

Abstract
The global decline in malaria has stalled1, emphasizing the need for vaccines that induce durable sterilizing immunity. Here we optimized regimens for chemoprophylaxis vaccination (CVac), for which aseptic, purified, cryopreserved, infectious Plasmodium falciparum sporozoites (PfSPZ) were inoculated under prophylactic cover with pyrimethamine (PYR) (Sanaria PfSPZ-CVac(PYR)) or chloroquine (CQ) (PfSPZ-CVac(CQ))—which kill liver-stage and blood-stage parasites, respectively—and we assessed vaccine efficacy against homologous (that is, the same strain as the vaccine) and heterologous (a different strain) controlled human malaria infection (CHMI) three months after immunization (https://clinicaltrials.gov/, NCT02511054 and NCT03083847). We report that a fourfold increase in the dose of PfSPZ-CVac(PYR) from 5.12 × 104 to 2 × 105 PfSPZs transformed a minimal vaccine efficacy (low dose, two out of nine (22.2%) participants protected against homologous CHMI), to a high-level vaccine efficacy with seven out of eight (87.5%) individuals protected against homologous and seven out of nine (77.8%) protected against heterologous CHMI. Increased protection was associated with Vδ2 γδ T cell and antibody responses. At the higher dose, PfSPZ-CVac(CQ) protected six out of six (100%) participants against heterologous CHMI three months after immunization. All homologous (four out of four) and heterologous (eight out of eight) infectivity control participants showed parasitaemia. PfSPZ-CVac(CQ) and PfSPZ-CVac(PYR) induced a durable, sterile vaccine efficacy against a heterologous South American strain of P. falciparum, which has a genome and predicted CD8 T cell immunome that differs more strongly from the African vaccine strain than other analysed African P. falciparum strains.
The morbidity and mortality induced by malaria were halved in little more than a decade using drug and anti-vector measures, but progress has stalled1. Vaccines that confer sterile protective immunity would prevent deaths and contribute to the elimination of malaria. Pre-erythrocytic-stage vaccines target the clinically silent sporozoite and/or liver stages of the life cycle of the parasite to induce sterile immunity that averts blood-stage infection, disease and transmission.
The most advanced malaria vaccine candidate, RTS,S (also known by the trade name Mosquirix), targets a major surface protein (circumsporozoite protein (CSP)) of P. falciparum sporozoites and confers partial protection against clinical malaria2–4. Another advanced candidate, Sanaria PfSPZ Vaccine, is composed of radiation-attenuated PfSPZs. PfSPZ Vaccine induces significant protection against homologous (that is, the same strain of P. falciparum in the vaccine and the challenge) and heterologous (a different strain) CHMI5–8 and against intense natural transmission in Africa9,10. Radiation-attenuated PfSPZs are non-replicating parasites and arrest early in the liver stage, as do first-generation genetically attenuated PfSPZs11,12,13.
For CVac with PfSPZs, non-attenuated PfSPZs (PfSPZ Challenge) are administered under antimalarial drug cover. In seminal studies, blood-stage schizonticides such as CQ14 or mefloquine15, given after exposure to mosquitoes carrying P. falciparum sporozoites, killed parasites after liver-stage development and brief blood-stage exposure16, and conferred a high level of homologous vaccine efficacy14,15 after an immunizing dose of up to 45 infected mosquito bites, much less than the more than 1,000 bites that are required for radiation-attenuated P. falciparum sporozoites17. This has been ascribed to parasite replication (up to 50,000-fold) in the liver. Similarly, three doses of 5.12 × 104 PfSPZs administered with CQ (Sanaria PfSPZ-CVac(CQ)) protected 100% of the participants (nine out of nine) against homologous CHMI 10 weeks later18. This is, to our knowledge, the highest level of durable vaccine efficacy against homologous CHMI that has been achieved. However, a dose of up to 45 infected mosquito bites given under CQ cover induced minimal protection against heterologous CHMI19,20.
Ideally, PfSPZ-CVac would induce protective immunity without exposure to blood-stage parasites, which cause clinical malaria. Mouse studies of CVac with CQ or PYR, which kills liver-stage parasites, showed that both regimens confer sterilizing homologous immunity21 with similar efficacy22. In humans, the antimalarial drug primaquine, which kills liver-stage parasites, reduced but did not eliminate the breakthrough of blood-stage infections during CVac administration, and CVac (primaquine) with 45 or fewer infected mosquito bites conferred minimal sterilizing immunity against homologous CHMI23.
Here, we examined PfSPZ-CVac(CQ) and PfSPZ-CVac(PYR) in human trials to assess the requirement for blood-stage parasite exposure and to examine the effects of PfSPZ dose for inducing durable sterilizing immunity.
Safety and efficacy of the low-dose study
In our first trial (https://clinicaltrials.gov/, ID NCT02511054; Extended Data Fig. 1), we compared three monthly doses (days 1, 29 and 57) of 5.12 × 104 PfSPZ-CVac (‘low-dose’ PfSPZ) and weekly treatment with 500 mg CQ (PfSPZ-CVac(CQ)) against three monthly doses (days 1, 29 and 57) of 5.12 × 104 PfSPZ-CVac (‘low-dose’ PfSPZ) and treatment with 50 mg PYR on days 2 and 3 (PfSPZ-CVac(PYR)) plus weekly CQ treatment as a safety measure against breakthrough parasitaemia. Vaccinations were safe and well-tolerated (Supplementary Information). PYR killed liver-stage parasites in all recipients (Extended Data Fig. 2). Similar to previous results18, four out of five PfSPZ-CVac(CQ) recipients (vaccine efficacy = 80%, P = 0.048; 95% confidence interval, 1–99%) were protected against homologous CHMI 3 months after the last dose; conversely, only two out of nine (vaccine efficacy = 22.2%, P = 0.8) PfSPZ-CVac(PYR) recipients were protected (Extended Data Fig. 3).
Safety and efficacy of the high-dose study
We hypothesized that higher PfSPZ doses would enhance immunity24 and performed a second study (https://clinicaltrials.gov/, ID NCT03083847). In the dose-escalating pilot phase, doses as high as 2 × 105 PfSPZ Challenge (NF54) (‘high dose’) were deemed safe for PfSPZ-CVac(CQ) and PfSPZ-CVac(PYR), which was given without weekly CQ (full safety information is provided in the Supplementary Information). Nonsteroidal anti-inflammatory drugs were given on days 7 and 8 after the first PfSPZ inoculation to PfSPZ-CVac(CQ) recipients to reduce symptoms related to brief blood-stage parasitaemia with higher PfSPZ Challenge doses.
In the efficacy phase, 42 participants were allocated to PfSPZ-CVac(PYR) (n = 20), PfSPZ-CVac(CQ) (n = 10) or no intervention (n = 12, infectivity control participants) (Extended Data Fig. 4 and Supplementary Information for further details regarding the study design). Vaccinations were safe and well-tolerated (Supplementary Information). One PfSPZ-CVac(CQ) recipient withdrew because of a serious adverse event that was possibly related to CQ (Extended Data Fig. 4). During the vaccination period, no participants had patent parasitaemia detected by microscopy. Subpatent parasitaemia was detected in all PfSPZ-CVac(CQ) recipients using quantitative PCR (qPCR) (which decreased with successive administrations, as described previously14,18), but after the third dose of 2 × 105 PfSPZs, only one out of seven vaccinated participants developed any detectable parasitaemia after receiving more than sixty 100% infectious doses, indicating thatsterile homologous immunity had developed after only two doses in six out of seven vaccinated participants. As expected, no PfSPZ-CVac(PYR) recipients had detectable subpatent parasitaemia during vaccination (Fig. 1a), due to PYR activity in the liver.
Fig. 1 |. Parasitaemia detected by qPCR after the first, second and third dose of 2 × 105 PfSPZ for the PfSPZ-CVac high-dose study and PYR activity against liver-stage parasites.
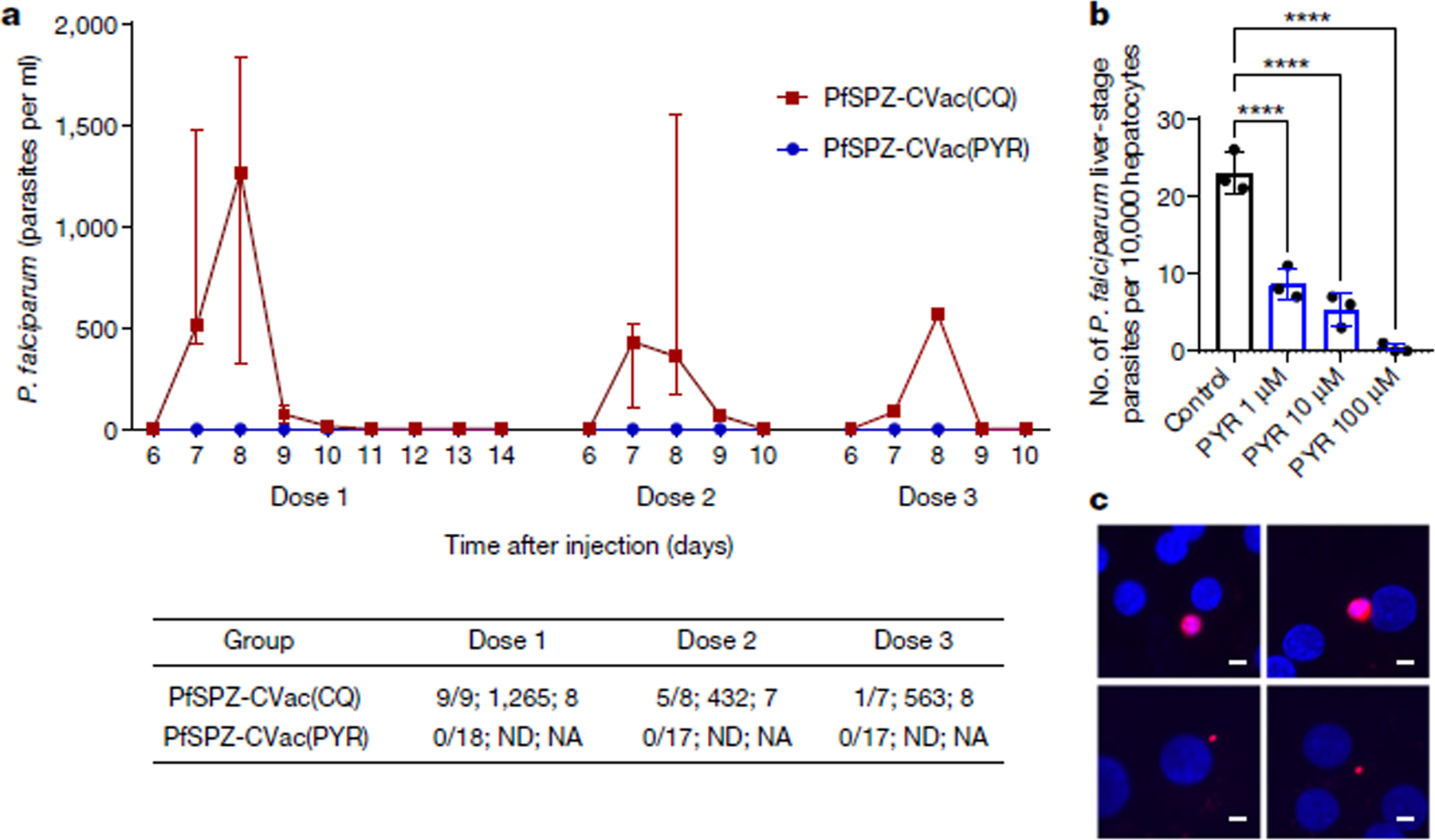
a, Median parasitaemia values and IQRs are shown for positive PfSPZ-CVac(CQ) participants. Dose 1, PfSPZ Challenge inoculation under CQ or PYR treatment cover with follow-up for 14 days; doses 2 and 3, PfSPZ Challenge inoculation under CQ or PYR treatment cover with follow-up for 10 days. The table shows (from left to right in each cell): the number of participants who were positive by qPCR/the number of injected participants; the median peak parasite density of positive participants (parasites per ml); and the mean day of peak parasite density (positive participants). NA, not applicable; ND, not detected. Individual data are shown in Extended Data Fig. 8b. b, c, PYR was added to cultures (1 μM, 10 μM or 100 μM) on days 2 and 3 after infection with daily medium changes until day 4, at which point cultures were fixed. b, On day 4, the remaining liver-stage parasites were counted. Data are mean ± s.e.m., n = 3 independent wells. Statistical significance was determined using a one-way ANOVA with post hoc pairwise comparisons. ****P < 0.0001. Parasite diameters are shown in Extended Data Fig. 9c. c, Representative images of P. falciparum from control (top) and PYR-treated (bottom left, 1 μM; bottom right, 10 μM) groups. Parasites (red) were identified by anti-HSP70. Scale bars, 5 μm.
To assess the effects of PYR on liver-stage parasites, we used an in vitro model with micropatterned primary human hepatocytes25–27. PYR (1, 10 or 100 μM) was added on days 2 and 3 after infection and significantly reduced the numbers of liver-stage parasites on day 4 in a dose-dependent manner, which suggests that PYR can kill parasites (Fig. 1b); the remaining parasites were significantly smaller, indicating that these parasites showed arrested development (Fig. 1c). Daily PYR 50 mg treatment given orally achieves plasma concentrations of around 300–600 μg ml−1 (around 1.2–2.4 μM)28.
Vaccine efficacy was assessed for homologous (NF54, vaccine strain) and heterologous (7G8, a South American parasite) CHMI with 3.2 × 103 PfSPZ Challenge 3 months after the third vaccination. All 12 unvaccinated infectivity control participants were diagnosed with P. falciparum infection by qPCR on days 9–11 (NF54; n = 4) or days 9–16 (7G8; n = 8). In the PfSPZ-CVac(PYR) groups, seven out of eight participants (87.5%, P = 0.003; 95% confidence interval, 42.5–100%) were protected against homologous and seven out of nine participants (77.8%, P = 0.001; 95% confidence interval, 39.8–100%) were protected against heterologous CHMI. In the PfSPZ-CVac(CQ) group, six out of six participants (100%, P = 0.001; 95% confidence interval, 54.1–100%) were protected against heterologous CHMI (Fig. 2).
Fig. 2 |. Vaccine efficacy of PfSPZ-CVac(PYR) against homologous and heterologous CHMI and PfSPZ-CVac(CQ) against heterologous CHMI, 3 months after the last immunization.
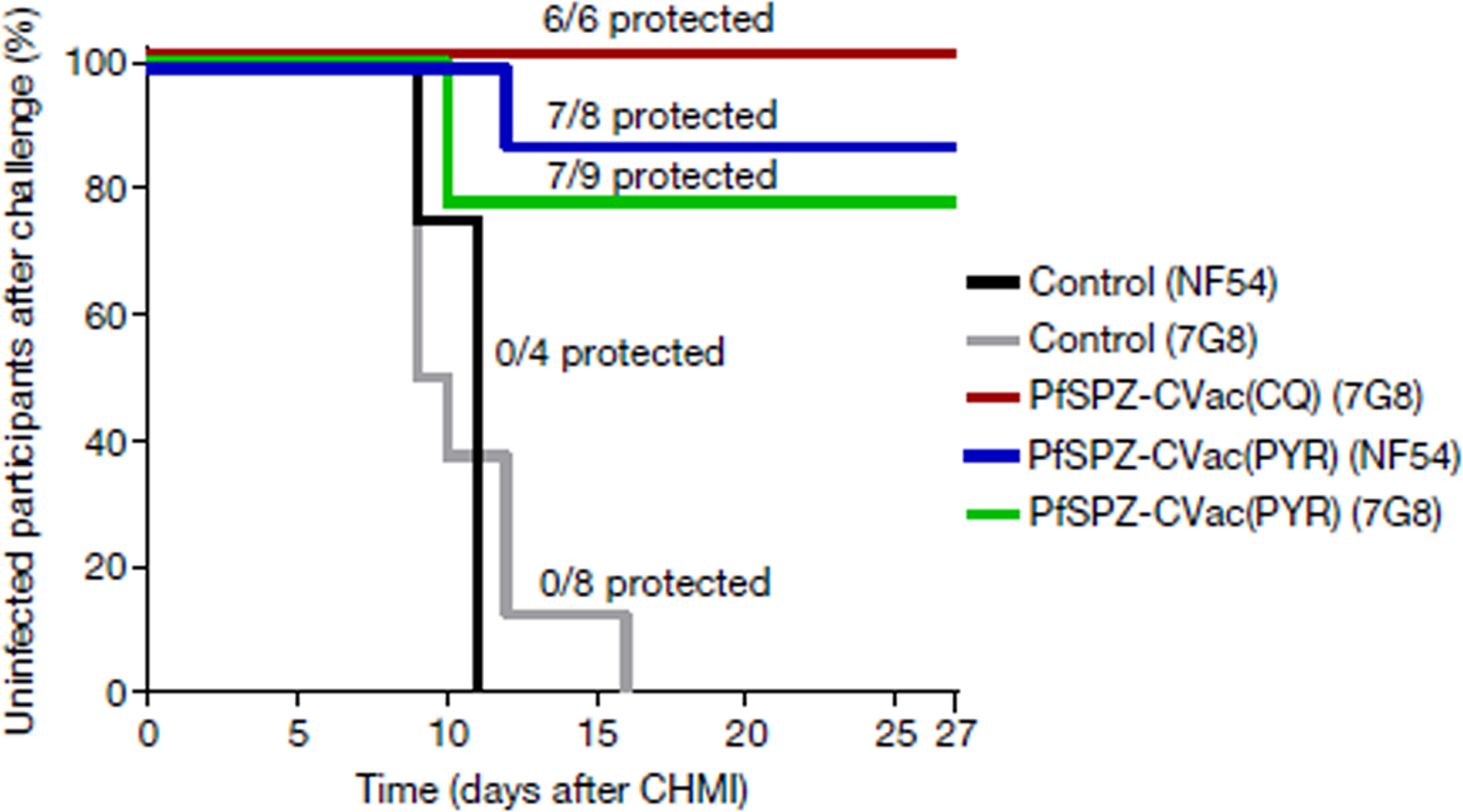
Survival curves display the percentage of study participants who remained protected throughout 27 days of follow-up after inoculation with a high dose of PfSPZ (2 × 105). Vaccine efficacy compared the vaccinated to unvaccinated groups for the time to first detectable parasitaemia after inoculation using a log-rank test. In the PfSPZ-CVac(PYR) group, 7/8 participants (vaccine efficacy = 87.5%, P = 0.003; 95% confidence interval, 42.5–100%) were protected from homologous CHMI and 7/9 participants (vaccine efficacy = 78%, P = 0.001; 95% confidence interval, 39.8–100%) from heterologous CHMI. In the PfSPZ-CVac(CQ) group, 6/6 participants (vaccine efficacy = 100%, P = 0.001; 95% confidence interval, 54.1–100%) were protected from heterologous CHMI.
Vδ2 γδ T cell expansion in vaccinated participants
PfSPZ Vaccine or PfSPZ-CVac administration expands Vδ2 γδ T cells (hereafter Vδ2 T cells)29,30. Vδ2 T cells were measured as a percentage of total T cells using ex vivo flow cytometry throughout the vaccination period (Fig. 3). At baseline, the median percentage of Vδ2 T cells was similar in the PfSPZ-CVac(PYR) and PfSPZ-CVac(CQ) groups (low dose, 2.59 (interquartile range (IQR) = 2.48–2.7) versus 1.42 (IQR: 1.33 – 4.65), P = 0.3127, Wilcoxon–Mann–Whitney test; high dose, 2.35 (IQR = 1.96–5.93) versus 2.36 (IQR = 0.88–3.67), P = 0.6187, Wilcoxon–Mann–Whitney test) (Extended Data Fig. 5). Subsequent data are expressed as fold change from baseline, either before (unadjusted) or after (adjusted) adding an offset of +1 to the numerator and denominator for calculations of fold change in %Vδ2 T cells (Fig. 3a, b present unadjusted values).
Fig. 3 |. Vδ2 γδ T cells related to protective immunity.
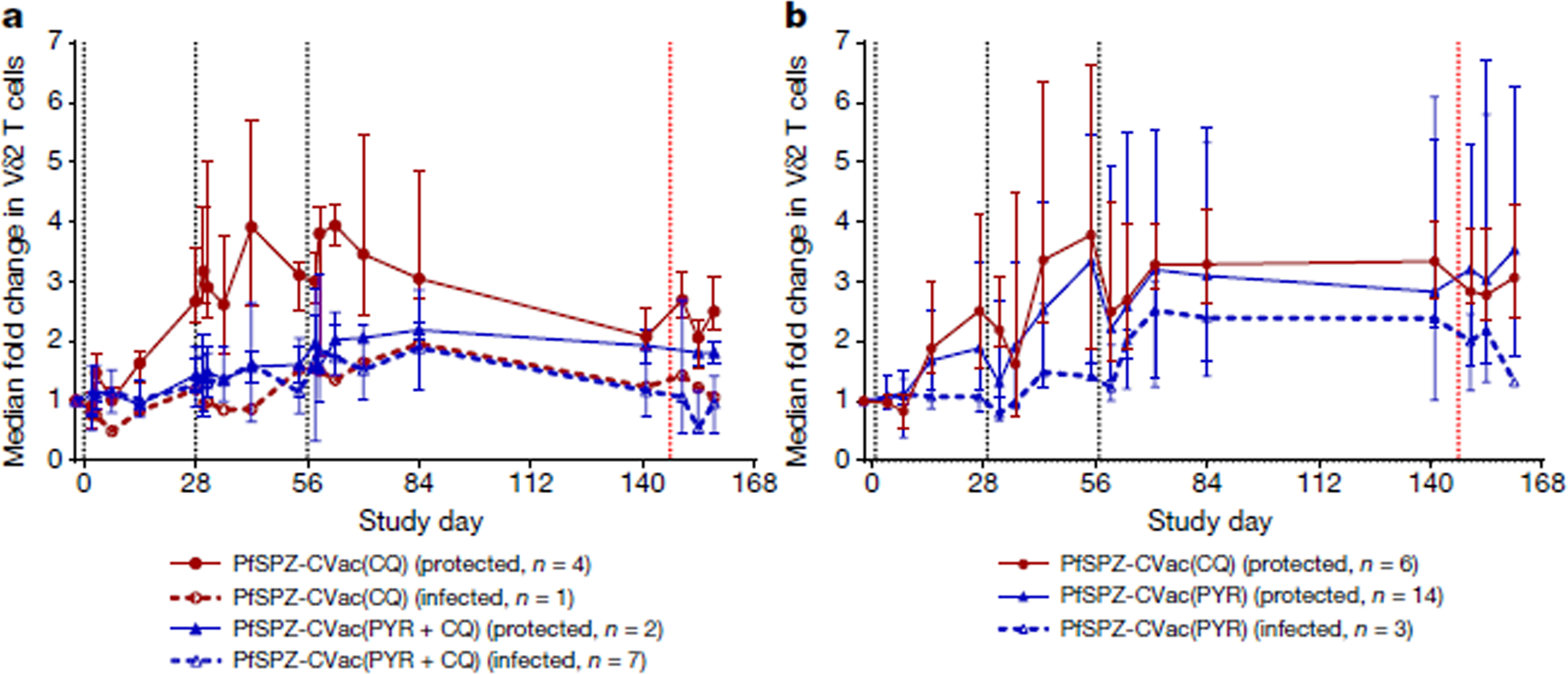
Vδ2 γδ T cells in vaccinated individuals stratified by protection status. Median fold changes and IQRs using unadjusted %Vδ2 values are shown. a, For the low-dose study, n = 4 PfSPZ-CVac(CQ) protected vaccinated participants; n = 1 PfSPZ-CVac(CQ) infected vaccinated participant; n = 2 PfSPZ-CVac(PYR + CQ) protected vaccinated participants; n = 7 PfSPZ-CVac(PYR + CQ) infected vaccinated participants. b, For the high-dose study, n = 6 PfSPZ-CVac(CQ) protected vaccinated participants; n = 14 PfSPZ-CVac(PYR) protected vaccinated participants; n = 3 PfSPZ-CVac(PYR) infected vaccinated participants. Black dashed lines indicate vaccination days; red dashed line indicates day of PfSPZ Challenge inoculation.
The fold change in the percentage of Vδ2 T cells during PfSPZ-CVac(CQ) was similar (P = 0.768, generalized estimating equation model using adjusted fold change values, see Methods) between the low dose (median = 2.01 (IQR = 1.9–2.98)) and the high dose (median = 2.84 (IQR = 2.21–3.04)) from dose 1 to day 84 (28 days after dose 3). All but one of the PfSPZ-CVac(CQ) vaccinated participants were protected during CHMI (in the low-dose study); the one infected vaccinated participant had the lowest fold increase in Vδ2 T cells among PfSPZ-CVac(CQ) recipients (adjusted fold change: 1.78, day 84).
Conversely, the fold change in Vδ2 T cells during the PfSPZ-CVac(PYR) study increased (P = 0.021, generalized estimating equation model) from the low dose (median = 1.6 (IQR = 1.18–1.96)) to the high dose (median = 2.02 (IQR = 1.68–3.94)) from dose 1 to day 84 (28 days after dose 3). In the high-dose study, protected PfSPZ-CVac(PYR) recipients (protected against NF54 and 7G8 challenges, n = 14) showed a significantly greater expansion of Vδ2 T cells from dose 1 to day 84 (28 days after dose 3) compared with infected vaccinated participants (P = 0.016, generalized estimating equation model), but did not differ immediately before CHMI (day 141; protected, median = 2.36 (IQR = 1.94–3.71); infected, median = 2.01 (IQR = 1.01–4.45); P = 0.45, Wilcoxon–Mann–Whitney test).
Antibody responses induced by PfSPZ-CVac
Two weeks after the last vaccination (day 70), all PfSPZ-CVac(PYR) or PfSPZ-CVac(CQ) recipients met the criteria for seroconversion according to IgG PfCSP levels analysed by enzyme-linked immunosorbent assay (ELISA). IgG levels 2 weeks after the last administration of 5.12 × 104 PfSPZ-CVac(CQ) (low-dose study) were similar in this trial (median net optical density (OD) 1.0 = 2,480) (Fig. 4a) and a previous trial (median net OD 1.0 = 3,844)18. IgG levels 2 weeks after vaccination were significantly higher in recipients of high-dose versus low-dose PfSPZ-CVac(PYR) (3,768 versus 255, respectively (P < 0.001)), but not in recipients of high-dose versus low-dose PfSPZ-CVac(CQ) (8,060 versus 2,480 (P = 0.178)) (Fig. 4a); IgG levels were higher in PfSPZ-CVac(CQ) than PfSPZ-CVac(PYR) participants for the low dose (2,480 versus 255 (P = 0.012)) but not the high dose (8,060 versus 3,768 (P = 0.280)). Similar relationships were seen before CHMI, 3 months after vaccination (Fig. 4b).
Fig. 4 |. Anti-PfCSP IgG levels by ELISA in vaccinated participants of the low-dose and high-dose studies.
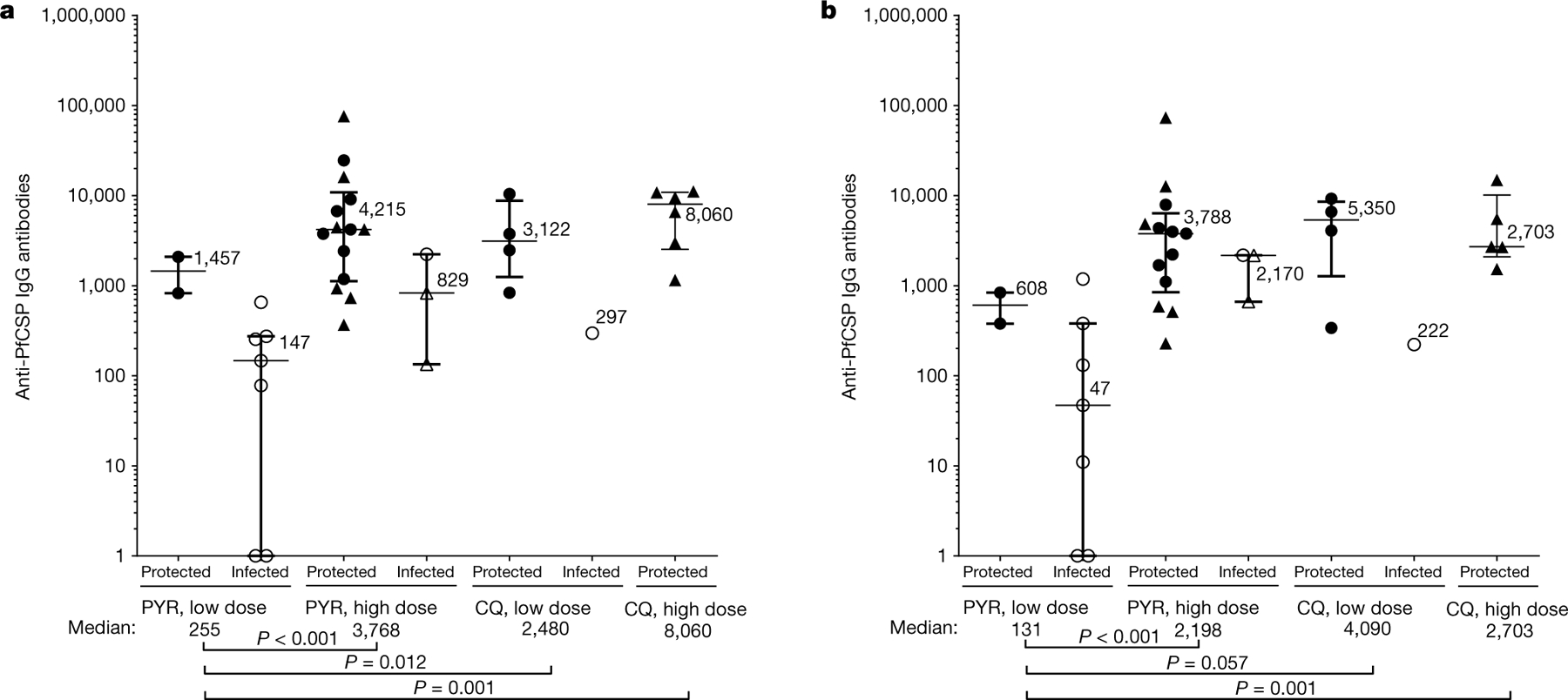
Filled circles are protected participants and open circles are infected participants in homologous CHMI; filled triangles are protected participants and open triangles are infected participants in heterologous CHMI. Median values and IQRs are displayed. P values were calculated using two-sided Wilcoxon–Mann–Whitney tests and group differences with P > 0.07 are not indicated. a, b, IgG antibodies against PfCSP (net OD 1.0) 2 weeks (day 70) (a) or approximately 3 months (before CHMI) after the third PfSPZ-CVac dose (b). PYR low-dose study, n = 2 protected and n = 7 infected vaccinated participants; PYR high-dose study, n = 14 protected and n = 3 infected vaccinated participants; CQ low-dose study, n = 4 protected and n = 1 infected vaccinated participant; CQ high-dose study, n = 6 protected vaccinated participants in a (day 70) and n = 5 protected vaccinated participants in b (before CHMI). No vaccinated participants were infected in the CQ high-dose study.
In general, median IgG levels were higher in protected versus infected vaccinated participants within study arms; small sample sizes precluded or limited subgroup statistical analyses. Within low-dose PfSPZ-CVac(PYR) participants, the two protected participants had higher median levels compared with the seven infected participants (median 1,457 versus 147 (P = 0.057)); within high-dose PfSPZ-CVac(PYR) participants, 14 protected participants had higher levels versus 3 infected participants (4,215 versus 829 (P = 0.065)) (Fig. 4). Among PfSPZ-CVac(PYR) participants, IgG levels 3 months after vaccination (before CHMI) were not related to protection in the low-dose (protected, 608; infected, 47; P = 0.306) or high-dose (protected, 3,788; infected, 2,170; P = 0.439) groups.
IgM PfCSP levels analysed by ELISAs 2 weeks after the third dose and before CHMI did not differ between high-dose PYR and CQ groups, with too few infected participants to assess the association with vaccine efficacy (Extended Data Fig. 6). In the automated immunofluorescence assay and automated inhibition of sporozoite invasion assay, the high-dose groups generally had higher levels of activity than low-dose groups (Extended Data Fig. 7).
Discussion
Progress in malaria control has plateaued, and new interventions are needed1. No human parasite vaccines have marketing authorization (licensure) in the USA or Europe. Furthermore, the complexity of the PfSPZ vaccines (like any eukaryotic cell vaccine) for which there are no suitable models in animals to test efficacy have required multiple human trials to assess the dose, schedule, route of administration and method of attenuation. Here, we report a culmination of that empirical process and show sterilizing immunity against heterologous P. falciparum CHMI for at least 3 months after vaccination. Although PfSPZ-CVac(CQ) was previously shown to induce long-lived protection, this was thought to be limited to homologous parasites19,20 and to require exposure to blood-stage parasites31,32. Our findings overturn both of these notions and strongly suggest that PfSPZ-CVac will confer broadly protective sterilizing immunity against P. falciparum in the field.
The P. falciparum 7G8 strain used for heterologous CHMI originates from Brazil. P. falciparum 7G8 is more divergent at the levels of the genome, proteome and CD8 T cell immunome than any of more than 400 P. falciparum isolates studied from east, west and central Africa, including the West African NF54 strain used for PfSPZ-CVac33. We previously reported that the field efficacy of a PfSPZ Vaccine regimen in adults in Mali against 6 months of intense P. falciparum transmission9 equalled or exceeded its vaccine efficacy at 6 months against CHMI with P. falciparum 7G8 in adults in the USA5. In a previous trial, participants who received chemoprophylaxis and sporozoites vaccination that induce high levels of homologous protection showed minimal heterologous protection against NF135.C10 (Cambodia) and NF166.C8 (Guinea) parasites20; future studies of different parasite challenge strains are warranted to predict field vaccine efficacy in Africa or other regions. The 100% vaccine efficacy against heterologous CHMI in PfSPZ-CVac(CQ) recipients here may have resulted from the fourfold increase in dosage above that required to achieve similar homologous protection, whereas CVac studies of individuals immunized by mosquito bite that conferred minimal heterologous protection had not increased the dosage above that required for homologous CHMI protection20. Whether doses above the 2 × 105 PfSPZ Challenge might further enhance sterilizing immunity requires further study, using more rigorous tests such as CHMI at later time points or field efficacy trials. The effect of the fourfold increase in dose was even more notable for PfSPZ-CVac(PYR): at 5.12 × 104 PfSPZs, vaccine efficacy was only 22% against homologous CHMI; at 2 × 105 PfSPZs, vaccine efficacy was 88% for homologous CHMI (P = 0.009, two-tailed Barnard’s test) and 78% for heterologous CHMI (P = 0.03, two-tailed Barnard’s test).
We show that durable, sterile vaccine efficacy against heterologous CHMI can be achieved without blood-stage parasite exposure during PfSPZ-CVac(PYR). The vaccine efficacy of PfSPZ-CVac(CQ) has been attributed in part to exposure to blood-stage parasites32, even though vaccinated participants were not protected against CHMI with blood-stage parasites34. Instead, blood-stage parasite exposure has been hypothesized to enhance pre-erythrocytic immunity by inducing protective cross-stage responses31. In a mouse model of Plasmodium yoelii infection, sterilizing immunity induced by CVac(CQ) included activity against liver- or blood-stage infection, and at higher doses was mediated primarily by activity against liver-stage parasites35. Our data do not exclude the possibility that blood-stage parasite exposure could enhance pre-erythrocytic immunity (PfSPZ-CVac(CQ) results), but do demonstrate that durable heterologous immunity is achieved without it (PfSPZ-CVac(PYR) results).
We were surprised that heterologous vaccine efficacy at 3 months of PfSPZ-CVac(PYR) with the 2 × 105 PfSPZ dose (78%) exceeded that of radiation-attenuated PfSPZs (PfSPZ Vaccine) with the 9 × 105 PfSPZ dose (20%)36. Radiation-attenuated PfSPZs are thought to develop normally for around 2 days before halting development, and never replicate. Using micropatterned primary human hepatocyte cultures, we found that PYR at clinically relevant concentrations28 on days 2–4 significantly reduced parasite numbers and size on day 4 (Fig. 1b, c), which suggests that the drug induces killing and developmental arrest of the parasite. The results are in agreement with our clinical finding that no parasites emerged in blood (Fig. 1a), and suggest that fine differences in attenuated PfSPZs, such as the timing of arrest and/or antigen repertoire, can substantially affect the efficacy of the vaccine.
We9 and others29 previously associated Vδ2 T cells with PfSPZ Vaccine protection. In mice that received SPZ vaccines, a subset of γδ T cells was required to induce protective CD8+ T cells that kill intrahepatocytic parasites, but γδ T cells did not directly mediate protection30. CD8+ T cells—presumably resident memory T cells—may eliminate parasite-infected hepatocytes directly or through IFNγ to kill the parasite37. These effector processes take place in the liver and have been difficult to study in humans38. In this study, Vδ2 T cell responses and vaccine efficacy increased with PfSPZ dose among PfSPZ-CVac(PYR) recipients. At the lower dosage (5.12 × 104), Vδ2 T cell increases (and vaccine efficacy) were significantly greater in PfSPZ-CVac(CQ) recipients than PfSPZ-CVac(PYR) recipients; at modestly higher dosage (2 × 105), Vδ2 T cell increases (and vaccine efficacy) were similar between PfSPZ-CVac groups. Among all vaccinated participants, those protected had significantly greater Vδ2 T cell expansion during immunizations. We surmise that vaccine-induced Vδ2 T cell responses may correspond to enhanced CD8 T cell responses in the liver that mediate protection against PfSPZ Challenge. More studies are needed to evaluate the immune responses including Vδ2 T cells that contribute to or correlate with protection.
Despite non-human data indicating that the protective immunity is mediated by T cells, several PfSPZ Vaccine trials associated PfCSP antibody levels and protection in malaria-naive8 and malaria-experienced9 individuals. Similarly, we find here a significant relationship between PfCSP levels and regimens that protect from CHMI. Notably, PfCSP antibody levels induced by the high-efficacy CVac regimens tested here (median net OD 1.0 levels of 8,060 and 4,215) were substantially lower than PfSPZ Vaccine regimens that conferred minimal heterologous protection5,36. We continue to think that the antibody responses are a correlate, but not a primary mediator of vaccine efficacy, although the antibodies probably have a role in the protection in some individuals.
PfSPZ-CVac(PYR) is attractive for future use in Africa for several reasons. Killing of the parasites during the clinically silent liver stages by PYR rather than during blood-stage development as occurs with CQ treatment offers a wider safety margin. In Africa and other areas of high malaria transmission, pregnant women already take PYR monthly (in combination with sulfadoxine) after the first trimester for malaria control. Millions of children in the Sahel receive PYR in a monthly drug combination (sulfadoxine–PYR + amodiaquine) during the malaria season39. The widespread use and excellent safety profile of PYR will facilitate the deployment of PfSPZ-CVac(PYR), in particular among pregnant women and children who bear the greatest burden of disease. Notably, antibody and T cell responses to PfSPZ Vaccine in children in Africa are comparable to those of non-immune individuals in the USA and Europe, and superior to those of adults in Africa40. A programme to assess PfSPZ-CVac(PYR) field efficacy has already begun by first assessing the vaccine efficacy against P. falciparum in adults in Mali (https://clinicaltrials.gov/, ID NCT03952650) before moving to children, and is exploring same-day administration of PYR and PfSPZ to improve feasibility and safety. PfSPZ-CVac could also be an excellent preventive vaccine for deployed military personnel and non-immune travellers: more than 90% of travellers to Africa stay fewer 10 weeks, and we have demonstrated up to 100% vaccine efficacy for 3 months against a 100% infectious dose in heterologous CHMI. Furthermore, it has been reported that all three doses can be administered within four weeks41.
Alternative whole-PfSPZ vaccine approaches are currently being developed, including genetically attenuated parasites that arrest at a specific lifecycle time point. This is hypothetically attractive as it obviates the need for antimalarial drugs, and late liver-stage-arresting parasites enhance sterilizing immunity compared with early-arresting parasites in mice11. However, breakthrough infections occurred with some genetically attenuated parasites in the clinic42. A recent genetically attenuated P. falciparum strain has been developed to arrest late in liver-stage development43 and therefore display a wider repertoire of antigens although many of these antigens are shared with blood-stage parasites and are highly variant. Future efficacy trials in malaria-experienced populations should examine the relative benefits of parasites that arrest earlier versus later in development, and PfSPZ-CVac(CQ) versus PfSPZ-CVac(PYR) models offer one such opportunity.
We have established that we can achieve a high level of protection for at least 3 months against heterologous P. falciparum parasites by immunizing with chemo-attenuated PfSPZ. Vaccines are the most efficient way to control any infectious disease. These data indicate that immunization can provide the level of protection against malaria needed for the control and elimination of malaria caused by P. falciparum.
Methods
The sample size was predetermined to identify safety concerns associated with investigational product administration, specifically severe adverse events. The main phase groups in both clinical studies were randomized: the CVac arms were randomized to either PfSPZ-CVac(PYR) or PfSPZ-CVac(CQ); the infectivity control participants were randomized to CHMI with either NF54 or 7G8 in the high-dose study (in the low-dose study, only CHMI NF54 was used). The pilot groups in both studies were not randomized. Laboratory investigators but not clinical investigators or participants were blinded to allocation during experiments and outcome assessment.
Clinical trial design
The clinical trial design for the initial low-dose study (https://clinicaltrials.gov/, ID NCT02511054) can be found in the Supplementary Information. The clinical trial design for the second, high-dose study is described below.
This was an open-label, randomized clinical trial conducted at the National Institutes of Health Clinical Center (NIH CC) in Bethesda, Maryland, USA (https://clinicaltrials.gov/, ID NCT03083847) from June 2017 to March 2019. The study adhered to Good Clinical Practice guidelines and US NIH guidelines and procedures. All participants provided written informed consent. The study was reviewed and approved by the US National Institute of Allergy and Infectious Diseases, NIH institutional review board and conducted under an FDA IND application.
The study objectives were to investigate the safety, tolerability, immunogenicity and vaccine efficacy of direct venous inoculation (DVI) with three doses at four-week intervals of aseptic, purified, cryo-preserved PfSPZ (Sanaria PfSPZ Challenge (NF54))44,45, combined with either PYR or CQ prophylaxis, known as PfSPZ chemoprophylaxis vaccination (PfSPZ-CVac). In addition, we sought to determine whether pre-erythrocytic stage immunity (induced by the PYR arm) was sufficient to protect against homologous (NF54) and heterologous (7G8) CHMI, or whether erythrocytic stage immunity (induced by the CQ arm) might be required for protection.
A total of 56 healthy malaria-naive adults were enrolled in this study (Extended Data Fig. 1). The study did not enrol women who were pregnant or nursing. Medical history, physical exams and safety laboratory (haematological, creatinine, alanine transferase and urinalysis) evaluations were performed at a screening visit. All volunteers had negative screening tests for human immunodeficiency virus (HIV), and hepatitis B and C. The study began with the dose-escalating pilot phase of the study to assess the safety and determine the optimal dosing of PfSPZ-CVac for the proposed regimens. In the PYR pilot study (n = 8), participants received one exposure of PfSPZ Challenge (NF54), in a dose-escalating manner of 5.12 × 104 (n = 2), 1 × 105 (n = 2) or 2 × 105 (n = 4) PfSPZ, along with PYR treatment 2 and 3 days after injection of the PfSPZ Challenge. All PYR pilot arms were safe and prevented detectable subpatent parasitaemia by sensitive qPCR; therefore, the highest dose (2 × 105 PfSPZ) was used during the main study. In the CQ pilot study (n = 6), participants received one exposure of the PfSPZ Challenge (NF54) either at 1 × 105 (n = 2) or 2 × 105 PfSPZ (n = 4) and CQ with a loading dose (1,000 mg) 2 days before the first DVI followed by a single maintenance dose (500 mg) administered 5 days after DVI. The CQ pilot group demonstrated that this regimen was safe, but tolerability in the higher dose pilot was less than expected, with more malaria-related symptoms experienced during days 7 and 8 after DVI. However, symptoms improved quickly with the co-administration of nonsteroidal anti-inflammatory drugs on the days of peak parasitaemia. Therefore, in consultation with an independent safety-monitoring committee, it was determined that the goal dose (2 × 105 SPZ) could be used during the main study with the addition of nonsteroidal anti-inflammatory drugs preemptively during days 7 and 8 after DVI for PfSPZ-CVac(CQ). Participants in the pilot group did not join the main study nor undergo CHMI.
In the main phase, the protocol was initially designed to enrol participants in either PfSPZ-CVac(PYR) with homologous CHMI with NF54 (n = 17) or PfSPZ-CVac(PYR) with heterologous CHMI with 7G8 (n = 10) or PfSPZ-CVac(CQ) with heterologous CHMI with 7G8 (n = 10). Owing to logistical reasons, the main study was conducted in two consecutive cohorts between January 2018 and March 2019, with a portion of each of the three PfSPZ-CVac groups enrolled in the first cohort while only the heterologous CHMI groups were enrolled in the second cohort. With the considerable evidence of the development of protective efficacy against both homologous and heterologous PfSPZ Challenge doses, the protocol was amended such that enrolment into a third cohort did not proceed, and the PYR arm undergoing homologous CHMI finished with 10 enrolled participants instead of the expected 17.
By the end of study, 17 of the 20 enrolled individuals who received PfSPZ-CVac(PYR) completed all three vaccinations (2 × 105 PfSPZ + 50 mg PYR on days 2 and 3 after DVI) (Fig. 1). Two participants withdrew before any receipt of the PfSPZ Challenge or PYR but had undergone enrolment (1 non-compliance, 1 secondary to acute illness) and 1 participant had to withdraw secondary to a scheduling conflict.
Ten participants were initially enrolled into the PfSPZ-CVac(CQ) arm, of which 7 completed all three vaccinations (2 × 105 PfSPZ + CQ loading dose of 1,000 mg 2 days beforehand + a weekly CQ maintenance dose of 500 mg for a total of 9 CQ doses). One individual who received PfSPZ-CVac(CQ) withdrew because of a serious adverse events (possibly related to CQ, mental status changes), one due to pregnancy, and one secondary to a scheduling conflict. One additional individual withdrew after completion of CVac, but before the CHMI phase, due to a serious adverse event unrelated to study participation (pneumothorax).
The CHMI phase was conducted with 3.2 × 103 PfSPZ of either PfSPZ Challenge NF54 or 7G846 administered by DVI approximately 13 weeks after the receipt of the third PfSPZ-CVac dose. Of those who received PYR, n = 8 underwent homologous NF54 CHMI, and n = 9 underwent heterologous 7G8 CHMI; from the CQ arm, n = 6 underwent heterologous 7G8 CHMI (one participant had withdrawn secondary to pregnancy before PfSPZ CHMI).
Infectivity controls were enrolled to undergo PfSPZ Challenge with 3.2 × 103 SPZ of either NF54 (n = 4) or 7G8 (n = 8). Participants were followed daily starting 6 days after CHMI for evaluation of the development of malaria parasitaemia defined as one positive NIH CC clinical diagnostic malaria qPCR (NIH CC qPCR) assay or one positive thick blood smear (TBS). Follow-up was completed either at the time of malaria diagnosis or at the protocol-defined end of study (27 days after CHMI).
All participants were treated with a 3-day standard treatment course of Malarone (also known as atovaquone/proguanil) either at the time of malaria diagnosis or at the end of study if they were not diagnosed with malaria during the follow-up period.
Research parasitaemia determination by qPCR
For the low-dose study, a modified DNA-based qPCR was used to detect subpatent parasitaemia starting days 6 to 10 (or day 14 for dose 1) after each DVI with PfSPZ Challenge (NF54) as previously described47. This method has been validated in our laboratory with the lowest limit of detection of approximately 200 parasites per ml.
For the high-dose study, starting days 6 to 10 (or day 14 for dose 1) after each DVI with PfSPZ Challenge (NF54), a modified RNA-based qPCR was used to detect subpatent parasitaemia as previously described48. This assay has been validated in our laboratory with the lowest limit of detection of approximately 15 parasites per ml based on a plasmid standard. Standard quantities were defined using reference samples with known microscopic values. Samples with resulting values of less than 15 parasites per ml are reported as negative (below the limit of detection/quantification) using this assay. More details of the primers and cycling conditions for both assays are reported in the Supplementary Information.
Malaria diagnostic qPCR (NIH CC qPCR)
The established NIH CC malaria diagnostic test in the CLIA-certified clinical laboratory, Malaria Genus Species (4-plex) PCR, was used for real-time safety monitoring during the vaccination period as well as for malaria diagnosis during CHMI8,49,50. NIH Malaria Genus Species (4-plex) PCR is a DNA-based test with a sensitivity of 500 parasites per ml of whole blood. NIH Malaria Genus Species 4-plex qRT–PCR was used for malaria diagnosis after administration of the Sanaria PfSPZ Challenge during vaccination (from 6 to 10 days (or 14 days after dose 1) after DVI) and CHMI phases (from 6 to 27 days after DVI or until malaria diagnosis) and for any visits during which a partipcipant presented as symptomatic. The turnaround was approximately 6 h. During the low-dose study, two consecutive positive NIH Malaria Genus Species (4-plex) PCR results or a single patent parasitaemia by TBS after PfSPZ Challenge were used for diagnosis and initiation of treatment with Malarone. For enhanced safety in the high-dose study, only one positive NIH Malaria Genus Species (4-plex) PCR result or a single patent parasitaemia by TBS after PfSPZ Challenge was used for diagnosis and initiation of treatment with Malarone.
TBS analyses
TBSs were prepared in duplicate and stained with Giemsa according to standard malaria CHMI procedures and evaluated by trained study microscopists from 6 to 10 days (or 14 days after dose 1) after each DVI during the vaccination phase. During the CHMI phase, TBSs were only performed when malaria diagnostic qPCR could not be reported on the same day or if a participant was symptomatic and met evaluation criteria per the study standard operating procedures. The results were reported to the study principal investigator within 24 h and were prioritized to be read immediately if a participant was symptomatic. At least 0.5 μl was scanned for the presence of malaria parasites, giving a theoretical limit of detection of 2 parasites per μl of blood. For symptomatic participants, at least 1.5 μl was evaluated before calling the TBS negative. Slides were considered positive if at least two unambiguous parasites per slide were identified and confirmed by a second microscopist.
Assessing in vitro PYR activity against liver-stage parasites
To assess in vitro PYR activity against liver-stage parasites, micropatterned co-cultures were generated as described previously26. In brief, glass-bottom 96-well plates were coated with rat-tail type-I collagen (50 mg ml−1) and subjected to soft lithographic techniques to pattern the collagen into microdomains (islands of 500 mm) that mediate selective hepatocyte adhesion. To create micropatterned co-cultures, cryopreserved primary human hepatocytes (Bioreclamation IVT, BGW lot, male, age 50) were pelleted by centrifugation and then seeded on collagen-micropatterned plates. 3T3-J2 mouse embryonic fibroblasts were seeded 1 day later.
The next day, 2 × 105 aseptic, purified cryopreserved PfSPZs were overlaid onto micropatterned co-cultures (that had been seeded the day before) in hepatocyte medium and kept at 37 °C and 5% CO2 for 3 h for infection to occur. After infection, wells were washed twice and fresh medium containing fibroblasts was added. Cultures were fixed on day 4 with ice-cold methanol for analysis by immunofluorescence.
On days 2 and 3, fresh medium containing 0μM, 1μM, 10μMor 100μMPYR was added to the cultures. Cultures were fixed on day 4 as described above.
The numbers of parasites per well were assessed by staining with a monoclonal antibody against the 70 kDa heat shock protein (HSP70) (clone 4C9, 5 μg ml−1, gift from F. Zavala, Johns Hopkins Universtiy) using a Nikon Eclipse Ti fluorescence microscope. Results are reported as the number of parasites per 104 hepatocytes.
Ex vivo flow cytometry
Freshly isolated whole blood was collected from each study participant and was used to enumerate the percentage and subsets of γδ T cells, Vδ2 T cells, CD4 T cells, CD8 T cells and NK cells (the gating strategy is shown in Extended Data Fig. 10). Activated CD4 and CD8 T cells were defined by co-expression of HLA-DR. Samples were collected at enrolment and at days 2, 3, 7, 14 and 28 after each vaccination. In addition, whole blood was also collected immediately before CHMI and days 3, 7 and 14 after CHMI. In brief, 200 μl of whole blood from each participant was used to stain with a cocktail of conjugated monoclonal antibodies. The samples were washed, red blood cells lysed and washed again before acquisition on a BD LSRII flow cytometer equipped with a blue, red and violet laser, using BD FACSDiva version 8 software. All events in the tubes were acquired and analysed using FlowJo v.10.4.1 software.
Antibody assays
IgG antibodies against PfCSP were assessed by ELISA, IgG antibodies against PfSPZs were assessed using an automated immunofluorescence assay and the capacity of sera to inhibit the invasion of hepatocytes (HC-04 cell line) by PfSPZs was assessed using an automated inhibition of sporozoite invasion assay, all of which have been described previously18. In addition, IgM antibodies against PfCSP were assessed by ELISA. In brief, 96-well plates (Nunc Maxisorp Immuno Plate) were coated overnight at 4 °C with 2.0 μg recombinant PfCSP in 50 μl coating buffer (KPL) per well. Plates were washed three times with 2 mM imidazole, 160 mM NaCl, 0.02% Tween-20, 0.5 mM EDTA and blocked with 1% bovine serum albumin blocking buffer (KPL) containing 1% non-fat dry milk for 1 h at 37 °C. Plates were washed three times and serially diluted serum samples (in triplicate) were added and incubated at 37 °C for 1 h. After three washes, a peroxidase-labelled goat anti-human IgM antibody (KPL) was added at a dilution of 2.0 μg ml−1 and incubated at 37 °C for 1 h. Plates were washed three times, ABTS peroxidase substrate was added for plate development, and the plates were incubated for 75 min at 22 °C. The plates were read with a Spectramax Plus384 microplate reader (Molecular Devices) at 405 nm. The data were collected using Softmax Pro GXP v.5 and fit to a four-parameter logistic curve to calculate the serum dilution at OD 1.0. A negative control (pooled serum from non-immune individuals from malaria-free area) was included in all assays. Serum from an individual with anti-PfCSP antibodies was used as a positive control. The antibody assays were conducted on sera obtained before immunization, 2 weeks after the third immunization and before CHMI. Definitions for a positive response were taken relative to the pre-dose 1 measurement. For ELISA, samples were considered positive if the difference between the post-immunization OD 1.0 and the pre-immunization OD 1.0 (net OD 1.0) was ≥50 and the ratio of the post-immunization OD 1.0 to pre-immunization OD 1.0 (ratio) was ≥3.0. For automated immunofluorescence assays, participants with a net reciprocal serum dilution for 2 × 105 arbitrary fluorescence units (AFU) of ≥150 and a ratio reciprocal serum dilution of ≥3.0 were considered positive. For the automated inhibition of sporozoite invasion assay, participants with a net reciprocal serum dilution for 80% inhibition of ≥10 in the inhibition of sporozoite invasion assay and a ratio reciprocal serum dilution for 80% inhibition of ≥3.0 in the inhibition of sporozoite invasion assay were considered positive. A non-parametric Wilcoxon–Mann–Whitney test was used to determine statistical significance for fold change values of antibody levels.
Statistical analysis
For the primary objective of assessing safety, the proportion of participants with at least one adverse event was compared by immunization schedule and dose group. Fisher’s exact tests were performed to assess whether groups differed with respect to these proportions. A Wilcoxon signed-rank test was performed to analyse differences in adverse events between initial immunizations and subsequent immunizations. To assess vaccine efficacy, the unvaccinated group was compared with the vaccine group for time to first detectable parasitaemia after the final challenge using a log-rank test, and the presence or absence of parasitaemia by a two-tailed Barnard’s test. To assess subpatent parasitaemia during the vaccination period, the two vaccine groups were compared using an unconditional exact test by the ORRT procedure51. Vaccine efficacy analyses were performed in R. The non-parametric Wilcoxon–Mann–Whitney test was used to determine statistical significance for group differences in levels or fold changes in Vδ2 T cells and in antibody levels. Subsequent Vδ2 data were expressed as fold change from baseline, with (adjusted) or without (unadjusted) addition of an offset of +1 to numerator and denominator values of the percentage of Vδ2 T cells. Generalized estimating equation models—using the log-transformed fold change in the adjusted percentage of Vδ2 T cells as the outcome variable—were employed to analyse the dynamics of Vδ2 T cells in the low-dose and high-dose trials in the PfSPZ-CVac(PYR) and PfSPZ-CVac(CQ) groups during the vaccination phase (study days −7 to 84) using Stata software v.16.1 (US).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Extended Data
Extended Data Fig. 1 |. Trial profile for PfSPZ-CVac low-dose study.
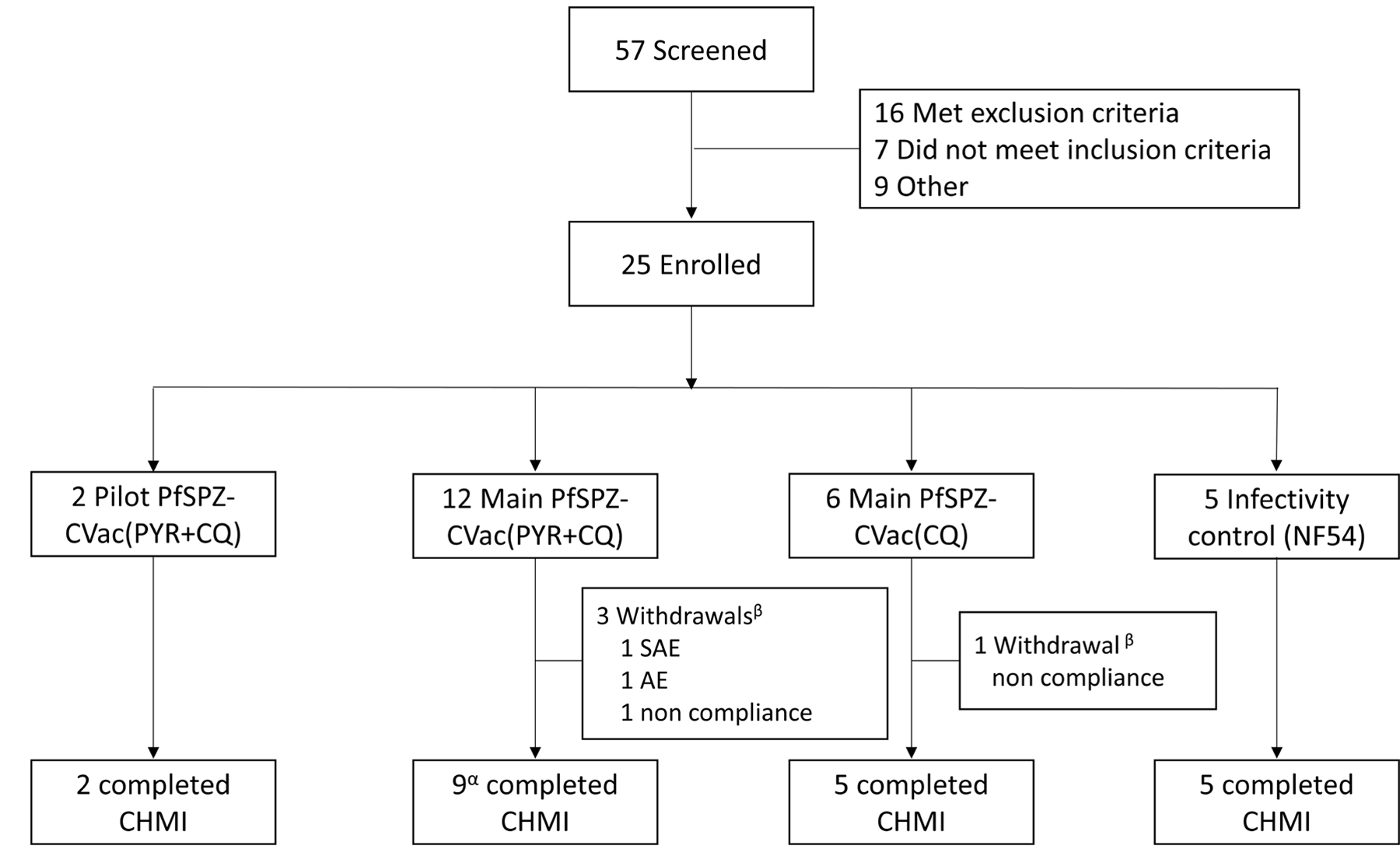
αOne participant was treated at day 16 after CHMI although that participant did complete the end-of-study visit. βThe serious adverse event (SAE) and adverse event (AE) in the PYR/CQ group were encephalopathy and eye irritation, respectively. The non-compliance incidents include a participant’s unwillingness to remain on pregnancy prevention (PYR/CQ group) and a work conflict preventing the participant from attending scheduled visits (CQ group).
Extended Data Fig. 2 |. Detection of parasitaemia in volunteers by qPCR after low-dose PfSPZ-CVac.
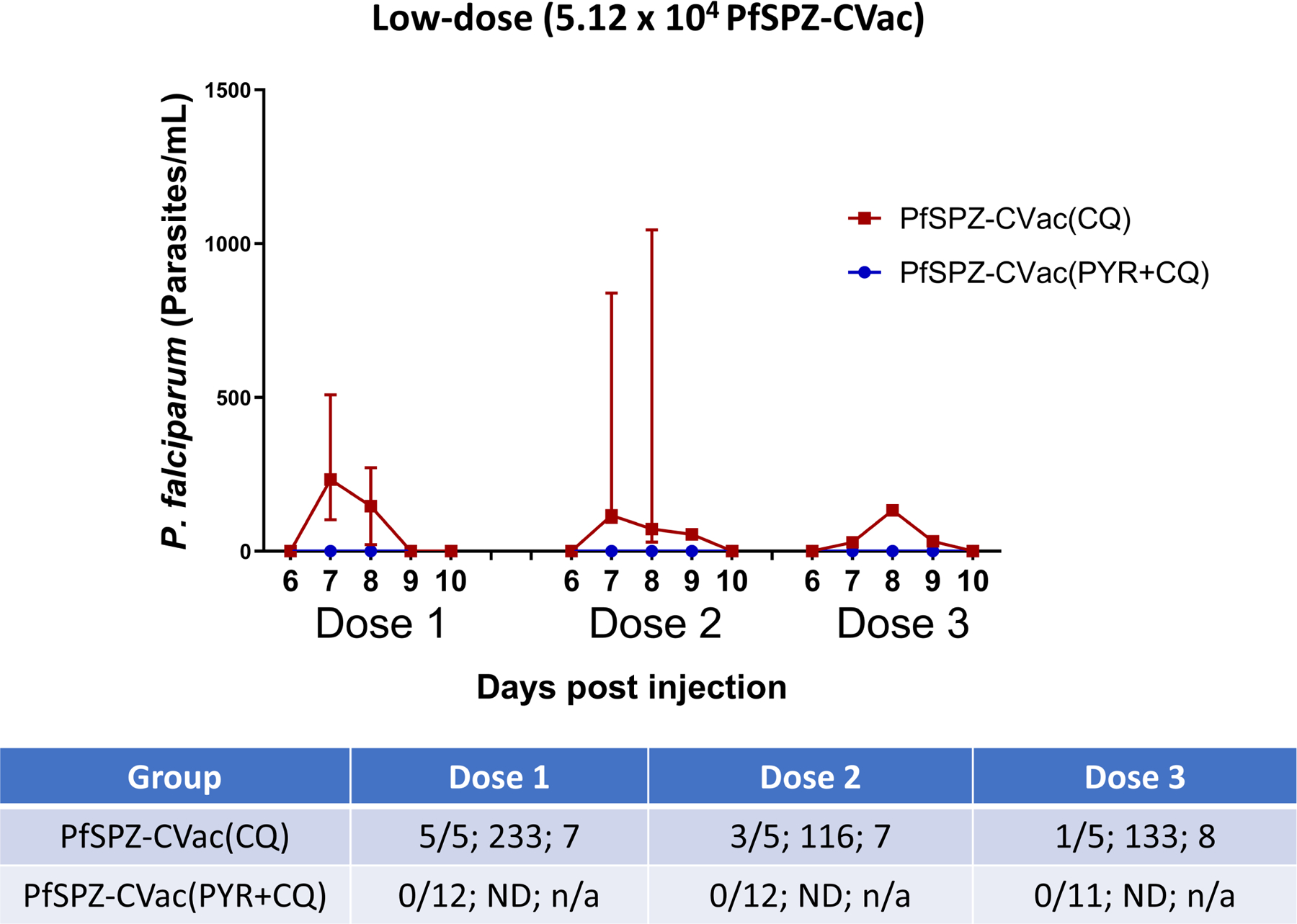
Parasitaemia was measured in participants on days 6–10 after each PfSPZ-CVac dose. The median parasitaemia is displayed for positive participants and error bars indicate the IQR. Vax, PfSPZ Challenge + CQ or PYR/CQ administration and follow-up until day 14 after PfSPZ Challenge 1 and day 10 after PfSPZ Challenge doses 2 and 3. The table shows (from left to right in each cell): the number of participants who were positive by qPCR/the injected number of participants; the median peak parasite density of positive participants (parasites per ml); and the average day of peak parasite density after each dose of PfSPZ-CVac. ND, not detected; n/a, not applicable.
Extended Data Fig. 3 |. Protective efficacy of PfSPZ-CVac(CQ) and PfSPZ-CVac(PYR + CQ) against homologous PfSPZ CHMI in the PfSPZ-CVac low-dose study.
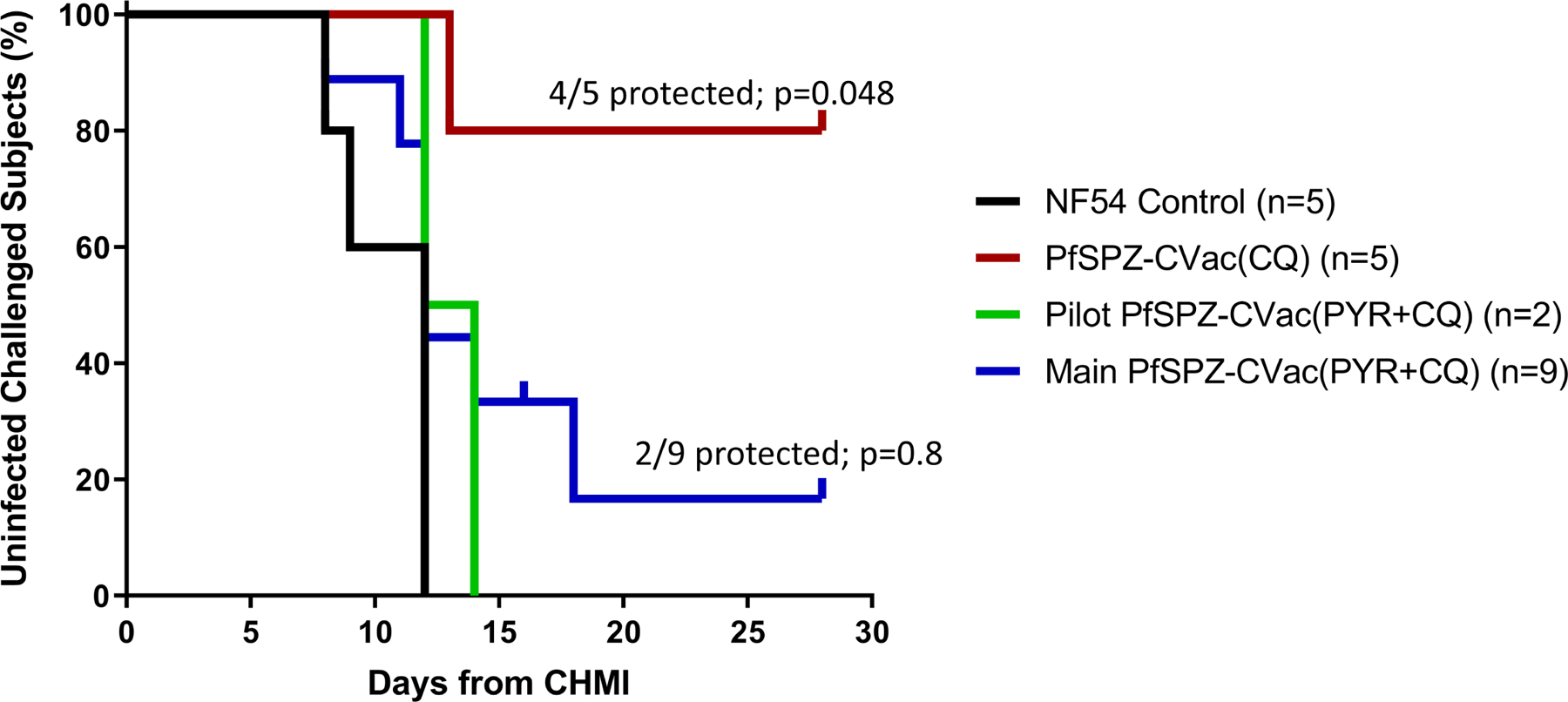
Participants were followed for 27 days after CHMI, and a survival curve is presented displaying the number of participants that remained protected throughout follow-up after homologous CHMI. To assess vaccine efficacy, the unvaccinated group was compared with the vaccine group for time to first detectable parasitaemia after the final challenge using a log-rank test, and the presence or absence of parasitaemia using a two-tailed Barnard’s test. In the pilot PfSPZ-CVac(PYR + CQ) group (to test safety), 0/2 participants were protected; in the main PfSPZ-CVac(PYR+CQ) group, 2/9 (22.2%, P = 0.8) participants were protected; in the PfSPZ-CVac(CQ) group, 4/5 (80%, P = 0.048; 95% confidence interval, 1–99%) participants were protected.
Extended Data Fig. 4 |. Trial profile for the PfSPZ-CVac high-dose study.
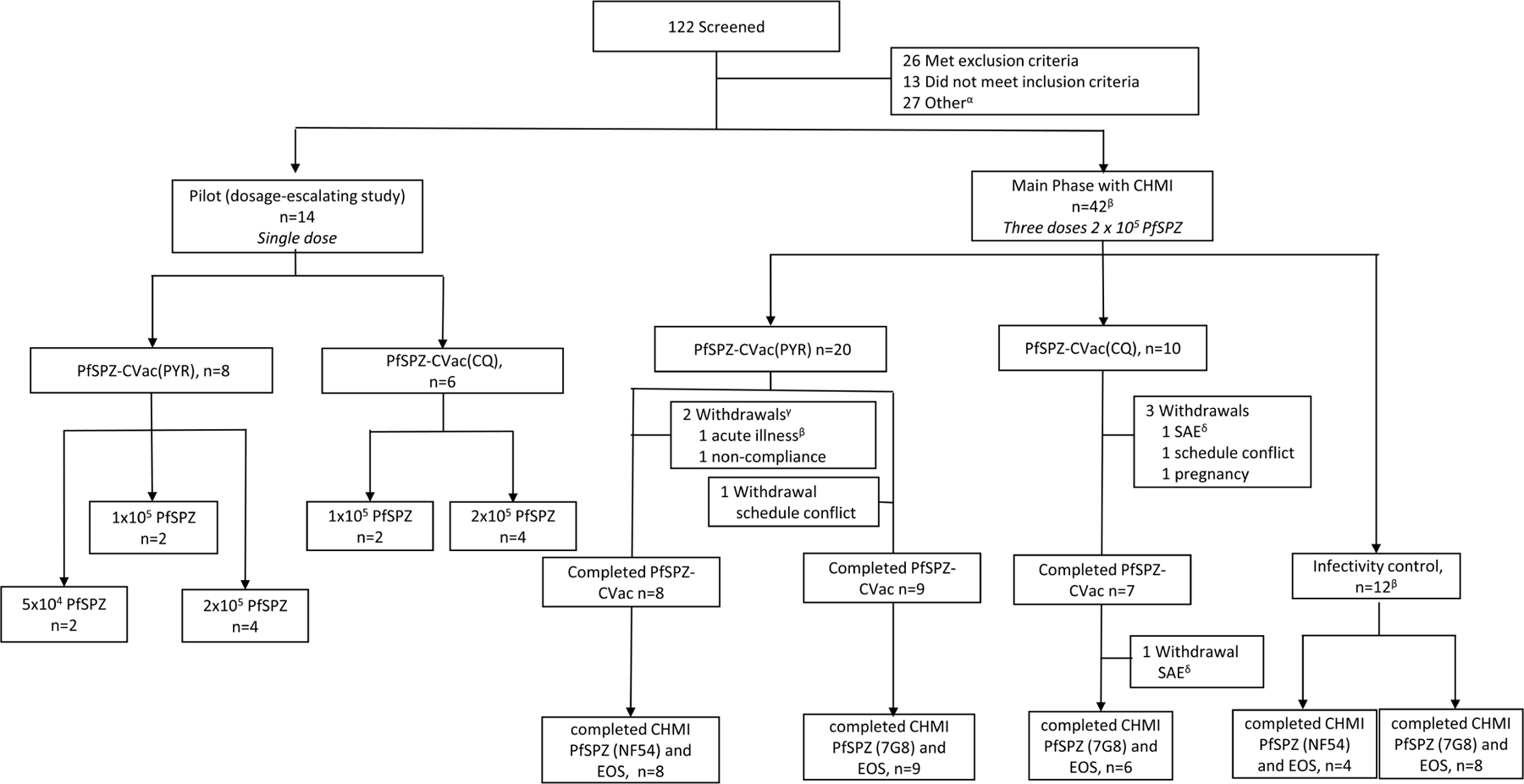
αOther, 15 withdrew consent, 6 deemed eligible after study was fully enrolled, 3 lost to follow-up, 2 schedule conflict, 1 withdrawn before randomization. βOne individual enrolled in the PYR group was withdrawn before receiving the first vaccination owing to acute illness, and enrolled in the control group. This person is counted twice. γBoth participants were withdrawn before receiving vaccination 1. δThe two serious adverse events in the CQ group were mental status change and pneumothorax.
Extended Data Fig. 5 |. Vδ2 γδ T cells at baseline in protected and infected individuals who received the vaccine.
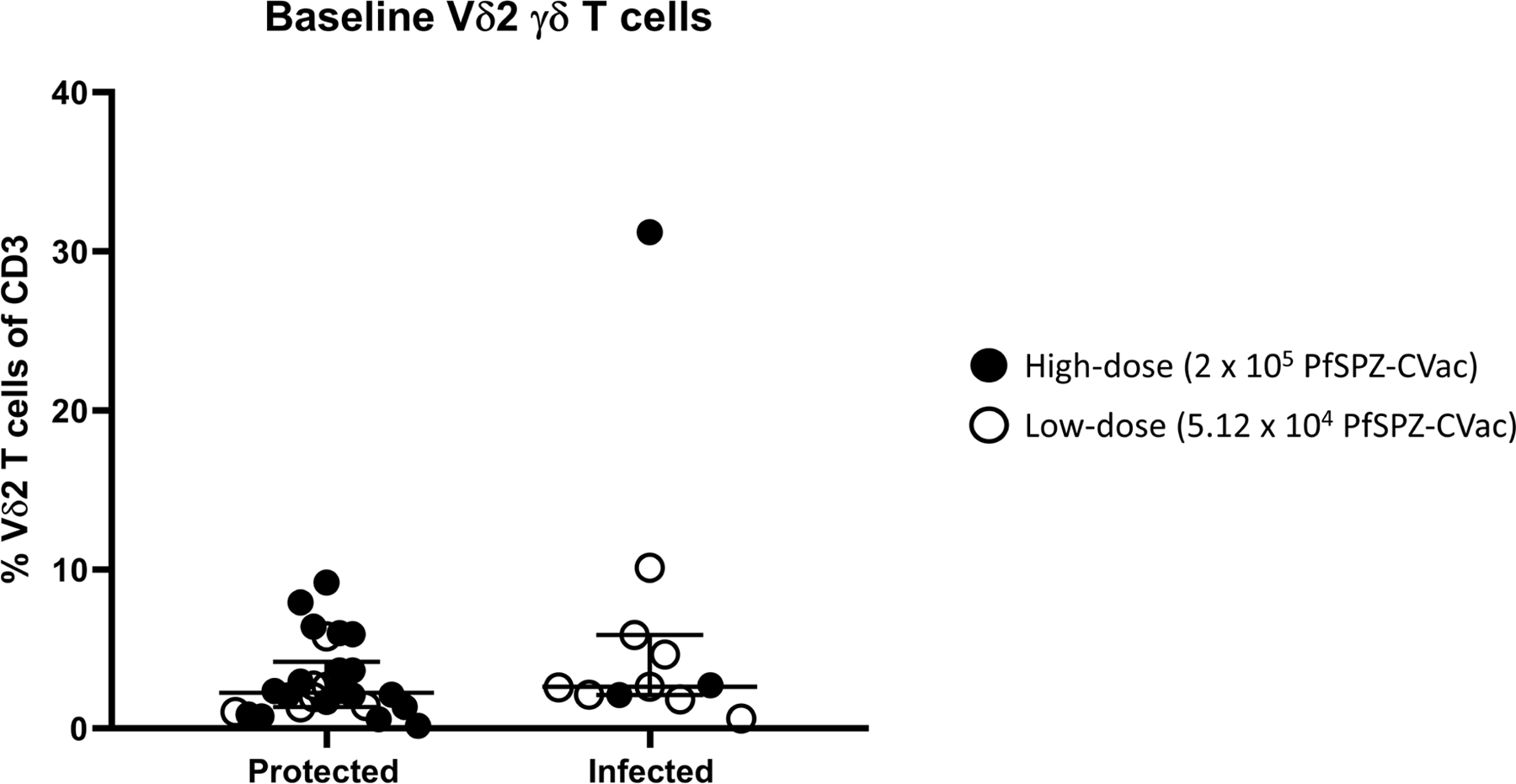
Filled circles indicate vaccinated participants in the high-dose study, and open circles indicate vaccinated participants in the low-dose study. Median values are displayed and error bars indicate the IQR. For protected individuals in the high-dose study, n = 20; for protected individuals in the low-dose study, n = 6. For infected individuals in the high-dose study, n = 3; for infected individuals in the low-dose study, n = 8.
Extended Data Fig. 6 |. Anti-PfCSP IgM antibodies in vaccinated participants in the PfSPZ-CVac high-dose study analysed using ELISA.
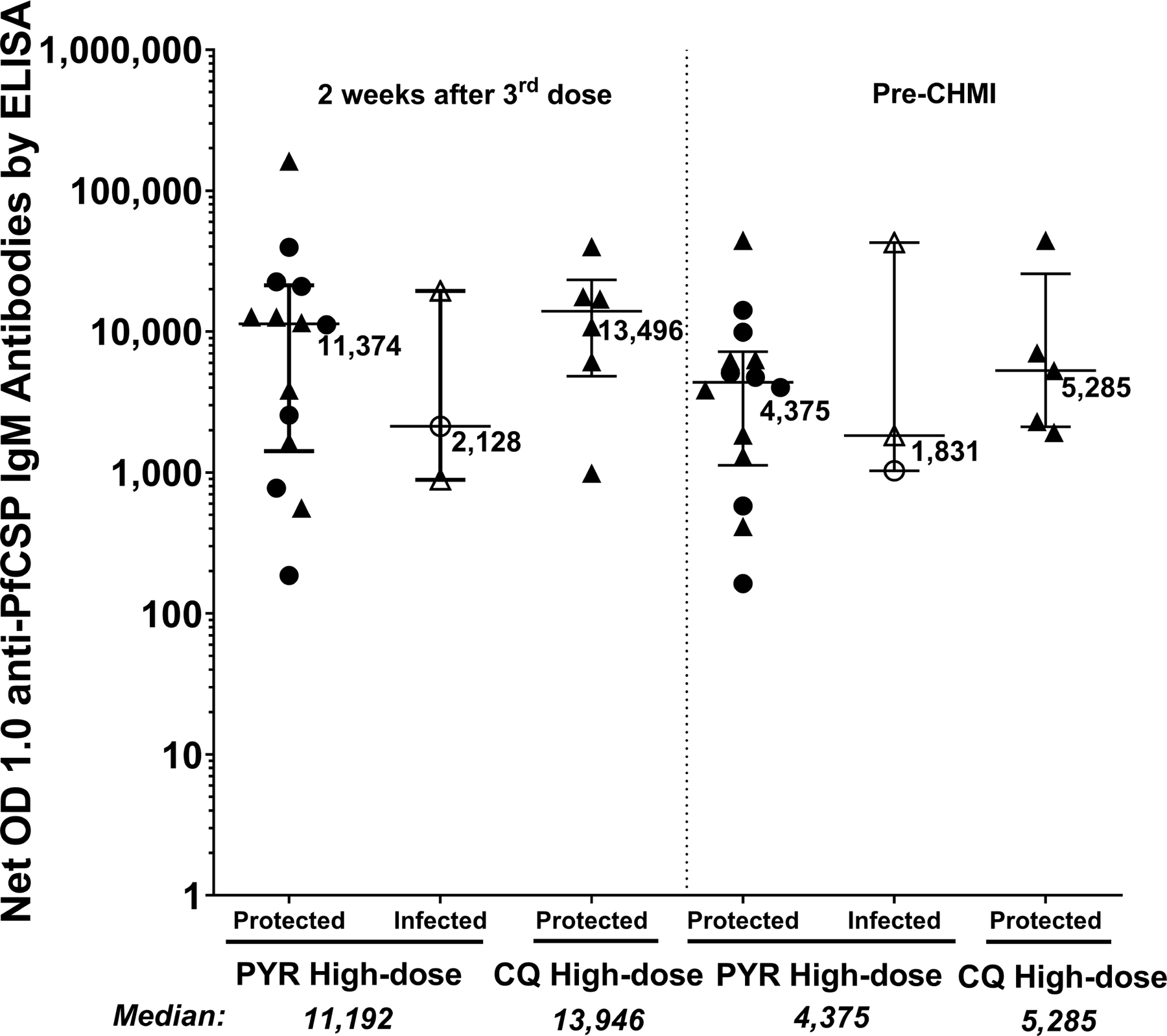
IgM antibodies against PfCSP (net OD 1.0) 2 weeks after the third dose of PfSPZ-CVac(PYR) and PfSPZ-CVac(CQ); and immediately before CHMI. Median values are displayed and error bars indicate the IQR. At both time points, sample sizes for the PYR high-dose study, n = 14 for protected and n = 3 for infected vaccinated participants; for the CQ high-dose study, n = 6 for protected vaccinated individuals at 2 weeks after the third CVac and n = 5 for protected vaccinated individuals at before CHMI. No vaccinated participants were infected in the CQ high-dose study.
Extended Data Fig. 7 |. Antibody responses in vaccinated participants of the PfSPZ-CVac low-dose and PfSPZ-CVac high-dose studies analysed using automated immunofluorescence and automated inhibition of sporozoite invasion assays.
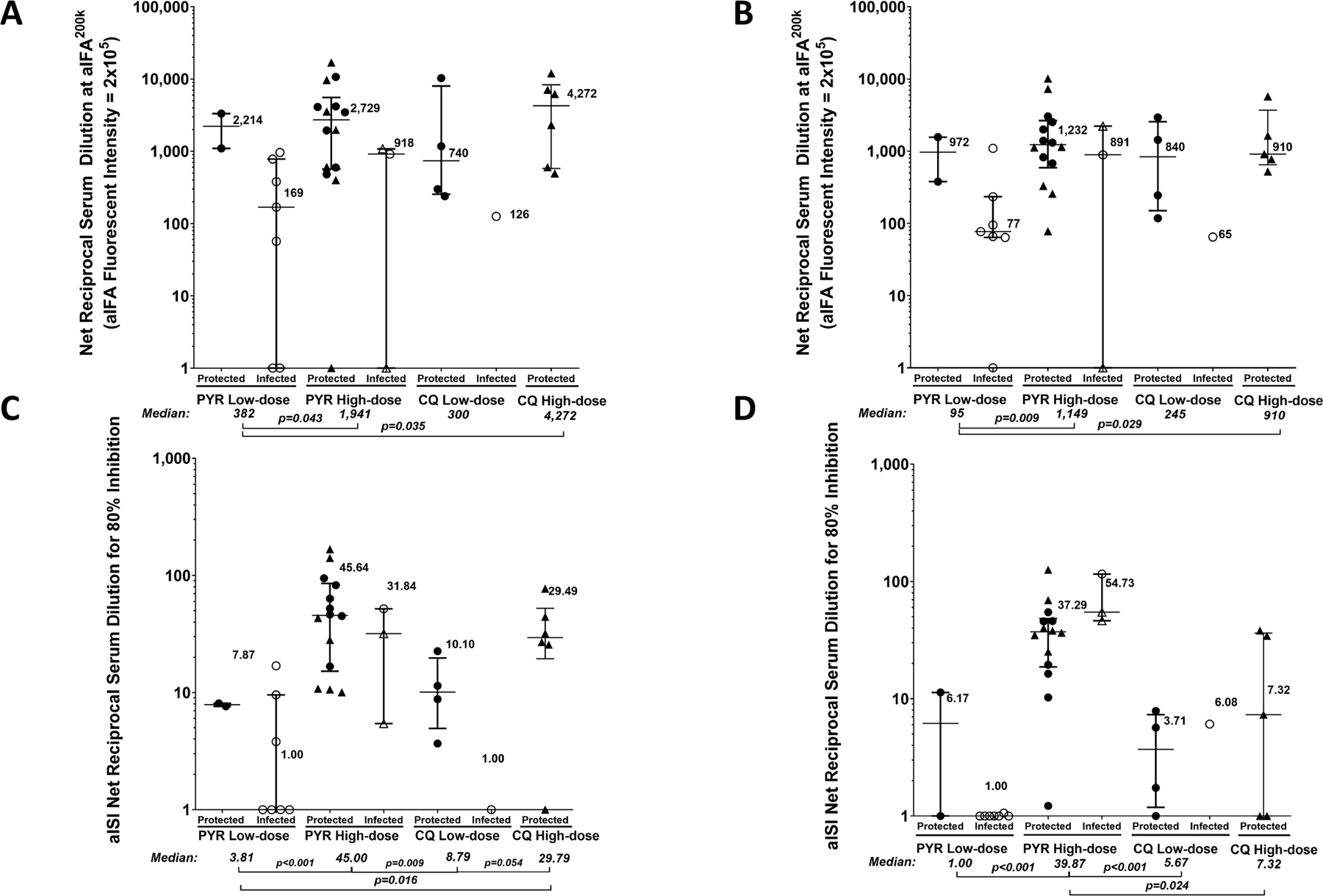
For each panel, filled circles are protected participants, open circles are infected participants who received homologous CHMI, filled triangles are uninfected (protected) participants and open triangles are infected participants who received heterologous CHMI. Median values are displayed and error bars indicate the IQR. P values were calculated using two-sided Wilcoxon–Mann–Whitney tests and group differences with P > 0.07 are not indicated. a, b, Antibodies against PfCSP by automated immunofluorescence assay were analysed 2 weeks after the third dose of PfSPZ-CVac(PYR) and PfSPZ-CVac(CQ) (a) and immediately before CHMI (b). c, d, Antibodies against PfCSP by automated inhibition of sporozoite invasion were analysed 2 weeks after the third dose of PfSPZ-CVac(PYR) and PfSPZ-CVac(CQ) (c) and immediately before CHMI (d). At both time points, sample sizes for the PYR low-dose study, n = 2 for protected and n = 7 for infected vaccinated participants; for the PYR high-dose study, n = 14 for protected and n = 3 for infected vaccinated participants; for the CQ low-dose study, n = 4 for protected and n = 1 for infected vaccinated participants; for the CQ high-dose study, n = 6 for protected participants in a and c (2 weeks after the third CVac), and n = 5 for protected participants in b and d (before CHMI). No vaccinated participants were infected in the CQ high-dose study. Anti-PfCSP antibodies at both 2 weeks after the last dose (high-dose 1,941 versus low-dose 382, P = 0.043) and immediately before CHMI (high-dose 1,149 versus low-dose 95, P = 0.009) were significantly higher in the high-dose PfSPZ-CVac(PYR) participants compared with the low-dose PfSPZ-CVac(PYR) participants. In PfPSZ-CVac(CQ) participants, the anti-PfSPZ antibodies were higher but not significant (2 weeks after the third dose, 300 versus 4,272, P = 0.176; before CHMI, 245 versus 910, P = 0.310). The inhibition of sporozoite invasion assay showed a statistically significant increase after vaccination for the high-dose PfSPZ-CVac(PYR) versus low-dose PfSPZ-CVac(PYR) when measured 2 weeks after the third dose (45.00 versus 3.81, P < 0.001) and immediately before CHMI (39.87 versus 1.00, P < 0.001). In PfPSZ-CVac(CQ) participants, the anti-PfCSP antibodies were significantly higher in the high-dose group when measured 2 weeks after the third dose (29.79 versus 8.79, P = 0.054) but not significantly different before CHMI (5.67 versus 7.32, P = 0.671).
Extended Data Fig. 8 |. Detection of parasitaemia by qPCR in each individual after the first, second and third dose of PfSPZ-CVac in the high-dose study.
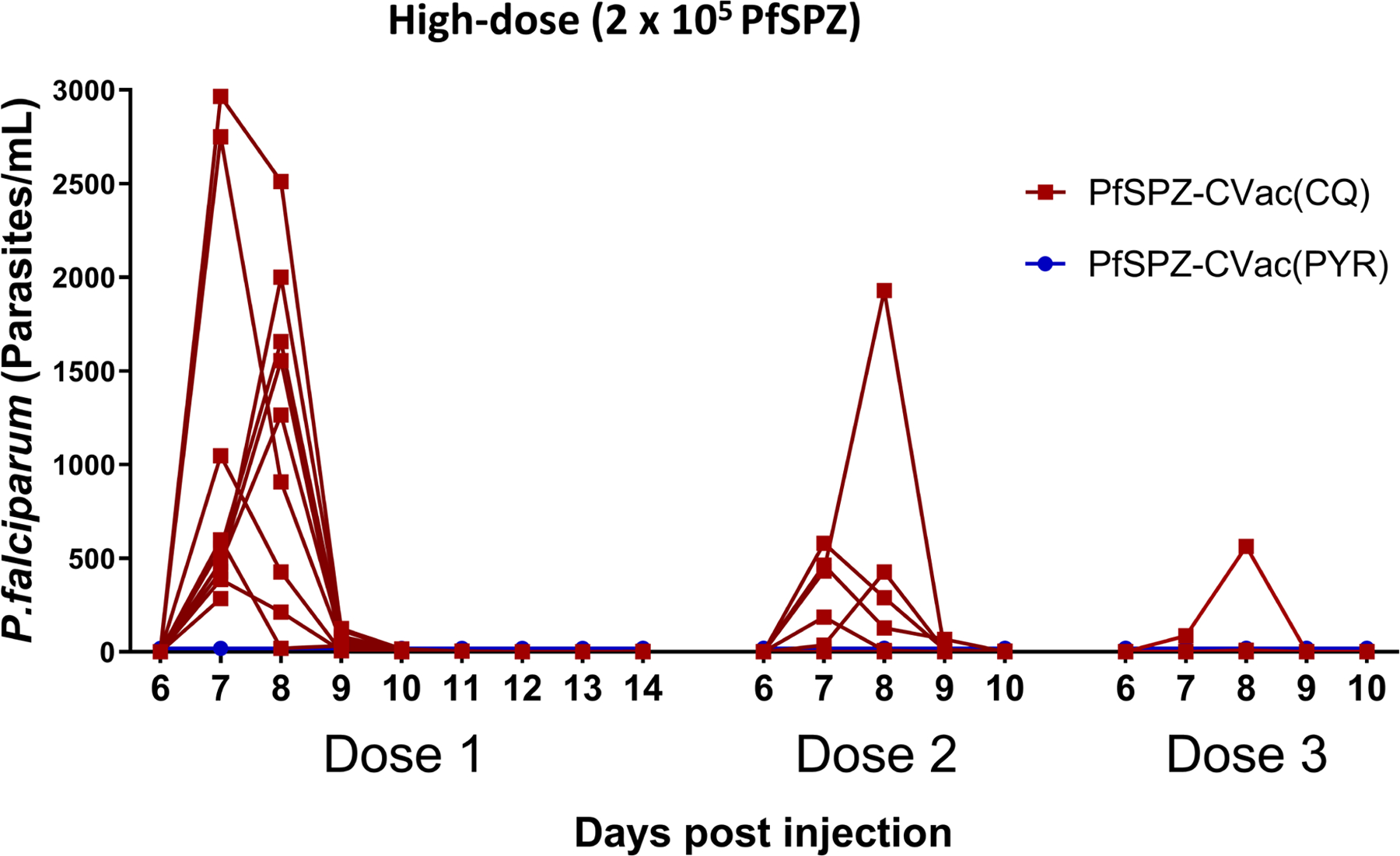
Parasitaemia was measured in participants on days 6–10 after each PfSPZ-CVac dose. Vax, PfSPZ Challenge + CQ or PYR administration and follow-up until day 14 after PfSPZ Challenge 1 and day 10 after PfSPZ Challenge doses 2 and 3.
Extended Data Fig. 9 |. In vitro PYR activity against liver-stage parasites.
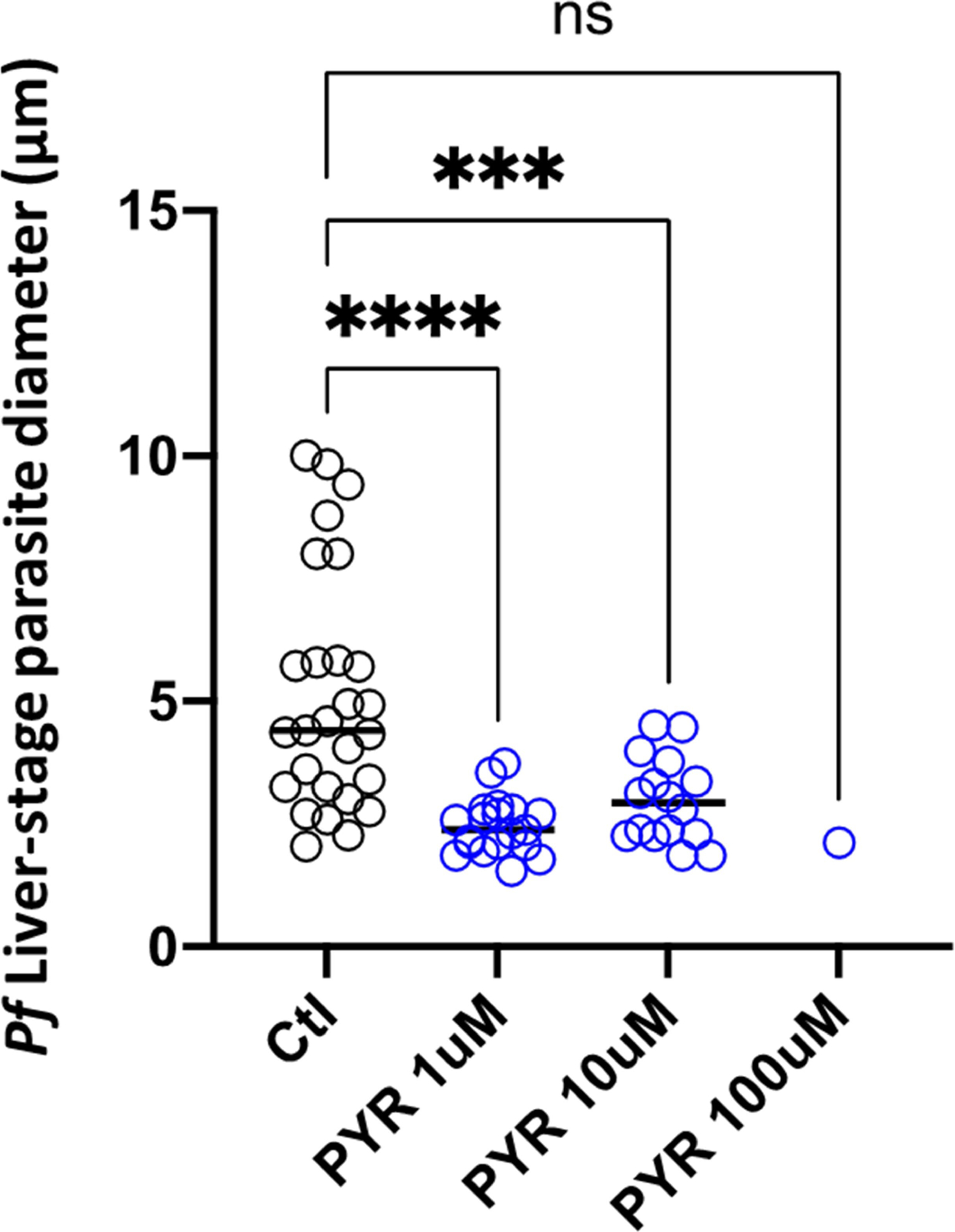
PYR was added to the cultures (blue) at three concentrations (1 μM, 10 μM and 100 μM), starting 2 days after infection and replaced with daily medium changes until day 4, at which point the cultures were fixed. The diameter of representative parasites of each group on day 4 are shown. Ctl, control. Statistical significance was determined by one-sided ANOVA. ***P < 0.001 (exact P = 0.0006); ****P < 0.0001; ns, not significant (exact P = 0.2335).
Extended Data Fig. 10 |. Flow cytometry gating strategy used to enumerate T cell subsets using flow cytometry ex vivo staining of whole blood.
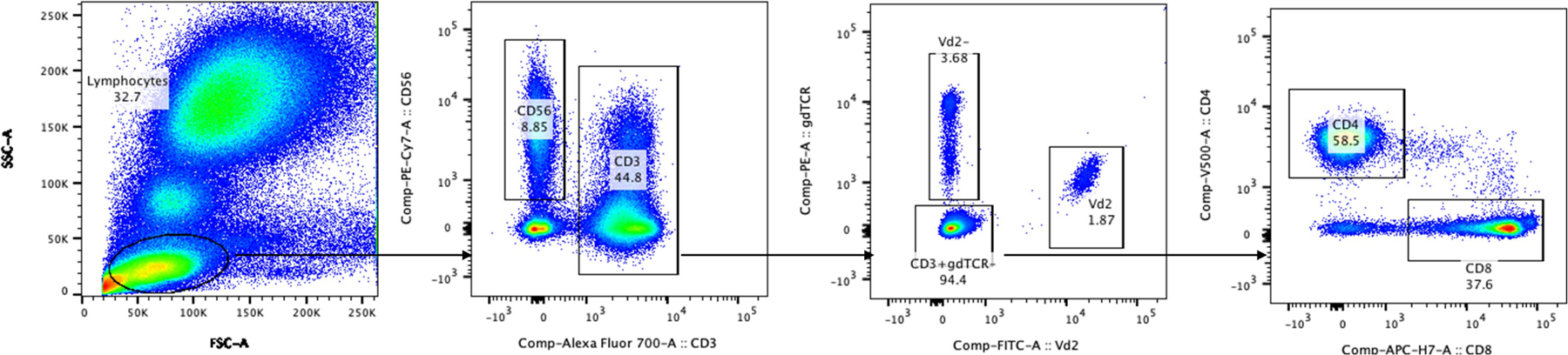
The FSC/SSC parameters were used to define the lymphocyte gate as the first gate in the whole blood ex vivo assay. Within the lymphocyte gate, CD3-Alexa-700-positive events (T cell gate) were gated against CD56-PE-Cy7-positive events (NK cells). In the T cell gate, plotting Vδ2-FITC versus gdTCR-PE identified discrete Vδ2+, Vδ2− and CD3+γδTCR− populations. The T cell gate was used to further delineate CD4- and CD8-positive events.
Supplementary Material
Acknowledgements
We thank the participants of the vaccine trial for their contribution and commitment to vaccine research. This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health and by the NIAID grants U01AI109700-01 and 2R44AI058375-11 to Sanaria for vaccine manufacture and trial execution. S.N.B. is a Howard Hughes Medical Institute Investigator. We thank the Sanaria and Protein Potential legal, administrative, manufacturing, quality, logistics, pharmaceutical operations, regulatory and clinical teams for their support of this project; T. Bauch, U. Desai, M. Mahoney, L. Walker, L. Williams of the Department of Laboratory Medicine, NIH CC for performing the malaria PCR diagnostic tests; N. Nerurkar for establishing and infecting the micropatterned co-cultures of hepatocytes; the LMIV and NIH CC clinical, pharmacy, immunology, laboratory, data, sample and project management teams and, in particular, NIH CC Outpatient Clinic 8 and Day Hospital Staffing; and J. Patrick Gorres for coordinating the preparation of the manuscript.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-021-03684-z.
Competing interests T.M., L.W.P.C., A.M., A.G., B.K.L.S., N.K.C., S.C., P.F.B., E.R.J., T.L.R. and S.L.H. are salaried, full-time employees of Sanaria, the developer and sponsor of Sanaria PfSPZ Vaccine. S.L.H. and B.K.L.S. also have financial interests in Sanaria. B.K.L.S. and S.L.H. are inventors on patents and patent applications that have been assigned to Sanaria. All other authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-021-03684-z.
Peer review information Nature thanks Stefan Kappe and Laurent Renia for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
The data supporting the findings of this study are available within the Article and its Supplementary Information and from the corresponding author upon reasonable request.
References
- 1.WHO. World Malaria Report 2019 (World Health Organization, 2019). [Google Scholar]
- 2.The RTS,S Clinical Trials Partnership. Efficacy and safety of the RTS,S/AS01 malaria vaccine during 18 months after vaccination: a phase 3 randomized, controlled trial in children and young infants at 11 African sites. PLoS Med. 11, e1001685 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.RTS,S Clinical Trials Partnership. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet 386, 31–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The RTS,S Clinical Trials Partnership. A phase 3 trial of RTS,S/AS01 malaria vaccine in African infants. N. Engl. J. Med. 367, 2284–2295 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epstein JE et al. Protection against Plasmodium falciparum malaria by PfSPZ vaccine. JCI Insight 2, e89154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jongo SA et al. Safety, immunogenicity, and protective efficacy against controlled human malaria infection of Plasmodium falciparum sporozoite vaccine in Tanzanian adults. Am. J. Trop. Med. Hyg. 99, 338–349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lyke KE et al. Attenuated PfSPZ vaccine induces strain-transcending T cells and durable protection against heterologous controlled human malaria infection. Proc. Natl Acad. Sci. USA 114, 2711–2716 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seder RA et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 341, 1359–1365 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Sissoko MS et al. Safety and efficacy of PfSPZ vaccine against Plasmodium falciparum via direct venous inoculation in healthy malaria-exposed adults in Mali: a randomised, double-blind phase 1 trial. Lancet Infect. Dis. 17, 498–509 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sissoko MS et al. Three dose regimen of PfSPZ vaccine protects adult Malians against Plasmodium falciparum through an intense transmission season: a randomised, controlled phase I trial. Lancet Infect. Dis. (in the press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butler NS et al. Superior antimalarial immunity after vaccination with late liver stage-arresting genetically attenuated parasites. Cell Host Microbe 9, 451–462 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kublin JG et al. Complete attenuation of genetically engineered sporozoites in human subjects. Sci. Transl. Med. 9, eaad9099 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Roestenberg M et al. A double-blind, placebo-controlled phase 1/2a trial of the genetically attenuated malaria vaccine PfSPZ-GA1. Sci. Transl. Med. 12, eaaz5629 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Roestenberg M et al. Protection against a malaria challenge by sporozoite inoculation. N. Engl. J. Med. 361, 468–477 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Bijker EM et al. Sporozoite immunization of human volunteers under mefloquine prophylaxis is safe, immunogenic and protective: a double-blind randomized controlled clinical trial. PLoS ONE 9, e112910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sahu T et al. Chloroquine neither eliminates liver stage parasites nor delays their development in a murine chemoprophylaxis vaccination model. Front. Microbiol. 6, 283 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffman SL et al. Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites. J. Infect. Dis. 185, 1155–1164 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Mordmüller B et al. Sterile protection against human malaria by chemoattenuated PfSPZ vaccine. Nature 542, 445–449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schats R et al. Heterologous protection against malaria after immunization with Plasmodium falciparum sporozoites. PLoS ONE 10, e0124243 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walk J et al. Modest heterologous protection after Plasmodium falciparum sporozoite immunization: a double-blind randomized controlled clinical trial. BMC Med. 15, 168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friesen J, Borrmann S & Matuschewski K Induction of antimalaria immunity by pyrimethamine prophylaxis during exposure to sporozoites is curtailed by parasite resistance. Antimicrob. Agents Chemother. 55, 2760–2767 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friesen J & Matuschewski K Comparative efficacy of pre-erythrocytic whole organism vaccine strategies against the malaria parasite. Vaccine 29, 7002–7008 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Healy SA et al. Chemoprophylaxis vaccination: phase I study to explore stage-specific immunity to Plasmodium falciparum in US adults. Clin. Infect. Dis. 71, 1481–1490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Epstein JE & Richie TL The whole parasite, pre-erythrocytic stage approach to malaria vaccine development: a review. Curr. Opin. Infect. Dis. 26, 420–428 (2013). [DOI] [PubMed] [Google Scholar]
- 25.March S et al. A microscale human liver platform that supports the hepatic stages of Plasmodium falciparum and vivax. Cell Host Microbe 14, 104–115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.March S et al. Micropatterned coculture of primary human hepatocytes and supportive cells for the study of hepatotropic pathogens. Nat. Protocols 10, 2027–2053 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gural N et al. In vitro culture, drug sensitivity, and transcriptome of Plasmodium vivax hypnozoites. Cell Host Microbe 23, 395–406.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White NJ Clinical pharmacokinetics of antimalarial drugs. Clin. Pharmacokinet. 10, 187–215 (1985). [DOI] [PubMed] [Google Scholar]
- 29.Ishizuka AS et al. Protection against malaria at 1 year and immune correlates following PfSPZ vaccination. Nat. Med. 22, 614–623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaidi I et al. γδ T cells are required for the induction of sterile immunity during irradiated sporozoite vaccinations. J. Immunol. 199, 3781–3788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nahrendorf W, Scholzen A, Sauerwein RW & Langhorne J Cross-stage immunity for malaria vaccine development. Vaccine 33, 7513–7517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nahrendorf W et al. Blood-stage immunity to Plasmodium chabaudi malaria following chemoprophylaxis and sporozoite immunization. eLife 4, e05165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moser KA et al. Strains used in whole organism Plasmodium falciparum vaccine trials differ in genome structure, sequence, and immunogenic potential. Genome Med. 12, 6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bijker EM et al. Protection against malaria after immunization by chloroquine prophylaxis and sporozoites is mediated by preerythrocytic immunity. Proc. Natl Acad. Sci. USA 110, 7862–7867 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belnoue E et al. Protective T cell immunity against malaria liver stage after vaccination with live sporozoites under chloroquine treatment. J. Immunol. 172, 2487–2495 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Lyke KE et al. Multidose priming and delayed boosting improve Plasmodium falciparum sporozoite vaccine efficacy against heterologous P. falciparum controlled human malaria infection. Clin. Infect. Dis. ciaa1294 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Hoffman SL & Doolan DL Malaria vaccines-targeting infected hepatocytes. Nat. Med. 6, 1218–1219 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Epstein JE et al. Live attenuated malaria vaccine designed to protect through hepatic CD8+ T cell immunity. Science 334, 475–480 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Issiaka D et al. Impact of seasonal malaria chemoprevention on hospital admissions and mortality in children under 5 years of age in Ouelessebougou, Mali. Malar. J. 19, 103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jongo SA et al. Safety and differential antibody and T-cell responses to the Plasmodium falciparum sporozoite malaria vaccine, PfSPZ vaccine, by age in Tanzanian adults, adolescents, children, and infants. Am. J. Trop. Med. Hyg. 100, 1433–1444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sulyok Z et al. Heterologous protection against malaria by a simple chemoattenuated PfSPZ vaccine regimen in a randomized trial. Nat. Commun. 12, 2518 (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spring M et al. First-in-human evaluation of genetically attenuated Plasmodium falciparum sporozoites administered by bite of Anopheles mosquitoes to adult volunteers. Vaccine 31, 4975–4983 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Goswami D et al. A replication-competent late liver stage-attenuated human malaria parasite. JCI Insight 5, e135589 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roestenberg M et al. Controlled human malaria infections by intradermal injection of cryopreserved Plasmodium falciparum sporozoites. Am. J. Trop. Med. Hyg. 88, 5–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mordmüller B et al. Direct venous inoculation of Plasmodium falciparum sporozoites for controlled human malaria infection: a dose-finding trial in two centres. Malaria J. 14, 117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laurens MB et al. Dose-dependent infectivity of aseptic, purified, cryopreserved Plasmodium falciparum 7G8 sporozoites in malaria-naive adults. J. Infect. Dis. 220, 1962–1966 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Divis PC, Shokoples SE, Singh B & Yanow SK A TaqMan real-time PCR assay for the detection and quantitation of Plasmodium knowlesi. Malar. J. 9, 344 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seilie AM et al. Beyond blood smears: qualification of Plasmodium 18S rRNA as a biomarker for controlled human malaria infections. Am. J. Trop. Med. Hyg. 100, 1466–1476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luethy PM et al. Diagnostic challenges of prolonged post-treatment clearance of Plasmodium nucleic acids in a pre-transplant autosplenectomized patient with sickle cell disease. Malar. J. 17, 23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rougemont M et al. Detection of four Plasmodium species in blood from humans by 18S rRNA gene subunit-based and species-specific real-time PCR assays. J. Clin. Microbiol. 42, 5636–5643 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gabriel EE, Nason M, Fay MP & Follmann DA A boundary-optimized rejection region test for the two-sample binomial problem. Stat. Med. 37, 1047–1058 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the Article and its Supplementary Information and from the corresponding author upon reasonable request.