

Abstract
Rhodaneses catalyze the transfer of the sulfane sulfur from thiosulfate or thiosulfonates to thiophilic acceptors such as cyanide and dithiols. In this work, we define for the first time the gene, and hence the amino acid sequence, of a 12-kDa rhodanese from Escherichia coli. Well-characterized rhodaneses are comprised of two structurally similar ca. 15-kDa domains. Hence, it is thought that duplication of an ancestral rhodanese gene gave rise to the genes that encode the two-domain rhodaneses. The glpE gene, a member of the sn-glycerol 3-phosphate (glp) regulon of E. coli, encodes the 12-kDa rhodanese. As for other characterized rhodaneses, kinetic analysis revealed that catalysis by purified GlpE occurs by way of an enzyme-sulfur intermediate utilizing a double-displacement mechanism requiring an active-site cysteine. The Kms for SSO32− and CN− were 78 and 17 mM, respectively. The apparent molecular mass of GlpE under nondenaturing conditions was 22.5 kDa, indicating that GlpE functions as a dimer. GlpE exhibited a kcat of 230 s−1. Thioredoxin 1 from E. coli, a small multifunctional dithiol protein, served as a sulfur acceptor substrate for GlpE with an apparent Km of 34 μM when thiosulfate was near its Km, suggesting that thioredoxin 1 or related dithiol proteins could be physiological substrates for sulfurtransferases. The overall degree of amino acid sequence identity between GlpE and the active-site domain of mammalian rhodaneses is limited (∼17%). This work is significant because it begins to reveal the variation in amino acid sequences present in the sulfurtransferases. GlpE is the first among the 41 proteins in COG0607 (rhodanese-related sulfurtransferases) of the database Clusters of Orthologous Groups of proteins (http://www.ncbi.nlm.nih.gov/COG/) for which sulfurtransferase activity has been confirmed.
Genes of known function belonging to the glp regulon of Escherichia coli encode proteins that are responsible for the metabolism of sn-glycerol 3-phosphate (glycerol-P) and its precursors, glycerol and glycerophosphodiesters (38). The genes comprising this regulon belong to five operons. Transcription of all but the glpEGR operon is negatively regulated by the glp repressor GlpR, a member of the DeoR family of transcriptional regulators (34, 65, 69–72). Operon glpACB, encoding the subunits of the anaerobic glycerol-P dehydrogenase, is located near min 51 of the E. coli genome (38). Divergently transcribed from glpACB is glpTQ. The genes glpT and glpQ encode glycerol-P permease and periplasmic glycerophosphodiesterase, respectively (38). The glpFKX operon, at min 89, encodes glycerol diffusion facilitator, glycerol kinase, and a fructose 1,6-bisphophatase (38; J. L. Donahue, J. L. Bownas, W. G. Niehaus, Jr., and T. J. Larson, unpublished data). The genes glpE and glpG, together with the gene encoding the transcriptional repressor, glpR, form a complex operon at min 77 that is divergently transcribed from glpD (71). The gene glpD encodes the aerobic glycerol-P dehydrogenase (38).
Prior to this study, the functions of the GlpE protein and the cytoplasmic membrane-associated GlpG protein were unknown (71, 72). The function of GlpG remains unknown. However, as reported by Tatusov et al. (58) and more recently by Hofmann et al. (23), GlpE exhibits sequence similarity to a superfamily of transfer proteins including the sulfurtransferases and the tyrosine and dual-specificity phosphatases. In this work we show that GlpE is a thiosulfate:cyanide sulfurtransferase (EC 2.8.1.1), an enzyme traditionally given the name rhodanese. Rhodaneses catalyze the transfer of the sulfane sulfur from thiosulfate to cyanide, forming thiocyanate and sulfite:
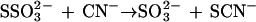 |
Although sulfurtransferases are present in many types of organisms from all three domains of life (57), their physiological roles are still in question. Proposed roles include cyanide detoxification (61), sulfur metabolism (15, 66), and mobilization of sulfur for iron-sulfur cluster biosynthesis or repair (9, 10, 51, 52).
At least two distinct rhodaneses and the related enzyme mercaptopyruvate sulfurtransferase have been described in E. coli, but the genes for these enzymes have not been mapped nor have the sequences for the proteins been determined (3, 60, 62). In this work, we define for the first time the gene, and hence the amino acid sequence, of a rhodanese from E. coli. In the database Clusters of Orthologous Groups of proteins (57), there are 41 proteins in COG0607 (rhodanese-related sulfurtransferases). To our knowledge, GlpE is the first of these proteins for which sulfurtransferase activity has been confirmed.
MATERIALS AND METHODS
Materials.
Unless listed below, the reagents used were purchased from Sigma Chemical Company or Fisher Scientific. Synthetic oligonucleotides were prepared with an Applied Biosystems DNA synthesizer (model 381A) using reagents supplied by Cruachem. New England Biolabs supplied restriction endonucleases and reagents for PCR and cloning. Ammonium thiosulfate and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) were purchased from Aldrich. Ferric nitrate and the sodium salt of mercaptopyruvate were purchased from ICN Pharmaceuticals, Inc. Promega supplied thioredoxin 1 from E. coli. Thioredoxin reductase purified from E. coli was a generous gift from C. H. Williams, Jr. (68).
Bacterial strains andplasmids.
The bacterial strains and plasmids used or constructed are listed in Table 1. BL21(DE3) harboring pGZ105 was used to overexpress GlpE for purification. Plasmids pGZ154 and pGZ132, in which glpE is controlled by the tetracycline-inducible PN25 promoter, were used to overexpress GlpE in DH5αZ1. To construct these plasmids, the glpE gene was amplified from pGZ105 by PCR with the primers 5′-acgAAttcccGctagCaat-3′ and 5′-tcactagtttgacagcttatc-3′, where uppercase letters indicate mismatches used for the creation of restriction sites. After cleavage with EcoRI and XbaI, the amplified product was cloned into the same sites of either pZA3 PN25-luc or pZE2 PN25-luc (42) to create pGZ154 and pGZ132, respectively. Plasmid pATCBOA2+6 (34) was used for constitutive expression of the periplasmic marker alkaline phosphatase.
TABLE 1.
E. coli strains and plasmids
| Strain or plasmid | Description | Reference |
|---|---|---|
| Strains | ||
| BL21(DE3) | hsdS gal (λcIts857 ind-1 Sam7 nin-5 lacUV5-T7 gene 1) | 55 |
| DH5αZ1 | (φ80d lacZΔM15) endA1 recA1 hsdR17 supE44 thi-1 gyrA relA1 Δ(lacZYA-argF)U169 (λatt lacIqtetR Spr) | 42 |
| Plasmids | ||
| pT7-7 | ColE1 origin Apr T7 promoter | 56 |
| pGZ105 | glpE in NdeI/HindIII fragment of pT7-7 | 72 |
| pZE2 PN25-luc | ColE1 origin Kmr PN25-luc | 42 |
| pZA3 PN25-luc | p15A origin Cmr PN25-luc | 42 |
| pGZ154 | glpE in EcoRI/XbaI fragment of pZA3 PN25-luc | This study |
| pGZ132 | glpE in EcoRI/XbaI fragment of pZE2 PN25-luc | This study |
| pATCBOA2+6 | ColE1 origin Apr Φ(glpT-phoA) | 34 |
Growth media and conditions.
Cultures were grown in Luria-Bertani broth (43) at 37°C. Antibiotics were included at 100 μg/ml for ampicillin, 50 μg/ml for kanamycin, 25 μg/ml for chloramphenicol, and 20 μg/ml for spectinomycin where appropriate. Overexpression of GlpE in cells carrying pGZ105 was induced in mid-log phase by the addition of 0.5 mM isopropylthio-β-d-galactopyranoside (IPTG) and in cells carrying pGZ154 or pGZ132 by 0.1 μg of tetracycline per ml. Cultures were grown for 2 to 4 h after induction and then harvested in late log phase as described below.
Cell fractionation procedures.
DH5αZ1 harboring pATCBOA2+6 (glpT-phoA transcriptional fusion) and pGZ154 was grown, and expression of GlpE was induced by using tetracycline as described above. Cells were harvested, and two fractionation procedures were used for preparation of cell extracts.
(i) Periplasmic fraction.
Spheroplasts were prepared by incubation of cells with lysozyme and EDTA essentially as described by Kaback (27). Cells were washed twice with 0.5 volume of 10 mM Tris-HCl (pH 8) and then resuspended in 30 mM Tris-HCl (pH 8)–20% sucrose–10 mM EDTA at an absorbance at 600 nm of approximately 3. Lysozyme was added (0.5 mg/ml) using a freshly prepared stock solution of 25 mg/ml in 10 mM Tris-HCl (pH 8). After incubation at room temperature for 30 min with gentle agitation, spheroplasts were removed by centrifugation for 5 min at 10,000 × g. The supernatant was decanted and saved as the periplasmic fraction. Cell pellets were resuspended in 10 mM Tris-HCl (pH 8) and lysed by sonication (cytoplasmic fraction).
(ii) Freeze-thaw fraction.
The second fractionation procedure combined freeze-thaw treatment (26) and incubation with EDTA to disrupt the outer membrane. Cells were harvested by centrifugation (6,000 × g for 5 min at 4°C) and washed with 0.5 volume of 25 mM Tris-acetate (pH 8.6)–10 mM ammonium thiosulfate precooled to 4°C. The cell pellet was stored at −70°C. The frozen cells were thawed on ice, resuspended in 1/50 the original volume of buffer A (50 mM Tris-HCl [pH 7.2], 3 mM EDTA), and incubated on ice for 30 min. Cells were collected by centrifugation, and the supernatant fraction was saved. Incubation in buffer A was repeated twice, and the three supernatant fractions were combined (freeze-thaw extract). After isolation of the freeze-thaw extract, the remaining cells were resuspended in 10 mM Tris-HCl (pH 8) and lysed by sonication (cytoplasmic fraction).
Rhodanese, alkaline phosphatase, and glucose 6-phosphate dehydrogenase activities were determined for the periplasmic, freeze-thaw, and cytoplasmic fractions. Total cellular enzyme activities were determined by using an aliquot of whole cells that was pelleted, resuspended in 10 mM Tris-HCl (pH 8), and sonicated as described above.
Protein purification.
BL21(DE3) harboring pGZ105 was grown in 500 ml of Luria-Bertani broth, and expression of GlpE was induced by the addition of IPTG as described above. Freeze-thaw extraction was performed as described above. The freeze-thaw extract was loaded onto a prepacked Waters quaternary methylamine (Q)-polymethacrylate anion-exchange column (10 by 100 mm; Protein-Pak Q15HR 1000Å) equilibrated at room temperature with buffer A. The flow rate was maintained at 1.5 ml/min. After being washed with 180 ml of buffer A, the column was developed with a 45-ml gradient from 0 to 105 mM NaCl in buffer A, followed by a 112.5-ml gradient from 105 to 255 mM NaCl in buffer A. Fractions containing rhodanese activity, eluted between 150 and 190 mM NaCl, were pooled. Rhodanese was concentrated by reapplying the pooled fractions diluted twofold with buffer A to the anion-exchange column. The enzyme was eluted with buffer A containing 750 mM NaCl. Fractions containing rhodanese activity were pooled, brought to 15% glycerol, and stored in aliquots at −70°C. Protein concentrations were determined by the method of Bradford (11) with bovine serum albumin as the standard, using materials purchased from Pierce Chemical Company and the manufacturer's microassay method.
Estimation of subunit and native molecular masses.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed as described by Laemmli (33) on 15% polyacrylamide gels. A modified sample loading buffer containing 62.5 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 0.02% bromophenol blue, and 1.5 M β-mercaptoethanol was prepared immediately prior to use. Use of the less-reducing buffer described by Laemmli (33) resulted in migration of GlpE as a doublet. Protein was visualized with Fast Stain from Zoion Biotech, Inc.
Gel filtration chromatography was used to determine the apparent molecular mass of GlpE under nondenaturing conditions. Samples of 0.1 ml containing from 40 to 80 μg of GlpE or various standards were applied to a Waters glass Protein-Pak 300SW column (8 by 300 mm). The column was equilibrated at ambient temperature with 20 mM Tris-HCl (pH 7.2)–1 mM EDTA–100 mM NaCl at a flow rate of 0.8 ml/min. Void volume and total volume were determined by using blue dextran and vitamin B12, respectively. The protein standards used for generation of the standard curve were bovine serum albumin, ovalbumin, soybean trypsin inhibitor, myoglobin, and RNase.
Assay of sulfurtransferase activity.
Two methods were used to measure the sulfurtransferase activity of GlpE. Assays were performed at ambient temperature (approximately 22°C).
(i) First method.
During purification of GlpE and for characterization of the enzyme, the assay used was essentially that described previously to quantify thiocyanate (3, 66). Assays contained 100 mM Tris-acetate (pH 8.6), 50 mM ammonium thiosulfate, 50 mM KCN, and enzyme in a final volume of 0.5 ml. Reactions were initiated by the addition of KCN and terminated, after 0.5 to 2 min, by the addition of 0.25 ml of 15% formaldehyde. Color was developed by the addition of 0.75 ml of ferric nitrate reagent[100 g of Fe(NO3)3 · 9H2O and 200 ml of 65% HNO3 per 1,500 ml]. Assays were clarified by centrifugation, and the absorbance at 460 nm was determined (66). One unit of enzyme is defined as the amount that catalyzes the production of 1 μmol of thiocyanate per minute and corresponds to an absorbance change at 460 nm of 2.8 in this system.
(ii) Second method.
Assays to measure the ability of GlpE to transfer sulfur from thiosulfate to thioredoxin 1 were performed essentially as described previously for the bovine liver rhodanese (48). Each assay (1 ml, final volume) contained 50 mM potassium phosphate (pH 8.2), 0.1 U of thioredoxin reductase per ml, 50 μM NADPH, and the indicated amount of thioredoxin 1. Cuvettes containing all reagents except NADPH were used as blanks. NADPH was added, and the reaction mixtures were allowed to equilibrate for at least 1 h. Periodic measurements of the absorbance at 340 nm were used to ensure that the mixtures had reached equilibrium. After equilibrium had been reached, either purified GlpE (4 μM monomer, 48 μg/ml), ammonium thiosulfate (80 mM), or both were added.
The basis for this assay is that when thioredoxin 1, which contains the active-site motif WCGPC, accepts a sulfane sulfur from rhodanese, a persulfide is formed at the N-terminal cysteine within the active site. The C-terminal cysteine within the active site then reacts with the persulfide, yielding sulfide and oxidized thioredoxin 1. Thioredoxin reductase acts to reduce the disulfide bond of oxidized thioredoxin 1, with the concomitant oxidation of NADPH (48). The decrease in absorbance at 340 nm was measured to determine the rate of NADPH oxidation.
Other enzyme assays.
Alkaline phosphatase activity was determined by utilizing the substrate p-nitrophenylphosphate and monitoring the increase in absorbance at 410 nm (34). Glucose 6-phosphate dehydrogenase activity was determined by following the increase in absorbance at 340 nm due to reduction of NADP in the presence of glucose 6-phosphate (21).
RESULTS
GlpE is a sulfurtransferase.
A PSI-BLAST search (4) using GlpE as the query revealed that GlpE exhibits sequence similarity to the family of sulfurtransferases that includes rhodaneses and mercaptopyruvate sulfurtransferases and to the family of tyrosine and dual-specificity phosphatases (23, 58). Together, these two families of proteins constitute a superfamily of transfer proteins (23, 58). Figure 1 illustrates alignment of GlpE with the conserved regions of several representative members of this superfamily. A comprehensive discussion of the superfamily, along with alignments for 37 members, has been published recently (23).
FIG. 1.

Sequence alignment of the conserved regions of GlpE with those of representative sulfurtransferases. A comprehensive alignment of sulfurtransferases has been published (23). The active site and two conserved structural motifs, designated CH2A and CH2B, are labeled. A consensus is indicated, where U signifies an uncharged residue. The solid triangle indicates the active-site cysteine, and asterisks highlight residues conserved in all four sequences. Basic residues in rhodanese important for binding of thiosulfate and/or anion inhibitors are underlined. Sud-Ws, sulfide dehydrogenase from W. succinogenes (32); Rhod-Bt, bovine liver rhodanese (54); MPST-Rn, rat mercaptopyruvate sulfurtransferase (45). Accession numbers: GlpE, M96795; Sud, X81642; Rhod, M58561; and MPST, D50564.
We found that overexpression of GlpE resulted in a 10- to 1,000-fold increase in thiosulfate:cyanide sulfurtransferase (rhodanese) activity depending on the expression system used (Table 2). The elevated level of rhodanese activity corresponded to an increased production of GlpE, as visualized by SDS-PAGE (Fig. 2). No increase in phosphatase activity was associated with overexpression of GlpE (data not shown).
TABLE 2.
Overexpression of GlpE and corresponding increase in rhodanese activity
| Strain (plasmid) | Rhodanese sp acta (U/mg) | Fold overexpression |
|---|---|---|
| DH5αZ1(pZE2 PN25-luc) | 0.03 | 1 |
| DH5αZ1(pGZ154) | 0.6 | 20 |
| DH5αZ1(pGZ132) | 0.7 | 23 |
| BL21(DE3)(pT7-7) | 0.04 | 1 |
| BL21(DE3)(pGZ105) | 21 | 530 |
Determined by using crude extracts of sonicated cells.
FIG. 2.

SDS-PAGE illustrating the overexpression and purification of GlpE. Lane 1, total cellular protein, BL21(DE3) harboring pT7-7 (10 μg). Lane 2, total cellular protein, BL21(DE3) harboring pGZ105 (10 μg). Lane 3, proteins released from intact cells [BL21(DE3) harboring pGZ105] by the modified freeze-thaw method (10 μg). Lane 4, pooled fractions following the first anion-exchange chromatographic step (2 μg). Lanes 5 and 6, pooled fractions following the second anion-exchange chromatographic step (2 and 4 μg, respectively). Lane 7, molecular mass markers, corresponding to 97, 66, 45, 30, 21, and 14 kDa.
Cellular location of GlpE.
Two distinct rhodanese activities have been described previously for E. coli. One is released from cells only after sonication (62). The other is “accessible” to substrates when intact cells are added to assay mixtures and is released from cells by freeze-thaw treatment (3, 62). Release of a protein by freeze-thaw treatment could indicate a periplasmic localization. However, some cytoplasmic proteins are released from cells treated with Tris-EDTA (8). Like the “accessible” enzyme, we found that GlpE was released from cells by freeze-thaw treatment (Table 3) and by most other methods that have been used for isolating periplasmic proteins, including treatment with chloroform (5) and osmotic shock (49) (data not shown). GlpE, however, has no N-terminal targeting sequence that would direct it to the periplasm.
TABLE 3.
Localization of GlpE to the cytoplasma
| Fraction | % of activity released
|
||
|---|---|---|---|
| Alkaline phosphatase | Glucose-6-phosphate dehydrogenase | Rhodanese | |
| Spheroplasts | 69 | 5 | 5 |
| Freeze-thaw | 76 | <1 | 97 |
Each cell fractionation procedure was performed at least twice. Reported results are from typical experiments. Total enzyme activities recovered by the preparation of spheroplasts from 33 ml of logarithmically growing culture were 0.03 U of alkaline phosphatase, 0.3 U of glucose-6-phosphate dehydrogenase, and 1 U of rhodanese. Total enzyme activities recovered by the freeze-thaw fractionation method from 25 ml of logarithmically growing culture were 12 U of alkaline phosphatase, 0.1 U of glucose-6-phosphate dehydrogenase, and 10 U of rhodanese.
To rigorously determine the cellular location of GlpE, the periplasmic fraction of cells overexpressing GlpE was isolated by preparation of spheroplasts with lysozyme and EDTA (27). Rhodanese activities in the periplasmic and cytoplasmic fractions were compared, with alkaline phosphatase and glucose-6-phosphate dehydrogenase activities serving as periplasmic and cytoplasmic markers, respectively (Table 3). Preparation of spheroplasts without a subsequent osmotic shock released most of the alkaline phosphatase activity but only 5% of the total rhodanese and glucose-6-phosphate dehydrogenase activities (Table 3). Preparation of spheroplasts by this method is considered a more stringent test for determining cellular location (7, 27). These results demonstrate that GlpE is a cytoplasmic protein.
Cells moderately overexpressing GlpE released most of the rhodanese and alkaline phosphatase activities but little glucose-6-phosphate dehydrogenase activity when subjected to freeze-thaw treatment (Table 3). It is likely that GlpE is released from the cytoplasm via the mechanosensitive channel, MscL, as has been described for thioredoxin, DnaK, and EF-Tu (2, 8).
Purification of GlpE.
The modified freeze-thaw treatment quantitatively released rhodanese activity from intact cells that overexpress GlpE, thereby facilitating purification of the enzyme (Table 3). The freeze-thaw extract was subjected to anion-exchange chromatography. Three peaks containing rhodanese activity were eluted. The relative amount of rhodanese in each peak varied, but two peaks eluting near 160 and 185 mM NaCl typically contained most of the total rhodanese activity. The third peak eluted in 1 M NaCl. The major protein found in each peak exhibited similar electrophoretic mobility (approximately 12 kDa) and was therefore tentatively identified as an isoform of GlpE. Rhodaneses can be isolated in a sulfur-free form, a persulfide-containing form (63, 67), and oxidized, less-active forms (3, 16, 24). The first two peaks were pooled and concentrated by rechromatography and step elution. The resulting protein was more than 95% homogeneous, as visualized by protein staining of an SDS–15% polyacrylamide gel (Fig. 2). From a 500-ml culture, 1.3 mg of GlpE was purified 25-fold to a specific activity of 270 U/mg (Table 4).
TABLE 4.
Purification of glpE-encoded rhodanese from E. coli
| Purification step | Volume (ml) | Amt of protein (mg) | Activity (U) | Sp act (U/mg) | Yield (%) |
|---|---|---|---|---|---|
| Whole cellsa | 500 | 120 | 1,300 | 11 | 100 |
| Freeze-thaw extract | 30 | 9.6 | 860 | 90 | 66 |
| Anion exchange 1 | 32 | 1.6 | 350 | 220 | 27 |
| Anion exchange 2 | 2 | 1.3 | 350 | 270 | 27 |
Activity was determined using sonicated samples of liquid cell culture.
Purified rhodanese stored at 4°C over several weeks lost activity, presumably due to oxidation, as described for other rhodaneses (3, 16, 24). Addition of micromolar concentrations of cysteine, dithiothreitol, or β-mercaptoethanol was found to reactivate oxidized enzyme. Loss of activity occurred less rapidly when 750 mM sodium chloride or ammonium sulfate was present.
Molecular mass of GlpE.
The molecular mass of the GlpE monomer calculated from the deduced amino acid sequence (12.1 kDa) was confirmed previously by SDS-PAGE (12.1 kDa) (72). Gel filtration chromatography was used to determine the apparent molecular mass of GlpE under nondenaturing conditions. GlpE chromatographed as a dimer (22.5 kDa) under a variety of buffer conditions (data not shown). Addition of cysteine or dithiothreitol, although able to activate oxidized enzyme, did not appreciably change the apparent size of the enzyme (data not shown).
Catalytic properties of GlpE.
The well-characterized bovine liver rhodanese utilizes a double-displacement (ping-pong) mechanism (63, 67). Data from activity measurements of purified GlpE with various concentrations of thiosulfate at fixed concentrations of cyanide were fit to the equation describing this type of mechanism. Figure 3A shows the data and the fit obtained when points exhibiting substrate inhibition by thiosulfate were excluded. Figure 3B is a secondary double-reciprocal plot of the apparent maximum velocities in Fig. 3A versus cyanide concentration. The regression of the data to the equation describing a double-displacement mechanism yielded Km values for thiosulfate and cyanide of 78 and 17 mM, respectively. The kcat based on GlpE functioning as a dimer was 230 s−1.
FIG. 3.

Kinetic characterization of the thiosulfate:cyanide sulfurtransferase reaction catalyzed by GlpE. Each assay, performed as described in the text, contained 10 μl (0.75 μg) of purified GlpE in 100 mM Tris-acetate (pH 8.6)–6 mM cysteine. (A) Double-reciprocal plot of the rate of thiocyanate formation versus thiosulfate concentration at various fixed concentrations of cyanide: 2.5 mM (■), 5 mM (▴), 10 mM (●), and 50 mM (⧫). (B) Secondary double-reciprocal plot of apparent Vmax from the data in panel A versus cyanide concentration. Data, excluding the points exhibiting substrate inhibition, were fit to the equation describing a double-displacement mechanism.
Although rhodaneses are related to mercaptopyruvate sulfurtransferases (Fig. 1) (45, 46), substitution of mercaptopyruvate in the standard sulfurtransferase assay resulted in less than 1% of the thiocyanate formation obtained with an equivalent concentration of thiosulfate. Cysteine, thioglycerol, dithiothreitol, and β-mercaptoethanol were unable to replace thiosulfate as the sulfur donor for GlpE (data not shown).
Several previously characterized rhodaneses, including the bovine liver enzyme (64) and the accessible rhodanese of E. coli, are specifically inhibited by certain anions (3). In contrast, little or no inhibition by anions was observed for GlpE. Addition of sodium phosphate, sodium acetate, sodium chloride, sodium sulfate, or potassium phosphate at an ionic strength of 0.3 resulted in 20% or less inhibition of rhodanese activity (data not shown). However, as shown in Fig. 3A, substrate inhibition by thiosulfate was observed, particularly at low cyanide concentrations. This type of inhibition was previously described for the bovine liver rhodanese (64) and the accessible rhodanese of E. coli (3).
Chemical inactivation of GlpE by DTNB.
Catalysis by the bovine liver rhodanese requires a cysteine residue. The cysteine forms a persulfide linkage to the sulfane sulfur from thiosulfate in an enzyme-sulfur intermediate (63, 67). GlpE contains two cysteine residues, one of which has been identified as the active site of GlpE, based on sequence alignment with other sulfurtransferases (Cys-65) (Fig. 1). GlpE was incubated with the cysteine-specific modifying reagent DTNB to verify that a cysteine residue is required for rhodanese activity. Incubation of GlpE with DTNB in a 1:2 molar ratio resulted in a greater-than-90% loss of activity (Fig. 4).
FIG. 4.

Inactivation of GlpE by DTNB. Purified GlpE (60 μg) was incubated overnight at an ambient temperature in 0.2 ml of 100 mM Tris-acetate (pH 8.6)–150 mM NaCl with various molar ratios of DTNB. Remaining rhodanese activity was determined and compared with that for GlpE incubated without DTNB.
Thioredoxin acts as a sulfur acceptor substrate for GlpE.
It has been shown that reduced dithiols such as dithiothreitol (1, 53), dihydrolipoate (50), and E. coli thioredoxin 1 (48) serve as sulfur-acceptor substrates for bovine liver rhodanese. Thioredoxin 1 acts as a substrate with an affinity near that determined for cyanide (apparent Kms of 18.5 μM [48]) and 63 μM [64], respectively). To test thioredoxin 1 as a sulfur acceptor substrate for GlpE, the assay method described for bovine rhodanese was used (48). (See Materials and Methods for the basis for this assay.) A mixture containing thioredoxin 1, thioredoxin reductase, and NADPH was allowed to equilibrate. Then GlpE, ammonium thiosulfate, or both were added. Addition of either GlpE or thiosulfate alone resulted in no significant oxidation of NADPH. However, addition of both GlpE and thiosulfate resulted in a marked increase in the rate of NADPH oxidation (Fig. 5A). Oxidation of NADPH was dependent on the presence of thioredoxin 1 and thioredoxin reductase as well as GlpE and thiosulfate. To quantify the affinity of thioredoxin 1 for the sulfur transferred from GlpE, the thioredoxin 1 concentration was varied while the thiosulfate concentration was held at 80 mM (Fig. 5B). Regression of the resulting data yielded an estimate of the apparent Km for thioredoxin 1 of 34 μM and an apparent Vmax of 1 nmol of NADPH oxidized per min, almost 15 times faster than the maximum rate of NADPH oxidation observed with the bovine liver rhodanese (48). This apparent maximum rate corresponds to a turnover number of 0.5 mol of NADPH oxidized per mol of GlpE dimer per min.
FIG. 5.

Thioredoxin 1 as a sulfur acceptor substrate for GlpE. (A) Assay mixtures containing 16.6 μM thioredoxin 1, 0.1 U of thioredoxin reductase per ml, and 50 μM NADPH were preequilibrated. At time zero, either 4 μM GlpE (■), 80 mM ammonium thiosulfate (⧫), or both (▴) were added. The absorbance at 340 nm was periodically measured to determine the rate of NADPH oxidation. (B) Double-reciprocal plot of the rate of NADPH oxidation measured as described for panel A versus thioredoxin 1 concentration.
DISCUSSION
Comparison of GlpE with other sulfurtransferases.
The well-characterized rhodanese from bovine liver mitochondria is approximately twice the size of GlpE and functions as a monomer. The bovine liver enzyme, however, contains two approximately 15-kDa domains. The two domains are structurally similar, although they exhibit only limited sequence similarity to each other (54). Hence, it is thought that duplication of an ancestral rhodanese gene has given rise to the genes that encode the two-domain rhodaneses. This hypothesis is supported by the identification of GlpE and Sud, a polysulfide sulfurtransferase from Wolinella succinogenes (29, 30), as single-domain sulfurtransferases having sequence similarity to the catalytic domain of the bovine liver enzyme (Fig. 1). Like the bovine enzyme, GlpE and Sud require a cysteine residue for activity and utilize a double-displacement reaction mechanism.
Although GlpE possesses some of the characteristics of the previously identified accessible rhodanese from E. coli (molecular weights of 12,000 and 14,000, accessibility of enzyme to substrates added to intact cells, and release from cells by freeze-thaw) (3), it is not likely that these two enzymes are identical. First, the catalytic properties of the two enzymes are distinct. The Kms for SSO32− are 78 and 5 mM for GlpE and the accessible rhodanese, respectively. The Kms for CN− are about the same (17 and 24 mM, respectively), but the kcats differ (115 s−1 per GlpE monomer versus 260 s−1 per monomer for the accessible rhodanese) (3). Also, GlpE does not exhibit anion-specific inhibition as described for accessible rhodanese and bovine liver rhodanese. Ki values for sulfate and acetate versus thiosulfate were 36 and 45 mM, respectively, for accessible rhodanese (3) and 40 and 167 mM, respectively, for bovine rhodanese (64). Specific inhibition of bovine rhodanese by anions has been linked to two positively charged residues (R248 and K249) located adjacent to the active-site cysteine and an arginine residue (R186) within the CH2A motif (36). None of these residues are conserved in GlpE (see Fig. 1). These basic residues have also been implicated in the recognition of thiosulfate as a substrate. Substitution of R186 in the bovine liver rhodanese with isoleucine increases the Km for thiosulfate from 4 to 70 mM. Substitution of K249 with alanine results in a protein that has almost no activity with thiosulfate as the substrate, but the variant can utilize a number of organic thiosulfonates almost as well as the wild-type protein (41). Lack of conservation of these residues in GlpE may be the basis for the lower affinity of GlpE for thiosulfate compared with other rhodaneses. The relatively low affinity of GlpE for both thiosulfate and cyanide suggests that these compounds are not the physiological substrates of the enzyme.
Finally, GlpE and accessible rhodanese differ in their response to reducing agents. Addition of cysteine not only activated oxidizedaccessible rhodanese but also resulted in a shift from dimeric to monomeric form (3). Although GlpE is activated by cysteine, it does not exhibit this shift to monomeric form in the presence of cysteine or dithiothreitol. The gene encoding GlpE is only one of a number of genes that have been identified in E. coli that encode rhodanese like proteins (23, 58). One of the other paralogs is likely the previously characterized accessible rhodanese. Deletion of the glpE gene did not significantly decrease total rhodanese activity measured in cell extracts, verifying that GlpE is not the only protein possessing rhodanese activity in E. coli (data not shown). Other organisms also contain multiple sulfurtransferase genes. For example, disruption of the rhdA (rhodanese) gene of Azotobacter vinelandii did not result in a discernible phenotype or abolish all rhodanese activity (13). Finally, among the 21 completely sequenced genomes in the COG database (57), 8 genomes contain multiple sulfurtransferase genes.
It is of interest that expression of both the accessible rhodanese and GlpE is subject to catabolite repression (3, 12, 71) and repressed during anaerobic growth (3) (data not shown).
GlpE and thioredoxin 1.
GlpE, like bovine liver rhodanese (48), is capable of transferring sulfur from thiosulfate to thioredoxin 1 with relatively high affinity. GlpE has an apparent Km for thioredoxin 1 of 34 μM when thiosulfate is present near its Km. Thus, the affinity for thioredoxin 1 is 500 times higher than that determined for cyanide (17 mM). Sulfurtransferases from other organisms may also have the ability to utilize dithiol proteins such as thioredoxin as sulfur acceptors. For example, a partially purified rhodanese isolated from Thiobacillus novellus was capable of producing sulfide from thiosulfate in the absence of cyanide. The physiological sulfur acceptor substrate present in the partially purified preparation had a molecular mass of between 10 and 20 kDa (22) and so may have been a protein similar to thioredoxin. Interestingly, the two structural motifs, designated CH2A and CH2B, that flank the active site in the superfamily of transfer proteins (Fig. 1) have been suggested to play a role in protein-protein interactions. These interactions may mediate recognition of protein substrates by the tyrosine and dual-specificity phosphatases (18). It is intriguing to speculate that the CH2A and CH2B motifs of the sulfurtransferases play a similar role in recognition of dithiol protein substrates. E. coli contains multiple dithiol proteins besides thioredoxin 1 that could fulfill this function. A second thioredoxin, the gene product of trxC, has recently been characterized (44). In addition to the two thioredoxins, E. coli possesses three isoforms of glutaredoxin, proteins structurally and functionally similar to the thioredoxins (6, 37). Given the overlap of function that has been demonstrated for the thioredoxin/glutaredoxin family of proteins, it is possible that these other proteins also serve as sulfur acceptor substrates for sulfurtransferases.
A number of cytoplasmic proteins in E. coli, including elongation factor Tu (25), DnaK (17), and the small dithiol proteins thioredoxin 1 and 2 (40, 44), and many others (39), are released from cells by most methods used for isolating periplasmic proteins. Retention of proteins such as DnaK and thioredoxins by lysozyme-generated spheroplasts, however, indicates a cytoplasmic location (17, 40, 44, 59). GlpE also is a cytoplasmic protein, since it is retained by spheroplasts.
Recently, it was shown that release of thioredoxin, DnaK, and elongation factor Tu by mild osmotic shock required the presence of the gated mechanosensitive ion channel MscL (2, 8). Preliminary results from our laboratory suggest that MscL also facilitates release of GlpE by osmotic shock (data not shown).
Physiological role of GlpE.
The physiological role of rhodaneses is still in question. Cyanide detoxification has been proposed as a possible role (61), but given the low affinity for cyanide (Km = 17 mM), this seems unlikely for GlpE.
Rhodaneses may play various roles in sulfur metabolism (66). The expression of RhdA, a rhodanese-like protein from Synechococcus sp. strain PCC7942, is induced by sulfur starvation (35). However, nothing has thus far implicated GlpE in metabolism of sulfur in E. coli. Growth on various sulfur sources or on limiting sulfur had no apparent effect on the expression of a single-copy glpE-lacZ transcriptional fusion (data not shown). To our knowledge, the only phenotype identified to date for a rhodanese-like enzyme is that of CysA from Saccharopolyspora erythraea (15). Disruption of the gene encoding CysA results in cysteine auxotrophy but, as described above for glpE, does not have a significant effect on the total rhodanese activity. The pathway for cysteine biosynthesis in S. erythraea differs from that established for E. coli and involves thiosulfate as an intermediate. Thus, it is possible that CysA functions to synthesize thiosulfate (15). Although thiosulfate is not an obligate intermediate in aerobic cysteine biosynthesis in enteric bacteria, thiosulfate might be an intermediate in anaerobic biosynthesis of cysteine (19, 31).
Rhodanese has also been proposed to mobilize sulfur for the formation or repair of iron-sulfur clusters. The iron-sulfur clusters of ferredoxins (10, 51), succinate dehydrogenase (9), and mitochondrial NADH dehydrogenase (52) could be reconstituted by incubation with various combinations of bovine liver rhodanese, thiosulfate, dihydrolipoate, and iron ions. Interestingly, thioredoxin 1 has also been proposed to play a role in the formation or repair of iron-sulfur clusters (14). The participation of sulfurtransferases in the formation of iron-sulfur clusters has been questioned, however, since there is strong evidence that NifS/IscS and associated proteins are involved in mobilization of sulfur from cysteine for synthesis of iron-sulfur clusters (20, 28, 47, 73).
Although GlpE has now been identified as a sulfurtransferase, its physiological function and possible association with the metabolism of glycerol-P remain to be elucidated. The relationship between GlpE and thioredoxin that was discovered during this study might help clarify these issues.
ACKNOWLEDGMENTS
We thank Ali T. van Loo-Bhattacharya and Janet Donahue for skillful technical assistance. We also thank E. V. Koonin for pointing out the similarity between GlpE and phospho- and sulfurtransferases and C. H. Williams, Jr., for providing thioredoxin reductase.
This work was supported by grant MCB-9118757 from the National Science Foundation.
REFERENCES
- 1.Aird B A, Horowitz P M. Acceptor substrate-potentiated inactivation of bovine liver rhodanese. J Biol Chem. 1988;263:15270–15276. [PubMed] [Google Scholar]
- 2.Ajouz B, Berrier C, Garrigues A, Besnard M, Ghazi A. Release of thioredoxin via the mechanosensitive channel MscL during osmotic downshock of Escherichia coli cells. J Biol Chem. 1998;273:26670–26674. doi: 10.1074/jbc.273.41.26670. [DOI] [PubMed] [Google Scholar]
- 3.Alexander K, Volini M. Properties of an Escherichia coli rhodanese. J Biol Chem. 1987;262:6595–6604. [PubMed] [Google Scholar]
- 4.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ames G F, Prody C, Kustu S. Simple, rapid, and quantitative release of periplasmic proteins by chloroform. J Bacteriol. 1984;160:1181–1183. doi: 10.1128/jb.160.3.1181-1183.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Åslund F, Beckwith J. The thioredoxin superfamily: redundancy, specificity, and gray-area genomics. J Bacteriol. 1999;181:1375–1379. doi: 10.1128/jb.181.5.1375-1379.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beacham I R. Periplasmic enzymes in gram-negative bacteria. Int J Biochem. 1979;10:877–883. doi: 10.1016/0020-711x(79)90117-4. [DOI] [PubMed] [Google Scholar]
- 8.Berrier C, Garrigues A, Richarme G, Ghazi A. Elongation factor Tu and DnaK are transferred from the cytoplasm to the periplasm of Escherichia coli during osmotic downshock presumably via the mechanosensitive channel MscL. J Bacteriol. 2000;182:248–251. doi: 10.1128/jb.182.1.248-251.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonomi F, Pagani S, Cerletti P, Cannella C. Rhodanese-mediated sulfur transfer to succinate dehydrogenase. Eur J Biochem. 1977;72:17–24. doi: 10.1111/j.1432-1033.1977.tb11219.x. [DOI] [PubMed] [Google Scholar]
- 10.Bonomi F, Pagani S, Kurtz D M J. Enzymic synthesis of the 4Fe-4S clusters of Clostridium pasteurianum ferredoxin. Eur J Biochem. 1985;148:67–73. doi: 10.1111/j.1432-1033.1985.tb08808.x. [DOI] [PubMed] [Google Scholar]
- 11.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 12.Choi Y-L, Kawase S, Kawamukai M, Sakai H, Komano T. Regulation of glpD and glpE gene expression by a cyclic AMP-cAMP receptor protein (cAMP-CRP) complex in Escherichia coli. Biochim Biophys Acta. 1991;1088:31–35. doi: 10.1016/0167-4781(91)90149-g. [DOI] [PubMed] [Google Scholar]
- 13.Colnaghi R, Pagani S, Kennedy C, Drummond M. Cloning, sequence analysis and overexpression of the rhodanese gene of Azotobacter vinelandii. Eur J Biochem. 1996;236:240–248. doi: 10.1111/j.1432-1033.1996.00240.x. [DOI] [PubMed] [Google Scholar]
- 14.Ding H, Demple B. Thiol-mediated disassembly and reassembly of [2Fe-2S] clusters in the redox-regulated transcription factor SoxR. Biochemistry. 1998;37:17280–17286. doi: 10.1021/bi980532g. [DOI] [PubMed] [Google Scholar]
- 15.Donadio S, Shafiee A, Hutchinson C R. Disruption of a rhodanese-like gene results in cysteine auxotrophy in Saccharopolyspora erythraea. J Bacteriol. 1990;172:350–360. doi: 10.1128/jb.172.1.350-360.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dungan J M, Horowitz P M. Thermally perturbed rhodanese can be protected from inactivation by self-association. J Protein Chem. 1993;12:311–321. doi: 10.1007/BF01028193. [DOI] [PubMed] [Google Scholar]
- 17.El Yaagoubi A, Kohiyama M, Richarme G. Localization of DnaK (chaperone 70) from Escherichia coli in an osmotic-shock-sensitive compartment of the cytoplasm. J Bacteriol. 1994;176:7074–7078. doi: 10.1128/jb.176.22.7074-7078.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fauman E B, Cogswell J P, Lovejoy B, Rocque W J, Holmes W, Montana V G, Piwnica-Worms H, Rink M J, Saper M A. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell. 1998;93:617–625. doi: 10.1016/s0092-8674(00)81190-3. [DOI] [PubMed] [Google Scholar]
- 19.Filutowicz M, Wiater A, Hulanicka D. Delayed inducibility of sulphite reductase in cysM mutants of Salmonella typhimurium under anaerobic conditions. J Gen Microbiol. 1982;128:1791–1794. doi: 10.1099/00221287-128-8-1791. [DOI] [PubMed] [Google Scholar]
- 20.Flint D H. Escherichia coli contains a protein that is homologous in function and N-terminal sequence to the protein encoded by the nifS gene of Azotobacter vinelandii and that can participate in the synthesis of the Fe-S cluster of dihydroxy-acid dehydratase. J Biol Chem. 1996;271:16068–16074. [PubMed] [Google Scholar]
- 21.Fraenkel D G, Levisohn S R. Glucose and gluconate metabolism in an Escherichia coli mutant lacking phosphoglucose isomerase. J Bacteriol. 1967;93:1571–1578. doi: 10.1128/jb.93.5.1571-1578.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukumori Y, Hoshiko K, Yamanaka T. Purification and some properties of thiosulphate-cleaving enzyme from Thiobacillus novellus. FEMS Microbiol Lett. 1989;53:159–163. doi: 10.1016/0378-1097(89)90384-4. [DOI] [PubMed] [Google Scholar]
- 23.Hofmann K, Bucher P, Kajava A V. A model of Cdc25 phosphatase catalytic domain and CDK-interaction surface based on the presence of a rhodanese homology domain. J Mol Biol. 1998;282:195–208. doi: 10.1006/jmbi.1998.1998. [DOI] [PubMed] [Google Scholar]
- 24.Horowitz P M, Hua S. Rhodanese conformational changes permit oxidation to give disulfides that form in a kinetically determined sequence. Biochim Biophys Acta. 1995;1249:161–167. doi: 10.1016/0167-4838(95)00037-u. [DOI] [PubMed] [Google Scholar]
- 25.Jacobson G R, Rosenbusch J P. Abundance and membrane association of elongation factor Tu in E. coli. Nature. 1976;261:23–26. doi: 10.1038/261023a0. [DOI] [PubMed] [Google Scholar]
- 26.Johnson B H, Hecht M H. Recombinant proteins can be isolated from E. coli cells by repeated cycles of freezing and thawing. Bio/Technology. 1994;12:1357–1360. doi: 10.1038/nbt1294-1357. [DOI] [PubMed] [Google Scholar]
- 27.Kaback H R. Bacterial membranes. Methods Enzymol. 1971;22:99–120. [Google Scholar]
- 28.Kispal G, Csere P, Prohl C, Lill R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999;18:3981–3989. doi: 10.1093/emboj/18.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klimmek O, Kreis V, Klein C, Simon J, Wittershagen A, Kroger A. The function of the periplasmic Sud protein in polysulfide respiration of Wolinella succinogenes. Eur J Biochem. 1998;253:263–269. doi: 10.1046/j.1432-1327.1998.2530263.x. . (Erratum, 260:568, 1999.) [DOI] [PubMed] [Google Scholar]
- 30.Klimmek O, Stein T, Pisa R, Simon J, Kroger A. The single cysteine residue of the Sud protein is required for its function as a polysulfide-sulfur transferase in Wolinella succinogenes. Eur J Biochem. 1999;263:79–84. doi: 10.1046/j.1432-1327.1999.00461.x. [DOI] [PubMed] [Google Scholar]
- 31.Kredich N M. Biosynthesis of cysteine. In: Neidhardt F C, Curtiss R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C.: ASM Press; 1996. pp. 514–527. [Google Scholar]
- 32.Kreis-Kleinschmidt V, Fahrenholz F, Kojro E, Kröger A. Periplasmic sulphide dehydrogenase (Sud) from Wolinella succinogenes: isolation, nucleotide sequence of the sud gene and its expression in Escherichia coli. Eur J Biochem. 1995;227:137–142. doi: 10.1111/j.1432-1033.1995.tb20369.x. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli U. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Larson T J, Cantwell J S, van Loo-Bhattacharya A T. Interaction at a distance between multiple operators controls the adjacent, divergently transcribed glpTQ-glpACB operons of Escherichia coli K-12. J Biol Chem. 1992;267:6114–6121. [PubMed] [Google Scholar]
- 35.Laudenbach D E, Ehrhardt D, Green L, Grossman A. Isolation and characterization of a sulfur-regulated gene encoding a periplasmically localized protein with sequence similarity to rhodanese. J Bacteriol. 1991;173:2751–2760. doi: 10.1128/jb.173.9.2751-2760.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lijk L J, Torfs C A, Kalk K H, De Maeyer M C, Hol W G. Differences in the binding of sulfate, selenate and thiosulfate ions to bovine liver rhodanese, and a description of a binding site for ammonium and sodium ions: an X-ray diffraction study. Eur J Biochem. 1984;142:399–408. doi: 10.1111/j.1432-1033.1984.tb08301.x. [DOI] [PubMed] [Google Scholar]
- 37.Lillig C H, Prior A, Schwenn J D, Aslund F, Ritz D, Vlamis-Gardikas A, Holmgren A. New thioredoxins and glutaredoxins as electron donors of 3′-phosphoadenylylsulfate reductase. J Biol Chem. 1999;274:7695–7698. doi: 10.1074/jbc.274.12.7695. [DOI] [PubMed] [Google Scholar]
- 38.Lin E C C. Dissimilatory pathways for sugars, polyols, and carboxylates. In: Neidhardt F C, Curtiss R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C.: ASM Press; 1996. pp. 307–342. [Google Scholar]
- 39.Link A J, Robison K, Church G M. Comparing the predicted and observed properties of proteins encoded in the genome of Escherichia coli K-12. Electrophoresis. 1997;18:1259–1313. doi: 10.1002/elps.1150180807. [DOI] [PubMed] [Google Scholar]
- 40.Lunn C A, Pigiet V P. Localization of thioredoxin from Escherichia coli in an osmotically sensitive compartment. J Biol Chem. 1982;257:11424–11430. [PubMed] [Google Scholar]
- 41.Luo G X, Horowitz P M. The sulfurtransferase activity and structure of rhodanese are affected by site-directed replacement of Arg-186 or Lys-249. J Biol Chem. 1994;269:8220–8225. [PubMed] [Google Scholar]
- 42.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 44.Miranda-Vizuete A, Damdimopoulos A E, Gustafsson J, Spyrou G. Cloning, expression, and characterization of a novel Escherichia coli thioredoxin. J Biol Chem. 1997;272:30841–30847. doi: 10.1074/jbc.272.49.30841. [DOI] [PubMed] [Google Scholar]
- 45.Nagahara N, Nishino T. Role of amino acid residues in the active site of rat liver mercaptopyruvate sulfurtransferase: cDNA cloning, overexpression, and site-directed mutagenesis. J Biol Chem. 1996;271:27395–27401. doi: 10.1074/jbc.271.44.27395. [DOI] [PubMed] [Google Scholar]
- 46.Nagahara N, Okazaki T, Nishino T. Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese: striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J Biol Chem. 1995;270:16230–16235. doi: 10.1074/jbc.270.27.16230. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura M, Saeki K, Takahashi Y. Hyperproduction of recombinant ferredoxins in Escherichia coli by coexpression of the ORF1-ORF2-iscS-iscU-iscA-hscB-hscA-fdx-ORF3 gene cluster. J Biochem (Tokyo) 1999;126:10–18. doi: 10.1093/oxfordjournals.jbchem.a022409. [DOI] [PubMed] [Google Scholar]
- 48.Nandi D L, Westley J. Reduced thioredoxin as a sulfur-acceptor substrate for rhodanese. Int J Biochem Cell Biol. 1998;30:973–977. doi: 10.1016/s1357-2725(98)00050-8. [DOI] [PubMed] [Google Scholar]
- 49.Neu H C, Heppel L A. The release of enzymes from Escherichia coli by osmotic shock and during the formation of spheroplasts. J Biol Chem. 1965;240:3685–3692. [PubMed] [Google Scholar]
- 50.Pagani S, Bonomi F, Cerletti P. The inhibition of rhodanese by lipoate and iron-sulfur proteins. Biochim Biophys Acta. 1983;742:116–121. doi: 10.1016/0167-4838(83)90366-7. [DOI] [PubMed] [Google Scholar]
- 51.Pagani S, Bonomi F, Cerletti P. Enzymic synthesis of the iron-sulfur cluster of spinach ferredoxin. Eur J Biochem. 1984;142:361–366. doi: 10.1111/j.1432-1033.1984.tb08295.x. [DOI] [PubMed] [Google Scholar]
- 52.Pagani S, Galante Y M. Interaction of rhodanese with mitochondrial NADH dehydrogenase. Biochim Biophys Acta. 1983;742:278–284. doi: 10.1016/0167-4838(83)90312-6. [DOI] [PubMed] [Google Scholar]
- 53.Pecci L, Pensa B, Costa M, Cignini P L, Cannella C. Reaction of rhodanese with dithiothreitol. Biochim Biophys Acta. 1976;445:104–111. doi: 10.1016/0005-2744(76)90163-7. [DOI] [PubMed] [Google Scholar]
- 54.Ploegman J H, Drent G, Kalk K H, Hol W G, Heinrikson R L, Keim P, Weng L, Russell J. The covalent and tertiary structure of bovine liver rhodanese. Nature. 1978;273:124–129. doi: 10.1038/273124a0. [DOI] [PubMed] [Google Scholar]
- 55.Studier F W, Moffatt B A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 56.Tabor S, Richardson C C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc Natl Acad Sci USA. 1985;82:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tatusov R L, Galperin M Y, Natale D A, Koonin E V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tatusov R L, Koonin E V, Lipman D J. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 59.Thorstenson Y R, Zhang Y, Olson P S, Mascarenhas D. Leaderless polypeptides efficiently extracted from whole cells by osmotic shock. J Bacteriol. 1997;179:5333–5339. doi: 10.1128/jb.179.17.5333-5339.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vachek H, Wood J L. Purification and properties of mercaptopyruvate sulfur transferase of Escherichia coli. Biochim Biophys Acta. 1972;258:133–146. doi: 10.1016/0005-2744(72)90973-4. [DOI] [PubMed] [Google Scholar]
- 61.Vennesland B, Castric P A, Conn E E, Solomonson L P, Volini M, Westley J. Cyanide metabolism. Fed Proc. 1982;41:2639–2648. [PubMed] [Google Scholar]
- 62.Volini M, Ogata K, Alexander K. Molecular forms of the rhodaneses. In: Liu T-Y, Sakakibara S, Schechter A N, Yagi K, Yajima H, Yasunobu K T, editors. Frontiers in biochemical and biophysical studies of proteins and membranes. New York, N.Y: Elsevier Science Publishing, Inc.; 1983. pp. 183–192. [Google Scholar]
- 63.Volini M, Westley J. The mechanism of the rhodanese-catalyzed thiosulfate-lipoate reaction: kinetic analysis. J Biol Chem. 1966;241:5168–5176. [PubMed] [Google Scholar]
- 64.Wang S F, Volini M. The interdependence of substrate and protein transformations in rhodanese catalysis. I. Enzyme interactions with substrate, product, and inhibitor anions. J Biol Chem. 1973;248:7376–7385. [PubMed] [Google Scholar]
- 65.Weissenborn D L, Wittekindt N, Larson T J. Structure and regulation of the glpFK operon encoding glycerol diffusion facilitator and glycerol kinase of Escherichia coli K-12. J Biol Chem. 1992;267:6122–6131. [PubMed] [Google Scholar]
- 66.Westley J. Thiosulfate:cyanide sulfurtransferase (rhodanese) Methods Enzymol. 1981;77:285–291. doi: 10.1016/s0076-6879(81)77039-3. [DOI] [PubMed] [Google Scholar]
- 67.Westley J, Heyse D. Mechanisms of sulfur transfer catalysis: sulfhydryl-catalyzed transfer of thiosulfonate sulfur. J Biol Chem. 1971;246:1468–1474. [PubMed] [Google Scholar]
- 68.Williams C H J. Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J. 1995;9:1267–1276. doi: 10.1096/fasebj.9.13.7557016. [DOI] [PubMed] [Google Scholar]
- 69.Yang B, Gerhardt S G, Larson T J. Action at a distance for glp repressor control of glpTQ transcription in Escherichia coli. Mol Microbiol. 1997;24:511–521. doi: 10.1046/j.1365-2958.1997.3651733.x. [DOI] [PubMed] [Google Scholar]
- 70.Yang B, Larson T J. Action at a distance for negative control of transcription of the glpD gene encoding sn-glycerol 3-phosphate dehydrogenase of Escherichia coli. J Bacteriol. 1996;178:7090–7098. doi: 10.1128/jb.178.24.7090-7098.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang B, Larson T J. Multiple promoters are responsible for transcription of the glpEGR operon of Escherichia coli K-12. Biochim Biophys Acta. 1998;1396:114–126. doi: 10.1016/s0167-4781(97)00179-6. [DOI] [PubMed] [Google Scholar]
- 72.Zeng G, Ye S, Larson T J. Repressor for the sn-glycerol 3-phosphate regulon of Escherichia coli K-12: primary structure and identification of the DNA-binding domain. J Bacteriol. 1996;178:7080–7089. doi: 10.1128/jb.178.24.7080-7089.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng L, Cash V L, Flint D H, Dean D R. Assembly of iron-sulfur clusters. Identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. J Biol Chem. 1998;273:13264–13272. doi: 10.1074/jbc.273.21.13264. [DOI] [PubMed] [Google Scholar]