

Abstract
IL-23 is a potent stimulus for Th17 cells. These cells have a distinct developmental pathway from Th1 cells induced by IL-12 and are implicated in autoimmune and inflammatory disorders including multiple sclerosis (MS). TGF-β, IL-6, and IL-1, the transcriptional regulator RORγt (RORC) and IL-23 are implicated in Th17 development and maintenance. In human polyclonally activated T cells, IL-23 enhances IL-17 production.
The aims of our study were: 1). To validate microarray results showing preferential expression of platelet activating factor receptor (PAF-R) on IL-23 stimulated T cells. 2). To determine whether PAF-R on activated T cells is functional, whether it is co-regulated with Th17-associated molecules, and whether it is implicated in Th17 function. 3). To determine PAF-R expression in MS.
We show that PAF-R is expressed on activated T cells, and is inducible by IL-23 and IL-17, which in turn are induced by PAF binding to PAF-R. PAF-R is co-expressed with IL-17 and regulated similarly with Th17 markers IL-17A, IL-17F, IL-22 and RORC. PAF-R is upregulated on PBMC and T cells of MS patients, and levels correlate with IL-17 and with MS disability scores. Our results show that PAF-R on T cells is associated with the Th17 phenotype and function.
Clinical Implications
Targeting PAF-R may interfere with Th17 function and offer therapeutic intervention in Th17-associated conditions, including MS.
Keywords: Interferon gamma, Interleukin 12, Interleukin 17, Interleukin 23, Multiple sclerosis, Platelet activating factor, Platelet activating factor receptor
1. Introduction
The role of Th1 cells, which produce interferon (IFN)-γ, Th17 cells (which produce IL-17), and Th1-17 cells (which produce both IFN-γ and IL-17 and are likely pathogenic) in multiple sclerosis (MS) in particular has been amply studied (Constantinescu and Gran, 2014; Edwards et al., 2010). Several immunotherapeutic agents for MS have been reported to reduce the levels of these cytokines (Balasa et al., 2017; Montes Diaz et al., 2018).
IL-12 and IL-23 are related proinflammatory cytokines involved in the development and maintenance of Th1 cells and Th17 cells, respectively. IL-12 and IL-23 share the p40 subunit which is covalently linked with a unique p35 or p19 subunit, respectively (Teng et al., 2015).
Although TGF-β, in a proinflammatory environment characterised by the presence of IL-6, and possibly IL-1, along with the master regulatory transcription factor, retinoic orphan receptor-γt (ROR-γt), or its human orthologue RORC, are required for Th17 development and differentiation from naïve T cells, the presence of IL-23 receptor is required for Th17 terminal differentiation and effector functions (Bettelli et al., 2006; O’Garra et al., 2008).
IL-23 stimulates the production of large amounts of IL-17 by supporting and expanding Th17 cells, a phenomenon observed both in the murine and the human immune system (Gaffen et al., 2014). In turn, IL-17 induces a wide range of cytokines, chemokines and metalloproteinases, which contribute to inflammation and tissue destruction (McGeachy et al., 2019).
The ability of IL-23 to induce IL-17 production in human polyclonally activated T cells and additional differential effects of IL-23 and IL-12 have been demonstrated (Hoeve et al., 2006). In this study, we investigated, using custom human microarrays, transcripts differentially induced by IL-12 and IL-23 in polyclonally activated human T cells (phytohemagglutinin-induced T cell blasts) These studies identified 205 genes significantly (over 8-fold relative to IL-12) up-regulated by IL-23 and 126 up-regulated over 8-fold relative to IL-23 by IL-12 (supplementary Table 1). We identified platelet activating factor receptor (PAF-R), a G-protein coupled 7-transmembrane domain receptor, to be significantly upregulated by IL-23 but down-regulated by IL-12. The enzyme, cytosolic phospholipase A2α, which catalyses the synthesis of three inflammatory mediators, including leukotrienes, prostaglandins and PAF, was also upregulated by IL-23 and downregulated by IL-12, suggesting that the PAF pathway is differentially regulated by these cytokines. We subsequently confirmed these findings in activated, purified T cell populations. Platelet activating factor (PAF), the ligand for PAF-R is a potent phospholipid inflammatory mediator that is associated with diverse effects on a variety of cells (Kihara, 2019; Honda et al., 2002).
PAF plays an important role in asthma and anaphylaxis, but also is elevated in the peripheral blood and cerebrospinal fluid of patients with MS (Callea et al., 1999), in other human autoimmune diseases (Edwards and Constantinescu, 2009), and in the central nervous system of mice with experimental autoimmune encephalomyelitis (EAE) (Lock et al., 2002), the most widely used model of MS (Constantinescu et al., 2011). PAF-R was also among the genes associated with allergies that were discovered by microarray analysis in brains of patients with MS, suggesting PAF/PAF-R may contribute to the MS pathology (Lock et al., 2002; Pedotti et al., 2003). This possibility was subsequently validated by the same group of investigators in EAE, where a PAF-R antagonist suppressed disease (Pedotti et al., 2003). This approach was successful in other EAE models as well (El Behi et al., 2007; Howat et al., 1989). Moreover, cPLA2a deficient mice are resistant to EAE and PAF-R deficient mice develop less severe disease (Marusic et al., 2005, 2008). In addition, PAF has been involved in neurodegeneration in EAE and PAF-R blockade is neuroprotective (Bellizzi et al., 2016).
Our microarray findings that PAF-R is expressed on activated T cells (which until now was not clearly known) and induced by IL-23 implicates PAF-R in the Th17 response and Th17 cell phenotype. This is also supported by recent findings showing a role for PAF-R in Th17 responses.
Here, we provide evidence to validate our microarray results and confirm that PAF-R is upregulated by IL-23 in part through IL-17, and show PAF-R to be a potential marker of Th17 cells and the PAF-PAF-R pathway to be potentially important for the development or maintenance of this pathogenic T cell population.
2. Materials and methods
2.1. Cell preparation
Peripheral blood mononuclear cells (PBMC) from healthy donors were isolated by gradient centrifugation with Histopaque 1077 (Sigma-Aldrich Dorset UK), prepared at 1 × 106 cells/ml in RPMI 1640 with 2 mM glutamine, 20 mM Hepes, 0.1 mg/ml penicillin and streptomycin and 10% fetal calf serum and cultured with 10μg/ml PHA at 37 °C and 5% CO2 for 72 h, then stimulated with 100U/ml IL-2 for 24 h and then rested 24 h in serum free media.
CD3+, CD4+ and CD8+ T cell populations were separated immunomagnetically using EasySep® (StemCell Technologies, UK).
PBMC were obtained as above from MS patients (20 females, 10 males, mean ± SD age, 43.8 ± 8.5) and matched controls (11 females, 9 males; mean ± SD age, 42.1. ± 7.8). Ethical approval was obtained from the Nottingham Research Ethics Committee 2 (NS090102). Subjects gave informed consent. MS was scored clinically using the Expanded Disability Status Scale (EDSS) (Kurtzke, 1983). PAF-R and IL-17 mRNA were measured by qRT-PCR (see below). In a separate group of 8 patients (6 females, 2 males, mean ± SD age, 43.1 ± 8.1) and 7 controls (4 females, 3 males, mean age 38 years), CD3± T cells were separated and PAF-R was measured in PBMC and T cells. The proportion of PAF-R RNA from T cells was consistently 50–60%.
2.2. Cell stimulation
Human PHA/IL-2T cell blasts or CD4+ cells stimulated with anti CD3/CD28, both at 1 × 106cells/ml, were either left untreated or treated as follows: incubated with IL-12 (100 ng/ml); IL-23 (10 ng/ml); IL-12 (100 ng/ml) & anti-IFN-γ (2.5 ng/ml); IL-23 & anti-IL-17 (10 ng/ml); IFN-γ (2.5 ng/ml); or IL-17 (0.5 ng/ml) for 24 h at 37 °C and 5% CO2. Rabbit and goat IgG were used as controls for anti-IFN-γ and anti-IL-17, respectively.
In other experiments T cells were also incubated with TGF-β (50 ng/ml) & IL-6 (20 ng/ml), or TGF-β & IL-6 & IL-23 for 24 h. The method was refined so optimum cytokine concentrations were used. PAF-R was inhibited using antagonist CV3988 (Biomol) (10 μM/ml) (Terashita et al., 1983). In other experiments, the PAF-R antagonist WEB2086 was used (Casals-Stenzel et al., 1987). Both inhibitors are reported to have similar effects (Hellewell and Williams, 1989). Cells were stimulated with PAF (Sigma) (3.6 nM) or IL-23 for 24 h. In some experiments, T cells were also stimulated in solution with 1 μg/ml each of anti-CD3 and anti-CD28 antibodies (Beckman Coulter, Paris Nord, Roissy, France) in the presence or absence of IL-12 (100 ng/ml), IL-23 (10 ng/ml), IL-12 (100 ng/ml) & anti-IFN-γ (2.5 ng/ml), IL-23 & anti-IL-17, IFN-γ (2.5 ng/ml) or IL-17 (0.5 ng/ml) for 24 h.
2.3. Intracellular staining
Brefeldin A was added for the last 4–6 h of a 24 h incubation with cytokines. After the 24 h, cells were fixed in 2% formaldehyde at room temperature for 5 min, washed by centrifugation once in PBA (PBS, 0.5% bovine serum albumin and 1% sodium azide), once in saponin buffer (PBA + 0.1% saponin) and once in 10% FCS in saponin buffer at 300g for 5 min. The supernatant was poured off and the cells resuspended in the residue, to which 10 μl anti-PAF-R (mouse anti-human monoclonal IgG2a antibody, Cayman Chemical) or isotyope control (Zymed) was added. Preliminary experiments on human PBMC showing that the adsorption of the antibody with the immunizing peptide LGFQDSKFHQ (Cayman Chemical) abolished the fluorescence on flow cytometry proved the specificity of the antibody. Cells were incubated, washed by centrifugation with saponin buffer, then incubated with 1 μg of FITC-conjugated goat anti mouse IgG (Zymed) for 30 min at room temperature. Staining with the directly PE-conjugated primary anti-IFN-γ and anti-IL-17 mAb (eBioscience) was as above except the second step (secondary antibody) was skipped. For double staining, cells were then blocked with unlabelled mouse Ig (Zymed) before staining with directly labeled mouse mAb. All cells were washed with 1 ml saponin buffer and resuspended in 0.5% formaldehyde for flow cytometry.
2.4. Quantitative real time PCR and non-quantitative PCR
qRT- PCR was used to assess PAFR, IL-17A and F, IL-22 and RORC mRNA abundance in human T cells. RNA was extracted using RNeasy kit (Qiagen). First-strand cDNA synthesis was initiated from 0.5 μg total RNA, using random hexamers and AMV reverse transcriptase (Promega). Oligonucleotide primer sequences were as follows: PAFR: forward 5′ CCTCCTTAGCACCAACTGTGTC 3′, reverse 5′ CAACCACTTCAGTGACCGTATCC 3′; β2microglobulin forward 5′ CTCCGTGGCCTTAGCTGTG 3′, reverse 5′ ATGTGTCTGGGTTTCATCCATC 3′; IL-17A forward 5′ GCACAAACTCATCCATCCC 3′, reverse 5′ CATAGTGAA GGCAGGAATCAC 3′; IL-17F forward 5′ TGCACAAAGTAAGCCACCAG 3′, reverse GCTTGCCTTTCTGAGTG AGG 3′; RORC forward 5′ TGCCAACAACCACACAGTCT 3′, reverse 5′ GATGGAAAGCCAGTTCCAAA 3′. IL-22 forward 5′ CTCCTTCTCTTGGCCCTCTT 3′, reverse 5′ GTTCAGCACCTGCTTCATCA 3′. qRT-PCR was carried out as described (Fahey et al., 2006).
Non-quantitative PCR was also carried out on purified CD3+ cells as follows: PBMC were isolated by gradient centrifugation with Histopaque 1077 (Sigma Aldrich, Dorset, and U.K) and stimulated with PHA for 72 h and recombinant human IL-2 for a further 24 h. The CD3+ human T cell enrichment kit (EasySep®) was utilized to obtain a purified population of CD3+ T cells. One million T cells were plated out into a 24 well plate and untreated or treated with 10 ng/ml of recombinant human IL-23 (Peprotech), or 2 ng phorbol dibutyrate (PDB)/1 μM Ionomycin for 18 h. The total RNA was extracted from the T cells by employing an RNeasy miniprepkit (Qiagen UK). DNase treatment of RNA samples was performed prior to RT-PCR (RQ1 RNase–free DNase, Promega, UK). The samples were analyzed by gel electrophoresis.
2.5. Measurement of intracellular calcium
Intracellular calcium was measured as previously described (Fox et al., 2004) on a Becton-Dickinson FACScan flow cytometer using Cellquest acquisition and analysis software. T cell blasts (5 × 10−6cells/ml) were loaded with the calcium fluorescent dye Fluo-3 AM (5 μM fc) at 37 °C for 30 min in the presence of the anion channel blocker probenecid (2.5 mM fc) to prevent leakage of the probe from the cells. 50 μl aliquots of the T cells were diluted in 940 μl of HEPES Tyrodes buffer containing 10 μl of calcium chloride to give a final concentration of 1 mM calcium. 250 μl of this suspension was applied to the flow cytometer to measure baseline fluorescence at time 0 s. A further 480 μl of the suspension was then added to 20 μl of PAF (3.6 nM fc) and median fluorescence measurements were recorded at 5, 15, 30, 60 and 120 s following addition of PAF. Further tests were performed in which no PAF was added and recordings were made at 5, 15, 30, 60 and 120 s. Untreated cells and cells that had been pre-treated with either IL-12 or IL-23 for 30 min were studied using this procedure.
2.6. ELISa
ELISA was performed to measure IL-17 in supernatants of stimulated T cells according to the manufacturer’s instructions (R&D, Abingdon, UK).
3. Results
3.1. PAF-R gene expression in T cells
In this study we validated our microarray analysis which had suggested that PAF-R is expressed by a subgroup of T cells and it was upregulated by IL-23 and down regulated by IL-12. PAF-R mRNA expression was assayed using quantitative real-time PCR (qPCR) in PHA/IL-2-derived T cell blasts and isolated CD4+ T cells from normal donors. Since IL-17 is a key cytokine induced by IL-23 we investigated the role of IL-17 in the IL-23 mediated induction of PAF-R mRNA.
Both IL-23 (p = 0.021, unpaired t-test) and IL-17 alone (p = 0.04, unpaired t-test) increased PAF-R mRNA expression in both T cell blasts (Fig. 1a) and purified CD4+ T cells stimulated with anti-CD3/anti-CD28 (Fig. 1b) (p = 0.001 for IL-23 and for IL-17, unpaired t-test) compared to cells not exposed to these cytokines. In the presence of a neutralizing IL-17 antibody (Fig. 1a and b), but not a control antibody (not shown), the induction of PAF-R by IL-23 was reduced to nearly baseline levels. Conversely, both IL-12 and IFN-γ decreased PAF-R mRNA expression (Fig. 1a & b) in both sets of stimulated cells (T cell blasts: IL-12, p = 0.021; IFN-γ, p = 0.03, unpaired t-test; CD4+ T cells: IL-12, p = 0.001; IFN-γ, p = 0.02, unpaired t-test). By adding a neutralizing IFN-γ antibody, but not a control antibody (not shown), the suppression induced by IL-12 was lost suggesting suppression is mediated by IFN-γ (Fig. 1a & b).
Fig. 1.
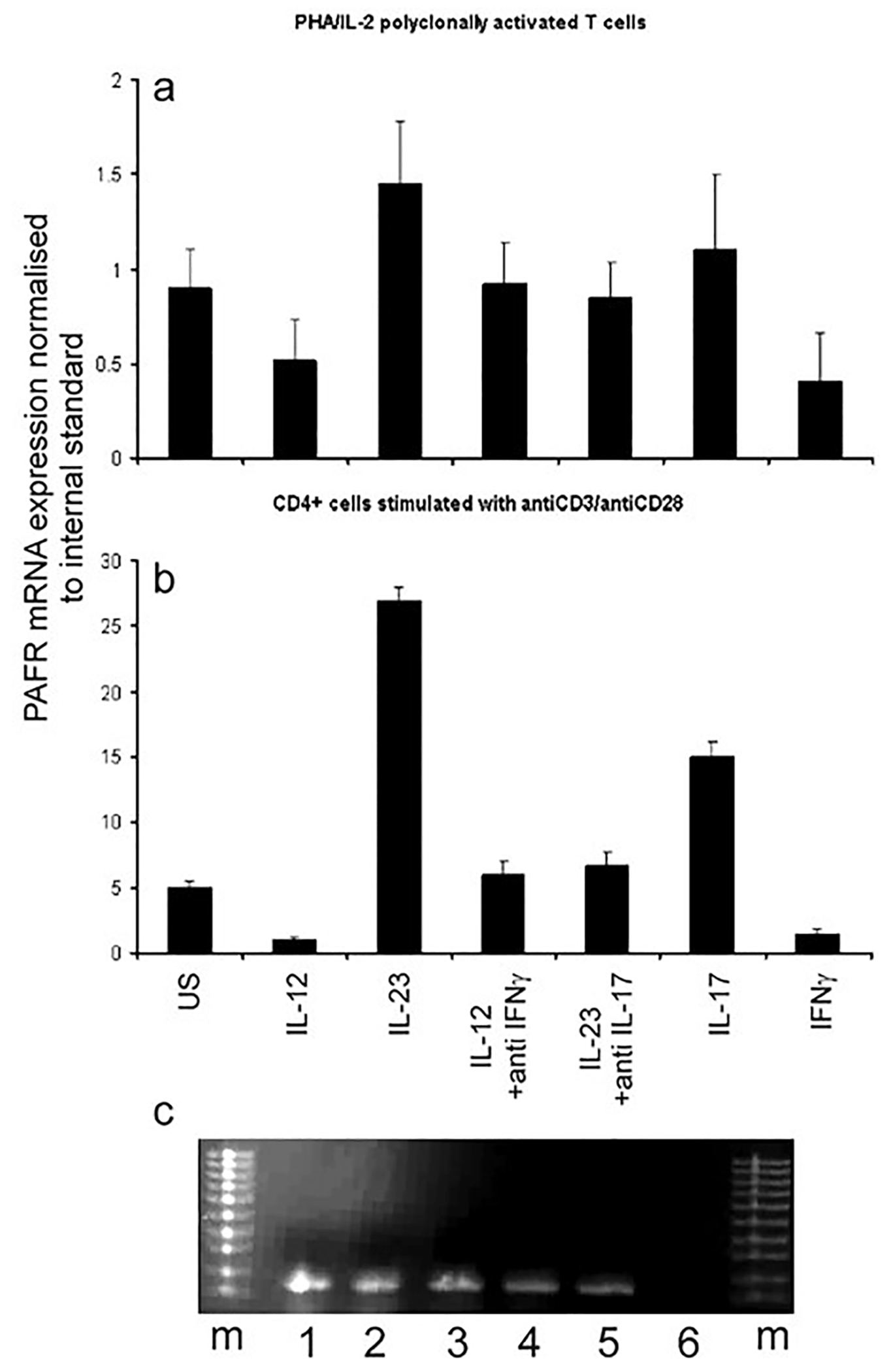
PAF-R gene expression in T cells. Quantitative reverse transcriptase real-time PCR was used to assess PAF-R abundance in human PHA/IL-2T cell blasts (a) and CD4+ cells stimulated with anti CD3/CD28 (b). Both sets of cells were either left untreated (US) or incubated with IL-12 (100 ng/ml), IL-23 (10 ng/ml), IL-12 (100 ng/ml) & anti-IFN-γ (2.5 ng/ml), IL-23 & anti-IL-17, IFN-γ (2.5 ng/ml) or IL-17 (0.5 ng/ml) for 24 h at 37 °C and 5% CO2. Rabbit and goat IgG were used as controls for anti-IFN-γ and IL-17, respectively and did not show an effect. Differences were as follows: a) means and SD are shown from 5 independent experiments. p = 0.023 US v IL-12; p = 0.021 US v IL-23; p = 0.03 US v IFN-γ; p = 0.04 US v IL-17. p values for US v the other conditions were not significant; p = 0.04 IL-12 v IL-12 v IL-12 + anti-IFN-γ; p = 0.03 IL-23 v IL-23 + anti-IL-17. b) p = 0.01 US v IL-12; p = 0.01 US v IL-23; p = 0.03 US v IFN-g; p = 0.03 US v IL-17; p = 0.01 IL-12 v IL-12 v IL-12 + anti-IFN-γ; p = 0.02 IL-23 v IL-23 + anti-IL-17. p values for US v the other conditions were not significant (c) PCR confirmation of PAF-R expression on purified CD3+ T cells. Agarose gel electrophoresis of PCR products as follows: m = molecular weight marker; 1: RNA from CD3+ cells stimulated with IL-23 (10 ng/ml) as described in materials and methods; 2: RNA from CD3+ cells stimulated with PDB/ionomycin; 3: DNAse treated RNA from CD3+ cells stimulated with PDB/ionomycin; 4: RNA from unstimulated CD3+ cells; 5: DNAse treated RNA from unstimulated CD3+ cells. 6. Lane loaded with the “no reverse transcriptase” negative control showing absence of genomic DNA. Representative gel of 3 separate experiments.
Because the PAF-R gene consists of a single exon, we took the following measures to rule out genomic DNA amplification by PCR: we used a RNA extraction kit that removes genomic DNA; we detected no product in the condition without reverse transcriptase; and we exposed the RNA to RNAse free DNAse without detecting differences in PCR products (Fig. 1c).
3.2. PAF-R protein expression in T cells
PAF-R expression was assessed at the protein level using flow cytometry. First we found very low level of expression of PAF-R protein in resting total CD3+ and CD3+ CD8–8 T cells when performing flow cytometry on whole unstimulated PBMC with gating on live lymphocytes. Positive fluorescence was also present in CD3–8 cells within the lymphocyte population (largely B cells) (Fig. 2a). The PAF-R protein level increases significantly in cells activated with PBD (20 ng/ml) and ionomycin (1 μg/ml) overnight. (Fig. 2a). We confirmed that PAF-R protein is also upregulated by IL-23 and downregulated by IL-12 in T cell blasts (Fig. 2b). The increase in fluorescence intensity compared to unstimulated cells ranged between 60 and 130% for IL-23. The fluorescence intensity reduction induced by IL-12 was between 40 and 100% (n = 5, p = 0.04 and 0.03; two-tailed unpaired t-test). The effects of IL-23 and IL-12 appear to be mediated, at least in part, through IL-17 and IFN-γ respectively. IL-23 up-regulation of PAF-R protein was reduced in the presence of a neutralising anti-IL-17 antibody (Fig. 2c) but not a control antibody (not shown). IL-12 suppression of PAF-R protein expression was reversed by adding a neutralizing IFN-γ antibody (Fig. 2d) but not a control antibody (not shown) (n = 3, p > 0.05 for IL-12 plus anti-IFN-γ and IL-23 plus anti-IL-17 compared to unstimulated cells; unpaired t-test). IFN-γ reduced the PAF-R protein by 43–120% (n = 5, p = 0.04), whereas IL-17 increased it by 50–100% (n = 4, p = 0.06). The results, expressed as percent change in mean fluorescence intensity, are shown in Fig. 2d. Combining IL-23 and IL-12 stimulation led to a slight (non-significant) reduction in the PAF-R level (n = 3, p = 0.1) (Fig. 2d).
Fig. 2.
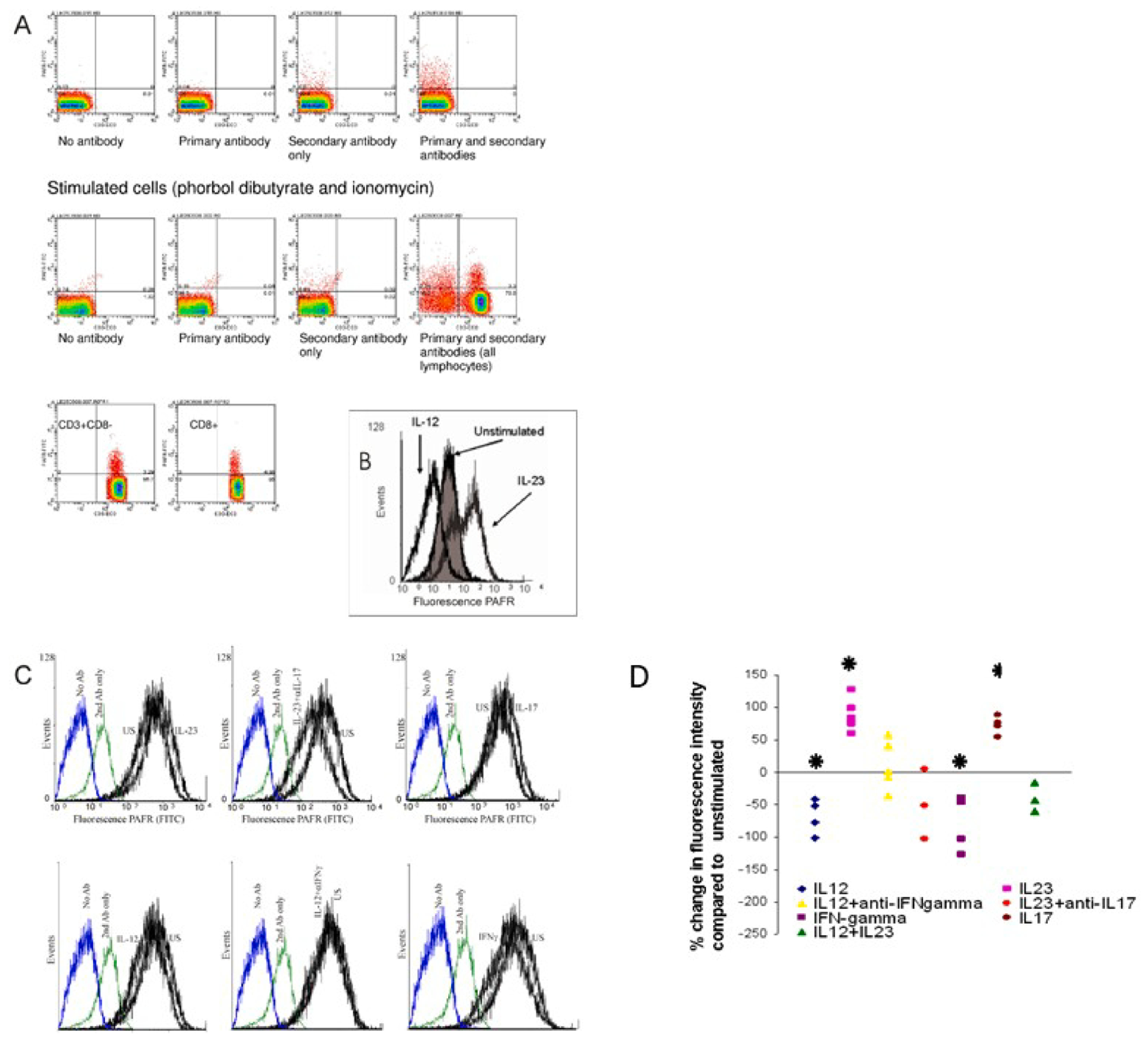
PAF-R protein expression in T cells. PAF-R protein expression was measured using flow cytometry. (A). This left panel shows unstimulated cells, gated on CD3+ CD8− lymphocytes, stained using anti-PAFR primary antibody with a FITC-labelled secondary antibody. Staining of PAFR on unstimulated CD4+ lymphocytes is <1%. The right panel shows cells have been stimulated with PDB/ionomycin overnight, stained and gated as above which significantly increases PAFR expression (6.17%). Representative scatterplot of 3 experiments; p < 0.01 for unstimulated vs stimulated. B) Histogram showing PAF-R expression on PHA/IL-2 T cell blasts stimulated with IL-12 vs IL-23. Results are shown from one representative experiment out of 5. (p = 0.029 for US vs IL-23; p = 0.04 for US vs IL-12). C) Involvement of IL-17 and IFN-γ in the effects of IL-23 and IL-12, respectively; and direct effects of IL-17 and IFN-γ on PAF-R protein expression. Representative results of 3 experiments. P = 0.04 for IL-17 and 0.045 for IFN-γD). Graph depicting percent changes in the expression of PAF-R in all the above stimulation conditions. Asterisk = p < 0.05; half-asterisk = p = 0.05.
3.3. Functionality of PAF-R on T cells
Although PAF-R is known to be expressed on a variety of cells, expression has not been extensively investigated on T cells. Generally, PAF-R is not thought to be expressed on resting T cells but our data indicated it could be up-regulated on activated T cells. Like many GPCR, PAF-R increases intracellular calcium, and PAF-R functional activity has been previously demonstrated in other cells by measuring intracellular calcium concentration. We found that 30 or 60 s stimulation with PAF 3.6 nM (Fig. 3a) or 7.2 nM (not shown) induced a significant increase in intracellular calcium concentration in the T cell blasts but this was not observed in cells exposed only to the PAF diluent and medium alone. Concentrations of PAF of 18 or 36 nM induced changes in size and granularity of the T cells within 120 s as noted on flow cytometry, suggesting decreased viability. There were no differences in the degrees of calcium increase in cells that had been briefly (30 min, unlikely to significantly modify PAF-R expression) pre-exposed to IL-12, IL-23 or no cytokine prior to stimulation with PAF 3.6 nM. (Fig. 3)
Fig. 3.
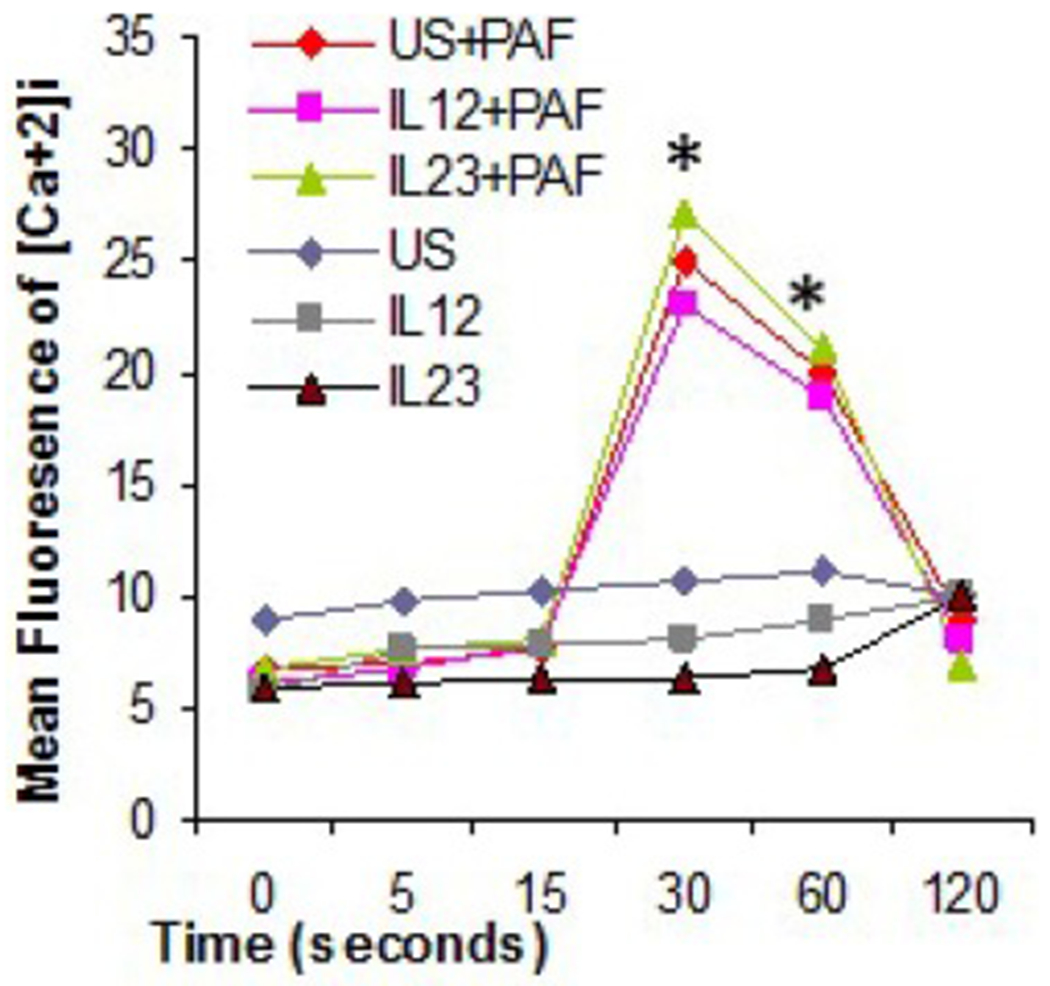
Functionality of PAF-R on T cells. Stimulation of T cells by PAF induces intracellular calcium release. Intracellular free calcium was measured using a FACScan flow cytometer after stimulation with 3.6 nM PAF (asterisk = p < 0.05). Results are shown as means of 3 independent experiments.
We have shown that IL-23 increases both PAF-R transcription and protein expression and that this effect is in part dependent on IL-17 and that IL-17 alone can enhance PAF-R expression. Investigating the time frame of these events using quantitative realtime PCR (qRT-PCR) we found that PAF-R mRNA expression is greater than IL-17 mRNA expression after 6 h of stimulation with IL-23 (1.6 vs 1.1 fold increase compared to unstimulated cells, respectively). However, by 12 h IL-17 message levels slightly exceed the PAF-R message levels (1.82 vs 1.78-fold induction) (Fig. 4a). We therefore hypothesised that PAF-R may not simply be induced in the development of Th17 cells but that PAF/PAF-R pathway may influence the IL-17 expression. We then examined the effect of PAF stimulation of T cell blasts on IL-17A production using ELISA. We found that PAF treatment induces IL-17A protein production, which can be blocked using PAF-R antagonist WEB2086 (Fig. 4b), suggesting a positive autocrine feedback loop inducing IL-17 expression and self-perpetuating Th17-mediated inflammation. In support of this we show that the addition of the synthetic competitive PAF-R antagonist, CV3988, also reduced the level of IL-17 induced by either IL-23 or PAF at both the mRNA (Fig. 4c) and protein level (data not shown), indicating that both IL-23 and PAF induce IL-17 and can jointly contribute to Th17 development.
Fig. 4.
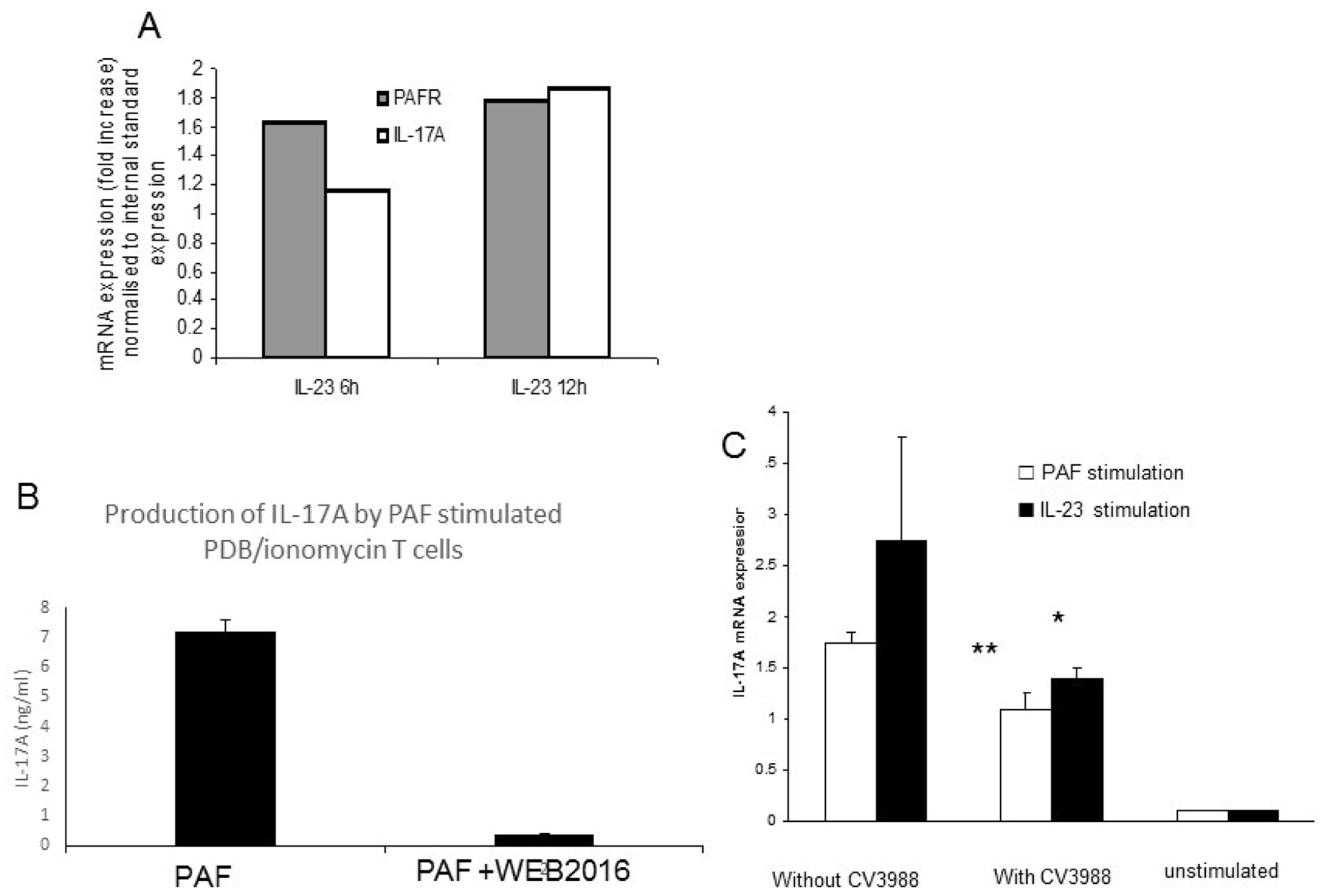
PAF/PAF-R interaction increases IL-17 expression in T cells blasts. (a) PAF increases IL-17 expression by T cell blasts as shown by intracellular staining (b). The addition of a competitive PAF-R antagonist (CV3988) reduced the level of IL-17 mRNA induced by both IL-23 and PAF.
3.4. PAF-R is co-regulated with IL-17 and other Th17 associated molecules
We used intracellular staining for IL-17 or IFN-γ followed by flow cytometry to determine whether PAF-R on T cell blasts co-expresses either or both of these cytokines. We show that PAF-R is co-expressed on cells producing IL-17 but not IFN-γ (Fig. 5). A population of cells in these experiments done on T cell blasts (>95% CD3+) appear to express PAF-R without co-expressing IL-17. We suspect they may represent CD8+ expressing PAF-R as shown in our experiments using PBD/ionomycin stimulation, and/or Th2 (PAF-R being involved in the allergic response, (Kasperska-Zajac et al., 2008)) or uncommitted activated T cells. Our results, however, suggest that PAF-R expression may distinguish Th17 cells from IFN-γ expressing Th1 cells.
Fig. 5.
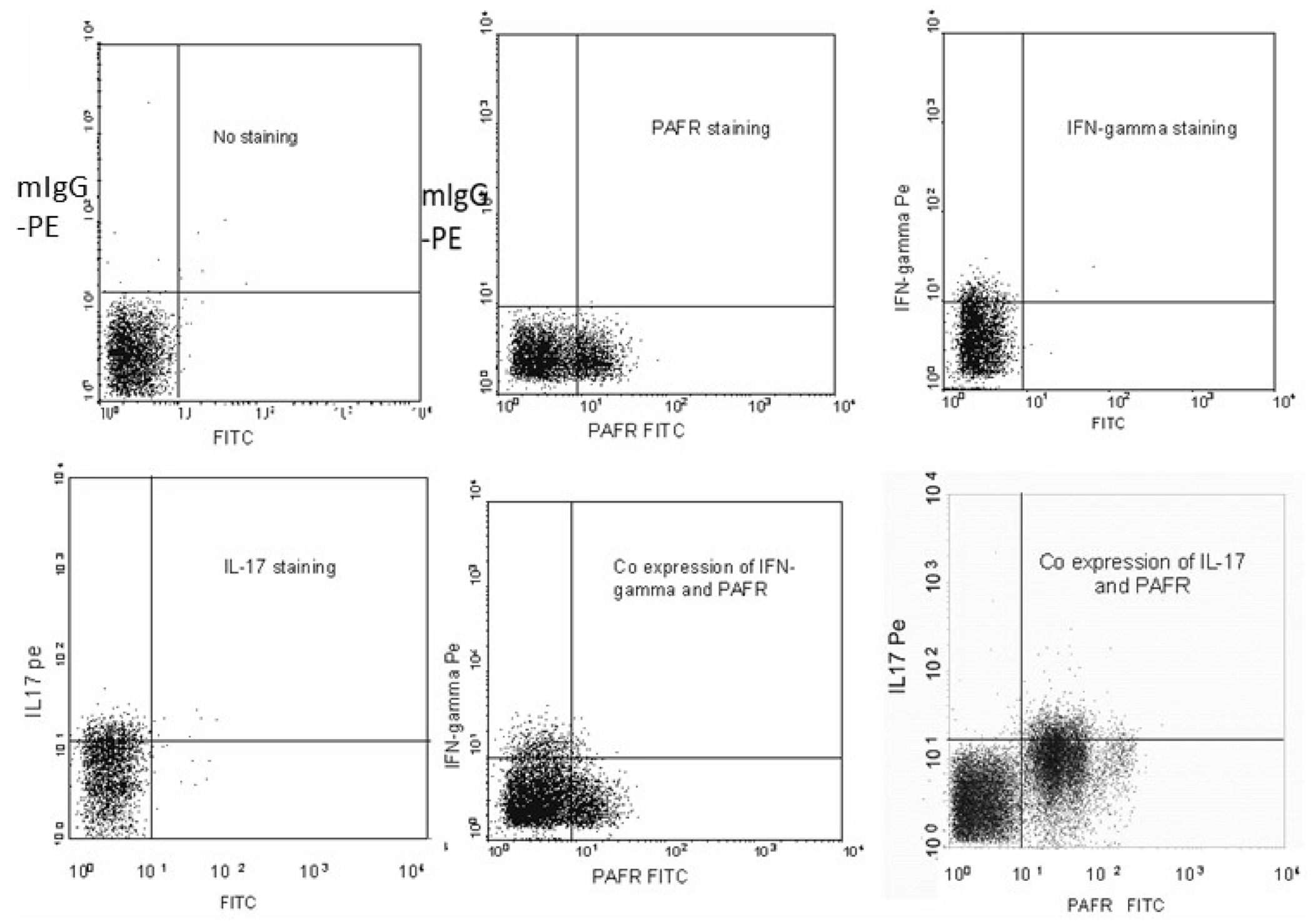
PAF-R is co-expressed with IL-17 but not with IFN-γ. T cell blasts were stained either alone with anti-IL-17 PE labelled antibody, anti-IFN-γ PE labelled antibody, and anti-PAF-R FITC labelled antibody or double stained with anti-IL-17 and anti-PAF-R or anti-IFN-gamma and anti-PAF-R antibodies. Expression and co-expression of the target proteins were measured using flow cytometry.
We investigated the expression of PAF-R in human T cells after stimulation with a combination of TGF-β, IL-6, and IL-23 as well as IL-23 and IL-12 alone. PAF-R mRNA expression was compared to that of IL-22, IL-17 and RORC. (see Fig. 6.) PAF-R expression followed a very similar trend in expression when compared to the other Th17 associated molecules, suggesting that PAF-R is co-regulated with other Th17 markers and molecules required for their development. In this serum-containing system, and using a mixed population of T cells including both naïve and memory cells, significant induction of Th17 markers was similar using IL-23 compared with TGF-β plus IL-6, or TGF-β, IL-6 and IL-23 in combination. Thus, compared to unstimulated cells, induction of PAF-R, RORC, IL-17A, IL-17F, and IL-22 was statistically significant (p < 0.05, unpaired t-test, n = 5 experiments). There were no differences between the induction methods using IL-23, TGF-β plus IL-6 or TGF-β plus IL-6 plus IL-23 (p > 0.05 for all other Th17 markers) with the exception of IL-22, which showed significantly less induction with IL-23 alone compared to the other stimuli (p < 0.05 for both TGF-β plus IL-6 and TGF-β plus IL-6 plus IL-23 compared to IL-23 alone; unpaired t-test); and for IL-17F which showed less induction with TGF-β plus IL-6 than with IL-23 alone (p = 0.045, unpaired t-test). Although we found significant induction of IL-17A, IL-17F, RORC and PAF-R in response to IL-22 (data not shown) this response was modest compared to that achieved with IL-23. IL-12 appeared to downregulate (PAF-R, RORC, IL-17A, IL-17F, p < 0.05) or fail to upregulate (IL-22, p > 0.05) these molecules (Fig. 6).
Fig. 6.
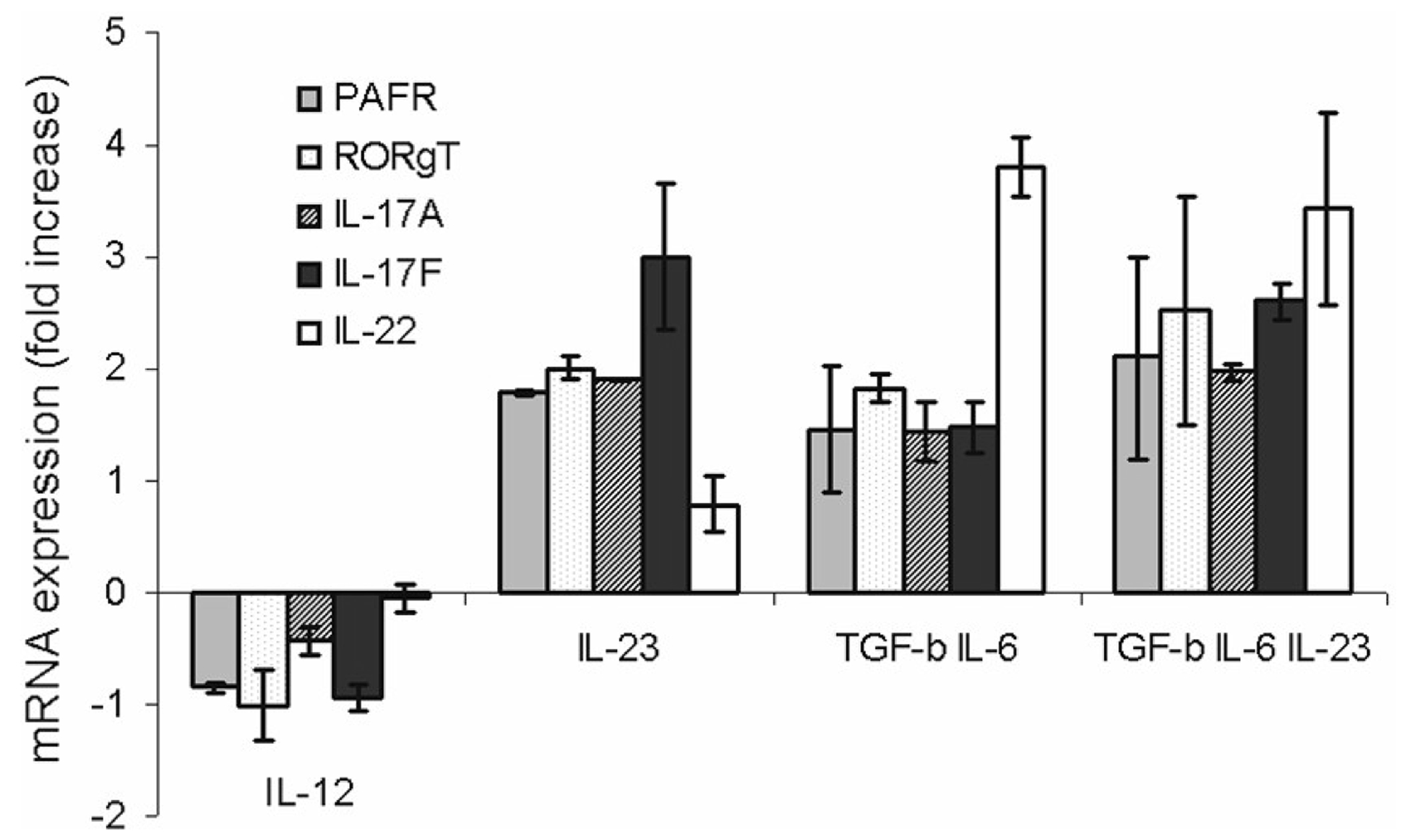
PAF-R expression is regulated in the same way as other Th17 associated molecules. PHA/IL-2 induced T cell blasts (1 × 106cells/ml) were either left untreated or incubated with IL-12 (100 ng/ml), IL-23 (10 ng/ml), TGF-β (50 ng/ml) & IL-6 (20 ng/ml) , or TGF-β (50 ng/ml) & IL-6 (20 ng/ml) & IL-23 (10 ng/ml) for 24 h at 37 °C and 5% CO2. RNA was extracted and PAF-R, IL-22, IL-17A&F and ROR γ T mRNA expression was measured using real time PCR.
3.5. Expression of PAF-R in multiple sclerosis patients
To determine the expression of IL-17 and PAF-R in peripheral blood of MS patients, we extracted RNA from peripheral blood mononuclear cells from 30 patients with relapsing MS and 20 age and sex matched controls. The demographic and clinical characteristics of these patients are listed in supplementary table 2. IL-17 and PAF-R mRNA expression was measured using real time PCR. MS patients have significantly higher PAF-R and IL-17A mRNA expression compared to controls; (Fig. 7a, b); p = 0.0001 and 0.02, respectively. PAF-R and IL-17A mRNA levels correlated well; Pearson’s r = 0.66; p = 0.023. Moreover, PAF-R mRNA correlated highly with MS disability scores as measured by the Expanded Disability Status Scale (EDSS) score; Pearson’s r = 0.61; p = 0.0003 (Fig. 7c).
Fig. 7.
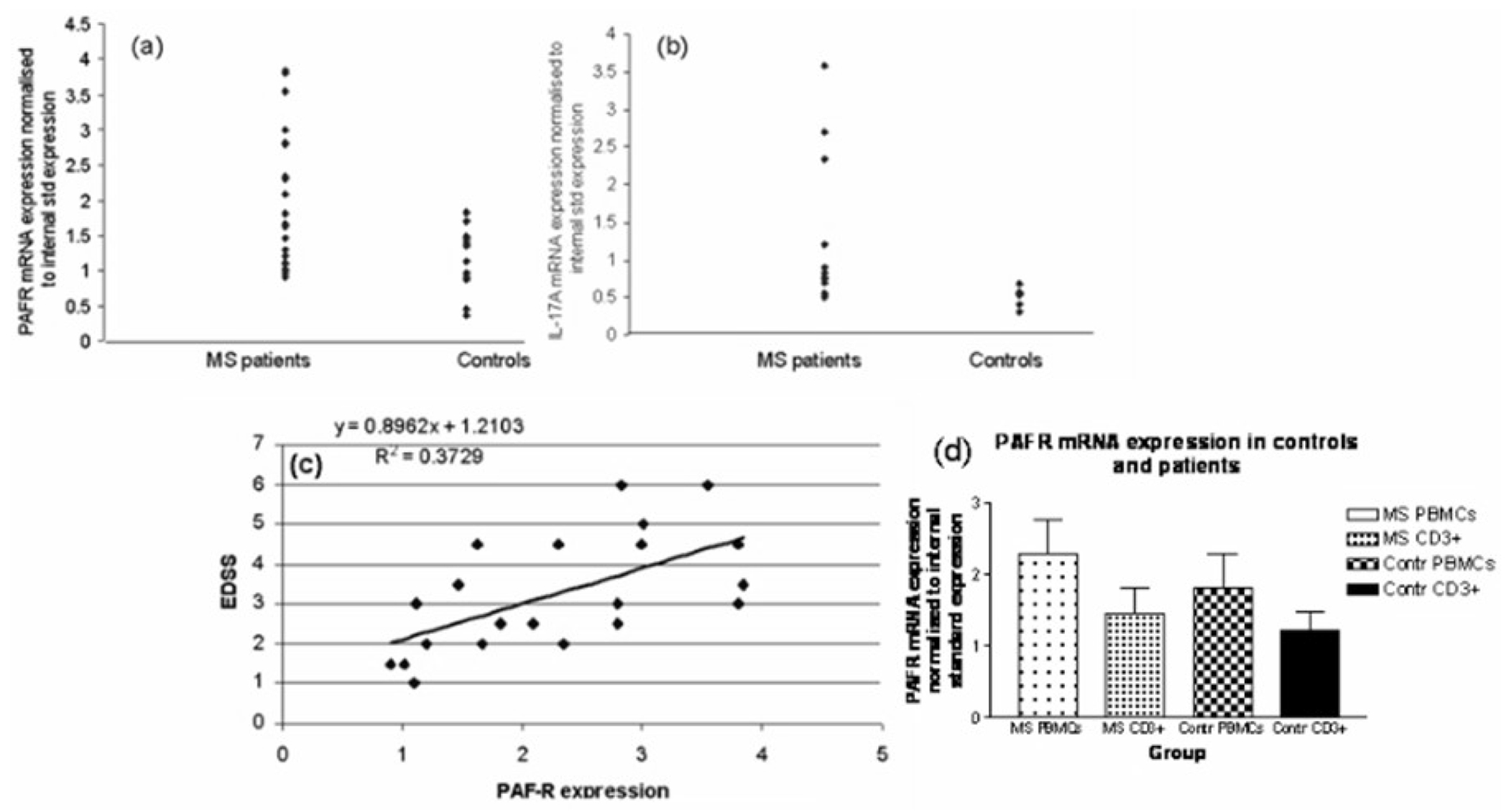
Expression of PAF-R in multiple sclerosis patients. RNA was extracted from peripheral blood mononuclear cells isolated from MS patients and age and sex matched controls. PAF-R (30 MS patients, 20 controls) (a) and IL-17A (12 MS patients, 7 controls) (b) mRNA expression was measured using real time PCR. EDSS scores were obtained during a standardised neurological examination at the time of blood collection. Pearson’s correlation coefficient was used to explore correlations between PAF-R and EDSS (c).
To determine the proportion of the PBMC PAF-R mRNA that is of T cell origin, we analysed a further group of 8 additional relapsing MS patients and 7 additional control subjects. The PBMC from each donor were divided in two samples. PAF-R RNA was measured from one sample (unfractionated PBMC) and from the other (CD3+ cells) after CD3+ cells were magnetically separated. The average proportion of RNA extracted from CD3+ cells represented 63% of the RNA extracted from total PBMC in MS patients and 67% of the PBMC RNA of controls (Fig. 7d). In this smaller group, the differences between PAF-R expression in MS and controls only showed a trend toward higher levels in total PBMC and in CD3+ cells in patients (p = 0.09 and 0.17, respectively, two-tailed unpaired t-test).
However, the results indicate that a substantial and comparable proportion of PAF-R mRNA in both MS patients and controls is of T cell origin.
4. Discussion
The aims of this study were 1) to validate our previous gene expression profiling results that identified PAF-R on activated T cells as a potential Th17 molecule; 2) to determine its functionality and coregulation with other Th17-associated molecules; and 3) to explore its role in MS. We confirmed the microarray results by qPCR and flow cytometry. Importantly, both the arrays and our T cell samples were validated in several ways. Some genes on the arrays have 2 clones, including PAF-R. The regulation by IL-12 and IL-23 is in the same direction, denoting internal consistency. Also, the classical IL-12 target IFN-γ was shown to be up-regulated as expected (Supplementary Table 1). In addition, we previously validated other array results, e.g. the regulation of glucocorticoid modulatory element binding proteins (GMEB) and went on to show its functional role in T cell survival (Kawabe et al., 2012). We also showed intracellular Ca increase indicating the functionality of PAF-R. We also provided evidence supporting a potential role in MS, in that its mRNA level expression is increased compared to controls and correlates with disability scores. A limitation of our study is that we did not expand our flow cytometry studies in normal cells into a more detailed examination of an MS cohort (although preliminary data, not reported here, suggest an up-regulation at protein level in MS as well). We took advantage of a collection of RNA samples from PBMC of untreated MS patients, in part collected as part of a study of interferon responsiveness (Tanasescu et al., 2017); but without corresponding PBMC. Future studies investigating more specifically MS and other autoimmune diseases are warranted. Another limitation of this study was that, in the flow cytometry experiments showing co-expression of PAF-R and IL-17 and mutual exclusion of PAF-R and IFN-γ, we only used PHA/IL-2 stimulation but did not use positive controls with strong inducers of these cytokines to show the magnitude of cytokine induction.
Increasing evidence implicates Th17 cells in the pathogenesis of autoimmune diseases. They produce chemokines, cytokines, metalloproteases and other inflammatory compounds that compromise the blood brain barrier, relevant to MS.
In this study we provide evidence for PAF-R expression in activated human T cells. Although not directly demonstrated (as most prior studies were on resting T cells), induction of PAF-R on T cells by CD2 or CD3 stimulation has already been suggested (Vivier et al., 1990), and canine T cells were also shown to express functional PAF-R that upregulate intracellular Ca after PAF stimulation (Calabresse et al., 1992). A previous study of human T cells that used unstimulated expression of HLA-DR as activation marker did not show PAF-R on T cells, but PAF RNA was measured by Northern blotting, a less sensitive method than the qPCR used here (Simon et al., 1994). Moreover, the similar parallel regulation of Th17-associated genes by IL-23, IL-17, IL-12, and IFN-γ strongly suggests that our results are not an artefact. We used strong stimuli (PHA/IL-2 followed by cytokines or PAF) which may explain our higher yield of Th17 cells compared with other studies. In addition, our findings are further validated by results on separated CD3+ and CD4+ cells simulated with anti-CD3. Th17 cells show considerable phenotypic and functional heterogeneity (Bystrom et al., 2019). Our findings do not allow a definitive conclusion regarding whether the PAF-R + Th17 cells belong to a specific subpopulation of Th17 cells, but in view of their associated cytokines and IL-23 responsiveness, they appear to be classical Th17 cells, and do not appear to be Th22 cells. The absent co-expression of IFN-γ indicates that PAF-R can distinguish them from the proinflammatory cells concomitantly expressing Th1 and Th17 markers (Th1-17 cells) which have also been associated with MS pathogenesis (Edwards et al., 2010)
We show that PAF-R on T cells increases intracellular Ca in response to PAF stimulation, and that PAF effects on T cells, including IL-17 induction, are blocked by PAF-R antagonists. We also show PAF-R up-regulation by IL-23, and demonstrate that its pattern of expression is similar to that of other Th17 associated molecules.
The PAF-R ligand, PAF, an inflammatory mediator with pleiotropic effects, appears early in inflammation. Here, we show that it induces IL-17, and that PAF-R expression is upregulated by IL-23 even before the upregulation of IL-17. Thus, PAF/PAF-R interactions may be involved in the early events leading to Th17 differentiation and trigger a self-amplification process similar to the CCR6/CCL20 loop described previously for Th17 cells (Acosta-Rodriguez et al., 2007a, 2007b).
Our results confirm two additional studies implicating PAF/PAF-R in Th17-mediated responses (Singh et al., 2011; Drolet et al., 2011). Both of these studies support our findings; however our study is the first to focus on the role of T cells in this new role of PAF/PAF-R pathway. We postulate that an inflammatory milieu containing PAF in addition to TGF-β may skew the T cell development towards Th17 and away from Treg commitment. This argument is strengthened by the fact that PAF itself induces IL-6, a key element in the induction of Th17 cells and suppression of Treg cell development (Hamel-Cote et al., 2019).
Besides IL-23, IL-1 is a potent Th17 stimulus (Acosta-Rodriguez et al., 2007a, 2007b). Interestingly, IL-1 has also been shown to upregulate the PAF pathway (Lee et al., 2000), and PAF plays an important role in asthma, and possibly also in MS and other autoimmune conditions. It is therefore plausible that PAF is an important member of an inflammatory network that enhances and perpetuates inflammation with Th17 predominant pathogenesis.
PAF/PAF-R pathway may be implicated in the optimal functioning of this T cell subset with crucial roles in MS, asthma, and other inflammatory disorders.
While it is still debatable whether the asthma and MS coexist more or less as frequently as expected based on the figures of their independent life time prevalence, the fact that they do coexist in a significant number of patients cannot be denied (Edwards and Constantinescu, 2004; Manouchehrinia et al., 2015). Therapeutic strategies that avoid exacerbation of one during treatment of the other should target shared pathogenic pathways. Notably, PAF inhibition has shown some promise for both MS and asthma (Brochet et al., 1995; Chu et al., 2011). Thus, targeting PAF/PAF-R pathway and, possibly through this, the Th17 pathway, may become a justified and worthwhile therapeutic approach in the treatment of these conditions.
Supplementary Material
Acknowledgments
We thank Prof T. Shimizu, Tokyo University, for helpful discussions. This work was supported in part by the Multiple Sclerosis Society of the Great Britain and Northern Ireland, the University Hospital Nottingham and The University of Nottingham.
Abbreviations:
- PAF
platelet activating factor
- PAF-R
platelet activating factor receptor
- ROR
retinoic orphan receptor
Footnotes
CRediT authorship contribution statement
Angela Midgley: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Writing - original draft, Visualization, Supervision, Writing - review & editing. Dina Barakat: Methodology, Validation, Formal analysis, Investigation, Writing - original draft, Writing - review & editing. Manjit Braitch: Methodology, Validation, Formal analysis, Investigation, Data curation, Visualization, Writing - review & editing. Calen Nichols: Methodology, Validation, Formal analysis, Software, Investigation, Data curation, Writing - review & editing. Mihailo Nebozhyn: Methodology, Validation, Formal analysis, Software, Data curation, Visualization, Writing - review & editing. Laura J. Edwards: Methodology, Validation, Formal analysis, Investigation, Writing - review & editing. Susan C. Fox: Methodology, Validation, Formal analysis, Resources, Investigation, Writing - review & editing. Bruno Gran: Conceptualization, Supervision, Writing - review & editing. R. Adrian Robins: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Supervision, Resources, Software, Writing - review & editing. Louise C. Showe: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Supervision, Resources, Software, Data curation, Writing - review & editing. Cris S. Constantinescu: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Supervision, Resources, Data curation, Visualization, Funding acquisition, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.imbio.2020.152023.
References
- Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G, 2007a. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol 8, 639. [DOI] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F, 2007b. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol 8, 942. [DOI] [PubMed] [Google Scholar]
- Balasa R, Maier S, Voidazan S, Hutanu A, Bajko Z, Motataianu A, Tilea B, Tiu C, 2017. Assessment of interleukin-17A, interleukin-10 and transforming growth factor-beta1 serum titers in relapsing remitting multiple sclerosis patients treated with avonex, possible biomarkers for treatment response. CNS Neurol. Disord.: Drug Targets 16, 93. [DOI] [PubMed] [Google Scholar]
- Bellizzi MJ, Geathers JS, Allan KC, Gelbard HA, 2016. Platelet-activating factor receptors mediate excitatory postsynaptic hippocampal injury in experimental autoimmune encephalomyelitis. J. Neurosci 36, 1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK, 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235. [DOI] [PubMed] [Google Scholar]
- Brochet B, Guinot P, Orgogozo JM, Confavreux C, Rumbach L, Lavergne V, 1995. Double blind placebo controlled multicentre study of ginkgolide B in treatment of acute exacerbations of multiple sclerosis. The Ginkgolide Study Group in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 58, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystrom J, Clanchy FIL, Taher TE, et al. , 2019. Functional and phenotypic heterogeneity of Th17 cells in health and disease. Eur. J. Clin. Invest 49, e13032. [DOI] [PubMed] [Google Scholar]
- Calabresse C, Nguer MC, Pellegrini O, Benveniste J, Richard Y, Thomas Y, 1992. Induction of high-affinity paf receptor expression during T cell activation. Eur. J. Immunol 22, 1349. [DOI] [PubMed] [Google Scholar]
- Callea L, Arese M, Orlandini A, Bargnani C, Priori A, Bussolino F, 1999. Platelet activating factor is elevated in cerebral spinal fluid and plasma of patients with relapsing-remitting multiple sclerosis. J. Neuroimmunol 94, 212. [DOI] [PubMed] [Google Scholar]
- Casals-Stenzel J, Muacevic G, Weber KH, 1987. Pharmacological actions of WEB 2086, a new specific antagonist of platelet activating factor [published correction appears in J Pharmacol Exp Ther 1988; 245 749]. J. Pharmacol. Exp. Ther 241, 974. [PubMed] [Google Scholar]
- Chu X, Ci X, He J, Wei M, Yang X, Cao Q, Li H, Guan S, Deng Y, Pang D, Deng X, 2011. A novel anti-inflammatory role for ginkgolide B in asthma via inhibition of the ERK/MAPK signaling pathway. Molecules 16, 7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu CS, Gran B, 2014. The essential role of T cells in multiple sclerosis: a reappraisal. Biomed. J 37, 34. [DOI] [PubMed] [Google Scholar]
- Constantinescu CS, Farooqi N, O’Brien K, Gran B, 2011. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol 164, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drolet AM, Thivierge M, Turcotte S, Hanna D, Maynard B, Stankova J, Rola-Pleszczynski M, 2011. Platelet-activating factor induces Th17 cell differentiation. Mediators Inflamm. 2011, 913802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LJ, Constantinescu CS, 2004. A prospective study of conditions associated with multiple sclerosis in a cohort of 658 consecutive outpatients attending a multiple sclerosis clinic. Mult. Scler 10, 575. [DOI] [PubMed] [Google Scholar]
- Edwards LJ, Constantinescu CS, 2009. Platelet activating factor/platelet activating factor receptor pathway as a potential therapeutic target in autoimmune diseases. Inflamm. Allergy Drug Targets 8, 182. [DOI] [PubMed] [Google Scholar]
- Edwards LJ, Robins RA, Constantinescu CS, 2010. Th17/Th1 phenotype in demyelinating disease. Cytokine 50, 19. [DOI] [PubMed] [Google Scholar]
- El Behi M, Zephir H, Lefranc D, Dutoit V, Dussart P, Devos P, Dessaint JP, Vermersch P, Prin L, 2007. Changes in self-reactive IgG antibody repertoire after treatment of experimental autoimmune encephalomyelitis with anti-allergic drugs. J. Neuroimmunol 182, 80. [DOI] [PubMed] [Google Scholar]
- Fahey AJ, Robins RA, Kindle KB, Heery DM, Constantinescu CS, 2006. Effects of glucocorticoids on STAT4 activation in human T cells are stimulus-dependent. J. Leukoc. Biol 80, 133. [DOI] [PubMed] [Google Scholar]
- Fox SC, Behan MW, Heptinstall S, 2004. Inhibition of ADP-induced intracellular Ca2+ responses and platelet aggregation by the P2Y12 receptor antagonists AR-C69931MX and clopidogrel is enhanced by prostaglandin E1. Cell Calcium 35, 39. [DOI] [PubMed] [Google Scholar]
- Gaffen SL, Jain R, Garg AV, Cua DJ, 2014. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol 14, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel-Cote G, Lapointe F, Veronneau S, Mayhue M, Rola-Pleszczynski M, Stankova J, 2019. Regulation of platelet-activating factor-mediated interleukin-6 promoter activation by the 48 kDa but not the 45 kDa isoform of protein tyrosine phosphatase non-receptor type 2. Cell Biosci. 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellewell PG, Williams TJ, 1989. Antagonism of Paf-induced oedema formation in rabbit skin: a comparison of different antagonists. Br. J. Pharmacol 197, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeve MA, Savage ND, de Boer T, Langenberg DM, de Waal Malefyt R, Ottenhoff TH, Verreck FA, 2006. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur. J. Immunol 36, 661. [DOI] [PubMed] [Google Scholar]
- Honda Z, Ishii S, Shimizu T, 2002. Platelet-activating factor receptor. J. Biochem 131, 773. [DOI] [PubMed] [Google Scholar]
- Howat DW, Chand N, Braquet P, Willoughby DA, 1989. An investigation into the possible involvement of platelet activating factor in experimental allergic encephalomyelitis in rats. Agents Actions 27, 473. [DOI] [PubMed] [Google Scholar]
- Kasperska-Zajac A, Brzoza Z, Rogala B, 2008. Platelet activating factor as a mediator and therapeutic approach in bronchial asthma. Inflammation 31, 112. [DOI] [PubMed] [Google Scholar]
- Kawabe K, Lindsay D, Braitch M, Fahey AJ, Showe L, Constantinescu CS, 2012. IL-12 inhibits glucocorticoid-induced T cell apoptosis by inducing GMEB1 and activating PI3K/Akt pathway. Immunobiology 217, 1118. [DOI] [PubMed] [Google Scholar]
- Kihara Y, 2019. Systematic understanding of bioactive lipids in neuro-immune interactions: lessons from an animal model of multiple sclerosis. Adv. Exp. Med. Biol 1161, 133. [DOI] [PubMed] [Google Scholar]
- Kurtzke JF, 1983. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 33, 1444. [DOI] [PubMed] [Google Scholar]
- Lee YM, Hybertson BM, Cho HG, Terada LS, Cho O, Repine AJ, Repine JE, 2000. Platelet-activating factor contributes to acute lung leak in rats given interleukin-1 intratracheally. Am. J. Physiol. Lung Cell. Mol. Physiol 279, L75. [DOI] [PubMed] [Google Scholar]
- Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L, 2002. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med 8, 500. [DOI] [PubMed] [Google Scholar]
- Manouchehrinia A, Edwards LJ, Roshanisefat H, Tench CR, Constantinescu CS, 2015. Multiple sclerosis course and clinical outcomes in patients with comorbid asthma: a survey study. BMJ Open 5, e007806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusic S, Leach MW, Pelker JW, Azoitei ML, Uozumi N, Cui J, Shen MW, DeClercq CM, Miyashiro JS, Carito BA, Thakker P, Simmons DL, Leonard JP, Shimizu T, Clark JD, 2005. Cytosolic phospholipase A2 alpha-deficient mice are resistant to experimental autoimmune encephalomyelitis. J. Exp. Med 202, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusic S, Thakker P, Pelker JW, Stedman NL, Lee KL, McKew JC, Han L, Xu X, Wolf SF, Borey AJ, Cui J, Shen MW, Donahue F, Hassan-Zahraee M, Leach MW, Shimizu T, Clark JD, 2008. Blockade of cytosolic phospholipase A2 alpha prevents experimental autoimmune encephalomyelitis and diminishes development of Th1 and Th17 responses. J. Neuroimmunol 204, 29. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Cua DJ, Gaffen SL, 2019. The IL-17 family of cytokines in health and disease. Immunity 50, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes Diaz G, Fraussen J, Van Wijmeersch B, Hupperts R, Somers V, 2018. Dimethyl fumarate induces a persistent change in the composition of the innate and adaptive immune system in multiple sclerosis patients. Sci. Rep 8, 8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Garra A, Stockinger B, Veldhoen M, 2008. Differentiation of human T(H)-17 cells does require TGF-beta! Nat. Immunol 9, 588. [DOI] [PubMed] [Google Scholar]
- Pedotti R, DeVoss JJ, Youssef S, Mitchell D, Wedemeyer J, Madanat R, Garren H, Fontoura P, Tsai M, Galli SJ, Sobel RA, Steinman L, 2003. Multiple elements of the allergic arm of the immune response modulate autoimmune demyelination. Proc. Natl. Acad. Sci. U. S. A 100, 1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon HU, Tsao PW, Siminovitch KA, Mills GB, Blaser K, 1994. Functional platelet-activating factor receptors are expressed by monocytes and granulocytes but not by resting or activated T and B lymphocytes from normal individuals or patients with asthma. J. Immunol 153, 364. [PubMed] [Google Scholar]
- Singh TP, Huettner B, Koefeler H, Mayer G, Bambach I, Wallbrecht K, Schon MP, Wolf P, 2011. Platelet-activating factor blockade inhibits the T-helper type 17 cell pathway and suppresses psoriasis-like skin disease in K5.hTGF-beta1 transgenic mice. Am. J. Pathol 178, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanasescu R, Midgley A, Robins RA, Constantinescu CS, 2017. Decreased interferon-β induced STAT-4 activation in immune cells and clinical outcome in multiple sclerosis. Acta Neurol. Scand 136, 233. [DOI] [PubMed] [Google Scholar]
- Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, Cua DJ, 2015. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med 21, 719. [DOI] [PubMed] [Google Scholar]
- Terashita Z, Tsushima S, Yoshioka Y, Nomura H, Inada Y, Nishikawa K, 1983. CV3988 - a specific antagonist of platelet activating factor (PAF). Life Sci. 32, 1975. [DOI] [PubMed] [Google Scholar]
- Vivier E, Deryckx S, Wang JL, Valentin H, Peronne C, de Vries JE, Bernard A, Benveniste J, Thomas Y, 1990. Immunoregulatory functions of paf-acether. VI. Inhibition of T cell activation via CD3 and potentiation of T cell activation via CD2. Int. Immunol 2, 545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.