

Abstract
Objectives
The goals of the study were: (1) to develop and evaluate non-replicating lentivirus vectors coding for feline coronavirus (FCoV)-specific micro (mi)RNA as a potential antiviral therapy for feline infectious peritonitis (FIP); (2) to assess the feasibility of transducing hematopoietic stem cells (HSCs) with ex vivo introduction of the miRNA-expressing lentivirus vector; and (3) to assess the ability of the expressed miRNA to inhibit FCoV replication in HSCs in vitro.
Methods
HSCs were obtained from feline bone marrow and replicated in vitro. Three lentiviruses were constructed, each expressing a different anti-FCoV miRNA. HSCs were stably transduced with the miRNA-expressing lentivirus vector that produced the most effective viral inhibition in a feline cell line. The effectiveness of the transduction and the expression of anti-FCoV miRNA were tested by infecting the HSCs with two different strains of FCoV. The inhibition of coronavirus replication was determined by relative quantification of the inhibition of intracellular viral genomic RNA synthesis using real-time, reverse-transcription PCR. The assessment of virus replication inhibition was determined via titration of extracellular virus using the TCID50 assay.
Results
Inhibition of FCoV was most significant in feline cells expressing miRNA-L2 that targeted the viral leader sequence, 48 h postinfection. miRNA-L2 expression in stably transduced HSCs resulted in 90% and 92% reductions in FIPV WSU 79-1146 genomic RNA synthesis and extracellular virus production, respectively, as well as 74% and 80% reduction in FECV WSU 79-1683 genomic RNA synthesis and extracellular virus production, respectively, as compared with an infected negative control sample producing non-targeting miRNA.
Conclusions and relevance
These preliminary results show that genetic modification of HSCs for constitutive production of anti-coronavirus miRNA will reduce FCoV replication.
Introduction
Feline infectious peritonitis (FIP) is caused by mutant feline coronavirus (FCoV) that can replicate efficiently in monocytes and macrophages with dysregulation of host cell-mediated immunity. This allows the virus to replicate unchecked to a high titer and efficient systemic viral replication appears to play a critical role in FIP pathogenesis.1,2 There is no treatment for FIP, which now accounts for the majority of infectious disease-related deaths in pet cats. 3 In a previous study, we proved that RNA interference (RNAi) that targets highly conserved viral genes can be used to inhibit FCoV replication in vitro. 4 RNAi, mediated by small interfering RNAs (siRNAs) or microRNAs (miRNAs), has therapeutic potential if the small RNA molecules can be delivered in sufficient quantity to monocytes/macrophages of an FIP-affected cat.
siRNAs and miRNAs share many similarities: both are short duplex RNA molecules that exert gene silencing effects at the post-transcriptional level by targeting messenger RNA, yet there is a difference in their therapeutic application. siRNA is exogenously delivered to mammalian cells and has a transient effect, with silencing only lasting a few days as the amount in the cell declines. Mammalian cells have not been shown to produce endogenous siRNA naturally. The amount of siRNA delivered to the cell is dependent on the success of the delivery vehicle used. Delivery of these molecules to the target cells in vivo is the biggest obstacle that faces RNAi therapeutic progress.
Unlike with siRNA, cells can be manipulated to produce miRNA. Introduction of miRNA expressing DNA into the host cell genome can be undertaken with use of lentivirus vectors. These vectors, like the viruses from which they are created, naturally incorporate into host DNA. The miRNA can then be constitutively expressed for the lifetime of the cell, in effect prolonging the silencing effect indefinitely. Use of RNAi in this manner has been applied for the treatment of viruses. Specifically, phase I and II clinical trials in which autologous genetically modified hematopoietic cells were transduced ex vivo to elicit an RNAi response against HIV, followed by infusion back into the patient, have already been performed successfully in people.5,6
In the current study, we describe the feasibility of generating an RNAi response against FCoV in feline hematopoietic stem cells (HSCs) by ex vivo introduction of DNA that codes for anti-FCoV miRNAs. The future therapeutic use of these stem cells, similar to HIV treatment, would be introduction of these cells into cats to produce monocytes and macrophages with the ability to reduce/inhibit coronavirus replication.
Materials and methods
HSC isolation, expansion and characterization
All experiments were carried out according to Institutional Animal Care and Use Committee (IACUC) protocol number 2181, which was reviewed and approved by the University of Tennessee IACUC. Bone marrow aspirates were obtained from the femurs of three young adult cats. Briefly, cats were sedated with a combination of ketamine and dexmedetomidine with torbugesic (intravenously). Also, lidocaine was administered at the aspiration sites to numb the area. Bone marrow was obtained by gentle aspiration with a syringe loaded with 300 IU/ml of heparin sulfate. Following the procedure and 24 h later, meloxicam was given orally for pain. Then, samples were processed as previously described with slight modifications.7,8 Bone marrow from each cat was diluted 1:4 with phosphate-buffered saline (PBS), and loaded onto 15 ml lymphocyte separation media (Ficoll; Thermo Fisher Scientific) in a 50 ml tube. The mix was centrifuged at 400 × g for 20 mins at 20°C to obtain the mononuclear fraction of cells. Next, cells were pelleted and washed in PBS buffer with 0.1% bovine serum albumin (BSA). The total number of mononuclear cells was counted, and the cells were seeded at a density of 2 × 105 cells per cm2 in Dulbecco’s Modified Eagle Medium/high glucose (DMEM/high glucose; Hylcone) containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin solution (Sigma Aldrich), recombinant human stem cell factor (50 ng/ml; R&D systems), and IL-3 (30 ng/ml; R&D systems). The cells were incubated at 37°C with 5% CO2. Adherent cells were observed daily using a Zeiss Axiovert 40CFL microscope (CarlZeiss MicroImaging) equipped with a Cannon Powershot A620 camera (Cannon USA) and analyzed using ZoomBrowser EX software (Cannon USA).
When enough cells were obtained, they were characterized using anti-CD antigens 34 antibodies (Abcam), as described below. These antigens are specifically expressed on hematopoietic cells. Cells were also cryopreserved in liquid nitrogen in cryopreservation medium (50% FBS, 5% dimethylsulfoxide 45% DMEM/high glucose) for future applications.
HSCs were characterized by flow cytometry and indirect immunofluorescence assay. For the flow cytometer analysis, cells were collected in flow buffer (60 ml 0.5% sodium azide solution, 87 ml PBS solution and 3 ml FBS), and incubated with 0.5 mg/ml Mouse BD Fc Block (BD Bioscience) for 20 mins at 4°C, and subsequently with anti-CD34 antibody (rabbit monoclonal CD34, 0.52 mg/ml; Abcam) for 60 mins. After washing, the cells were incubated with Alexa 488 goat antirabbit antibody (2 mg/ml; Invitrogen) for 20 mins at 4°C in the dark. Cells were collected by centrifugation at 200 × g at 4°C for 5 mins at each step and were washed thoroughly with flow buffer. Staining was assessed on a flow cytometer (FACS Aria; Becton Dickson) by electronic gating on a dual parameter dot plot of forward and side scatter. For each sample, 10,000 events were measured. Raw data of fluorescence representing the number of positive cells was measured and analyzed by Cell Quest software.
For the indirect immunofluorescence assay (IFA), cells grown in 35 × 10 mm cell culture dishes were rinsed with PBS and fixed in 4% paraformaldehyde in PBS. Cells were incubated with 1% BSA in PBS for 40 mins and stained with anti-CD34 antibody at a dilution of 1:100 in PBS for 2 h at room temperature. The cells were washed three times with PBS (5 mins each wash). Then the cells were incubated with Alexa 488 goat antirabbit antibody (Thermo Fisher Scientific) for 1 h at room temperature in the dark. The cells were washed three times with PBS (5 mins each wash). Cells were counterstained with DAPI (Thermo Fisher Scientific) and examined and analyzed with a Zeiss Axiovert 40CFL microscope.
Design of FCoV-specific miRNA double-stranded DNA and generation of expression vector
miRNA double-stranded DNA sequences were designed based on our previously developed siRNAs that produced >80% viral RNA inhibition and sequence data for FCoVs available in the GeneBank public database, using an RNA interference designer (Thermo Fisher Scientific). 4 Three miRNA sequences were chosen and developed for testing, including miRNA-L1 and L2, which targeted the viral leader sequence, and miRNA-N, which targeted the nucleocapsid gene (Table 1). The sequences of these miRNAs were further compared with FCoV sequences published in the GenBank database to confirm that target regions were highly conserved among FCoV strains. Each designed miRNA sequence, as well as a negative non-targeting miRNA (control) sequence (Thermo Fisher Scientific), was cloned into an expression plasmid (BLOCK-iT Pol II miR RNAi Expression Vector; Thermo Fisher Scientific) according to the manufacturer’s protocol. This expression plasmid contained emerald green fluorescent protein (GFP) to allow visual tracking of the successfully transduced cells with a Zeiss Axiovert 40CFL microscope.
Table 1.
Sequences of the designed miRNAs
| miRNA | Sequence (3′- 5′) |
|---|---|
| miRNA-L1 | TGCTGATTTCGTTTAGTTCGAGTTGGGTTTTGGCCACTGACTGACCCAACTCGCTAAACGAAAT |
| miRNA-L2 | TGCTGTTTAGTTCGAGTTGGTGTCCGGTTTTGGCCACTGACTGACCGGACACCCTCGAACTAAA |
| miRNA-N | TGCTGTATGCAATAGGGTTGCTTGTAGTTTTGGCCACTGACTGACTACAAGCACCTATTGCATA |
Next, these sequences were transferred into destination plasmids, one plasmid per miRNA sequence, following the manufacturer’s protocol. The destination plasmid used in this study contained an antibiotic (blasticidin) resistance gene that allowed for the selection of stably transfected cell lines for long-term RNAi studies.
Generation of non-replicating transducing lentivirus stock
The 293FT cell line (Thermo Fisher Scientific) was used to generate the lentivirus stocks. This cell line was derived from human embryonal kidney cells that have been transformed to allow production of high levels of lentivirus RNA and the gag/pol and rev proteins required for virus packaging when the cells are co-transfected with expression plasmids that contain these virus genes. Lentiviruses were produced according to the manufacturer’s instructions, and each virus expressed a different one of the miRNAs. A total of 6 × 106 293FT cells were used to produce each lentivirus. Forty-eight to 72 h following introduction of the plasmids into the cells, virus-containing supernatants were harvested, aliquoted and stored at −80°C.
Selection of stably transfected Crandell Rees feline kidney cells using blasticidin
Crandell Rees feline kidney (CrFK) cells (American Type Culture Collection) were propagated in DMEM and Ham’s F-12 (DMEM-F12; Lonza) supplemented with 10% heat-inactivated FBS and maintained at 37°C and 5% CO2 in an incubator. The cells were plated in 25 cm2 tissue culture flask. When the cells reached >80% confluence the culture media were removed and 3 ml lentivirus stock was added. On the following day (day 2), the medium containing virus was removed and replaced with fresh complete medium (DMEM-F12 supplemented with 10% heat-inactivated FBS). On the following day (day 3), the medium was removed and replaced with fresh, complete medium containing the appropriate amount of blasticidin (10 μg/ml; Thermo Fisher Scientific) to select for stably transduced cells. This was undertaken to ensure that all cells used for the virus challenge experiment were producing the miRNAs. Transduced cells were determined via observing intracellular GFP by the EVOS FL cell imaging system (Thermo Fisher Scientific). Then, the stably transduced cells were harvested and re-plated in 25 cm2 tissue culture flasks. When cells reached approximately 80% confluence, another 3 ml lentivirus stock was added to increase the number of integrated copies of the lentivirus. After 24 h, the media containing virus was removed and replaced with fresh media. Next, a subset of cells that were transfected once, as well as cells that were transfected twice, were collected and assessed via quantitative real-time PCR, using lentivirus-specific primers and probe to determine the relative increase in the copy number of the integrated lentivirus per cell following the second transfection. Further, the relative increase of the integrated lentivirus per cell was re-evaluated via intracellular GFP observation by the EVOS FL Cell Imaging System (Thermo Fisher Scientific) and/or a Zeiss Axiovert 40CFL microscope.
Selection of stably transfected HSCs using blasticidin
Selection of stably transfected HSCs was prepared as described previously for CrFK cells with some modification (2 μg/ml blasticidin was added instead of 10 μg/ml).
Viral challenge assay in CrFK cell line
Virus challenge studies were initially carried out in stably transduced CrFK cells to assess the ability of the designed miRNAs to inhibit viral replication. This allowed for selection of the best constructed vector prior to transduction of the feline HSCs. Stably transduced CrFK cells plated in 12-well plates at approximately 80% confluence were either infected with FIPV WSU 79-1146 or FECV WSU 79-1683 (American Type Culture Collection) at an multiplicity of infection (MOI) of 0.1. One hour after incubation, the cells were washed with DMEM-F12, and fresh complete medium was added to each well. After 48 h, samples of cell culture medium as well as cultured cells were collected for viral titration and relative quantification of intracellular FCoV genomic RNA. Control samples used in each experiment included untreated FCoV infected CRFK cells and FCoV-infected cells treated with a negative control non-targeting miRNA to test for potential non-specific effects. Each miRNA was tested in duplicate, and each experiment was performed twice.
Viral challenge assay in feline HSCs
Stably transduced feline HSCs, which express miRNA L2, were plated in 35 × 10 mm cell culture dishes (Primaria; Corning) at approximately 80% confluence. These cells were then either infected with FIPV WSU 79-1146 or FECV WSU 79-1683 at an MOI of 0.1, as mentioned previously for CrFK cells.
Quantitative real-time reverse transcriptase PCR
For cellular viral RNA quantification, cells were harvested 48 h following virus infection and nucleic acid was extracted using the RNeasy Plus Mini kit (Qiagen), according to manufacturer recommendations. Reverse transcription and quantitative PCR were undertaken as previously described, using primers and a carboxyfluorescein-labeled probe targeting the viral 7b coding region, as this region is highly conserved in FCoV. 9 Reactions were carried out using Superscript III Platinum One Step qRT-PCR Kit (Thermo Fisher Scientific) in a SmartCycler II (Cepheid). The total amount of RNA in each reaction was standardized according to mRNA expression of the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase, which was used for relative quantitation of the viral RNA by the 2−ΔΔCt method. 10
TCID50 assay
Forty-eight hours postinfection with FCoV, 500 µl tissue culture medium was collected from each well and stored at −80°C prior to viral titration using the TCID50 assay. In brief, cultured media were serially diluted 10-fold with DMEM. Diluted virus suspensions were added to monolayer CRFK cells cultured in 96-well plates (six wells per dilution). Wells were monitored for cytopathic effect 72 h postinfection using an inverted phase-contrast microscope. The TCID50 endpoint values were calculated according to the method of Reed and Muench. 11
Statistical analysis
All statistical analyses were conducted using the t-test procedure. A P value of 0.05 was used for statistical significance in all tests.
Results
Lentiviruses were used successfully to integrate antifeline coronavirus miRNA-coding DNA into the genome of CrFK cells, as determined via intracellular GFP observation by fluorescence microscope, as shown in Figure 1. Further, re-transfection of stably transduced cells resulted in a four-fold increase in the amount of integrated virus per cell as assessed by quantitative real-time PCR. These stably transduced CrFK cells were infected with FCoV and the ability of each constitutively expressed miRNA to inhibit coronavirus replication was determined by comparing the amount of viral RNA produced in the treated cells vs the amount produced in cells expressing scrambled miRNA (non-targeting) by real-time reverse transcriptase PCR. Three miRNAs – miRNA-L1 and miRNA-L2, which targeted the viral leader sequence, and miRNA-N, which targeted the nucleocapsid gene – exhibited variable inhibitory effects on viral replication in vitro. miRNA-L1 and miRNA-N resulted in <50% reduction in FIPV WSU 79-1146 and FECV WSU 79-1683 genomic RNA synthesis compared with the negative control sample, while miRNA-L2 resulted in 63 ± 2.8% and 73 ± 3.5% reductions in FIPV WSU 79-1146 and FECV WSU 79-1683 genomic RNA, respectively, as compared with the negative control sample. Based on these results, further experiments were performed for miRNA-L2 only. In order to study whether silencing of viral genomic RNA by miRNA-L2 had any effect on the yield of progeny virus, TCID50 assays were performed. miRNA-L2 resulted in 60 ± 3.5% and 55 ± 4.2% reduction in the extracellular virus titer of FIPV WSU 79-1146 and FECV WSU 79-1683, respectively, as compared with the negative-control miRNA sample.
Figure 1.
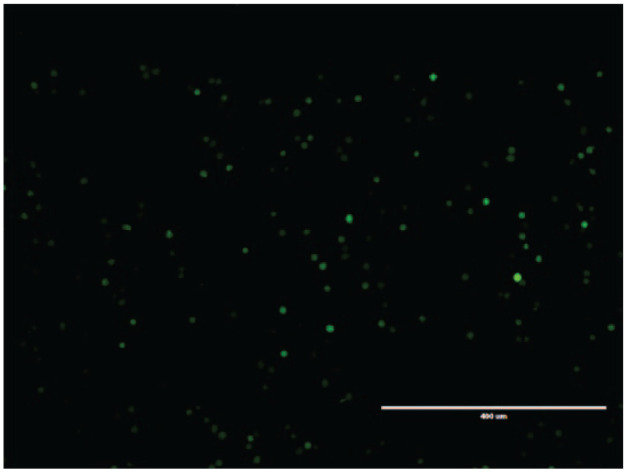
Successfully transduced Crandell Rees feline kidney cells as observed by the EVOS FL Cell Imaging System
CD34-positive HSCs were obtained. This stem cell population represented approximately 2–5% of the total cell population from the bone marrow, based on flow cytometry results. The stem cells were expanded in vitro, as shown in Figure 2. Following cell expansion, cells were confirmed to be stem cells by IFA testing with the CD34 antibody. Expanded HSCs were stably transduced with the constructed lentivirus that expressed miRNA-L2. Microscopic evaluation of cell viability (by means of an inverted phase-contrast microscope) revealed that the number of viable cells among stably transduced HSCs was comparable with the number of viable cells among untreated HSCs using trypan blue stain. Inhibition of coronavirus replication by constitutive expression of the miRNA-L2 in infected HSCs was 90 ± 2.8% and 74 ± 4.2% in FIPV WSU 79-1146 and FECV WSU 79-1683 genomic RNA, respectively, as compared with the negative control sample. Further, miRNA-L2 resulted in 92 ± 2.8% and 80 ± 1.4% reduction in the extracellular virus titer of FIPV WSU 79-1146 and FECV WSU 79-1683, respectively, as compared with the negative control HSCs that expressed non-targeting miRNA (Figure 3).
Figure 2.
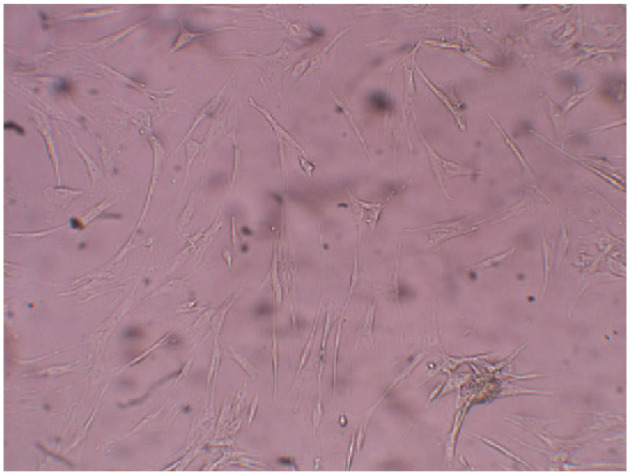
Expanded feline hematopoietic stem cells, as observed with a Zeiss Axiovert 40CFL microscope
Figure 3.
The effect of miRNA-L2 on extracellular virus production in hematopoietic stem cells
Discussion
In the current study, we assessed the feasibility of transducing HSCs with FCoV-specific, miRNA-coding DNA by ex vivo introduction with a non-replicating lentivirus vector as a means of delivery of this method of viral inhibition in a manner that could potentially be used in vivo.
Recent advances in the development of stem cell-based therapeutic approaches, as well as the development of technologies that allow the genetic modification of these cells, make these cells a potential delivery system for small RNA molecules. HSCs are capable of self-renewal and differentiation into all hematopoietic lineages, including monocytes and macrophages. 12 The stable insertion of genes in hematopoietic progenitor cells could have a significant impact. As progenitor cells, their replication would produce daughter cells with their genomic heritage, potentially resulting in a considerable number of genetically engineered cells.
In this study, lentiviruses were used successfully to integrate stably miRNA of interest into HSC genomes as determined via intracellular GFP observation by fluorescence microscopy. Lentiviral vectors are effective tools for gene transfer and integrate variable numbers of proviral DNA copies in variable proportions of cells. While there are concerns with the use of retroviruses and cancer development, lentiviruses, unlike retroviruses, tend to integrate distally from promoters in introns, potentially limiting their overall oncogenicity. 13 Retroviral delivery systems have been used safely in dogs and cats, and the absence of malignant tumors in any of >20 dogs or >15 cats that received neonatal intravenous injection of a retrovirus vector for gene therapy and followed for 1–11 years, suggests that the risk of malignant transformation with this gene therapy approach is low. 14 This approach has also been used in HSCs for humans in clinical trials for treatment of HIV. 13 An added biosafety feature of the lentivirus used in this study is its inability to replicate. The portions of the genome that code for structural and packaging proteins are not included in the virus that will be delivered to the stem cells, and the long terminal repeat portion is self-inactivated following insertion into the genome.
Recently, HSCs have been reported to be permissive to the integration of multiple copies of proviral DNA into the genome. 15 As many as an average of six integrations per cell have been revealed in populations of human hematopoietic cells engrafted in the bone marrow of immunoincompetent mice. In the present study, we found that feline HSCs are permissive for integration of proviral DNA. The effectiveness of the integrated miRNA to inhibit the targeted gene is expected to correlate positively with the frequency of transduced cells and also with the number of integrations per cell. Therefore, in this study, we used the antibiotic resistance property of the constructed lentivirus to select stably transduced cells. Further, we re-transduced the stably transduced cells with lentivirus to increase the number of integrated virus per cell, which was observed via fluorescence microscopy, based on the intensity of observed intracellular GFP expression in transduced cells and as assessed by real-time PCR to determine lentiviral copy number at an integrated proviral DNA level.
The significant reduction in FCoV replication in HSCs indicates an effective integration of miRNA-L2 into feline HSC genomes. The ability of the lentivirus to integrate and/or the number of integrated virus per cell differed according to the cell type. Because of the high mutation rate of FCoV, the miRNA-L2 used in this study was designed to target a highly conserved region of the viral leader sequence, which plays a pivotal role in virus gene expression and replication. The virus strains tested in this study were both serotype II isolates, but the designed miRNA should be able recognize and target various strains of FCoVs circulating in the field, including both serotypes I and/or II, based on comparisons of this target region among FCoV sequences of both serotypes in GenBank.
Interestingly, we also discovered in this study that feline HSCs are easily infected with FCoV in vitro, unlike differentiated monocytes and macrophages, as reported in the literature. 16 This suggests that FCoV infection of bone marrow may play a role in the development of FIP, but nothing to this effect has been reported in the literature.
Conclusions
The preliminary results in this study suggest that genetic modification of HSCs for constitutive production of anticoronavirus miRNAs will reduce FCoV replication. This proof-of-concept work will potentially lead to in vivo introduction of these genetically modified HSCs for the prevention or treatment of FIP. Successful reinfusion of these modified FCoV-protected cells may have the potential to minimize viral loads. Reducing the viral load in an FIP-diseased cat may, in part, restore the host antiviral immune response and thereafter improve virus clearance. In addition to treating FIP, these modified HSCs may protect cats with a high risk of developing FIP. The progeny-modified monocytes/macrophages will inhibit FCoV replication and thereby reduce the likelihood of mutation and reduce the expansion of any virus population containing the mutation of importance in the development of FIP.
Acknowledgments
We thank Ms Gina Galyon for assistance with bone marrow harvesting, and Ms Nancy Neilsen for assistance with flow cytometry.
Footnotes
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: This work was funded by San Francisco Foundation Miller Trust/Winn Feline Foundation (grant number MT 13-008).
Accepted: 24 May 2016
References
- 1. De Groot-Mijnes JDF, Van Dun JM, van der Most RG, et al. Natural history of a recurrent feline coronavirus infection and the role of cellular immunity in survival and disease. J Virol 2005; 79: 1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Takano T, et al. A ‘possible’ involvement of TNF-alpha in apoptosis induction in peripheral blood lymphocytes of cats with feline infectious peritonitis. Vet Microbiol 2007; 119: 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hartmann K. Feline infectious peritonitis. Vet Clin North Am Small Anim Pract 2005; 35: 39–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anis EA, Wilkes RP, Kania SA, et al. Effect of small interfering RNAs on in vitro replication and gene expression of feline coronavirus. Am J Vet Res 2014; 75: 828–834. [DOI] [PubMed] [Google Scholar]
- 5. Amado RG, Mitsuyasu RT, Rosenblatt JD, et al. Anti-human immunodeficiency virus hematopoietic progenitor cell-delivered ribozyme in a phase I study: myeloid and lymphoid reconstitution in human immunodeficiency virus type-1-infected patients. Hum Gene Ther 2004; 15: 251–262. [DOI] [PubMed] [Google Scholar]
- 6. Mitsuyasu RT, Merigan TC, Carr A, et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat Med 2009; 15: 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Munoz JL, Greco SJ, Patel SA, et al. Feline bone marrow-derived mesenchymal stromal cells (MSCs) show similar phenotype and functions with regards to neuronal differentiation as human MSCs. Differentiation 2012; 84: 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martin DR, Cox NR, Hathcock TL, et al. Isolation and characterization of multipotential mesenchymal stem cells from feline bone marrow. Exp Hematol 2002; 30: 879–886. [DOI] [PubMed] [Google Scholar]
- 9. Gut M, Leutenegger CM, Huder JB, et al. One-tube fluorogenic reverse transcription-polymerase chain reaction for the quantitation of feline coronaviruses. J Virol Methods 1999; 77: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 2008; 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- 11. Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg 1938; 27: 493–497. [Google Scholar]
- 12. Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med 2010; 2: 640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gavrilov K, Saltzman WM. Therapeutic siRNA: principles, challenges, and strategies. Yale J Biol Med 2012; 85: 187–200. [PMC free article] [PubMed] [Google Scholar]
- 14. Morris KV, Rossi JJ. Lentiviral-mediated delivery of siRNAs for antiviral therapy. Gene Ther 2006; 13: 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Charrier S, Ferrand M, Zerbato M, et al. Quantification of lentiviral vector copy numbers in individual hematopoietic colony-forming cells shows vector dose-dependent effects on the frequency and level of transduction. Gene Ther 2011; 18: 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dewerchin HL, Cornelissen E, Nauwynck HJ. Replication of feline coronaviruses in peripheral blood monocytes. Arch Virol 2005; 150: 2483–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]