

Abstract
Current cancer vaccines using T cell epitopes activate antitumor T cell immunity through dendritic cell/macrophage-mediated antigen presentation, but they lack the ability to promote B/CD4 T cell crosstalk, limiting their anticancer efficacy. We developed antigen-clustered nanovaccine (ACNVax) to achieve long-term tumor remission by promoting B/CD4 T cell crosstalk. The topographic features of ACNVax were achieved using an iron nanoparticle core attached with an optimal number of gold nanoparticles, where the clusters of HER2 B/CD4 T cell epitopes were conjugated on the gold surface with an optimal intercluster distance of 5–10 nm. ACNVax effectively trafficked to lymph nodes and cross-linked with BCR, which are essential for stimulating B cell antigen presentation-mediated B/CD4 T cell crosstalk in vitro and in vivo. ACNVax, combined with anti-PD-1, achieved long-term tumor remission (>200 days) with 80% complete response in mice with HER2+ breast cancer. ACNVax not only remodeled the tumor immune microenvironment but also induced a long-term immune memory, as evidenced by complete rejection of tumor rechallenge and a high level of antigen-specific memory B, CD4, and CD8 cells in mice (>200 days). This study provides a cancer vaccine design strategy, using B/CD4 T cell epitopes in an antigen clustered topography, to achieve long-term durable anticancer efficacy through promoting B/CD4 T cell crosstalk.
Keywords: Cancer vaccine, B cell antigen presentation, B/CD4 T cell crosstalk, tumor immune microenvironment, anti-PD-1/PD-L1 immunotherapy, long-term antigen-specific memory B cells and T cells, Tertiary lymphoid structures
Graphical Abstract
Current anticancer vaccines are designed to activate T cell immunity against tumor-associated antigens (TAAs) or neoantigens,1–3 which have achieved promising clinical anticancer efficacy in patients with certain cancer types, such as melanoma.4–12 However, these vaccines have shown only limited efficacy in improving overall survival in patients with various other cancer types, although they successfully activated T cell immunity.1–3,5,6,13–19
Typically, a vaccine against infectious diseases needs to stimulate both B cell and T cell immunity in a healthy body to generate long-lasting efficacy.20–24 However, current cancer therapeutic vaccines using T cell epitopes are not designed to activate B cell immunity because the role of B cell immunity in cancer vaccines has been a subject of debate for several decades.25–33 In fact, if the purpose of a cancer vaccine is solely to activate B cell immunity for antibody secretion,34 it will neither generate durable anticancer efficacy nor have a clinical need, since monoclonal antibody therapy is readily available for cancer treatment. In addition, there is controversy over whether activation of B cell immunity may have protumor activity, although recent studies suggest this may be related to regulatory B cells rather than other forms of B cell activations.25–33
However, the latest clinical studies suggest B cell activation is critical for achieving durable anticancer efficacy in patients using immune checkpoint blockade (ICB) therapy.35–39 B/CD4 T cell crosstalk, especially B cell and follicular T cell (Tfh) crosstalk, is critical to drive anticancer immunity and is associated with the clinical response to ICB therapy.35–45 B cell activation in tumors improves antigen presentation, driving the expansion of tumor-specific T cells for anticncer efficacy. Activated B cells in tumors also secrete inflammatory cytokines (including TNF, IL-2, IL-6, and IFNγ) to activate and recruit other immune effector cells (e.g., CD 4 and CD 8 T cells) to promote antitumor immunity.35,37,41,42 More importantly, B cell activation may be required to achieve long-term T cell memory during immunotherapy.46–49 Furthermore, when B cell tumor infiltration is present in a tertiary lymphoid structure (TLS) structure, which are organized lymphoid aggregates consisting of mainly B cells and CD4 T cells, it tends to show favorable clinical repsonse to ICB therapy.35–45
Since the presence of B cell immunity is correlated with enhanced long-term survival rates in patients treated with anti-PD-1 therapy,35–39 it is imperative to investigate whether incorporating B cell immunity to enhance B cell antigen presentation-mediated B/CD4 T cell crosstalk in current cancer vaccine designs can improve long-term efficacy. However, current cancer vaccines using T cell epitopes (without B cell epitopes) only activate T cell immunity through dendritic cell/macrophage-mediated antigen presentation.19,50 Furthermore, current vaccine delivery systems using nanoparticle encapsulated antigen inside or simply mixing antigen with adjuvant are unable to bind and cross-link with B cell receptor (BCR), and thus they are unable to activate B cell receptor-mediated antigen uptake, processing, and presentation to CD4 T cells.51
To design a nanovaccine for promoting B cell antigen presentation-mediated B/CD4 T cell crosstalk,52,53 three critical challenges must be addressed.52,53 First, the vaccine needs to efficiently penetrate the lymph nodes and directly engage with B cells. While nanoparticles smaller than 200 nm can enter the lymph nodes, they tend to accumulate in the subcapsular sinus macrophages positioned above the B-cell follicles, rather than reaching the follicles themselves.54 Second, the antigens must be displayed on the nanoparticle surface with a high antigen density comparable to immunogenic viruses in order to induce robust B cell activation.55 Third, repetitive antigens in a cluster with an optimal distance between clusters is crucial for efficient cross-linking with the BCR.56–58 This cross-linking triggers effective internalization of the antigen, decreases the threshold required to activate the BCR, and overcomes tolerance in antitumor B cell immunity.59–61 Unfortunately, the commonly used nanoplatforms (<200 nm) with peptide epitopes uniformly distributed on the surface cannot achieve the optimal distance of 5–10 nm62 required for effective BCR cross-linking while maintaining a high antigen density simultaneously.63 Several studies have demonstrated that antigen cluster structures, such as a glycoprotein on virus-like nanoparticles,62,64 with an ideal spacing between antigen clusters, can induce a high level of BCR cross-linking while preserving a high surface antigen density. However, such nanoplatforms with antigen cluster structure have not yet been realized in peptide nanovaccine design.
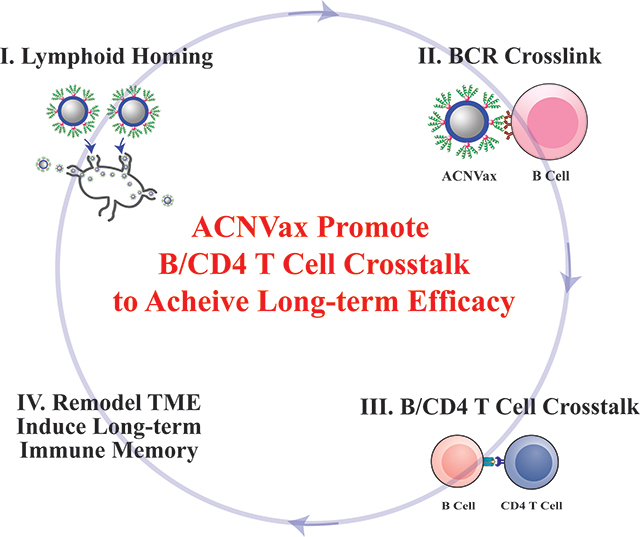
In this study, we designed a nanovaccine using HER2 B/CD4 T cell epitopes in an antigen-cluster topography (ACNVax) to achieve long-term tumor remission by promoting B cell antigen presentation-mediated B/CD4 T cell crosstalk We engineered the ACNVax with an antigen-cluster topography on its surface, including a high density of HER2 B/CD4 cell epitopes and optimal distance (5–10 nm) between two antigen clusters. These topographic features of ACNVax promote lymph node trafficking and cross-linking with BCR,65 which are essential for stimulating B cell antigen presentation-mediated B/CD4 T cell crosstalk in vitro and in vivo.50,66 To further stimulate TLS formation tumors, we also generated ACNVax with LIGHT, a member of the tumor necrosis factor (TNF) ligand family tested previously reported to promote TLS formation.67,68 We tested ACNVax/LIGHT in combination with anti-PD-1 antibody to achieve long-term tumor remission (>200 days) with a complete response (CR) rate of 80% in mice with HER2 breast cancer. We used CyTOF and single-cell RNA-seq to evaluate how ACNVax/LIGHT remodeled the tumor immune microenvironment. Finally, we monitored how ACNVax/LIGHT generates long-term antigen-specific B/CD4/CD8 T cell immune memory for its long-term efficacy. Our data provide a cancer vaccine design strategy, using B/CD4 T cell epitopes in an antigen clustered topography, to achieve long-term durable anticancer efficacy through promoting B cell antigen presentation-mediated B/CD4 T cell crosstalk and remodeling the tumor immune microenvironment (Scheme 1).
Scheme 1. Antigen-Clustered Nanovaccine (ACNVax), Combined with anti-PD-1, Achieves Long-Term Tumor Remission by Promoting B Cell Antigen Presentation-Mediated B/CD4 T Cell Crosstalka.

aACNVax efficiently penetrates the lymph node, crosslinks with B cell receptor (BCR), and promotes B cell antigen presentation-mediated B/CD4 T cell crosstalk. ACNVax vaccination, in combination with anti-PD-1, achieved long-term tumor remission through remodeling immune microenvironment with increased memory B/CD4/CD8 immunity.
RESULTS/DISCUSSION
Engineer ACNVax with Clustered-Antigen Topography to Effectively Bind/Cross-Link with B Cell Receptor (BCR).
To prepare a nanoparticle with clustered antigen topography for efficient BCR cross-linking, we first prepared the iron nanoparticle core coated with poly(siloxane)- and poly(ethylene glycol)-containing diblock copolymer (IONP-Polymer, 15 nm), as we reported previously69–71 (Figure S1a). Ultrasmall gold nanoparticles (AuNPs) (2 nm) were then attached to the surface of IONP-Polymer, which is used to form an antigen-clustered nanoparticle (ACN) (Figure S1b). The antigen-duster topography of ACNVax was achieved by conjugating antigen peptides with cysteine to the surface AuNPs of the ACN through a thiol-Au reaction (Figure 1a,b).69–71 The human HER2 CD4 T cell and B cell epitope (CDDD-PESFDGDPASNTAPLQPEQLQ-GGK) was used as the antigen to promote T cell-dependent B cell activation.72–74
Figure 1.

Engneering ACNVax with antigen-clustered topography to effectively cross-link with B cell receptor (BCR). (a) Size of the ACN and ACN with antigen (ACNVax) by dynamic light scattering (DLS) analysis. (b) Scanning transmission electron microscopy (STEM) high-angle annular dark-field (HAADF) images of ACN with different ratios of AuNP/IONP from 0–30%; scale bar is 50 nm at 0% condition; scalebar is 20 nm at 5–30% conditions. STEM image of a single ACNVax (bottom right of b). (c) Number of surface AuNPs per ACN. (d) Distances between AuNPs on ACN surfaces when different Au:Fe ratios were used to generate the ACN as calculated by mathematical modeling (Supporting Methods, Figure S1, and Tab1es S1–S4). (e) Peptide loading of ACN standardized on a per gold nanoparticle basis (R = 0.95). (f) Number of peptides on ACNVax with variable AuNP number (0 AuNP, black; 4 AuNPs, blue; 12 AuNPs, red) as determined by a modified fluorescamine fluorescence detection assay. Data for quantification are shown as mean ± SD, n = 3. (g) Schematic illustration of B cell receptor cross-link experiments and results as shown in panels h and i. (h) Confocal image of Cy3 and hapten labeled ACNVax (red, 20 nM antigens) binding/cross-linking (yellow) with B cell receptor (antibody staining, green) in hapten-specific B cells from QM mice splenocytes, compared with other control groups (20 nM antigens): Cy3 and hapten labeled CD4/B antigen (PepVax), Cy3 and hapten labeled IONPVax, Cy3 and hapten labeled AuNPVax, Cy3 and hapten labeled ACNVax with longer distance (~15 nm) between clusters (ACNVax-LC), and Cy3 and hapten labeled ACNVax with low density of antigen (2% of peptide loading, ACNVax-LD). Blue, phalloidin stain of actin filaments; green, B cell receptor staining using Alexa Fluor 488-AffniPure Fab Fragment Goat Anti-Mouse IgM (μ Chain Specific) antibody; red: Cy3-labeled CD4/B-hapten epitope. The scale bar is 2.5 μm. (i) Quantification of cross-linking signals from panel h. Statistical comparisons were conducted between ACNVax versus all other groups. Statistical comparisons are based on one-way ANOVA, followed by post hoc Tukey’s pairwise comparisons or by Student’s unpaired t test. The asterisks denote statistical significance at the level of **** p < 0.0001. ANOVA, analysis of variance; SD, standard deviation; n.s., no statistical significance.
To control the optimal distance between antigen clusters on ACNVax at 5–10 nm, which is an ideal distance for BCR cross-linking56,57 we adjusted the ratio of AuNP to IONP-Polymer and calculated the distances between AuNPs on the ACNVax surface based on two methodologies (the arc length equation for a circle and the triangulation equation). The weight ratios of AuNP to IONP-Polymer at 0–30% generated ACNVax with 0–12 AuNP on each IONP-Polymer surface which correlated to 5–20 nm distances between antigen clusters on ACN (Figures 1b–d, S1c–e, and Tables S1–S4).
To achieve a high antigen density, we adjusted the peptide to ACN (calculated by Fe) weight ratio. The maximum peptide loadings were 232 ± 73 (0 AuNPs), 888 ± 42 (4 AuNPs), and 1954 ± 157 (12 AuNPs) peptides per ACNVax (Figure 1e,f). The nonspecific peptide binding to IONP-Polymer surface (without AuNP) was ~12% (Figure 1e,f), suggesting that most peptides were conjugated on the AuNPs of ACNVax but not on the IONP-Polymer core, which provided the antigen cluster topography.
The ACNVax was optimized with overall antigen density (~2000 peptides/ACN) and distance (~5 nm) between antigen dusters (12 AuNPs per ACN), which equates to a peptide density of 20,000–25,000 per μm2 (Figure S1f), comparable to the antigen density reported for virus-like particles (e.g., hepatitis B virus, ~20,OOO antigens). ACNVax with conjugated antigen was 44 ± 2 nm by dynamic light scattering (DLS) (Figure 1a) with a polydispersity index (PDI) of 0.087 ± 0.03 and a zeta potential of −16 mV ± 4 (Table S5). This size and surface charge are optimal for efficient lymph node draining and penetration.53 ACNVax was stable under in vivo relevant serum conditions (Figure S2a).
The high density of B cell epitopes cross-links BCRs to trigger strong B cell-mediated antigen uptake.59–61 To test if ACNVax facilitates BCR cross-linking, we used hapten-specific B cells from the spleen of QM transgenic mice to incubate with Cy3- and hapten-labeled ACNVax where hapten was conjugated at die end of HER2 B cell epitope.75 The results showed that hapten-labeled ACNVax (red) efficiently bound to BCRs (stained with FITC-labeled antibody, green) on the hapten-specific B cells from 1 to 5 min to generate cross-linked BCR clusters on the B cell surface (Figures 1g and S3).76
To investigate if the antigen-clustered topogaphy of ACNVax is indeed critical for BCR cross-linking, we prepared different control groups using the same HER2 B/CD4 antigen and labeled with hapten and Cy3: (a) IONPVax using the same IONP core and surface conjugation with similar overall density of B/CD4 antigen and similar overall size, but without antigen cluster mimicry (2323 ± 394 peptides per IONPVax, 48 ± 5 nm, PDI: 0.074 ± 0.02) (Figure S2b–d and Table S6); (b) AuNPVax using the same surface AuNPs (2 nm) and same B/CD4 antigen, but without antigen cluster mimicry; (c) ACNVax-LC with longer antigen cluster distance (~20 nm between antigen cluster); (d) ACNVax-LD with lower peptide density (2% of peptide loading); (e) soluble peptide (PepVax); (f) lipoVax (the liposome, prepared according to previous reports,63 was modified to have the same antigen uniformly conjugated on its surface with a similar overall density, but without antigen cluster topography). The image and quantification data showed that all these groups had significantly less BCR cross-link compared to ACNVax (Figures 1h,i and S3). Specifically, ACNVax with longer antigen cluster distance and lower peptide density reduced BCR cross-link significantly compared to ACNVax with antigen density (150 peptides per AuNP, 2000 peptides/ACN) and distance (~5 nm) between antigen clusters (12 AuNPs per ACN). These data suggest that antigen cluster topography is critical to achieving highly efficient BCR cross-link. In addition, we also showed that longer distance between antigen clusters and/or low peptide density of ACNVax particles decreased the production of HER2 specific IgG, indicating a lower level of B cell activation (Figure S1g).
ACNVax Promoted B Cell Antigen Presentation-Mediated B/CD4 T Cell Crosstalk.
To test if BCR cross-linking by ACNVax promotes B cell antigen presentation, we compared the in vitro B cell uptake of antigen following incubation of hapten-specific B cells with ACNVax (hapten), IONPVax (hapten), or with PepVax (hapten) for 15, 30, and 60 min (Figure S4). Results showed that more ACNVax was internalized by B cells than IONPVax and PepVax, which suggests that antigen-clustered topography of ACNVax increases uptake.
To study if ACNVax could promote B cell antigen presentation mediated B/CD4 T cell crosstalk, we engineered ACNVax with HER2-CD4/B epitope-hapten and OT-II CD4 epitope (chicken ovalbumin323–339), incubated with hapten-specific B cells from the splenocytes of QM mice75 (B cells labeled with CFSE tracker), then mixed with OT-II specific CD4 T cells from the spleen of OT-II transgenic mice (labeled with CFSE tracker)77 for 24–96 h (Figures 2a S5). IONPVax and PepVax with the same antigens were used for comparison.
Figure 2.

ACNVax promoted B cell antigen presentation to CD4 T cells resulting in antigen-specific T cell activation and proliferation. (a) Diagram of experimental design. ACN with HER2-B/CD4 antigen-hapten and OT-II CD4 epitope was incubated with splenocytes from QM mice (with a portion of B cells labeled with CFSE) and OT-II specific CD4 T cells (labeled with CFSE) from the splenocytes of OT-II transgenic mice. IONPVax and PepVax with the same amount of antigen as ACNVax particles were used as controls. (b) Representative flow cytometry analysis and quantification (96 h) of hapten-specific B cell activation by measuring geometric mean intensity of CD86 marker. (c) Representative flow cytometry analysis and quantification (96 h) of hapten-specific B cell proliferation, measured by the percentage of decrease in CFSE+ hapten-specific B cells compared to controls. (d) Representative flow cytome&y analysis and quantification (96 h) of OT-II specific CD4 T cell activation with B cell incubation by measuring geometric mean intensity of CD69 marker. (e) Representative flow cytometry analysis and quantification (96 h) of OT-II specific CD4 T cell proliferation with B cell incubation by measuring the percentage of decreased CFSE+ OT-II specific CD4 T cells compared to control. (f) Representative flow cytometry analysis and quantification (96 h) of OT-II specific CD4 T cell activation without B cell incubation, measured by the geometric mean intensity of CD69 marker. (g) Representative flow cytometry analysis and quantification (96 h) of OT-II specific CD4 T cell proliferation without B cell incubation were measured by measuring the percentage of decreased CFSE+ OT-II specific CD4 T cells compared to control. Data for quantification are shown as mean ± SD, n = 3. For panels b–g, statistical comparisons were conducted among ACNVax and LPS with other groups. Statistical comparisons are based on one-way ANOVA, followed by post hoc Tukey’s pairwise comparisons or by Student’s unpaired t test. The asterisks denote statistical significance at the level of * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. ANOVA, analysis of variance; SD, standard deviation; n.s., no statistical significance.
To test if ACNVax could activate antigen-specific B cells, we monitored the activation and proliferation of hapten-specific B cells. The results showed that ACNVax (with HER2-CD4/B antigen-hapten and OT-II CD4 epitope) increased the activation of hapten-specific B cell by 3.5-fold (as monitored by CD86 and CD69 intensity in hapten-specific B cell; Figures 2b and S6) and proliferation of hapten-specific B cell by 32.7-fold (as monitored by CFSE staining) after 96 h (Figures 2c and S7) compared to other control groups with the same antigens. These data suggest ACNVax could effectively activate antigen-specific B cells.
To test if these B cells activated by ACNVax could present CD4 T cell epitope to activate antigen-specific CD4 T cells, we monitored the activation and proliferation of OT-II specific CD4 T cells. The results showed that ACNVax (with HER2-CD4/B antigen-hapten and OT-II CD4 epitope) increased activation of OT-II-specific CD4 T cell by 28-fold (as monitored by CD69 and CD25 after 96 h) and proliferation by 90-fold after 96 h compared to other control groups with the same antigens (Figures 2d,e, S8, and S9). To confirm that ACNVax did not enhance nonspecific CD4 T cell activation, we also monitored CD4 T cell activation from splenocytes of QM mice (not specific to OT-II CD4 epitope). The data showed that ACN (with HER2-CD4/B antigen-hapten and OT-II CD4 epitope) did not activate nonspecific CD4 T cells from QM mice (Figure S10). These data suggested that the T cell epitope on ACNVax was efficiently processed by B cells and presented to activate antigen specific CD4 T cells.
Furthermore, to confirm if ACNVax induced B/CD4 T cell crosstalk owing to B cell antigen presentation but not owing to other antigen presenting cells such as dendritic cells or macrophages, we depleted B cells from splenocytes of QM using CD19 positive selection kit and then coincubated with OT-II-specific CD4 T cells from OT-II transgenic mice (Figure S5). The results showed that depletion of B cells impaired ACNVax activation of OT-II-specific CD4 T cells (Figures 2f,g and S11–S13). These suggest that B cell and CD4 T cell crosstalk by ACNVax is indeed mediated by B cell antigen presentation.
ACNVax Efficiently Penetrated into Lymph Node and Induced a Robust Tfh-Dependent B Cell Response.
A robust Tfh-dependent B cell response requires efficient delivery of antigen to lymph nodes to encounter B cells We employed 2D and 3D confocal fluorescent imaging to assess the homing efficiency of ACNVax to the draining lymph nodes. The results showed that FITC-labeled ACNVax penetrated deep inside lymph nodes where B cells reside in both B cell zone and cortex (Figures 3a and S14 and Movies S1–S6). As a comparison, IONPVax without antigen-clustered topography was mainly localized on the surface of the lymph nodes (Figure S14 and Movies S7–S12). We also tested antigen uptake by B cells in the lymph nodes in mice by IVIS following injection (s.c.) (Figure S15a,b). Results showed that more ACNVax was internalized by B cells than other control groups, which suggests that antigen-clustered topography of ACNVax increases B cell uptake.
Figure 3.

ACNVax penetrated efficiently into the lymph node and induced a robust Tfh cell-supported germinal center (GC) response in vivo. (a) Confocal imaging of ACNVax penetration into lymph nodes. Scale bar is 200 μm in whole-lymph-node images and 50 μm in magnified images. (b) CyTOF analysis of immune cells from lymph nodes (n = 3). SPADE analysis. Node sizes indicate absolute number of cells. Nodes are colored based on the log ratio of the relative number of immune cells in the ACNVax group to that in the PepVax or IONPVax group in the same lymph node. Red indicates a higher relative number of immune cells in the ACNVax group, and blue indicates a lower relative number. (c) Quantification of GC B cells and Tfh cells by CyTOF. CyTOF markers for each immune cell population are shown in Figure S16. (d–f) GC B cells, antigen-specific GC B cells, and Tfh cells in the lymph nodes after three vaccinations in BALB/c mice using control (PBS), HER2-B/CD4 peptide, IONPVax, or ACNVax (14.6 nmol antigen and 13.9 nmol 2′3′-cGAMP as adjuvant) at days 0, 7, and 14 and analyzed at day 24. (d and e) Representative flow cytometry analysis and quantification of germinal center B cells (d) and HER2-specific germinal center B-cells (e) in lymph nodes using B-cell receptor tetramer staining. CD3−B220+ CD9S+ GL-7+ populations were identified as GC B cells. Data for quantification are shown as mean ± SD, n = 3. (f) Flow cytometry quantification of Tfh cells in lymph nodes. B220−CD4+CXCRS+PD-1+ populations were identified as Tfh cells. (g) Diagram of experimental design for activation-induced markers assay (AIM) for measuring antigen specific Tfh and antigen experienced CD4 T cells. (h and i) Representative flow cytometry analysis and quantification of AIM+ Tfh cells (h) and AIM+ antigen experienced CD4 T cells (i). B220−CD4+CXCRS+PD-1+ populations were identified as Tfh cells. CD69+CD40L+/− populations from Tfh cells were identified as AIM+ Tfh cells. B220−TD4+CD62L+CD69+CD40L+/− populations were identified as AIM+ antigen experienced CD4 T cells. Data for quantification are shown as mean ± SD, n = 3. Statistical comparisons were conducted between ACNVax and IONPVax, PepVax, and control groups. Statistical comparisons are based on one-way ANOVA, followed by post hoc Tukey’s pairwise comparisons or by Student’s unpaired t test. The asterisks denote statistical significance at the level of * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. ANOVA, analysis of variance; SD, standard deviation.
To study how ACNVax stimulates the immune responses in vivo, we used CyTOF to investigate immune cell composition in the draining lymph nodes of normal BALB/c mice 10 days after three vaccinations (Table S7 and Figure S16).78 Global analysis using spanning-tree progression analysis of density-normalized (SPADE) confirmed that ACNVax induced a higher level of GC B, Tfh, and plasma cells in the lymph nodes and HER2 Specific antibody titer serum than IONPVax and PepVax groups (Figure 3b,c), whereas CD8 T cell frequency was not changed after vaccination (Figure 3b). This result ACNVax induced a robust Tfh-supported GC response.
To confirm the results by flow cytometry and further evaluate the antigen specific GC B and Tfh cells, we enumerated GC B cells and CD4 T cells in the lymph nodes of BALB/c mice after three vaccinations with ACNVax (at day 24) and in comparison with mice vaccinated with IONPVax or with PepVax of the same antigens. Flow cytometry analysis showed that ACNVax generated a robust GC B cell response (16.21% of all B cells) and antigen-specific GC B cells (7.6% of all GC B cells), which is 2- to 5-fold higher than PepVax or IONPVax groups (Figures 3d,e and S17a,b) ACNVax vaccinations also increased Tfh cells by 4.7-fold and 3-fold compared to PepVax and IONPVax (Figures 3f and S17c).
To determine if ACNVax vaccination activated antigen-specific Tfh cells, we used the activation-induced markers (AIM) assay to measure antigen-specific Tfh cell activation. ACNVax vaccination increased the frequency of antigen-specific Tfh cells by 4.2-fold (Figures 3g,h and S18) and the frequency of antigen-specific CD4 T cells by 4.5-fold (Figures 3g,i and S19) compared to IONPVax and PepVax vaccinations.
Since robust GC responses generate robust antigen-specific IgG, we examined the HER2-specific IgG in serum after three vaccinations on day 38. ACNVax vaccination generated a 4-fold higher HER2-specific IgG titer, a 3-fold higher HER2-specific IgG1 titer, and a 5-fold higher HER2-specific IgG2a titer than vaccination with IONPVax (Figures 3c and S20a–d) The HER2 antibody specificity was confirmed by testing antibody binding to D2F2/E2 HER2+ cell line by flow cytometry (Figure S20e–g). These data suggest that ACNVax with the antigen cluster structure uniquely enhanced the Tfh-dependent B cell response and antigen-specific IgG production comparison with nanoparticles the same core but uniformly distributed antigen on their surface (IONPVax) or PepVax.
ACNVax Combined with anti-PD-1 Antibody Achieved Long-Term Remission of HER2+ Breast Cancer.
To compare the efficacy of ACNVax combined with anti-PD-1 antibody to current clinically approved treatment options for HER2+ cancer, we used three vaccinations (day 4, 11, 18) in BALB/c mice with subcutaneous HER2+ breast cancer using HER2 overexpression mouse cancer cell line (D2F2/E2).79 The results showed ACNVax plus anti-PD-1 had much better tumor inhibition effect compared to murine HBR2 antibody (Table S8) plus anti-PD-1, clinically tested Neu-Vax vaccine (composed of E75 as HER2 CD8 epitope and GM-CSF) plus anti-PD-1, and anti-PD-1 alone (Figure 4a).80–83
Figure 4.

ACNVax combined with anti-PD-1 antibody achieved long-term remission of HER2+ breast cancer. A HER2 breast cancer model was established by inoculating BALB/c mice s.c. with 2.5 × 105 D2F2/E2 mouse mammary tumor cells encoding human ErbB-2 (HER2). Four days after tumor inoculation, mice were treated with different vaccine formulations in combination with anti-PD-1 antibody. For each vaccine, we used 14.6 nmol of HER2 CD4 and B cell epitope ((CDDD-PESFDGDPASNTAPLQPEQLQ-(GGK)) with 13.9 nmol 2′3′-cGAMP as the adjuvant. ACNVax-T had the same ACN but conjugated with the HER2 CD8 T cell epitope (E75). (a) Antitumor efficacy of ACNVax clinical for treatment HER2 breast cancer. Average tumor volume changes of HER2 breast cancer mice after treatment with PBS (control), anti-PD-1 antibody (100 μg), murine HER2 antibody (100 μg, Table S8) plus any-PD-1 antibody (100 μg), NeuVax (E75, 14.6 nmol combined with GM-CSF, 5 μg) plus and-PD-1 antibody (100 μg) or ACNVax (14.6 nmol HER2 epitope, 13.9 nmol 2′3′-cGAMP) plus anti-PD-1 antibody (100 μg). The data represent the mean ± SD; n = 7 for the control, anti-PD-1, HER2 antibody plus anti-PD-1 and NeuVax plus anti-PD-1 groups; n = 8 for the ACNVaxplus anti-PD-1. (b) Antitumor efficacy of ACNVax compared with different nanovaccine after three vaccinations. IONPVax: core component of ACN (35 nm) with antigen uniformly conjugated on the nanoparticle surface; AuNPVax: the surface single antigen cluster component of ACN (2–5 nm); and LipoVax: liposome with antigen uniformly conjugated on the nanoparticle surface. For each vaccine, we used 14.6 nmol of epitopes with 13.9 nmol of 2′3′-cGAMP as the adjuvant. n = 7 for each group. (c) Antitumor efficacy of ACNVax with LIGHT (50 ng) plus anti-PD-1 antibody compared with other vaccine groups. Data represent mean ± SD, n = 8 for the control, PepVax + anti-PD-1, IONPVax + anti-PD-1, ACNVax-T + anti-PD-1, and PepVax/LIGHT + anti-PD-1 groups; n = 9 for the ACNVax + anti-PD-1 and ACNVax&LIGHT + anti-PD-1 groups. (d) Antitumor efficacy of ACNVax LIGHT (50 ng) plus anti-PD-1 compared with IONPVax with LIGHT plus anti-PD-1 after five vaccinations. Data represent the mean ± SD, n = 10 for all groups. Statistical comparisons were conducted between ACNVax + anti-PD-1 and other groups in panels a and b and between ACNVax&LIGHT + anti-PD-1 and other groups in panels c and d. Statistical comparisons are based on one-way ANOVA, followed by post hoc Tukey’s pairwise comparisons or by Student’s unpaired t test. The asterisks denote statistical significance at the level of * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. ANOVA, analysis of variance; SD, standard deviation; n.s., no statistical significance.
To investigate how antigen-clustered topography in ACNVax influenced its anticancer efficacy, we compared its efficacy with IONPVax (the core component of ACN 35 nm with antigen uniformly conjugated on the nanoparticle surface, but without antigen cluster structure), the AuNP-B/CD4 (the surface component of ACN 2–5 nm, but without antigen cluster structure), and lipoVax (liposome with antigen uniformly conjugated on the nanoparticle surface, but without antigen cluster topography). The data showed that ACNVax after 3 vaccinations induced significantly better tumor inhibition with 57% complete response (CR) compared to other nanovaccine groups, which only slightly inhibited tumor growth with no CR (0%) (Figure 4b). These data suggest that the antigen-clustered structure of ACNVax indeed plays a critical role in its superior anticancer efficacy in comparison to IONPVax and AuNPVax
Since the presence of TLS, along with B/CD4 T cell crosstalk, is associated with better responses to anti-PD-1 immunotherapy,35–39 we combined a peptide of LIGHT, a member of the tumor necrosis factor (TNF) ligand family that can stimulate immune cell tumor infiltration and TLS formation,67,68 with ACNVax to test whether it could enhance anticancer activity further. LIGHT has been previously reported to induce robust de novo formation of TISs in tumors and to greatly enhance antitumor immune responses and efficacy.67,68 ACNVax (with LIGHT) combined with anti-PD-1 antibody (three vaccination doses) inhibited tumor growth by 96% and achieved long-term cancer remission (>200 days) with a complete response (CR) rate of 44% (Figure 4c), which is more significant than ACNVax (without LIGHT) plus anti-PD-1 antibody (cancer growth inhibition by 85%, CR rate of 11%). Following a clinical used dose regimen (five vaccinations at day 4, 11, 18, 32, 46), ACNVax (with LIGHT), combined with anti-PD-1 antibody, enhanced the complete response (CR) rate to 80% (>200 days, Figure 4d). In contrast, both IONPVax and PepVax plus anti-PD-1 only slightly inhibited tumor growth. The addition of LIGHT did not significantly improve the anticancer efficacy in either the IONPVax plus anti-PD-1 or PepVax plus anti-PD-1 groups (Figure 4d). In addition, ACNVax-T, which used the same ACN but conjugated with the HER2 CD8 T cell epitope (E75), did not significantly inhibit tumor growth (vs control) (Figure 4c). results demonstrated that ACNVax with LIGHT achieves long-term anticancer efficacy in HER2 positive breast cancer.
To test if ACNVax would show therapeutic efficacy in HER2-low breast cancer, we tested the efficacy of ACNVax on two mouse breast cancer models with low HER2 levels: transgenic PyMT-MMTV breast cancer and 4T1 subcutaneous breast cancer (Figure S21).84,85 The data showed that ACNVax had significantly better anticancer efficacy than the control and PepVax groups on PyMT-MMTV and 4T1 tumor mouse model (Figure S21). Because 70% of breast cancer patients show a low-level expression of HER2 (HER2-low) despite not being defined as HER2 positive tumor,86 these data suggest that ACNVax may offer therapeutic advantages in a broad range of breast cancer populations.
ACNVax Induces B/T Lymphocytes Infiltration into the Tumor.
To understand how ACNVax influences immune cell infiltration in tumors, we used CyTOF to profile all immune cells in tumor tissues from BALB/c mice with subcutaneous HER2+ breast cancer (D2F2/E2) at 28 days after vaccination. The cell composition in tumor tissues was compared in mice treated with anti-PD-1 antibody, anti-PD-1 and murine HER2 antibody, and a CD8 T cell vaccine NeoVax (E75and GM-CSF). Results showed that ACNVax combined with anti-PD-1 antibody dramatically increased the frequency of tumor-infiltrating B cells by 13.7-fold, CD4 T cells by 9.5-fold, and CD8 T cells by 5.2-fold among total cells in the tumor compared to the other treatment groups (Figures 5, S16, and S22 and Table S9).
Figure 5.

ACNVax induced robust B/CD4/CD8 immune cell infiltration in tumors. (a) Cytometry by time-of-flight (CyTOF) analysis of the immune cell population from tumor samples of mice 10 days after different treatments (3 vaccinations). Global analysis using SPADE unsupervised clustering analysis of tumor-infiltrating immune cells. Nodes are colored based on the log ratio of the relative number of immune cells in the ACNVax group to that in the anti-PD-1 antibody group in the same node: red indicates higher and blue indicates lower numbers in the ACNVax than the comparison groups. (Marker information can be found in Figure S16 and Table S9). (b) Qantification of immune cells among total cells in tumor after different treatments, expressed as a fraction of total cells from the samples.
ACNVax Remodels the Tumor Immune Microenvironment.
To comprehensively analyze how ACNVax remodels the tumor immune microenvironment, we performed single-cell RNA-seq of tumor immune cells from BALB/c mice with HER2+ breast cancer after vaccination with ACNVax, ACNVax with LIGHT, or IONPVax in combination with anti-PD-1 antibody.
Single-cell RNA-seq analysis results suggest that ACNVax (w/o LIGHT) enhances B and CD4 T cell interaction in tumors in comparison to IONPVax and control group. For instance, ACNVax (B cells: 14.99%; CD4 T cells: 14.74%) and ACNVax/LIGHT (B cells: 16.53%; CD4 T cells: 10.41%) showed significantly higher proportions of B cells and CD4 T cells than those of the IONPVax group (B cells: 10.33%; CD4 T cells: 7.15%) (Figure 6a,b). ACNVax induced higher levels of CD40 in B cell and CD40L gene expression in T cell (Figures 6c and S23a,b) than vaccination with IONPVax or control treatments. CD40 and CD40L are implicated in effective B and CD 4 T cell interactions. ACNVax vaccination also induced significantly higher expression of Aicda in B cells in tumors than IONPVax vaccination or control treatments. Aicda encodes the enzyme needed to initiate somatic hypermutation and Ig gene class switch recombination, features of effective germinal center responses.87,88 Furthermore, ACNVax increased the expression of genes related to GC-Tfh responses, including BCL-6 and IL21 compared to IONPVax or control groups (Figures 6c and S23a,b). These data suggest that ACNVax induced a potent CD4 T cell-dependent B cell activation in tumors compared to IONPVax and control groups.
Figure 6.

ACNVax remodeled the tumor immune microenvironment. Control (PBS), IONPVax (14.6 nmol HER2 epitope with 13.9 nmol 2′3′-cGAMP), ACNVax (14.6 nmol HER2 epitope with 13.9 nmol 2′3′-cGAMP), and ACNVax/LIGHT (14.6 nmol antigen, 13.9 nmol 2′3′-cGAMP, 50 ng LIGHT). Vaccines were given on days 4, 11, and 18 in combination with 100 μg of anti-PD-1 antibody biweekly for 3 weeks in D2F2/E2 tumor-bearing mice. (a) Uniform manifold approximation and projection (UMAP) plot of tumor-infiltrating immune cells. (b) Stacked bar charts show the quantified ratio of different phenotypes of immune cells in different treatment groups. (c) Boxplot of selected gene expression levels in tumor infiltrated B and T cells from single-cell RNA sequencing. Statistical comparisons were conducted between ACNVax&LIGHT and ACNVax with IONPVax and control. (d and e) Uniform manifold approximation and projection (UMAP) plot of CD4 T cell subclusters (d) and CD8 T cell subclusters (e) from tumor-infiltrating immune cells. (f) Heatmap of selected gene expression levels related to organized aggregates of immune cells in total tumor infiltrated immune cells from single-cell RNA sequencing. (g) Immunofluorescent staining of tumor lymphoid follicle from mouse tumor samples in the ACNVax&LIGHT + anti-PD-1 antibody group (14.6 nmol HER2 B/CD4 epitope, 13.9 nmol 2′3′-cGAMP, 50 ng of LIGHT, 3 times every 7 days, 100 μg of anti-PD-1 antibody biweekly for 3 weeks). Primary antibodies used are rabbit antimouse CD20 antibody (Invivogen), goat antimouse CD3 antibody (R&D Systems), mouse antimouse CD23 antibody (Invivogen), and rat antimouse PNAd antibody (Biolegend). Secondary antibodies (Abcam) used are donkey antirabbit antibody-AF488, donkey antigoat antibody-AFS94, donkey antimouse antibody-AF647, and donkey antirat antibody-AB405. Scale bar is 100 μm. Statistical comparisons are based on one-way ANOVA, followed by post hoc Tukey’s painwise comparisons or by Student’s unpaired t test. The asterisks denote statistical significance at the level of **** p < 0.0001. ANOVA, analysis of variance; SD, standard deviation.
Since anticancer immune response relies on CD4 and CD8 T cell activation, and previous reports suggested Tfh-dependent B cell activation promote anticancer CD4 and CD8 T cell immunity,35,37,41,42 we further investigated if ACNVax altered the subpopulations of CD4 T cells (memory, regulatory, and Tfh) and CD8 T cells (memory, cytotoxic, and exhausted) in tumors, in comparison with IONPVax and control group. Among different subpopulations of CD4 T cells, vaccination with ACNVax (combined with anti-PD-1 ) increased 1,5 to 3-fold memory CD4 T cells, increased 1.5 to 5-fold Tfh, and decreased 1.5 to 2-fold Treg in tumors compared to vaccination with IONPVax (combined with anti-PD-1) or control group (Figures 6d and S24a). Among different subpepulations of CD8 T cells, ACNVax vaccination (combined with anti-PD-1) increased memory CD8 T cells by 9-fold, cytotoxic CD8 T cells by 3.5-fold, and decreased exhausted CD8 T cells by 20-fold compared to control groups, ACNVax increased memory CD8 T cells by 1.5-fold in comparison with vaccination with IONPVax (Figures 6e and S24b). In addition, ACNVax also induced higher expression of genes encoding antitumor cytokines, such as interferon-gamma, tumor necrosis factor, interleukins, and granzyme B, in immune cells and T cells in comparison to IONPVax or control groups (Figures 6f and S23b,c). These data demonstrated the effect of ACNVax to promote CD4 and CD 8 T cells response and anticancer cytokine production, which may explain the potent anticancer efficacy of ACNVax.
Single-cell RNA-seq analysis results also suggested that ACNVax increased gene expression related to organized aggregates of immune cell formation in tumors compared to the IONPVax and control treatments (Figures 6f and S23e). ACNVax increased the levels of the chemotaxss factors CCL19, CCL21a, CXCL13, and CCL2 compared with those of the IONPVax and control (Figure S23a,fe). ACNVax also induced increased gene expression of receptors for chemotaxis factors and adhesion receptors, such as CXCR4, CXCR5, CCR7 and L-selectin, CD11a and VLA-4, in tumor infiltrating B and T cells compared with those of the IONPVax and control groups (Figure S23a,b), similar to literature reports.35–37
To confirm the formation of organized aggregates of immune cells, we use both fluorescent staining to analyze tumor tissues after vaccination (Figures 6g, S25, and S26). Fluorescent staining showed that ACNVax plus anti-PD-1 antibody (with LIGHT) increased the presence of TLS-like structure, where B cells (CD20, Figures 6g, S25, and S26) were surrounded by T cells (CD3, Figures 6g, S25, and S26). We also observed the presence of follicular dendritic cells (CD23, CD21/CD35 (CR1/CR2), Figures 6g and S25) with scattered high endothelial venules (PNAd, Figure 6g) in ACNVax plus anti-PD-1 antibody (with LIGHT) group in comparison with other treatment groups, similar to the literature TLS.35,36,89
ACNVax Induced Long-Term Antigen-Specific Memory B Cells, CD4 T Cells, and CD8 T Cells.
To test whether ACNVax vaccination induced a persistent immune memory against cancer growth, we rechallenged five mice whose tumors were eliminated after three vaccinations at 200 days by subcutaneous injection of 2.5 × 105 D2F2/E2 HER2+ cancer cells. The data showed that each of the five mice completely rejected tumors whereas every unvaccinated mouse rapidly grew tumors (Figures 4 and 7a). The results demonstrated that ACNVax vaccination generated long-term, persistent immune memory against cancer growth.
Figure 7.

ACNVax induced long-term immune memory against tumor rechallenge and increased long-term antigen-specific memory B, CD4, and CD8 T cells, (a) Tumor rechallenge at 200 days for mice with long-term remission after three times of vaccination (ACNVax). D2F2/E2 HF.R2 cancer cells (2.5 × 105) were s.c. injected into the mice with long-term remission and control mice (normal BALB/c mice). Average and individual tumor growth curves were measured. Data represent the mean ± SD, n = 5. (b and c) Representative flow cytometry analysis and quantification of class switched memory B cells (b) and antigen specific class switched memory B cells (c) from spleen, lymph node, bone marrow and peripheral blood of the mice in panel a. B220+ CD38+ GL-7−IgD− IgM− populations were identified as class switched memory B cells. HER2 tetramer positive class switched memory B cells were identified as antigen specific class switched memory B cells. Data for quantification are shown as mean ± SD, n = 3. (d) Quantification of flow cytometry results of CD4 T effector memory (TEM) and CD4 T central memory (TCM) cells from lymph node and peripheral blood of the mice in panel a. CD8− CD4+ CD44+ CD62L− populations were identified as CD4 TEM cells; CD8− CD4+ CD44+ CD62L+ populations were identified as CD4 TCM cells, (e) Diagram of experimental design for activation-induced markers assay (AIM) for measuring antigen specific CD4 TEM/TCM cells from spleen. Representative flow cytometry analysis and quantification of AIM+ CD4 TEM/TCM cells. CD69+ CD40L+/− populations from CD4 TEM/TCM cells were identified as AIM+ CD4 TEM/TCM cells. (f) Quantification of flow cytometry results of CD8 TEM and CD8 TCM cells from lymph node and peripheral blood of the mice in panel a. CD4− CD8+ CD44+ CD62L− populations were identified as CD8 TEM cells; CD4− CD8+ CD44+ CD62L+ populations were identified as CD8 TCM cells. (g) Diagram of experimental design for activation-induced markers assay (AIM) for measuring antigen specific CD8 TEM/TCM cells from spleen. Representative flow cytometry analysis and quantification of AIM+ CD8 TEM/TCM cells. CD25+ OX40+/− populations from CD8 TEM/TCM cells were identified as AIM+ CD8 TEM/TCM cells. Data for quantification are shown as mean ± SD, n = 3. Statistical comparisons were conducted between ACNVax and control. Statistical comparisons are based on one-way ANOVA, followed by post hoc Tukey’s pairwise comparisons or by Student’s unpaired t test. The asterisks denote statistical significance at the level of * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. ANOVA, analysis of variance; SD, standard deviation.
To further examine whether ACNVax vaccination could generate long-term antigen-specific memory B, CD4, and CD8 T cells, we used flow cytometry and activation-induced markers (AIM) assay to enumerate these cells in the mice that rejected tumors following rechallenge, ACNVax vaccination increased class switch memory B cells (B220+ CD38+ GL-7−IgD− IgM−) by 3.5-, 2.5-, 2-, 1.5-fold in spleen, lymph nodes, bone marrow, and peripheral blood, respectively (Figures 7b and S27); increased antigen-specific class switch memory B cells (B220+ CD38+ GL-7− IgD− IgM− Tetramer+) by 10-, 13-, 7-, and 5.6-fold in spleen, lymph nodes, bone marrow, and peripheral blood, respectively (Figures 7c and S27); and increased antigen-specific plasma cells (markers) by 20-fold in the bone marrow of the mice (Figure S28), in comparison with control mice.
ACNVax vaccination induced CD4 T effector memory cells (CD4 TEM: CD8− CD4+ CD44+ CD62L−) by 2-fold in the lymph nodes and peripheral blood (Figures 7d and S29); increased antigen-specific CD4 T effector memory cells (AIM+ CD4 TEM: CDS− CD4+ CD44+ CD62L− CD69+CD40L+/−) by 14-fold in the spleen (Figures 7e and S30a); increased CD4 T central memory cells (CD4 TCM: CD8− CD4+ CD44+ CD62L+) by 10- and 7-fold in the lymph nodes and peripheral blood, respectively (Figures 7d and S29); increased antigen-specific CD4 T central memory cells (AIM+ CD4 TCM: CD8− CD4+ CD44+ CD62L+ CD69+ CD40L+/−) by 12.5-fold in the spleen (Figures 7e and S30a); increased tissue resident memory CD4 T cells (CD4 TRM: CD8−CD4+ CD69+CD103+) by 3.5- and 9.8-fold in the fat pad and lungs of the mice, respectively, (Figure S31a–c), in comparison with control mice.
ACNVax vaccination also increased CD8 T effector memory cells (CD8 TEM: CD4−CD8+ CD44+ CD62L−) by 3.6-fold and 3.3-fold in the lymph nodes and peripheral blood, respectively (Figures 7f and S29); increased antigen-specific CD8 T effector memory cells (AIM+ CD8 TEM: CD4− CD8+ CD44+ CD62L− CD2S+ OX40+/−) by 6-fold in the spleen (Figures 7g and S30b); increased CD8 T central memory cells (CD8 TCM: CD4− CD8+ CD44+ CD62L+) by 5-fold in the lymph nodes and peripheral blood (Figures 7f and S29); increased antigen-specific CD8 T central memory (AIM+ CD8 TCM: CD4−CD8+ CD44+ CD62L+CD2S+OX40+/−) by more than 10-fold in the spleen (Figures 7g and S30b); increased tissue resident memory CD8 T cells (CD8 TRM: CD4− CD8+ CD69+ CD103+) by 2.5- and 19.3-fold in the fat pads and lungs of mice, respectively (Figure S31a,b,d), in comparison with control mice.
Our results suggest that ACNVax induced a persistent anticancer immune memory through promoting long-term antigen-specific memory B, CD4 T, and CD8 T cells. Since ACNVax vaccinations do not contain CD8 T cell epitope, the increased CD8 memory cells are likely as a secondary consequence of antitumor CD4 T and B cells activation in response to tumor antigens.
The development of therapeutic cancer vaccines has mainly focused on T cell cancer vaccines, but these have had limited clinical success. Inspired by recent findings that B cell tumor infiltration and B/CD4 T cell crosstalk are strongly associated with better clinical efficacy of checkpoint blockade immunotherapy, we designed an antigen-clustered nanovaccine (ACNVax) that achieved long-term tumor remission with a complete response rate of 80% (>200 days, Figure 4) when combined with anti-PD-1 antibody. Unlike other T cell cancer vaccines, ACNVax achieved superior anticancer efficacy by promoting B cell antigen presentation-mediated B/CD4 T cell crosstalk. (Figure 2),50,52,53,56,57,59–61,66,90–92 Tfh-dependent B cell activation (Figures 2 and 3), and remodeling tumor immune microenvironment (Figures 5 and 6). The long-term efficacy of ACNVax was mediated by a concerted long-term antigen-specific memory B cell, CD4 T, and CD8 T cell immunity (Figure 7 and Scheme 1). The ACNVax has an antigen-clustered topography on its surface, featuring a high density of HER2 B and CD4 T cell epitopes on the duster (150 peptides/duster) and optimal distance (5–10 nm) between antigen dusters for efficient cross-link with BCR (Figure 1).56,57,59–61,65,90–92 This unique topography promoted lymph nodes trafficking (Figure 3), cross-linking with B cell receptor (BCR) (Figure 1), and B cell antigen presentation-mediated B/CD4 T cell crosstalk (Figure 2).50,52,53,56,57,59–61,66,90–92 Subseqnently, ACNVax promoted very strong Tfh-dependent GC responses in lymphoid organs as measured by both CyTOF and flow cytometry. More importantly, ACNVax remodeled the tumor immune microenvironment by enhancing infiltration of B cells, CD4 and CDS T cells as measured by both CyTOF and single-cell RNA-seq. In addition, ACNVax, combined with anti-PD-1, not only activated long-term antigen-specific B and CD4 T cell memory bat also generated long-term antigen-specific CD8 T cell memory (Figures 6 and 7), which contributed to its efficacy in complete rejection of tumor rechallenges at 222 days in cured mice with HEB2+ breast cancer (Figure 7).
CONCLUSIONS
The findings in our study have two important implications. First, our study suggests that incorporating B cell immunity in the cancer vaccine design by promoting B cell antigen presentation-mediated B/CD4 T cell crosstalk achieves long-term durable anticancer efficacy when combined with anti-PD-1 immunotherapy. Second, the cancer vaccine design for this purpose needs an optimal delivery system, which facilitates lymph node penetration allowing antigen to directly interact with B cells for BCR cross-finking and B cell antigen presentation-mediated B/CD4 T cell crosstalk Most other nanoparticle systems, which encapsulate the antigen inside, would enhance uptake and antigen presentation by DCs or macrophages since the function of these APCs is to uptake nanoparticles, but those nanoparticle delivery systems would not be able to enhance the uptake and antigen presentation by B cells since B cell antigen presentation requires antigen binding to BCR to trigger cross-link.19,50 Our ACNVax with antigen-clustered topography, which has a high density of antigens on the duster and optimal distance (5–10 nm) between antigen clusters, exhibits more efficient cross-link with BCR, promoting B cell antigen presentation-mediated B/CD4 T cell crosstalk compared to IONPVax without the surface antigen duster structure. Furthermore, the ACNVax viral-like structure might also enhance lymph node draining and penetration to induce B/CD4 T cell crosstalk.50,93
In conclusion, our data provide a cancer nanovaccine design with B/CD4 epitopes in an antigen-clustered topography to achieve long-term durable anticancer efficacy by promoting B cell antigen presentation-mediated B/CD4 T cell crosstalk and remodeling of the tumor immune microenvironment.
METHODS
Materials.
All reagents were obtained from commercial sources without further purification, except γ-methacryloxypropyltrimethoxysilane (98%), which was purified by distillation under reduced pressure, and 2,2-azobis(isobutyronitrile) (98%), which was purified by recrystallization in ethanol. Iron oxide (III) (FeO(OH), hydrated, catalyst grade, 30–50 mesh), oleic add (technical grade, 90%), ammonium iron(II) sulfate hexahydrate (ACS reagent, 99%), 1-octadecene (technical grade, 90%), anhydrous tetrahydrofuran (THF, 99.8%), carbon disulfide (99.9%), magnesium turnings (>99.5%), 2-chloro-2-phenylacetyl chloride (CPAC, 90%), poly(ethylene axide) monomethyl ether (PEO), anhydrous dioxane (99.8%), dimethylformamide (DMF, 99.9%), dimethyl sulfoxide (DMSO, 99.9%), o-phenanthroline monohydrate (ACS reagent, 99%), hydroquinone (ACS reagent, 99%), sodium sulfide, chloroauric acid, nitric acid (ACS reagent, 70%), and hydrochloric acid (ACS reagent, 37%) were purchased from Sigma-Aldrich. Dulbecco’s phosphate-buffered saline (DPBS) and Hank’s buffered salt solution (HBSS) were obtained from Fisher Scientific. Mouse uncoated IgG and IgM Total ELISA Kits, 1-Step Ultra TMB-ELISA substrate solution, HRF-conjugated goat antimouse IgG2a secondary antibody, HRP-conjugated goat antimouse IgG2a secondary antibody, Nunc Immobilizer Amino 96-well ELISA plates, BupH carbonate bicarbonate buffer packs (coating buffer), Pierce protein-free PBS-tween blocking buffer, 20x PBS-Tween wash buffer, Geneticin. (G418) selective antibiotic, Invitrogen eBiosdence fixable viability dye eFluor 780, and Molecular Probes streptavidin Alexa Fluor 647 conjugate were obtained from Thermo Fisher Scientific. The EasySep Mouse B Cell Isolation Kit, EasySep Mouse CD4 T Cell Isolation Kit, and EasySep Mouse CD19 Positive Selection Kit II were purchased from StemCell Technologies. Fluo-4 AM was purchased from Thermo Fisher Scientific. AF488-AffiniFure Fab Fragment Goat Anti-Mouse IgM and μ Chain Specific were purchased from Jackson Immuno Research laboratory Inc. Alexa Fluor Plus 405 phalloidin was purchased from Thermo Scientific. Antimeuse PD-1 antibody (CD279) was purchased from Bio X Cell. Mouse GM-CSF was obtained from SHENANDOAH Biotechnology Inc. LIGHT (TNFSF14) was purchased from Sino Biological HRP-conjugated goat antimouse IgG secondary antibody, Zombie UV fixable viability kit, FITC antimouse CD19, PE/Dazzle. 594 antimouse IgD, Alexa Fluor 647 antimouse/house GL7 antigen, Brilliant Violet 421 and PE/Dazzle 594 antimouse/human CD45R/B220, FITC antimouse CD95, Brilliant Violet 421 antimouse/human CD11b, Alexa Fluor 647 antimouse CD21/CD35 (CR2/CR1), Alexa Fluor 594 antimouse CD169, FITC antimouse CD169 and PE goat antimouse IgG secondary antibody, FITC antimouse CD19, Brilliant Violet 605 antimouse CD19, Alexa Fluor 594 antimouse CD19, APC/Cyanme7 antimouse CD86, and FITC antimouse CD3 were purchased from BioLegend. HER2 peptides (CDDDPESFDGDPAS-NTAPLQPEQLQ (Human HER2 CD4/B epitope), Biodn-PESFD-GDPASNTAPLQPEQLQ, CDDDPESFDGDPASNTAPLQPEQ-LQGGGK, CDDDPESFDGDPASNTAPLQPEQLQ-GGG-Lys (NP), CDDDPESFDGDPASNTAPLQPEQLQ-(Lys(N3)-DBCO- -Cy3) -GGG-(Lys (NP)), CDDDPESFDGDPASNTAPLQPEQLQ-EDFITC), CDDDKIFGSLAFL (Human/Mouse HER2 CD 8 epitope), and E75 (KIFGSLAFL, Human/Mouse HER2 CD 8 epitope), OVA323–339 (CISQAVHAAHAEINEAGR, recognize by CD4 T cell from OT-II mice) were custom synthesized by LifeTein. Iron oxide nanoparticles (30 nm) stabilized by oleic acid in chloroform were purchased from Ocean Nanotech, Cyclic [G(2′,S′)-pA(3′,5′)p] (2′3′-cGAMP) was acquired from InvSvoGen, Fluorescamine was purchased from MP Biomedicals. Suifo-Cy5.5 NHS ester was acquired from Lumiprobe. Microvette 500 Z-Gel serum collection vials with dotting factors were obtained from Sarstedt Matrigel basement membrane matrix was purchased from Corning. Gold and iron standards were purchased from Fluka Analytical. Murine HER2 antibody (Murine 2C4):94 Mouse IgG2a constant chain chimeric with same variable region as human pertuzumab (Perjeta) were purchased from GenScript. The amino add sequences of murine 2C4 are listed in Table S8.
Mice.
All animal experiments were conducted according to protocols approved by the University of Michigan Committee on Use and Care of Animals (UCUCA). BALB/c mice aged 5–7 weeks were purchased from Charles River Laboratories (Wilmington, MA).
Cells.
All cells were maintained at 37 °C in a 5% CO2/95% air atmosphere and approximately 85% relative humidity. D2F2/E2 cells were generated by cotransfection with pRSV/neo and pCMV/E2 encoding human ErbB-2 (HER2) (provided by Dr. Wei-Zen Wei).79 The cells were cultured in complete high-glucose DMEM supplemented with 10% NCTC 109 media, 1% l-glutamine, 1% MEM nonessential amino adds, 0.5% sodium pyruvate, 2.5% sodium bicarbonate, 1% pen/strep, 5% cosmic calf serum, and 5% fetal bovine D2F2/E2 cells generated by cotransfection with pRSV/neo and pCMV/E2 encoding human ErbB-2 (HER2), 500 μg/mL Geneticin and 50 μM 2-mercaptoethanol. RAW264.7 macrophages were cultured in complete RPMI-1640 media supplemented with 10% fetal bovine serum, 1% l-glutamine, 1% MEM nonessential amino acid solution, 1% sodium pyruvate, and 1% pen/strep. Primary B-cells, CD4 T cells, and splenocytes were cultured in RPMI-1640 media supplemented with 10% fetal bovine serum, 2-Mercaptoethanol (50 μM), and 1% pen/strep. 4T1 cells were cultured ATCC-formulated RPMI-1640 Medium with 10% fetal bovine serum and 1% pen/strep.
Preparation and Characterization of ACN.
ACN was prepared based on previously reported protocols.70 IONF-Polymer was made by thermal decomposition and further coated with a polysiloxane-containing copolymer. AuNPs (2 nm) were synthesized by reacting sodium sulfide (Na2S) as the reducing reagent with gold in the form of chloroauric add (HAuCl4). AuNPs were then attached onto the surface of IONP-Polymer to produce a spiky topography. The final Au:Fe ratio of the formulated ACN was quantified by inductively coupled plasma–mass spectrometry (ICP-MS) using a PerkinElmer Nexion 2000 based on previously reported protocols modified from the analysis by inductively coupled plasma–optical emission spectroscopy (ICP-OES).95 ACN formulations were imaged by STEM using a JEOL 2100F with a CEOS probe corrector. The true particle sizes of AuNPs, IONP-Polymer, and ACN were quantified using ImageJ software. The volume-weighted hydrodynamic particle size, polydispersity index, and zeta potential of all formulations in Milli-Q water at 25 °C were evaluated with a Malvern Zetasizer Nano-ZS using DLS and phase analysis light scattering, respectively.
Preparation and Characterization of Lipid-Coated IONP.
Lipid-coated iron-oxide nanoparticles were prepared by the thin-film hydration method based on previously reported methods.96 DSPE-PEG (2000)-maleimide (10 mg) was added to 1 mg of 30 nm iron-oxide nanoparticles stabilized by oleic add in chloroform and gently mixed. The resulting solution was subjected to solvent rotary evaporation to remove all chloroform and form a thin film. Simultaneously, this film and 100 mM PBS pH 7.4 were heated to 75 °C in an oven. When it reached 75 °C, hot PBS was rapidly added to the film and mixed immediately and vigorously to facilitate thin-film hydration. The resulting nanoparticle solution was stored at 4 °C to promote lipid self-assembly. Free phospholipids were removed by magnetic separation overnight at 4 °C using an EasySep magnetic separator device (StemCell Technologies). The volume-weighted hydrodynamic particle size, polydispersity index and zeta potential of all formulations In Milli-Q water at 25 °C were evaluated with a Malvern Zetasizer Nano-ZS using DLS and phase analysis light scattering, respectively.
Preparation and Characterization of ACNVax and lONPVax.
HER2 peptides were conjugated to ACNVax via a gold–thiol linkage. HER2 peptide was added to ACNVax at a 5× weight ratio excess in Milli-Q. water and incubated overnight at 4 °C. HER2 peptides were conjugated to lipid-coated IONP via maleimide–thiol chemistry. Both materials were purified either by magnetic separation overnight at 4 °C or by centrifugal separation at 10,000g for 30 min at 4 °C. Peptide loading was determined by fluorescence quantification using a modified fiuorescamine peptide quantification assay in the presence of ACN or IONP-Lipid (Ex/Em: 390/465 nm, Biotek Cytation 5).97 Quantification was performed using a standard curve with increasing peptide concentration with a standardized concentration of nanoparticles (IONP-Lipid or ACN) to account for quenching effects.
Investigation of BCR Cross-Link.
Cross-Linking Activation Imaging.76
B cells were isolated from splenocytes of QM mice through negative selection using the EasySep Mouse B Cell Isolation Kit (STEMCELL: 19854).
B cells isolated from QM mice (5 × 106 cells/mL) were then incubated with 20 μg/mL Alexa Fluor 488-AffiniPure Fab Fragment Goat Anti-Mouse IgM (μ Chain Specific) on ice for 30 min in the dark (Jackson: 115–167-020). Cells (2 × 106 cells/mL) were washed and then incubated with antigen (equal amount of epitope, 20 nM, Cy3 labeled, and NP conjugated) in a total volume of 400 μL for 1 and 5 min at 37 °C. After antigen incubation, cells were fixed with 6% paraformaldehyde (800 μL) for 10 min immediately at 37 °C, permeabilized with a 0.1% Triton X HBSS solution (800 μL) for 10 min, and then incubated with Alexa Fluor Plus 405 phalloidin in staining buffer (200 μL, 5 mg/mL BSA, 0.1% Triton X in HBSS) on ice for 2 h. After two washes, the cells were plated onto eight-well glass chambers pretreated with 0.1% poly l-lysine (LabTech II) on ice for at least 4 h before confocal imaging.
Calculation of Cross-Linking Ratio.
Cells with significant red (B cell receptors, (BCRs)) fluorescence were circled and quantified in ImageJ (NTH) to calculate the fluorescence intensity of different channels (blue, phalloidin; red, BCRs; green, antigen) in all images. The intensity of green (antigen) fluorescence was divided by the intensity of red (BCR) fluorescence to calculate the cross-linking ratio of the cells. Up to 50 cells (10 cells from each comer of images and 10 cells from middle) in each image (or all cells if there were fewer than 50 cells) were used to calculate the cross-linking ratio for each time point of each sample;
Investigation of B/CD4 T Cell Crosstalk In Vitro.
CD4 T cells were isolated from splenocytes of OT-II mice (The Jackson Laboratory, Strain, #004194) through negative selection using the EasySep Mouse CD4 T Cell Isolation Kit (STEMCELL: 19852). B cells were isolated from splenocytes of QM mice through negative selection using the EasySep Mouse B Cell Isolation Kit (STEMCELL: 19854), Splenocytes from QM mice deplete B cells through positive selection using the EasySep Mouse CD19 Positive Selection Kit II (STHMCELL: 18954). For activation and proliferation, markers, CD4-PE; B220-Alexa Fluor 594, CD69-Brifliant violet 421, CD86-Alexa Fluor 647, CD25-APC/Fire750 and CFSE are used. All antibody markers are from Biolegend.
Cell mixtures within each well of a 24-well plate, including CD4 T cells (0.5 million, labeled with CFSE) isolated from splenocytes of OT-II mice, B cells (1 million, labeled with CFSE) isolated from splenocytes of QM mice, full slpeenocytes (1.5 million, no CFSE label) from QM mice. Cell mixtures are then coincubated with soluble peptides, IONPVax, and ACNVax for 24, 72, and 96 h (All with the same amount of antigens, OT-II CD4 epitope, OVA323–339 (CISQA-VHAAHAEINEAGR), 2 μM and hapten conjugated HER2 B/CD4 epitope, CDDDPESFDGDPASNTAPLQPEQLQ-GGG-(Lys (hapten), 2 μM). After each time point, cells are collected for flow cytometry analysis.
Immunofluorescence Staining of Lymph Nodes.
To determine the ACNVax (conjugated to ED-FITC labeled HER2-B/CD4 peptide, 233.6 nmol HER2 epitope) distribution, lymph nodes were harvested 12 h after subcutaneous injections. Harvested tissues were immediately fixed in 1% paraformaldehyde for 1 h and then immersed in 30% sucrose in 0.1% NaN3 in PBS overnight Treated tissues were then embedded in optical coherence tomography (OCT) compound and frozen in a CO2(s) + EtOH bath. Tissue sections (15 μm) were prepared and dried for 0.5 h before staining. After incubation with blocking buffer and staining solution, slides were mounted with VECTASHIELD Mounting Medium for confocal imaging. Brilliant Violet 421 B220, Alexa Fluor 594 CD 3, and Alexa Fluor 647 CD169 were used for lymph node immune fluorescence staining.
CyTOF Analysis of Immune Patterns from Lymph Nodes and Tumors.
Lymph node and tumor samples were harvested and dissociated into single-cell suspensions 10 days after the second booster vaccination. CyTOF antibody conjugation and data acquisition were done as previously described.98,99 Briefly, antibodies were conjugated to lanthanide metals (Fluidigm) using the Maxpar Antibody Labeling Kit (Fluidigm), Unstimulated single-cell suspensions were washed once with heavy-metal-free PBS and stained with 1. 25 μM Cell-ID cisplatin-195Pt (Fluidigm) at room temperature for 5 min. Fc receptors were blocked with TruStain FcX (antimouse CD16/32, Biolegend), and surface staining was done on ice for 60 min. Cells were then fixed with 1.6% paraformaldehyde and permeabilized with Invitrogen permeabilization buffer before intracellular antibody staining. Cells were left in 62.5 nM Cell-ID intercalator iridium-191/193 (Fluidigm) in 1.6% paraformaldehyde in PBS overnight at 4 °C before acquisition on a CyTOF Helios system (Fluidigm). A signal-correction algorithm based on the calibration bead signal was used to correct for any temporal variation in detector sensitivity.
CyTOF data were analyzed as previously described.99 All events were gated to remove noncellular events (negative for DNA intercalator), dead cells (negative for uptake of cisplatin), and doublets. SPADE clustering and the viSNE algorithm were applied using the Cytobank platform, SPADE nodes were manually bubbled based on phenotypic markers, and viSNE populations were manually gated using the same markers.
Analysis of Germinal Center B Cells, Antigen-Specific Germinal Center B Cells, and T Follicular Helper Cells by Flow Cytometry.
Mice were immunized as described previously. At day 10 after the second booster, mice were sacrificed, and lymph nodes were dissected for ex vivo analysis by flow cytometry. CD3−B220+ CD95+GL-7+ populations were identified as germinal center B cells. Germinal center derived antigen-specific B-cell analysis was accomplished using tetramer staining based on previously established protocols with minor modifications.100 HER2/neu peptide tetramers were prepared by mixing biotin-labeled HER2 peptide with brilliant violet 421-labeled streptavidin at an 8:1 molar ratio at room temperature for 1 h without further purification. Markers used are CD3-Alexa Fluor 647, B220-APC/Fire750, CD95-brflliant violet 605, and GL-7-FITC. B220−CD4+ CXCR5+ PD-1+ populations were identified as Tfh cells. Markers used are CD4-FITC, B220-Alexa Fluor 594, CXCR5-brilliant violet 421, and PD-1-APC/Fire750.
Analysis of Activation Induced Marker Assay for T Cells by Flow Cytometry.
Mice were immunized as described in previous work.101,102 At day 10 after the second booster, mice were sacrificed and spleens were harvested for single cell suspension. Spleen cells (2 million) from different vaccination groups were then incubated with HER2-B/CD4 peptide (2 μg/mL) for 20 h in 24-well plate. After incubation, cells were then collected and measured by flow cytometry. B220− CD4+ CXCR5+ PD-1+ populations were identified as Tfh cells. CD69+ CD40L+/− populations from Tfh cells were identified as AIM+ Tfh cells. Markers, CD4-FITC, B220-Alexa Fluor 594, CXCR5-brilliant violet 421, PD-l-APC/Fire750, CD69-PE, and CD40L-APC are used. B220− CD4+ CD62L+ CD69+ CD40L+/− populations were identified as AIM+ antigen experienced CD4 T cells. B220− CD4+ CD62L− CD69+ CD40L+/− populations were identified as AIM+ naive CD4 T cells. Markers, CD4-FTTC, B220-Alexa Fluor 594, CD44-brilliant violet 421, CD62L− APC/Fire750, CD69-PE, and CD40L-APC are used.
Enzyme-Linked Immunosorbent Assay (ELISA) for Antibody Titer Measurements.
At day 0, mice were immunized with the equivalent of 14.6 nmol or 1.46 nmol of HER2 peptide plus 13.9 nmol of 2′3′-cGAMP regardless of formulation type. Starting at day 14, mice were boosted twice at two-week intervals with 50% of the original dosage of both antigen and adjuvant (days 14 and 28). To evaluate serum antibody titers, blood was collected by submandibular puncture 10 days after each immunization (days 10, 24, and 38). Serum was separated from whole blood by centrifugal separation at 10,000g for 5 min at 25 °C using Microvette 500 Ser-Gel collection vessels with a clotting activator.
Absolution quantification of total IgG and total IgM antibody analysis was performed using the mouse uncoated total IgG and total IgM ELISA kits based on manufacturer-recommended protocols (Thermo Fisher). Antigen-specific IgG, IgG1, and IgG2a antibody titers were quantified based on previously established protocols for indirect ELISA, with minor modifications.103 Specifically, HER2 peptides (200 μL, 100 μg/mL in 100 mM carbonate buffer, pH 9.4) were chemically conjugated to ELISA plates through the terminal amine group utilizing Nunc Immobilizer Amino immunoassay plates by overnight incubation with exposure to light at room temperature. Following overnight incubation, ELISA plates were washed three times with 100 mM PBS pH 7.4 with 2% Tween-20. The ELISA plates were then blocked overnight at 4 °C with 300 μL of ELISA blocked (Pierce Protein-Free PBS Blocking Buffer) and washed three times. Serum samples containing primary antibodies were serially diluted (101–108-fold) using 100 mM PBS pH 7.4 containing 10% ELISA blocker reagent and were added to each well (to200 μL total volume) for 2 b incubation at room temperature. After three washes, 100 μL of 500-fold diluted anti-IgG-HRP, anti-IgG1-HRP, or anti-IgG2a-HRP was added to each well and incubated for 1 h at room temperature. The ELISA plates were washed five times, and then 100 μL of 1-Step Ultra TMB Substrate Solution was added to each well. The solution was allowed to incubate and develop color for 15–20 min at room temperature with gentle agitation. Color development was stopped by the addition of 100 μL of 100 mM sulfuric add. Colorimetric development was quantified by absorbance spectroscopy at 450 nm using a BioTek Cytation 5 plate reader. Antibody titers were determined by any absorbance signal at a given dilution factor that was greater than the PBS control absorbance signal plus three standard deviations.
Anticancer Efficacy in BALB/c Mice with HER2+ Breast Cancer.
BALB/c mice were inoculated with 2.5 × 105 D2F2/E2 cells subcutaneously in the right flank. D2F2/E2 cells were prepared at a concentration of 2.5 × 106 cells/mL in 100 μL and were mixed with an equal volume with Matrigel matrix. Mice were subcutaneously immunized with different vaccine formulations, some in combination with anti-PD-1 antibody, via intraperitoneal injection. Tumor size was measured every 2 days. Tumor volumes were calculated as . End points were determined by using the End-Stage Illness Scoring System; mice receiving an End-Stage Illness Score greater than 6 were euthanized by CO2 asphyxiation. Survival rates were calculated by the Kaplan–Meier method and were compared by the log-rank test.
Single-Cell Sequencing for Immune Fingerprints of Tumors and Data Processing.
Tumor samples were harvested at 42 days after inoculation and dissociated into single-cell suspensions using a MA900 Cell Sorter (Sony). Single-cell suspensions were subjected to final cell counting on a Countess II Automated Cell Counter (Thermo Fisher) and diluted to a concentration of 700–1000 nuclei/μL. We built 3′ single-nucleus libraries using the 10x Genomics Chromium Controller and following the manufacturer’s protocol for 3′ V3.1 chemistry with NextGEM Chip G reagents (10x Genomics). Final library quality was assessed using TapeStation 4200 (Agilent), and libraries were quantified by Kapa qPCR (Roche). Pooled libraries were subjected to 150 bp paired-end sequencing according to the manufacturer’s protocol (Illumina NovaSeq 6000). Bcl2fastq2 Conversion Software (Illumina) was used to generate demultiplexed Fastq files, and aCellRanger Pipeline (10x Genomics) was used to align reads and generate count matrices.
CellRanger output and single-cell RNA-seq data were analyzed using the R package Seurat version 4.0. Quality control parameters were utilized to filter out dead cells, doublets, and cells without the minimal number of expressed genes. Raw unique molecular identifier (UMI) counts were log-normalized. Various genes were identified using the standard deviation from the mean (using only nonzero values). Data were scaled and centered by regressing library size and mitochondrial mRNA counts. Principal component analysis (PCA) was performed using various genes. The first 15 principal components were used as the input for uniform manifold approximation and projection (UMAP) to reduce the dimensionality of the single-cell data and project them onto two-dimensional graphs. Clusters were identified using a shared nearest neighbor (SNN) modularity optimization-based clustering algorithm. Marker genes defining each cluster were identified using Seurat’s FindAllMarkers function, which employs a Wilcoxon rank sum test to determine significant genes. The SingleR package104 and the ImmGen reference (http://rstatsjmmgen.org/DataPage/)105 were used to assign cluster identity to individual cell types. The top 50 genes were analyzed in established gene expression data of immune cells, which can be obtained from ImmGen Data sets. In addition, cell clusters and markers were analyzed using the CellMarker database (http://bio-bigdata.hrbmu.edu.cn/CellMarker/index.jsp)106 and published signatures.42
Gene expression: For each cell, the gene expression measurement was normalized by its total expression, multiplied by a scale-factor of 10,000, and log-transformed. The collapsed gene signature score for each sample was computed as follows: The sum of the normalized count values of each gene in the signature was used to identify B/T cell clusters or all cells in a given sample. The median gene expression signature was scored for each sample to draw heatmaps and boxplots.
Immunofluorescence Staining of Tumor Lymphoid Follicle.
For immunofluorescence staining, tumor tissues were harvested at the end point. Harvested tissues were immediately fixed in 1% paraformaldehyde for 1 h and then immersed in 30% sucrose in 0.1% NaN3 in PBS overnight Treated tissues were then embedded in optical coherence tomography (OCT) compound and frozen in a CO2(s)+EtOH bath. Tissue sections (15 μm) were prepared and dried for 0.5 h before staining. Sections were then incubated with blocking solution for 1L For Figure 6g, secondary antibody staining methods were used. Primary antibodies used are rabbit antimouse CD20 antibody (invivogen). goat antimouse CD3 antibody (R&B Systems), mouse antimmise CD23 antibody (Invivogen), and rat antimouse PNAd antibody (Biolegend), Secondary antibodies (Abcam) used are donkey antirabbit antibody-AF488, donkey antigoat antibody-AF594, donkey antimouse antibody-AF647j and donkey antirat antibody-AF405. For Figure S25, primary staining with antibody conjugated to fluorophores were used. Markers used are Alexa Fluor 594 CD19 (B cells), FITC CD3 (T cells) and Aleia Fluor 647 antimouse CD21/CD35 (CR2/CR1). After staining, slides were mounted with VECTASHIELD Mounting Medium or DAPI Fluoromount-G for confocal imaging.
Immunohistochemistry (IHC) on Tumor Tissues.
Tumor tissues were harvested at the end point and immediately fixed in paraformaldehyde. The fixed tissues were then embedded in paraffin for preparation of 5 μm tissue sections. After deparaffinization, rehydration and antigen unmasking, slides were incubated with primary and secondary antibodies for immunostaining. Antibodies were used to identify different populations of immune cells: CD20 (B cells) and CD3 (T cells).
Tumor Rechallenge and Analysis of Immune Memory Cells and Activation Induced Marker Assay by Flow Cytometry.
At day 200, the mice with complete tumor remission after treatment were rechallenged by subcutaneous inoculation of 2.5 × 105 D2F2/E2 cancer cells. The control group was normal BALB/c mice. The mice were observed for 40 more days without any additional treatment. At 40 days after the rechallenge, mice were sacrificed. The lymph nodes, spleen, bone marrow, and peripheral Hood were harvested for flow cytometry, Tetramer staining optimization are same as antigen specific germinal center B cells. B220+ CD38+ GL-7− IgD− IgM− populations were identified as class switched memory B cells. HER2 tetramer positive class switched memory B cells were identified as antigen specific class switched memory B cells. Markers used are B220-AIexa Fluor 594, CD38-APC/Fire650, GL-7-FITC, IgD-Alexa Fluor700, and IgM-PE. CD3−R22GlowIg(H+L)+tetramer+ were identified as antigen specific plasma cells. Markers used are CD3-Alexa Fluor647, B220-APC/Fire750, and Ig(H+L)-PE. CDS− CD4+ CD44+ CD62L− populations were identified as CD4 T effector memory cells; CD8− CD4+ CD44+ CD62L+ populations were identified as CD4 T central memory cells. CD4− CD8+ CD44+ CD62L− populations were identified as CD8 T effector memory cells, CD4− CD8+ CD44+ CD62L+ populations were identified as CD8 T central memory cells. Markers used are CD4-PE, CD8-brilliant violet421, CD44-Alexa Fluor 647, CD62L-FTTC. CDS− CD4+ CD69+ CD103−/+ populations were identified as CD4 Tissue resident memory T cells. CD4− CD8+ CD69+ CD103+ populations were identified as CDS Tissue resident memory T cells. Markers used are CD4-PE, CDS-brilliant violet 421, CD69-APC/Fire750, and CD103-Alexa Fluor 488.
Spleen cells (2 million) from different vaccination groups were then incubated with HER2-B/CD4/CD8 peptides (2 μg/mL) for 20 h at 24 well plate. After incubation, cells were then collected and measured by flow cytometry. CD8− CD4+ CD44+ CD62L+ populations were identified as CD4 TCM cells. CD69+ CD40L+/− populations from CD4 TEM/TCM cells were identified as AIM+ CD4 TEM/TCM cells. Markers used are CD8-Alexa Fluor 594, CD4-FITC, CD44-brilliant violet 421, CD62L-APC/Fire750, CD69-PE, and CD40L-APC. CD4− CD8+ CD44+ CD62L+ populations were identified as CD8 TCM cells. CD25+ OX40+/− populations from CD8 TEM/TCM cells were identified as AIM*’ CD8 TEM/TCM cells. Markers used are CD8-Alexa Fluor 594, CD4-FITC, CD44-brilliant violet 421, CD62L-APC/Fire750, CD25-PE, and OX4G-APC.
Statistics.
Data are expressed as the mean ± standard deviation (SD), unless otherwise specified. Two groups were compared using the unpaired Student’s t-test. Means of multiple groups were compared with one-way analysis of variance (ANOVA) followed by Tukey’s post hoc pairwise comparison. AH probability values are two-sided, and p < 0.05 was considered statistically significant. Statistical analyses were carried out using the GraphPad Prism 9 software package.
Supplementary Material
ACKNOWLEDGMENTS
This work is partially supported by NIH grants to DS (R01 AI154072) and YLL (R01 DE026728) and a Cancer Center Support Grant (NIH P30 CA046592) for shared resource facility use (Immunology, Advanced Genomics, and Pharmacokinetics shared resources).
Footnotes
The authors declare the following competing financial interest(s): The University of Michigan has submitted a patent application, in which some authors are listed as inventors.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.3c13038.
Addtional nanoparticle characterization; in vitro crosslink test; B cell uptake; gating strategies for flow cytometry; additional results for B/CD4 crosstalk; confocol image of lymph node distribution and delivery efficiency; in vivo measurement of GC and Tfh in lymph node and spleen; in vivo measurement of antigen specific CD4 T cells; HER2-specific antibody production; efficacy on PyMT and 4T-1 tumor; additional results for single-cell analysis; evaluate TLS formation (PDF)
Contributor Information
Chengyi Li, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Ryan Clauson, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Luke F. Bugada, Department of Chemical Engineering, College of Engineering, University of Michigan, Ann Arbor, Michigan 48109, United States
Fang Ke, Department of Microbiology and Immunology, Medical School\ University of Michigan, Ann Arbor, Michigan 48109, United States.
Bing He, Department of Computational Medicine & Bioinformatics, Medical School, University of Michigan, Ann Arbor, Michigan 48109, United States.
Zhixin Yu, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Hongwei Chen, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Binyamin Jacobovitz, Microscopy Core, Medical School, University of Michigan, Ann Arbor, Michigan 48109, United States.
Hongxiang Hu, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Polina Chuikov, Department of Microbiology and Immunology, Medical School, University of Michigan, Ann Arbor, Michigan 48109, United States.
Brett Dallas Hill, Department of Chemical Engineering College of Engineering University of Michigan, Ann Arbor, Michigan 48109, United States.
Syed M. Rizvi, Department of Chemical Engineering College of Engineering University of Michigan, Ann Arbor, Michigan 48109, United States
Yudong Song, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Kai Sun, Department of Materials Science and Engineering College of Engineering University of Michigan, Arm Arbor, Michigan 48109, United States.
Pasieka Axenov, Department of Microbiology and Immunology, Medical School, University of Michigan, Ann Arbor, Michigan 48109, United States.
Daniel Huynh, Department of Microbiology and Immunology, Medical School, University of Michigan, Ann Arbor, Michigan 48109, United States.
Xinyi Wang, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Lana Garmire, Department of Computational Medicine & Bioinformatics, Medical School University of Michigan, Ann Arbor, Michigan 48109, United States.
Yu Leo Lei, Departments of Head and Neck Surgery, Cancer Biology, and Translational Molecular Pathology, the University of Texas M.D. Anderson Cancer Center, Houston, Texas 77054, United States.
Irina Grigorova, Department of Microbiology and Immunology, Medical School University of Michigan, Ann Arbor, Michigan 48109, United States.
Fei Wen, Department of Chemical Engineering College of Engineering, University of Michigan, Ann Arbor, Michigan 48109, United States.
Marilia Cascalho, Department of Microbiology and Immunology, Medical School University of Michigan, Ann Arbor, Michigan 48109, United States.
Wei Gao, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Duxin Sun, Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Data Availability Statement
The data that support the plots in this paper and other findings of this study are available from the corresponding author upon reasonable request. The code for single cell RNA-Seq analysis is available at: https://github.com/hebinghb/Tumor_B_Cell_Vaccine.
REFERENCES
- (1).Saxena M; van der Burg SH; Melief CJM; Bhardwaj N Therapeutic cancer vaccines. Nature Reviews Cancer 2021, 21 (6), 360–378. [DOI] [PubMed] [Google Scholar]
- (2).Hollingsworth RE; Jansen K, Turning the comer on therapeutic cancer vaccines. NPJ. vaccines 2019, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Jiang T; Shi T; Zhang H; Hu J; Song Y; Wei J; Ren S; Zhou C Tumor neoantigens: from basic research to clinical applications. J. Hematol Oncol 2019, 12 (1), 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hu Z; Leet DE; Allesøe RL; Oliveira G; Li S; Luoma AM; Liu J; Forman J; Huang T; Iorgulescu JB; et al. Personal neoantigen vaccines induce persistent memory T ceil responses and epitope spreading in patients with melanoma. Nat. Med. 2021, 27 (3), 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ott PA; Hu Z; Keskjn DB; Shukla SA; Sun J; Bozym DJ; Zhang W; Luoma A; Giobbie-Hurder A; Peter L; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547 (7662), 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ott PA, Hu-Lieskovan S Chmielowski B; Govindan R; Naing A; Bhardwaj N; Margolin K; Anrad MM; Hellmann MD; Lin JJ; et al. A Phase lb Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 2020,183 (2), 347–362. [DOI] [PubMed] [Google Scholar]
- (7).Sahin U; Oehm P; Derhovanessian E; Jabulowsky RA; Vormehr M; Gold M; Maurus D; Schwarck-Kokarakis D; Kuhn AN; Omokoko T; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585 (7823), 107–112. [DOI] [PubMed] [Google Scholar]
- (8).Modems and Merck announce mTNA-4157/V940, an investigational personalized mTNA cancer vaccine, in combination with Keytruda, was granted breakthrough therapy designation by the FDA for adjuvant treatment of patients with high-risk melanoma following complete resection. https://investors.modernatx.com/news/news-details/2023/Moderna-and-Merck-Announce-mRNA-4157V940-an-Investigational-Personaiized-mRNA-Cancer-Vaccine-in-Combination-With-KEYTRUDAR-pembroIizumab-was-Granted-Breakthrough-Therapy-Designation-by-the-FDA-for-Adjuvant-Treatment-of-Patients-With-Hjgh-Risk-Melanom/defaultaspx February 22, 2023.
- (9).MSK mRNA Pancreatic Cancer Vaccine Trial Shows Promising Results, https://www.mskcc.org/news/can-mrna-vaccines-fight-pancreatic-cancer-msk-clinical-researchers-are-trying-find-out May 31, 2022.
- (10).Cafri G; Gartner JJ; Zaks T; Hopson K; Levin N; Paria BC; Parkhurst MR; Yossef R; Lowery FJ; Jafferji MS; et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin Invest 2020, 130 (11), 5976–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Palmer CD; Rappaport AR; Davis MJ; Hart MG; Scallan CD; Hong SJ; Gitlin L; Kraemer LD; Kounlavouth S; Yang A; et al. Individualized, heterologous chimpanzee adenovirus and self-amplifying mRNA neoantigen vaccine for advanced metastatic solid tumors: phase 1 trial interim results. Net Med. 2022, 28 (8), 1619–1629. [DOI] [PubMed] [Google Scholar]
- (12).Platten M; Bunse L; Wick A; Bunse T; Le Cornet L; Harting L; Sahm F; Sanghvi K; Tan CL; Poschke l.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592 (7854), 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Awad MM; Govindan R; Balogh KN; Spigel DR; Garon EB; Bushway ME; Poran A; Sheen JH; Kohler V; Esaulova E; et al. Personalized neoantigen vaccine NEO-PV-01 with chemotherapy and anti-PD-1 as first-line treatment for non-squamous non-small cell lung cancer. Cancer Cell 2022, 40 (9), 1010–1026. [DOI] [PubMed] [Google Scholar]
- (14).Keskin DB; Anandappa AJ; Sun J; Tirosh I; Mathewson ND; Li S; Oliveira G; Giobbie-Hurder A; Felt K; Gjini E; et al. Neoantigen vaccine generates intratumoral T cell responses in phase lb glioblastoma trial. Nature 2019, 565 (7738), 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kissick HT Is It Possible to Develop Cancer Vaccines to Neoantigens, What Are the Major Challenges, and How Can These Be Overcome? Neoantigens as Vaccine Targets for Cancer. Cold Spring Harb Perspecf BioL 2018, 10 (11), a033704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kissick HT; Sanda MG The role of active vaccination in cancer immunotherapy: lessons from clinical trials. Curr. Opin Immunol 2015, 35, 15–22. [DOI] [PubMed] [Google Scholar]
- (17).Hailemichael Y; Dai ZM; Jaffarzad N; Ye Y; Medina MA; Huang XF; Dorta-Estremera S,M; Greeley N,R; Nitti G; Peng W,Y; et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Not Med. 2013, 19 (4), 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Finn OJ; Rammensee HG Is It Possible to Develop Cancer Vaccines to Neoantigens, What Are die Major Challenges, and How Can. These Be Overcome? Neoantigens: Nothing New in Spite of the Name. Cold Spring Harb Perspect Biol 2018, 10 (ll), a028829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sellars MC; Wu CJ; Fritsch EF Cancer vaccines: Building a Bridge over troubled waters. Cell 2022, 185 (15), 2770–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sette A; Crotty S Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184 (4), 861–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Heitmann JS; Bilich T; Tandler C.j Nelde A; Malinger Y; Marconato M; Rsusch J; Jäger S; Denk M; Richter M; et al. A COVID-19 peptide vaccine for the induction of SARS-CoV-2 T cell immunity. Nature 2022, 601 (7894), 617–622, [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Keeton R; Tincho MB; Ngomti A; Baguma R; Benede N; Suzuki A; Khan K; Cele S; Bernstein M; Karim F; et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature 2022, 603 (7901), 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tarke A; Coelho CH; Zhang Z; Dan JM; Yu ED; Methot N; Bloom NL; Goodwin B; Phillips E; Mallal S; et al. SARS-CoV-2 vaccination induces immunological T cell memory able to cross-recognize variants from Alpha to Omicron. Cell 2022, 185 (5), 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang Z; Mateus J; Coelho CH; Dan JM; Moderbacher CR; Gálvez RL; Cortes FH; Grifoni A; Tarke A; Chang J; et al. Humoral and cellular immune memory to four COVID-19 vaccines. Cell 2022, 185 (14), 2434–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yuen GJ; Demissie E; Pillai S B lymphocytes and cancer: a love-hate relationship. Trends Cancer 2016, 2 (12), 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Qin Z; Richter G; Schuler T; Ibe S; Cao X; Blankenstein T B cells inhibit induction of T cell-dependent tumor immunity. Not Med. 1998, 4 (5), 627–630. [DOI] [PubMed] [Google Scholar]
- (27).Shah S; Divekar AA; Hilchey SP; Cho HM; Newman CL; Shin SU; Nechustan H; Challita-Eid PM; Segal BM; Yi KH; et al. Increased rejection of primary tumors in mice lacking B cells: inhibition of anti-tumor CTL and TH1 cytokine responses by B cells. Inf. J. Cancer 2005, 117 (4), 574–586. [DOI] [PubMed] [Google Scholar]
- (28).Brodt P; Gordon J, Anti-tumor immunity in B lymphocyte-deprived mice, I. Immunity to a chemically induced tumor, J Immunol 1978, 121 (1), 359–362. [PubMed] [Google Scholar]
- (29).Barbera-Guillem E; Nelson MB; Barr B; Nyhus JK; May KF Jr; Feng L; Sampsel JWB lymphocyte pathology in human colorectal cancer. Experimental and clinical therapeutic effects of partial B cell depletion. Cancer Immunol Immunother 2000, 48 (10), 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shen P; Fillatreau S Antibody-independent functions of B cells: a focus on cytokines. Nat Rev. Immunol 2015, 15 (7), 441–451. [DOI] [PubMed] [Google Scholar]
- (31).DiLillo DJ; Yanaba K; Tedder TF B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J. Immunol 2010, 184 (7), 4006–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).de Jonge K; Tille L; Loureaco J; Maby-El Hajjami H; Nassiri S; Racle J; Gfeller D; Delorenzi M; Verdeil G; Baumgaertner P; et al. Inflammatory B cells correlate with failure to checkpoint blockade in melanoma patients. Oncoimmunobgji 2021, 10 (1), 1873585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).de Visser KE; Korets LV; Coussens LM De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent Cancer Cell 2005, 7 (5), 411–423. [DOI] [PubMed] [Google Scholar]
- (34).Kanmaya PT B-cell epitope peptide cancer vaccines: a new paradigm for combination immunotherapies with novel checkpoint peptide vaccine. Future Oncol 2020, 16 (23), 1767–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Helmink B. A Reddy;, S. M.j Gao J; Zhang S; Basar R; Thakur R; Yizhak K; Sade-Feldman M; Blando J; Han G; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577 (7791), 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Cabrita R; Lauss M; Sanna A; Donia M; Skaarup Larsen M; Mitra S; Johansson l.; Phung B; Harbst K; Vallon-Christersson J; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577 (7791), 561–565. [DOI] [PubMed] [Google Scholar]
- (37).Petitprez F; de Reyniés A; Keung EZ; Chen TW,.; Sun CM; Calderaro J; Jeng YM; Hsiao LP; Lacroix L; Bougoüin A; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577 (7791), 556–560. [DOI] [PubMed] [Google Scholar]
- (38).Kroeger DK; Milne K; Nelson BH Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin. Cancer Res. 2016, 22 (12), 3005–3015, [DOI] [PubMed] [Google Scholar]
- (39).Garaud S; Buisseret L; Solinas C; Gu-Trantien C; de Wind A; Van den Eynden G; Naveaux C; Lodewyckx JN; Boisson A; Duvillier H, Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight 2019, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sagiv-Barfi L; Czerwinski DK; Shree T; Lohmeyer JJK; Levy R Intratumoral immunotherapy relies on B and T cell collaboration. Sci Immunol 2022, 7 (71), No. eabn5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cui C; Wang J; Fagerberg E; Chen PM; Connolly KA; Damo M; Cheung JF; Mao T; Askari AS; Chen S; et al. Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell 2021, 184 (25), 6101–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Hollern DP,.; Xu N; Thennavan; Glodowski C; Garcia-Recio.; Mott KR;He X; Garay JP; Carey Ewend K; Marron D; et al. B Cells and T Follicular Helper Cells Mediate Response to Checkpoint Inhibitors in High Mutation Burden Mouse Models of Breast Cancer. Cell 2019, 179 (5), 1191–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wiehnd A; Patel MR; Cardenas MA; Eberhardt CS; Hudson WH; Obeng R C; Griffith CC; Wang X; Chen ZG; Kissick HT; et al. Defining HFV-specific B cell responses in patients with head and neck cancer. Nature 2021, 597, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chaurio RA; Anadon CM; Lee Costich T; Payne KK; Biswas S; Harro CM; Moran C; Ortiz AC; Cortina C; Rigolizzo KE,.; et al. TGF-β-mediated silencing of genomic organizer SATB1 promotes Tfh cell differentiation and formation of intra-tumoral tertiary lymphoid structures. Immunity 2022, 55 (1), 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Overacre-Delgoffe AE; Bumgarner.; Cillo A,R; Burr AH,P; Tometich j. T.; Bhattacharjee A; Bruno TC; Vignali DAA; Hand TW Microbiota-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. Immunity 2021, 54 (12), 2812–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Graalmann T; Borst K; Manchanda H; Vaas L; Bruhn M; Graalmann L; Koster M; Verboom M; Hallensleben M; Guzman CA; et al. B cell depletion impairs vaccination-induced CD8(+) T cell responses in a type I interferon-dependent manner. Ann. Rheum Din 2021, 80 (12), 1537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Guo L; Kapur R; Aslam R; Speck ER; Zufferey A; Zhao Y; Kim M; Lazarus A H.; Ni, H.; Semple, J. W. CD20+ B-cell depletion therapy suppresses murine CD8+ T-cell-mediated immune thrombocytopenia. Blood 2016, 127 (6), 735–738. [DOI] [PubMed] [Google Scholar]
- (48).Klarquist J; Cross EW; Thompson SB; Willett B; Aldridge DL; Caffrey-Carr AK; Xu Z; Hunter CA; Getahun A; Kedl RMB cells promote CD8 T cell primary and memory responses to subunit vaccines. Cell Rep 2021, 36 (8), 109591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Tanchot C; Rocha B CD8 and B cell memory: same strategy, same signals. Nat Immunol 2003, 4 (5), 431–432. [DOI] [PubMed] [Google Scholar]
- (50).Schudel A; Francis DM; Thomas SN Material design for lymph node drug delivery. Not rev Mater. 2019, 4 (6), 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wang J; Hu X; Xiang D Nanoparticle drug delivery systems: an excellent carrier for tumor peptide vaccines. Drug delivery 2018, 25 (1), 1319–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Fries CN j Curvino EJ; Chen JL; Permar SR; Fouda GG; Collier JH Advances in nanomaterial vaccine strategies to address infectious diseases impacting global health. Not Nanotechnol 2021, 16, 1. [DOI] [PubMed] [Google Scholar]
- (53).Bachmann MF,.; Jennings GT, Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Not Rev. Immunol 2010, 10 (11), 787–796. [DOI] [PubMed] [Google Scholar]
- (54).Singh A Eliciting B cell immunity against infectious diseases using nanovaccines. Not Nanotechnol 2021, 16 (1), 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Cheng W The Density Code for the Development of a Vaccine? J. Pharm. Sd. 2016, 105 (11), 3223–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hinton HJ; Jegerlehner A; Bachmann MF Pattern recognition by B cells: the role of antigen repetitiveness versus Toll-like receptors. Curr. Top Microbiol Immunol 2008, 319, 1–15. [DOI] [PubMed] [Google Scholar]
- (57).Pierce SK; Liu WL The tipping points in the initiation of B cell signalling: how small changes make big differences. Not Rev. Immunol 2010, 10 (11), 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zabel F; Kundig TM; Bachmann M, Virus-induced humoral immunity F: on how B cell responses are initiated. Curr. Opin Virol 2013, 3 (3), 357–362. [DOI] [PubMed] [Google Scholar]
- (59).Kim YM; Pan JY; Korbel GA; Peperzak V; Boes M; Ploegh HL Monovalent ligation of the B cell receptor induces receptor activation but fails to promote antigen presentation. Proc. Natl. Acad. Sci. U.S.A 2006, 103 (9), 3327–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Turner JS; Ke F; Grigorova IL B Cell Receptor Crosslinking Augments Germinal Center B Cell Selection when T Cell Help Is Limiting. Cell Rep 2018, 25 (6), 1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Maeda FY; van Haaren JJ; Langley DB; Christ D; Andrews NW; Song W Surface-associated antigen induces permeabilization of primary mouse B-cells and lysosome exocytosis facilitating antigen uptake and presentation to T-cells. eLije 2021, 10, e66984 DOl: 10.7S54/eLife.66984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Mohsen MO; Augusto G; Bachmann MF The 3Ds in virus-like particle based-vaccines: “Design, Delivery and Dynamics”, Immunol Rev. 2020, 296 (1), 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Chen Z; Wholey WY; Hassani Najafabadi A; Moon JJ; Grigorova l.; Chackerian B; Cheng W. Self-Antigens Displayed on Liposomal Nanoparticles above a Threshold of Epitope Density Elicit Class-Switched Autoreactive Antibodies Independent of T CeB Help. /. Immunol 2020, 204 (2), 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Ingale J; Stano A; Guenaga J; Sharma SK; Nemazee D; Zwick MB; Wyatt RT High-Density Array of Well-Ordered HIV-1 Spikes on Synthetic Liposomal Nanopattid.es Efficiently Activate B Cells. Ce 11 Rep 2016, 15 (9), 1986–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Cheng W The Density Code for the Development of a Vaccine? J. Pharm. Sci 2016, 105 (11), 3223–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Junt T; Moseman EA; Iannacone M; Massberg S; Lang PA; Boes M; Fink K; Henrickson SE; Shayakhmetov DM; Di Paolo NC; et al. Subcapsular sinus macrophages in lymph nodes dear lymph-borne viruses and present them to antiviral B cells. Nature 2007, 450 (7166), 110–114. [DOI] [PubMed] [Google Scholar]
- (67).Lu TT; Browning JL Role of the Lymphotoxin/LIGHT System in the Development and Maintenance of Reticular Networks and Vasculature in Lymphoid Tissues. Front immunol 2014, 5, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Johansson-Percival A; He B; Li ZJ; Kjellén A; Russell K; Li J; Larma I; Ganss R, De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat. Immunol 2017, 18 (11), 1207–1217. [DOI] [PubMed] [Google Scholar]
- (69).Tan YS; Sansanaphongpricha K; Xie Y; Donnelly CR; Luo X; Heath BR; Zhao X; Bellile E; Hu H; Chen H; et al. Mitigating SOX2-potentiated Immune Escape of Head and Neck Squamous Cell Carcinoma with a STING-inducing Nanosatellite Vaccine, din. Cancer Res. 2018, 24 (17), 4242–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Chen H, W.; Ren XQ; Paholak H,LJ; Burnett J; Ni F;Fang XL; Sun DX Facile Fabrication of Near-Infrared-Resonant and Magnetic Resonance Imaging-Capable Nanomediators for Photothermal Therapy. Acs Appl Mater. Inter 2015, 7 (23), 12814–12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Chen HW; Burnett J; Zhang FX; Zhang JM; Paholak H; Sun DX Highly crystallized iron oxide nanoparticles as effective and biodegradable mediators for photo thermal cancer therapy, J. Mater. Chem. B 2014, 2 (7), 757–765. [DOI] [PubMed] [Google Scholar]
- (72).Rockberg J; Sdhwenk JM j Uhlén, M. Discovery of epitopes for targeting the human epidermal growth, factor receptor 2 (HER2) with antibodies. Mol. oncol 2009, 3 (3), 238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Shukla S; Wen AM; Commandeur U; Steinmetz NF Presentation of HER2 epitopes using a filamentous plant virus-based vaccination platform.J. Mater. Chem. B 2014, 2 (37), 6249–6258. [DOI] [PubMed] [Google Scholar]
- (74).Jasmska J; Wagner S; Radauer C; Sedivy R; Brodowicz T; Wiltschke C; Breiteneder H; Pehamberger H; Scheiner O; Wiedermann U; et al. Inhibition of tumor cell growth by antibodies induced after vaccination with peptides derived from the extracellular domain of Her-2/neu. Int J. Cancer 2003, 107 (6), 976–983. [DOI] [PubMed] [Google Scholar]
- (75).Cascalbo M; Ma A; Lee S; Masat L; Wabl M A quasi-monoclonal mouse. Science (New York, N.Y.) 1996, 272 (5268), 1649–1652. [DOI] [PubMed] [Google Scholar]
- (76).Veneziano R; Moyer TJ; Stone MB,.; Wamhoff EC; Read BJ; Mukherjee S; Shepherd T,R; Das J; Schief WR; Irvine DJ; et al. Role of nanoscale antigen organization on B-cell activation probed using DNA origami. Nat. Nanotechnol 2020,15 (8), 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Bamden MJ; Allison J; Heath WR; Carbone FR Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunology and cell biology 1998, 76 (1), 34–40. [DOI] [PubMed] [Google Scholar]
- (78).Galluzzi L; Yamazaki T; Demaria S Heavy Metal to Rock the Immune Infiltrate. Trends Immunol 2017, 38 (8), 539–541. [DOI] [PubMed] [Google Scholar]
- (79).Whittington PJ; Radkevich-Brown O; Jacob JB; Jones R, F.; Weise, A M.; Wei, W. Z. Her-2 DNA versus cell vaccine: immunogenicity and anti-tumor activity. Cancer Immunol Immunother 2009, 58 (5), 759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Clifton GT; Gall V; Peoples GE; Mittendorf E A Clinical Development of the E75 Vaccine in Breast Cancer. Breast care (Basel, Switzerland) 2016, 11 (2), 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Peoples GE; Holmes JP; Hueman MT; Mittendorf EA; Amin A; Khoo S; Dehqanzada ZA; Gurney JM; Woll MM; Ryan GB; et al. Combined clinical trial results of a HER2/neu (E75) vaccine for die prevention of recurrence in high-risk breast cancer patients: U.S. Military Cancer Institute Clinical Trials Group Study 1–01 and 1–02. Clin. Cancer Res. 2008, 14 (3), 797–803. [DOI] [PubMed] [Google Scholar]
- (82).Mittendorf EA; Clifton GT; Holmes JP; Schneble E; van Echo D; Fonniah S; Peoples GE Final report of the phase I/II clinical trial of the E7S (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann. Oncol 2014, 25 (9), 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Costa RLB; Soliman H; Czerniecki BJ The clinical development of vaccines for HER2(+) breast cancer: Current landscape and future perspectives. Cancer Treat Rev. 2017, 61, 107–115. [DOI] [PubMed] [Google Scholar]
- (84).Schrörs B; Boegel S; Albrecht C; Bukur T; Buknr V; Holtsträter C; Ritzel C; Manninen K; Tadmor AD; Vormehr M; et al. Multi-Omics Characterization of the 4T1Murtie Mammary Gland Tumor Model. Front Oncol 2020, 10, 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Lin EY; Jones JG; Li P; Zhu L; Whitney KD; Muller W,J; Pollard JW Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. AJP 2003, 163 (5), 2113–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Tarantino P; Curigliano G; Tolaney SM Navigating the HER2-Low Paradigm in Breast Oncology: New Standards, Future Horizons. Cancer Discov 2022, 12 (9), 2026–2030. [DOI] [PubMed] [Google Scholar]
- (87).Muramatsu M; Sankaranand VS; Anant S; Sugai M; Kinoshita K; Davidson NO; Honjo T Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. JBC 1999, 274 (26), 18470–18476. [DOI] [PubMed] [Google Scholar]
- (88).Klein U; Dalla-Favera R Germinal centres: role in B-cell physiology and malignancy. Nat Rev. Immunol 2008, 8 (1), 22–33. [DOI] [PubMed] [Google Scholar]
- (89).Schumacher TN; Thommen DS Tertiary lymphoid structures in cancer. Science (New York, N.Y.) 2022, 375 (6576), No. eabf9419. [DOI] [PubMed] [Google Scholar]
- (90).Senapati S; Darling RJ; Ross KA; Wannemeuhler MJ; Narasimhan B; Mallapragada SK Self-assembling synthetic nanoadjuvant scaffolds cross-link B cell receptors and represent new platform technology for therapeutic antibody production. Sci. Adv. 2021, 7 (32), eabjl691 DOI: 10.1126/sciadv.abj1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Srinivas Reddy A; Tsuurkas P K; Raychaudhuri, S. Monte Carlo study of B-cell receptor clustering mediated by antigen crosslinking and directed transport. Cell Mol. Immunol 2011, 8 (3), 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Bonasia CG; Abdulahad WH; Rutgers A; Heeringa P; Bos NA B Cell Activation and Escape of Tolerance Checkpoints: Recent Insights from Studying Autoreactive B Cells. Cells 2021, 10 (5), 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Thierry GR; Kuka M; De Giovanni M; Mondor I; Brouilly N; Iannacone M; Bajénoff M The conduit system exports locally secreted IgM from lymph nodes, J. Exp Med. 2018, 215 (12), 2972–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Franklin MC; Carey KD; Vajdos FF; Leahy DJ; de Vos AM; Sliwkowski MX Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex Cancer Cell 2004, 5 (4), 317–328. [DOI] [PubMed] [Google Scholar]
- (95).Clauson RM; Chen M; Scheetz LM; Berg B; Chertok B Size-Controlled Iron Oxide Nanoplatforms with Lipidoid-Stabilized Shells for Efficient Magnetic Resonance Imaging-Trackable Lymph Node Targeting and High-Capacity Biomolecule Display. ACS Appl. Mater. Interfaces 2018, 10 (24), 20281–20295. [DOI] [PubMed] [Google Scholar]
- (96).Tong S; Hou S; Ren B; Zheng Z; Bao G Self-assembly of phospholipid-PEG coating on nanoparticles through dual solvent exchange. Nano Lett. 2011, 11 (9), 3720–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Udenfriend S; Stein S; Böhlen P; Dairman W; Leimgruber W; Weigele M Fluorescamine: a reagent for assay of amino acids, peptides, proteins, and primary amines in the picomole range. Science 1972, 178 (4063), 871–872. [DOI] [PubMed] [Google Scholar]
- (98).Mingueneau M; Krishnaswamy S; Spitzer MH; Bendall SC; Stone EL; Hedrick SM; Pe’er D; Mathis D; Nolan GP; Benoist C Single-cell mass cytometry of TCR signaling: amplification of small initial differences results in low ERK activation in NOD mice. Proc. Natl. Acad. Sci. U.S.A. 2014, 111 (46), 16466–16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Billi AC; Gharaee-Kermani M; Fullmer J; Tsoi LC; Hill BD; Gruszka D; Ludwig J; Xing X; Estadt S; Wolf SJ The female-biased factor VGLL3 drives cutaneous and systemic autoimmunity. JCI Insight 2019, 4 (8), el27291 DOI: 10.1172/jci.insight.127291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Franz B; May KF, Jr,; Dranoff G; Wucherpfennig K Ex vivo characterization and isolation of rare memory B cells with antigen tetramers. Blood 2011, 118 (2), 348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).DiPiazza AT; Leist SR; Abiona OM; Moliva JI; Werner A; Minai M; Nagata BM; Bock KW; Phung E; Schäfer A; et al. COVID-9 vaccine mRNA-1273 elicits a protective immune profile in mice that is not associated with vaccine-enhanced disease upon SARS-CoV-2 challenge. Immunity 2021, 54 (8), 1869–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Reiss S; Baxter AE; Cirelli K,M; Dan JM; Moron A; Daigneault A; Brassard N; Silvestri G; Routy JP; Havenar-Daughton C; et al. Comparative analysis of activation induced marker (AIM) assays for sensitive identification of antigen-specific CD4 T cells. PloS one 2017, 12 (10), No. e0186998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Sanchez AB; Nguyen T; Dema-Ala R; Kummel AC; Kipps TJ; Messmer BT A general process for the development of peptide-based immunoassays for monoclonal antibodies. Cancer Chemothera Pharmacol 2010, 66 (5), 919–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Aran D; Looney AP; Liu L; Wu E; Fong V; Hsu A; Chak S; Naikawadi RP; Wolters PJ; Abate A R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage, Nat Immunol 2019, 20 (2), 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Heng TS; Painter MW; et al. The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol 2008, 9 (10), 1091–1094. [DOI] [PubMed] [Google Scholar]
- (106).Zhang X; Lan Y; Xu J; Quan F; Zhao E; Deng C; Luo T; Xu L; Liao G; Yan M; et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Adds Res. 2019, 47 (D1), D721–d728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the plots in this paper and other findings of this study are available from the corresponding author upon reasonable request. The code for single cell RNA-Seq analysis is available at: https://github.com/hebinghb/Tumor_B_Cell_Vaccine.