

Abstract
Multiple sclerosis (MS) is a chronic disease with an underlying pathology characterized by inflammation-driven neuronal loss, axonal injury, and demyelination. Bruton’s tyrosine kinase (BTK), a nonreceptor tyrosine kinase and member of the TEC family of kinases, is involved in the regulation, migration, and functional activation of B cells and myeloid cells in the periphery and the central nervous system (CNS), cell types which are deemed central to the pathology contributing to disease progression in MS patients. Herein, we describe the discovery of BIIB129 (25), a structurally distinct and brain-penetrant targeted covalent inhibitor (TCI) of BTK with an unprecedented binding mode responsible for its high kinome selectivity. BIIB129 (25) demonstrated efficacy in disease-relevant preclinical in vivo models of B cell proliferation in the CNS, exhibits a favorable safety profile suitable for clinical development as an immunomodulating therapy for MS, and has a low projected total human daily dose.
Introduction
Multiple sclerosis (MS) is a chronic disabling neuro-inflammatory disease shown to have a higher prevalence in women1 of northern European ancestry.2 This condition, which is typically diagnosed between the ages of 20 and 40, affects 2.5 million people worldwide. The relatively high incidence of this condition in the young population of developed countries has prompted the development of a number of disease-modifying therapies.3 Among these, several B cell depleting anti-CD20 monoclonal antibodies have been developed, including ocrelizumab,4,5 the first treatment to be approved for activated inflammatory relapsing multiple sclerosis (RMS) and primary progressive multiple sclerosis (PPMS).6
Despite significant advances in the treatment of this disease, MS patients still face a significant unmet need for medicines that offer higher efficacy and improved safety profiles.7,8 Therefore, there is continued interest in identifying new mechanisms and pathways that facilitate selective modulation of B cell activity without rendering patients immune-compromised and prone to opportunistic infections. Bruton’s tyrosine kinase (BTK), a nonreceptor protein-tyrosine kinase of the TEC kinase family, is a drug target of high interest since mutations on the BTK gene validated its functional importance in B cell biology. These mutations were demonstrated to cause a rare but severe genetic disease known as X-linked agammaglobulinemia (XLA).9 The inability of these young male patients to develop protective autoantibodies is attributed to the critical role that BTK plays in B cell receptor signaling during B cell development in bone marrow. This deleterious effect of BTK genetic deficiency is not expected to be recapitulated by pharmacological inhibition of BTK since treated individuals will have a pre-existing B lymphocyte pool and antibody repertoire. In contrast, pharmacological inhibition of BTK is anticipated to block proliferation, differentiation, migration, and effector functions of normal and malignant B cells.10,11 In innate myeloid cells such as monocytes, macrophages, and CNS-resident microglia, BTK is involved in regulating the activation of Fc receptor (FcR) signaling, leading to the generation of reactive oxygen species (ROS), secretion of cytokines, and degranulation resulting from binding of FcRs to immune complexes.12,13 Due to the plethora of immune responses linked to BTK function, it has remained a drug target of high interest to treat numerous autoimmune indications where B cells and innate immune cells are primary drivers of disease progression.14
BTK is a unique kinase due to its role in selectively modulating B cell activation via B cell receptor (BCR) signaling and its propensity to be targeted utilizing covalent and noncovalent orthosteric small molecule inhibitors. The use of irreversible inhibitors to attenuate the activity of BTK is facilitated by the presence of a cysteine residue within the ATP binding pocket, which is considered a rare primary structure, with only eight other kinases reported to have this novel feature. The FDA’s 2013 approval of ibrutinib (1, Figure 1), a covalent BTK inhibitor for the treatment of mantle cell lymphoma (MCL), stimulated a revival in the pursuit of targeted covalent inhibitors (TCIs)15−18 as a therapeutic strategy to develop clinical candidates for the treatment of chronic nonlife-threatening diseases. To date, there have been six covalent BTK drugs approved globally for the treatment of oncology indications, and currently, more than 20 BTK inhibitors19 are in clinical trials evaluating both covalent and reversible modes of action (MOA) as potential new autoimmune therapies.20 Within the context of MS,21 several highly anticipated late-stage clinical readouts are pending. As part of those, EMD Serono recently announced that their covalent BTK-inhibitor evobrutinib22 (4) failed to meet primary end points in a Phase 3 clinical trial for relapsing-remitting multiple sclerosis (RRMS). This result shifts the spotlight onto tolebrutinib23 (2), Sanofi/Principia’s covalent BTK inhibitor, scheduled to complete Phase 3 for RRMS and primary progressive multiple sclerosis (PPMS) in 2024.
Figure 1.

Selected BTK inhibitors of different binding modes are reported in the literature.
Additionally, Novartis’ remibrutinib23 (5), a highly selective covalent BTK inhibitor, will complete a Phase 3 clinical trial in RRMS in 2026. As an alternative MOA to covalent inhibition, Roche continues to develop fenebrutinib (3), a selective, reversible BTK inhibitor, which met the primary and secondary end points in a Phase 2 RRMS trial and is anticipated to complete the Phase 3 trial in PPMS in 2026. While Biogen’s peripherally restricted reversible BTK-inhibitor BIIB091 (6) is currently in Phase 2 clinical trials as both mono and combination therapy with dimethyl fumarate (DMF) is scheduled to readout at the end of 2026.24 It is hypothesized that targeting both B cell24 and other antigen-presenting cells (APC)25 via combo therapy can achieve superior efficacy than monotherapy, thus potentially minimizing the daily dose body burden, which has been attributed to observed safety findings observed with covalent inhibitors26 evobrutinib 1, tolebrutinib 2, and recently reported for fenebrutinib 3. The nature of the safety findings (drug-induced liver injury, DILI)27 appears to be idiosyncratic; however, it can occur via the formation of reactive metabolites; thus, our mitigation strategy has been to optimize both the potency and drug-like-properties to minimize the clinical dose to achieve high efficacy and potency for our clinical candidates.
Although the competitive landscape for BTK inhibitors appears crowded, significant differences exist in the MOA being evaluated in the clinic. For example, tolebrutinib (2) and evobrutinib28 (4) are second-generation BTK covalent inhibitors designed to demonstrate superior kinome selectivity to address adverse events (AEs), such as rash, diarrhea, cardiovascular effects, and an increased risk for bleeding observed with ibrutinib.29 While fenebrutinib30 (3) is a selective, reversible BTK inhibitor designed to extend into the “H3” selectivity pocket to block the phosphorylation of Tyr551,31 an essential residue required for BTK activation. Furthermore, all BTK inhibitors, regardless of their MOA, currently being developed to treat neuroinflammation, demonstrate CNS MPO values32,33 in a similar range (<3.6 out of a maximum of 6) considered suboptimal for CNS drugs (Table 1). In addition to the high molecular weight (MW) and the number of hydrogen bond donors, “H3” pocket binders (3, 5, and 6) score low on the topological polar surface area (tPSA) with values of 110 and above. Similarly, Type I binders (1, 2, 4, among others), while having lower tPSA, feature unfavorable central nervous system multiparameter optimization (CNS MPO) scores due to the contribution of the characteristic diphenyl ether moiety negatively impacting both molecular weight (170 g/mol or ∼38% of MW) and lipophilicity. With a desire to identify a differentiated covalent BTK inhibitor to treat MS, our medicinal chemistry campaign was directed toward identifying a molecule with low lipophilicity, favorable kinome selectivity, and improved blood–brain barrier (BBB) permeability to achieve efficacious concentrations in the CNS at low human total daily doses.34
Table 1. Comparison of the CNS MPO Scores and Physicochemical Properties of Selected BTK Inhibitors.
| compound name | CNS MPO scorea | MW [g/mol] | tPSA [Å] | HBD | clog Pb,c | clog D (pH 7.4)b,c |
|---|---|---|---|---|---|---|
| Ibrutinib (1) | 3.52 | 440.2 | 99 | 2 | 4.1 | 3.6 |
| Tolebrutinib (2) | 3.44 | 455.2 | 95 | 2 | 4.7 | 3.7 |
| Fenebrutinib (3) | 3.35 | 664.4 | 121 | 2 | 2.4 | 2.3 |
| Evobrutinib (4) | 3.11 | 429.2 | 93 | 3 | 4.8 | 3.9 |
| Remibrutinib (5) | 2.19 | 507.2 | 110 | 3 | 3.2 | 3.8 |
| BIIB091 (6) | 3.30 | 542.3 | 128 | 2 | 1.9 | 2.4 |
| Optimal for CNS MPO score | 6 (max.) | <350 | 40 < x < 90 | 0 | <3.0 | <2.0 |
Basic pKa contribution is 1 across all entries.
MoKa was used to calculate clog P, clog D, and pKa.
Results and Discussion
From our experience developing peripheral BTK inhibitors, we understood the importance of kinome selectivity and its impact on the tolerability and safety profiles observed in preclinical species and humans. Although the exquisite selectivity demonstrated with “H3” binders such as fenebrutinib30 (4), remibrutinib35 (5), and BIIB09136 (6) is ideal from a clinical safety perspective, the physicochemical properties37 were deemed unfavorable for targeting BTK in the CNS (Table 1).38 To address this issue, we had chosen to employ TCI as a strategy to differentiate against known covalent BTKi’s in the clinic (1, 2, 4; Table 1) based on a combination of improved kinome selectivity, physicochemical properties, and brain penetration. Utilizing both structure-based design (SBD) and pharmacophore ligand-based drug design, we successfully identified a novel series of covalent BTK inhibitors to target both B cells and myeloid cells, including CNS-resident microglia located in the meninges, perivascular spaces, and CNS parenchyma of the brain and spinal cord of MS patients.
Hit to Lead Identification Efforts
To identify a brain-penetrant TCI, we first focused on identifying scaffolds with improved CNS-drug-like properties. Thus, we sought to restrict the use of structural motifs that could negatively impact the molecules’ physicochemical properties,37 leading to an increase in the tPSA and/or lipophilicity (clog P, clog, see above, Table 1). We hypothesized that this could be achieved by focusing our design efforts on analogues that neither bind to the gatekeeper nor the “H3” pocket while maintaining crucial interactions with the hinge region to achieve acceptable binding. Second, we pursued hinge and linker motifs that would limit the number of hydrogen bond donors to increase the likelihood of identifying a suitable brain-penetrant inhibitor. With these design parameters in mind, we hypothesized that selectivity could be obtained by targeting unique interactions with side chain residues within the hinge region and ribose pocket to leverage sequence differences between BTK and other cysteine-containing kinases.
Our initial medicinal chemistry approach was centered around designing and evaluating compounds with a minimum pharmacophore consisting of three components: (a) a hinge-binder motif, (b) an sp3-rich linker, and (c) a Michael acceptor as a covalent warhead to engage the reactive cysteine (Cys481) in the active site of BTK. This hit-finding campaign led to the discovery of compound 7 featuring a pyrazolo[1,5-a]pyrazine hinge binding motif and an O-linked 4-hydroxy-piperidyl linked to an acrylamide covalent warhead, which was later confirmed by X-ray cocrystallography. Hit 7 exhibited desirable CNS-drug-like properties with low molecular weight (352 amu), tPSA (78 Å2), and zero H-bond donors, which resulted in a maximal CNS MPO score of 6.0.32,33 Despite the favorable properties, this initial hit exhibited modest inhibition of BTK via a biochemical assay (BTK IC50 = 63 nM) but was able to confirm that the improved properties, provided MDR1 efflux ratios expected to lead to increased CNS exposure. Further optimization of the sp3-rich linker, while maintaining favorable MPO scores, led to chiral pyrrolidine 8 and piperidine 9 with increased biochemical potency (BTK IC50 = 4.6 and 3.4 nM). Since covalent binding is a two-step mechanism, first requiring reversible binding defined by the Ki (disassociation constant measured from the koff/kon), and subsequent irreversible covalent bond formation defined by the kinact, it was necessary to develop a novel continuous-read kinetic enzyme assay to drive the medicinal chemistry effort. Gratifyingly, both inhibitors also exhibited inactivation of BTK as measured in this kinact/Ki assay, while compound 7 did not demonstrate any covalent inhibition of BTK in this assay (8 and 9 log kinact/Ki = 3.33 and 3.50, 7 log kinact/Ki = < 2.4). Optimization of the linker led to the identification of chiral azepane 10, which exhibited biochemical inhibitory potency in the picomolar range (BTK IC50 = 0.5 nM) and efficient covalent binding (BTK log kinact/Ki = 4.43) while maintaining favorable CNS-drug-like properties and low MDR1 efflux (Table 2). Kinome profiling39 of compound 10 indicated favorable selectivity by only inhibiting seven of 403 kinases tested (>90% inhibition at 1 μM concentration of 10). (Figure 2a) Furthermore, compound 10 also exhibited improved selectivity for BTK relative to the nine active site cysteine-containing kinases as compared to 2 and 4 (see Table 5).
Table 2. Hit and Lead Identification in Improved CNS-Property Space.

| compound | CNS MPO score | MW [g/mol] | tPSA [Å2] | cLog P | log kinact/Ki | BTK IC50 [nM] | MDCK-MDR1 Papp A2B [ER] | RLM/HLM [mL/min/kg] |
|---|---|---|---|---|---|---|---|---|
| 7 | 6.0 | 352.4 | 77.6 | 1.19 | <2.4 | 63 | 73 [6] | 71.7/<4.74 |
| 8 | 6.0 | 338.4 | 77.6 | 1.37 | 3.33 | 4.6 | 39 [6] | 53.1/8.18 |
| 9 | 6.0 | 352.4 | 77.6 | 1.93 | 3.50 | 3.4 | 96 [5] | 908/17 |
| 10 | 5.86 | 366.4 | 77.6 | 1.75 | 4.43 | 0.5 | 59 [5] | 820/74.2 |
Figure 2.

(a) Kinome selectivity (KINOMEscan, Eurofins DiscoverX) for compound 10 at 1 μM concentration; 7 out of 403 kinases were inhibited with S(10) selectivity scores40 of 0.017. (b) X-ray cocrystal structure of 10 (magenta; PDB 8TU3) bound to the kinase domain of human BTK. Hydrogen bond interactions are shown as dotted lines.
Table 5. Selectivity of Selected BTK Inhibitors over Kinases with a Reactive Cysteine at the 481 Position.
| compound |
|||||||
|---|---|---|---|---|---|---|---|
| amino acid @484 position | tolebrutinib (2) Kd [nM] | evobrutinib (4) Kd [nM] | 10Kd [nM] | 25Kd [nM] | 27Kd [nM] | ||
| BTK Kd [nM] | Asn | 1.4 | 4.6 | 0.6 | 0.63 | 0.31 | |
| Kinase with Cys481 | Fold Selectivity for BTK (off-target kinase Kd/BTK Kd) | ||||||
| BMX | Asn | 3 | 4 | 43 | 43 | 14 | |
| TEC | Asn | 1 | <1 | 9 | 9 | 7 | |
| TXK | Asn | 5 | 37 | 27 | 27 | 6 | |
| JAK3 | Asp | 847 | 1217 | 157 | 157 | 390 | |
| BLK | Asp | 3 | 133 | 460 | 460 | 839 | |
| ITK | Asp | 671 | 587 | 1200 | 1206 | 274 | |
| EGFR | Asp | 9 | 2000 | >10 000 | >10 000 | 9355 | |
| ERBB2 | Asp | 2 | 1152 | >10 000 | >10 000 | 7419 | |
| ERBB4 | Glu | 4 | 137 | 700 | 714 | 7741 | |
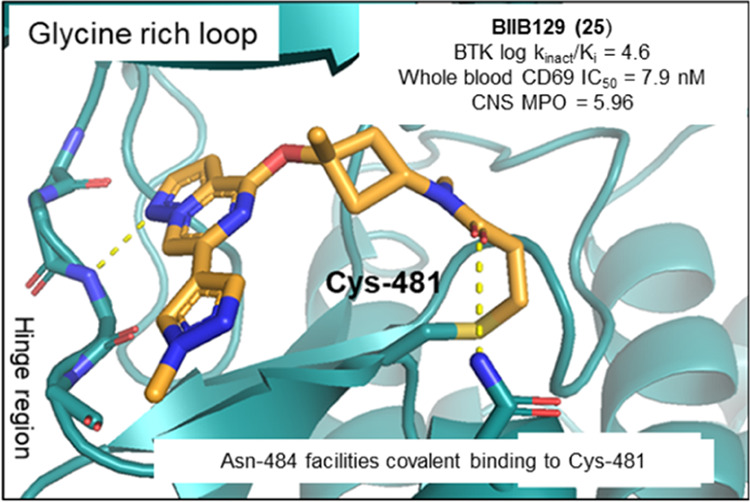
An analysis of the BTK cocrystal structure with 10 (Figure 2b) revealed that the pyrazolo[1,5-a]pyrazine hinge binding motif formed a hydrogen bond with the backbone nitrogen of Met477. This interaction oriented the azepane linker of compound 10 toward the glycine-rich loop and positioned the carbonyl oxygen of the carboxamide favorably to engage the side chain amide of Asn484 in a hydrogen bond. We speculated that this unique electrostatic interaction may be a prerequisite for covalent modification and may infer additional kinome selectivity as Asn484 was a nonconserved residue in off-target kinases, such as BLK, EGFR, ERBB2, ERBB4, ITK, and JAK3, and compounds enabling this novel interaction were hypothesized to preferentially covalently bind in the BTK active site.41
Further profiling confirmed that the biochemical and covalent potency of compound 10 was found to translate to a sustained functional effect on B cells as measured by CD69 inhibition in an in vitro human whole blood (WB) assay using anti-IgD stimulation13 (WB CD69 IC50 = 0.33 μM). Additionally, this functional potency correlated well with activity measured in an in vitro BTK target occupancy (TO) assay in Ramos cells (IC50 = 2.3 nM).42 Compound 10 demonstrated moderate to low P-gp efflux as measured in Madin-Darby canine kidney cells transfected with human MDR1 gene (MDCK-MDR1 ER = 5.4)43 and a breast cancer resistance protein44 (BCRP ER = 1.9) transporter assay, which translated into acceptable brain exposure34 as measured in a constant infusion rat study resulting in a Kp,uu = 0.37. However, the pharmacokinetic (PK) profile of 10 in both rat and dog showed rapid clearance (CL [mL/min/kg] rat/dog = 133/79), leading to low bioavailability (%F rat/dog = 3.0/4.5). To further elucidate the cause for the poor oral bioavailability, a monkey portal vein PK study confirmed that 10 exhibited high first-pass hepatic clearance (%F < 0.1), while the portal vein plasma AUC indicated Fg*Fa [%] = 38. A follow-up rat bile duct-cannulated (BDC) study indicated low renal CL (9.1 mL/min/kg) and negligible biliary CL (0.52 mL/min/kg). Moreover, when rats were predosed with the known CYP inhibitor 1-aminobenzotriazole,45 there was a significant increase in the plasma AUC (8.5 fold), leading to an improvement in the oral exposure (see the Supporting Information (SI)). In vitro metabolite identification (Met-ID) studies of 10 in rat and human liver microsomes (LM) and rat, dog, monkey, and human hepatocytes revealed CYP-mediated oxidation of the azepane linker and oxidation and nucleophile addition of glutathione (GSH = l-γ-glutamyl-l-cysteinyl-glycine)46 to the acrylamide warhead (see the SI) as major metabolic soft spots. This finding was verified with an in vivo Met-ID of 10 in BDC rat bile and urine samples (see the SI). Despite these shortcomings, the identification of compound 10 and the discovery of its novel binding mode and favorable kinome selectivity marked the entry of the structure-based lead-optimization (LO) stage of the program.
Lead-Optimization Efforts
A survey of the covalent BTK inhibitors in the clinic or on the market revealed that besides branebrutinib47 and acalabrutinib,48 an acrylamide is commonly utilized as a covalent warhead.49 This electrophile appears to provide the necessary balance between reactivity and stability to enable dosing in humans. Thus, we focused our lead-optimization efforts on modifying the amino-ether linker to improve the CYP-mediated metabolic stability while maintaining desirable potency, kinome selectivity, and CNS-like properties.
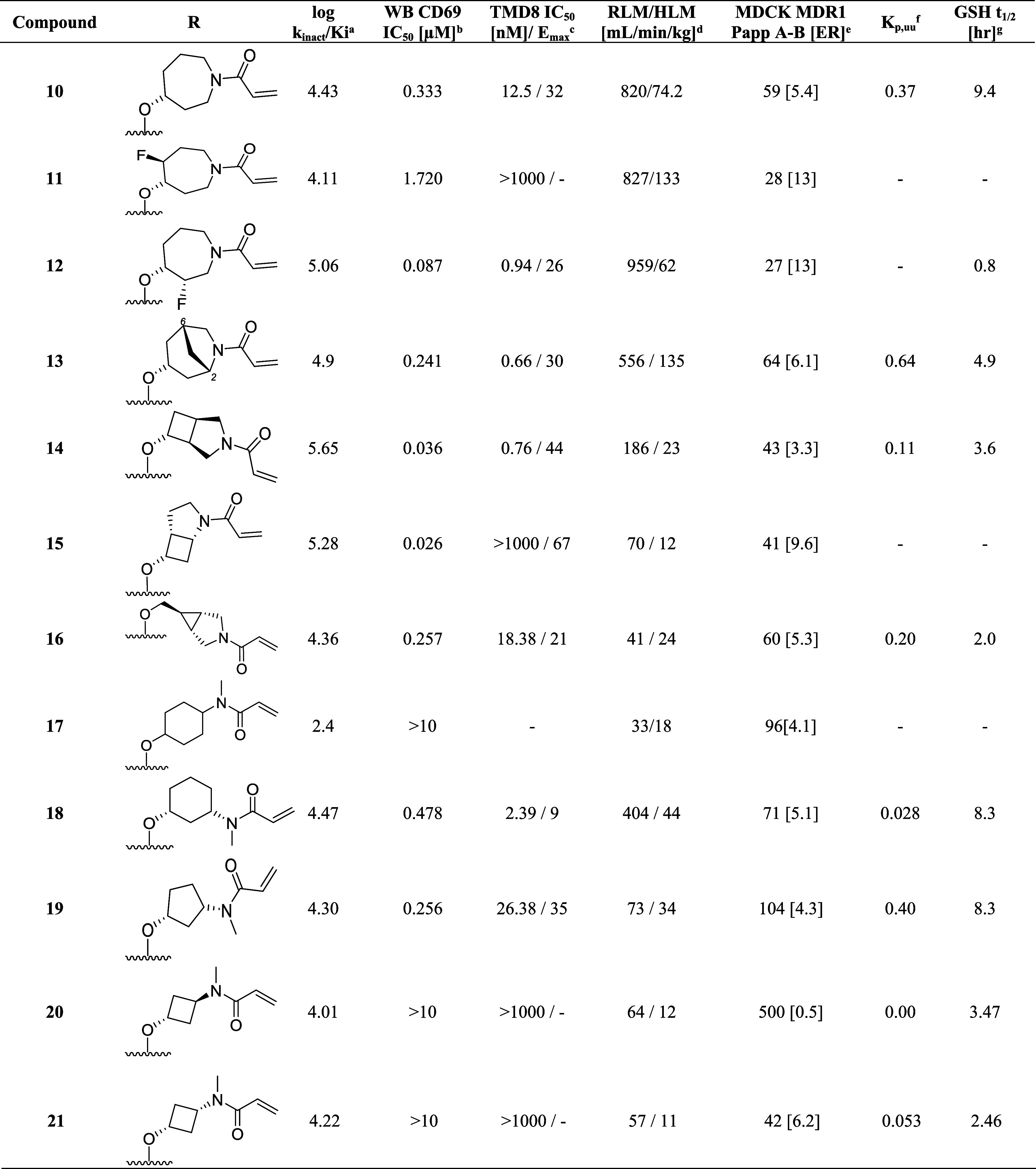
To implement this strategy, we first introduced methyl substituents on the azepane ring to impose a conformational constraint, which was anticipated to improve the potency by locking the warhead into a favorable low-energy orientation and simultaneously blocking the metabolic oxidation of the azepane core. Unfortunately, these analogues with improved microsomal stability typically had inferior potency (data not shown). Additional attempts to reduce clearance by introducing a fluorine atom adjacent to ether linkage afforded 11 (Table 3) did not improve the liver microsome stability and showed a disconnect between inhibitory activity and whole blood potency (log kinact/Ki = 4.11; WB CD69 IC50 = 1.72 μM;). Moving the fluorine substituent to the C3 position on the azepane resulted in 12, which exhibited a significant improvement in both biochemical and cellular potency while affording a modest improvement in human liver microsome stability (log kinact/Ki = 5.06; WB CD69 IC50 = 0.086 μM; HLM = 62 mL/min/kg). The increase in potency of 12 was attributed to the inductive properties of the fluorine group, which was hypothesized to increase the electrophilicity of the acrylamide warhead. However, the enhanced reactivity also contributed to greater susceptibility toward glutathione conjugation (GSH adduct formation), leading to a short half-life as measured in the in vitro GSH-stability assay (GSH t1/2 < 1 h for 12).50
Table 3. Bridged Bicyclic Amine and Exocyclic Amine Replacements for the Azepane Linker.
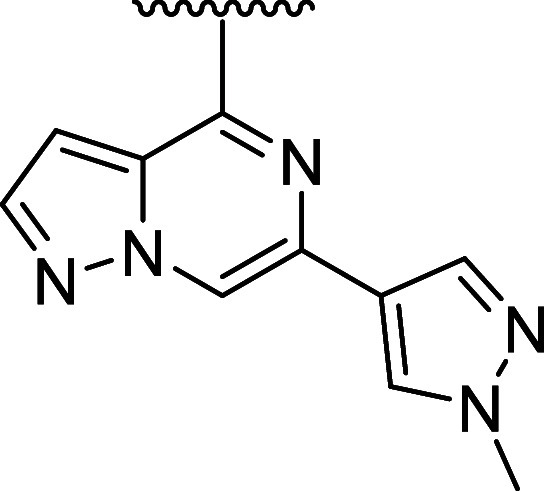
kinact/Ki values were determined using the nonphosphorylated BTK protein in a continuous-read kinetic enzyme assay, with the mean value for replicates shown in parentheses.
Mean IC50 values for the inhibition of CD69 expression on CD19+ B cells in human whole blood upon stimulation with anti-IgD.
Mean IC50 for the inhibition of TMD8 B cell proliferation.
Metabolic stability in rat and human liver microsomes (Intrinsic Clearance (Clint)).
Efflux ratio = Papp(B – A)/Papp(A – B) MDCK-MDR1.
Free unbound brain-to-plasma ratio as determined in a 24 h IV infusion rat PK study.
In vitro reactivity assessment using a GSH conjugation assay.
Utilizing the cocrystal structure containing compound 10, a series of analogues were designed containing conformationally constrained bicyclic azepane linkers with the intent to block metabolism by introducing steric bulk. This strategy51 showed promise as compound 13, containing a methylene bridge between carbon 2 and 6 on the azepane, exhibited a modest improvement in the intrinsic clearance in rodents (Rat liver microsome RLM = 556 mL/min/kg) and biochemical as well as cellular whole blood potency (log kinact/Ki = 4.90, WB CD69 IC50 = 0.24 μM) when compared with 10. In a continued effort to establish an in vitro–in vivo correlation (IVIVC) for this lead series, compound 13 was evaluated in vivo in rat PK experiments. Despite exhibiting high in vivo clearance (rat CL = 132 mL/min/kg), this modification did result in improved brain exposure (Kp,uu = 0.64) and oral bioavailability (F = 15%).
While the majority of the compounds tested in the biochemical and WB cellular assays had shown a good correlation in potency, we identified outliers for which the loss in cellular potency could not be explained by the physicochemical properties or the log kinact/Ki value (i.e., compound 11). We also observed that when tested using immortalized TMD8 cells, some compounds were unexpectedly inactive or exhibited only partial maximum effect (Emax)52,53 in preventing cellular proliferation (i.e., compound 13, TMD8 IC50 = 0.60 nM, Emax = 30%). These findings were rationalized by a reduction in cell viability,54 although experiments were not performed to establish the structure–activity relationship (SAR) contributing to the differences between a reduction in cellular growth versus an increase in cell death. Instead, we opted to include the TMD8 assay as an additional filter to exclude compounds exhibiting Emax < 100%, resulting in the deprioritization of compounds such as 13 and the investigation of other constrained bicyclic linkers. Among these, incorporation of the 3-azabicyclo[3.2.0]heptane linker in 14, led to significant improvement in the biochemical and whole blood potency (log kinact/Ki = 5.65; WB CD69 IC50 = 0.036 μM) but significant cell death was observed in the TMD8 assay (TMD8 IC50 = 0.76 nM; Emax = 44%). Although the in vivo clearance of 14 was above liver blood flow (rat CL = 247 mL/min/kg), this result was considered a significant improvement compared to 10, which warranted further investigation. Repositioning the acrylamide adjacent to the cyclobutyl moiety in 15 led to a drastic improvement in intrinsic clearance; however, the modification resulted in only partial inhibition in the whole blood assay and a complete loss in activity in the TMD8 assay. While attempting to mimic the three-dimensional pharmacophore55 necessary for achieving high potency, it was discovered that replacing the azepane core with the azabicyclic[3.1.0]hexyl moiety resulted in a decrease in the intrinsic clearance of 16 (RLM/HLM CL = 41/24 mL/min/kg). To further refine the predictive power of the physiologically based pharmacokinetic (PKPB) models, 16 was dosed in the rat infusion study, resulting in a reasonable rat brain exposure with a Kp,uu of 0.2.
Unfortunately, when screened in the GSH-stability assay, 16 demonstrated a relatively short half-life (GSH t1/2 2.0 h), which, combined with the low Emax in the TMD8 assay, resulted in the deprioritization of this molecule from further profiling. Despite achieving good biochemical potency and an improvement in the metabolic stability when tested in the liver microsome assay, the low Emax in the TMD8 assay and short half-lives in the GSH-stability assay observed with the structurally complex bicyclic ring systems led to the pursuit of alternative strategies.
Based on the in vitro liver microsomes data, we concluded that restricting the flexibility of the linker contributed to improvements in the potency and the metabolic stability of this lead series. Since our initial attempts to rigidify the linker between the hinge-binder motif and covalent warhead using fused rings were met with limited success, we approached the problem from a slightly different angle. Appreciating the need to maintain the optimal conformation to achieve covalent binding in combination with minimizing the number of HBDs to achieve satisfactory CNS exposure, we initiated a series of docking studies to identify alternative linkers as potential azepane replacements.56
A virtual screening effort returned several alternative linkers, including the 4-aminocyclohexanol moiety illustrated in 17 (mix of cis/trans isomers). While this compound exhibited moderate potency in the biochemical assay (log kinact/Ki = 2.4), it had favorable in vitro absorption, distribution, metabolism, and excretion (ADME) properties such as microsomal stability (RLM/HLM = 33/18 mL/min/kg) and efflux in the MDR1-MDCK cell line (ER = 4.1). However, a dramatic improvement in the biochemical activity was observed by moving the N-methyl acrylamide to the 3-position on the cyclohexyl ring (18); furthermore, docking predicted the cis-configuration to be the preferred stereoisomer for binding. This compound was active in the human whole blood assay (WB CD69 IC50 = 0.48 μM) and demonstrated improved half-life in GSH buffer over previous compounds (GSH t1/2 = 8.3 h). Despite these improvements, analogue 18 suffered from high intrinsic clearance above liver blood flow (RLM/HLM = 404/44 mL/min/kg) and minimal brain exposure (Kp,uu = 0.03). Fortunately, restricting the flexibility of the linker with the cis-3-aminocyclopentanol moiety in compound 19 led to an improvement in whole blood potency (WB CD69 IC50 = 0.26 μM), in vitro microsomal stability (RLM/HLM = 73/34 mL/min/kg), and lower efflux (MDR1-MDCK [ER] = 4.3), which also translated into improved brain exposure (Kp,uu = 0.40).
The encouraging potency data of compounds 17, 18, and 19, in combination with results from molecular modeling, led to the conclusion that the optimal linker length required for the acrylamide warhead to interact with Cys481 and Asn484 was equivalent to a distance of four bond lengths between the nitrogen and oxygen atoms. To test this hypothesis, we synthesized 20 and 21, which were predicted to have the correct spatial orientation to facilitate the covalent binding of acrylamide with Cys481. Additionally, the inherent plane of symmetry dissecting the cyclobutane ring eliminated chirality and simplified the synthesis by removing the need for chiral resolution of racemic material or sourcing of chiral building blocks. Interestingly, the trans- and cis-isomers 20 and 21 showed comparable potency (20 log kinact/Ki = 4.01 and 21 log kinact/Ki = 4.22) and suggested that the Cys481 and Asn484 side chain residues were tolerant of small changes in the binding conformation of the covalent warhead to the protein. Although 20 was not a P-glycoprotein (P-gp) substrate (MDR1-MDCK ER = 0.5), the in vivo brain exposure was below the level of quantification (BLQ), while the cis-analogue 21 despite a higher efflux ratio (MDR1-MDCK ER = 6.2) demonstrated detectable CNS exposure (Kp,uu = 0.05). Unfortunately, both cyclobutyl analogues were inactive in WB CD69 and TMD857 assays. Regardless, we continued their optimization due to their synthetic tractability and reduced molecular weight, focusing on improving whole blood and TMD8 activity and BBB-permeability.
It is worth mentioning that a thorough warhead screen58,59 to replace the acrylamide moiety in compound 21 with an alternative electrophile that maintained high activity for Cys481 and reduced promiscuity over other nucleophiles, such as glutathione, was performed at this stage. However, the screen, which comprised alternative Michael acceptors such as substituted acrylamides, but-2-ynamides, and sulfonamides motifs, was unsuccessful in identifying an alternative warhead with improved GSH chemical stability and comparable potency as seen with the acrylamide. These results were consistent with the docking studies, which indicated that minor differences in the orientation of the electrophile resulted in a less optimal geometry required for irreversible binding. Thus, the decision was made to continue utilizing the acrylamide electrophile in the design of future analogues as it offered the best compromise between on- and off-target reactivity.
To systematically explore the SAR of the linker in 21, we synthesized a set of constrained cyclobutyl-containing analogues. Among these, compound 22 (Table 4) had excellent inhibitory activity and exhibited good potency in both CD69 and TMD8 assays (log kinact/Ki = 4.59, WB CD69 IC50 = 0.736 μM, TMD8 IC50 = 2.94 nM) and a long half-life in GSH buffer of almost 10 h (GSH t1/2 = 9.6 h). Reducing the length of the carbon bridge by one carbon in analogue 23 resulted in further improvement in potency (log kinact/Ki = 5.11, WB CD69 IC50 = 0.16 μM, TMD8 IC50 = 1.79 nM). Unfortunately, while an Emax of 90% (mean over 10 runs; 100% inhibition for 3 runs) was observed in the whole blood assay, the Emax in the TMD8 assay plateaued at 42%, which was attributed to low cell viability. To assess whether the disconnect in the cellular assays correlated to the physicochemical properties of the molecules, we decided to focus on deconstructing the fused bicyclic ring to establish any trends in the SAR. As part of this effort, introducing a methyl substituent on the cyclobutane ring α to the amide nitrogen afforded analogue 24, which, although maintaining preferred cis-conformation, resulted in a loss of potency in the biochemical (log kinact/Ki = 3.59) and whole blood assay (WB CD69 IC50 = 0.91 μM). However, this analogue demonstrated outstanding GSH stability in buffer (t1/2 = 15 h) and good human microsomal stability (HLM = 15 mL/min/kg). Switching the position of the methyl group α to the ether linker afforded cis-isomer 25 with good potency in the biochemical assay (log kinact/Ki = 4.60) and moderate intrinsic clearance (RLM/HLM = 125/38 mL/min/kg). Despite having a relatively high efflux ratio in the MDR1-MDCK cell line (ER = 8.7), 25 was moderately brain penetrant in a rat constant infusion experiment (Kp,uu = 0.11). More importantly, methylation of the cyclobutyl ring α to the ether linkage addressed the previously observed disconnect between biochemical and cellular assays, and compound 25 was found to inhibit whole blood CD69 activation and cellular proliferation in the TMD8 cells with Emax = 100% and IC50 values in the low and subnanomolar range, respectively (WB CD69 IC50= 0.079 μM, TMD8 IC50 = 0.82 nM). Based on this finding and to address the biochemical to cellular potency disconnect, we devised synthetic routes to prepare the α-methyl analogues of 14 and 15 since these compounds had previously demonstrated the highest log kinact/Ki potency. While 26 had biochemical and whole blood potency comparable to the unmethylated congener 14, the disconnect remained with an Emax of 45% in the TMD8 assay. In contrast to 26, incorporation of the methyl substituent on 15 led to compound 27 and the desired improvement in efficacy in the cell-based assays with complete inhibition in human whole blood and TMD8 assays and double-digit nanomolar and picomolar IC50 (WB CD69 IC50 = 0.036 μM; TMD8 IC50 = 0.0006 μM), respectively. In vitro ADME screening showed that 27 had comparable intrinsic clearance (RLM/HLM = 180/36 mL/min/kg) to 25 and was found to have superior brain exposure in rats (Kp,uu = 0.44) despite being a P-gp substrate (MDR1-MDCK [ER] = 12.8).
Table 4. Cyclobutyl-Containing Amino-alcohol Linkers and Identification of the Crucial α-Tertiary Ethers.
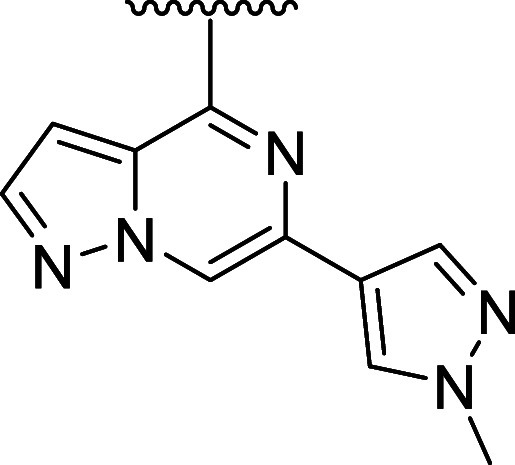
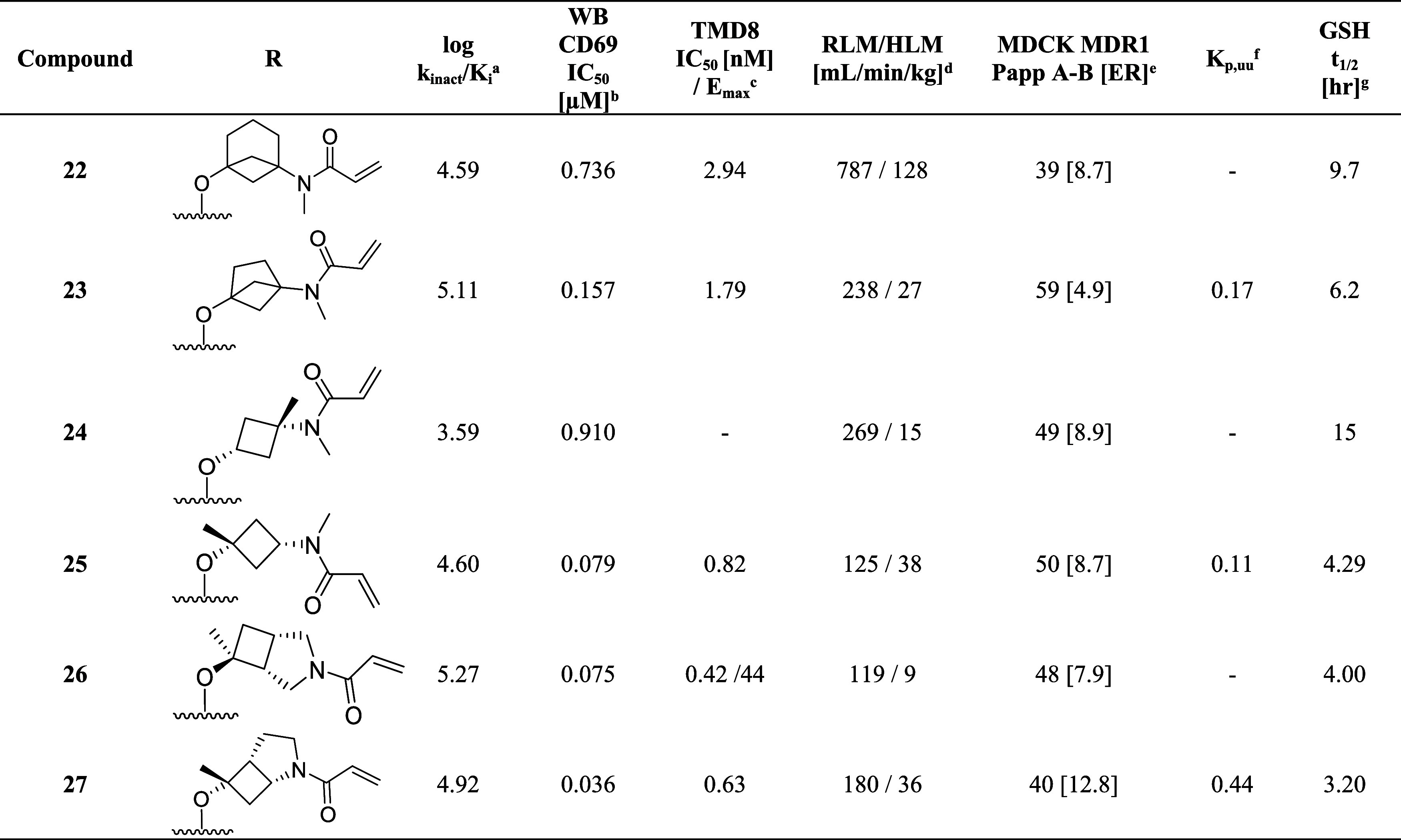
kinact/Ki values were determined using the nonphosphorylated BTK protein in a continuous-read kinetic enzyme assay, with the mean value for replicates shown in parentheses.
Mean IC50 values for the inhibition of CD69 expression on CD19+ B cells in human whole blood upon stimulation with anti-IgD.
Mean IC50 for the inhibition of TMD8 B cell proliferation.
Metabolic stability in rat and human liver microsome (Intrinsic Clearance (Clint)).
Efflux ratio = Papp(B – A)/Papp(A – B) MDCK-MDR1.
Free unbound brain-to-plasma ratio as determined in a 24 h IV infusion rat PK study.
In vitro reactivity assessment using a GSH conjugation assay.
The X-ray cocrystal structures of 25 and 27 obtained with full-length human BTK revealed an almost perfect overlay of the binding poses and conformations of both compounds. As depicted in Figure 3a,b, the pyrazolo[1,5-a]pyrazine hinge binding motif of 25 and 27 engages the backbone nitrogen of Met477 in a hydrogen bond (2.9 Å distance), while the α-tertiary ether and the newly introduced methyl groups project toward the glycine-rich P-loop. The N-methyl acrylamide substituent in 25 overlays perfectly with the methylene carbon in the 3-position of the azabicyclo[3.2.0]heptane ring in 27 (Figure 3b), which lines up the amide carbonyl to form an interaction with the side chain amide of Asn484 (3.5 Å distance), while the terminal end of the acrylamide warhead forms a carbon–carbon bond with Cys481 (1.8 Å length). Based on the two molecules demonstrating almost identical binding mode and good overlap, compound 27 can be regarded as a conformationally restricted analogue of 25. Furthermore, this explains the improvement in potency of compound 27 relative to that of 25, as the former seems to be locked into the favorable conformation for binding to Asn484 and Cys481. These cocrystal structures confirmed that the binding mode of initial lead compound 10 could be maintained and set the stage to test the hypothesis that the newly formed hydrogen bond with Asn484 was crucial for kinome selectivity.
Figure 3.

(a) X-ray cocrystal structure and binding mode of 25 (cyan; PDB 8TU4) in the active site of BTK. Hydrogen bond interactions are shown as gray dotted lines. (b) Superposition of the binding modes of compound 25 (cyan) and compound 27 (orange; PDB 8TU5) in the BTK ATP binding site was done from a top-down view with Met477 to the bottom left and Asn848 to the right (Cys481 in the back).
When profiled in a selectivity panel60 of 403 kinases (Figure 4), both 25 and 27 were found to have high selectivity scores (S(10)-score = 0.025 and 0.012, respectively), and significant activity was only observed against 10 and 5 kinases, respectively at 1 μM concentration (see the SI). Furthermore, 25 and 27 were profiled along with other covalent BTK inhibitors (evobrutinib (4) and tolebrutinib (2)) for their potential to inhibit other kinases bearing a reactive cysteine in the active site (Table 5) by the measurement of Kd. Both compounds showed superior selectivity for BTK when compared with evobrutinib (4) and tolebrutinib (2), which showed significant off-target activity against tyrosine-protein kinase (TEC) and bone marrow kinase on chromosome X (BMX). In particular, 25 showed greater than 10-fold selectivity for BTK over all but one (9-fold against TEC) of the 10 kinases tested. Compound 27 demonstrated a selectivity pattern comparable to that of 25 with activity against TEC (6.5-fold) and TXK tyrosine kinase (5.5-fold) within 10-fold of BTK and in good accordance with the kinome-scan data. This finding clearly illustrates the importance and influence of the amino acid residue at position 484 on kinome selectivity, as off-target activity (<100-fold) was only observed for kinases containing Asn484, while superior selectivity (>150 fold) was observed against kinases having either the Asp484 or Glu484 residue. On the other hand, tolebrutinib (2) was promiscuous within the 10 profiled kinases with double-digit nanomolar potency against all but two of them (JAK3 and ITK). This unique interaction with Asn484 and the resulting improvement in selectivity over other covalent inhibitors utilizing the back pocket behind gatekeeper Thr474 is remarkable, given the reduced size and the improved CNS properties (CNS MPO scores) of this new class of BTK inhibitors (Tables 1 and 6).
Figure 4.
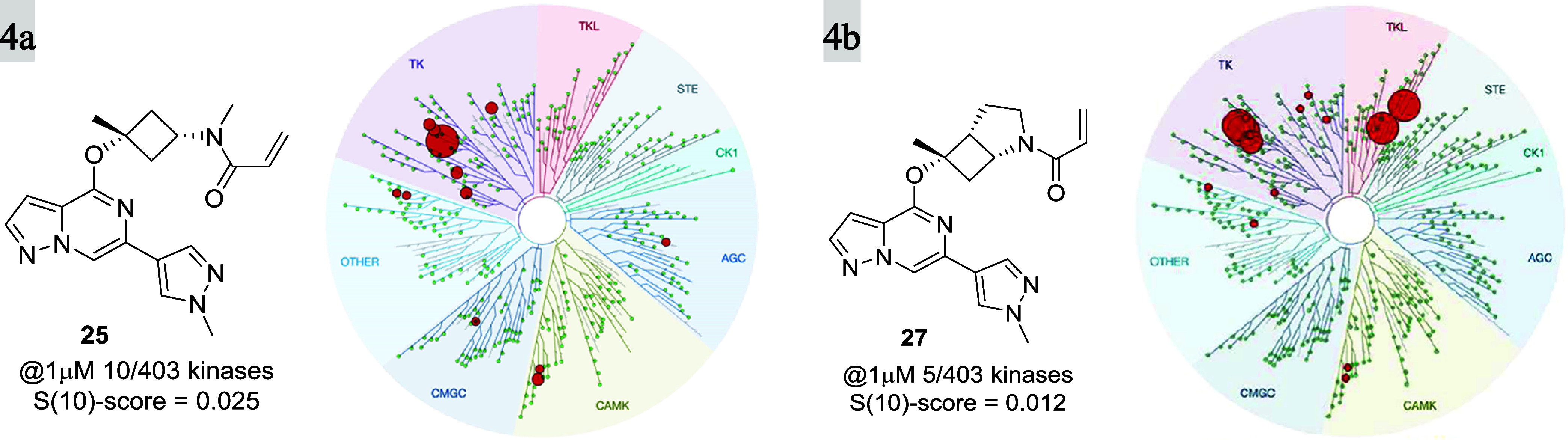
KINOMEscan, Eurofins DiscoverX, for compounds 25 (a) and 27 (b) at 1 μM concentration. Ten and 5 out of 403 kinases were inhibited with S(10) selectivity scores of 0.025 and 0.012, respectively.
Table 6. Comparison of Profiles of Initial Hit 10 and Compounds 25 and 27.

| specific measure | 10 | 25 | 27 |
|---|---|---|---|
| In Vitro | |||
| log kinact/Kia | 4.43 | 4.60 | 4.92 |
| WB CD69 IC50 [μM]b | 0.333 | 0.079 | 0.036 |
| TMD8 IC50 [nM]c | 12.5 | 0.817 | 0.630 |
| RLM/HLM [mL/min/kg]d | 820/74 | 125/38 | 180/36 |
| GSH t1/2 [h]e | 9.4 | 4.3 | 3.2 |
| fu (hPPB/rPPB/rbrainPB)f | 0.13/0.29/0.16 | 0.10/0.08/0.04 | 0.05/0.12/0.13 |
| MDR1-MDCK (A-B/ER)g | 59/5.4 | 50/8.7 | 40/12.8 |
| BCRP (A-B/ER)h | 1.9 | 1.1 | 1.0 |
| solubility pH 1.5, 7.5 [μg/mL]i | 48 | 46 | 40 |
| kinome selectivityj @1 μMk (S(10)-score)l | 7/403 (0.017) | 10/403 (0.025) | 5/403 (0.012) |
| MW [g/mol]/Elog D/CNS MPOm | 366.2/2.4/5.86 | 366.2/2.5/5.96 | 378.2/2.4/5.82 |
| In Vivo | Rodentn | ||
| Kp,uu | 0.37 | 0.11 | 0.44 |
| Vdss [L/kg] | 19.2 | 4.3 | 7.5 |
| IV T1/2 [h] | 10.3 | 3.59 | 0.74 |
| CL [mL/min/kg] | 133 | 87 | 121 |
| F [%] | 3 | 25 | 33 |
| In Vivo | Nonrodento | ||
| Vdss [L/kg] | 0.73 | 1.19 | 1.07 |
| IV T1/2 [h] | 0.21 | 0.29 | 0.37 |
| CL [mL/min/kg] | 34.2 | 53.5 | 37.0 |
| F [%]v | <0.05 | 21 | 16 |
kinact/Ki values were determined using the nonphosphorylated BTK protein in a continuous-read kinetic enzyme assay, with the mean value for replicates shown in parentheses.
Mean IC50 values for the inhibition of CD69 expression on CD19+ B cells in human whole blood upon stimulation with anti-IgD.
Mean IC50 for the inhibition of TMD8 B cell proliferation.
Metabolic stability in rat and human liver microsome (Intrinsic Clearance (Clint)).
In vitro reactivity assessment using a GSH conjugation assay.
% Fraction unbound was measured at a concentration of 2 μM in human plasma via rapid equilibrium dialysis (RED).
Efflux ratio = Papp(B – A)/Papp(A – B) MDCK-MDR1.
Efflux ratio = Papp(B – A)/Papp(A – B) BCRP.
Kinetic solubility was determined using an Analiza shake-flask solubility assay.
DiscoverX KINOMEScan Kinase Screen.
Total number of kinases screened (403) excludes BTK.
MoKa was used to calculate clog P, clog D, and pKa.
Rodent PK studies were completed in male Sprague-Dawley (SD) rats, measured in triplicate.
Nonrodent PK studies were completed in cynomolgus monkeys, measured in triplicate.
As illustrated in Table 6, compounds 25 and 27 exhibited improved potencies across all BTK-specific assays and intrinsic clearance in human and rat liver microsomes compared to that of the original lead 10. All three compounds have comparable molecular weights, low lipophilicity (Elog D < 2.5), similar solubility (>40 μg/mL at pH 6.8), and moderate binding in human/rat plasma and rat brain homogenate (hPPB, rPPB, rat brainPB). While 25 and 27 are both MDR1 substrates, BCRP-mediated efflux was low, and both compounds maintained brain exposure as measured by rat Kp,uu, which was consistent with the high CNS MPO scores of >5.8. Despite in vivo clearances above liver blood flow (Table 6) and relatively short half-lives in rat IV-bolus PK, significantly improved bioavailability of 25 and 33%, respectively, was determined for 25 and 27, which was a significant improvement over the 3% bioavailability observed for 10. Furthermore, relatively high maximal plasma concentrations in rat PO experiments were reported, increasing our confidence that we could achieve significant target coverage in vivo (see below). Similar to its behavior in rats, 10 had extremely low bioavailability (%F = <0.1%) with virtually no plasma exposure (Cmax < 0.5 ng/mL) in a cynomolgus monkey (cyno) PK study (5 mg/kg PO). Interestingly, despite the short half-live (T1/2 = 0.29 and 0.37 h) and high clearance (cyno CL = 55 and 37 mL/min/kg, respectively), 25 and 27 were found to have acceptable bioavailability in cyno at 21 and 16%, respectively. As part of the preclinical safety in vitro screening, both compounds were tested for genotoxicity using the Ames and micronucleus test. Unfortunately, 27 showed a positive signal in the Ames test, whereas 25 was negative in both Ames and micronucleus tests, indicating a low risk for having genotoxicity potential (Table 7). With the discovery of 25, a potent and selective covalent BTK-inhibitor with good ADME, an in vitro safety profile, and kinase selectivity, we decided to continue further characterization of the compound as a potential clinical candidate.
Table 7. Preclinical Toxicology Summary of Compound 25.
| 25 (BIIB129) | |
|---|---|
| In Vitro Safety Profile | |
| Cyp DDI IC50 [μM] | 2C9 = 5.2 |
| Cyp TDI (fold change over control) | 3A4 1-fold |
| 2C9 0.5-fold | |
| hERG IC50 [μM] | >30/>30 |
| radio ligand/patch clamp | |
| NaV 1.5/CaV 1.2 | >30/>30 |
| IC50 [μM] | |
| cytotox. Glu/Gal | >30/>30 |
| IC50 [μM] | |
| Ames | negative |
| micronucleus | negative |
| chromosomal aberration | negative |
| 3T3 NRU phototoxicity | negative |
| hepatotoxicity (HUREL human primary hepatocyte microliver) | minimal toxicity (ATP) |
| In Vivo Toxicology Studies | |
| rat micronucleus | negative |
| cynomolgus monkey cardiovascular | no adverse findings |
| 10, 30, 100 mg/kg | |
| rat 28-day | NOAEL = 200 mg/kg/day (365× male, 689× female safety marginsa) |
| 30, 100, 300 mg/kg BID | |
| cynomolgus monkey 28-day | NOAEL = 60 mg/kg/day (140× male, 136× female safety marginsa) |
| 10, 30, 100 mg/kg BID | |
Free peripheral AUC margin based on the projected human efficacious exposure.
Biological Evaluation of Compound 25
A comprehensive array of cellular screens was devised to investigate both on- and off-target activities (Table 8). Since EGFR inhibition has been implicated with AEs61 observed in the clinic with covalent BTK inhibitors, we devised a biochemical covalent binding assay which confirmed that 25 exhibited minimal activity (kinact/Ki = 6 M–1 s–1) when tested against the recombinant human EGFR protein. Furthermore, 25 was shown to bind only 4 of 190 kinases (BTK TO50 = 6.8 nM, TEC TO50 = 18 nM, ITK TO50 = 308 nM and BLK TO50 = 3.9 nM) among 3500 total proteins detected when tested in a cellular matrix comprising of human Burkitt’s lymphoma cell line (Ramos), B cell lymphoma line (TMD8), and human T cell leukemia line (Jurkat) as a 1:1:1 mixture using kinomebead competitive chemoproteomics.62,63 Additional in vitro target occupancy studies in Ramos/TMD8/Jurkat cells using a clickable probe of 25 and biotin-TAMRA azide to detect labeled protein by in-gel fluorescence and mass spectrometry confirmed covalent binding to BTK and TEC (TO50 = 5.5 nM and TO50 = 1.2 nM), with only marginal activity observed for BLK and ITK at the highest concentration tested.64 To evaluate the effectiveness of covalent binding to BTK at blocking BCR and FcγRs signaling in cellular systems, Ramos B cells were activated with anti-IgM and treated in a dose–response with 25, resulting in the inhibition of PLCγ2 phosphorylation with an IC50 = 1.2 nM. In TMD8 cells 25, it was shown to inhibit the activation and translocation of NF-κB to the nucleus65 with an IC50 = 1.9 nM, and in human peripheral blood mononuclear cells (PBMCs) afforded an IC50 = 13 nM at inhibiting anti-IgD-induced B cell activation. To quantify the ability of 25 to prevent TMD8 proliferation, we employed the CellTiter-Glo Luminescent Cell viability assay using ATP levels as an indirect readout of cell number leading to a cellular IC50 = 7.1 nM. Further transcriptomic and flow-cytometry profiling of TMD8 cells implicated CD19 and CD36 expression as biomarkers of BTK inhibition, which yielded IC50 = 4.2 nM and IC50 = 1.8 nM, respectively, when tested in a dose–response manner with 25. To ascertain the impact of BTK inhibition on FcR signaling in innate immune cells,66,6725 was tested in neutrophils, leading to the inhibition of FcγR-mediated ROS production with IC50 = 3.2 nM. When dosed in monocytes, 25 blocked FcγR-induced TNF secretion (IC50 = 3.0 nM), while, in human whole blood, an IC50 = 124 nM was observed for the inhibition of FcεR-induced basophil degranulation (CD63).68 Since anti-CD20 B cell depleting antibodies are thought to drive clinical efficacy in MS patients primarily by preventing B cell-mediated antigen presentation to T cells,69 it was necessary to determine the potency of 25 at blocking this MOA. Accordingly, in a previously described assay designed to test B cell-mediated antigen presentation to T cells,1325 reduced the proliferation of OVA323–339-specific T cells in response to OVA targeted to and presented by B cells with an IC50 = 1.9 nM. With a comprehensive understanding of the downstream and functional effects associated with covalent inhibition of BTK in vitro, it was necessary to examine how the potency translated to in vivo efficacy in preclinical studies.
Table 8. In vitro Biological Profile of BIIB129 (25).
| assay format (cell type, stimulus) | readout |
|---|---|
| kinact/Ki (purified nonphosphorylated BTK protein) | 6 M–1 s–1 |
| TO50 (TMD8 cells) | 6.8 nM |
| IC50 PLCγ2 phosphorylation (Ramos cells, IgM) | 1.2 nM |
| IC50 activation and translocation of NF-κB (TMD8) | 1.9 nM |
| IC50 B cell activation (PBMCs anti-IgD/BCR) | 13 nM |
| IC50 TMD8 proliferation (CD19 and CD36 expression) | 4.2 and 1.2 nM |
| IC50 blocking FcγR-induced TNF secretion (monocytes) | 3.0 nM |
| IC50 B cell-mediated antigen presentation to T cells | 1.9 nM |
In Vivo Efficacy, ADME, and Preclinical Toxicology
The evaluation of the in vivo efficacy of 25 in blocking BCR-mediated activation of CNS-compartmentalized B cells and inhibition of FcγR-induced microglia functions required the development of two novel preclinical in vivo models. The first model utilized surgical implantation of TMD8 cells in the cerebral ventricular space of immunodeficient rat NOD.SCID mice. After an optimized resting period, the animals were dosed orally with 25 (BID at 2.5, 10, and 25 mg/kg) and vehicle for 2.5 days. At the end of the study, the TMD8 cells were isolated from the CNS of the treated animals and analyzed for CD36 and CD19 expression using flow cytometry, resulting in a dose–response with maximal inhibition at 25 mg/kg. To assess the efficacy of 25 on blocking FcγR-induced microglia proliferation in mice, we utilized a previously reported mouse model.12 In this model, microglia reactivity and proliferation can be induced by the peripheral presentation of anti-MOG antibodies, which accumulate and bind to myelin in the CNS via the Fab region and to microglia FcR via the Fc region. Mice were injected intraperitoneally (IP) with a 30 mg/kg dose of FcR-binding anti-MOG antibodies, which was demonstrated to trigger microglia reactivity and proliferation in an FcR- and BTK-dependent manner. The treated animals were dosed orally with 25 (BID at 0.1, 0.3, 1, 3.2, 10, and 31.7 mg/kg) and demonstrated a dose-dependent reduction in Ki67+ microglia proliferation with an EC50 = 1.5 mg/kg. Having demonstrated BTK-dependent in vivo efficacy of 25 by inhibiting both B cell and microglia activity in the CNS in two distinct preclinical models, additional studies were conducted to interrogate potential off-target activities, as this would impact the overall safety profile of the potential candidate. Compound 25 did not inhibit CD4+ or CD8+ T cell activation or proliferation in response to anti-CD3/CD8 stimulation up to 1 μM, the highest concentration tested in the assay. This observation is consistent with the results from the BioMAP system,70 which examines the correlation between binding to recombinant protein in the KINOMEscan versus the functional effect in a cellular system (Figure 5). Although 25 did exhibit effects on pathways in the T cell system (Sag system), endothelium/PBMCs (LPS and Mphg systems), epithelial cells (BE3C system), and smooth muscle cells and fibroblasts (HDF3CGF and MyoF systems) at concentrations of 3.3 and 10 μM, minimal activity was detected outside of the BT culture system at 0.37 μM, which was projected to be the human therapeutic exposure needed for full target occupancy over the dosing interval.
Figure 5.

Phenotypic BioSeek study data of compound 25 at 0.37, 1.1, 3.3, and 10 μM. The gray area shows the 95% confidence area of each biomarker response in the absence of the drug. Data points that fall outside the gray area signify inhibition (below) or augmentation (above) of the biomarker response at the indicated concentration of compound 25.
The overall in vitro and in vivo preclinical safety profile of 25 was favorable (Table 7). No signals were detected (IC50 > 30 μM) in the Nav 1.5 and Cav 1.2 ion channels and hERG assays, indicating a low risk for cardiovascular toxicity. These results were supported by no adverse effects in the cardiovascular systems of telemetered cyno dosed up to 100 mg/kg, providing a 397-fold exposure multiple to the total daily exposure (AUC) at the predicted human efficacious dose exposure. The low genotoxic potential of the compound was demonstrated by negative results in the Ames test (with and without rat liver S9 metabolic activation), in vitro micronucleus and chromosomal aberration tests (human peripheral lymphocytes with and without S9), and a rodent (Sprague–Dawley rat) micronucleus study. Additionally, the 3T3 Neutral Red Uptake (NRU) phototoxicity test was negative, and the hepatotoxicity test using a coculture model of primary human hepatocytes and stromal cells (HUREL microliver) resulted in minimal toxicity (ATP measurement after 14 days of treatment). The preclinical repeat-dosing toxicology studies were performed in rodent (Sprague–Dawley rat) and nonrodent (cynomolgus monkey) species for 28 days at 30, 100, and 300 mg/kg in rats and 10, 30, and 100 mg/kg in cyno dosed BID via oral gavage. The cynomolgus monkey was selected as the nonrodent species due to high CL and poor oral bioavailability found in dogs (Table 6) owing to the dog-specific predominance and extensiveness in the pyrazole N-demethylation (SI). The use of cyno was also aided by the extensive safety knowledge of BTK inhibitors acquired from the development of BIIB091.36 The 28-day studies, involving assessments of clinical signs, body weight, food consumption, ECG, clinical pathology, organ weight, gross pathology, and histopathology, demonstrated that 25 was well tolerated with no observed adverse effect level (NOAEL) of 200 mg/kg/day for rat and 60 mg/kg/day for cyno. These corresponded with large safety margins in rats (male 365-fold, female 689-fold) and cyno (male 140-fold, female 136-fold) based on the projected human efficacious exposure.
A comprehensive assessment of the ADME properties of 25 (Table 6) was conducted, leading to high plasma CL across all animal species tested (CL was high for rat, dog, and cyno, 87, 74.2, and 54 mL/min/kg % liver blood flow %QH 158, 240, and 122% respectively), with a Vss between 1.2 and 4.3 L/kg, T1/2 of 0.29–3.4 h, and low to moderate oral bioavailability (25% rat, 4.5% dog, and 21% cyno) when dosed as a free base. The in vitro ADME screening confirmed that 25 was a moderate substrate for the P-gp efflux transporter (MDR1-MDCK efflux ratio 11); however, it is not a substrate of the BCRP transporter. These data are consistent with the in vivo free brain and free plasma concentration ratio (Kp,uu) of 0.11 in rats (obtained by IV infusion) and 0.27 in cyno (obtained at a single time point when dosed orally). The in vitro metabolism was investigated in rat, dog, cyno, and human hepatocytes and showed no unique human metabolites (SI). The prominent site of metabolism across all species was at the acrylamide warhead, including oxidation and diol formation at the acrylamide, as well as direct glutathione conjugation and its subsequent metabolites. In cyno and dog, the amide N-desmethyl and the pyrazole N-desmethyl were major metabolites, respectively, both of which are biologically active and observed in human hepatocytes but to a lesser extent. When screened for potential drug–drug interaction (DDI) risk in vitro, 25 did not exhibit TDI for CYP3A4, CYP2D6, CYP2C9, or CYP1A2 in human liver microsomes up to the highest concentration tested (10 μM), nor did it induce CYP1A2, CYP2B6, or CYP3A4 mRNA expression in the cultured human hepatocyte assay. To understand the route of elimination, male bile duct-cannulated Sprague–Dawley rats were dosed IV at 1.5 mg/kg with 25. Cumulative amounts of unchanged 25 excreted in rat bile and urine over 24 h post dose in comparison to the IV administered amount have indicated that biliary and urinary clearance was negligible, accounting for 0.34 and 0.02% of the total systemic clearance, respectively. In addition, Met-ID in bile and urine samples following a PO dose of 30 mg/kg to BDC rats has confirmed the acrylamide being the predominant site of metabolism of 25 in rats in vivo (see the SI).
PK/pharmacodynamic (PD) modeling of 25 in mice was performed to describe the time course of the BTK target occupancy in various tissues. The time course of BTK inactivation by the irreversible inhibitor was modeled by using a differential equation (see the SI). The in vivo potency kinact/Ki estimated in mice was approximately 5- to 6-fold higher than the in vitro potency kinact/Ki against mouse and human BTK. Human PK prediction of 25 was performed using a combination of allometric scaling and in vitro–in vivo extrapolation (SI). A summary of prediction methods and the predicted values for volume of distribution (Vss), bioavailability (F), first-order absorption rate constant (Ka), unbound brain-to-plasma concentration ratio (Kp,uu), and plasma clearance (CL) are shown in Table 9.
Table 9. Summary of Predicted Human Pharmacokinetics Parameters of BIIB129 (25) and Prediction Methods.
| PK parameter | predicted value | prediction method |
|---|---|---|
| Vss | 1.18 L/kg | single species (monkey) scaling with unbound fraction correction |
| F | 18% | averaging nonclinical F across species rat, dog, and monkey |
| Ka | 4 h–1 | averaged from absorption rate constants estimated for rat, dog, and monkey. |
| Kp,uu | 0.3 | In vitro–in vivo extrapolation of transporter activity71 |
| CL | 44.5 mL/min/kg | averaged from two methods: (1) allometric scaling from mouse, rat, dog, and monkey; (2) in vitro–in vivo extrapolation with a correction factor averaging from observed CL vs. predicted CL in rat, dog, and monkey. |
The human efficacious dose was defined as the dose associated with approximately 90% BTK occupancy in the CNS at the end of the dosing interval, i.e., at Ctrough. Human BTK occupancy (peripheral and CNS) following BID dosing was predicted using the projected human PK parameters and the PK/PD relationship established in mice. PK/PD simulations were performed with two BTK half-life values (48 and 96 h), based on the range from published clinical data,48,72−74 with predicted time courses to achieve desired steady-state BTK occupancy in human CNS at different doses, plotted in Figure 6. The efficacious clinical dose of BIIB129 (25) to achieve 90% target occupancy of BTK in human CNS is predicted to be in the 40 to 80 mg daily dose range following BID dosing.
Figure 6.

Prediction of BTK target occupancy in the human CNS following BID dosing of BIIB129 (25) by translational quantitative PK/PD modeling and simulation.
Chemistry
An efficient and modular synthetic route toward these covalent BTK inhibitors was developed with the goal of being highly tunable for rapid interrogation of the SAR. Depicted in Scheme 1 is the medicinal chemistry route toward BIIB129 (25), which commenced with commercially available 4,6-dichloropyrazolo[1,5-a]pyrazine (28). An efficient 2 step, one-pot procedure, including a nucleophilic aromatic substitution75 with commercially available tert-butyl ((1s,3s)-3-hydroxy-3-methylcyclobutyl)carbamate (29) and a palladium-mediated Suzuki-coupling76 with boron pinacolate 30, was developed giving rapid access to intermediate 31, which allowed exploration of different substituents on the Boc-protected nitrogen. For the synthesis of 25, however, filtration of the crude material over a silica column and treatment of intermediate 31 with KHMDS, followed by the addition of iodomethane, was used to install the N-methyl group in 32. To avoid cleavage of the ether linkage in intermediate 32, the removal of the Boc protecting group was carried out with trifluoroacetic acid (TFA) in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) as the solvent, resulting in secondary amine 33. Installation of the acrylamide as covalent warhead in the last step with acryloyl chloride and N,N-diisopropylethylamine (DIPEA) as the base furnished the desired compound 25via five steps and just three isolated synthetic intermediates and good overall yield of 27%.
Scheme 1. Synthesis of BIIB129 (25) from Commercial Building Blocks.

Conditions: (a) 29, KHMDS (1 M in tetrahydrofuran (THF)) then 28, 1,4-dioxane, rt to 0 °C; (b) 30, Pd-PEPPSI-IPr catalyst, rt to reflux; (c) KHMDS (1 M in THF), iodomethane, THF, rt; (d) TFA, HFIP, rt; and (e) DIPEA, acryloyl chloride, THF, 0 °C.
Conclusions
Herein, we describe the discovery of brain penetrant and selective covalent BTK inhibitors in an improved CNS-property space over known TCIs for the same target. These BTK inhibitors feature a novel binding mode omitting relatively lipophilic substituents to extend behind the Thr477 gatekeeper (i.e, compounds 1, 2, 4) or moieties of high molecular weight and increased polarity to engage the “H3″ pocket (i.e., compounds 3, 5, 6). The hydrogen bond of the acrylamide carbonyl of compounds such as 25 with the side chain nitrogen of Asn484, an interaction that, to our knowledge, has not been previously described, is believed to be responsible for the excellent kinome selectivity. This hypothesis is supported by the improved selectivity profile of our lead compounds (10, 25, and 27) over clinical assets evobrutinib (4) and tolebrutinib (2) against kinases bearing reactive Cys in the active site. Only kinases BMX, TEC, and TXK, which feature an Asn in the 484 position, were inhibited with <50-fold selectivity over BTK in this assessment (Table 5). Our structure-based lead-optimization efforts of compound 10 revealed that the incorporation of a methyl group at the α-tertiary ether position of a cyclobutane-containing linker between the hinge binding motif and the acrylamide warhead was critical for CD69 and TMD8 potency and to reach complete Emax in particular.77 Compounds 25 and 27, due to their excellent potencies in all assays against BTK and favorable brain exposure in rat (Kp,uu), were selected for in vitro preclinical toxicology screening, which cleared 25 for further in vivo profiling, whereas 27 was deprioritized due to a positive signal in the Ames genotoxicity test. 25 was found to have sufficient bioavailability in rats and cyno, and PKPB modeling suggested low projected total human doses to cover the IC90 in the brain over the dosing interval (20 and 40 mg BID, respectively; Figure 6). Additionally, 25 demonstrated BTK-dependent in vivo efficacy in the CNS in two distinct preclinical models by inhibiting both TMD8 B cells transplanted in the CNS of immunodeficient mice and microglial activity in an anti-MOG mouse model. With the improved physicochemical and CNS properties, excellent selectivity, and potency, contributing to a decrease in the projected human daily dose, 25 has the potential to mitigate the risk78 observed with covalent BTK drugs and be a best-in-class CNS immune-modulating therapy for MS.
Experimental Section
General Procedures
All solvents and chemicals used were reagent grade. Anhydrous solvents were used as purchased from commercial sources. Analytical thin-layer chromatography (TLC) and silica gel column chromatography were performed on a Merck silica gel 60 (230–400 mesh). The removal of solvents was conducted by using a rotary evaporator, and residual solvents were removed from nonvolatile compounds using a vacuum manifold maintained at approximately 1 Torr. NMR spectra were recorded on a Bruker Avance 400 and 500 MHz NMR spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) relative to residual undeuterated solvent as an internal reference, and coupling constants (J) are reported in hertz (Hz). Splitting patterns are indicated as follows: s = singlet; d = doublet; t = triplet; q = quartet; qn = quintet; dd = doublet of doublets; dt = doublet of triplets; tt = triplet of triplets; m = multiplet; and br = broad peak. Silica gel column chromatography was performed using 20–40 μM (particle size), 250–400 mesh, or 400–632 mesh silica gel using either a Teledyne ISCO Combiflash RF or a Grace Reveleris X2 with ELSD purification systems using prepacked silica gel columns. All final compounds were purified to ≥95% purity, as determined by LC/MS analysis: a sample is dissolved in a suitable solvent such as MeCN, dimethyl sulfoxide (DMSO), or MeOH and is injected directly into the column using an automated sample handler. The analysis used one of the following methods: (A) an acidic method (1.3 or 3.5 min runs) conducted on a Waters Acquity UPLC BEH (MS ionization: ESI) instrument equipped with a C18 column (2.1 mm × 30 mm, 3.0 mm, or 2.1 mm × 50 mm, C18, 1.7 μm), eluting with 1.5 mL/4 L TFA in water (solvent A) and 0.75 mL/4 L TFA in acetonitrile (solvent B) or (B) a basic method (1.3 or 3.5 min runs) conducted on a Waters Acquity UPLC BEH (MS ionization: ESI) instrument equipped with XBridge Shield RP18, 5 μm column (2.1 mm × 30 mm, 3.0 mm i.d.) or 2.1 mm × 50 mm, C18, 1.7 μm column, eluting with 2 mL/4 L NH3·H2O in water (solvent A) and acetonitrile (solvent B). Preparative high-performance liquid chromatography (HPLC) purifications were performed using two methods: (A) an acidic method on a Waters Sunfire LC-MS OBD C18 PREP (MS ionization: ESI; UV detection at 220/254 nm, waters automatic collection) eluting with 1.5 mL/4 L TFA in water (solvent A) and 0.75 mL/4 L TFA in acetonitrile (solvent B) or (B) a basic method on a Waters XSELECT CSH C18 PREP (MS ionization: ESI; UV detection at 220/254 nm, waters automatic collection) eluting with 2 mL/4 L NH3·H2O in water (solvent A) and acetonitrile (solvent B). All reported yields are isolated yields.
tert-Butyl ((1s,3s)-3-Methyl-3-((6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrazin-4-yl)oxy)cyclobutyl)carbamate (31) and tert-Butyl Methyl((1s,3s)-3-methyl-3-((6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrazin-4-yl)oxy)cyclobutyl)carbamate (32)
Step 1
In a 500 mL three-neck round-bottom flask equipped with a condenser under a nitrogen atmosphere, KMHDS (1 M in THF, 2.2 mL) was added to a solution of tert-butyl ((1s,3s)-3-hydroxy-3-methylcyclobutyl)carbamate (29) (5.0 g, 24.8 mmol) in 1,4-dioxane (150 mL) at rt. After 15 min, a solution of 4,6-dichloropyrazolo[1,5-a]pyrazine (28) (4.3 g, 22.58 mmol) in 1,4-dioxane (50 mL) was added dropwise to the suspension upon cooling of the reaction mixture in an ice/water bath. The resulting reaction mixture was degassed by being purged with nitrogen for 30 min. At rt, a degassed solution of K3PO4 (9.6 g, 45.2 mmol) in water (45 mL) was added, followed by a degassed solution of 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (30) (9.4 g, 45.2 mmol) in 1,4-dioxane (50 mL) and a solid Pd-PEPPSI-IPr catalyst (93 mg, 0.14 mmol). After the resulting reaction mixture was purged with nitrogen for an additional 30 min, it was heated at reflux for 90 min. The vigorously stirred reaction mixture was allowed to cool to rt, and EtOAc (200 mL) and water (100 mL) were added. After 30 min, the organic phase was separated, washed with water (2 × 50 mL) and brine (50 mL), dried over Na2SO4, filtered, and concentrated in vacuo to afford a residue that was purified by silica gel chromatography (80 g silica gel, 0–80% [3:1 EtOAc/EtOH] with 2% NH4OH modifier in heptanes) to yield 31 (11.9 g, assumption 100% yield), which was used without further purification in the next step. LCMS m/z = 399.1 (M + H)+; tr = 0.83 min.
Step 2
To a solution of crude 31 (10.7 g, 20.3 mmol) dissolved in THF (150 mL), KHMDS (1 M in THF, 30.5 mL) was added dropwise at rt for 15 min. After an additional 15 min, iodomethane (5.4 g, 35.6 mmol, 2.2 mL) was added dropwise to the above reaction mixture at rt. After 1 h, ice water (50 mL) was added to the reaction mixture, followed by EtOAc (50 mL) and brine (5 mL) at rt. The aqueous phase was separated and extracted with EtOAc (2 × 50 mL), and the combined organic phase was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo to afford crude material, which was purified by silica column chromatography (120 g silica gel, 0–70% [3:1 EtOAc/EtOH] with 2% NH4OH modifier in heptanes) to yield the desired compound 32 (4.1 g, 48% over two steps) as a pale-yellow oil. LCMS m/z = 413.1 (M + H)+. 1H NMR (500 MHz, methanol-d4): δ (ppm) 8.42 (s, 1H), 8.05 (s, 1H), 7.93 (s, 1H), 7.91 (d, J = 2.4 Hz, 1H), 6.77 (d, J = 1.2 Hz, 1H), 4.45–4.10 (m, 1H), 3.95 (s, 3H), 2.82 (s, 3H), 2.81–2.74 (m, 2H), 2.67 (br s, 2H), 1.81 (s, 3H), 1.46 (s, 9H).
(1s,3s)-N,3-Dimethyl-3-((6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrazin-4-yl)oxy)cyclobutan-1-amine (33)
To a solution of tert-butyl methyl((1s,3s)-3-methyl-3-((6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrazin-4-yl)oxy)cyclobutyl)carbamate (32) (4.1 g, 9.8 mmol) in HFIP (45 mL) was added TFA (2.2 g, 19.6 mmol, 1.5 mL) at rt. The resulting reaction mixture was stirred for 12 h and diluted with EtOAc (50 mL), followed by a sat. NaHCO3 solution (25 mL) and brine (10 mL). After vigorous stirring for 30 min, the organic phase was separated, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting residue was purified by column chromatography (24 g of silica gel, 80–100% [3:1 EtOAc/EtOH] with 2% NH4OH modifier in heptanes) to yield the title compound 33 (2.53 g, 82% yield). LCMS m/z = 313.1 (M + H)+. 1H NMR (500 MHz, methanol-d4): δ (ppm) 8.41 (d, J = 1.2 Hz, 1H), 8.05 (s, 1H), 7.97–7.86 (m, 2H), 6.81–6.72 (m, 1H), 3.95 (s, 3H), 3.11–2.96 (m, 1H), 2.90–2.76 (m, 2H), 2.31 (s, 3H), 2.31–2.25 (m, 2H), 2.31–2.25 (m, 2H), 1.80 (s, 3H).
N-Methyl-N-((1s,3s)-3-methyl-3-((6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrazin-4-yl)oxy)cyclobutyl)acrylamide (25)
To a solution of (1s,3s)-N,3-dimethyl-3-((6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyrazin-4-yl)oxy)cyclobutan-1-amine 33 (4.1 g, 9.8 mmol) and DIPEA (2.8 g, 21.7 mmol, 3.8 mL) in THF (50 mL) was added acryloyl chloride (819 mg, 9.04 mmol, 740 μL) at 0 °C. After 30 min, the reaction mixture was diluted with EtOAc (50 mL) and a sat. aq. NaHCO3 solution (50 mL) was added. The vigorously stirred biphasic mixture was brought to rt, and stirring was continued for another 30 min. The organic phase was separated, washed with water (25 mL) and brine (25 mL), dried over Na2SO4, filtered, and concentrated in vacuo to afford a residue, which was purified by column chromatography (80 g silica gel, 0–100% [3:1 EtOAc/EtOH] with 2% NH4OH modifier in heptanes). The colorless solid was recrystallized from EtOAc/heptanes (1/3, 45 mL) to afford the title compound as a freely flowing crystalline solid (1.8 g, 68% yield). Melting point: 137.5 °C. HRMS (ESI+): m/z calcd for C19H23N6O2 [M + H]+, 367.1877; found, 367.1885. 1H NMR (500 MHz, methanol-d4): δ (ppm) 8.44 (s, 1H), 8.06 (s, 1H), 7.98–7.85 (m, 2H), 6.85–6.67 (m, 2H), 6.26–6.12 (m, 1H), 5.74 (br d, J = 9.2 Hz, 1H), 4.77–4.45 (m, 1H), 3.95 (s, 3H), 3.12–2.94 (m, 3H), 2.94–2.62 (m, 4H), 1.86 (s, 3H).
Acknowledgments
The authors thank the chemistry team members at WuXi AppTec for synthetic support, especially during the COVID-19 pandemic. Additionally, the authors thank the parallel medicinal chemistry (PMC) and purification service request (PSR) teams as well as compound management at Biogen, in particular Michael Dechantsreiter, Kevin Barry, Peter Vargo, and Sam Cziria for their contribution in delivering high-quality material.
Glossary
Abbreviations Used
- ADME
absorption, distribution metabolism and excretion
- BID
bis in die
- DDI
drug–drug interactions
- DIPEA
N,N-diisopropylethylamine
- EtOH
ethanol
- EtOAc
ethyl acetate
- HBD
hydrogen bond donor
- IVIVC
in vitro in vivo correlation
- KHMDS
potassium bis(trimethylsilyl)amide
- PPB
plasma protein binding
- tPSA
topological Polar Surface Area
- rt
room temperature
- TDI
time-dependent inhibition
- TFA
trifluoroacetic acid
- TO
target occupancy
- WB
whole blood
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00220.
Experimental procedures for the synthesis and characterization of intermediates and selected compounds, including LCMS and NMR spectra; ADME data for lead compounds, Bioseek data for lead compound 25; crystal structure of compounds 10, 25, and 27 bound to BTK (PDF)
Molecular formula strings (CSV)
Kinome selectivity (compounds 10, 25, and 27) (XLSX)
Funding was provided by Biogen, who employed all authors at the time this research was conducted.
The authors declare no competing financial interest.
Special Issue
Published as part of Journal of Medicinal Chemistryvirtual special issue “Exploring Covalent Modulators in Drug Discovery and Chemical Biology”.
Supplementary Material
References
- Harbo H. F.; Gold R.; Tintore M. Sex and gender issues in multiple sclerosis. Ther. Adv. Neurol. Disord. 2013, 6 (4), 237–248. 10.1177/1756285613488434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrie W.; Yang Y.; Irving-Pease E. K.; Attfield K. E.; Scorrano G.; Jensen L. T.; Armen A. P.; Dimopoulos E. A.; Stern A.; Refoyo-Martinez A.; Pearson A.; Ramsoe A.; Gaunitz C.; Demeter F.; Jorkov M. L. S.; M?ller S. B.; Springborg B.; Klassen L.; Hyldgard I. M.; Wickmann N.; Vinner L.; Korneliussen T. S.; Allentoft M. E.; Sikora M.; Kristiansen K.; Rodriguez S.; Nielsen R.; Iversen A. K. N.; Lawson D. J.; Fugger L.; Willerslev E. Elevated genetic risk for multiple sclerosis emerged in steppe pastoralist populations. Nature 2024, 625 (7994), 321–328. 10.1038/s41586-023-06618-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piehl F. Current and emerging disease-modulatory therapies and treatment targets for multiple sclerosis. J. Intern. Med. 2021, 289 (6), 771–791. 10.1111/joim.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch E. S.; Pretzsch R.; Weber M. S. A Milestone in Multiple Sclerosis Therapy: Monoclonal Antibodies Against CD20-Yet Progress Continues. Neurotherapeutics 2021, 18 (3), 1602–1622. 10.1007/s13311-021-01048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser S. L.; Bar-Or A.; Comi G.; Giovannoni G.; Hartung H. P.; Hemmer B.; Lublin F.; Montalban X.; Rammohan K. W.; Selmaj K.; Traboulsee A.; Wolinsky J. S.; Arnold D. L.; Klingelschmitt G.; Masterman D.; Fontoura P.; Belachew S.; Chin P.; Mairon N.; Garren H.; Kappos L. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376 (3), 221–234. 10.1056/NEJMoa1601277. [DOI] [PubMed] [Google Scholar]
- Roach C. A.; Cross A. H. Anti-CD20 B Cell Treatment for Relapsing Multiple Sclerosis. Front. Neurol. 2020, 11, 595547 10.3389/fneur.2020.595547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garjani A.; Patel S.; Bharkhada D.; Rashid W.; Coles A.; Law G. R.; Evangelou N. Impact of mass vaccination on SARS-CoV-2 infections among multiple sclerosis patients taking immunomodulatory disease-modifying therapies in England. Multiple Scler. Relat. Disord. 2022, 57, 103458 10.1016/j.msard.2021.103458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Wang L.; Shen L.; Tang K. Response of COVID-19 vaccination in multiple sclerosis patients following disease-modifying therapies: A meta-analysis. EBioMedicine 2022, 81, 104102 10.1016/j.ebiom.2022.104102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey A. E.; Ahmad F.. X-linked Agammaglobulinemia. In StatPearls; StatPearls Publishing LLC: Treasure Island (FL), 2023. [Google Scholar]
- de Rooij M. F. M.; Kuil A.; Geest C. R.; Eldering E.; Chang B. Y.; Buggy J. J.; Pals S. T.; Spaargaren M. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012, 119 (11), 2590–2594. 10.1182/blood-2011-11-390989. [DOI] [PubMed] [Google Scholar]
- Smith C. E.; Islam T. C.; Mattsson P. T.; Mohamed A. J.; Nore B. F.; Vihinen M. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. BioEssays 2001, 23 (5), 436–446. 10.1002/bies.1062. [DOI] [PubMed] [Google Scholar]
- Pellerin K.; Rubino S. J.; Burns J. C.; Smith B. A.; McCarl C. A.; Zhu J.; Jandreski L.; Cullen P.; Carlile T. M.; Li A.; Rebollar J. V.; Sybulski J.; Reynolds T. L.; Zhang B.; Basile R.; Tang H.; Harp C. P.; Pellerin A.; Silbereis J.; Franchimont N.; Cahir-McFarland E.; Ransohoff R. M.; Cameron T. O.; Mingueneau M. MOG autoantibodies trigger a tightly-controlled FcR and BTK-driven microglia proliferative response. Brain 2021, 144 (8), 2361–2374. 10.1093/brain/awab231. [DOI] [PubMed] [Google Scholar]
- Bame E.; Tang H.; Burns J. C.; Arefayene M.; Michelsen K.; Ma B.; Marx I.; Prince R.; Roach A. M.; Poreci U.; Donaldson D.; Cullen P.; Casey F.; Zhu J.; Carlile T. M.; Sangurdekar D.; Zhang B.; Trapa P.; Santoro J.; Muragan P.; Pellerin A.; Rubino S.; Gianni D.; Bajrami B.; Peng X.; Coppell A.; Riester K.; Belachew S.; Mehta D.; Palte M.; Hopkins B. T.; Scaramozza M.; Franchimont N.; Mingueneau M. Next-generation Bruton’s tyrosine kinase inhibitor BIIB091 selectively and potently inhibits B cell and Fc receptor signaling and downstream functions in B cells and myeloid cells. Clin. Transl. Immunol. 2021, 10 (6), e1295 10.1002/cti2.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torke S.; Weber M. S. Inhibition of Bruton s tyrosine kinase as a novel therapeutic approach in multiple sclerosis. Expert Opin. Invest. Drugs 2020, 29 (10), 1143–1150. 10.1080/13543784.2020.1807934. [DOI] [PubMed] [Google Scholar]
- Boike L.; Henning N. J.; Nomura D. K. Advances in covalent drug discovery. Nat. Rev. Drug Discovery 2022, 21 (12), 881–898. 10.1038/s41573-022-00542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer R. A. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discovery Today 2015, 20 (9), 1061–1073. 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Baillie T. A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem., Int. Ed. 2016, 55 (43), 13408–13421. 10.1002/anie.201601091. [DOI] [PubMed] [Google Scholar]
- Singh J. The Ascension of Targeted Covalent Inhibitors. J. Med. Chem. 2022, 65 (8), 5886–5901. 10.1021/acs.jmedchem.1c02134. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Gong H.; Meng F. Recent Advances in BTK Inhibitors for the Treatment of Inflammatory and Autoimmune Diseases. Molecules 2021, 26 (16), 4907. 10.3390/molecules26164907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringheim G. E.; Wampole M.; Oberoi K. Bruton’s Tyrosine Kinase (BTK) Inhibitors and Autoimmune Diseases: Making Sense of BTK Inhibitor Specificity Profiles and Recent Clinical Trial Successes and Failures. Front. Immunol. 2021, 12, 662223 10.3389/fimmu.2021.662223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geladaris A.; Torke S.; Weber M. S. Bruton’s Tyrosine Kinase Inhibitors in Multiple Sclerosis: Pioneering the Path Towards Treatment of Progression?. CNS Drugs 2022, 36 (10), 1019–1030. 10.1007/s40263-022-00951-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalban X.; Wallace D.; Genovese M. C.; Tomic D.; Parsons-Rich D.; Le Bolay C.; Kao A. H.; Guehring H. Characterisation of the safety profile of evobrutinib in over 1000 patients from phase II clinical trials in multiple sclerosis, rheumatoid arthritis and systemic lupus erythematosus: an integrated safety analysis. J. Neurol., Neurosurg. Psychiatry 2023, 94 (1), 1–9. 10.1136/jnnp-2022-328799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D. S.; Arnold D. L.; Vermersch P.; Bar-Or A.; Fox R. J.; Matta A.; Turner T.; Wallstrom E.; Zhang X.; Mares M.; Khabirov F. A.; Traboulsee A.; et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: a phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20 (9), 729–738. 10.1016/S1474-4422(21)00237-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi I.; Jeon D.; Moseman J. E.; Muralidhar A.; Potluri H. K.; McNeel D. G. Role of B cells as antigen presenting cells. Front. Immunol. 2022, 13, 954936 10.3389/fimmu.2022.954936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S. M.; Kim W. U. Targeted Immunotherapy for Autoimmune Disease. Immune Network 2022, 22 (1), e9 10.4110/in.2022.22.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama S.; Atsumi R.; Takakusa H.; Kobayashi Y.; Kurihara A.; Nagai Y.; Nakai D.; Okazaki O. A zone classification system for risk assessment of idiosyncratic drug toxicity using daily dose and covalent binding. Drug Metab. Dispos. 2009, 37 (9), 1970–1977. 10.1124/dmd.109.027797. [DOI] [PubMed] [Google Scholar]
- Atallah E.; Wijayasiri P.; Cianci N.; Abdullah K.; Mukherjee A.; Aithal G. P. Zanubrutinib-induced liver injury: a case report and literature review. BMC Gastroenterol. 2021, 21 (1), 244 10.1186/s12876-021-01825-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell R. D.; Qiu H.; Askew B. C.; Bender A. T.; Brugger N.; Camps M.; Dhanabal M.; Dutt V.; Eichhorn T.; Gardberg A. S.; Goutopoulos A.; Grenningloh R.; Head J.; Healey B.; Hodous B. L.; Huck B. R.; Johnson T. L.; Jones C.; Jones R. C.; Mochalkin I.; Morandi F.; Nguyen N.; Meyring M.; Potnick J. R.; Santos D. C.; Schmidt R.; Sherer B.; Shutes A.; Urbahns K.; Follis A. V.; Wegener A. A.; Zimmerli S. C.; Liu-Bujalski L. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J. Med. Chem. 2019, 62 (17), 7643–7655. 10.1021/acs.jmedchem.9b00794. [DOI] [PubMed] [Google Scholar]
- Paydas S. Management of adverse effects/toxicity of ibrutinib. Crit Rev. Oncol. Hematol. 2019, 136, 56–63. 10.1016/j.critrevonc.2019.02.001. [DOI] [PubMed] [Google Scholar]
- Crawford J. J.; Johnson A. R.; Misner D. L.; Belmont L. D.; Castanedo G.; Choy R.; Coraggio M.; Dong L.; Eigenbrot C.; Erickson R.; Ghilardi N.; Hau J.; Katewa A.; Kohli P. B.; Lee W.; Lubach J. W.; McKenzie B. S.; Ortwine D. F.; Schutt L.; Tay S.; Wei B.; Reif K.; Liu L.; Wong H.; Young W. B. Discovery of GDC-0853: A Potent, Selective, and Noncovalent Bruton’s Tyrosine Kinase Inhibitor in Early Clinical Development. J. Med. Chem. 2018, 61 (6), 2227–2245. 10.1021/acs.jmedchem.7b01712. [DOI] [PubMed] [Google Scholar]
- Kurosaki T. Molecular mechanisms in B cell antigen receptor signaling. Curr. Opin. Immunol. 1997, 9 (3), 309–318. 10.1016/S0952-7915(97)80075-1. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Chandrasekaran R. Y.; Hou X.; Troutman M. D.; Verhoest P. R.; Villalobos A.; Will Y. Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci. 2010, 1 (6), 420–434. 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1 (6), 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loryan I.; Reichel A.; Feng B.; Bundgaard C.; Shaffer C.; Kalvass C.; Bednarczyk D.; Morrison D.; Lesuisse D.; Hoppe E.; Terstappen G. C.; Fischer H.; Di L.; Colclough N.; Summerfield S.; Buckley S. T.; Maurer T. S.; Friden M. Unbound Brain-to-Plasma Partition Coefficient, K(p,uu,brain)-a Game Changing Parameter for CNS Drug Discovery and Development. Pharm. Res. 2022, 39 (7), 1321–1341. 10.1007/s11095-022-03246-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer M.; Berger W.; Gimenez-Arnau A.; Hayama K.; Jain V.; Reich A.; Haemmerle S.; Lheritier K.; Walsh P.; Xia S.; Storim J. Remibrutinib, a novel BTK inhibitor, demonstrates promising efficacy and safety in chronic spontaneous urticaria. J. Allergy Clin. Immunol. 2022, 150 (6), 1498–1506 e2. 10.1016/j.jaci.2022.08.027. [DOI] [PubMed] [Google Scholar]
- Hopkins B. T.; Bame E.; Bajrami B.; Black C.; Bohnert T.; Boiselle C.; Burdette D.; Burns J. C.; Delva L.; Donaldson D.; Grater R.; Gu C.; Hoemberger M.; Johnson J.; Kapadnis S.; King K.; Lulla M.; Ma B.; Marx I.; Magee T.; Meissner R.; Metrick C. M.; Mingueneau M.; Murugan P.; Otipoby K. L.; Polack E.; Poreci U.; Prince R.; Roach A. M.; Rowbottom C.; Santoro J. C.; Schroeder P.; Tang H.; Tien E.; Zhang F.; Lyssikatos J. Discovery and Preclinical Characterization of BIIB091, a Reversible, Selective BTK Inhibitor for the Treatment of Multiple Sclerosis. J. Med. Chem. 2022, 65 (2), 1206–1224. 10.1021/acs.jmedchem.1c00926. [DOI] [PubMed] [Google Scholar]
- Pajouhesh H.; Lenz G. R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2 (4), 541–553. 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson J. T.; Piquette-Miller M. Blood-brain barrier: an impediment to neuropharmaceuticals. Clin. Pharmacol. Ther. 2015, 97 (4), 308–313. 10.1002/cpt.77. [DOI] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H.; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lélias J.-M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23 (3), 329–336. 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26 (1), 127–132. 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Tonge P. J. Drug-Target Kinetics in Drug Discovery. ACS Chem. Neurosci. 2018, 9 (1), 29–39. 10.1021/acschemneuro.7b00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daryaee F.; Zhang Z.; Gogarty K. R.; Li Y.; Merino J.; Fisher S. L.; Tonge P. J. A quantitative mechanistic PK/PD model directly connects Btk target engagement and in vivo efficacy. Chem. Sci. 2017, 8 (5), 3434–3443. 10.1039/C6SC03306G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartzke D.; Fricker G. Establishment of optimized MDCK cell lines for reliable efflux transport studies. J. Pharm. Sci. 2014, 103 (4), 1298–1304. 10.1002/jps.23901. [DOI] [PubMed] [Google Scholar]
- Engdahl E.; van Schijndel M. D. M.; Voulgaris D.; Di Criscio M.; Ramsbottom K. A.; Rigden D. J.; Herland A.; Ruegg J. Bisphenol A Inhibits the Transporter Function of the Blood-Brain Barrier by Directly Interacting with the ABC Transporter Breast Cancer Resistance Protein (BCRP). Int. J. Mol. Sci. 2021, 22 (11), 5534. 10.3390/ijms22115534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montellano P. R. O. d. 1-Aminobenzotriazole: A Mechanism-Based Cytochrome P450 Inhibitor and Probe of Cytochrome P450 Biology. Med. Chem. 2018, 08 (3), 38–65. 10.4172/2161-0444.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potęga A. Glutathione-Mediated Conjugation of Anticancer Drugs: An Overview of Reaction Mechanisms and Biological Significance for Drug Detoxification and Bioactivation. Molecules 2022, 27 (16), 5252. 10.3390/molecules27165252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson S. H.; Liu Q.; Beaudoin Bertrand M.; Batt D. G.; Li L.; Pattoli M. A.; Skala S.; Cheng L.; Obermeier M. T.; Moore R.; Yang Z.; Vickery R.; Elzinga P. A.; Discenza L.; D’Arienzo C.; Gillooly K. M.; Taylor T. L.; Pulicicchio C.; Zhang Y.; Heimrich E.; McIntyre K. W.; Ruan Q.; Westhouse R. A.; Catlett I. M.; Zheng N.; Chaudhry C.; Dai J.; Galella M. A.; Tebben A. J.; Pokross M.; Li J.; Zhao R.; Smith D.; Rampulla R.; Allentoff A.; Wallace M. A.; Mathur A.; Salter-Cid L.; Macor J. E.; Carter P. H.; Fura A.; Burke J. R.; Tino J. A. Discovery of Branebrutinib (BMS-986195): A Strategy for Identifying a Highly Potent and Selective Covalent Inhibitor Providing Rapid in Vivo Inactivation of Bruton’s Tyrosine Kinase (BTK). J. Med. Chem. 2019, 62 (7), 3228–3250. 10.1021/acs.jmedchem.9b00167. [DOI] [PubMed] [Google Scholar]
- Barf T.; Covey T.; Izumi R.; van de Kar B.; Gulrajani M.; van Lith B.; van Hoek M.; de Zwart E.; Mittag D.; Demont D.; Verkaik S.; Krantz F.; Pearson P. G.; Ulrich R.; Kaptein A. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363 (2), 240–252. 10.1124/jpet.117.242909. [DOI] [PubMed] [Google Scholar]
- Huang F.; Han X.; Xiao X.; Zhou J. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules 2022, 27 (22), 7728. 10.3390/molecules27227728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cee V. J.; Volak L. P.; Chen Y.; Bartberger M. D.; Tegley C.; Arvedson T.; McCarter J.; Tasker A. S.; Fotsch C. Systematic Study of the Glutathione (GSH) Reactivity of N-Arylacrylamides: 1. Effects of Aryl Substitution. J. Med. Chem. 2015, 58 (23), 9171–9178. 10.1021/acs.jmedchem.5b01018. [DOI] [PubMed] [Google Scholar]
- Fang Z.; Song Y.; Zhan P.; Zhang Q.; Liu X. Conformational restriction: an effective tactic in ’follow-on’-based drug discovery. Future Med. Chem. 2014, 6 (8), 885–901. 10.4155/fmc.14.50. [DOI] [PubMed] [Google Scholar]
- Fallahi-Sichani M.; Honarnejad S.; Heiser L. M.; Gray J. W.; Sorger P. K. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat. Chem. Biol. 2013, 9 (11), 708–714. 10.1038/nchembio.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks E. A.; Galarza S.; Gencoglu M. F.; Cornelison R. C.; Munson J. M.; Peyton S. R. Applicability of drug response metrics for cancer studies using biomaterials. Philos. Trans. R. Soc., B 2019, 374 (1779), 20180226 10.1098/rstb.2018.0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S. Y.; Guan N.; Yan R.; Warner K.; Taylor S. D.; Meyer A. S. Measurement and models accounting for cell death capture hidden variation in compound response. Cell Death Dis. 2020, 11 (4), 255 10.1038/s41419-020-2462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach A. R.; Gillet V. J.; Lewis R. A.; Taylor R. Three-dimensional pharmacophore methods in drug discovery. J. Med. Chem. 2010, 53 (2), 539–558. 10.1021/jm900817u. [DOI] [PubMed] [Google Scholar]
- Aldeghi M.; Malhotra S.; Selwood D. L.; Chan A. W. Two- and three-dimensional rings in drugs. Chem. Biol. Drug Des. 2014, 83 (4), 450–461. 10.1111/cbdd.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohda S.; Sato T.; Kogoshi H.; Fu L.; Sakano S.; Nara N. Establishment of a novel B-cell lymphoma cell line with suppressed growth by gamma-secretase inhibitors. Leuk. Res. 2006, 30 (11), 1385–1390. 10.1016/j.leukres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Burgess J.; Colclough N.; Davies N. L.; Lenz E. M.; Orton A. L.; Ward R. A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf. Model. 2017, 57 (12), 3124–3137. 10.1021/acs.jcim.7b00553. [DOI] [PubMed] [Google Scholar]
- Péczka N.; Orgován Z.; Ábrányi-Balogh P.; Keserű G. M. Electrophilic warheads in covalent drug discovery: an overview. Expert Opin. Drug Discovery 2022, 17 (4), 413–422. 10.1080/17460441.2022.2034783. [DOI] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29 (11), 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Ghasoub R.; Albattah A.; Elazzazy S.; Alokka R.; Nemir A.; Alhijji I.; Taha R. Ibrutinib-associated sever skin toxicity: A case of multiple inflamed skin lesions and cellulitis in a 68-year-old male patient with relapsed chronic lymphocytic leukemia – Case report and literature review. J. Oncol. Pharm. Pract. 2020, 26 (2), 487–491. 10.1177/1078155219856422. [DOI] [PubMed] [Google Scholar]
- Dittus L.; Werner T.; Muelbaier M.; Bantscheff M. Differential Kinobeads Profiling for Target Identification of Irreversible Kinase Inhibitors. ACS Chem. Biol. 2017, 12 (10), 2515–2521. 10.1021/acschembio.7b00617. [DOI] [PubMed] [Google Scholar]
- Eberl H. C.; Werner T.; Reinhard F. B.; Lehmann S.; Thomson D.; Chen P.; Zhang C.; Rau C.; Muelbaier M.; Drewes G.; Drewry D.; Bantscheff M. Chemical proteomics reveals target selectivity of clinical Jak inhibitors in human primary cells. Sci. Rep. 2019, 9 (1), 14159 10.1038/s41598-019-50335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Jesson M. I.; Seneviratne U. I.; Lin T. H.; Sharif M. N.; Xue L.; Nguyen C.; Everley R. A.; Trujillo J. I.; Johnson D. S.; Point G. R.; Thorarensen A.; Kilty I.; Telliez J. B. PF-06651600, a Dual JAK3/TEC Family Kinase Inhibitor. ACS Chem. Biol. 2019, 14 (6), 1235–1242. 10.1021/acschembio.9b00188. [DOI] [PubMed] [Google Scholar]
- Petro J. B.; Rahman S. M.; Ballard D. W.; Khan W. N. Bruton’s tyrosine kinase is required for activation of IκB kinase and nuclear factor κB in response to B cell receptor engagement. J. Exp. Med. 2000, 191 (10), 1745–1754. 10.1084/jem.191.10.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casserly C. S.; Nantes J. C.; Whittaker Hawkins R. F.; Vallieres L. Neutrophil perversion in demyelinating autoimmune diseases: Mechanisms to medicine. Autoimmun. Rev. 2017, 16 (3), 294–307. 10.1016/j.autrev.2017.01.013. [DOI] [PubMed] [Google Scholar]
- Waschbisch A.; Schroder S.; Schraudner D.; Sammet L.; Weksler B.; Melms A.; Pfeifenbring S.; Stadelmann C.; Schwab S.; Linker R. A. Pivotal Role for CD16+ Monocytes in Immune Surveillance of the Central Nervous System. J. Immunol. 2016, 196 (4), 1558–1567. 10.4049/jimmunol.1501960. [DOI] [PubMed] [Google Scholar]
- Schäfer T.; Starkl P.; Allard C.; Wolf R. M.; Schweighoffer T. A granular variant of CD63 is a regulator of repeated human mast cell degranulation. Allergy 2010, 65 (10), 1242–1255. 10.1111/j.1398-9995.2010.02350.x. [DOI] [PubMed] [Google Scholar]
- Palanichamy A.; Apeltsin L.; Kuo T. C.; Sirota M.; Wang S.; Pitts S. J.; Sundar P. D.; Telman D.; Zhao L. Z.; Derstine M.; Abounasr A.; Hauser S. L.; von Büdingen H. C. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci. Transl. Med. 2014, 6 (248), 248ra106 10.1126/scitranslmed.3008930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg E. L.; O’Mahony A.. Complex Primary Human Cell Systems for Drug Discovery. In Human-based Systems for Translational Research; Coleman R., Ed.; The Royal Society of Chemistry, 2014. [Google Scholar]
- Trapa P. E.; Matthew D. T.; Thomas Y. L.; Travis T. W.; Tristan S. M.; Nandini C. P.; Mark A. W.; John P. U.; Anthony A. C.; Bo F.; Jennifer L. L. In Vitro–In Vivo Extrapolation of Key Transporter Activity at the Blood–Brain Barrier. Drug Metab. Dispos. 2019, 47 (4), 405. 10.1124/dmd.118.083279. [DOI] [PubMed] [Google Scholar]
- Becker A.; Martin E. C.; Mitchell D. Y.; Grenningloh R.; Bender A. T.; Laurent J.; Mackenzie H.; Johne A. Safety, Tolerability, Pharmacokinetics, Target Occupancy, and Concentration-QT Analysis of the Novel BTK Inhibitor Evobrutinib in Healthy Volunteers. Clin. Transl. Sci. 2020, 13 (2), 325–336. 10.1111/cts.12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. K.; Xing J.; Catlett I. M.; Adamczyk R.; Griffies A.; Liu A.; Murthy B.; Nowak M. Safety, pharmacokinetics, and pharmacodynamics of BMS-986142, a novel reversible BTK inhibitor, in healthy participants. Eur. J. Clin. Pharmacol. 2017, 73 (6), 689–698. 10.1007/s00228-017-2226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb D. J.; Wollin S. L.; Schnapp A.; Bischoff D.; Erb K. J.; Bouyssou T.; Guilliard B.; Strasser C.; Wex E.; Blum S.; Thaler E.; Nickel H.; Radmacher O.; Haas H.; Swantek J. L.; Souza D.; Canfield M.; White D.; Panzenbeck M.; Kashem M. A.; Sanville-Ross M.; Kono T.; Sewald K.; Braun A.; Obernolte H.; Danov O.; Schaenzle G.; Rast G.; Maier G.-M.; Hoffmann M. BI 1002494, a Novel Potent and Selective Oral Spleen Tyrosine Kinase Inhibitor, Displays Differential Potency in Human Basophils and B Cells. J. Pharmacol. Exp. Ther. 2016, 357 (3), 554–561. 10.1124/jpet.116.233155. [DOI] [PubMed] [Google Scholar]
- Williamson A. Ueber die Theorie der Aetherbildung. Justus Liebigs Ann. Chem. 1851, 77 (1), 37–49. 10.1002/jlac.18510770104. [DOI] [Google Scholar]
- Suzuki A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576 (1), 147–168. 10.1016/S0022-328X(98)01055-9. [DOI] [Google Scholar]
- Schönherr H.; Cernak T. Profound Methyl Effects in Drug Discovery and a Call for New C-H Methylation Reactions. Angew. Chem., Int. Ed. 2013, 52 (47), 12256–12267. 10.1002/anie.201303207. [DOI] [PubMed] [Google Scholar]
- Dahal U. P.; Obach R. S.; Gilbert A. M. Benchmarking in Vitro Covalent Binding Burden As a Tool To Assess Potential Toxicity Caused by Nonspecific Covalent Binding of Covalent Drugs. Chem. Res. Toxicol. 2013, 26 (11), 1739–1745. 10.1021/tx400301q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.