

Abstract
This study identified tumorigenic processes most dependent on murine heat shock protein 72 (HSP72) in the mouse mammary tumor virus-PyMT mammary tumor model, which give rise to spontaneous mammary tumors that exhibit HSP72-dependent metastasis to the lung. RNA-seq expression profiling of Hspa1a/Hspa1b (Hsp72) WT and Hsp72−/− primary mammary tumors discovered significantly lower expression of genes encoding components of the extracellular matrix (ECM) in Hsp72 knockout mammary tumors compared to WT controls. In vitro studies found that genetic or chemical inhibition of HSP72 activity in cultured collagen-expressing human or murine cells also reduces mRNA and protein levels of COL1A1 and several other ECM-encoding genes. In search of a possible mechanistic basis for this relationship, we found HSP72 to support the activation of the tumor growth factor-β–suppressor of mothers against decapentaplegic-3 signaling pathway and evidence of suppressor of mothers against decapentaplegic-3 and HSP72 coprecipitation, suggesting potential complex formation. Human COL1A1 mRNA expression was found to have prognostic value for HER2+ breast tumors over other breast cancer subtypes, suggesting a possible human disease context where targeting HSP72 may have a therapeutic rationale. Analysis of human HER2+ breast tumor gene expression data using a gene set comprising ECM-related gene and protein folding-related gene as an input to the statistical learning algorithm, Galgo, found a subset of these genes that can collectively stratify patients by relapse-free survival, further suggesting a potential interplay between the ECM and protein-folding genes may contribute to tumor progression.
Keywords: Heat shock protein 72 (HSP72), HSP70, Extracellular matrix (ECM), Collagen, COL1A1, HER2
Introduction
The stress-inducible heat shock protein 72 (HSP72) has been implicated as an important modulator of tumorigenesis in multiple animal tumor models.1, 2 Consistent with these findings, HSP72 has been reported to regulate numerous tumorigenic processes, including cell growth, cell survival, oncogenic signaling, and overcoming oncogene-induced senescence.2, 3, 4, 5, 6, 7, 8, 9 These properties of HSP72, when considered with the viability of Hsp72−/− (Hspa1a/Hspa1b null) mice, frequent upregulation of HSP72 in human tumor tissue, and the potential for simultaneous inactivation of multiple signaling pathways have made HSP72 an attractive anticancer target.4, 7, 10, 11, 12 We have previously found that Hsp72−/− mice are protected against mammary tumor metastasis driven by the PyMT oncogene, suggesting that in addition to the above-mentioned processes, HSP72 also supports tumor metastasis.1 To gain a further understanding of HSP72′s role in tumorigenesis, we sought to identify the tumorigenic pathways most dependent upon HSP72 and their underlying mechanistic association.
HSP72 is encoded by the stress-inducible human HSPA1A/HSPA1B and mouse Hspa1a/Hspa1b genes of the HSP70 gene/protein family.13 Like other HSP70 family proteins, HSP72 is a molecular chaperone and functions in concert with HSP90 and a series of co-chaperones to coordinate protein folding and protein quality control.14 Previous HSP70 protein interaction studies indicate that HSP72 has many binding partners and subsequently has the potential to regulate the signaling capacity of many cellular processes.14, 15, 16, 17, 18 The mechanisms that mediate HSP72′s regulation of specific signaling pathways continue to be investigated. However, key mechanisms appear to include canonical and noncanonical protein–protein interactions, complex formation with different co-chaperones and their interacting partners, and additional regulatory control by different combinations of post-translational modifications.15, 16, 19, 20 While previous studies have shown HSP72 to be linked to several tumorigenic cellular processes, which of these cellular systems are most dependent on HSP72 and underlie the apparent importance of HSP72 to tumorigenesis remains to be fully understood.
To address the study’s aim of identifying tumorigenic processes most acutely dependent upon HSP72 within the mouse mammary tumor virus (MMTV)-PyMT model, an RNA-seq approach was utilized that quantitated the relative transcript abundance within metastatic primary murine mammary tumors with and without HSP72 expression. The mRNA of proteins that constitute the extracellular matrix (ECM) such as collagen, type I, alpha 1 (Col1a1) were found to be the most significantly altered HSP72-dependent gene signature in murine mammary tumors in vivo. The dependence of human COL1A1 and mouse Col1a1 mRNA expression upon HSP72 was also observed in cultured mammalian cells in vitro. Given the association between altered tumor ECM composition and formation of invasive and metastatic tumors, the identified reduced expression of ECM-related biomolecules, hereinafter referred to as “ECM,” in contexts of inhibited HSP72 activity suggest a potential contributing basis for the limited metastatic potential of Hsp72−/− mammary tumors.1, 21, 22, 23, 24, 25, 26 Analysis of human tumor gene expression datasets using the statistical algorithm, Galgo, found that expression of a subset of genes associated with protein folding and the ECM could stratify HER2+ breast cancer transcriptomic signatures by patient survival, further suggesting a potential interplay of these processes in tumor progression. This is the first study to our knowledge to examine the mechanisms by which HSP72 influences the transcriptomic landscape of tumors and to describe a role for HSP72 in the modulation of tumor “ECM” production in vivo.
Results
HSP72 modulates the expression of mammary tumor “ECM” genes in vivo
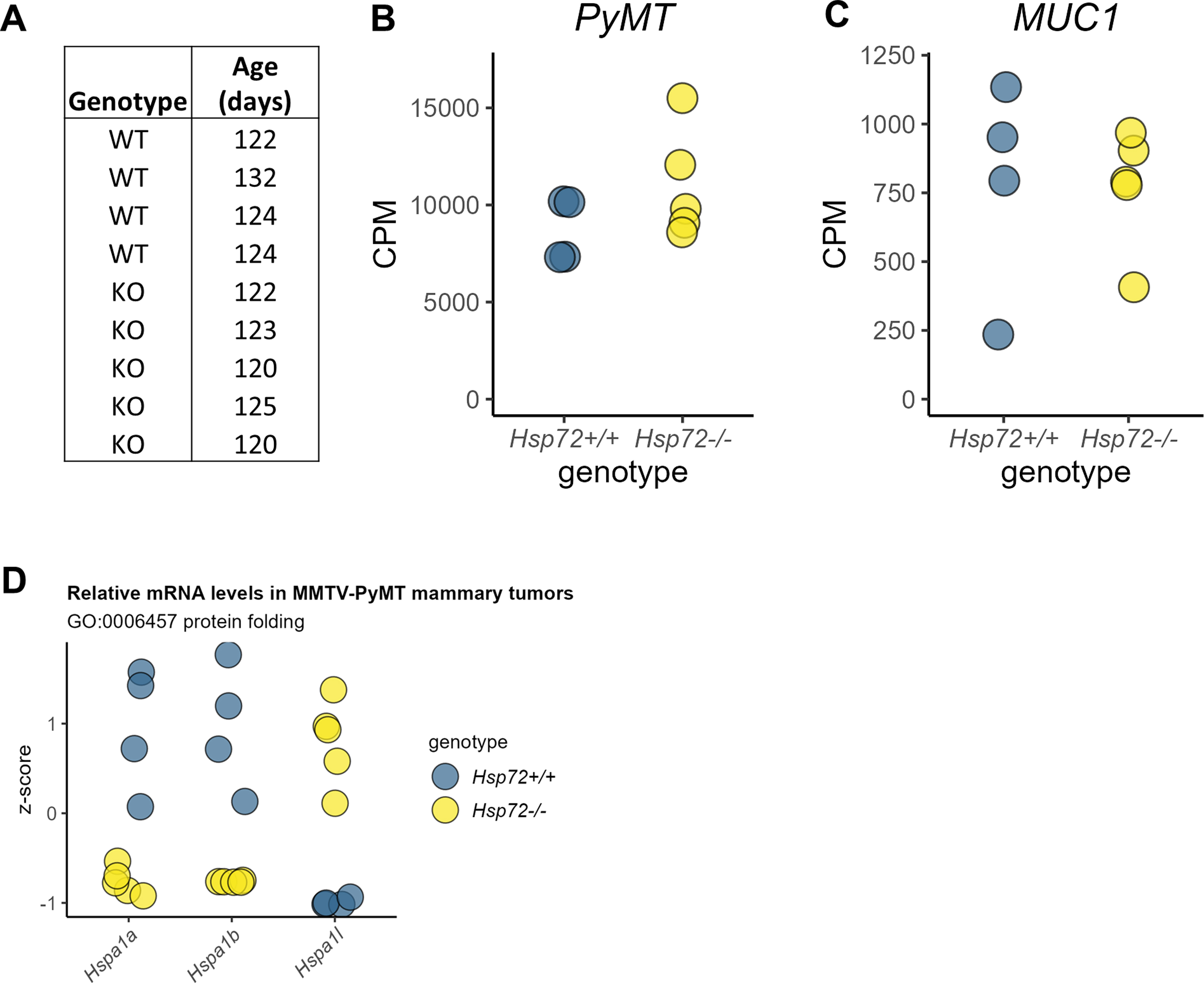
We have previously reported HSP72 to support PyMT-driven mammary tumor metastasis to the lung in the murine MMTV-PyMT mammary tumor model.1 MMTV-PyMT transgenic mice develop multifocal mammary tumors that metastasize to the lung in immune-competent animals.27 PyMT-driven mammary tumors also exhibit several histological and molecular features characteristic of human breast tumors, including stage-wise development of metastatic carcinomas that exhibit loss of estrogen receptor and progesterone receptor expression and expression of HER2 at advanced stages.27, 28, 29, 30 We, therefore, considered this model to be a valuable context in which to study and identify tumorigenic processes supported by HSP72 that may also occur in human tumors. To identify potential processes by which HSP72 supports mammary tumor metastasis in this model, we performed bulk RNA-seq analysis on primary mammary tumors derived from 4 WT and 5 Hsp72−/− animals at approximately 123 days of age (Supplementary Figure 1(a)). Differential gene expression analysis identified 73 genes to be altered between WT and Hsp72−/− mammary tumors (false discovery rate [FDR] < 0.05) (Figure 1(a) and (b), Supplementary Table 4). No significant difference in the expression of the PyMT or human MUC1 transgenes was observed (Supplementary Figure 1(b) and (c)). As expected, tumor samples from HSP72−/− animals had significantly fewer normalized read counts for the Hspa1a and Hspa1b genes (Supplementary Figure 1(d)). Reads aligning to Hspa1a and Hspa1b were found to be limited to the 5′ regions indicating substantial and functional deletion of these genes, illustrated in Lang et al.31 Interestingly, the relative expression of the HSP70 gene family member Hspa1l was significantly increased in Hsp72−/− tumors (FDR < 0.001), suggesting a possible compensatory expression mechanism upon functional deletion of both Hspa1a and Hspa1b (Supplementary Figure 1(d)).
Fig. 1.

HSP72 modulates the expression of “ECM” genes in metastatic mammary tumors. (a) Heatmap representation of the directional change of differentially expressed genes (DEGs) from bulk RNA-seq analysis of Hsp72 WT and Hsp72−/− MMTV-PyMT primary metastatic mammary tumors shown as gene-wise z-scores of TMM-normalized counts per million (CPM) following edgeR differential gene expression analysis (DGE). (b) Volcano plot representation of DEGs representing false discovery rate (FDR) v log2 fold change. The dotted lines indicate FDR 0.05 and log2 fold change 0.6. (c) Gene ontology over-representation analysis applied to the DEGs (FDR < 0.05) using the enrichGO function of the clusterprofiler package. Genes with at least 3 CPM across at least four samples were used as the background gene list. (d) Relative mRNA levels in Hsp72 WT and Hsp72−/− tumors of DEGs belonging to the GO term “extracellular matrix organization.” Values represent gene-wise z-scores across samples based on TMM-normalized CPM. Abbreviations used: BP, biological process; CC, cellular components; GO, gene ontology; HSP72, heat shock protein 72; MF, molecular functions; MMTV, mouse mammary tumor virus.
Over-representation analysis of the differentially expressed genes (DEGs) identified terms associated with ECM-related biological process (BP) and protein folding-related BP, cellular components, and molecular functions to be significantly altered in Hsp72−/− mammary tumors compared to WT tumors (Figure 1(c)). DEGs genes categorized within the gene ontology (GO) BP term “extracellular matrix organization” (GO:0030198) included Col1a1, Col1a2, Col5a1, Col5a2, and Mmp2 (Figure 1(d)). Altered ECM composition and organization are recognized as important progressive factors for several cancers, including breast cancer.22, 24, 25, 26 Of note, altered expression of the ECM component collagen I, has previously been shown to promote lung metastasis in PyMT-driven mammary tumors.24 Given the prominence of the “ECM” signature within the identified DEGs, we hypothesized that altered expression and/or organization of the tumor ECM may be one basis for the differential metastatic potential exhibited by the Hsp72 WT and Hsp72−/− MMTV-PyMT mammary tumors.
Genetic or chemical inhibition of HSP72 decreases collagen expression in vitro
To further investigate a relationship between HSP72 and the expression of collagens and other ECM-related molecules, we next examined the effect of perturbing HSP72 activity on the mRNA and protein levels of “ECM” genes identified to be dependent upon murine HSP72 in MMTV-PyMT murine mammary tumors. We focused on collagen I as it has previously been shown to have an important role in murine mammary tumor metastasis.24 Knockdown of human HSPA1A by stable expression of HSPA1A-targeting shRNAs in cultured Hs578T breast cancer cells was associated with reduced levels of COL1A1 mRNA and COL1A1 protein levels (Figure 2(a) and (b)). Protein levels of COL1A1 were also lower in Hsp72−/− mouse embryonic fibroblasts (MEFs) compared to WT counterparts (Figure 2(c)). Notably, levels of the constitutively expressed HSP family A (Hsp70) member 8 (HSC70/HSPA8) detected by an HSP70 polyclonal antibody were maintained in Hsp72−/− MEFs suggesting the observed effects upon COL1A1 protein expression are specific to HSP72. Several “ECM” mRNAs associated with HSP72 in the MMTV-PyMT murine mammary tumors were also found to have reduced levels in Hsp72−/− MEFs, including Col1a1, Col1a2, Col5a1, Mmp2, and Sparc (Figure 2(d)). We next examined whether chemical inhibitors of HSP72 activity also influenced the expression of “ECM” mRNAs and proteins. The allosteric HSP70 inhibitor, JG98, stabilizes the ADP-bound conformation of HSP70 and perturbs binding of the nucleotide exchange factor BAG family molecular chaperone regulator 3, leading to inhibition of the HSP70 functional cycle of substrate binding and release that is coupled to its ATPase activity.32, 33 JG98 was found to reduce COL1A1 protein levels in Hs578T cells at concentrations in the order of 100 nM (Figure 2(e)). Of note, these effects appeared to be independent of any altered levels of HSP72 (Figure 2(e)). JG98 treatment of Hs578T cells was also observed to reduce mRNA levels of human COL1A1 with a general trend for reduced levels of other “ECM” mRNAs (Figure 2(f)). Considered together, these data indicate that inhibition of HSP72 activity leads to reduced expression of several “ECM” mRNAs and proteins across several experimental systems.
Fig. 2.

Genetic or chemical inhibition of HSP72 decreases collagen expression in vitro. (a) Western blot analysis of Hs578T cell lysates stably expressing HSPA1A-targeting shRNAs or a non-silencing control (shCtrl) showing the relative levels of the indicated proteins. Densitometry analysis represents fold-change in COL1A1 and HSP72 levels normalized to β-ACTIN relative to shRNA ctrl, n = 3. (b) RT-qPCR analysis of Hs578T cell lysates stably expressing HSPA1A-targeting shRNAs or a shCtrl showing the relative levels of the indicated mRNAs normalized to TMEM11, n = 1 biological replicate, 3 technical replicates shown. (c) Western blot analysis of cultured WT and Hsp72−/− immortalized MEF cell lysates. Densitometry analysis shows fold change in COL1A1 levels normalized to β-ACTIN relative to respective WT MEF samples, n = 4. (d) RT-qPCR analysis of cultured WT and Hsp72− immortalized MEFs showing the relative levels of the indicated mRNAs normalized to Tmem11 relative to WT samples, n = 3. (e) Western blot analysis of cell lysates of Hs578T cells treated with a titration of JG98 for 72 h, the relative expression represents values normalized to the DMSO control sample. The number of replicates represented in the densitometry analysis is n = 3 (DMSO), 1 (0.01 µM), 1 (0.1 µM), 1 (0.25 µM), 2 (0.5 µM), 3 (1.0 µM). (f) RT-qPCR analysis of Hs578T cells treated with 1 µM JG98 for 72 h, the relative expression represents values normalized to TMEM11 relative to respective DMSO control samples, n = 3. A one-sample t-test was performed for all data normalized to control samples, *P < 0.05, **P < 0.01, ***P < 0.001. Error bars represent ± SD. Abbreviations used: COL1A1, collagen, type I, alpha 1; HSP72, heat shock protein 72; MEF, mouse embryonic fibroblasts.
HSP72 modulates the fibrogenic tumor growth factor-β pathway
As the data indicated a role for HSP72 in the modulation of mRNA expression of “ECM” genes, we sought to identify a potential relationship between HSP72 and regulatory pathways upstream of “ECM” production. As the tumor growth factor-β (TGF-β)/suppressor of mothers against decapentaplegic (SMAD) pathway is a well-established regulator of “ECM” production and tumor metastasis,34 we investigated a possible relationship between HSP72 and TGF-β/SMAD signaling as one possible mechanism underlying the reduced “ECM” gene expression observed in the Hsp72−/− MMT tumors. At concentrations of JG98 or JG231 observed to reduce COL1A1 protein levels, ~20% lower levels of SMAD2/3 were observed after 24 h of treatment (Figure 3(a)). As JG98 and JG231 also have activity towards HSC70,32 we examined levels of SMAD2/3 in WT and Hsp72−/− MEFs and found Hsp72−/− MEFs have reduced SMAD3 levels compared to WT MEFs despite comparable HSC70 protein levels (Figure 3(b)). Immunoblots using a SMAD3 antibody reactive to phosphorylated S423 and S425, the residues at which SMAD3 is phosphorylated by transforming growth factor beta receptor 1 (TβRI) upon activation by TGF-β,35 indicated reduced phosphorylation of SMAD3 at these sites in Hsp72−/− MEFs cultured in growth media containing 10% fetal bovine serum (FBS) (Figure 3(b)). We next further examined whether HSP72 supports activating phosphorylation of SMAD3 by TβRI in response to recombinant human TGF-βI. Treatment of serum-starved MEFs with TGF-βI led to increased phospho-SMAD3 S423/S425 levels in WT MEFs, however total levels of TGF-βI-induced SMAD3 phosphorylation were markedly lower in Hsp72−/− MEFs (Figure 3(c)). The relative levels of p-SMAD3/total SMAD3 were comparable between TGF-β treated WT and Hsp72−/− MEFs, while total levels of SMAD3 were lower in Hsp72−/− MEFs.
Fig. 3.

HSP72 modulates the fibrogenic TGF-β pathway. (a) Western blot analysis of cultured Hs578T cells treated with the indicated concentrations of JG98 and JG231 for 24 h. The densitometry analysis represents levels of SMAD3 normalized to β-ACTIN relative to the respective DMSO control sample, n = 1. (b) Western blot analysis of cultured WT and Hsp72−/− immortalized MEFs cell lysates showing the relative levels of the immunoblotted proteins. The densitometry analysis represents levels of phospho-SMAD3 S423/S425 normalized to total SMAD3 and total SMAD3 normalized to β-ACTIN as indicated, relative to WT levels, n = 5. Note that a high contrast adjustment was applied to the HSP72 immunoblot for improved clarity. (c) Western blot analysis of cell lysates of immortalized cultured WT and Hsp72−/− MEFs. MEFs were serum starved overnight and then treated with or without 5 ng/mL TGF-βI for 30 min in serum-free media, at which point cell lysates were collected for Western blot analysis. The densitometry analysis represents levels of phospho-SMAD3 S423/S425 normalized to total SMAD3 relative to the non-TGF-βI-treated WT MEF sample, n = 3. (d) Immunoprecipitation (IP) of SMAD3 in HEK293T cells and immunoblot of indicated proteins, n = 1. Note that high contrast was applied to the HSP72 and HOP immunoblots for improved clarity. (e) RT-qPCR analysis of Hs578T cells treated with 1 µM JG98 for 72 h, the relative expression represents values normalized to TMEM11 relative to respective DMSO control samples, n = 3. (f) RT-qPCR analysis of cultured WT and Hsp72−/− immortalized MEFs showing the relative levels of the indicated mRNAs normalized to Tmem11 relative to WT samples, n = 3. (g) RT-qPCR analysis of cultured WT and Hsp72−/− immortalized MEFs showing the relative levels of the indicated mRNAs normalized to Tmem11 relative to WT samples, n = 3 for all but Tgfb3, n = 2. A one-sample t-test was performed for all data normalized to control samples, *P < 0.05, **P < 0.01, ***P < 0.001. Error bars represent ±SD. Abbreviations used: HOP, HSP70/HSP90 organizing protein; HSP72, heat shock protein 72; MEF, mouse embryonic fibroblasts; SMAD3, suppressor of mothers against decapentaplegic-3; TGF-β, tumor growth factor-β.
To further examine the possibility of HSP72 functionally supporting SMAD3 activity, we performed immunoprecipitation (IP) assays to determine whether HSP72 co-precipitates with SMAD3 IP. HSP72 was found to coprecipitate with SMAD3 along with the HSP72 co-chaperone HSP70/HSP90 organizing protein (HOP), also known as stress-induced phosphoprotein 1 (Figure 3(d)).
To investigate the relationship between HSP72 and elements of the TGF-β pathway, we examined TGFB1 and TGFB2 mRNA levels of Hs578T cells when treated with JG98 and observed a statistically significant 50% reduction in TGFB2 mRNA levels in samples treated with the HSP72 inhibitor (Figure 3(e)). Similarly, mouse Tgfb2 mRNA levels were found to be substantially lower in Hsp72−/− MEFs compared to WT MEFs (Figure 3(f)). Taken together, these findings indicate that in addition to supporting levels of phosphorylated SMAD3, HSP72 also supports gene expression of human TGFB2 and mouse Tgfb2.
We next assessed the expression of other mouse gene targets of interest found to mediate altered “ECM” expression (C-X-C motif chemokine ligand 12 (Cxcl12)36) or encode ECM components (Biglycan (Bgn), Lysyl oxidase (Lox)). Each of these mRNAs was found to be reduced in Hsp72−/− MEFs compared to WT MEFs, providing further evidence of a relationship between HSP72 activity and the expression of ECM components (Figure 3(g)). Previously we demonstrated phosphorylation of the oncogene MET proto-oncogene, receptor tyrosine kinase (c-MET) to be reduced in MMTV-PyMT Hsp72−/− mammary tumors with evidence of reduced expression of mouse hepatocyte growth factor (Hgf) mRNA expression.1 Consistent with this finding, we also found Hgf mRNA levels to be substantially reduced in Hsp72−/− MEFs relative to WT MEFs (Figure 3(g)). Thus, while we have provided evidence here for reduced activity of the TGFβ-“ECM” pathway in MMTV-PyMT Hsp72−/− mammary tumors, additional mechanisms including and beyond HGF/c-MET and CXCL12 appear to also be altered and may also contribute to the reduced metastatic potential of MMTV-PyMT Hsp72−/− mammary tumors.
High levels of “ECM” expression are associated with shorter relapse-free survival in HER2+ breast cancer
As altered ECM composition has been identified as an important protumorigenic and prometastatic factor across multiple experimental and clinical studies,22, 24, 25, 26, 37 we next sought to identify a human context in which targeting a protumorigenic ECM, potentially via inhibition of HSP72, may have a therapeutic rationale. As our studies focused on metastatic mammary tumors, we examined the relationship between COL1A1 expression and clinical outcomes across human breast cancers using a publicly available large microarray-based gene expression dataset linked to patient survival.38 Using this approach, we did not identify a significant relationship between COL1A1 expression and relapse-free survival (RFS) across the entire human breast cancer cohort (Figure 4(a)). However, when we examined the relationship of COL1A1 expression with RFS across breast cancer PAM50 subtypes individually, we found that COL1A1 showed a distinctly prognostic relationship for HER2+ breast cancer, where high levels of COL1A1 predicted reduced RFS with a hazard ratio of 1.33, P = 0.021 (Figure 4(b)). To further investigate the relationship between COL1A1 expression and patient outcome for HER2+ breast cancer, in addition to other “ECM” genes, we utilized Galgo. Galgo is a statistical learning algorithm that identifies robust transcriptomic classifier signatures associated with patient survival from gene expression profiles of tumor samples.39 To evaluate the potential prognostic value of the expression of “ECM” and HSP-related genes in HER2+ breast cancer, we applied the Galgo algorithm to transcriptomic and clinical HER2+ breast cancer data using genes associated with the GO terms “GO_EXTRACELLULAR_MATRIX” and “GO_PROTEIN_FOLDING” as filters to identify genes within these gene sets that are most associated with patient outcome in HER2+ breast cancer (Supplementary Figure 2). From a total of 707 HER2+ tumor expression profiles, 472 samples were used as the training set, with a remaining 235 as the test set (Figure 5(a)). Galgo identified a refined gene signature of 102 genes that separated the HER2+ cohort into 2 further subcohorts with distinct risk profiles (Supplementary Table 5). Patient survival analysis shows the refined gene signature identified by Galgo stratifies the HER2+ breast tumor cohort into two subtypes of lower and higher risk that are consistent across both the training and test data sets (Figure 5(b)). Genes within the refined classifier gene signature included genes such as COL1A1, COL1A2, LAMB3, and MMP2 that were found to exhibit dependency upon HSP72 (Fig. 1, Fig. 2). GAGE gene enrichment analysis of higher-risk versus lower-risk transcriptomic signatures demonstrated that ECM-related terms to be featured amongst GO terms increased in the higher-risk HER2 tumors (Figure 5(c)). We next estimated the degree of fibroblast enrichment in the analyzed tumors using the “MCP-Counter” method, which computes the estimated abundance of eight immune and two stromal cell populations (including fibroblasts) in heterogeneous tissues from transcriptomic data.40 Consistent with the enrichment of ECM-encoding genes in the higher-risk HER2+ tumors, the application of the MCP counter algorithm to the distinct gene expression profiles predicted higher-risk HER2+ tumors to have a significantly increased fibroblast signature (Figure 5(d)). Differential expression analysis of transcriptomic profiles classified into high and low risk confirmed higher levels of “ECM” genes in the high-risk HER2+ cohort (Supplementary Table 6). Together, these findings indicate that high levels of “ECM” expression are associated with worse outcomes in HER2+ breast cancer, and that a gene signature combining “ECM” and genes of the protein folding pathway can further stratify high and low-risk HER2 breast tumor expression profiles.
Fig. 4.

High COL1A1 mRNA expression is associated with poor relapse-free survival in HER2+ human breast cancer. (a) Kaplan–Meier plots of relapse-free survival (RFS) for breast cancer patients across all subtypes stratified by COL1A1 mRNA expression assessed by microarray. (b) RFS of HER2+ breast cancer patients exhibiting high or low expression of COL1A1 assessed by microarray. Abbreviations used: COL1A1, collagen, type I, alpha 1; HR, hazard ratio.
Fig. 5.

Gene expression analysis using the Galgo statistical algorithm indicates high levels of “ECM” expression are associated with shorter RFS in HER2 + breast cancer. (a) Heat maps showing the scaled expression of the “ECM”-folding gene and protein-folding gene signature within the training and test datasets. The high (red) and low-risk (blue) classification identified by Galgo is indicated in the top panel above the heat maps. (b) Patient survival analysis of patients classified into the low or high-risk subtypes by the Galgo algorithm. (c) GAGE pathway enrichment analysis indicating the molecular pathways enriched in the low and high-risk subtypes identified by Galgo. (d) MCP-counter analysis of HER2+ breast tumor expression profiles classified as low and high-risk by Galgo showing the estimated fibroblastic infiltration score in the respective tumor samples. Abbreviations used: ECM, extracellular matrix; RFS, relapse-free survival.
Discussion
Our experiments indicate that HSP72 is a key modulator of “ECM” expression in MMT-PyMT mammary tumors and multiple collagen-expressing mammalian cultured cells of different origins. When these findings are considered with past studies that have described altered “ECM” production and organization to be a key prometastatic factor, our studies suggest altered “ECM” production may underlie the reduced metastatic potential of Hsp72−/− MMT mammary tumors.21, 22, 23, 24, 25, 26 Higher levels of tumor “ECM” have been associated with tumor invasion, higher infiltration of macrophages, lower lymphocyte infiltration, increased tumor pressure and vessel compression, poor macromolecule permeation, increased mechanosignaling, hypoxia, and influence responses to chemical inhibition of oncogenic pathways.21, 36, 41, 42 Studies such as these and others that have described tumor desmoplasia to be associated with tumor properties that are adverse to treatment have led to the targeting of tumor desmoplasia as a treatment strategy to promote tumor responses.43, 44 In support of this strategy, past studies have shown anti-desmoplastic approaches to improve lymphocyte infiltration to metastatic mammary xenograft tumors and to sensitize tumors to immune checkpoint blockade in vivo.36 Our findings suggest that targeting HSP72 may provide a novel approach to inhibiting protumorigenic “ECM” production by tumors.
Our analysis of a publicly available gene expression and patient survival database found that COL1A1 expression has distinctly greater prognostic value for HER2+ breast cancer than other breast cancer subtypes. Using the Galgo algorithm, we further identified a selection of “ECM”-related gene and protein folding-related gene that can stratify HER2+ breast cancer patient cohorts for higher and lower risk based on RFS. This analysis further suggests altered “ECM” production to be an important tumorigenic factor for HER2+ breast cancer. When the relationship between the expression of genes encoding ECM components and HSP72 reported here is considered with previous studies that have shown HSP72 to be essential for HER2-driven tumorigenesis in vivo, these observations together suggest targeting HSP72 in HER2+ breast tumors warrants further investigation as a potential treatment strategy.2
We explored the TGF-β pathway as a potential mechanism by which HSP72 may act to support “ECM” production. In support of a possible regulatory role for HSP72 in the TGF-β pathway, we found targeting HSP72 to reduce active levels of SMAD3 in cultured MEFs. HSP72 was also found to coprecipitate with SMAD3 in cultured HEK293T cells, however, the mechanism by which HSP72 supports SMAD3 activity was not determined. Evidence for HOP in a co-complex with HSP72 and SMAD3 led us to pursue a previously described HSP72-HOP-HSP90 facilitated signaling hypothesis, where the association of an HSP72–SMAD3 complex with an HSP90:TβRI complex was hypothesized to facilitate phosphorylation of SMAD3 by TβRI.45, 46 However, this remains to be fully tested by further studies. The mRNA encoding the profibrogenic chemokine CXCL12 was almost quantitatively silenced by Hsp72 gene inactivation, while Tgfb2 mRNA levels were also reduced in Hsp72−/− MEFs, indicating HSP72 to regulate levels of several profibrogenic “ECM” molecules. In addition, mRNA levels encoding HGF, the ligand for oncogene c-MET was reduced in Hsp72−/− MEFs. Our previous studies had shown that phospho-c-MET was almost totally depleted from Hsp72−/− MMT tumors. In these tumors, phospho-c-MET was found exclusively in clusters enriched in cells bearing cancer stem cell markers, indicating a role for an HSP72 > HGF > c-MET pathway in tumor initiation and metastasis.1 While we found supportive evidence for HSP72 to have regulatory activity upon the upstream TGF-β–SMAD3 pathway as a potential mechanism connecting HSP72 to transcriptional regulation of “ECM” genes, the level at which HSP72 activity influences the expression of “ECM” mRNAs remains to be investigated further. Given that HSP72 interacts with many different proteins and RNA species,47 both protein-based regulatory mechanism and RNA-based regulatory mechanism are possible methods by which HSP72 may regulate the expression of “ECM” genes. Collectively, these findings suggest that HSP72 may have a key role in the capacity of cancer-associated fibroblasts to produce protumorigenic “ECM”-stimulatory molecules as well as the “ECM” molecules themselves.
We found that inhibition of “ECM” expression upon genetic deletion of Hsp72 could not be compensated for by the major constitutive HSP72 paralog HSC70. This would suggest that modulation of “ECM” production is a somewhat exclusive pro-oncogenic property of HSP72. Our findings add to previous studies that have identified roles for HSP72 and other components of the heat shock response system to have roles in ECM regulation. For example, previous studies have linked HSP72 and the HSP72 co-chaperone BAG family molecular chaperone regulator 3 to collagen expression.48, 49 The major regulator of stress-induced HSP72 expression, heat shock factor 1, has also been linked to “ECM” production and/or ECM organization by multiple studies.50, 51, 52 Our findings suggest one mechanism by which heat shock factor 1 may influence “ECM” expression is potentially through its regulation of HSP72 expression. Of note, we also reported three miRNAs that are predicted to target both HSP and “ECM” mRNAs, suggesting the potential for regulatory mechanisms that dually regulate both “ECM” production and expression of HSPs.53 While the initial tumorigenic processes found to be regulated by HSP72 were largely intracellular, our findings further contribute to the increasing evidence of regulatory roles for HSPs acting at the level of the tumor microenvironment.
We have reported evidence to indicate that HSP72 has a key role in supporting “ECM” production within MMT tumors. Based on studies that have linked desmoplasia to tumorigenesis, we suggest this role of HSP72 may be a basis for the reduced metastatic potential observed in MMT Hsp72−/− mice.1 While we set out to determine which tumorigenic processes are most dependent on HSP72, and/or underlie HSP72's importance for tumorigenesis, this may prove to be dependent on the genetic context of the tumor and the related processes driving tumorigenesis. For example, a previous study reported that the apparent importance of HSP72 for HER2-driven tumorigenesis was overcoming oncogene-induced senescence.2 Contexts where multiple HSP72-dependent protumorigenic processes converge simultaneously, such as the apparent importance of HSP72 for HER2-driven mammary tumors and “ECM” production, and the apparent association of “ECM” production with poor outcome for HER2+ breast cancer, may prove to be the most suitable for targeting HSP72 as an anticancer treatment strategy.
Methods
Cell culture
Hs578T (ATCC, HTB-126) cells were cultured in DMEM (Gibco, 11965-092) with 10% FBS (Gibco, 10082-147) supplemented with 0.01 mg/mL human insulin (Sigma, I9278). HEK293T cells (GenHunter, Q401) were cultured in DMEM + 10% FBS. Hsp72 (Hspa1a/Hspa1b) WT and Hsp72−/− MEFs were established in a previous study11 and cultured in DMEM supplemented with 1× non-essential amino acids (Gibco, 11140-050). All cell cultures were maintained at 37 °C in a humidified atmosphere and 5% CO2.
Chemicals, plasmids, and reagents
JG98 and JG231 were produced by the Jason Gestwicki Laboratory.32, 33 JG98 was also sourced from Selleckchem (cat. no. S6721). These compounds were solubilized in DMSO (Sigma, D2650), aliquoted to minimize freeze-thaw, and stored at −80 °C or −20 °C. Recombinant human TGF-β1 (R&D Systems, 7754-BH/CF) was solubilized in reconstitution buffer (R&D Systems, RB04) at 100 µg/mL, aliquoted, and stored at −80 °C.
Animals
Animal studies were carried out in accordance with the animal protocols approved by the Institutional Animal Care and Use Committee of Boston University Medical Center. Hspa1a/Hspa1b−/− (Hsp72−/−) C57BL/6 MUC1. Tg MMTV-PyMT (MMT) animals were generated by crossing Hspa1a/Hspa1b−/− (Hsp72−/−) C57BL/6 mice,11 with C57BL/6 mice double transgenic for the polyomavirus middle T antigen (MMTV) driven by the MMTV-long terminal repeat and the human MUC1 transgenes (a kind gift from Sandra J Gendler, Mayo Clinic, Scottsdale, AZ, USA) as previously described.1
RNA-seq analysis
RNA-seq analysis was performed using a previously described workflow.31 MMT Hsp72+/+ and Hsp72−/− female mice were housed for approximately 122 days (Supplementary Figure 1(a)), at which time RNA was isolated from spontaneous mammary tumors. Mammary tumors were excised from euthanized animals, dissected into small 1–3 mm pieces, and transferred to a 50 mL tube containing 2 mL serum-free DMEM. A collagenase I/1× trypsin solution was added (3 mg/mL collagenase I) to make a 5 mL solution that was then incubated in a water bath at 37 °C for 35–50′ to facilitate cell dissociation. The mammary tumor solution was then mashed through a 100 µm nylon cell strainer into a 50 mL centrifuge tube. The cell dissociate solution was then made to 50 mL with DMEM/10% FBS, and dissociated cells were pelleted by centrifugation. The supernatant was discarded, and the cell pellet was transferred to a microcentrifuge tube with 1 mL DMEM/10% FBS. Cells were pelleted at 4 °C and then washed in ice-cold 1× PBS. The cells were again pelleted, and the supernatant discarded, cell pellets were resuspended in 700 µL of Qiazol lysis buffer and homogenized by passing through a Qiashredder column. RNA was extracted as per the miRNeasy kit protocol (Qiagen). Purified RNA samples were stored at −80 °C until the full sample set was collected. RNA integrity of samples was assessed by Bioanalyzer (Agilent) with samples of RIN ≥6.5 used for cDNA library synthesis. cDNA library synthesis was performed on ribosome RNA-depleted total RNA using a KAPA Stranded RNA-Seq with RiboErase (KAPA Biosystems, KK8483) and Illumina adapters. cDNA libraries were pooled and sequenced with an Illumina HiSeq 2500 (50 cycles, paired-end) to give 10–20 million reads per sample. RNA integrity analysis, cDNA library pooling, and NGS services were provided by Harvard Biopolymers Facility. Raw RNA-seq data was processed in a high-performance computing environment using Trimmomatic 0.36 to remove adapters and filter for high-quality reads.54 Read quality was confirmed using FastQC 0.11.5,55 and aligned to the Mus musculus annotated genome GRCm38 (release 103),56 using STAR 2.5.4a aligner with the --quantMode GeneCounts option.57 Read alignment and feature counts assigned to the PyMT (coordinates sourced from J02288.1) and human MUC1 (Gene ID 4582) transgenes were included in the workflow by addition of the PyMT and MUC1 coding sequences to the GRCm38 primary assembly genome files created with STAR --runMode genomeGenerate using an edited Gencode vM25 annotation file to include appropriate coordinates for the two transgenes. Using R 4.1.0, feature counts were then filtered for features with at least 3 counts per million across 4 tumor samples, which gave 13,386 remaining features. Feature counts were then TMM-normalized and DEGs identified and fold changes quantified using the exactTest function within the edgeR statistical package.58 GO analysis was applied to the DEG list using the clusterprofiler and msigdbr packages,59, 60 which included the usage of the enrichGO function and the org.Mm.eg.db mouse database61 for over-representation analysis, with a selection of BP, molecular function, and cellular component ontologies. RNA-seq data from this study has been deposited in NCBI's Gene Expression Omnibus62 and is accessible through GEO Series accession number GSE255056 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE255056).
RT-qPCR analysis
RNA samples from cultured cells were isolated using the RNeasy kit (Qiagen, 74104) with QIAshredder columns (Qiagen, 79654) for homogenization. RNA was quantified with a Nanodrop spectrophotometer (ThermoFisher Scientific), and 1 µg of RNA was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad, 1708891) according to the manufacturer’s instructions. cDNA was diluted to 5 ng/µL with nuclease-free water, and 10 ng of cDNA was quantified by qPCR. Assays were run in a MicroAmp Fast Optical 96-Well Reaction Plate (Applied Biosystems, 4346906) using the PowerUP SYBR Green Master Mix (Applied Biosystems, A25742) and primers (Supplementary Table 1) at a final reaction concentration of 0.5 µM. Primers were sourced from cited references, designed using Primer-BLAST, or selected from PrimerBank,63 and the sequences verified for the predicted target using Primer-BLAST to the Homo sapiens or M musculus RefSeq databases. Amplification was performed using a StepOne Plus Instrument or 7500 Real Time PCR System (Applied Biosystems) with the cycling conditions 50 °C for 2 min, 95 °C for 2 min, [95 °C for 3 s, 60 °C for 30 s] for 40 cycles. Amplification efficiency and Cq values were determined using the LinRegPCR software.64 PCR reactions were carried out in triplicate, and mean values were calculated with the mean ± SD of the triplicates presented.
Gene expression analysis using the Galgo algorithm
For the analysis of expression profiles obtained from microarray technology, normalized Log2 values of fluorescence intensity were used to obtain relative gene expression values. For each hybridization probe, the corresponding Entrez GeneID was used to identify genes when available, and if not available, probe IDs were programmatically matched to their corresponding Entrez GeneID according to the annotations provided by the manufacturers. When several probes corresponded to the same gene, the probe with the highest variance in the cohort was considered as representative of the gene. Finally, when training the Galgo algorithm, only genes that were present in all the platforms evaluated were used.39
In all cases, progression-free survival was used as the clinical endpoint, i.e., the time from diagnosis to the documentation of disease progression or death. This approach was used as some studies reported RFS and others, overall survival data. Considering the small percentage of HER2 patients in the Breast Cancer population (between 10% and 20%), to train Galgo, gene expression data from a total of 4608 patients from 13 different cohorts was compiled from the Synapse platform (for the METABRIC and OSLO cohorts, https://www.synapse.org) and from the “MetaGxBreast” package (for the rest of the cohorts, https://bioconductor.org/packages/release/data/experiment/html/MetaGxBreast.html) (detailed in Supplementary Table 2). Once the data was obtained, the samples were classified into the different molecular subtypes according to the PAM50 algorithm,65 obtaining a total of 707 samples classified as HER2.
Using the “msigdbr” package,59 a list of genes associated with the GO terms “GO_PROTEIN_FOLDING” and “GO_EXTRACELLULAR_MATRIX” was compiled, which totaled 442 genes associated with these molecular pathways (Supplementary Table 3). Using this gene set of ECM-related genes and protein folding-related genes, the Galgo algorithm was seeded with 100 initial random solutions and run for 100 generations. To train the model, the transcriptomic data was filtered by the target genes and the population of HER2 patients was split into a training (472 samples) and a test set (235 samples). After running Galgo with the 442 genes associated with protein folding processes or “ECM,” two HER2-ECM subtypes with high and low survival risk were obtained. To visualize the survival trends of the patient subgroups according to the HER2-ECM signature, Kaplan–Meier plots were plotted for the training set and the test sets, and the log-rank test was used to assess the significance of the separation between the curves. Differential expression analysis was applied to the identified high and low-risk cohorts using edgeR.58
To evaluate the different molecular pathways associated with the different HER2-ECM subtypes, a GSEA analysis was performed.66 Due to its ability to test for coordinated alteration on a predefined set of functionally related genes, the GAGE method was used using 17,202 gene sets derived from GO terms.67 From this analysis, the up-regulated and down-regulated sets for each of the HER2-ECM subtypes analyzed in the training sets were obtained, which allowed the most relevant molecular pathways associated with the different subtypes to be identified. Fibroblast tumor infiltration was estimated for each of the HER2-ECM subtypes identified by Galgo using the “MCP-Counter” method.40 The relative abundance of fibroblasts in each of the samples was estimated, and the estimated values were compared between the different HER2-ECM subtypes using a Student's t-test.
Western blot analysis
Cell lysates were prepared by removing culture media and washing two times with ice-cold PBS. RIPA lysis buffer (Boston Bioproducts, BP-115) was added containing a Halt Protease & Phosphatase inhibitor cocktail (Pierce, 78441). Cell lysates were clarified by centrifugation in a benchtop centrifuge at 15,000g and the protein content within supernatants was quantified using BCA Protein Assay Kit (Pierce, 23227). Equal protein mass across cell lysate samples (typically 10–15 µg) was prepared for SDS/PAGE by adding LDS Sample Buffer (GenScript, M00676-10) containing 10% β-mercaptoethanol (Sigma, M3148) and heating samples at 70 °C for 10′. Samples were then run on SurePAGE, Bis-Tris, 4–12% gradient gels (GenScript, M00654) with MOPS running buffer (GenScript, M00138) for 40–50′ at 200 V with the inclusion of Chameleon Duo Pre-stained Protein Ladder (LI-COR, 928-60000). Proteins were transferred to Immobilon-FL PVDF Transfer membrane (Merck Millipore, IPFL00010) at 4 °C by electrophoresis using 1× Transfer buffer (Boston Bioproducts, BP-190) at 100 V for 45′ when detecting small-mid size proteins or 90′ for larger proteins (~120 kDa). Membranes were blocked with Intercept Blocking Buffer (LI-COR, 927-60001) at RT for 1 h. Primary antibodies were added overnight in a blocking buffer with 0.2% Tween-20 (Boston Bioproducts, P-934). Unbound primary antibodies were washed away with four 5′ washes of 1× TBST (Boston Bioproducts, IBB-181X) and secondary antibodies IRDye 800CW Goat anti-Rabbit (LI-COR, 926-32211) and IRDye 680RD Goat anti-Rabbit (LI-COR, 926-68070) added for 30′ at RT in blocking buffer with 0.2% Tween-20% and 0.02% SDS (Boston Bioproducts, BM-230). Unbound secondary antibody was removed with four 5′ washes of 1× TBST and one wash of 1× TBS (Boston Bioproducts, BM-301X). Membranes were imaged using an Odyssey CLx instrument (LI-COR) on auto-mode, and signals were quantified using ImageStudio (LI-COR). When necessary, membranes were stripped with NewBlot PVDF Stripping Buffer (LI-COR, 928-40032). The primary antibodies used in this study were β-ACTIN (Sigma, A5441), COL1A1 (Cell Signaling Technology (CST), 84336 & 91144S), COL1A2 (Santa Cruz Biotechnology, sc-376350), HOP (Thermo Fisher, PA5-21473), HSP72 (Stressmarq, SMC-100), HSP70 polyclonal (Novus Biologicals, NBP2-46806), SMAD2/3 (Santa Cruz, sc-133098), phospho-SMAD3 S423/S425 (CST 9520, Abcam ab25903).
IP and protein interaction assays
HEK293T cells were grown on 10 cm culture dishes to 90% confluency with DMSO (1:18,710 dilution) at which time media was removed from the dishes and cells were washed twice with ice-cold PBS and lysed in HEPES buffer with CHAPS (Boston Bioproducts, BP-453) with 1× Halt protease and phosphatase inhibitors (Pierce, 78441) (IP buffer). Lysates were clarified by centrifugation at 13,000g on a benchtop centrifuge and lysate protein content was quantified using the BCA method (Pierce, 23227). Exactly 1000 µg of protein was added to each IP in microcentrifuge tubes and made to 500 µL with IP buffer. For 1% input, 5 µL of IP lysate preparations were stored at −20 °C. Equal amounts (0.5 µg) of antibodies, SMAD3 (CST9523S) and Rabbit IgG control (CST2729) were added to each respective IP and incubated overnight with rotation at 4 °C. The next day, 25 µL of Protein A/G Magnetic Beads (Pierce, 88802) were added to each IP and incubated at RT for 1 h with rotation. Immunoprecipitate was isolated using a magnetic rack, and non-bound material was discarded. The immunoprecipitate was then washed 3 times for 5′ with rotation at RT with IP buffer. Immunoprecipitates were eluted off the Protein A/G magnetic beads by the addition of 40 µL of LDS buffer with 10% β-mercaptoethanol (Sigma, M3148) and incubation at RT for 10′ with occasional vortex mixing. The eluate was collected and analyzed by Western blot using the above protocol with 15 µL of eluate added to the SDS/PAGE gels.
Lentiviral shRNA
Stable knockdown of HSP72 in Hs578T cells was achieved by transduction of lentiviral expression vectors targeting the coding region of human HSPA1A (NM_005345.5) mRNA (Genecopoeia, cat. No. HSH009077-33-LVRU6P, HSH009077-34-LVRU6P). To generate lentivirus, HEK293T cells (GenHunter, Q401) were transfected with a mix of the packaging vectors 2.5 µg psPAX2 and 2.5 µg pMD2. G (Addgene), and with 5 µg of a human HSPA1A-targeted (shHSP72-33 or shHSP72-34) vector or a non-silencing control shRNA transgene vector using the Fugene 6 transfection reagent. The morning following transfection, cells were gently washed with 1× PBS, and the growth media was replenished. Lentivirus-conditioned medium was then collected 24 h later, passed through a 0.45 µm filter (Millipore, SLHP033RS), and stored at −80 °C. Cultured Hs578T cells were then transduced with the lentivirus with polybrene. Transduced cells were selected with the inclusion of puromycin (Invivogen, ANT-PR-1) in the culture media.
Statistical analysis
A Student’s t-test was applied for statistical inference between paired conditions. A one-sample t-test was applied for statistical inference to normalized values. P-values < 0.05 were considered statistically significant. Microsoft Excel and R 4.1.0 were used for statistical testing.
Declarations of interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Jason E. Gestwicki has patent (related to HSP70 inhibitors) issued to Regents of the University of California. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors wish to thank Jianlin Gong for providing the MMT primary tumor samples used for this study. The authors wish to thank Clayton Hunt for kindly reviewing the manuscript and for providing the HSP72 WT and HSP72−/− MEF cells. The authors wish to thank Eva Csizmadia for her valuable support of animal studies. Synthesis of HSP72-targeting chemical compounds was supported by grant no. NS059690 (J. Gestwicki). The authors wish to thank our colleagues Mary Ann Stevenson, Ayesha Murshid, Thiago Borges, Michael Sherman, Thomas Prince, Robert Matts, John Price, Vera Evtimov, Takanori Eguchi, Vanessa Bret-Mounet, and Yu Jing J. Heng for their support and discussion of this work. The authors wish to thank Shelli McAlpine, Didier Picard, and Adrienne Edkins for kindly providing research materials.
Footnotes
Supplementary data associated with this article can be found online at doi:10.1016/j.cstres.2024.04.006.
Contributor Information
Benjamin J. Lang, Email: ben.lang.170@gmail.com.
Stuart K. Calderwood, Email: stukcalderwood@gmail.com.
Appendix A. Supplementary material
Supplementary material.
.
Supplementary material.
.
Supplementary material.
.
Supplementary material.
.
Supplementary material.
.
(a) Ages of animals at the time of tumor RNA sample collection. (b) Relative mRNA levels of the PyMT oncogene in mammary tumors of Hsp72+/+ and Hsp72−/− animals in TMM-normalized counts per million (CPM). (c) Relative mRNA levels of the human MUC1 gene in mammary tumors of Hsp72+/+ and Hsp72−/− animals in TMM-normalized CPM. (d) Relative tumor mRNA levels of DEGs associated with the GO term “protein folding” by genotype. Values represent gene-wise z-scores across samples based on TMM-normalized CPM. Abbreviations used: DEGs, differentially expressed genes; GO, gene ontology.
{kind=link}
.
Workflow of analysis of HER2+ human breast tumor gene expression data and application of the Galgo algorithm using a gene set input comprised of genes related to protein folding and the extracellular matrix.
{kind=link}
.
Data availability statement
Data will be made available on request.
References
- 1.Gong J., Weng D., Eguchi T., et al. Targeting the hsp70 gene delays mammary tumor initiation and inhibits tumor cell metastasis. Oncogene. 2015;34:5460–5471. doi: 10.1038/onc.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meng L., Hunt C., Yaglom J.A., Gabai V.L., Sherman M.Y. Heat shock protein Hsp72 plays an essential role in Her2-induced mammary tumorigenesis. Oncogene. 2011;30:2836–2845. doi: 10.1038/onc.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beere H.M., Wolf B.B., Cain K., et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 4.Budina-Kolomets A., Balaburski G.M., Bondar A., Beeharry N., Yen T., Murphy M.E. Comparison of the activity of three different HSP70 inhibitors on apoptosis, cell cycle arrest, autophagy inhibition, and HSP90 inhibition. Cancer Biol Ther. 2014;15:194–199. doi: 10.4161/cbt.26720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cesa L.C., Shao H., Srinivasan S.R., et al. X-linked inhibitor of apoptosis protein (XIAP) is a client of heat shock protein 70 (Hsp70) and a biomarker of its inhibition. J Biol Chem. 2018;293:2370–2380. doi: 10.1074/jbc.RA117.000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabai V.L., Yaglom J.A., Waldman T., Sherman M.Y. Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol Cell Biol. 2009;29:559–569. doi: 10.1128/MCB.01041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lang B.J., Guerrero-Gimenez M.E., Prince T.L., Ackerman A., Bonorino C., Calderwood S.K. Heat shock proteins are essential components in transformation and tumor progression: cancer cell intrinsic pathways and beyond. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20184507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powers M.V., Clarke P.A., Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell. 2008;14:250–262. doi: 10.1016/j.ccr.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Yaglom J.A., Gabai V.L., Sherman M.Y. High levels of heat shock protein Hsp72 in cancer cells suppress default senescence pathways. Cancer Res. 2007;67:2373–2381. doi: 10.1158/0008-5472.CAN-06-3796. [DOI] [PubMed] [Google Scholar]
- 10.Ciocca D.R., Calderwood S.K. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones. 2005;10:86–103. doi: 10.1379/csc-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunt C.R., Dix D.J., Sharma G.G., et al. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol Cell Biol. 2004;24:899–911. doi: 10.1128/MCB.24.2.899-911.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yun C.W., Kim H.J., Lim J.H., Lee S.H. Heat shock proteins: agents of cancer development and therapeutic targets in anti-cancer therapy. Cells. 2019;9 doi: 10.3390/cells9010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kampinga H.H., Hageman J., Vos M.J., et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lang B.J., Guerrero M.E., Prince T.L., Okusha Y., Bonorino C., Calderwood S.K. The functions and regulation of heat shock proteins; key orchestrators of proteostasis and the heat shock response. Arch Toxicol. 2021;95:1943–1970. doi: 10.1007/s00204-021-03070-8. [DOI] [PubMed] [Google Scholar]
- 15.Colvin T.A., Gabai V.L., Gong J., et al. Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014;74:4731–4740. doi: 10.1158/0008-5472.CAN-14-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nitika, Zheng B., Ruan L., et al. Comprehensive characterization of the Hsp70 interactome reveals novel client proteins and interactions mediated by posttranslational modifications. PLoS Biol. 2022;20 doi: 10.1371/journal.pbio.3001839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryu S.W., Stewart R., Pectol D.C., et al. Proteome-wide identification of HSP70/HSC70 chaperone clients in human cells. PLoS Biol. 2020;18 doi: 10.1371/journal.pbio.3000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willmund F., del Alamo M., Pechmann S., et al. The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell. 2013;152:196–209. doi: 10.1016/j.cell.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson O.T., Gestwicki J.E. Multivalent protein-protein interactions are pivotal regulators of eukaryotic Hsp70 complexes. Cell Stress Chaperones. 2022;27:397–415. doi: 10.1007/s12192-022-01281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nitika, Porter C.M., Truman A.W., Truttmann M.C. Post-translational modifications of Hsp70 family proteins: expanding the chaperone code. J Biol Chem. 2020;295:10689–10708. doi: 10.1074/jbc.REV120.011666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acerbi I., Cassereau L., Dean I., et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol. 2015;7:1120–1134. doi: 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naba A., Clauser K.R., Lamar J.M., Carr S.A., Hynes R.O. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. Elife. 2014;3 doi: 10.7554/eLife.01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Provenzano P.P., Eliceiri K.W., Campbell J.M., Inman D.R., White J.G., Keely P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Provenzano P.P., Inman D.R., Eliceiri K.W., et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schedin P., Keely P.J. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb Perspect Biol. 2011;3:a003228. doi: 10.1101/cshperspect.a003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L., Wang L., Yang H., Li C., Fang C. Identification of potential genes related to breast cancer brain metastasis in breast cancer patients. Biosci Rep. 2021;41 doi: 10.1042/BSR20211615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guy C.T., Cardiff R.D., Muller W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954-961.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Attalla S., Taifour T., Bui T., Muller W. Insights from transgenic mouse models of PyMT-induced breast cancer: recapitulating human breast cancer progression in vivo. Oncogene. 2021;40:475–491. doi: 10.1038/s41388-020-01560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin E.Y., Jones J.G., Li P., et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weng D., Penzner J.H., Song B., Koido S., Calderwood S.K., Gong J. Metastasis is an early event in mouse mammary carcinomas and is associated with cells bearing stem cell markers. Breast Cancer Res. 2012;14 doi: 10.1186/bcr3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lang B.J., Holton K.M., Gong J., Calderwood S.K. A workflow guide to RNA-seq analysis of chaperone function and beyond. Methods Mol Biol. 2018;1709:233–252. doi: 10.1007/978-1-4939-7477-1_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X., Srinivasan S.R., Connarn J., et al. Analogs of the allosteric heat shock protein 70 (Hsp70) inhibitor, MKT-077, as anti-cancer agents. ACS Med Chem Lett. 2013;4:1042–1047. doi: 10.1021/ml400204n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shao H., Li X., Moses M.A., et al. Exploration of benzothiazole rhodacyanines as allosteric inhibitors of protein-protein interactions with heat shock protein 70 (Hsp70) J Med Chem. 2018;61:6163–6177. doi: 10.1021/acs.jmedchem.8b00583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hao Y., Baker D., Ten Dijke P. TGF-beta-mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20112767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X., Sun Y., Constantinescu S.N., Karam E., Weinberg R.A., Lodish H.F. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc Natl Acad Sci U SA. 1997;94:10669–10674. doi: 10.1073/pnas.94.20.10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen I.X., Chauhan V.P., Posada J., et al. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc Natl Acad Sci USA. 2019;116:4558–4566. doi: 10.1073/pnas.1815515116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robertson C. The extracellular matrix in breast cancer predicts prognosis through composition, splicing, and crosslinking. Exp Cell Res. 2016;343:73–81. doi: 10.1016/j.yexcr.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Gyorffy B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput Struct Biotechnol J. 2021;19:4101–4109. doi: 10.1016/j.csbj.2021.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guerrero-Gimenez M.E., Fernandez-Munoz J.M., Lang B.J., et al. Galgo: a bi-objective evolutionary meta-heuristic identifies robust transcriptomic classifiers associated with patient outcome across multiple cancer types. Bioinformatics. 2020;36:5037–5044. doi: 10.1093/bioinformatics/btaa619. [DOI] [PubMed] [Google Scholar]
- 40.Becht E., Giraldo N.A., Lacroix L., et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanker A.B., Estrada M.V., Bianchini G., et al. Extracellular matrix/integrin signaling promotes resistance to combined inhibition of HER2 and PI3K in HER2(+) breast cancer. Cancer Res. 2017;77:3280–3292. doi: 10.1158/0008-5472.CAN-16-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Netti P.A., Berk D.A., Swartz M.A., Grodzinsky A.J., Jain R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 43.Jain R.K. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205–2218. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nandi T., Pradyuth S., Singh A.K., Chitkara D., Mittal A. Therapeutic agents for targeting desmoplasia: current status and emerging trends. Drug Discov Today. 2020 doi: 10.1016/j.drudis.2020.09.008. [DOI] [PubMed] [Google Scholar]
- 45.Lang B.J., Prince T.L., Okusha Y., Bunch H., Calderwood S.K. Heat shock proteins in cell signaling and cancer. Biochim Biophys Acta Mol Cell Res. 2022;1869 doi: 10.1016/j.bbamcr.2021.119187. [DOI] [PubMed] [Google Scholar]
- 46.Wrighton K.H., Lin X., Feng X.H. Critical regulation of TGFbeta signaling by Hsp90. Proc Natl Acad Sci USA. 2008;105:9244–9249. doi: 10.1073/pnas.0800163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leone S., Srivastava A., Herrero-Ruiz A., et al. HSP70 binds to specific non-coding RNA and regulates human RNA polymerase III. Mol Cell. 2024;84:687–701. doi: 10.1016/j.molcel.2024.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Du F., Li S., Wang T., et al. BAG3 regulates ECM accumulation in renal proximal tubular cells induced by TGF-beta1. Am J Transl Res. 2015;7:2805–2814. [PMC free article] [PubMed] [Google Scholar]
- 49.Shin J.U., Lee W.J., Tran T.N., Jung I., Lee J.H. Hsp70 knockdown by siRNA decreased collagen production in keloid fibroblasts. Yonsei Med J. 2015;56:1619–1626. doi: 10.3349/ymj.2015.56.6.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dhanani K.C.H., Samson W.J., Edkins A.L. Fibronectin is a stress responsive gene regulated by HSF1 in response to geldanamycin. Sci Rep. 2017;7 doi: 10.1038/s41598-017-18061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Levi-Galibov O., Lavon H., Wassermann-Dozorets R., et al. Heat shock factor 1-dependent extracellular matrix remodeling mediates the transition from chronic intestinal inflammation to colon cancer. Nat Commun. 2020;11:6245. doi: 10.1038/s41467-020-20054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scherz-Shouval R., Santagata S., Mendillo M.L., et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158:564–578. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okusha Y., Guerrero-Gimenez M.E., Lang B.J., et al. MicroRNA-570 targets the HSP chaperone network, increases proteotoxic stress and inhibits mammary tumor cell migration. Sci Rep. 2022;12 doi: 10.1038/s41598-022-19533-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andrews S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 56.Frankish A., Diekhans M., Ferreira A.M., et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dobin A., Davis C.A., Schlesinger F., et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dolgalev I. msigdbr: MSigDB Gene Sets for Multiple Organisms in a Tidy Data Format. R package version 7.4.1. https://CRAN.R-project.org/package=msigdbr.
- 60.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carlson M. org.Mm.eg.db: Genome wide annotation for Mouse. R package version 3.14.0.
- 62.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang X., Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003;31 doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ruijter J.M., Ramakers C., Hoogaars W.M., et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37 doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parker J.S., Mullins M., Cheang M.C., et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Subramanian A., Tamayo P., Mootha V.K., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo W., Friedman M.S., Shedden K., Hankenson K.D., Woolf P.J. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009;10:161. doi: 10.1186/1471-2105-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.
Supplementary material.
Supplementary material.
Supplementary material.
Supplementary material.
(a) Ages of animals at the time of tumor RNA sample collection. (b) Relative mRNA levels of the PyMT oncogene in mammary tumors of Hsp72+/+ and Hsp72−/− animals in TMM-normalized counts per million (CPM). (c) Relative mRNA levels of the human MUC1 gene in mammary tumors of Hsp72+/+ and Hsp72−/− animals in TMM-normalized CPM. (d) Relative tumor mRNA levels of DEGs associated with the GO term “protein folding” by genotype. Values represent gene-wise z-scores across samples based on TMM-normalized CPM. Abbreviations used: DEGs, differentially expressed genes; GO, gene ontology.
Workflow of analysis of HER2+ human breast tumor gene expression data and application of the Galgo algorithm using a gene set input comprised of genes related to protein folding and the extracellular matrix.
Data Availability Statement
Data will be made available on request.