

Abstract
Cardiac amyloidosis is a group of diseases characterized by the deposition of amyloid fibers in cardiac tissue. Two forms are mainly reported: light chain (AL) and transthyretin (ATTR) amyloidosis. Among the complications of transthyretin amyloidosis there are thrombotic events and, to a lesser extent, hemorrhagic events. The latter are likely caused by perivascular amyloid deposition resulting in capillary fragility, in addition to INR lability during anticoagulant therapy. The onset of thrombotic events may be caused by the high prevalence of atrial fibrillation (AF), mechanical cardiac dysfunction and atrial myopathy observed in patients with transthyretin amyloidosis. It remains unclear why thromboembolic events occur even in patients with sinus rhythm or adequate anticoagulation, though a hypercoagulable state or underlying inflammation may be involved. We report a case of cryptogenic ischemic stroke in an 86-year-old woman with transthyretin amyloidosis and sinus rhythm. Traditional coagulation tests, whole blood rotational thromboelastometry and impedance aggregometry did not show a hypercoagulable state. The thrombin generation assay did not reveal a prothrombotic state. However, the study of extracellular vesicles highlighted underlying immune-mediated endothelial damage likely responsible for the thrombotic diathesis. It could be hypothesized that inflammation plays a role in the hypercoagulability of patients with transthyretin amyloidosis. Larger prospective studies are needed to validate our hypothesis.
Keywords: transthyretin amyloidosis, thrombotic complications, thromboelastometry thrombin generation, extracellular vesicles, case report
Introduction
Cardiac amyloidosis is a group of diseases caused by the deposition of amyloid fibers in heart tissue. There are two main forms of this disease: immunoglobulin light chain amyloidosis (AL); and transthyretin amyloidosis (ATTR) which is subdivided into hereditary transthyretin (TTR) amyloidosis (ATTRv) and wild-type TTR amyloidosis (ATTRwt) (1, 2). Among the most important complications of ATTR are thrombotic events and less frequently, hemorrhagic episodes (3). Although the latter appear to stem from perivascular amyloid deposition and consequent capillary fragility as well as labile INR during anticoagulant therapy, little is known about the mechanisms underlying thromboembolic events in patients with ATTR (4). The most common thrombotic events in ATTR amyloidosis are intracardiac thrombi and cerebral ischemic events, and less frequently, peripheral and splanchnic thrombosis. Amyloidosis fibers infiltrate the atrium and cause mechanical dysfunction resulting in blood stasis as well as endothelial damage, both elements of Virchow's triad that promote thrombosis. Hence the high prevalence of atrial fibrillation (AF), mechanical cardiac dysfunction and atrial myopathy in patients with ATTR. It remains unclear why thrombosis may occur even in patients with sinus rhythm and, albeit rarely, in adequately anticoagulated patients (5). Transthyretin may also be involved in the activation and regulation of the coagulation and fibrinolytic systems (6), which may contribute to the onset of thrombosis. We report a case of cryptogenic ischemic stroke in an 86-year-old female patient with transthyretin amyloidosis and sinus rhythm who underwent extensive coagulation, vascular and inflammatory workups to identify the possible pathogenetic mechanisms underlying the thrombotic event.
Methods
After obtaining written informed consent and following overnight fasting, blood samples were collected 37 days after the acute event, by venipuncture directly into 5 BD Vacutainer® Citrate Tubes containing 0.109 M (3.2%) sodium citrate (9:1 blood to anticoagulant ratio) using a 21 gauge needle with light tourniquet, after discharging the first mL of venous blood. Platelet-poor plasma (PPP) was prepared within 1 h of blood collection by double centrifugation at 2,500 × g for 15 min at room temperature. Aliquots (0.5 mL) were immediately frozen and stored at −80 °C until analysis.
Traditional coagulation tests
We measured prothrombin time/international normalized ratio, activated partial thromboplastin time, FXI, FX, FIX, FVIII, FII, fibrinogen, antithrombin (AT), protein C (PC), and protein S (PS), plasminogen, plasminogen activator inhibitor-1 (PAI-1) antigen, alpha2-antiplasmin, lupus anticoagulant (LA), anticardiolipin (aCL) antibodies (IgG and IgM), and anti-beta-2 glycoprotein 1 (anti-β2GP1) antibodies (IgG and IgM). Moreover, the presence of FV Leiden mutation and prothrombin G20210A variant was assessed. Prothrombin time (PT, n.v. 70%–100%), international normalized ratio (INR) and activated partial thromboplastin time (aPTT, n.v. 22.8–31 s) were measured according to standard procedures. Activity levels of FXI (n.v. 80%–120%), FX (n.v. 80%–120%), FIX (n.v. 80%–120%), FVIII (n.v. 60%–160%), and FII (n.v. 80%–120%) were measured using specific factor-deficient plasma (Siemens Healthcare Diagnostics, Milan, Italy). Fibrinogen concentration (n.v. 150–450 mg/dl) was measured by the Clauss method using a BCT-Analyzer (Dade Behring, Marburg, Germany) according to the manufacturer's recommendations. AT activity (n.v. 80%–120%) was measured using a thrombin-based chromogenic substrate assay (Berichrom® Antithrombin III, Siemens Healthcare Diagnostics) and PC activity (n.v. 80%–120%) was measured using a commercial kit (Protein C Reagent, Siemens Healthcare Diagnostics, Milan, Italy). AT and PC were performed using a BCS XP coagulation analyser (Siemens Healthcare Diagnostics, Milan, Italy). PS activity (n.v. 70%–130%) was assessed using the ProS kit (Instrumentation Laboratory, Milan, Italy) on an ACL TOP 300 CTS coagulation analyser (Instrumentation Laboratory, Milan, Italy). Coagulation inhibitors were measured according to the standard protocols supplied by the manufacturer, as previously reported (7). Plasminogen (n.v. 75%–140%), PAI-1 antigen (1.0–25.0 ng/ml) and alpha2-antiplasmin (n.v. 80%–120%) were measured according to standard's procedures. LA (dRVVT, n.v. 26–45 s and aPTT-LA, n.v. 32–43 s), aCL antibodies (IgG and IgM) (n.v. < 8 U/ml, in both cases) and anti-β2GP1 antibodies (IgG and IgM) (n.v. < 10 U/ml, in both cases) were measured as previously reported (8). Finally, genetic polymorphisms were determined using ABI Prism 3100 Genetic Analyzer (Applied Biosystem, Waltham, MA, USA) according to the manufacturer's instructions.
Whole blood coagulation tests
Rotational thromboelastometry (ROTEM® apparatus, Werfen, Bedford, MA, USA) is a whole blood (WB) viscoelastic test that evaluates changes in the viscoelastic resistance of the blood by applying a constant rotational force, and globally assesses the coagulation process from clot formation and stabilization to lysis (9). We performed the ROTEM® analysis according to the standard protocols supplied by the manufacturer, as previously reported (10, 11). The following tests were performed: INTEM (to assess intrinsic coagulation pathway), EXTEM (to assess the extrinsic coagulation pathway) and FIBTEM (to assess fibrinogen contribution to clot formation and stability). For EXTEM and INTEM assays, the following ROTEM® parameters were considered: (i) clotting time (CT, sec), the time from the beginning of the coagulation analysis until an increase in amplitude of 2 mm; (ii) clot formation time (CFT, sec), the time between an increase in amplitude of the thromboelastogram from 2 to 20 mm; (iii) maximum clot firmness (MCF, mm), the maximum amplitude reached in the thromboelastogram; (iv) alpha-angle (α, degrees), the angle between the horizontal axis and tangential line to the thromboelastogram through the 2 mm amplitude; (v) maximum lysis (ML, %), a measure of the percent of decrease in amplitude of the thromboelastogram at the end of the test. For FIBTEM assay CT, MCF and ML were considered. The CT reflects the initiation phase of the clotting process, CFT and alpha angles describe clot kinetics, MCF reflects clot stiffness and ML indicates the speed of fibrinolysis.
Platelet aggregation was tested using the Multiplate® function analyzer (Roche Diagnostics GmbH, Mannheim, Germany). Impedance aggregometry is a platelet function test in WB used to diagnose platelet alterations, in the monitoring of antiplatelet therapy and as a potential predictor of the need for transfusion and bleeding risk during major surgical procedures. The instrument continuously measures the changes in the electrical resistance (called “impedance”) between two copper wires. The greater the area, the more platelets aggregate. Briefly, 300 μl of citrated WB were added to an equal amount of saline solution, preheated at 37 °C, and platelet aggregation was tested after specific activation with arachidonic acid (ASPItest), adenosine diphosphate (ADPtest) and a thrombin analogue (TRAPtest). Platelet aggregation was electronically measured for 6 min and expressed as units of area under the curve (AUC) plotted over time in arbitrary units (U) (1 U = 10 AU × min) (12).
Thrombin generation (TG) was assessed using the calibrated automated thrombogram method (Thrombinoscope BV, Diagnostica Stago, Maastricht, The Netherlands) according to manufacturer's instructions (13). The test was performed on citrated WB within 4 h of collection, with and without the addition of thrombomodulin 20 nM (TM; Synapse Research Institute, Maastricht, the Netherlands) as previously described (14, 15). Citrated WB was firstly mixed with the fluorogenic thrombin substrate Z-Gly-Gly-Arg-aminomethylcoumarin (ZGGR-AMC; Bachem, Basel, Switzerland) and incubated for 10 min at 37 °C. Subsequently, WB was mixed with a solution containing tissue factor (TF) (Innovin®, Siemens Healthcare Diagnostics, Marburg, Germany) CaCl2 and MgCl2 in the presence or absence of TM. The volume ratio of WB, substrate solution, and TF-containing solution was 3:1:2. Sixty-five μl of the resulting mixture was transferred into wells of a 96-well plate. The final concentrations in the well were 50% WB, 1 pmol/L TF, 6 mmol/L CaCl2, 3 mM MgCl2, and 416.7 μmol/L ZGGR-AMC in the absence or presence of 20 nmol/L TM. The concentration of TM was chosen based on dose-response experiments in healthy controls, to inhibit TG by approximately 50%. All blood samples were measured in triplicate and calibrated by replacing the TF-containing solution with α2-macroglobulin-thrombin complex (α2M-T, corresponding with 300 nmol/L thrombin activity). Fluorescence signals were recorded at 37 °C with an interval time of 6 s by a microplate fluorometer (Fluoroskan AscentTM, Thermo Labsystems, Helsinki, Finland) with λex = 485 nm and λem = 538 nm using Fluoroskan Ascent Software (version 2.6). TG curves were described in terms of endogenous thrombin potential (ETP, nM*min), peak (nM), lag time (min), time to peak (min) and velocity index (vel index, nM/min). The results obtained were compared to those of a small (n. 20) historical group of healthy subjects previously described (16).
Extracellular vesicles
The pre-analytic phase of Extracellular vesicles (EV) analysis has been reported previously (17). We thawed PPP in a water bath for 5 min at 37 °C and after isolating large extracellular vesicles (L-EVs) by centrifugation at 14,000 g for 30 min at 4 °C, samples were immediately processed for immunolabeling. Samples were analyzed after a single freeze-thaw cycle. Flow cytometry analysis was performed using a CytoFLEX S flow cytometer (Beckman Coulter, United States), as previously reported (17, 18). For EV size calibration of the flow cytometer, fluorescent polystyrene beads Gigamix a mix 1:1 of Megamix FSC & SSC Plus (BioCytex, Marseille, France) were used in sizes of 0.1, 0.16, 0.2, 0.24, 0.3, 0.5, and 0.9 μm. Violet side scatter (VSSC) and FL1 channel gain were set to visualize the beads. The side scatter (SSC) from the 405 nm violet laser (VSSC) was used as a trigger signal to discriminate the noise with a size detection limit of 80 nm. Gigamix bead solution was gated to exclude background noise (from the solution itself). After turning the set in VSSC and forward scatter (FSC), a rectangular gate was set between the 0.2 and 0.9 μm bead populations and defined as L-EVs gate. Prior to staining, the antibody mixtures were centrifuged at 20,000 g for 30 min to remove fluorescent particles. The final concentration of the antibodies used was 1–5 μg/ml. The dilution buffer used, PBS and Annexin V buffer (Abcam, ab214484), were sterilized through a 0.2 µm mesh filter to reduce background noise. L-EVs were labeled with calcein-AM (referred to calcein-green, EMD Millipore Corp., cod.CI206700-1MG), Alexa Fluor 647-labeled Annexin V (Abcam, cod.ab214484), phycoerythrin (PE)-labeled anti-CD62E antibody (clone HAE-1f; BioLegend, San Diego, CA, cod.336008), mouse anti-human-TF (1:100 diluted, #4509, American Diagnostica) followed by anti-mouse IgG secondary antibody Alexa Fluor 750 (1:300 diluted, Sigma-Aldrich, cod.SAB4600211-250UL), PE-labeled anti-CD62P (clone CLB-Thromb/6; Beckman Coulter, Marseille, France, cod.IM1759U) PE/Cyanine 7-labeled anti-CD41 (clone P2; Beckman Coulter, cod.6607115), ECD labeled anti-CD45 (clone J33; Beckman Coulter, cod.A07784), PE-labeled anti-CD14 (clone 61D3; Invitrogen, cod.12-0149-42). Calcein-AM staining and the non-conjugated primary antibodies were incubated for 30 min at 37 °C. Subsequently, the conjugated antibodies and the secondary antibodies (i.e., anti-mouse IgG Alexa Fluor 750), were incubated for 30 min in the dark at RT. Finally, L-EVs were resuspended in 120 μl of sterile filtered Annexin V buffer using a 96-well polypropylene flat plate and analyzed within 1 h of immunolabeling. Parallel incubation was performed with isotype-matched control antibodies and with the secondary antibody alone to exclude non-specific staining. Fluorescence measured with the respective isotype negative control antibody were subtracted in order to avoid non-specific background signal. Unstained samples were used as negative control. Flow cytometer acquisition settings were maintained for all samples, including triggering threshold, voltages and flow rate. No substantial changes in scatter or fluorescence signals were observed between unstained and matched isotype controls. We expressed EVs as events/μl (absolute count) with the volume measurement of the CytoFLEX. Files were exported and data was evaluated by CytExpert (Software Version 2.3, Beckman Coulter). True EV events were defined as double-positive stained for calcein-AM and one of the other specific antibodies. Triple-positive EVs were also evaluated for calcein-AM, anti-CD62E and anti-TF, and namely, E-Selectin+TF+. Calcein-AM is a non-fluorescent marker for EVs which becomes fluorescent only after absorption due to the esterase present in the double phospholipid membrane of EVs (calcein+). Annexin V is an index of apoptosis which binds to phosphatidylserine, a membrane phospholipid whose presence on the external surface after activation of platelets promotes coagulation and thrombosis. CD62E/E-Selectin+ is a marker of activated endothelial cells; CD62P/P-Selectin+, marker of activated platelets; CD41, marker of platelet-derived EVs; CD45, marker of leukocyte-derived EVs and CD14, marker of monocyte/macrophage-derived EVs. The levels of EVs in our patient were compared to those observed in 20 healthy age- (±2 years) and sex-matched controls.
Case report
The patient was admitted to our Emergency Department (ED) due to a sudden onset of left hemisyndrome and deviation of gaze to the right, as witnessed by family members. Past medical history included a previous tuberculosis with right-sided cavitation and transthyretin cardiac amyloidosis diagnosed in May 2022 — i.e., suggestive echocardiographic findings with left ventricular hypertrophy without cardiac kinetic abnormalities; absence of monoclonal serum component in blood chemistry and urinary tests; supported by a bone scintigraphy with cardiac hyperuptake (Perugini score 3). Moreover, the patient was undergoing antihypertensive and anxiolytic therapies. White blood cell count and hemoglobin were within the normal ranges. Renal and liver function tests were normal. Pre-stroke mRS score was 1. A computed tomography (CT) scan of the brain showed a malacic area limited to the left mesial frontal region, traceable to an ischemic event in the region of the distal branches of the ipsilateral anterior cerebral artery (Alberta stroke program early CT score, ASPECTS 9). A CT angiography of the cerebral-afferent vessels combined with a perfusion study showed no recognizable opacification in the M1 section of the right middle cerebral artery, due to thrombotic occlusion. The perfusion study revealed abnormal Tmax and cerebral blood flow (CBF) values in the volume of parenchyma studied with a mismatch of approximately 21.3, due to the presence of a large area of ischemic penumbra in the region of the right middle cerebral artery (Figure 1). The neurological exam performed in the ED showed a lack of leveling of the left upper limb and sagging of the left lower limb, and fluent speech albeit with dysarthria (National Institutes of Health Stroke Scale, NIHSS 12). Since there were no contraindications, systemic thrombolysis was performed for a weight of 73 kg, total dose 65.7 mg (10% bolus, 1 hour of continuous infusion). The patient was later transferred to the angiography suite to undergo mechanical thrombectomy, with manual thromboaspiration of the organized red thrombus. The final control angiography showed complete recanalization of the right middle cerebral artery (modified thrombolysis in cerebral infarction, mTICI 3). Finally, the patient was admitted to the Stroke Unit. During hospitalization the patient remained haemodynamically stable, with good respiratory exchange and afebrile. The electrocardiogram (ECG) showed a first-degree atrioventricular block, right bundle branch block and left anterior fascicular block. The patient was under continuous electrocardiography monitoring throughout her stay (4 days) in the Stroke Unit after the acute event, and no atrial fibrillation was detected. Vital parameters remained normal and stable during hospitalization. The control brain CT scan performed on the first day of hospitalization confirmed an ischemic lesion in the right internal capsule. A neurosonological study of the intracranial and cerebral-afferent vessels to ascertain its etiology, confirmed the patency of the right middle cerebral artery whereas there was a significant stenosis before the procedure. Furthermore, a resting echocardiography also revealed only moderate left atrial dilatation and mild mitral regurgitation. Finally, a 24-h Holter monitor detected no occult arrhythmias. A full cardiological evaluation recommended continuation of the ongoing follow-up for cardiac amyloidosis. Acetylsalicylic acid was initiated as secondary prevention therapy and atorvastatin was also introduced at low dosage due to the vascular pleiotropic effect. A Doppler ultrasound of the supra-aortic trunks revealed an extracranial finding of mild stenosis of the carotid bifurcation and at the origin of the right external carotid artery and the Doppler ultrasound of the intracranial vessels instead was within normal limits. The patient underwent cycles of kinesiotherapy with good response. The discharge neurological exam showed a clear improvement: a slight weakness of the left lower extremity and a slight impairment of the left seventh cranial nerve persisted. The patient walked unassisted with the aid of a walker and was fed orally with a soft diet and free fluids.
Figure 1.
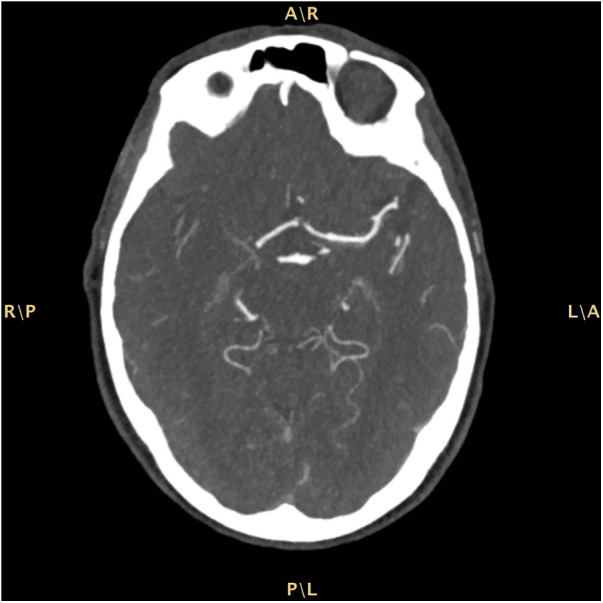
Computed tomography (CT) scan of the brain.
The coagulation parameters of our patient are reported in Table 1, and they were all normal. The search for aCL antibodies and anti-β2GP1 antibodies was negative. Genetic testing excluded the presence of the FV Leiden mutation and prothrombin G20210A variant. The thromboelastometry profiles and Multiplate® aggregometry results are reported in Figure 2. All parameters considered were within the normal ranges, except the AUC value in ASPItest which was significantly reduced vs. the lower limit of the normal range, likely due to ongoing antiplatelet therapy at the time of blood sample collection. The results of WB-TG are reported in Table 1. The values observed were within the range (25th-75th percentile) obtained in a small cohort (n. 10) of healthy subjects. Our patient's L-EVs measurements are reported in Table 2. We found that endothelium-derived (CD14+ and E-selectin) and leukocyte-derived (CD45+) L-EVs, as well as E-selectin+TF+ L-EVs were higher vs. healthy controls. We also observed an increase in the absolute count of L-EVs vs. healthy controls. Conversely, platelet-derived L-EVs (CD41+, P-selectin and CD41+P-selectin+) were lower in our patient vs. healthy controls.
Table 1.
Traditional coagulation tests and thrombin generation.
| Value | Reference range | |
|---|---|---|
| Prothrombin time, PT (%) | 80.4 | 70–100 |
| International normalized ratio, INR | 1.11 | – |
| Activated partial thromboplastin time, aPTT (sec) | 30.7 | 22.8–31.0 |
| FXI (%) | 80.6 | 80–120 |
| FX (%) | 83.6 | 80–120 |
| FIX (%) | 88.2 | 80–120 |
| FVIII (%) | 119.6 | 60–120 |
| FII (%) | 84.9 | 80–120 |
| Fibrinogen (mg/dl) | 238 | 150–450 |
| Antithrombin (%) | 83.3 | 80–120 |
| Protein C coagulometric activity (%) | 80.9 | 80–120 |
| Protein C chromogenic activity (%) | 85.7 | 80–120 |
| Protein C antigen (%) | 84 | 80–120 |
| Protein S coagulometric activity (%) | 81.6 | 70–130 |
| Protein S free antigen (%) | 102 | 80–120 |
| Protein S total antigen (%) | 91 | 80–120 |
| Plasminogen activity (%) | 73.6 | 75–140 |
| Plasminogen activator inhibitor-1 antigen (ng/ml) | 11.6 | 1.0–25.0 |
| Alpha2-antiplasmin (%) | 80.9 | 80–120 |
| aPTT—Lupus Anticoagulant (sec) | 33.7 | 32.0–43.0 |
| dRVVT (sec) | 32.8 | 26.0–45.0 |
| Anticardiolipin antibodies IgG (U/ml) | 1.2 | <10 |
| Anticardiolipin antibodies IgM (U/ml) | 2.3 | <10 |
| Anti-beta-2 glycoprotein 1 IgG (U/ml) | 1.4 | <8 |
| Anti-beta-2 glycoprotein 1 IgM (U/ml) | 2.0 | <8 |
| APC sensitivity ratio | 3.71 | >2.00 |
| APC sensitivity ratio normalization | 1.23 | >0.84 |
| FV Leiden mutation | Wild type | – |
| Prothrombin G20210A variant | Wild type | – |
| Thrombin Generation | ||
| ETP (nM*min) | 1,525 | 1,374–1,804 |
| ETP + TM (nM*min) | 893 | 755–1,138 |
| Peak (nM) | 182.9 | 175.6–228.9 |
| Peak + TM (nM) | 119.9 | 109.6–132.7 |
| Lag time (min) | 3.7 | 3.2–4.3 |
| Lag time + TM (min) | 5.7 | 5.1–6.5 |
| Time to peak (min) | 8.8 | 8.3–10.0 |
| Time to peak + TM (min) | 9.9 | 9.5–11.5 |
| Vel index (nM/min) | 38.0 | 29.3–39.7 |
| Vel index + TM (nM/min) | 28.2 | 24.5–37.6 |
dRVVT, diluted Russel viper venom time; APC, activated protein C; ETP, endogenous thrombin potential; TM, thrombomodulin, Vel, velocity.
Figure 2.

(A) Multiplate aggregometry profiles. ASPI-test, arachidonic acid-induced aggregation; ADP-test, adenosine-diphosphate-induced aggregation; TRAP-test, thrombin receptor activating peptide-induced aggregation. (B) Thromboelastometry profiles. EXTEM, extrinsic coagulation pathway assay; INTEM, intrinsic coagulation pathway assay; FIBTEM, contribution of fibrinogen to blood clotting assay; CT, clotting time; CFT, clot formation time; ɑ, ɑ-angle; MCF, maximum clot firmness; ML, maximum lysis.
Table 2.
Large EV subgroups levels.
| Patient | Controls | |
|---|---|---|
| Calcein + Annexin V+ | 113 | 328 |
| Calcein + CD41+ | 492 | 1,052 |
| Calcein + P-selectin+ | 614 | 2,216 |
| Calcein + CD41 + P-selectin+ | 41 | 259 |
| Calcein + CD45+ | 1,443 | 1,673 |
| Calcein + CD14+ | 102 | 73 |
| Calcein + E-selectin | 1,540 | 1,261 |
| Calcein + TF+ | 107 | 79 |
| Calcein + E-selectin + TF+ | 56 | 31 |
Data shown as median in events/µl with the volume measurement of the.
CytoFLEX (Beckman Coulter). EV, extracellular vesicle; TF, tissue factor.
Discussion
Thrombotic events are among the most severe complications of amyloidosis, particularly in ATTR and mainly as it pertains to intracardiac thrombi and cerebrovascular events. In a recently published review (19), we endeavored to identify studies that investigated the incidence of and the risk factors associated with thromboembolism in patients with ATTR — there were few and comprising small cohorts. The study by Bukari S et al. (20) confirmed a high thromboembolic risk in patients with ATTRwt-CA and AF, considering the latter was present on average in 88% of patients. It has been noted that even ATTR patients in sinus rhythm or in atrial fibrillation with adequate anticoagulant therapy may develop both atrial and cerebrovascular thromboembolic events, as in our case, and more rarely, peripheral thrombosis. In fact, a study by Vilches et al. (21) found a 1.3 incidence rate among subjects with normal sinus rhythm without oral anticoagulant therapy (OAT), 1.7 among those with AF undergoing OAT, and 4.8 among those with AF without OAT. It is of the utmost importance to understand the mechanisms underlying thromboembolism in these patients, particularly whether the coagulation system is involved, to identify who might benefit from early anticoagulant treatment. Thus, our patient underwent an extensive coagulation workup (i.e., the extrinsic and intrinsic coagulation pathways, fibrinolysis, antiphospholipid antibody syndrome and inherited thrombophilia testing). All these tests excluded the presence of a prothrombotic state. Moreover, those tests performed in WB, and in particular thromboelastometry, impedance aggregometry and TG all failed to detect a hypercoagulable state which may have explained the thrombotic episode suffered by our patient. It bears noting that the ASPI test highlighted a markedly reduced platelet function, traceable to the fact that our patient was taking antiplatelet drugs at the time of blood sampling. Finally, we performed an analysis of EVs which are cell-derived vesicles delimited by a phospholipid bilayer, with a diameter of 30–1,000 nm, released by extrusion of the cytoplasmic membrane of activated cells in normal (physiological) conditions and during diseases (pathological conditions), and considered as mediators of intercellular communication (22). There have been reports that L-EVs may have procoagulant properties due to the presence of negatively charged membrane phospholipids (i.e., phosphatidylserine) on their surface, which provide a substrate for the protagonists of coagulation (intrinsic pathway) but can also trigger coagulation (extrinsic pathway) through the expression of tissue factor (23). The origin of L-EVs can be identified by studying surface antigens (e.g., activated endothelial cells, platelets, leukocytes) and the different subtypes can be quantified (24). Our main endeavor was to measure EV levels once the patients had recovered or overcome the active phase of the stroke, to ascertain whether there was any correlation between “baseline” EV levels and cardiac amyloidosis. In our case, it had been over a month since the acute event (37 days) which placed our patient in the early subacute phase according to the traditional classification of ischemic stroke. Furthermore, the EV trend according to the study by Lundström et al. (25) would place us towards the end of the convalescence phase, hence the reason why we deemed this time window appropriate for blood sampling. We compared our patient's EV profile to that observed in 20 healthy age- and sex-matched controls. Interestingly, both endothelium-derived and leukocyte-derived L-EVs measured in our patient were higher vs. control group. We hypothesized that these findings may suggest an underlying immune-mediated endothelial damage responsible for the thrombotic diathesis, and therefore, inflammation may contribute to the coagulopathy in patients with ATTR. To the best of our knowledge there are no other studies published so far in the literature that have evaluated the circulating levels of L-EVs in patients with ATTR. However, it bears noting that lower levels of platelet-derived EVs in our patient vs. controls is not an unexpected finding. There have been reports in the literature that antiplatelet drugs, as effectively administered in our patient given the ASPItest results, may reduce plasma levels of platelet-derived L-EVs (26). Bulut D et al. (27) demonstrated a reduction in the number of circulating L-EVs derived from endothelium and platelets in 15 male patients with coronary artery disease being treated with aspirin. Another interesting case was reported by Kim HJ et al. (28) wherein after the discontinuation of aspirin in 17/46 patients presenting with a chronic inflammatory disease such as Kawasaki syndrome, there was a renewed increase in circulating platelet-derived L-EVs. The hypothesis is that there may be an inflammatory substrate in ATTR amyloidosis as well. The study by Lv Y at al (29). also suggests that treatment with aspirin can reduce the number of circulating endothelial cells and platelet-derived L-EVs in patients with cardiovascular diseases. On the other hand, we would argue that ascertaining the effect of statins on endothelium-derived EVs is still a matter of debate, and it has been evaluated mostly in cultured endothelial cells (30). Statins have been shown to reduce levels of circulating EVs derived from endothelium, platelets and inflammatory cells (31). Although there are several studies in the literature demonstrating that statins downregulate the release of endothelium-derived EVs by human coronary artery endothelial cells (32–34), few studies have investigated the effects of statins on platelet function. These studies demonstrate that there are pathways potentially responsible for interactions between statins and platelets, and support the hypothesis that statin treatment may reduce not only the number of platelet-derived EVs but markers of activated platelets as well (35).
We would be remiss not to mention some of the limitations of our study. We did not perform a high-resolution brain Magnetic Resonance Imaging (MRI) and thus were not able to exclude the presence of cerebral small vessel disease—such as, cerebral amyloid angiopathy (CAA)—as a cause of ischemia. Nevertheless, our patient did not present a clinical history compatible with CAA and the acute event occurred in a large vessel (i.e., the right middle cerebral artery, on cerebral CT angiography) requiring thrombolysis, without hemorrhagic complications, as seen on subsequent CT scans. Therefore, we deemed the hypothesis of CAA unlikely. Moreover, we could not definitively exclude a cardioembolic etiology for the cerebral ischemic event in our patient. Throughout hospitalization, we found no evidence of atrial fibrillation and the echocardiogram revealed no intracardiac thrombi. Nevertheless, we cannot definitively rule it out. The use of fluorescent polystyrene beads for EV size calibration constitutes another limitation as they do not have the same refractive index as EVs, which may also vary by size. Another limitation of the study, according to the latest minimal information for studies of extracellular vesicles (MISEV) recommendations, is that no single measurement or method is able to satisfy all EV characterization requirements, and the use of different orthogonal techniques is recommended. Unfortunately, we did not perform electron microscopy or nanoparticle tracking analysis (NTA) as these techniques require large volumes of plasma sample.
In conclusion, our findings are preliminary and did not allow us to draw any definitive conclusions on the possible use of L-EVs as predictive biomarkers in clinical practice. Larger prospective studies are needed to confirm our findings and to clarify the role, if any, of L-EVs as a possible risk factor of thrombotic events in patients with ATTR.
Funding Statement
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Azienda Ospedaliera Università di Padova. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
AN: Writing – original draft, Writing – review & editing. ST: Writing – original draft, Writing – review & editing. CB: Writing – review & editing, Writing – original draft. EC: Writing – review & editing, Writing – original draft. PS: Writing – review & editing, Writing – original draft. LS: Writing – original draft, Writing – review & editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- 1.Rapezzi C, Merlini G, Quarta C, Riva L, Longhi S, Leone O, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. (2009) 120:1203–12. 10.1161/CIRCULATIONAHA.108.843334 [DOI] [PubMed] [Google Scholar]
- 2.Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. (2016) 387:2641–54. 10.1016/S0140-6736(15)01274-X [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. (2021) 42:1554–68. 10.1093/eurheartj/ehab072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicol M, Siguret V, Vergaro G, Aimo A, Emdin M, Dillinger JG, et al. Thromboembolism and bleeding in systemic amyloidosis: a review. ESC Heart Fail. (2022) 9:11–20. 10.1002/ehf2.13701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cappelli F, Tini G, Russo D, Emdin M, Del Franco A, Vergaro G, et al. Arterial thrombo-embolic events in cardiac amyloidosis: a look beyond atrial fibrillation. Amyloid. (2021) 28:12–8. 10.1080/13506129.2020.1798922 [DOI] [PubMed] [Google Scholar]
- 6.Wieczorek E, Ożyhar A. Transthyretin: from structural stability to osteoarticular and cardiovascular diseases. Cells. (2021) 10:1768. 10.3390/cells10071768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campello E, Spiezia L, Radu CM, Bulato C, Gavasso S, Tormene D, et al. Circulating microparticles and the risk of thrombosis in inherited deficiencies of antithrombin, protein C and protein S. Thromb Haemost. (2016) 115:81–8. 10.1160/TH15-04-0286 [DOI] [PubMed] [Google Scholar]
- 8.Rossetto V, Spiezia L, Franz F, Salmaso L, Pozza LV, Gavasso S, et al. The role of antiphospholipid antibodies toward the protein C/protein S system in venous thromboembolic disease. Am J Hematol. (2009) 84:594–6. 10.1002/ajh.21466 [DOI] [PubMed] [Google Scholar]
- 9.Zanetto A, Senzolo M, Vitale A, Cillo U, Radu C, Sartorello F, et al. Thromboelastometry hypercoagulable profiles and portal vein thrombosis in cirrhotic patients with hepatocellular carcinoma. Dig Liver Dis. (2017) 49:440–5. 10.1016/j.dld.2016.12.019 [DOI] [PubMed] [Google Scholar]
- 10.Shalaby S, Simioni P, Campello E, Spiezia L, Gavasso S, Bizzaro D, et al. Endothelial damage of the portal vein is associated with heparin-like effect in advanced stages of cirrhosis. Thromb Haemost. (2020) 120:1173–81. 10.1055/s-0040-1713169 [DOI] [PubMed] [Google Scholar]
- 11.Spiezia L, Bertini D, Boldrin M, Radu C, Bulato C, Gavasso S, et al. Reference values for thromboelastometry (ROTEM®) in cynomolgus monkeys (Macaca fascicularis). Thromb Res. (2010) 126:e294–7. 10.1016/j.thromres.2010.07.016 [DOI] [PubMed] [Google Scholar]
- 12.Campello E, Spiezia L, Zabeo E, Maggiolo S, Vettor R, Simioni P. Hypercoagulability detected by whole blood thromboelastometry (ROTEM®) and impedance aggregometry (MULTIPLATE®) in obese patients. Thromb Res. (2015) 135:548–53. 10.1016/j.thromres.2015.01.003 [DOI] [PubMed] [Google Scholar]
- 13.Campello E, Bulato C, Spiezia L, Boscolo A, Poletto F, Cola M, et al. Thrombin generation in patients with COVID-19 with and without thromboprophylaxis. Clin Chem Lab Med. (2021) 59:1323–30. 10.1515/cclm-2021-0108 [DOI] [PubMed] [Google Scholar]
- 14.Li L, Roest M, Sang Y, Remijn JA, Fijnheer R, Smit K, et al. Patients with multiple myeloma have a disbalanced whole blood thrombin generation profile. Front Cardiovasc Med. (2022) 9:919495. 10.3389/fcvm.2022.919495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan J, Konings J, Yan Q, Kelchtermans H, Kremers R, de Laat B, et al. A novel assay for studying the involvement of blood cells in whole blood thrombin generation. J Thromb Haemost. (2020) 18:1291–301. 10.1111/jth.14786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zanetto A, Campello E, Bulato C, Willems R, Konings J, Roest M, et al. Whole blood thrombin generation shows a significant hypocoagulable state in patients with decompensated cirrhosis. J Thromb Haemost. (2024) 22:480–92. 10.1016/j.jtha.2023.10.008 [DOI] [PubMed] [Google Scholar]
- 17.Campello E, Radu CM, Duner E, Lombardi AM, Spiezia L, Bendo R, et al. Activated platelet-derived and leukocyte-derived circulating microparticles and the risk of thrombosis in heparin-induced thrombocytopenia: a role for PF4-bearing microparticles? Cytometry. (2018) 94:334–41. 10.1002/cyto.b.21507 [DOI] [PubMed] [Google Scholar]
- 18.Campello E, Radu CM, Simion C, Spiezia L, Bulato C, Gavasso S, et al. Longitudinal trend of plasma concentrations of extracellular vesicles in patients hospitalized for COVID-19. Front Cell Dev Biol . (2022) 9:770463. 10.3389/fcell.2021.770463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Napolitano A, De Michieli L, Sinigiani G, Berno T, Cipriani A, Spiezia L. Thromboembolic and bleeding events in transthyretin amyloidosis and coagulation system abnormalities: a review. J Clin Med. (2023) 12:6640. 10.3390/jcm12206640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bukhari S, Khan SZ, Bashir Z. Atrial fibrillation, thromboembolic risk, and anticoagulation in cardiac amyloidosis: a review. J Card Fail. (2023) 29:76–86. 10.1016/j.cardfail.2022.08.008 [DOI] [PubMed] [Google Scholar]
- 21.Vilches S, Fontana M, Gonzalez-Lopez E, Mitrani L, Saturi G, Renju M, et al. Systemic embolism in amyloid transthyretin cardiomyopathy. Eur J Heart Fail. (2022) 24:1387–96. 10.1002/ejhf.2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owens AP, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. (2011) 108:1284–97. 10.1161/circresaha.110.233056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campello E, Spiezia L, Radu CM, Simioni P. Microparticles as biomarkers of venous thromboembolic events. Biomarkers Med. (2016) 10:743–55. 10.2217/bmm-2015-0063 [DOI] [PubMed] [Google Scholar]
- 24.Mooberry MJ, Key NS. Microparticle analysis in disorders of hemostasis and thrombosis. Cytometry. (2016) 89:111–22. 10.1002/cyto.a.22647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lundström A, Mobarrez F, Rooth E, Thålin C, von Arbin M, Henriksson P, et al. Prognostic value of circulating microvesicle subpopulations in ischemic stroke and TIA. Transl Stroke Res. (2020) 11:708–19. 10.1007/s12975-019-00777-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taus F, Meneguzzi A, Castelli M, Minuz P. Platelet-derived extracellular vesicles as target of antiplatelet agents. What is the evidence? Front Pharmacol. (2019) 10:1256. 10.3389/fphar.2019.01256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulut D, Becker V, Mügge A. Acetylsalicylate reduces endothelial and platelet-derived microparticles in patients with coronary artery disease. Can J Physiol Pharmacol. (2011) 89:239–44. 10.1139/y11-013 [DOI] [PubMed] [Google Scholar]
- 28.Kim HJ, Choi EH, Lim YJ, Kil HR. The usefulness of platelet-derived microparticle as biomarker of antiplatelet therapy in kawasaki disease. J Korean Med Sci. (2017) 32:1147–53. 10.3346/jkms.2017.32.7.1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lv Y, Tan J, Miao Y, Zhang Q. The role of microvesicles and its active molecules in regulating cellular biology. J Cell Mol Med. (2019) 23:7894–904. 10.1111/jcmm.14667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montoro-García S, Lip GY, Shantsila E. Atorvastatin and its collateral effects on microparticles. Thromb Haemost. (2011) 106:185–6. 10.1160/TH11-05-0335 [DOI] [PubMed] [Google Scholar]
- 31.Suades R, Padró T, Alonso R, Mata P, Badimon L. Lipid-lowering therapy with statins reduces microparticle shedding from endothelium, platelets and inflammatory cells. Thromb Haemost. (2013) 110:366–77. 10.1160/TH13-03-0238 [DOI] [PubMed] [Google Scholar]
- 32.Badimon L, Suades R, Arderiu G, Peña E, Chiva-Blanch G, Padró T. Microvesicles in atherosclerosis and angiogenesis: from bench to bedside and reverse. Front Cardiovasc Med. (2017) 4:77. 10.3389/fcvm.2017.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res. (2017) 120:229–43. 10.1161/CIRCRESAHA.116.308537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tramontano AF, O'Leary J, Black AD, Muniyappa R, Cutaia MV, El-Sherif N. Statin decreases endothelial microparticle release from human coronary artery endothelial cells: implication for the rho-kinase pathway. Biochem Biophys Res Commun. (2004) 320:34–8. 10.1016/j.bbrc.2004.05.127 [DOI] [PubMed] [Google Scholar]
- 35.Nenna A, Nappi F, Lusini M, Satriano UM, Schilirò D, Spadaccio C, et al. Effect of statins on platelet activation and function: from molecular pathways to clinical effects. Biomed Res Int. (2021) 2021:6661847. 10.1155/2021/6661847 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.