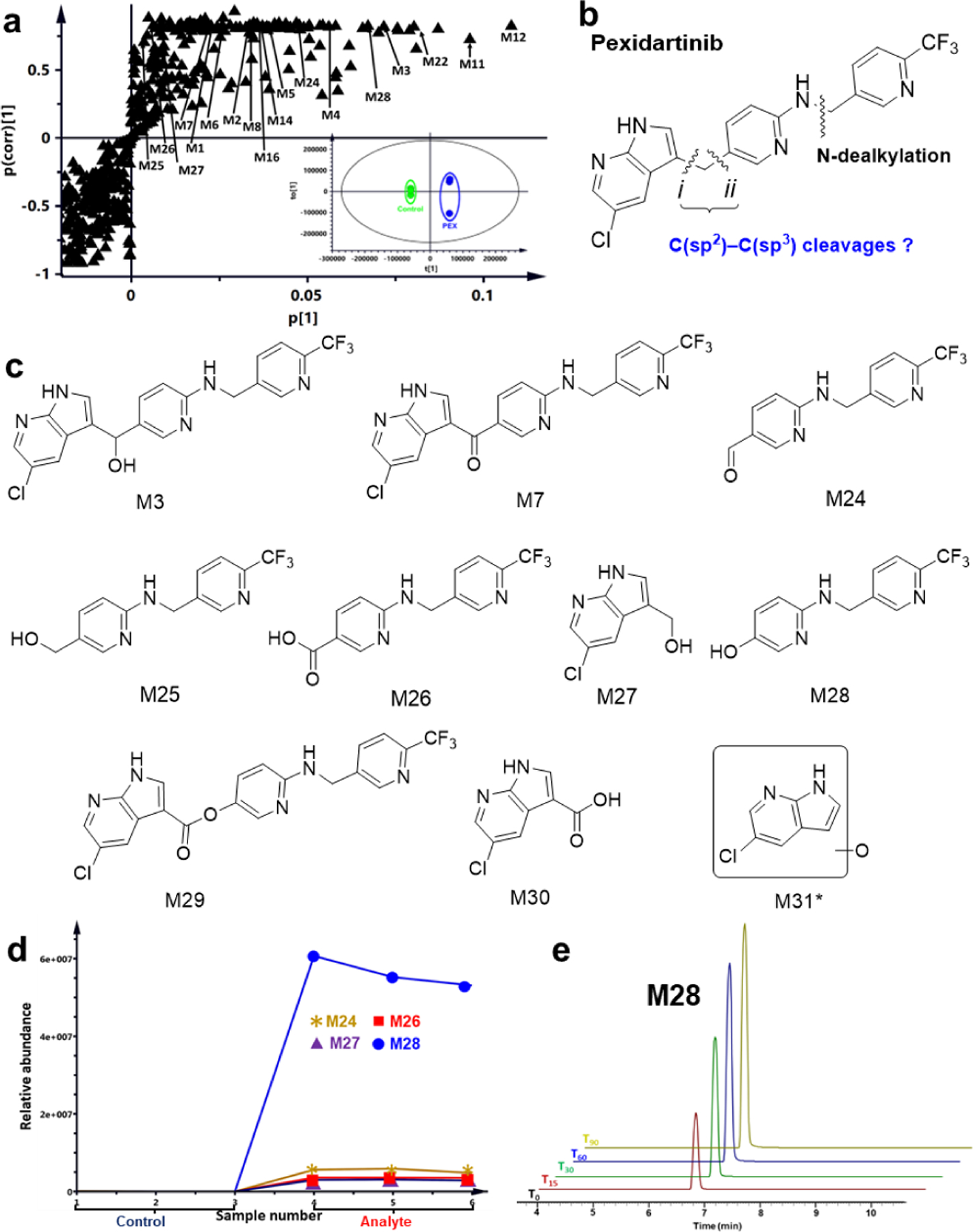
Metabolomics reveals C(sp2)-C(sp3) bond cleavages in pexidartinib (PEX) metabolism. Metabolomic analysis was conducted on control and PEX samples incubated with human liver microsomes (HLM). Incubation conditions are detailed in the method section (n = 3 for each group). (a) Loading S-plot from OPLS-DA analysis. Top-ranking ions associated with PEX metabolites are labeled. The separation of the control and PEX groups is shown in Fig. 1a inset, where t[1] and to[1] are the score of each sample in principal component 1 and 2, respectively. (b) Positions of inferred C(sp2)-C(sp3) bond cleavages in PEX. (c) Structures of alcohol M3, ketone M7, and metabolites resulting from C(sp2)-C(sp3) bond cleavages. (d) Trend plots for M24, M26, M27 and M28. (e) Extracted ion chromatograms showing time-dependent production of M28. Samples at the indicated time points were analyzed by LC-Q Exactive MS; the relative abundance of each ion was based on peak area. Experiments were performed in triplicate. *, M31 was detected only in samples that had been concentrated prior to LC-MS analysis.
CYP3A Isozymes Mediate PEX C(sp2)-C(sp3) Bond Cleavages.
Metabolites identified in HLM may be generated by a single P450 isoform, or by multiple P450s acting sequentially, or by other detoxifying enzymes.[25] To identify P450 isozymes directly involved in generating these PEX metabolites, we used reaction profiling with recombinant human P450s and chemical inhibition experiments in HLM. All ten human P450s tested could generate M24 and M25, with CYP3A4 and CYP3A5 showing the highest activity (Table S2); these products result from C-C bond cleavage at position i of PEX (Fig. 1b). M24 is made in significant amounts in CYP3A4 or in HLM in the presence of H2O2 (green bars in Figs. S2c and S2d); interestingly, it is one of the four metabolites detected when PEX reacts with H2O2 in solution (Fig. S2b). No P450 tested in isolation generated detectable M26, M30, or M31; these may require the action of multiple P450s in succession, or of other enzymes. However, both CYP3A4 and CYP3A5 generated M27, M28, and M29 (Table S2), which are consistent with C-C bond cleavage at position ii as shown in Fig. 1b. These products are not seen when CYP3A4 uses H2O2 (Fig. S2c), or when PEX reacts with H2O2 in solution (Fig. S2b). We note that hydrolysis of ester M29 would give rise to M28 and M30. CYP3A4 and CYP3A5 are the major enzymes contributing to the formation of most PEX metabolites. The time-courses of producing these metabolites by recombinant CYP3A4 and CYP3A5 are presented in Figs. 2a and 2b, respectively, with CYP3A4 exhibiting higher activity. Co-incubation of PEX in HLM with ketoconazole (KCZ), a potent and selective CYP3A inhibitor, decreases the generation of M26, M27, and M28 by 93%, 88%, 87%, and 90%, respectively (Fig. 2c), indicating that CYP3A4 and CYP3A5 contribute strongly to the formation of these metabolites in HLM. To understand the physical basis for how PEX might be metabolized by CYP3A, we performed computational modeling to explore potential binding poses of CYP3A:PEX complexes. Our modeling results revealed that when PEX accesses the catalytic heme iron center of both human CYP3A4 and CYP3A5 isozymes, the indicated carbons involved in C-C bond cleavage approach the heme iron (Figs. 2d, 2e, and 2f). Collectively, these findings provide strong evidence that the CYP3A enzymes contribute to the cleavage of these PEX C-C bonds.
Figure 2.
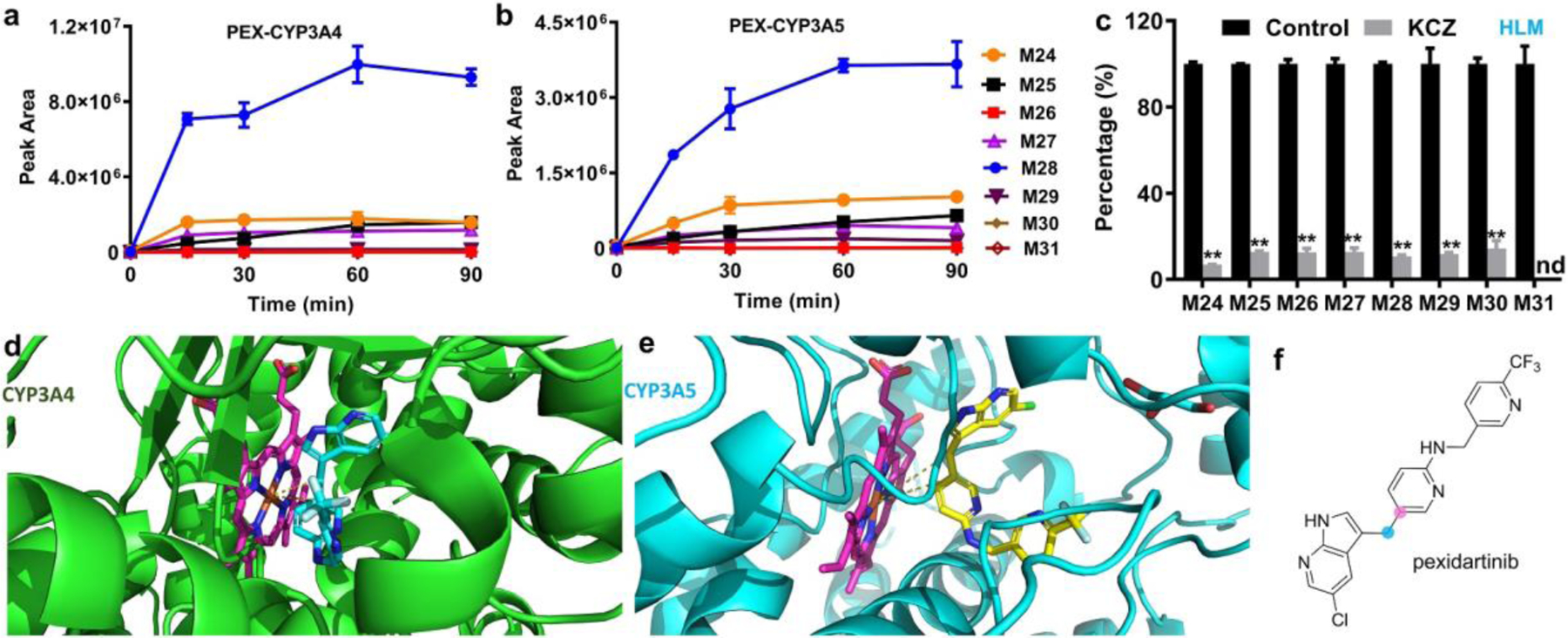
CYP3A mediated C-C bond cleavages to form M24, M26, M27 and M28. (a, b) The time-dependent production of ions corresponding to M24-M31 by CYP3A4 and CYP3A5, respectively. (c) Ketoconazole (KCZ), a selective CYP3A inhibitor, inhibits the formation of M24-M31 in HLM. The percentages of controls were set as 100%. Incubation conditions for (a–c) are described in the method section; The data in (a–c) are expressed as mean ± s.e.m. (n = 3). Statistical analysis for (c) was conducted using two-tailed Student’s independent t-test. **P<0.01; nd, not detected. (d) DOCK model of CYP3A4:PEX interaction. CYP3A4 is shown as green cartoon; PEX is shown as cyan sticks; heme is shown as magenta sticks. (e) DOCK model of CYP3A5:PEX interaction. CYP3A5 is shown as cyan cartoon; PEX is shown as yellow sticks; heme is shown as magenta sticks. (f) The carbons involved in C-C bond cleavages are indicated.
CYP3A isozymes are the most abundant P450s in human liver, and are responsible for the metabolism of approximately 50% of marketed drugs.[26] Documented cases of C–C bond cleavage reactions are rare for these isozymes, or indeed for any human P450s that mediate xenobiotic metabolism, and the mechanisms of such C-C bond cleavages remain poorly understood. We therefore investigated the basis for CYP3A-mediated cleavages of two C(sp2)–C(sp3) bonds in PEX.
P450 metabolic diversity stems from an interplay of the iron-oxygen species formed at the active site and substrate molecular recognition.[2, 19b] P450s activate molecular oxygen using a thiolate-ligated heme iron cofactor to produce the short-lived iron(III)-peroxo, iron-(III)-hydroperoxo, and iron (IV)-oxo intermediates in catalytic cycles.[12, 27] The iron(III)-peroxo species (known as Compound 0) is considered a super-nucleophile; the iron-(III)-hydroperoxo species could act as a nucleophile or an electrophile; and the iron (IV)-oxo species (Compound I) is a cation radical that behaves as an electrophile.[11b, 27–28] All these species have been proposed to contribute to P450-mediated C–C bond cleavage. The mechanism of non-drug metabolizing P450-mediated C(sp3)–C(sp3) bond cleavage (e.g., C-demethylation) in steroids and fatty acids has been extensively investigated, and generally requires oxidations at two adjacent carbon atoms.[11b] C–C bond cleavage does not usually occur with unfunctionalized methylene, olefin or aromatic carbons and requires prior functionalization (e.g., hydroxylation).
PEX Ketone M7 Does Not Undergo Baeyer-Villiger Oxidation in HLM or CYP3A4.
PEX metabolites we identified from HLM include ketone M7 and M29, an ester consistent with Baeyer–Villiger (B-V) oxidation[29] of M7 (Fig. 3). Phenol M28 and acid M30 could be hydrolysis products of M29, and acid M26 could be one hydrolysis product of the other possible B-V ester product, IM1 (we do not detect that ester itself, but the mass of M31 is consistent with the alcohol hydrolysis product of that ester). To determine if B-V oxidations are involved in the formation of the unusual products, we synthesized ketone M7 (Scheme S1) and incubated it with HLM or recombinant CYP3A isozymes. Ketone M7 cannot be converted to M29, M28, M26 or M24 by HLMs or by recombinant enzymes (Figs. 4 and S9), eliminating M7 as an intermediate on the pathway to these metabolites. These findings completely rule out the involvement of a B-V mechanism in these unusual PEX C-C bond cleavages.
Figure 3.
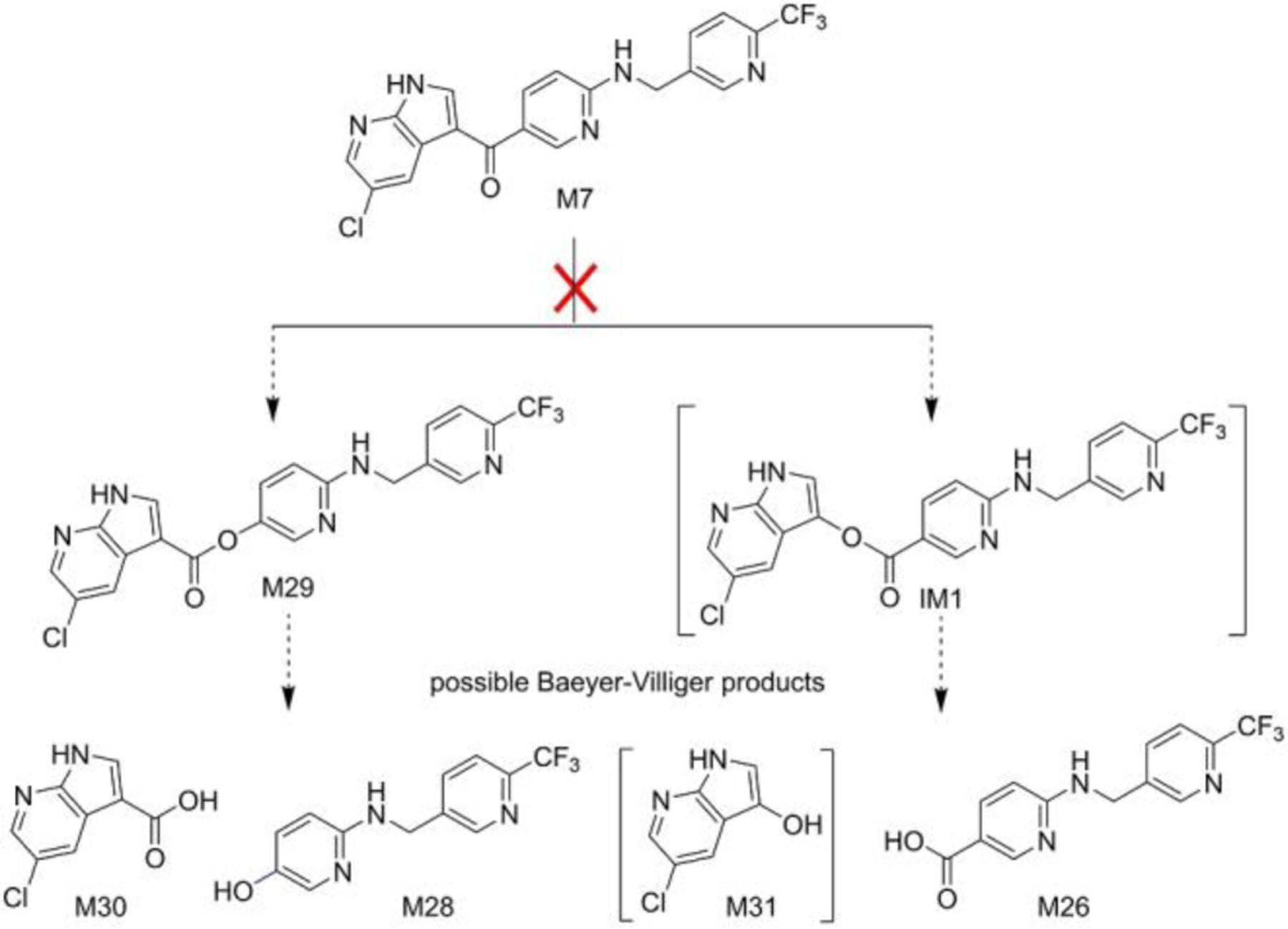
Ketone M7 is not converted to PEX C-C cleavage products, ruling out a Baeyer-Villiger mechanism for generating these metabolites. Species that have not been confirmed by synthesis are in brackets. IM, intermediate.
Figure 4.
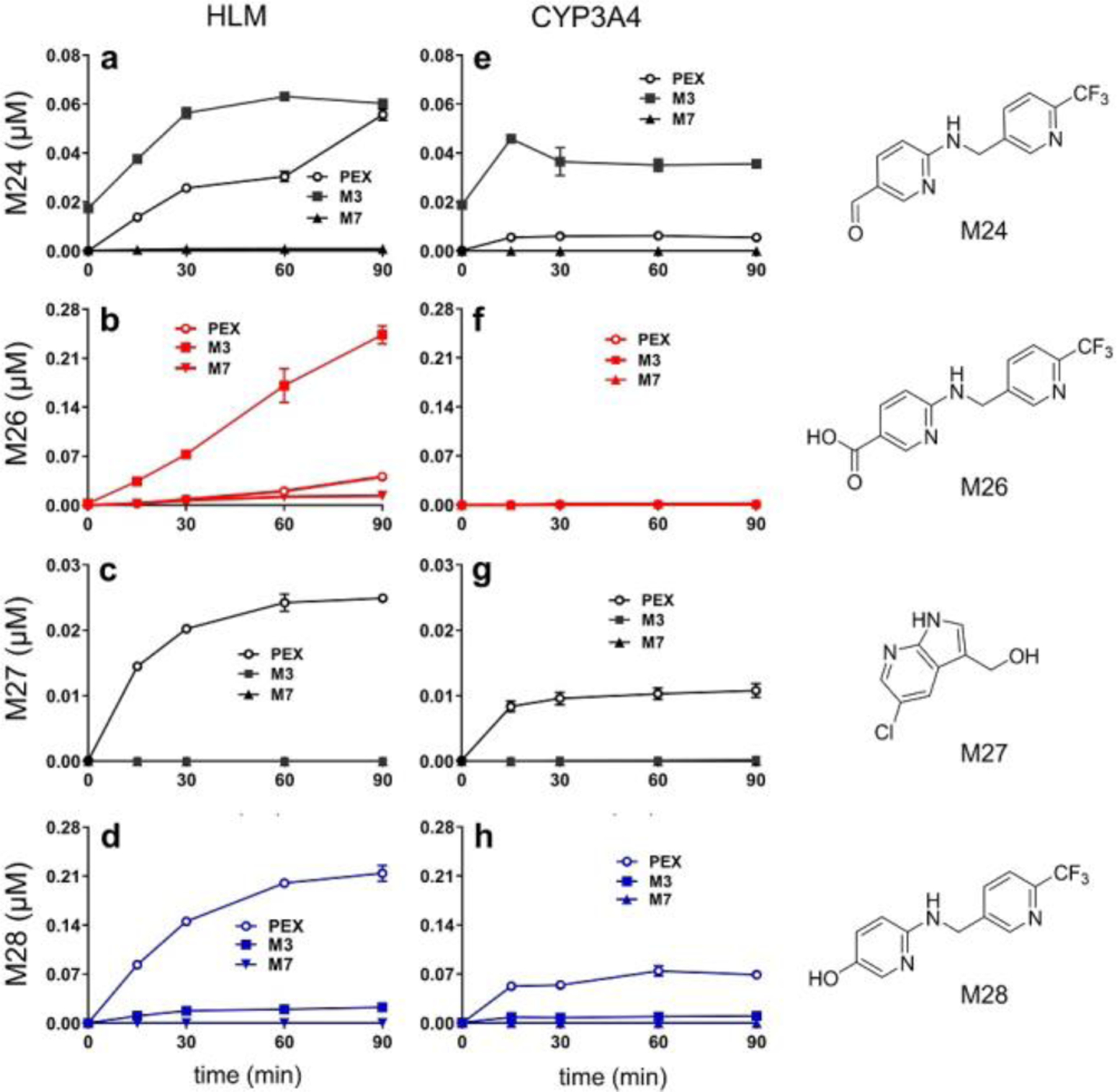
Time courses of C-C cleavage product formation using PEX, M3 or M7 as substrates. (a–d) Time courses in HLM. (e–h) Time courses with CYP3A4. Incubation conditions are detailed in experimental procedures. Metabolites were quantified based on standard curves. The formation of M26 in HLM is linear, but time-dependent inhibition may happen for the formation of M24, M27, and M28 in HLM and CYP3A4. The data are expressed as mean ± s.e.m. (n = 3).
We then examined whether alcohol M3 might be subject to P450-mediated C-C bond cleavage. Identifying the metabolic products of M3 is complicated by its poor stability under our acidic methanol/water LC-MS conditions: M3 reacts with methanol to form the methyl ether, and the isotope from 18O water exchanges into M3 (Fig. S10). Quenching enzyme reactions with acetonitrile and using acetonitrile/water LC gradients prevent these complications. Incubating M3 with HLM generates both acid M26 and phenol M28 (Fig. 4), but M3 gives these products in a M26:M28 ratio of 6:1 whereas PEX gives a ratio of 1:5. Time courses of metabolite production in HLM indicate that M26 production from PEX exhibits a kinetic lag, whereas M26 production from M3 does not (Fig. 4b). From the six-fold higher rate of M26 production using M3 compared to PEX as a substrate, we infer that M3 is on the kinetic pathway from PEX to M26. From the five-fold lower rate of M28 production with M3 compared to PEX, we infer that M3 is not on the kinetic pathway from PEX to M28. The ability of M3 to yield M28 at a low rate could be explained by M3 being first converted to PEX, which then converts to M28. Time courses show that PEX is produced on incubating synthetic M3 with NADPH in the presence or absence of CYP3A (Fig. S11), supporting our inference that M3 is not on the pathway to M28 and further demonstrating the reactivity of this PEX metabolite.
PEX Alcohol M3 Undergoes a Pseudo Retro-Aldol Reaction.
Alcohol M3 is converted more efficiently than PEX to aldehyde M24 by HLMs or CYP3A (Figs. 4a and 4e), suggesting that M3 is on the kinetic pathway for M24 formation. We propose that the beta-hydroxy enamine moiety of M3 (circled in Fig. 5) can undergo a pseudo-retro-aldol reaction to give aldehyde M24 and 5-chloro-7-azaindole (CAI, Fig. 5).
Figure 5.
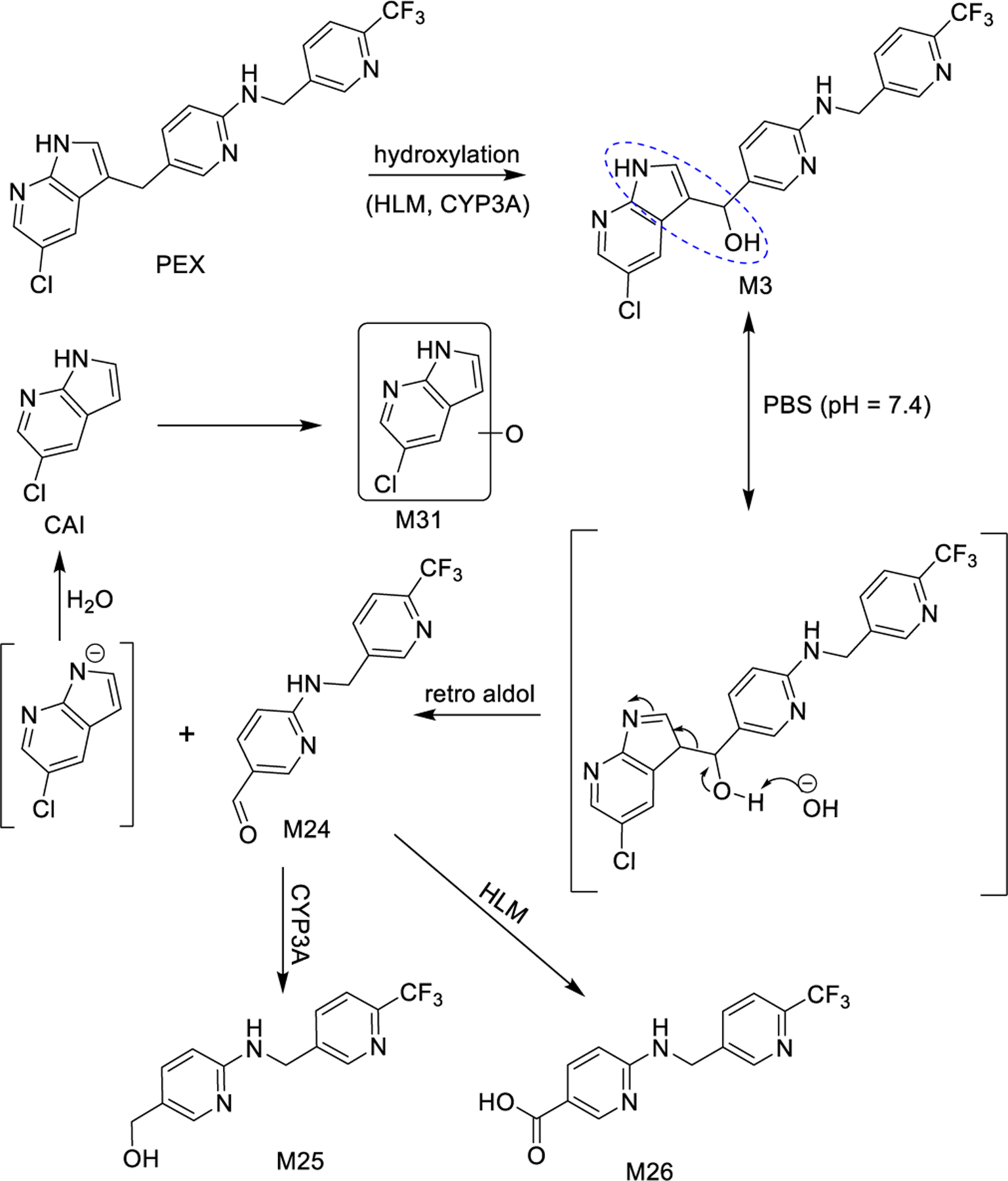
A pseudo retro-aldol reaction of M3 generates M24 and CAI. M24 can be further metabolized to M25 or M26, and CAI can be oxidized to M31.
Aldehyde M24 is generated from PEX by each recombinant P450 tested and in the control reaction (Table S2), consistent with the retro-aldol reaction occurring spontaneously in solution (Table S3). The M24 produced from M3 by the CYP3A enzymes is two-fold higher than the other P450s or the control reaction, which likely reflects their ability to convert PEX to M3 (Table S2), since under the conditions of Table S3 a competing reaction is M3 reduction to PEX by NADPH (Fig. S11). The detection of aldehyde M24 when PEX is exposed to H2O2 (Fig. S2b) is consistent with the spontaneous retro-aldol cleavage of some of the M3 that is generated by reaction of PEX with peroxide.
We observe CAI at low levels when PEX is incubated with HLM, along with two mono-oxygenated CAI metabolites, one of which we initially detected as M31 (Fig. S8). The LC-MS/MS mono-oxygenation patterns of CAI in HLM are very similar to those observed from PEX incubation in HLM (Fig. S8e–h), suggesting that CAI released from PEX by the retro-aldol can be oxidized to give M31. (We cannot rule out the possibility that the 7-azaindole might in some instances be oxidized prior to the retro-aldol.)
When incubated in HLM, aldehyde M24 primarily undergoes oxidation to acid M26, while a small fraction is converted to alcohol M25 (Fig. S12). When incubated with CYP3A4, however, M24 is predominantly reduced to M25 (Fig. S12). This explains the robust production of M26 from PEX in HLM (Fig. S2) and the failure of CYP3A4 alone to make M26 from PEX (Table S2). Both HLM and CYP3A4 also generate trace quantities of M28 (Fig. S12). Incubating PEX with HLM in the presence of methoxyamine, an aldehyde trapping reagent, decreases the amount of M26 but not M28 that is generated (Figs. S13–S14), establishing that M26 derives at least in part from M24 and that M28 does not significantly derive from M24. We conclude that PEX metabolite M3 is susceptible to spontaneous C-C bond cleavage in aqueous solution at site i of Fig. 1b by a pseudo-retro-aldol mechanism to give aldehyde M24 and CAI.
CYP3A Mediates Oxygen Ipso-Addition to PEX.
We next sought to understand the mechanism of CYP3A-mediated C-C bond cleavage at the ii position in PEX (Fig. 1b). To determine the source of the M28 phenolic oxygen, we used an H218O- and/or 18O2-enriched incubation system for PEX metabolism. Interpretation of data from H218O enriched buffer is complicated by exchange into precursors or products, as we showed for M3 (Fig. S10), and as we detect for aldehyde M24, acid M26, alcohol M27, and acid M30. M28 shows trace incorporation of 18O when generated from PEX in the presence of H218O, but >75% incorporation of 18O when generated in the presence of 18O2 in HLM or CYP3A4 (Fig. 6), consistent with incorporation from P450-activated molecular oxygen.
Figure 6.
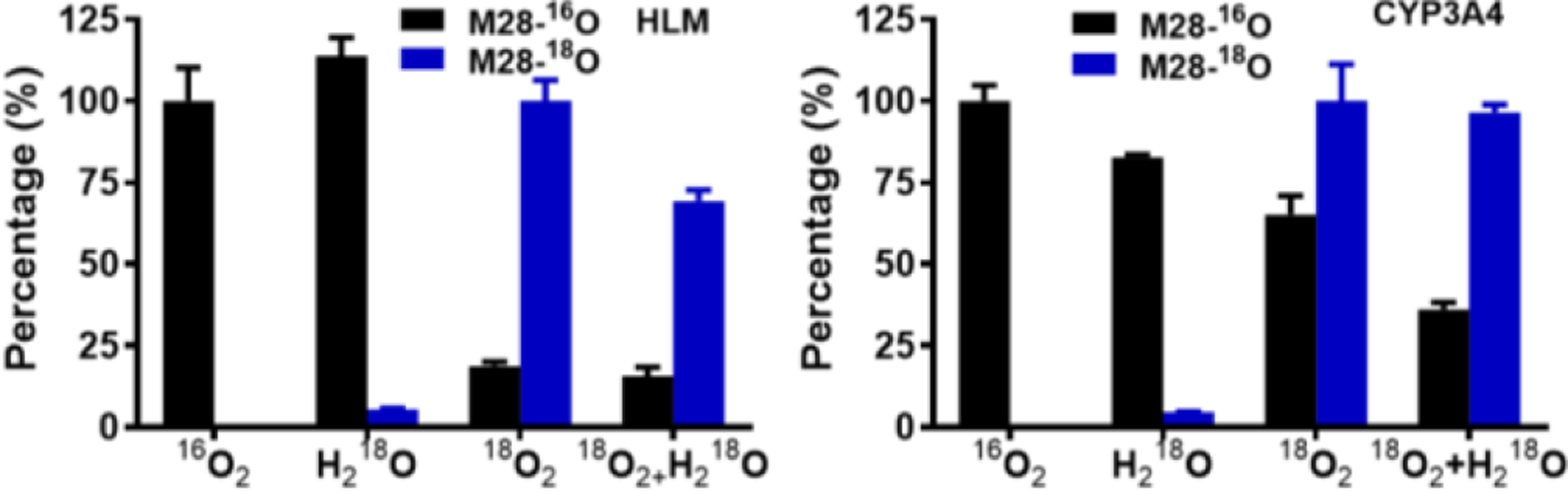
Relative abundances of M28-16O and M28-18O generated in HLM or by CYP3A4. The percentage of 16O2 group was set as 100%. Data are expressed as mean ± s.e.m. (n = 3). Samples were analyzed by UHPLC-Q Exactive MS, peaks for 16O or 18O products were extracted based on their exact mass (error < 3 ppm), and peak area was normalized to the largest value in any of the 4 groups.
We therefore hypothesize that CYP3A mediates oxygen ipso-addition at the 5-alkylated position of the N-protected pyridine-2-amine moiety to give intermediate IM2 (Fig. 7), which undergoes substituent elimination to cleave the C(sp2)-C(sp3) bond at the PEX ii position (Fig. 1b) and give M28. This mechanism predicts oxygen incorporation from O2 into phenol M28 and the formation of alcohol M27 from the resonance stabilized cation released by C-C bond scission (IM3/IM4; Fig. 7). Consistent with this, we observe M27 and M28 when PEX is incubated with either HLM or CYP3A (Fig. 4); moreover, in an incubation system fortified with glutathione (GSH), we detect adduct M32 (Fig. S15), which this mechanism predicts would result from reaction of GSH with IM3 or IM4 (Fig. 7). CYP3A4 and NADPH-dependent production of M28 from PEX is not significantly decreased by catalase, and CYP3A4 with H2O2 does not give M28 using PEX as substrates (Fig. S2c).
Figure 7.
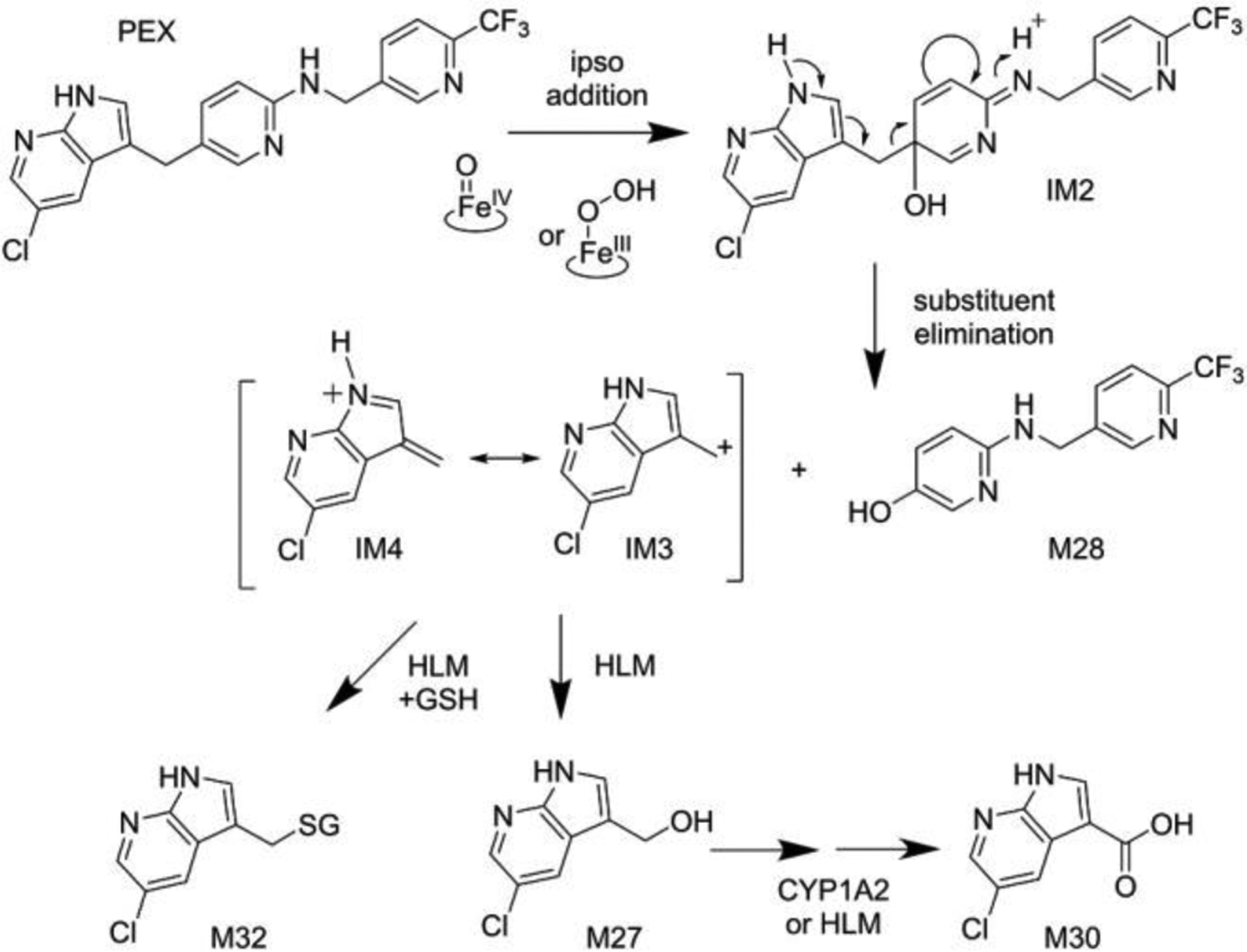
CYP3A mediated ipso-addition of activated oxygen to PEX. Intermediate IM2 undergoes substituent elimination to gives M28 and a stable cation that leads to adducts M32 or M27. M27 can be further metabolized to M30. GSH, reduced glutathione; IM, intermediate.
Previously, Ohe et al reported that P450s catalyze oxygen ipso-addition to para-substituted phenol compounds to form quinols[30]: when the para-substituent is carboxyl, acetyl, or hydroxymethyl, the quinol undergoes C-C bond cleavage to yield hydroquinone, but when the para-substituent is an alkyl group, the quinol is usually stable. However, industrial chemical bisphenol A undergoes CYP3A-mediated ipso-addition with scission of the expected quinol to hydroxycumyl alcohol and hydroquinone due to C(sp2)-C(sp3) bond cleavage that releases a stable tertiary carbocation (Scheme S6).[31] These reports further support our proposed ipso-addition mechanism. Demonstrating CYP3A-mediated ipso-addition of oxygen to a 5-alkylated N-protected pyridin-2-amine, for the first time, establishes that ipso-addition reactions contribute to P450-mediated drug metabolism and occur for substrates other than para-alkylated phenols.
Acid M30 arises from C-C cleavage at the PEX ii position (Fig. 1b), but it is not directly interpreted by the ipso-addition mechanism. Although M30 is generated from PEX in HLM (Fig. S2a), it is not detected with CYP3A4 (Table S2). Using synthetic alcohol M27 as a substrate, we found that CYP1A2 converts M27 to M30 rapidly whereas CYP3A does so very slowly (Table S4), providing a path in HLM from the ipso-addition products to M30 and explaining why M30 is formed in the PEX metabolism with HLM but not in reactions with recombinant CYP3A.
CYP3A4 Mediates C(sp2)-C(sp3) Bond Cleavage of PEX Analogs via Ipso-Addition.
To our knowledge, PEX is the only drug for which P450-mediated C(sp2)-C(sp3) bond cleavage occurs via ipso-addition. To enhance our understanding of this reaction, we assessed the behavior of CYP3A in the ipso addition reaction using commercially available PEX analogs (Fig. 8). From FLT3-IN-2, PLX-647, and PLX5622 we detect ipso-addition products (phenols M33 and M35, as well as alcohols M27, M34 and M36; see Fig. 8), establishing that neither the 5-chloro of the 7-azaindole nor the pyridine nitrogen of the trifluoromethyl picolylamine are essential to this mechanism, and that other diversifications of these rings are compatible with CYP3A-mediated ipso-addition of oxygen. The structures of M33, M34, M35 and M36 were determined based on their exact masses and MS/MS fragment analyses (Figs. S16–S18).
Figure 8.
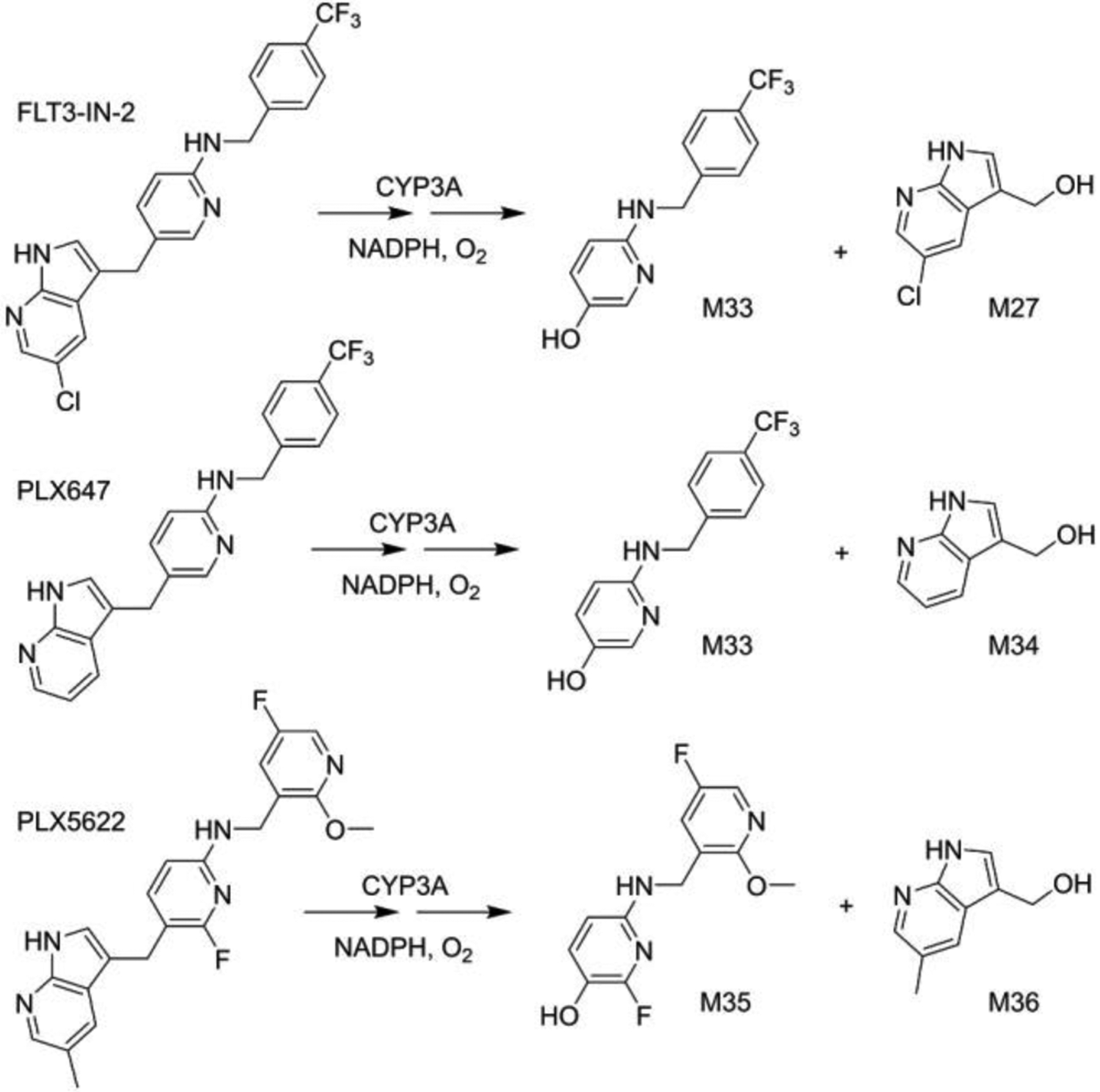
CYP3A mediated C(sp2)–C(sp3) bond cleavages of PEX analogs. The cleavage products are consistent with the proposed ipso-addition mechanism.
Although p-benzoylphenol can undergo P450-mediated ipso-addition,[30b] PEX ketone M7 does not undergo analogous ipso-addition in HLM or with CYP3A isozymes (Fig. 4). PEX has four modifiable rings, so a deeper understanding of factors that impact the efficiency and regioselectivity of CYP3A-mediated ipso-addition of oxygen and C(sp2)-C(sp3) bond cleavages could be achieved from reactivity studies with variants on the PEX scaffold.
Efforts to engineer P450 catalytic activities usually focus on bacterial metabolic P450s because of their solubility, stability, and substrate specificity. Consistent with their evolutionary role detoxifying xenobiotics, human P450s that act in drug metabolism show high substrate promiscuity, but a given P450 may perform activities such as hydroxylation of unactivated C-H bonds with quite good regio- and stereo-selectivity.[32] The products of C-C bond cleavages via CYP3A mediated ipso-addition reported here are abundant, indicating that this rare reaction is a major contributor to PEX metabolism. Our findings extend the reaction profile of CYP3A, the most abundant human P450. Additionally, the enzymatic ipso oxygenation reaction is highly regiospecific, often occurring at positions that might be less accessible in traditional chemical synthesis. Further studies to elucidate the substrate scope and detailed mechanism of ipso-additions may inform the engineering of P450s for catalysis[33] or the development of biomimetic heme-like iron porphyrin catalysts.[34]
Conclusion
We report the CYP3A-mediated uncommon cleavages of two C(sp2)-C(sp3) bonds in the FDA-approved drug PEX, which proceed by very different mechanisms. One bond is cleaved via an ipso-addition of oxygen to PEX and the other is cleaved by the non-enyzme dependent pseudo-retro-aldol reaction of a monohydroxylated PEX (M3). C-C bond cleavages are rare for human drug-metabolizing P450s, and two such C-C cleavages occurring in a single drug is without precedent. This study is the first documented instance of CYP3A-mediated ipso-addition of oxygen to 5-alkylated N-protected pyridin-2-amines, and the similar reactivity of the tested analogs extends the substrates subject to this reaction beyond simple para-substituted phenols. Cleavage of a C(sp2)-C(sp3) bond in a PEX metabolite (M3) via a pseudo-retro-aldol mechanism does not expand the range of known P450 activities but could serve to inform the optimization or design of drugs. This work also highlights the synthetic and degradative potential of CYP3A-catalyzed reactions under mild and environmentally friendly conditions and may inspire biomimetic or P450-reprogramming methods for addressing the challenging task of C-C bond cleavage.
Supplementary Material
Acknowledgements
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R61/R33 HD099995) and the National Institute of Diabetes and Digestive and Kidney (R01 DK121970) to Dr. Feng Li; Feng Li is supported in part by National Institute on Aging (P01 AG066606) to Dr. Hui Zheng and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (P01 HD087157) to Dr. Martin M. Matzuk.
Footnotes
Supporting Information
The authors have cited additional references within the Supporting Information.[35]
References
- [1].Allpress CJ, Grubel K, Szajna-Fuller E, Arif AM, Berreau LM, J Am Chem Soc 2013, 135, 659–668. [DOI] [PubMed] [Google Scholar]
- [2].Grant JL, Mitchell ME, Makris TM, Proc Natl Acad Sci U S A 2016, 113, 10049–10054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Roque JB, Kuroda Y, Gottemann LT, Sarpong R, Science 2018, 361, 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Dai PF, Ning XS, Wang H, Cui XC, Liu J, Qu JP, Kang YB, Angew Chem Int Ed Engl 2019, 58, 5392–5395; [DOI] [PubMed] [Google Scholar]; b) Chen F, Wang T, Jiao N, Chem Rev 2014, 114, 8613–8661. [DOI] [PubMed] [Google Scholar]
- [5].Dai P-F, Wang H, Cui X-C, Qu J-P, Kang Y-B, Organic Chemistry Frontiers 2020, 7, 896–904. [Google Scholar]
- [6].Zhu J, Xue Y, Zhang R, Ratchford BL, Dong G, Journal of the American Chemical Society 2022, 144, 3242–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Qin C, Zhou W, Chen F, Ou Y, Jiao N, Angew Chem Int Ed Engl 2011, 50, 12595–12599. [DOI] [PubMed] [Google Scholar]
- [8].a) Chen K, Arnold FH, J Am Chem Soc 2020, 142, 6891–6895; [DOI] [PubMed] [Google Scholar]; b) Liu Z, Calvó-Tusell C, Zhou AZ, Chen K, Garcia-Borràs M, Arnold FH, Nature Chemistry 2021, 13, 1166–1172. [DOI] [PubMed] [Google Scholar]
- [9].a) Cheng Q, Lamb DC, Kelly SL, Lei L, Guengerich FP, J Am Chem Soc 2010, 132, 15173–15175; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) MacKenzie KR, Zhao M, Barzi M, Wang J, Bissig KD, Maletic-Savatic M, Jung SY, Li F, Eur J Pharm Sci 2020, 153, 105488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].He F, Mori T, Morita I, Nakamura H, Alblova M, Hoshino S, Awakawa T, Abe I, Nat Chem Biol 2019, 15, 1206–1213. [DOI] [PubMed] [Google Scholar]
- [11].a) Bolleddula J, Chowdhury SK, Drug Metab Rev 2015, 47, 534–557; [DOI] [PubMed] [Google Scholar]; b) Guengerich FP, Yoshimoto FK, Chem Rev 2018, 118, 6573–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Meunier B, de Visser SP, Shaik S, Chem Rev 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- [13].Hargrove TY, Wawrzak Z, Guengerich FP, Lepesheva GI, J Biol Chem 2020, 295, 9998–10007; K. D. McCarty, Y. Tateishi, T. Y. Hargrove, G. I. Lepesheva, F. P. Guengerich, Angew Chem Int Ed Engl. 2024, 63:e202317711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Henry HL, Steroids 2001, 66, 391–398. [DOI] [PubMed] [Google Scholar]
- [15].Mak PJ, Duggal R, Denisov IG, Gregory MC, Sligar SG, Kincaid JR, J Am Chem Soc 2018, 140, 7324–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hackett JC, Brueggemeier RW, Hadad CM, J Am Chem Soc 2005, 127, 5224–5237. [DOI] [PubMed] [Google Scholar]
- [17].Sivaramakrishnan S, Ouellet H, Matsumura H, Guan S, Moenne-Loccoz P, Burlingame AL, Ortiz de Montellano PR, J Am Chem Soc 2012, 134, 6673–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Shoji O, Fujishiro T, Nakajima H, Kim M, Nagano S, Shiro Y, Watanabe Y, Angew Chem Int Ed Engl 2007, 46, 3656–3659; [DOI] [PubMed] [Google Scholar]; b) Matsunaga I, Sumimoto T, Ueda A, Kusunose E, Ichihara K, Lipids 2000, 35, 365–371; [DOI] [PubMed] [Google Scholar]; c) Fang B, Xu H, Liu Y, Qi F, Zhang W, Chen H, Wang C, Wang Y, Yang W, Li S, Sci Rep 2017, 7, 44258; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cryle MJ, De Voss JJ, Chem Commun 2004, 1, 86–87. [DOI] [PubMed] [Google Scholar]
- [19].a) Rittle J, Green MT, Science 2010, 330, 933–937; [DOI] [PubMed] [Google Scholar]; b) Denisov IG, Makris TM, Sligar SG, Schlichting I, Chem Rev 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]
- [20].a) Kagan M, Dain J, Peng L, Reynolds C, Drug Metab Dispos 2012, 40, 1712–1722; [DOI] [PubMed] [Google Scholar]; b) Harrell AW, Siederer SK, Bal J, Patel NH, Young GC, Felgate CC, Pearce SJ, Roberts AD, Beaumont C, Emmons AJ, Pereira AI, Kempsford RD, Drug Metab Dispos 2013, 41, 89–100. [DOI] [PubMed] [Google Scholar]
- [21].Mayol RF, Cole CA, Luke GM, Colson KL, Kerns EH, Drug Metab Dispos 1994, 22, 304–311. [PubMed] [Google Scholar]
- [22].a) Monestime S, Lazaridis D, Drugs R D 2020, 20, 189–195; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lamb YN, Drugs 2019, 79, 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Lewis JH, Gelderblom H, van de Sande M, Stacchiotti S, Healey JH, Tap WD, Wagner AJ, Pousa AL, Druta M, Lin CC, Baba HA, Choi Y, Wang Q, Shuster DE, Bauer S, Oncologist 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gelderblom H, Wagner AJ, Tap WD, Palmerini E, Wainberg ZA, Desai J, Healey JH, van de Sande MAJ, Bernthal NM, Staals EL, Peterfy CG, Frezza AM, Hsu HH, Wang Q, Shuster DE, Stacchiotti S, Cancer 2021, 127, 884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Qin X, Hakenjos JM, MacKenzie, Barzi M, Chavan H, Nyshadham P, Wang J, Jung SY, Guner JZ, Chen S, Guo L, Krishnamurthy P, Bissig KD, Palmer S, Matzuk MM, Li F, Drug Metab Dispos 2022, 50, 128–139; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu X, Lu YF, Guan X, Zhao M, Wang J, Li F, Biochem Pharmacol 2016, 109, 70–82. [DOI] [PubMed] [Google Scholar]
- [25].Fukami T, Yokoi T, Nakajima M, Annu Rev Pharmacol Toxicol 2022, 62, 405–425. [DOI] [PubMed] [Google Scholar]
- [26].Gibbs MA, Hosea NA, Clin Pharmacokinet 2003, 42, 969–984. [DOI] [PubMed] [Google Scholar]
- [27].Vatsis KP, Coon MJ, Arch Biochem Biophys 2002, 397, 119–129. [DOI] [PubMed] [Google Scholar]
- [28].Kim H, Rogler PJ, Sharma SK, Schaefer AW, Solomon EI, Karlin KD, J Am Chem Soc 2020, 142, 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].ten Brink GJ, Arends IW, Sheldon RA, Chem Rev 2004, 104, 4105–4124. [DOI] [PubMed] [Google Scholar]
- [30].a) Ohe T, Mashino T, Hirobe M, Tetrahedron Letters 1995, 36, 7681–7684; [Google Scholar]; b) Ohe T, Mashino T, Hirobe M, Drug Metab Dispos 1997, 25, 116–122. [PubMed] [Google Scholar]
- [31].Nakamura S, Tezuka Y, Ushiyama A, Kawashima C, Kitagawara Y, Takahashi K, Ohta S, Mashino T, Toxicology Letters 2011, 203, 92–95. [DOI] [PubMed] [Google Scholar]
- [32].Guengerich FP, Chem Res Toxicol 2008, 21, 70–83. [DOI] [PubMed] [Google Scholar]
- [33].a) Zhou Q, Chin M, Fu Y, Liu P, Yang Y, Science 2021, 374, 1612–1616; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miller JC, Lee JHZ, McLean MA, Chao RR, Stone ISJ, Pukala TL, Bruning JB, De Voss JJ, Schuler MA, Sligar SG, Bell SG, J Am Chem Soc 2023, 145, 9207–9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu W, Lavagnino MN, Gould CA, Alcazar J, MacMillan DWC, Science 2021, 374, 1258–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang C, Zhang J, Ibrahim PN, Artis DR, Bremer R, Wu G, Zhu H, Nespi M, Patent, WO 2008/064265, 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.