

Summary
As therapies, oncolytic viruses regress tumors and have the potential to induce antitumor immune responses that clear hard-to-treat and late-stage cancers. Despite this promise, clearance from the blood prevents treatment of internal solid tumors. To address this issue, we developed virus-delivering Salmonella (VDS) to carry oncolytic viruses into cancer cells. The VDS strain contains the PsseJ-lysE delivery circuit and has deletions in four homologous recombination genes (ΔrecB, ΔsbcB, ΔsbcCD, and ΔrecF) to preserve essential hairpins in the viral genome required for replication and infectivity. VDS delivered the genome for minute virus of mice (MVMp) to multiple cancers, including breast, pancreatic, and osteosarcoma. Viral delivery produced functional viral particles that are cytotoxic and infective to neighboring cells. The release of mature virions initiated new rounds of infection and amplified the infection. Using Salmonella for delivery will circumvent the limitations of oncolytic viruses and will provide a new therapy for many cancers.
Subject areas: Microbiology, Bacteriology, Virology, Cell biology
Graphical abstract
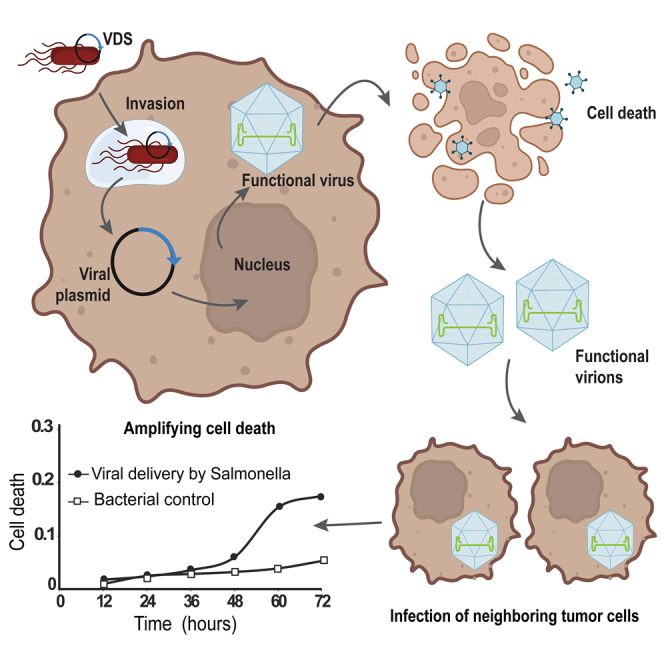
Highlights
-
•
Salmonella delivered the oncolytic virus, minute virus of mice (MVMp), to cancer cells
-
•
Deleting recombination genes was required to maintain the hairpin of the virus
-
•
Delivered viral genomes produce functional virions that kill multiple cancer cell types
-
•
Produced virus particles infect neighboring cells to initiate new rounds of infection
Microbiology; Bacteriology; Virology; Cell biology
Introduction
Many of the current problems associated with cancer treatment, e.g., metastatic disease and refractory tumors, could be overcome with microbial therapies. Oncolytic viruses (OVs) have the potential to treat many tumors by directly lysing cancer cells and stimulating immune responses that eliminate cancer cells regardless of their location in the body.1,2,3,4 In tumors, OVs induce cell lysis, which releases tumor-associated antigens (TAAs) and forms antitumor immunity.1,2,4,5,6 By inducing this abscopal effect, OVs have been shown to clear metastatic lesions.3,4 Because of this efficacy, the US Food and Drug Administration (FDA) approved a modified herpes simplex virus (Talimogene laherparepvec [T-VEC]) for the treatment of melanoma.1,2,3,7,8,9,10,11,12,13,14,15 The utility of treatment with other viruses is currently being explored in over 400 clinical trials.6 Despite this potential, OVs have not been effective at treating internal solid tumors.4,7,12,14,16 When injected systemically, OVs are cleared from the blood and do not effectively reach tumors.1,4,7,10,11,12,14,15,16,17,18,19,20,21,22,23 After intravenous injection, viruses (1) are cleared away by innate anti-viral immunity or neutralizing antibodies,1,4,7,10,12,14,15,16,17,18,19,20,21,22 (2) are sequestered in other organs, reducing the viral load in tumors,1,11,14,19,21,23 and (3) weakly escape vascular compartments into cancer cells.5,19 These problems could be overcome with a carrier with tumor-specific tropism. Our group and others have shown that bacterial therapies predominantly accumulate in tumors after intravenous injections.24,25,26,27,28,29,30,31,32,33 Developing a bacterial system to deliver viruses into cancer cells would couple the benefits of these two microbial therapies and focus treatment specifically to cancers.
Here we report, for the first time, the development of intracellular-delivering (ID) Salmonella to carry OVs into cancer cells. ID Salmonella is a bacterial system we recently developed to deliver macromolecules into cancer cells in tumors.34 These bacteria preferentially accumulate 3,000 times more in tumors than the clearance organs (liver and spleen) and over 100,000 times more than other healthy organs.24,25,26,27,28 ID Salmonella contain a genetic circuit (PsseJ-lysE) that controls the release of plasmids and macromolecules.34 In tumors, Salmonella naturally invades cancer cells.34 After cell invasion, the PsseJ promoter activates lysE, which lyses the bacteria and releases the molecular cargo into the cytoplasm of infected cells.34 Compared to other reported cellular carriers (e.g., exosomes, T cells, and mesenchymal stem cells), Salmonella have a higher therapeutic index. For these other carriers, tumor accumulation is rate limiting and most have accumulation rates below 10%.20,21
Minute virus of mice (MVM) is a small single-stranded DNA (ssDNA) virus of the Protoparvovirus genus that naturally lyses cancer cells.35,36,37,38,39,40 The prototype strain (MVMp) does not integrate into the genome, is non-infectious, and is non-pathogenic in humans.39,41,42 Because of this lack of pathogenicity, and their natural oncolytic activities, parvoviruses are good candidates for viral therapy.23,43 After MVMp infects cells, host polymerase produces NS1 from a double-stranded template of the viral DNA.11,41,44,45 NS1 is the initiator protein that creates a site-specific nick in the ssDNA and serves as a helicase to initiate DNA replication.37,41,44,46 This DNA template is used to produce more single-stranded viral genomes, which are assembled in the nucleus into new virus particles.39,47,48 Cell lysis kills cells and releases new viral particles that infect neighboring cells and initiate new rounds of infection.4,9,11,14,49 Because of its small size (5 kb), the MVMp genome can be included in a standard cloning plasmid.50 When mammalian cells are transfected with an MVMp-containing plasmid, they transcribe NS1 and produce viral particles, similar to natural infection.50,51
Bacterial delivery of viral DNA is complicated by hairpins in the genomes of many viruses. These hairpins are crucial for DNA replication, packaging, and viral infectivity. Palindromic sequences are not tolerated in Salmonella and are subject to partial or complete deletion.52,53 MVMp has two imperfect palindromes at the 3′ and 5′ termini that form hairpins in the DNA of the viral genome.35,37,45,47,50,54,55,56 These palindromic regions are primers for replication and are essential for virus formation and function.36,45,47,50,54,55,56 When transformed into cloning strains of E. coli, homologous recombination forms site-specific deletions in the right-end hairpin of plasmids containing the MVMp genome.54 This deletion is prevented in SURE 2 (“stop unwanted rearrangement events” 2) E. Coli (ΔrecB, ΔrecJ, and ΔsbcC), which is restriction minus, endonuclease deficient, and recombination deficient.
Salmonella contains two systems for repairing DNA damage and regulating homologous recombination: one for double-stranded breaks (DSBs) and another for single-stranded gap (SSG) repairs.57,58,59 These systems maintain genetic stability but also modify foreign DNA.57 A key component of the DSB repair system is RecBCD, which is a helicase-nuclease complex that initiates repair by unwinding double-stranded DNA (dsDNA) and creating a single-stranded tail for RecA to bind.57,60,61,62,63,64 RecA is a central protein in homologous recombination that compares and exchanges complementary DNA sequences.57,63 Two additional components of the DSB repair system are SbcB and SbcCD.65 SbcCD is a nuclease that cleaves double-stranded hairpins.62,63,64 The primary component of SSG repair system is RecFOR.57,60 Similar to RecBCD, RecFOR mediates the binding of RecA to single-stranded regions of DNA.57,62,64,66 There is considerable redundancy between these pathways. RecFOR can repair DSBs in the absence of RecBCD, where RecJ functions as an exonuclease to create a single-stranded overhang to facilitate the loading of RecA.57,62,64 SbcB, in concert with SbcCD, can activate the RecFOR pathway to repair the DSBs in the absence of RecBCD.57,60,61,62,64,67
The goal of this work was to create virus-delivering Salmonella (VDS, Figure 1). To be a suitable carrier for OVs, the bacterial vector must (1) preserve the viral genome and (2) deposit it intact into cancer cells. We hypothesized that 1) DNA delivered by ID Salmonella is expressed by cancer cells, 2) removal of homologous recombination genes from Salmonella prevents hairpin deletion and stabilizes the MVMp genome, 3) Salmonella delivery of the MVMp genome produces functional viral particles, and 4) the produced viral particles are infective and kill cancer cells. To test these hypotheses, we generated seven strains of Salmonella with deletions in six genes: recA, recB, recF, sbcB, sbcCD, and recJ. We formed VDS by transforming these homologous-recombination-deficient Salmonella (HRDS) with a plasmid containing the MVMp genome and a second plasmid containing the PsseJ-lysE delivery circuit. We used fluorescence microscopy to measure the delivery of plasmid DNA to cancer cells with ID Salmonella. We used the gentamicin invasion assay to determine the viability and invasiveness of the HRDS strains. We treated cancer cells with VDS to measure the production of NS1, the initiation of virus formation, and the cytotoxicity of the delivered virus. Finally, we retreated cells with conditioned media to quantify the infectiveness of released virions. By delivering OVs, VDS has the potential to expand the effectiveness of microbial therapy to include more solid tumors and be effective for a broader range of cancer patients.
Figure 1.

Bacterial delivery of viruses into the cancer cells
(1) VDS, carrying the genome of MVMp, invades cancer cells. (2) Once intracellular, Salmonella lysis releases the viral plasmid into Salmonella-containing vacuoles (SCVs). SCV breakdown releases the plasmid into the cytoplasm. (3) After transport into the nucleus, cellular machinery converts the viral DNA into double-stranded DNA that is transcribed and translated to form structural (i.e., capsid) and non-structural proteins. (4) The double-stranded DNA is also a template for synthesizing new viral genomes. (5) The virus progeny is assembled in the nucleus. (6) The viral infection causes cell lysis, releasing new virions that infect the neighboring cells and initiate new rounds of infection.
Results
ID Salmonella delivers functional plasmid DNA into cancer cells
To measure DNA delivery, we applied ID Salmonella enterica serovar Typhimurium (henceforth referred to as Salmonella) to cultures of cancer cells (Figure 2). The delivery of functional DNA is a critical component of a virus carrier system. To visualize DNA delivery and expression, we transformed ID Salmonella with a plasmid containing GFP under control of the cytomegalovirus (CMV) promoter. This promoter only initiates transcription in eukaryotic cells.68 Salmonella with CMV-GFP did not express GFP. In addition to CMV-GFP, ID-CMV-GFP Salmonella contained the PsseJ-lysE circuit, which induces lysis after cell invasion.34 Control Salmonella contained CMV-GFP, but not PsseJ-lysE. When incubated with murine 4T1 mammary cancer cells, ID-CMV-GFP produced GFP (arrows, Figure 2A). Compared to controls, ID-CMV-GFP produced significantly more GFP (p < 0.001, Figure 2B). This difference indicates that the lysis circuit is critical for delivering plasmids into cancer cells. GFP expression also indicates that, after bacterial lysis, released plasmids were transported to the nucleus and delivered genes were translated by recipient cells.
Figure 2.

ID Salmonella delivers encoding plasmids into cells
(A) Administration of ID Salmonella with a eukaryotic-expression plasmid (CMV-GFP) and the intracellular-delivering gene circuit (PsseJ-lysE) induced GFP expression (left, arrows). Bacteria (ID-CMV-GFP) were co-cultured with 4T1 cancer cells for 2 h. Extracellular bacteria were cleared with 50 μg/mL gentamicin. Live images were acquired 24 h after invasion. There was no GFP expression in parental Salmonella containing only the CMV-GFP plasmid but not the lysis circuit (right). Scale bar is 50 μm.
(B) Bacteria with PsseJ-lysE (ID-CMV-GFP) induced significantly more GFP expression than control CMV-GFP bacteria (∗∗∗, p < 0.001).
Parental Salmonella disables MVMp replication
When not modified, parental Salmonella (ΔmsbB, ΔpurI, Δxyl) deletes 97 bp from the right-end hairpin of MVMp (Figure 3). The genome of MVMp contains two palindromes at either end that are primers for replication and are essential for virus formation. The genome for MVMp is on a plasmid that also contains a ColE1 prokaryotic origin of replication, and ampicillin resistance (Figure 3A). Transcription of MVMp is initiated by the P4 promoter within its genome in a mammalian nucleus.48 Replicating this plasmid in SURE 2 recombination-deficient E. coli (ΔrecB, ΔrecJ, and ΔsbcC) maintained both hairpins (Figure 3B). SSPI digestion of the MVMp plasmid produced three fragments: 5,078, 1,395 and 732 bp (Figure 3B, left). The smallest fragment contains the right-end hairpin. When replicated in parental Salmonella, a 97 bp section was deleted from the right hairpin, reducing the third fragment to 635 bp (Figure 3B, right). The location of this deletion from the center of the palindrome in the right-end hairpin was confirmed by sequencing (Figure 3C). Without the right-end hairpin, the MVMp genome is inactive and NS1 is not produced (Figure 3D). To measure virus initiation, MVMp plasmids were maintained in either SURE 2 or parental Salmonella and transfected into HEK-293T cancer cells. The plasmids with intact hairpins formed the replication-initiating NS1 protein (Figure 3D). Plasmids that were maintained in parental Salmonella did not form NS1 (Figure 3D). Taken together, these results indicate that parental Salmonella deletes the right-end hairpin, which renders the virus replication incompetent.
Figure 3.

Parental Salmonella deleted the right-end hairpin and disabled viral replication
(A) The MVMp plasmid contains the full MVM genome including the left- and right-end hairpins. This plasmid replicates in SURE 2 and produces viruses when transfected into mammalian cells.
(B) An SSPI restriction digest of the MVMp plasmid produced three fragments (left), 5,078, 1,395, and 732 bp, when maintained in SURE 2. Propagation in parental Salmonella deleted 97 bp from the right hairpin, reducing the third fragment to 635 bp.
(C) After sequencing, the MVMp plasmid that was propagated in SURE 2 (top) contained the full MVM genome. Propagation in parental Salmonella deleted 97 bp from the center of the palindrome in the right-end hairpin (bottom).
(D) The NS1 protein (83 kDa) was produced when HEK-293T cells were transfected with MVMp after propagation in SURE 2. No NS1 was produced in cells transfected with MVMp propagated in parental Salmonella. The lower band is non-phosphorylated NS1 that is less prevalent in cells.69
Deletion of homologous recombination genes stabilizes the right-end hairpin of MVMp
To create a bacterial strain to deliver functional virus, we deleted homologous recombination genes from Salmonella (Figure 4). We generated seven strains with different combinations of deletions of six genes (recA, recB, recF, sbcB, sbcCD, and recJ) that are critical components of the homologous-recombination machinery. We termed these HRDS strain A (ΔrecA), strain B (ΔrecB), strain F (ΔrecF), strain CD (ΔsbcCD), strain BB’F (ΔrecB, ΔsbcB, and ΔrecF), strain BCDJ (ΔrecB, ΔsbcCD, and ΔrecJ), and strain BB’CDF (ΔrecB, ΔsbcB, ΔsbcCD and ΔrecF). The BCDJ strain has the same deletions as SURE 2 E coli (ΔrecB, ΔsbcCD, and ΔrecJ).
Figure 4.

Deletion of homologous recombination genes maintains the right-end hairpin of MVMp
(A) SSPI restriction digest of the MVMp plasmid, when maintained in (left to right) SURE 2 (lane 1), parental Salmonella (lane 2), A-HRDS (lane 3), B-HRDS (lane 4), F-HRDS (lane 5), BB’F (lane 6), CD (lane 7), BB’CDF (lane 8), and BCDJ (lane 9). Strains CD, BB’CDF, and BCDJ maintained the complete right-end hairpin similar to SURE 2. All of the other strains had a 97 bp deletion resulting in a shorter third fragment (635 bp).
(B) The sequence of plasmids maintained in parental Salmonella, or strains A, B, F, and BB’F had a 97 bp deletion in the center of the palindrome of the right-end hairpin. The sequence of plasmids maintained in strains CD, BB’CDF, and BCDJ had intact right-end hairpins.
(C) After HEK-293T cells were transfected with MVMp plasmids that were maintained in SURE 2, BB’CDF, and CD, the cells produced NS1 protein (83 KDa). Uninfected cells did not produce NS1.
(D) The MVMp plasmid was maintained in SURE 2, BB’CDF, and CD for three generations. For each passage, the plasmid maintained the hairpin for all Salmonella strains.
When the MVMp plasmid was replicated in each of these seven bacterial strains, four of the strains deleted 97 bp from the right-end hairpin (A, B, F, and BB’F), as shown by SSPI restriction digest (Figure 4A) and DNA sequencing (Figure 4B). Three of the knockout stains maintained the hairpin (CD, BCDJ, and BB’CDF; Figures 4A and 4B).
Two of strains that maintained the right-end hairpin (CD and BB’CDF) also maintained virus function (Figure 4C). The MVMp plasmid was replicated in CD and BB’CDF, purified, and transfected into HEK-293T cells. After transfection, all of these cells formed NS1 (Figure 4C), demonstrating the initiation of virus formation. To show the durability of virus replication, the MVMp plasmid was maintained in CD, BB’CDF, and SURE 2 for three passages. After each of the generations, both CD and BB’CDF maintained the full-length plasmid and the right-end hairpin (similar to SURE 2, Figure 4D).
Deletion of homologous recombination genes does not impair growth or invasion
For strains CD and BB’CDF, which stabilized the right-end hairpin, deletion of homologous recombination genes did not affect bacterial growth or invasiveness into cancer cells (Figure 5). In culture, the growth of strain CD and BB’CDF was comparable with that of parental Salmonella (Figure 5A). The growth of strain BCDJ, however, was severely impaired. The deletions in BCDJ delayed growth by more than 6 h (Figure 5A). At 9 h after inoculation, the density of BCDJ was 4.3 times lower than that of parental Salmonella (p < 0.0001, Figure 5B). The densities of CD and BB’CDF were comparable to the density of parental Salmonella at 9 h (Figure 5B).
Figure 5.

Deletion of homologous recombination genes does not impair growth or cell invasion
(A) Bacterial growth was measured for 16 h for strains BB’CDF, CD, BCDJ, and parental Salmonella. Two strains (BB’CDF and CD) had similar growth patterns compared to parental Salmonella. The growth of BCDJ was delayed by 6 h.
(B) At 9 h, the bacterial density of BCDJ was significantly lower compared to parental Salmonella (∗∗∗∗, p < 0.0001). Strains BB’CDF and CD had similar densities to parental Salmonella.
(C) After 2 h of invasion, the number of intracellular bacteria (green, arrows) for BB’CDF (top right) and CD (bottom left) was similar to parental Salmonella (top left). Incubation with strain BCDJ (bottom right) produced less intracellular bacteria. Some cells contained regions of hyper-replicating bacteria (black arrows). Scale bar is 50 μm.
(D‒F) There was no significant difference in the invasion levels of BB’CDF (D) or CD (E) compared to parental Salmonella. Invasion levels were significantly compromised for the BCDJ strain (F) compared to parental Salmonella (p < 0.05).
(G) Cells invaded with BB’CDF had less hyper-replicating Salmonella than parental (∗, p < 0.05) or CD (∗∗, p < 0.01) Salmonella.
Similar to growth, the CD and BB’CDF strains invaded into cancer cells at comparable rates to parental Salmonella (Figures 5C–5F). To measure invasiveness, the Salmonella strains (CD, BB’CDF, BCDJ, and parental) were co-cultured with 4T1 murine mammary cancer cells for 2 h and extracellular bacteria were cleared with gentamicin. After 4 h, bacteria were inside cells (arrows) for the CD, BB’CDF, and parental strains (Figure 5C). Few BCDJ were present inside cells (Figure 5C). After co-culture with parental and CD Salmonella, some cells contained hyper-replicated bacteria in their cytoplasm (black arrows, Figure 5C). There was no difference in the percentage of cells invaded by CD and BB’CDF compared with parental Salmonella (Figures 5D and 5E). The percentage of invaded cells was significantly lower for BCDJ compared to parental Salmonella (p < 0.05, Figure 5F). Because of its poor ability to grow and invade cancer cells, the BCDJ was not used. The number of cells containing hyper-replicating Salmonella was less after invasion in BB’CDF than either parental (p < 0.05) or CD (p < 0.01) Salmonella (Figure 5G).
Engineered Salmonella delivers MVMp to cancer cells and induces lysis
To create VDS we transformed HRDS with two plasmids: one that contains the macromolecular delivery system (PsseJ-lysE) and a second that contains the MVMp genome. Transforming the two strains that maintained the right-end hairpin (BB’CDF and CD) created two strains: VDS-B (BB’CDF) and VDS-C (CD). To test the delivery of MVMp, both strains of VDS were applied to cultures of 4T1 cells (Figure 6A). After 2 h of co-culture, extracellular bacteria were removed with gentamicin (Figure 6A). After a further 36 h to allow for plasmid release, transport to the nucleus, and gene expression, the cells cultured with VDS-B produced the viral NS1 protein (Figure 6B). NS1 production was comparable to positive control cells that were directly transfected with the MVMp plasmid using lipofectamine (Figure 6B). As expected, no NS1 was produced by uninfected cells. Cells co-cultured with VDS-C did not produce NS1 (Figure 6B).
Figure 6.

VDS delivers functional virions that kill cancer cells
(A) To measure virus delivery and induced lysis, VDS was applied to cultures of cancer cells (top). Murine mammary carcinoma cells (4T1) were incubated (blue arrows) with VDS-B or bacterial controls (BB’CDF). VDS-B contains both the MVMp and PsseJ-lysE plasmids; BB’CDF contains only the PsseJ-lysE plasmid. Similarly, VDS-C was compared to strain CD, its bacterial control. After 2 h, external bacteria were removed with gentamicin (green arrow). In parallel, positive control cells were directly transfected with MVMp using lipofectamine (bottom). After 36 h, NS1 expression was measured by immunoblot. After a further 36 h, cell viability and cell death were measured using crystal violet, ethidium homodimer, and MTT.
(B) After VDS-B invaded cancer cells in culture, the cells produced viral NS1, similar to direct transfection with the MVMp plasmid. Cell invaded with VDS-C did not produce NS1.
(C) Cells delivered MVMp with VDS-B formed dead-cell plaques (arrow). Cells were stained with crystal violet. Bacterial infection alone formed few plaques. Scale bar is 1 mm.
(D) The area of cell death area was significantly higher in cells invaded with VDS-B than in cells invaded with BB’CDF (∗, p < 0.05).
(E) Over time, treatment with VDS-B killed more cancer cells than bacterial controls. At 60 h after invasion (or transfection), more dead cells (arrows, ethidium homodimer+) started to appear in cultures treated with VDS-B or directly transfected with MVMp. Scale bar is 50 μm.
(F) With time, the area of cell death increased for VDS-B and MVMp transfection but remained constant for BB’CDF.
(G) Death caused by VDS-B was greater than BB’CDF (∗, p < 0.05).
(H) Cell viability, measured by MTT assay, decreased for VDS-B and MVMp transfection compared to BB’CDF (∗, p < 0.05).
(I) VDS-C did not increase cell death compared to CD (∗∗∗, p < 0.001).
Bacterial delivery of MVMp with VDS-B caused cell death (Figures 6C–6H). To measure cell death, either VDS-B or control bacteria were applied to 4T1 cells (Figure 6A). These cells were compared to cells that were directly transfected with the MVMp plasmid. The bacterial control was the BB’CDF strain, which did not contain the MVMp plasmid but was transformed with the delivery (PsseJ-lysE) plasmid, and had the same knockouts (ΔrecB, ΔsbcB, ΔsbcCD, and ΔrecF) as VDS-B. Seventy-two hours after bacterial invasion, cells treated with VDS-B formed virus-infected, dead-cell plaques (Figure 6C). The area of the plaques for VDS-B was significantly more than that of bacterial controls (p < 0.05, Figure 6D). Over time, VDS-B caused death similarly to transfection controls (arrows in Figures 6E and 6F). In comparison, there was minimal death in bacterial controls (Figures 6E and 6F). The area of cell death increased at a similar rate after infection with VDS-B or transfection with MVMp (Figure 6F). At 72 h, MVMp, delivered by VDS-B, significantly increased the area of cell death compared to control bacteria (p < 0.05, Figure 6G). A secondary method, MTT, also demonstrated that VDS-B caused more cell death than bacterial controls (p < 0.05, Figure 6H)).
Unlike VDS-B, VDS-C did not cause cell death (Figure 6I). Over time, the area of cell death did not increase for cells treated with VDS-C (Figure S1). At 72 h, there was no difference in cell death between VDS-C and strain HRDS-CD, its bacterial control (Figure 6I). Direct transfection of MVMp caused more cell death than VDS-C (p < 0.001, Figure 6I). Lack of virus formation and undetectable cell death shows limited utility for VDS-C (compared to VDS-B).
VDS creates functional virus particles that infect new cells
Delivery of MVMp by VDS forms functional and infective virions (Figure 7). To measure the functionality of produced viruses, VDS was applied to cultures of 4T1 cancer cells for 2 h (Figure 7A). After this time for invasion, the cells were washed and fed with complete media containing gentamicin. After an additional 72 h, the culture medium was removed, filtered, and added to fresh media with gentamicin, at a 40:60 ratio. The 0.2 μm filter prevented passing bacteria to the secondary cultures, while enabling transfer of formed viruses. Even in the absence of filtration, the addition of gentamicin killed all bacteria in these media (Figure S2B). The media mixture was added to a secondary culture of HEK-293T cells (Figure 7A). After 48 h in this conditioned media, the HEK-293T cells that received VDS-B media produced NS1 (Figure 7B). Conditioned media from primary cultures that had been directly transfected with MVMp also produced NS1. Media from cells treated with VDS-C did not form NS1 (Figure 7B). Two replicates of this experiment with gentamicin-treated media also showed that VDS-B formed viruses (Figures S2C and S2D). Together, these results show that delivery of the MVMp plasmid with VDS-B forms full virion particles that are infective to naive cells.
Figure 7.

VDS delivers function virions that spread to neighboring cells
(A) To measure the infectivity of the produced virus particles, conditioned media from VDS-treated cells were applied to cultures of naive cells. Cancer cells (4T1) were incubated (blue arrows) with either (VDS-B or VDS-C) or (VDS-B or BB’CDF bacterial controls). After 2 h external bacteria were cleared with gentamicin (green arrow, top). Positive controls were cells directly transfected with the MVMp plasmid using lipofectamine (bottom). After 72 h, the culture medium was collected and filtered to eliminate all contaminating bacteria. The filtered media were added to fresh media at a 40:60 ratio, and 50 μg/mL gentamicin was added. This conditioned medium was added to naive cultures of (1) HEK-293T cells to measure virus formation and (2) 4T1 cancer cells to measure cytotoxicity.
(B) Secondary cultures of HEK-293T cells that received media (and virus) from primary cultures treated with VDS-B (or transfected with MVMp) produced NS1. Uninfected controls and primary cultures treated with VDS-C did not produce NS1.
(C) Virions in conditioned media from VDS-B cultures killed more 4T1 cells than bacterial controls (BB’CDF) that did not produce virus (∗∗∗, p < 0.001). Death was equivalent to viruses from directly transfected cells.
The viral particles produced by VDS-B are cytotoxic to cancer cells (Figure 7C). A similar procedure was used to measure induced cell death. There were a couple of key differences. Primary cells were treated with VDS-B or control bacteria (strain BB’CDF) to control for compounds released by bacteria into the conditioned media. Recipient cells in the secondary cultures were 4T1 cancer cells not HEK-293T. The conditioned media from cells treated with VDS-B killed 50% of the recipient cells, which was significantly more than bacterial controls (p < 0.001, Figure 7C). Cell death was equivalent to cells treated with conditioned media from MVMp-transfected cultures (Figure 7C). A similar serial-dilution experiment showed that diluting the conditioned media ten times killed all cancer cells that received the produced viruses (n = 6, (Figure S2E). These results show that the virions produced by treating cells with VDS-B are cytotoxic to cancer cells.
VDS-mediated cell death is independent of cancer type
When applied to multiple cancer cell types, VDS induced the formation of cytotoxic viral particles that kill cells and infect neighboring cells (Figure 8). To measure virus formation, VDS-B was applied to HEK-293T human embryonic kidney cells, U2OS human osteosarcoma cells, and KPCY murine pancreatic ductal adenocarcinoma cells (Figure 8A). A similar procedure was used as Figure 6A earlier. As with 4T1 cells, MVMp is infective in these cells; NS1 was produced by cells that were directly transfected with the MVMp plasmid using lipofectamine (Figure 8A). When VDS-B was applied for 2 h, these cells also produce NS1 (Figure 8A). VDS-C did not produce NS1 in any of the cell types.
Figure 8.

Salmonella-mediated delivery of MVMp is independent of cell type
(A) Treatment with VDS-B produced NS1 when applied to human embryonic kidney cells (HEK-293T), human osteosarcoma cells (U2OS), and murine pancreatic ductal adenocarcinoma cells (KPCY). No NS1 was expressed in any of the cell lines after being treated with VDS-C.
(B) Treatment with VDS-B significantly decreased viability in KPCY (∗∗, p < 0.01) and U2OS (∗∗∗∗, p < 0.0001) cells, compared to bacterial controls (BB’CDF).
(C) Re-infection of HEK-293T cells with conditioned media from primary cultures of KPCY and U2OS cells that were treated with VDS-B produced NS1. Conditioned media from cells directly transfected with the MVMp plasmid also produced NS1. Untreated controls and cultures treated with VDS-C did not produce NS1.
The delivery of MVMp with VDS killed osteosarcoma and pancreatic ductal adenocarcinoma cells (Figure 8B). At 72 h after infection of KPCY cells, VDS-B significantly reduced viability compared to bacterial controls (strain BB’CDF; p < 0.01, Figure 8B). In U2OS cell, MVMp delivered by VDS-B also significantly reduced viability compared to bacteria controls (p < 0.0001, Figure 8B). As with results in Figure 6, VDS-C did not induce death in either of these cell lines (Figure S3).
When applied to osteosarcoma and pancreatic ductal adenocarcinoma cells, VDS produced infective viral particles (re-infection, Figure 8C). A similar procedure was used as Figure 7A earlier. VDS was added to cell cultures for 2 h, extracellular bacterial were killed with gentamicin, and conditioned media was added to cultures of HEK-293T cells. After 48 h in conditioned media, the HEK-293T cells that received media from VDS-B cultures produced NS1 (Figure 8C). Virus was also present in the culture media from cells transfected with the MVMp plasmid (Figure 8C). No virus was produced by cultures treated with VDS-C. Viewed together, these data demonstrate that MVMp delivery by VDS resulted in the production of viable virus particles that kill infected cancer cells, independent of cell type.
Discussion
We have created a bacterial vector that delivers OVs into cancer cells. The genome for the virus (MVMp) was contained on a plasmid that the bacteria propagated without deleting palindromic sections that are essential for virus infection and replication (Figures 3, 4, and 5). When these VDSs encounter cancer cells, they invade and deposit the plasmid into the cellular cytoplasm. After the delivered plasmid reaches the nucleus, the cell expresses the viral genes (Figures 2 and 6). Production of initiating proteins (here NS1) begins the process of forming and assembling viral particles in the cells (Figure 6). Once formed, these virions kill the invaded cancer cells (Figure 6) and spread into the environment. There the viruses invade and kill naive cells (Figure 7). The release of virions by infected cells perpetuates and amplifies the infection.
Our results show that deletion of homologous recombination genes enables Salmonella to carry and maintain the MVMp genome (Figures 3, 4, and 5). This parvovirus genome contains a right-end hairpin that is unstable in Salmonella (Figure 3). We rationally designed VDS to prevent this deletion. Of the seven knockout stains we created, one (BB’CDF) performed the best. The remaining six had deficiencies that made them poor candidates for viral delivery. The first deletion (ΔrecA, strain A) was the most obvious because of the central role of RecA in homologous recombination. Previous research has shown that deletion of recA stabilized the herpesvirus genome,70 suggesting that this deletion may stabilize MVMp. However, deletion of the recA gene did not prevent hairpin deletion (Figure 4). This may have occurred because RecA-independent mechanisms affect homologous sequences less than 1 kb in length.63
It has also been reported that E. coli lacking recB, sbcB, and recF stably propagates the MVMp plasmid.54 In Salmonella, however, neither this combined deletion (strain BB’F) nor the individual ΔrecB (strain B) and ΔrecF (strain F) deletions maintained the right-end hairpin. This difference suggests that there are differences in homologous recombination between Salmonella and E. coli that affect palindromic DNA.
All the remaining strains (CD, BCDJ, and BB’CDF) did not delete the right-end hairpin. The common knockout in each of these strains (ΔsbcCD) suggests that this nuclease is primary cause of hairpin deletion. Although these strains maintained the full MVMp genome, two (CD and BCDJ) were not good candidates for viral delivery. Strain BCDJ, which has the same knockouts as SURE 2 E coli, grew poorly and did not efficiently invade into cells (Figure 5). Strain CD lacked the ability to deliver viral DNA into cancer cells and propagate a virus infection (Figures 6, 7, and 8). The combination of ΔsbcCD with ΔrecB, ΔsbcB, and ΔrecF (BB’CDF) enabled DNA delivery and propagation of parvovirus within several cancer cell lines (Figures 6, 7, and 8).
There are several possible explanations for why VDS-B is more effective than VDS-C for delivering virus. One reason for this difference might be that the deletion of recB makes the BB’CDF strain more attenuated, which would increase its sensitivity to lysis and increase plasmid delivery once intracellular. Another possible explanation is that VDS-B remains in Salmonella-containing vacuoles (SCVs) after invasion. Residence in SCVs is required for lysis and virus delivery.34 Because Salmonella only hyper-replicate in the cytoplasm, the number of cells with hyper-replicating clusters (Figure 5C) is an indication of vacuole escape.71,72 After invasion, VDS-B formed fewer hyper-replicating clusters than parental and CD Salmonella (Figures 5C and 5G), suggesting that these bacteria remained in SCVs and delivered more viral plasmids into cells.
The amount of virus delivered appeared to vary between cell lines (Figure 8). This difference could be caused by several factors, including susceptibility to bacterial invasion and replication rate. For example, MVMp replication depends on cell division. Within the three cell lines in Figure 8, KPCY cells grow faster (t½ = 16 h) than 4T1 (t½ = 20 h) and U2OS (t½ = 29 h) cells, suggesting that more virus could form in KPCY cells. Rate of invasion could also affect the rate of virus production. We have yet to find an epithelial cell line that Salmonella does not invade, but the rates of invasion could depend on cell type and location.
Because of its broad applicability, VDS has the potential to be a universal platform for virus delivery to tumors. A strength of OVs as a cancer therapy is that the mechanism of cell lysis is independent of the biology of the infected cell and therefore appropriate for treating multiple cancer types. Here, we showed that VDS delivers functional viruses to multiple cancer types, including breast carcinoma, pancreatic carcinoma, and osteosarcoma (Figure 8). In addition, the elimination of homologous recombination genes from VDS makes it suitable for delivering multiple types of viruses. As demonstrated here, we used VDS to deliver MVMp, which is an ssDNA virus. VDS, however, could also deliver dsDNA, single-stranded RNA (ssRNA), and double-stranded RNA (dsRNA) viruses.
As a cargo vector, VDS would enable OV treatment of solid tumors. After systemic injection, the Salmonella would protect the viral DNA from clearance by neutralizing antibodies in the blood. The bacterial vector will protect the viral genome because the virus will only exist as a DNA sequence on the contained plasmid during transportation to the tumor. While in the blood, there will be no viral proteins for antibodies or complement to detect. In addition, we have shown that bacteria preferentially accumulate 100,000 times more than most organs,34,73 thus preventing sequestration in non-tumor organs.
After preferential accumulation in tumors, we envision that VDS will deliver viral genomes into local cancer cells in tumors. There, the genomic material would localize to the nucleus and form functional virus particles (Figure 7). Cell lysis would release the viruses into the local environment to infect neighboring cancer cells (Figure 7). Because of OV replication, even small doses will expand the infection, amplify the immune response, and trigger a potent bystander effect. The generated anticancer immunity would suppress tumor recurrence and prevent formation of metastases. By combining the benefits of Salmonella delivery and OV therapy, VDS has the potential to provide an effective new therapy for many cancers.
Limitations of the study
This is the first published study to show that bacteria can be used to deliver the genetic code of a virus and initiate a self-propagating infection in cancer cells. We chose MVMp because of its size and non-pathogenic nature, as guided by Dr. Peter Tattersall, an expert in the field. One limitation of MVMp is that it is highly contagious and prohibited in animal colonies, limiting work to in vitro studies. Another limitation was that viral replication and spread were quantified using western blot analysis of the NS1 protein coupled with re-infection assays to quantify viral propagation. These methods demonstrated the ability of engineered Salmonella to deliver and initiate MVMp infection and spread in vitro. However, these methods only quantified population level, rather than single-cell delivery and infection dynamics. Moreover, this study’s results were limited to viral delivery, infection, and death dynamics within multiple cancer cell lines but did not investigate viral propagation within non-transformed stromal and/or immune cells that would be found within tumors.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-NS1 | Majumder et al.44 | N/A |
| HRP-conjugated goat anti-mouse IgG | R&D Systems | Cat# HAF007;RRID: AB_357234 |
| Bacterial and virus strains | ||
| E. coli SURE 2 | Agilent | 200152 |
| Salmonella enterica serovar Typhimurium (VNP20009) | Vion Pharmaceuticals, Inc. | N/A |
| Salmonella strain A | This paper | N/A |
| Salmonella strain B | This paper | N/A |
| Salmonella strain F | This paper | N/A |
| Salmonella strain CD | This paper | N/A |
| Salmonella strain BB’F | This paper | N/A |
| Salmonella strain BCDJ | This paper | N/A |
| Salmonella strain BB’CDF | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Fetal bovine serum | Cytiva | SH3091003 |
| Dulbecco’s modified Eagle medium-low glucose | Sigma Aldrich | D5523-10X1L |
| Roswell Park Memorial Institute 1640 | Sigma Aldrich | R8755-10L |
| MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide | Themo Fisher | Cat# M6494 |
| Ethidium Homodimer-1 (EthD-1) | Thermo Fisher | Cat# E1169 |
| Kanamycin sulfate | Fisher Scientific | Cat# J17924.06 |
| Carbenicillin disodium salt | Chem-Impex | Cat# 00049 |
| Gentamicin sulfate | Fisher Scientific | Cat# BP918-1 |
| Chloramphenicol | Fisher Scientific | Cat# 227921000 |
| SSPI-HF restriction enzyme | New England Biolabs | Cat# R3132S |
| Laemmli SDS sample buffer, reducing (6X) | Fisher Scientific | Cat# J61337.AC |
| Crystal Violet | Acros organics | 229641000 |
| Hoechst 33342 Solution | Invitrogen | Cat# H1399 |
| Oxoid™ Hektoen Enteric Agar | Thermo Fisher | Cat# CM0419B |
| Immobilon Western Chemiluminescent HRP Substrate | Millipore Sigma | WBKLS |
| Critical Commercial Assays | ||
| Lipofectamine 3000 Transfection Reagent | Fisher Scientific | Cat# L3000008 |
| Zyppy plasmid miniprep kit | Zymo | Cat# D4037 |
| Deposited Data | ||
| Raw and analyzed data | This paper | Mendeley Data: https://doi.org/10.17632/rk36fh29ks.1 |
| Experimental models: Cell lines | ||
| Human embryonic kidney cells (HEK-293T) | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| Human osteosarcoma cells (U2OS) | ATCC | Cat# HTB-96; RRID: CVCL_0042 |
| Murine pancreatic ductal adenocarcinoma cells (KPCY) | Kerafast | Cat# EUP001-FP; RRID: CVCL_YM32 |
| Murine mammary carcinoma cells (4T1) | ATCC | Cat# CRL-2539; RRID: CVCL_0125 |
| Oligonucleotides | ||
| Primers for deleting sbcB-Forward (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting sbcB-Reverse (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting sbcCD-Forward (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting sbcCD-Reverse (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting recF-Forward (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting recF-Reverse (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting recB-Forward (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting recB-Reverse (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting recJ-Forward (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting recJ-Reverse (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting recA-Forward (SeeTable S1) | Thermo fisher | N/A |
| Primers for deleting recA-Reverse (SeeTable S1) | Thermo fisher | N/A |
| Recombinant DNA | ||
| ColE1-Amp-MVMp | Yale School of Medicine, New Haven, CT | N/A |
| p15A-Chlor-PsseJ-lysE-ptac-GFP | Raman et al.34 | N/A |
| ColE1-Amp-CMV-GFP | This paper | N/A |
| pKD46 | Yale CGSC E. Coli stock center | 7669 |
| pKD4 | Yale CGSC E. Coli stock center | 7632; GenBank: AY048743.1 |
| pCP20 | Yale CGSC E. Coli stock center | 7629 |
| Software and algorithms | ||
| ImageJ | Schneider et al.74 | https://ij.imjoy.io/ |
| Prism | GraphPad | https://www.graphpad.com/ |
| Snapgene | Dotmatics | https://www.snapgene.com/ |
| Biorender | N/A | https://www.biorender.com/ |
| Other | ||
| ImageQuant LAS4000 imaging system | GE Healthcare | N/A |
| Axio Observer Z.1 microscope | Zeiss | N/A |
| Spectramax ID3 | Molecular devices | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Neil S. Forbes (forbes@umass.edu).
Materials availability
Seven new Salmonella strains (A: ΔrecA; B: ΔrecB; F: ΔrecF; CD: ΔsbcCD; BB’F: ΔrecB, ΔsbcB and ΔrecF; BCDJ: ΔrecB, ΔsbcCD and ΔrecJ; and BB’CDF: ΔrecB, ΔsbcB, ΔsbcCD and ΔrecF) were generated for this study and were derived from the parental strain VNP20009 (ΔmsbB, ΔpurI, Δxyl). There are restrictions to the availability of these strains and can be made available with an MTA.
Data and code availability
-
•
All raw data reported in this paper have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. All the original western and microscopy images will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Cell lines
Human embryonic kidney cells (HEK-293T, RRID:CVCL_0063; ATCC, Manassas, VA), human osteosarcoma cells (U2OS, RRID:CVCL_0042), and murine pancreatic ductal adenocarcinoma cells (KPCY, RRID:CVCL_YM32;) were maintained in low glucose Dulbecco’s modified Eagle medium (DMEM; Sigma Aldrich, St. Louis, MO) with sodium bicarbonate (pH 7.4) and 10% fetal bovine serum (FBS; Cytiva, Marlborough, MA) at 37°C and 5% CO2. Murine mammary carcinoma cells (4T1, RRID:CVCL_0125) were maintained in Roswell Park Memorial Institute (RPMI 1640; Sigma Aldrich, St. Louis, MO) medium supplemented with 2 g/l of sodium bicarbonate and 10% FBS at 37°C and 5% CO2. For live-cell microscopy, cells were incubated with DMEM with 20 mM HEPES buffering agent and 10% FBS.
Bacterial culture
All bacterial cultures (SURE 2, Salmonella) were grown in LB (10 g/L sodium chloride, 10 g/L tryptone, and 5 g/L yeast extract). Resistant strains of bacteria were grown in the presence of 100 μg/mL of carbenicillin, 33 μg/mL chloramphenicol, or 50 μg/mL of kanamycin.
Bacterial strains and plasmids
The MVMp plasmid was provided by Dr. Peter Tattersall (Yale School of Medicine, New Haven, CT). The plasmid is ampicillin resistant and has a ColE1 origin of replication. The ptac-GFP plasmid contains green fluorescent protein (GFP) under control of the constitutive bacterial promoter ptac and a Lysin gene E from ΦX174 bacteriophage under the control of intracellular responsive Salmonella promoter PsseJ.34 This plasmid is chloramphenicol resistant and has a p15A origin of replication.
Method details
GFP expression from a eukaryote promoter after delivery with Salmonella
To measure plasmid and gene delivery, two plasmids for delivery and eukaryotic GFP expression were transformed into parental Salmonella (ΔmsbB, ΔpurI, Δxyl) and termed as ID-CMV-Sal. The GFP expression plasmid contained the gene for green fluorescent protein (GFP) under control of the cytomegalovirus (CMV) mammalian expression promoter. The delivery plasmid contained the PsseJ-lysE circuit. Control bacteria were parental Salmonella transformed with CMV-GFP but not the PsseJ-lysE lysis plasmid. Salmonella strains were grown to an OD600 between 0.8 and 1.0 and added to cultures of 4T1 cells for 2 h. During this period, the bacteria invaded into the cancer cells. Following the invasion, cells were rinsed with phosphate-buffered saline (PBS) five times and incubated in RPMI with 2 g/L sodium bicarbonate, 10% FBS, and 50 μg/mL gentamicin. The gentamicin in the media eliminates extracellular bacteria. Live cells were imaged 24 h later to quantify for GFP expression by the cells. The cells were also stained with 10 μg/mL Hoechst 33342 (Invitrogen, Waltham, MA) to identify nuclei. Three regions in each well were randomly selected, and 20 cells per regions were randomly selected and scored based on GFP expression to determine the percentage of the cells expressing GFP.
Bacterial gene deletions
Seven strains of Salmonella enterica serovar Typhimurium were created to prevent homologous recombination. All genetic deletions (ΔrecA, ΔrecB, ΔsbcCD, ΔsbcB, ΔrecF, and ΔrecJ) were derived from the parental strain VNP20009 (ΔmsbB, ΔpurI, Δxyl) using a modified lambda red recombination protocol with primers of specific homology regions.75 These genes were deleted to create seven strains (A: ΔrecA; B: ΔrecB; F: ΔrecF; CD: ΔsbcCD; BB’F: ΔrecB, ΔsbcB and ΔrecF; BCDJ: ΔrecB, ΔsbcCD and ΔrecJ; and BB’CDF: ΔrecB, ΔsbcB, ΔsbcCD and ΔrecF).
Salmonella was transformed with pKD46 (Yale CGSC E. Coli stock center), grown to OD600 0.1, and induced with 10 mM arabinose. Once the OD600 reached 0.6–0.8, the bacteria were centrifuged at 3000 × g for 15 min. The pellet was washed twice with ice-cold water. A PCR product for the in-frame deletion of the gene using specific primers (Table S1) was amplified from the pKD4 plasmid (GenBank: AY048743.1) containing the FRT-KAN-FRT sequence. The PCR product, which contains 50 bp homology for the gene, was transformed into Salmonella using electroporation. The recovery was plated on kanamycin plates (50 μg/mL) and grown overnight. The colonies were screened for deletion by performing a colony PCR of the junction sites of the inserted PCR amplified products. Colonies with a successful knockout were grown overnight at 43°C to eliminate pkd46.
To create another knockout in the Salmonella strain with a gene deletion, the antibiotic resistance was removed by transforming the pCP20 plasmid into the deleted Salmonella. The colonies were grown overnight at 43°C to eliminate pcp20. After the elimination of the plasmid, the overnight culture was diluted to approximately 100 CFU/mL and plated on hektoen plates. The colonies which were dark green were confirmed as Salmonella. The colonies were sequentially streaked on hektoen, kanamycin, and chloramphenicol agar to verify the removal of the antibiotics.
Hairpin stability and bacterial growth
To test hairpin stability, the MVMp plasmid was transformed into each knockout strain by electroporation. Electroporation was performed in 1 mm cuvettes at 1800 V and 25 μF with a time constant of 5 msec. Plasmids were extracted using the ZymoPURE plasmid miniprep kit per the manufacturer’s instructions (Irvine, CA) and sent for sequencing (Massachusetts General Hospital CCIB DNA core facility) To further test hairpin stability, isolated plasmids were digested with SSPI restriction enzyme (New England Biolabs, Ipswich, MA). Strains BB’CDF and CD strains were transformed with a plasmid containing the PsseJ-lysE cassette to deliver the MVMp plasmid and were termed virus-delivering Salmonella (VDS-B and VDS-C, respectively). The growth rate of the knockout strains was determined by inoculating cultures at 5 × 105 CFU/mL, growing at 37°C, and measuring OD600 every 30 min for 16 h on a plate reader.
Intracellular invasion
To determine the effect of gene deletions on cell invasion, the knockout strains were co-cultured with 4T1 cancer cells. Cells were seeded in a 12-well plate at 30% confluence and incubated for a 24 h in RPMI. Once the cells were 70% confluent, the BB’CDF, CD and BCDJ strains were added to the cells at a multiplicity of infection (MOI) of 20. All Salmonella strains were transformed with a plasmid containing EGFP under a constitutive promoter and grown to an OD600 between 0.8 and 1.0. Salmonella were centrifuged at 13,000 rpm for a minute. The media was removed and the pellet was suspended in PBS. The bacteria were then added to the cell cultures for 2 h. Following this invasion period, cultures were rinsed with 1X PBS five times and incubated in RPMI with 2 g/L sodium bicarbonate, 10% FBS, and 50 μg/mL gentamicin. Cells were stained with 10 μg/mL Hoechst 33342 (Invitrogen, Waltham, MA). Multiple regions in a well were randomly selected, and 30 cells/regions were randomly selected and scored based on EGFP expression to quantify the percentage of the cells that were invaded.
Virus delivery
To measure initiation of virus production, VDS-B and VDS-C were added to the HEK-293T cells at an MOI of 20. After 2 h of invasion, extracellular bacteria were removed with gentamicin. The positive control was the MVMp plasmid transfected into naive cells using Lipofectamine 3000 per manufacturer’s protocol (Thermo Fisher, Waltham, MA). Thirty-six hours after infection or transfection, cells were scraped from the flasks. Cells were collected and pelleted by centrifugation at 1000g for 5 min. Cells were suspended in Laemmli SDS 6x sample buffer (Alfa Aesar, Haverhill, MA). Samples were loaded onto NuPage 4–12% Bis-Tris gels (Invitrogen, Waltham, MA) and separated using electrophoresis. Proteins were transferred to polyvinylidene difluoride membranes (Millipore Sigma, Burlington, MA) and probed using mouse monoclonal anti-NS1 (gift from Dr. Kinjal Majumder) at a 1/500 dilution. Blots were incubated with HRP-conjugated goat anti-mouse IgG (R&D Systems, Catalog # HAF007, RRID:AB_357234; 1/1000 dilution) and visualized using Immobilon Western chemiluminescent HRP substrate (Millipore Sigma, Burlington, MA) on an ImageQuant LAS4000 imaging system (GE Healthcare, Chicago, IL). Delivery was also measured in 4T1, KPCY, and U2OS cells at an MOI of 100. For KPCY and U2OS cells, the anti-NS1 antibody was diluted 1/250 and the anti-mouse IgG was diluted 1/500.
Cell viability and death
Cell death was quantified after adding VDS to cancer cells. Bacteria (VDS-B or VDS-C) were added to cultures of 4T1 cells at an MOI of 100. Strains without the MVMp plasmid (CD or BB’CDF) were negative controls. The positive control was 4T1 cells transfected with the MVMp plasmid using Lipofectamine 3000. After 2 h of invasion, bacteria were cleared with gentamicin. Seventy-two hours after infection/transfection, the cells were fixed with 10% formaldehyde in water for 1 h at room temperature. Dead cell plaques were stained with crystal violet staining, dead cells were stained with ethidium homodimer and cell viability was determined by MTT assay.
For crystal violet staining, the formaldehyde was removed and the cells were stained with 1% crystal violet stain (Acros Organics, Waltham, MA). Cells were incubated in crystal violet for 15 min and rinsed with water. Cells were imaged on a Zeiss Axio Observer Z.1 microscope using a 5× objective lens. Images were tiled to image the entire surface. The area of unstained cells was analyzed using ImageJ.
To quantify cell death, 1 μg/mL ethidium homodimer (Invitrogen, Waltham, MA) was added to cells 72 h after infection or transfection. Live cell images were acquired every 10 min. The experiment was performed in triplicate, and death was determined in three areas per well. The extent of cell death was determined in ImageJ by measuring the area with red fluorescence compared to the area covered by cells at the time of invasion/transfection.
To measure cell viability, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Themo Fisher, Waltham, MA) was added to cultures treated with VDS. Cells (4T1, KPCY or U2OS) were seeded on 12-well plates at 30% confluence. Cells were infected with strains VDS-B or BB’CDF (bacterial negative control) at an MOI of 100. The positive control was cells transfected with the MVMp plasmid using lipofectamine. Seventy-two hours after infection/transfection, cells were incubated with 5 mg/mL MTT-solution for 2 h. After discarding the supernatant, 500 μL isopropanol with 25 N HCl was added to each well. Absorbance was measured at 570 nm (Spectramax ID3). Cell viability was calculated relative to uninfected cells. This assay was performed in triplicate wells for each condition.
Re-infection
The infectivity of virions formed by VDS was determined by re-infecting cells with conditioned media. Bacteria (VDS-B or BB’CDF) were added to cultures of 4T1 cells for 2 h and cleared with gentamicin. Positive control cells were transfected with the MVMp plasmid. Media was collected from infected cells 72 h following bacterial invasion or transfection. Media was stored at −80°C until use. Media was thawed at room temperature. The conditioned media was filtered with 0.2 μm filter and was added to fresh media with 50 μg/mL gentamicin, at a 40:60 to HEK-293T cells. The cells were analyzed 48 h after re-infection for NS1 protein as described above. The conditioned media, with 50 μg/mL gentamicin, was also added to 4T1 cells plated on 12-wells at 30% confluence. Viability was analyzed 48 h after re-infection using MTT, as described above.
Quantification and statistical analysis
A comparison of two populations was performed in Excel (Microsoft Office Professional Plus 2016) using two-tailed, unpaired Student’s t-tests. Comparisons of multiple conditions were performed in GraphPad Prism 9.2.0 using ANOVA with a Bonferroni correction. Values are reported as means ± standard errors (SEMs). Statistical significance was confirmed when p < 0.05.
Acknowledgments
We gratefully acknowledge financial support from the National Cancer Institute of the National Institutes of Health, grant R01CA188382, and the Department of Defence (United States), grants W81XWH1910602 and HT94252310067. We would also like to thank Dr. Kinjal Majumder for providing us with the NS1 antibody and Dr. Peter Tattersall for providing the MVMp plasmid and his valuable guidance on this subject.
Declaration of interests
V.R. and N.S.F. are co-founders and shareholders of Ernest Pharmaceuticals, Inc. V.R. is an employee, and N.S.F. is a member of its scientific advisory board.
Published: April 25, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109813.
Supplemental information
References
- 1.Zou H., Mou X.Z., Zhu B. Combining of Oncolytic Virotherapy and Other Immunotherapeutic Approaches in Cancer: A Powerful Functionalization Tactic. Glob. Chall. 2023;7 doi: 10.1002/gch2.202200094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi T., Song X., Wang Y., Liu F., Wei J. Combining Oncolytic Viruses With Cancer Immunotherapy: Establishing a New Generation of Cancer Treatment. Front. Immunol. 2020;11:683. doi: 10.3389/fimmu.2020.00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaufman H.L., Kohlhapp F.J., Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu P.Y., Fan X.M., Zhang Y.N., Wang S.B., Wan W.J., Pan H.Y., Mou X.Z. The limiting factors of oncolytic virus immunotherapy and the approaches to overcome them. Appl. Microbiol. Biotechnol. 2020;104:8231–8242. doi: 10.1007/s00253-020-10802-w. [DOI] [PubMed] [Google Scholar]
- 5.Lin D., Shen Y., Liang T. Oncolytic virotherapy: basic principles, recent advances and future directions. Signal Transduct. Target. Ther. 2023;8:156. doi: 10.1038/s41392-023-01407-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li K., Zhao Y., Hu X., Jiao J., Wang W., Yao H. Advances in the clinical development of oncolytic viruses. Am. J. Transl. Res. 2022;14:4192–4206. [PMC free article] [PubMed] [Google Scholar]
- 7.Mondal M., Guo J., He P., Zhou D. Recent advances of oncolytic virus in cancer therapy. Hum. Vaccin. Immunother. 2020;16:2389–2402. doi: 10.1080/21645515.2020.1723363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.SM O.B., Mathis J.M. Oncolytic Virotherapy for Breast Cancer Treatment. Curr. Gene Ther. 2018;18:192–205. doi: 10.2174/1566523218666180910163805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goradel N.H., Baker A.T., Arashkia A., Ebrahimi N., Ghorghanlu S., Negahdari B. Oncolytic virotherapy: Challenges and solutions. Curr. Probl. Cancer. 2021;45 doi: 10.1016/j.currproblcancer.2020.100639. [DOI] [PubMed] [Google Scholar]
- 10.Fukuhara H., Ino Y., Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373–1379. doi: 10.1111/cas.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bretscher C., Marchini A. H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future. Viruses. 2019;11 doi: 10.3390/v11060562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahman M.M., McFadden G. Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers. 2021;13 doi: 10.3390/cancers13215452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seymour L.W., Fisher K.D. Oncolytic viruses: finally delivering. Br. J. Cancer. 2016;114:357–361. doi: 10.1038/bjc.2015.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwan A., Winder N., Muthana M. Oncolytic Virotherapy Treatment of Breast Cancer: Barriers and Recent Advances. Viruses. 2021;13 doi: 10.3390/v13061128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shalhout S.Z., Miller D.M., Emerick K.S., Kaufman H.L. Therapy with oncolytic viruses: progress and challenges. Nat. Rev. Clin. Oncol. 2023;20:160–177. doi: 10.1038/s41571-022-00719-w. [DOI] [PubMed] [Google Scholar]
- 16.Mahasa K.J., de Pillis L., Ouifki R., Eladdadi A., Maini P., Yoon A.R., Yun C.O. Mesenchymal stem cells used as carrier cells of oncolytic adenovirus results in enhanced oncolytic virotherapy. Sci. Rep. 2020;10:425. doi: 10.1038/s41598-019-57240-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis J.J., Fang B. Oncolytic virotherapy for cancer treatment: challenges and solutions. J. Gene Med. 2005;7:1380–1389. doi: 10.1002/jgm.800. [DOI] [PubMed] [Google Scholar]
- 18.Reale A., Calistri A., Altomonte J. Giving Oncolytic Viruses a Free Ride: Carrier Cells for Oncolytic Virotherapy. Pharmaceutics. 2021;13 doi: 10.3390/pharmaceutics13122192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferguson M.S., Lemoine N.R., Wang Y. Systemic delivery of oncolytic viruses: hopes and hurdles. Adv. Virol. 2012;2012 doi: 10.1155/2012/805629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy D.G., Bell J.C. Cell carriers for oncolytic viruses: current challenges and future directions. Oncolytic Virother. 2013;2:47–56. doi: 10.2147/OV.S36623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willmon C., Harrington K., Kottke T., Prestwich R., Melcher A., Vile R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol. Ther. 2009;17:1667–1676. doi: 10.1038/mt.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Power A.T., Wang J., Falls T.J., Paterson J.M., Parato K.A., Lichty B.D., Stojdl D.F., Forsyth P.A.J., Atkins H., Bell J.C. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol. Ther. 2007;15:123–130. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 23.Allaume X., El-Andaloussi N., Leuchs B., Bonifati S., Kulkarni A., Marttila T., Kaufmann J.K., Nettelbeck D.M., Kleinschmidt J., Rommelaere J., Marchini A. Retargeting of rat parvovirus H-1PV to cancer cells through genetic engineering of the viral capsid. J. Virol. 2012;86:3452–3465. doi: 10.1128/JVI.06208-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kasinskas R.W., Forbes N.S. Salmonella typhimurium specifically chemotax and proliferate in heterogeneous tumor tissue in vitro. Biotechnol. Bioeng. 2006;94:710–721. doi: 10.1002/bit.20883. [DOI] [PubMed] [Google Scholar]
- 25.Forbes N.S. Engineering the perfect (bacterial) cancer therapy. Nat. Rev. Cancer. 2010;10:785–794. doi: 10.1038/nrc2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W.K., Lu M.F., Kuan Y.D., Lee C.H. The treatment of mouse colorectal cancer by oral delivery tumor-targeting Salmonella. Am. J. Cancer Res. 2015;5:2222–2228. [PMC free article] [PubMed] [Google Scholar]
- 27.Raman V., Van Dessel N., O'Connor O.M., Forbes N.S. The motility regulator flhDC drives intracellular accumulation and tumor colonization of Salmonella. J. Immunother. Cancer. 2019;7:44. doi: 10.1186/s40425-018-0490-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arrach N., Cheng P., Zhao M., Santiviago C.A., Hoffman R.M., McClelland M. High-throughput screening for salmonella avirulent mutants that retain targeting of solid tumors. Cancer Res. 2010;70:2165–2170. doi: 10.1158/0008-5472.CAN-09-4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J.E., Phan T.X., Nguyen V.H., Dinh-Vu H.V., Zheng J.H., Yun M., Park S.G., Hong Y., Choy H.E., Szardenings M., et al. Salmonella typhimurium Suppresses Tumor Growth via the Pro-Inflammatory Cytokine Interleukin-1beta. Theranostics. 2015;5:1328–1342. doi: 10.7150/thno.11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phan T.X., Nguyen V.H., Duong M.T.Q., Hong Y., Choy H.E., Min J.J. Activation of inflammasome by attenuated Salmonella typhimurium in bacteria-mediated cancer therapy. Microbiol. Immunol. 2015;59:664–675. doi: 10.1111/1348-0421.12333. [DOI] [PubMed] [Google Scholar]
- 31.Kawaguchi K., Miyake K., Zhao M., Kiyuna T., Igarashi K., Miyake M., Higuchi T., Oshiro H., Bouvet M., Unno M., Hoffman R.M. Tumor targeting Salmonella typhimurium A1-R in combination with gemcitabine (GEM) regresses partially GEM-resistant pancreatic cancer patient-derived orthotopic xenograft (PDOX) nude mouse models. Cell Cycle. 2018;17:2019–2026. doi: 10.1080/15384101.2018.1480223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffman R.M. Tumor-Targeting Salmonella typhimurium A1-R: An Overview. Methods Mol. Biol. 2016;1409:1–8. doi: 10.1007/978-1-4939-3515-4_1. [DOI] [PubMed] [Google Scholar]
- 33.Igarashi K., Kawaguchi K., Kiyuna T., Miyake K., Miyake M., Li S., Han Q., Tan Y., Zhao M., Li Y., et al. Tumor-targeting Salmonella typhimurium A1-R combined with recombinant methioninase and cisplatinum eradicates an osteosarcoma cisplatinum-resistant lung metastasis in a patient-derived orthotopic xenograft (PDOX) mouse model: decoy, trap and kill chemotherapy moves toward the clinic. Cell Cycle. 2018;17:801–809. doi: 10.1080/15384101.2018.1431596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raman V., Van Dessel N., Hall C.L., Wetherby V.E., Whitney S.A., Kolewe E.L., Bloom S.M.K., Sharma A., Hardy J.A., Bollen M., et al. Intracellular delivery of protein drugs with an autonomously lysing bacterial system reduces tumor growth and metastases. Nat. Commun. 2021;12:6116. doi: 10.1038/s41467-021-26367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cotmore S.F., Tattersall P. Parvoviruses: Small Does Not Mean Simple. Annu. Rev. Virol. 2014;1:517–537. doi: 10.1146/annurev-virology-031413-085444. [DOI] [PubMed] [Google Scholar]
- 36.Cotmore S.F., Tattersall P. Genome packaging sense is controlled by the efficiency of the nick site in the right-end replication origin of parvoviruses minute virus of mice and LuIII. J. Virol. 2005;79:2287–2300. doi: 10.1128/JVI.79.4.2287-2300.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cotmore S.F., Tattersall P. Parvovirus diversity and DNA damage responses. Cold Spring Harb. Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a012989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blechacz B., Russell S.J. Parvovirus vectors: use and optimisation in cancer gene therapy. Expert Rev. Mol. Med. 2004;6:1–24. doi: 10.1017/S1462399404008026. [DOI] [PubMed] [Google Scholar]
- 39.Ros C., Bayat N., Wolfisberg R., Almendral J.M. Protoparvovirus Cell Entry. Viruses. 2017;9 doi: 10.3390/v9110313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herrero Y Calle M., Cornelis J.J., Herold-Mende C., Rommelaere J., Schlehofer J.R., Geletneky K. Parvovirus H-1 infection of human glioma cells leads to complete viral replication and efficient cell killing. Int. J. Cancer. 2004;109:76–84. doi: 10.1002/ijc.11626. [DOI] [PubMed] [Google Scholar]
- 41.Cornelis J.J., Lang S.I., Stroh-Dege A.Y., Balboni G., Dinsart C., Rommelaere J. Cancer gene therapy through autonomous parvovirus--mediated gene transfer. Curr. Gene Ther. 2004;4:249–261. doi: 10.2174/1566523043346228. [DOI] [PubMed] [Google Scholar]
- 42.Brandenburger A., Russell S. A novel packaging system for the generation of helper-free oncolytic MVM vector stocks. Gene Ther. 1996;3:927–931. [PubMed] [Google Scholar]
- 43.Raykov Z., Grekova S., Leuchs B., Aprahamian M., Rommelaere J. Arming parvoviruses with CpG motifs to improve their oncosuppressive capacity. Int. J. Cancer. 2008;122:2880–2884. doi: 10.1002/ijc.23472. [DOI] [PubMed] [Google Scholar]
- 44.Majumder K., Boftsi M., Whittle F.B., Wang J., Fuller M.S., Joshi T., Pintel D.J. The NS1 protein of the parvovirus MVM Aids in the localization of the viral genome to cellular sites of DNA damage. PLoS Pathog. 2020;16 doi: 10.1371/journal.ppat.1009002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cotmore S.F., Tattersall P. Parvoviral host range and cell entry mechanisms. Adv. Virus Res. 2007;70:183–232. doi: 10.1016/S0065-3527(07)70005-2. [DOI] [PubMed] [Google Scholar]
- 46.Op De Beeck A., Caillet-Fauquet P. The NS1 protein of the autonomous parvovirus minute virus of mice blocks cellular DNA replication: a consequence of lesions to the chromatin? J. Virol. 1997;71:5323–5329. doi: 10.1128/JVI.71.7.5323-5329.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marchini A., Bonifati S., Scott E.M., Angelova A.L., Rommelaere J. Oncolytic parvoviruses: from basic virology to clinical applications. Virol. J. 2015;12:6. doi: 10.1186/s12985-014-0223-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mattola S., Hakanen S., Salminen S., Aho V., Mäntylä E., Ihalainen T.O., Kann M., Vihinen-Ranta M. Concepts to Reveal Parvovirus-Nucleus Interactions. Viruses-Basel. 2021;13 doi: 10.3390/v13071306. ARTN 1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill C., Carlisle R. Achieving systemic delivery of oncolytic viruses. Expert Opin. Drug Deliv. 2019;16:607–620. doi: 10.1080/17425247.2019.1617269. [DOI] [PubMed] [Google Scholar]
- 50.Merchlinsky M.J., Tattersall P.J., Leary J.J., Cotmore S.F., Gardiner E.M., Ward D.C. Construction of an infectious molecular clone of the autonomous parvovirus minute virus of mice. J. Virol. 1983;47:227–232. doi: 10.1128/JVI.47.1.227-232.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lang S.I., Boelz S., Stroh-Dege A.Y., Rommelaere J., Dinsart C., Cornelis J.J. The infectivity and lytic activity of minute virus of mice wild-type and derived vector particles are strikingly different. J. Virol. 2005;79:289–298. doi: 10.1128/JVI.79.1.289-298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hagan C.E., Warren G.J. Viability of palindromic DNA is restored by deletions occurring at low but variable frequency in plasmids of Escherichia coli. Gene. 1983;24:317–326. doi: 10.1016/0378-1119(83)90092-6. [DOI] [PubMed] [Google Scholar]
- 53.Leach D.R. Long DNA palindromes, cruciform structures, genetic instability and secondary structure repair. Bioessays. 1994;16:893–900. doi: 10.1002/bies.950161207. [DOI] [PubMed] [Google Scholar]
- 54.Boissy R., Astell C.R. An Escherichia coli recBCsbcBrecF host permits the deletion-resistant propagation of plasmid clones containing the 5'-terminal palindrome of minute virus of mice. Gene. 1985;35:179–185. doi: 10.1016/0378-1119(85)90170-2. [DOI] [PubMed] [Google Scholar]
- 55.Tattersall S.F.C.a.P. Cold Spring Harbor Laboratory Press; 1996. Parvovirus DNA Replication. DNA Replication in Eukaryotic Cells. [Google Scholar]
- 56.Li L., Cotmore S.F., Tattersall P. Parvoviral left-end hairpin ears are essential during infection for establishing a functional intranuclear transcription template and for efficient progeny genome encapsidation. J. Virol. 2013;87:10501–10514. doi: 10.1128/JVI.01393-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buljubasic M., Hlevnjak A., Repar J., Dermic D., Filic V., Weber I., Zahradka K., Zahradka D. RecBCD- RecFOR-independent pathway of homologous recombination in Escherichia coli. DNA Repair. 2019;83 doi: 10.1016/j.dnarep.2019.102670. [DOI] [PubMed] [Google Scholar]
- 58.Cano D.A., Pucciarelli M.G., García-del Portillo F., Casadesús J. Role of the RecBCD recombination pathway in Salmonella virulence. J. Bacteriol. 2002;184:592–595. doi: 10.1128/JB.184.2.592-595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rocha E.P.C., Cornet E., Michel B. Comparative and evolutionary analysis of the bacterial homologous recombination systems. PLoS Genet. 2005;1 doi: 10.1371/journal.pgen.0010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Galitski T., Roth J.R. Pathways for homologous recombination between chromosomal direct repeats in Salmonella typhimurium. Genetics. 1997;146:751–767. doi: 10.1093/genetics/146.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith G.R. Homologous recombination in E. coli: multiple pathways for multiple reasons. Cell. 1989;58:807–809. doi: 10.1016/0092-8674(89)90929-x. [DOI] [PubMed] [Google Scholar]
- 62.Spies M., Kowalczykowski S.C. ASM Press; 2004. Homologous Recombination by the RecBCD and RecF Pathways. 2005. [DOI] [Google Scholar]
- 63.Lloyd R.G., Low K.B. 2 Edition. ASM Press; 1996. Homologous Recombination. [Google Scholar]
- 64.Kowalczykowski S.C., Dixon D.A., Eggleston A.K., Lauder S.D., Rehrauer W.M. Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee J., Jo I., Ahn J., Hong S., Jeong S., Kwon A., Ha N.C. Crystal structure of the nuclease and capping domain of SbcD from Staphylococcus aureus. J. Microbiol. 2021;59:584–589. doi: 10.1007/s12275-021-1012-0. [DOI] [PubMed] [Google Scholar]
- 66.Zhang X., Wanda S.Y., Brenneman K., Kong W., Zhang X., Roland K., Curtiss R. Improving Salmonella vector with rec mutation to stabilize the DNA cargoes. BMC Microbiol. 2011;11:31. doi: 10.1186/1471-2180-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miesel L., Roth J.R. Evidence that SbcB and RecF pathway functions contribute to RecBCD-dependent transductional recombination. J. Bacteriol. 1996;178:3146–3155. doi: 10.1128/jb.178.11.3146-3155.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zuniga R.A., Gutierrez-Gonzalez M., Collazo N., Sotelo P.H., Ribeiro C.H., Altamirano C., Lorenzo C., Aguillon J.C., Molina M.C. Development of a new promoter to avoid the silencing of genes in the production of recombinant antibodies in chinese hamster ovary cells. J. Biol. Eng. 2019;13:59. doi: 10.1186/s13036-019-0187-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muharram G., Le Rhun E., Loison I., Wizla P., Richard A., Martin N., Roussel A., Begue A., Devos P., Baranzelli M.C., et al. Parvovirus H-1 induces cytopathic effects in breast carcinoma-derived cultures. Breast Cancer Res. Treat. 2010;121:23–33. doi: 10.1007/s10549-009-0451-9. [DOI] [PubMed] [Google Scholar]
- 70.Cicin-Sain L., Brune W., Bubic I., Jonjic S., Koszinowski U.H. Vaccination of mice with bacteria carrying a cloned herpesvirus genome reconstituted in vivo. J. Virol. 2003;77:8249–8255. doi: 10.1128/jvi.77.15.8249-8255.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Finn C.E., Chong A., Cooper K.G., Starr T., Steele-Mortimer O. A second wave of Salmonella T3SS1 activity prolongs the lifespan of infected epithelial cells. PLoS Pathog. 2017;13 doi: 10.1371/journal.ppat.1006354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knodler L.A., Nair V., Steele-Mortimer O. Quantitative Assessment of Cytosolic Salmonella in Epithelial Cells. PLoS One. 2014;9 doi: 10.1371/journal.pone.0084681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Forbes N.S., Munn L.L., Fukumura D., Jain R.K. Sparse initial entrapment of systemically injected Salmonella typhimurium leads to heterogeneous accumulation within tumors. Cancer Res. 2003;63:5188–5193. [PubMed] [Google Scholar]
- 74.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mosberg J.A., Lajoie M.J., Church G.M. Lambda red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics. 2010;186:791–799. doi: 10.1534/genetics.110.120782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All raw data reported in this paper have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table. All the original western and microscopy images will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.