

Abstract
Autoimmune vasculitis of the medium and large elastic arteries can cause blindness, stroke, aortic arch syndrome, and aortic aneurysm. The disease is often refractory to immunosuppressive therapy and progresses over decades as smoldering aortitis. How the granulomatous infiltrates in the vessel wall are maintained and how tissue-infiltrating T cells and macrophages are replenished is unknown. Single cell and whole tissue transcriptomic studies of immune cell populations in vasculitic arteries identified a CD4+ T cell population with stem cell-like features. CD4+ T cells supplying the tissue-infiltrating and tissue-damaging effector T cells survived in tertiary lymphoid structures around adventitial vasa vasora, expressed the transcription factor T cell factor 1 (TCF1), had high proliferative potential, and gave rise to two effector populations, Eomesodermin (EOMES)+ cytotoxic T cells and B-cell lymphoma 6 (BCL6)+ T follicular helper-like cells. TCF1hiCD4+ T cells expressing the interleukin 7 receptor (IL-7R) sustained vasculitis in serial transplantation experiments. Thus, TCF1hiCD4+ T cells function as disease stem cells and promote chronicity and autonomy of autoimmune tissue inflammation. Remission-inducing therapies will require targeting stem-like CD4+ T cells instead of only effector T cells.
One Sentence Summary:
Stem-like CD4+ T cells replicating in tertiary lymphoid structures supply effector T cells in autoimmune vasculitis.
INTRODUCTION
The aorta and its major branch vessels are considered immunoprivileged sanctuary spaces, protected from aggressive inflammation by layers of immunosuppressive mechanisms. This immunoprivilege is lost in giant cell arteritis (GCA), a prototypic autoimmune disease manifesting with destructive aortitis and vaso-occlusive vasculitis of the 1st to 5th aortic branches (1, 2). In GCA, T cells and macrophages are arranged in granulomatous infiltrates, and vasculitic lesions occupy the intima, media, and adventitia. This vasculitis causes blindness, stroke, aortic arch syndrome, and aortic aneurysm and is often resistant to current immunosuppressive therapy (3). Smoldering vasculitis resulting in progressive aortic wall destruction, aneurysm formation, and dissection is emerging as the major challenge in the long-term management of GCA. A better understanding of the tissue tropism of the disease for the ascending aorta, of immune effector cell populations in early and chronic disease, and of signaling pathways resisting conventional immunosuppression is needed to improve diagnosis and management. Definition of these disease risk factors will conceptually inform the paradigms relevant for other autoimmune diseases, which also persist for decades.
The identification of shared T cell clonotypes in independent arteries of patients with GCA has supported the concept that GCA is an antigen-specific disease (4, 5). The granulomatous infiltrates are dominated by CD4+ T cells, and lesional macrophages can differentiate into multinucleated giant cells that secrete tissue-injurious metalloproteinases (6). Autoantibodies are distinctly infrequent in GCA (1, 2). The disease affects individuals older than 50 years, but in contrast to patients with other autoimmune diseases, signatures of accelerated immune aging have not been reported in patients with GCA (7). How effector CD4+ T cells mediate disease, in particular persistent disease, is not understood. Notably, the vascular lesions are occupied by an array of T cell effector populations, including T helper (Th)1, Th17, Th9 and Th21 cells (1). Th1 cells appear to be particularly resistant to immunosuppressive therapy (3), but it remains unclear whether they are chronically recruited to the vessel wall or whether they are generated in situ. The diversity of functional T cell subsets has challenged the notion of a single vasculitogenic antigen and instead has drawn attention to endogenous abnormalities in T cell reactivity. In support, CD4+ and CD8+ T cells isolated from patients with GCA aberrantly express NOTCH receptors (8), which guide tissue-invasive CD4+ T cells through NOTCH ligands expressed on vasa vasorum endothelial cells (EC) (9). Another critical component of GCA pathogenesis is a defect in inhibitory immune checkpoints (10, 11) due to the low expression of programmed cell death ligand 1 (PD-L1) and CD155 on lesional antigen presenting cells, leaving lesional T cells with insufficient negative signaling.
Pathogenic concepts of autoimmune disease have focused on autoantigen recognition as the driving force. Yet, the pathogenesis of autoimmune diseases spans over decades, often including long periods of clinically silent autoimmunity. Pathogenic immune recognition events happen in distinct tissue environments, with the host immune system evolving over time. In population-based cohorts, a unique metabolic phenotype consisting of low blood glucose, low body mass index, and low lipids has been identified as a risk factor preceding GCA onset by decades (12). Beyond metabolic conditions, structural elements related to the ascending aorta may predispose older individuals to develop aortitis. Tissue-specific risk factors are indicated by the preferential occurrence of therapy-induced aortitis in checkpoint inhibitor treated cancer patients (13). Improved treatment of GCA will require understanding of its tissue tropism and of the molecular determinants through which the disease gains lasting immune memory.
In current work, we have defined the interplay between the aortic wall tissue environment and the induction of durable T cell immunity, sustaining aortitis, disease chronicity and therapeutic resistance. All patients with aortitis studied here had evidence of highly organized lymphoid aggregates in the aortic wall. Such tertiary lymphoid structures (TLS) provided shelter for TCF1hi CD4+ T cells that served as a local source for multiple TCF1lo effector populations, including tissue-damaging Eomesodermin (EOMES)+ cytotoxic CD4+ T cells and structure-maintaining interleukin (IL)-21-producing T follicular helper (Tfh)-like CD4+ T cells. TLS-residing TCF1hi CD4+ T cells had self-renewal capacity and sustained disease in serial transfer experiments; assigning them as in situ disease stem cells that need to be targeted to disrupt the autonomy and chronicity of autoimmune vasculitis.
RESULTS
Aortic aneurysms resected from patients with aortitis contain adventitial perivascular TLS.
In the panarteritic lesions of GCA, T cells and macrophages typically form granulomatous infiltrates in the media, with multinucleated giant cells accumulating at the media-intima border, along the fragmented internal elastic lamina. Dense lymphocytic infiltrates accrue outside of the external lamina elastica at the adventitia-media border. To examine the diversity and the dynamics of disease-inducing T cells, we proceeded with a comprehensive spatial analysis in 42 ascending aortic wall samples, collected from 22 cases of aortitis and 20 cases of non-inflamed ascending aortic aneurysms (inherited aortopathy and mild atherosclerosis) (Table 1). Irrespective of etiology, all tissues had common features of aneurysm formation (loss of elastin fibers, medial degeneration) (fig. S1A) and some of the aortitis cases had typical giant cells in medial infiltrates (fig. S1A). All aortitis samples exhibited dense mononuclear cell aggregates around adventitial microvessels (Fig. 1A), a phenotype restricted to aortas with vasculitis and only minimally present in disease controls and normal aorta (Fig. 1B). Immunofluorescence imaging of the mononuclear aggregates identified T cells and B cells (Fig. 1C) and Ki67+ proliferating cells (fig. S1B), indicating that the highly organized lymphoid architectures in the adventitia functioned as tertiary lymphoid structures (TLS), inducible ectopic lymphoid tissues under chronic inflammatory conditions (14, 15). Aortic TLS were composed of three distinct zones: T cell zones, B cell zones, and T/B mixed zones. B cell zones were identified through CD21+ follicular dendritic cell (FDC) networks, some of which contained germinal centers (fig. S1C and D). Half of the aortic TLS were mainly composed of T and B cells, but about 25% contained predominantly T cells (Fig. 1D, fig. S1E and F). Most TLS were in advanced maturational stages with distinct T or B cell areas (Fig. 1E), mimicking the architecture of lymph nodes. TLS formed in the adventitia, infrequently in the media (fig. S1G to I) and contained mostly CD4+ T cells (Fig. 1F). Aortic TLS were rich in CD11c+ dendritic cells (DCs), often arranged in interconnected networks (Fig. 1G). T cells had close contact to antigen-presenting HLA-DR+ cells (Fig. 1H). Analysis of TLS-residing cells revealed Podoplanin+ lymphatic vessels, known to contribute to T cell survival through IL-7 production (16, 17) (Fig. 1I). A distinguishing feature of aortic TLS was the high density of microvessels (Fig. 1J), pronounced in the T cell areas (Fig. 1K). Unexpectedly, both arterioles and venules could function as TLS-guiding vessels and many of them expressed PNAd (Fig. 1L), a marker for high endothelial venules (HEV). PNAd+ endothelial cells encased clumps of extravasating T cells, next to subendothelial pockets of T cells arranged between smooth muscle cell layers of the vessel wall (Fig. 1M to P, fig. S2A to C). TLS grew as an extension of subendothelial lymphocyte pockets, identifying specialized EC formations and dedicated T cells as early elements in the process of aortic wall lymphoid organogenesis.
Table 1.
Clinical characteristics of patients with GCA aortitis or non-inflamed ascending aortic aneurysm.
| Non-inflamed ascending | Aortitis (n = 22) | |
|---|---|---|
| aortic aneurysm (n = 20) | ||
| Age (mean ± SD) | 72.5 ± 6.5 | 74.0 ± 6.1 |
| Male:Female | 8:12 | 10:10 |
| ESR (mean ± SD, mm/hr) | 4.95 ± 9.2 | 6.3 ± 8.5 |
| CRP (mean ± SD, mg/L ) | 0.79 ± 1.5 | 3.3 ± 3.7 |
| Treatment | ||
| Prednisone | 0 (0%) | 1 (4.5%) |
| Prednisone dose (mg/day) | 4mg | |
| Methotrexate | 0 (0%) | 0 (0%) |
| Active Systemic Disease | 0 (0%) | 0 (0%) |
| Anti-platelet | 10 (50%) | 13 (59%) |
| Statin | 12 (60%) | 9 (40.9%) |
| ACE-I/ARB | 15 (75%) | 11 (50%) |
| History of Polymyalgia Rheumatica | 0 (0%) | 7 (31.8%) |
| History of congenital heart disease | 10 (50%) | 0 (0%) |
| Smoking | ||
| Current | 0 (0%) | 6 (27.3%) |
| Former | 7 (35%) | 7 (31.8%) |
Values are n (%) unless otherwise noted. ESR: Erythrocyte sedimentation time, CRP: C-reactive protein, ACE-I: angiotensin-converting enzyme inhibitor, ARB: angiotensinⅡ receptor blocker.
Fig. 1. TLS are formed in large vessel vasculitis.
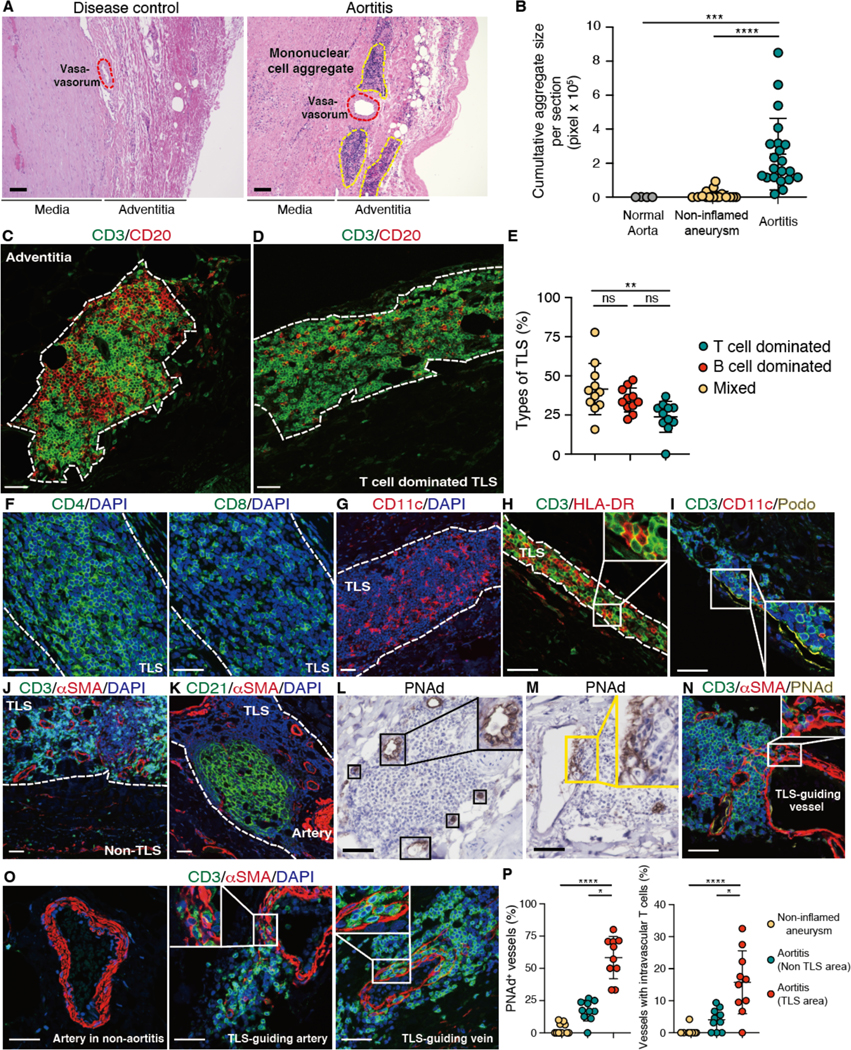
Tissue samples were collected from the ascending aorta of patients with GCA and patients with non-inflammatory aneurysm (disease control). Tissue sections were analyzed by multi-color immunofluorescence (IF); nuclei were stained with DAPI. (A) In GCA aortitis, mononuclear cells formed organized lymphoid aggregates around adventitial blood vessels. Tissues were stained with hematoxylin and eosin. (B) Cumulative size of mononuclear cell aggregates per tissue section (aortitis: n=22, disease control: n=20, normal aorta: n=4). Each dot represents one tissue. (C and D) Tissues were stained for CD3 and CD20; representative images of a mixed TLS (C) and a T cell dominated TLS (D) are shown. (E) Depending on the dominant cell type, TLS were categorized as T cell dominated, B cell dominated or mixed. Proportions are shown for 11 cases of aortitis. (F to K) Tissues were stained for CD4 and CD8 (F); CD11c (G); CD3 and HLA-DR (H); CD3, CD11c, and Podoplanin (Podo) (I); CD3 and αSMA (J); or CD21 and αSMA (K). (L and M) Peripheral lymph node addressin (PNAd) expression was examined by immunohistochemistry. PNAd+ high endothelial cells are shown in (L). Subendothelial immune cell pockets are indicated in (M). (N and O) Tissue were stained for CD3, αSMA and PNAd (N) and for CD3 and αSMA (O). (P) Frequencies of PNAd+ vessels and vessels with intravascular T cells in aortitis and in non-inflamed aneurysms (n=10) are compared. TLS and T cell zones are marked by dotted lines. Data are presented as mean ± SD with individual values indicated. Data were analyzed by Kruskal-Wallis test with post-hoc (B and P), one-way ANOVA with Tukey post hoc test (E). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bars, 100 μm (A) or 50 μm (C and D, F to O).
A TLS gene signature separates aortitis from non-inflamed aneurysm.
To characterize immune responses relevant for aortic autoimmunity, we proceeded with global tissue RNA-sequencing (RNA-seq) analysis of 42 aortic aneurysm samples (n=22 Aortitis, n=20 non-inflammatory aneurysms). Unsupervised principal component analysis (PCA) showed clear separation of the aortitis group from the disease control group (Fig. 2A). A total of 1070 genes were differentially expressed between the two groups (adjusted P value <0.05) (Fig. 2B, fig. S3A). Gene ontology analysis of the 1070 differentially expressed genes (DEGs) demonstrated the enrichment of adaptive immune activation pathways in aortitis (Fig. 2C). DEGs highly enriched in the aortitis tissues pointed towards ongoing T cell- and B cell-dependent immune responses: T cells (CD3E, CD4, CD8A, GZMB, LTB), B cells (CD19, CD38, SDC1, FCRL3, FCRL5, CR2, LTB, JCHAIN, IGLL5, CD79B), antigen presentation (HLA-DRA, HLA-DMA, CD86), homeostatic chemokines (CXCL13, CCL19), proliferation and activation (MKI67, BTLA) (Fig. 2B). Unbiased hierarchical clustering of these genes provided a clear separation of the aortitis group from the disease control group and from normal aortic tissue (Fig. 2D, fig. S3B). The 22 gene transcripts that divided the aortic tissues into aortitis versus non-inflammatory aneurysms have previously been associated with the formation of TLS in non-lymphoid tissue sites (18–20).
Fig. 2. Inflamed aortic wall expresses a TLS transcriptomic signature.

Human ascending aortic tissues from patients with giant cell aortitis (n=22) or non-inflamed aortic aneurysms (n=20 disease controls) were analyzed by bulk RNA-seq. (A and B) A principal component analysis (PCA) (A) and a volcano plot showing DEGs between aortitis and non-inflamed aneurysms (B) are presented. (C) Shown are the top 10 enriched pathways in GCA aortitis. Statistical significance was determined by enrichr (48). (D) The hierarchical clustering heatmap indicates 22 transcripts relevant in TLS formation (TLS gene signature) in aortitis and non-inflamed aneurysms. (E) Subgroup analysis comparing the TLS gene signature in the indicated clinical subtypes. (F) TLS size (Fig. 1C) and TPM values of CXCL13, CCL19 and CCL21 (n=22 aortitis cases, n=20 disease controls) are correlated. (G) Shown are comparisons of TPM values of TLS-related genes (CXCL13, CCL19, CCL21, IL-7, IL15, TNFSF13B, TNFSF8 and TNFRSF8) in aortitis and disease controls. (H) CIBERSORT analysis of RNA-seq data is used for a quantitative evaluation of immune cell infiltrates in aortitis (n=22) and disease controls (n=20). (I) Estimated proportions of resting and activated CD4+ memory T cells in aortitis are shown. Data are presented as mean ± SD. Data were analyzed by Mann-Whitney U test (G, I). Correlations were determined by Pearson’s correlation analysis (F). **P < 0.01, ****P < 0.0001; ns, not significant.
The aortitis cases included patients with limited and widespread GCA and the control aneurysms resulted from atherosclerosis or inherited aortopathies. Underlying disease processes did not affect expression patterns of the 22 TLS-associated gene transcripts (Fig. 2E) (21). Expression of CXCL13 and CCL19, but not of CCL21, was upregulated in aortitis and closely associated with the size of the mononuclear cell aggregates (Fig. 2F and G). Transcripts for lymphocyte survival factors, such as IL7, IL15 and the gene encoding B-cell activating factor, BAFF (TNFSF13B), were all significantly higher in the aortitis versus the control tissues (p>0.0001) (Fig. 2G). Gene Set Enrichment Analysis (GSEA) confirmed enrichment of the human tissue resident memory T cell (Trm) signature (fig.S3C) (22). TNFSF8 and TNFRSF8, facilitating interaction between Tfh-like T cells and age-associated B cells within TLS (23), were also highly elevated in the aortitis tissues (Fig. 2G).
Tissue-infiltrating CD45+ immune cells were highly enriched in aortitis (fig. S3D). To capture cellular elements relevant in the formation of structured lymphoid follicles, we performed CIBERSORT analysis on bulk RNA-seq data (Fig. 2H, fig. S3E to F). Both tissue sources expressed gene patterns indicative of resting CD4+ memory T cells and monocytes. However, gene expression profiles for some cell types appeared enriched or even unique for the vasculitic tissues. Activated CD4+ memory T cells and memory B cells were exclusively seen in aortitis (Fig. 2H). Unexpectedly, most tissue-infiltrating T cells were in a resting state in aortitis (Fig. 2I, fig. S3F). Together, tissue transcriptomics confirmed the histomorphologic data placing TLS into the aortic wall affected by autoimmune disease and provided evidence for in situ activation of a small subset of tissue-residing T cells.
Tissue-residing CD4+ T cells in vasculitis are heterogenous.
To unravel the heterogeneity of CD4+ T cells involved in autoimmune vasculitis, we sought to isolate tissue CD4+ T cells from arteries unexposed to immunosuppressive therapy. We induced vasculitis in arteries engrafted into immuno-deficient NOD scid gamma (NSG) mice that were reconstituted with peripheral blood mononuclear cells from patients with active GCA (fig. S4A) (9, 12). In the inflamed arteries, tissue-infiltrating T cells formed dense aggregates in the adventitia (fig. S4B). We isolated CD4+ T cells from the explanted arteries for single cell RNA-seq studies. Following stringent quality control, we retained 680 CD4+ T cells with a median of 2122 genes per cell. To directly compare the scRNA-seq data from induced vasculitis with the bulk RNA-seq data from the aortic samples, we combined the scRNA-seq data into a pseudobulk data set. 85% of the genes were overlapping in the two data sets (fig. S4C), indicating a high degree of similarity in the T cell populations of the experimental murine vasculitis versus spontaneous human vasculitis.
Clustering of the scRNA-seq transcripts yielded 5 clusters and differential gene expression analysis among the 5 clusters revealed notable differences in their transcriptomic profiles (Fig. 3A to C). Cluster 0 was characterized by the expression of the transcription factors (TF) TCF7 (encoding TCF1), LEF1 and FOXO1 and exhibited higher expression of survival related genes (IL7R), co-stimulatory molecules (CD28, CD27, FAS, CD226), but not CD38. This signature is reminiscent of transcriptional profiles obtained from the long-lived human CD8+ memory population known as CD8+ memory stem T cells (Tsmc) (24). Cluster 0 also expressed the primary chemokine receptors relevant for lymphoid tissue homing (CCR7, CXCR3, CXCR5) as well as GPR183, a gene involved in dendritic cell recruitment (Fig. 3D and E, fig. S4D).
Fig. 3. Single cell RNA sequencing defines multiple subsets of vasculitogenic CD4+ T cells.
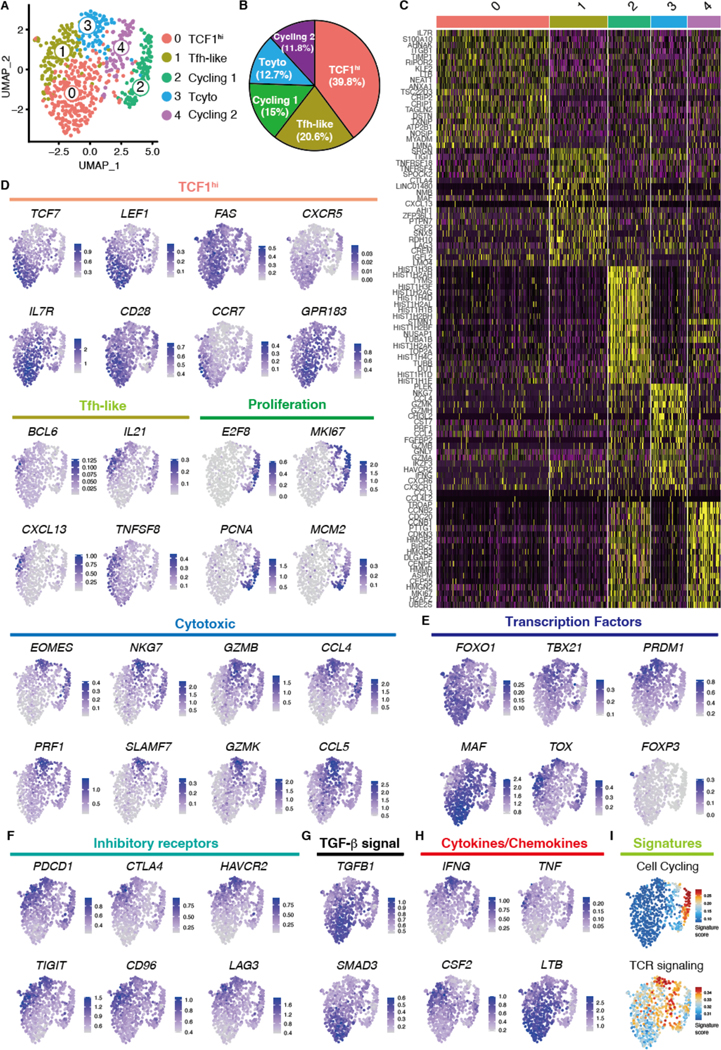
Vasculitis was induced in human arteries engrafted into NSG mice by adoptively transferring PBMCs from patients with GCA. CD4+ T cells were isolated from digested inflamed arteries and analyzed by scRNA-seq. (A) Uniform manifold approximation and projection (UMAP) representation of the CD4+ T cell dataset of 680 cells from the inflamed arteries (n=3); each dot represents a single T cell, and cells are colored as 1 of 5 discrete subsets. (B) A circle graph of the relative frequencies of each CD4+ T cell subset is shown. (C) Shown is a heatmap of the top 20 differentially expressed genes driving heterogeneity of vessel wall infiltrating CD4+ T cells. (D to H) UMAP visualizations of gene expression in CD4+ T cells are shown; Cluster defining signature genes (D), key transcriptional factors (E), inhibitory receptors (F), genes involved in TGF-β signaling (G), and cytokines and chemokines (H) are presented. (I) UMAP presentation shows cell cycling and TCR signaling gene signature scores for individual CD4+ T cells.
Distinguishing gene transcripts defining Cluster 2 included the TFs BCL6, MAF, and TOX2, and genes implicated in B cell helper functions such as IL21, CXCL13, and TNFSF8, identifying them as Tfh-like cells (Fig. 3D and E, fig. S4D) (23, 25, 26). Gene profiles distinguishing Cluster 2 and 4 contained the cell cycle genes ME2F8, MKI67, PCNA, and MCM2. Genes defining Cluster 3 included EOMES and SLAMF7, as well as genes involved in cytotoxicity such as PRF1, NKG7, GZMB, and GZMK. Cluster 3 expressed transcripts for the chemokines CCL4 and CCL5 (Fig. 3D, fig. S4D) (27).
Based on these findings, we annotated the clusters as TCF1hi CD4+ T cells (cluster 0), Tfh-like T cells (cluster 1), cycling (cluster 2 and 4), and cytotoxic CD4+ T cells (cluster 3), respectively. TCF1hi CD4+ T cells lacked transcriptional evidence for TOX-driven exhaustion as well as an EOMES-driven cytotoxic program (Fig. 3E). The expression of PRDM1 (encoding B-lymphocyte-induced maturation protein 1 or BLIMP-1) and TCF7 was mutually exclusive (Fig. 3E), consistent with BLIMP-1 repressing TCF1 and IL7R (28–30). Despite the high expression of inhibitory markers, such as TOX, PDCD1 and TIGIT (Fig. 3E and F), cytotoxic CD4+ T cells transcribed higher amounts of TBX21 and IFNG, enabling them to provide pathogenic effector functions (Fig. 3E and F) (27). FOXP3-expressing CD4+ regulatory T (Treg) cells were distinctly rare (Fig. 3E). TCF1hi CD4+ T cells expressed TGFB1 and SMAD3 transcripts (Fig. 3G), consistent with the notion that transforming growth factor (TGF)-β signaling can promote development of Tstem and of tissue resident memory T cells (31). In support, TCF1hi CD4+ T cells were enriched for integrin family genes (ITGA4, ITGB1, ITGB7) (fig. S4D), whereas markers of tissue residence (CX3CR, ITGAE) were mainly found in the transcriptome of other CD4+ T cells (fig. S4D). TCF1hi CD4+ T cells expressed LTB, which is essential for TLS development (32) (Fig. 3H).
Tfh-like cells isolated from the inflamed arteries had high expression of CSF2, which encodes granulocyte-macrophage colony-stimulating factor (GM-CSF), a proinflammatory cytokine specialized in monocyte activation (33) (Fig. 3H), suggesting that they may promote aortitis through an in situ GM-CSF supply. Gene signature analysis revealed that Cluster Cycling 2 exhibited a stronger T cell receptor (TCR) signaling signature (Fig. 3I), suggesting that Cluster Cycling 1 and 2 differ in activation status.
TCF1hi CD4+ T cells give rise to TCF1lo effector and cycling CD4+ T cells.
To explore the potential lineage relationship between CD4+ T cell subpopulations described above, we performed pseudotime analysis using Monocle3. The cell trajectory started from TCF1hiCD4+ T cells and subsequently divided into 3 branches, two of which progressed towards two distinct effector cell populations, Tfh-like and cytotoxic CD4+ T cells. The third branch gave rise to cycling CD4+ T cells (Fig. 4A). Along the projected pseudotime course, TCF7 expression gradually decreased (Fig. 3D), providing strong support for the concept that TCF1hiCD4+ T cells may differentiate into TCF1lo effector CD4+ T cells and serve as the source of in situ proliferating CD4+ T cells.
Fig. 4. Different vasculitogenic CD4+ T cell subpopulations share TCR clones.

Cluster assignment of scRNA-seq was performed as in Fig. 3. (A) Single-cell trajectories of vasculitogenic CD4+ T cells were generated with Monocle3 pseudotime analysis. CD4+ T cells are colored by clusters and the black line traces the trajectory (left panel). The pseudotime trajectory marks three branch nodes; the split of cycling and differentiating CD4+ T cells, which then both separate into two subsets (middle panel). In the right panel, the root of the trajectory is represented by TCF1hi CD4+ T cells, which transition into three TCF1lo branches: cycling CD4+ T cells, cytotoxic (cyto) CD4+ T cells, and Tfh-like T cells. (B to G) Tissue-derived CD4+ T cells were analyzed by single cell T cell receptor sequencing (scTCR-seq). (B) Recovered CDR3 sequences for TRA and TRB are projected on the same UMAP as the scRNA-seq data. (C) Proportions of successful CDR3 recovery for TRA and TRB are shown for each T cell subset. (D and E) Expanded TCRα (D) and TCRβ (E) clonotypes (clonotype frequency > 1 cell) are shown. (F) Shown are examples of expanded clonotypes that share TCRα or TCRβ across distinct T cell subsets. (G and H) Proportion of clonal overlap and TCR repertoire similarity score (TRSS) amongst tissue-infiltrating CD4+ T cell subpopulations for TCRα (G) and TCRβ (H).
To validate these results, we performed single cell TCR sequence (scTCR-seq) analysis from the same cDNA starting material as used for scRNA-seq and examined whether TCR clonotypes were shared between the 5 distinct T cell populations. We recovered complementarity determining region 3 (CDR3) sequences for TRA and TRB from 70.7%, TRA only from 10.2% and TRB only from 13.8% of the CD4+ T cells (Fig. 4B and C). We defined a clone as 2 or more T cells that shared the same CDR3 sequences for either α or β TCR chains. More than 40% of both TRA and TRB were clonally expanded and expanded clonotypes were present in all 5 T cell clusters (Fig. 4D and E, fig. S5A and B). Typically, expanded clonotypes were shared between distinct CD4+ T cell subsets (Fig. 4F). TCF1hi CD4+ T cells shared TCR clonotypes with all other CD4+ T cell clusters (Fig. 4G and H, fig. S5C). These results lend support for the concept that CD4+ TCF1hi T cells exhibit high plasticity and self-renewal capacity and give rise to more differentiated effector T cells.
We next explored whether the distinction between TCF7-expressing and effector molecule-expressing T cells was maintained in the tissue lesions. Expression of TCF7 in tissue-derived CD4+ T cells was negatively correlated with those of several effector molecules, including TBX21, EOMES, IFNG, and GZMB (Fig. 5A). In peripheral blood mononuclear cells (PBMCs), TBX21 (also known as T-bet) expression, typically associated with effector differentiation, correlated inversely with TCF1 protein expression in both CD4+ and CD8+ T cells of aortitis patients and controls (Fig. 5B and C).
Fig. 5. TCF1 expression correlates with the T cell differentiation state.

(A) Gene expression of TCF7 was correlated with the transcription factors TBX21 and EOMES and the effector molecules IFNG and GZMB in scRNA-seq data of individual CD4+ T cells isolated from vasculitic lesions. Color coding reflects the cluster assignment in Fig. 3A. (B to H) PBMCs were collected from patients with GCA aortitis and age-matched controls and analyzed by flow cytometry. (B and C) Expression of the transcription factors TCF1 and TBX21 (also known as T-bet) in CD4+ (B) and CD8+ T cells (C) (n=6 each) is shown. Representative dot plots and quantification of TBX21 mean fluorescence intensities (MFI) in TCF1hi, TCF1int and TCF1lo T cells are presented. (D) TCF1 expression was quantified in healthy CD4+ and CD8+ T cells isolated from healthy older adults subdivided into naïve, central memory (TCM), effector memory (TEM) and terminally differentiated effector memory CD45RA+ T cells (TEMRA) (n=8 each). (E and F) Naïve (E) and memory (F) CD4+ T cells were isolated, labeled with CellTrace and stimulated in vitro. TCF1 expression was measured 6 days after stimulation by flow cytometry. Representative dot plots and data from three donors are shown. (G and H) CD4+ (G) and CD8+ (H) T cells from patients with GCA aortitis (n=6) and age-matched controls (n=8) were subdivided into naïve, TCM, TEM, and TEMRA. Representative dot plots, frequencies of the T cell subsets and combined proportions of (naïve plus TCM) and (TEM plus TEMRA) are shown. Data are presented as mean ± SD with individual values indicated. Data were analyzed by unpaired two-tailed t-test (B to D, G and H). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To functionally characterize TCF1hi and TCF1low CD4+ T cells in the autoimmune disease process, we examined TCF1 expression across the T cell subsets of older healthy adults (64 to 83 years old). In both CD4+ and CD8+ T cells, naïve and central memory T cells (TCM) typically expressed higher concentrations of TCF1 protein than effector memory T cells (TEM) and effector memory CD45RA+ T cells (TEMRA) (Fig. 5D), highlighting that TCF1 expression is a characteristic feature of less differentiated T cells, even in older individuals. We probed the response of TCF1hi T cells to TCR stimulation. Naïve and memory CD4+ T cells from older adults were separated, labeled with CellTrace Violet and stimulated to proliferate. The offspring of naïve CD4+ T cells maintained high TCF1 expression in the proliferating cells during early divisions. Eventually, TCF1lo populations emerged (Fig. 5E and F). In contrast, the proportion of TCF1hiCD4+ T cells amongst the memory population was under 50% prior to stimulation and progressively decreased with each division (Fig. 5E and F). Thus, naïve CD4+ T cells were a superior source of TCF1hi progeny, repopulating T cells with stem-like characteristics.
We asked the question whether patients that accumulate TCF1hi T cells in the adventitial TLS of the ascending aorta have a higher pool of such T cells in the periphery. We examined the distribution of peripheral blood T cell subpopulations in patients with aortitis versus age-matched controls. CD4+:CD8+ T cell ratios were indistinguishable (fig. S6). Unexpectedly, patient-derived T cells were biased towards non-effector T cells. The proportion of non-effector CD4+ T cells (naïve plus central memory T cells) was higher and frequencies of TEMs and TEMRAs were lower in patients compared to healthy controls (Fig. 5G). A similar skew was observed in CD8+ T cells (Fig. 5H), whereas this was not detected in T cells derived from patients with non-inflammatory aortic aneurysm (fig. S7). These findings raise the possibility that accumulation of TCFhi T cell populations and delayed immune aging is a risk factor for aortitis.
TCF1hiCD4+ T cells preferentially reside within TLS and might act as a reservoir for effector memory T cells.
To verify the results obtained by scRNA-seq of tissue-derived CD4+ T cells, we returned to aortic tissues and located the functional T cell subsets identified by transcriptomics. Immunofluorescence analysis identified TCF1hi T cells within the TLS (Fig. 6A, fig. S8A). TCF1 expression in T cells was variable, ranging from high signal expression to undetectable (Fig. 6A). To define the relationship between T cell location and TCF1 expression, we determined the proportion of TCF1hi and TCF1low T cells in 3 anatomical locations: adventitial TLS, adventitial non-TLS area, and medial compartment. More than 50% of the TLS-residing T cells were strongly positive for TCF1 (Fig. 6A to C). More than 80% of the T cells in other areas were TCF1 low (Fig. 6A to C). Consistently, TCF1 expression was associated with the size of the TLS (Fig. 6D).
Fig. 6. TCF1hi CD4+ T cells preferentially localize to TLS.
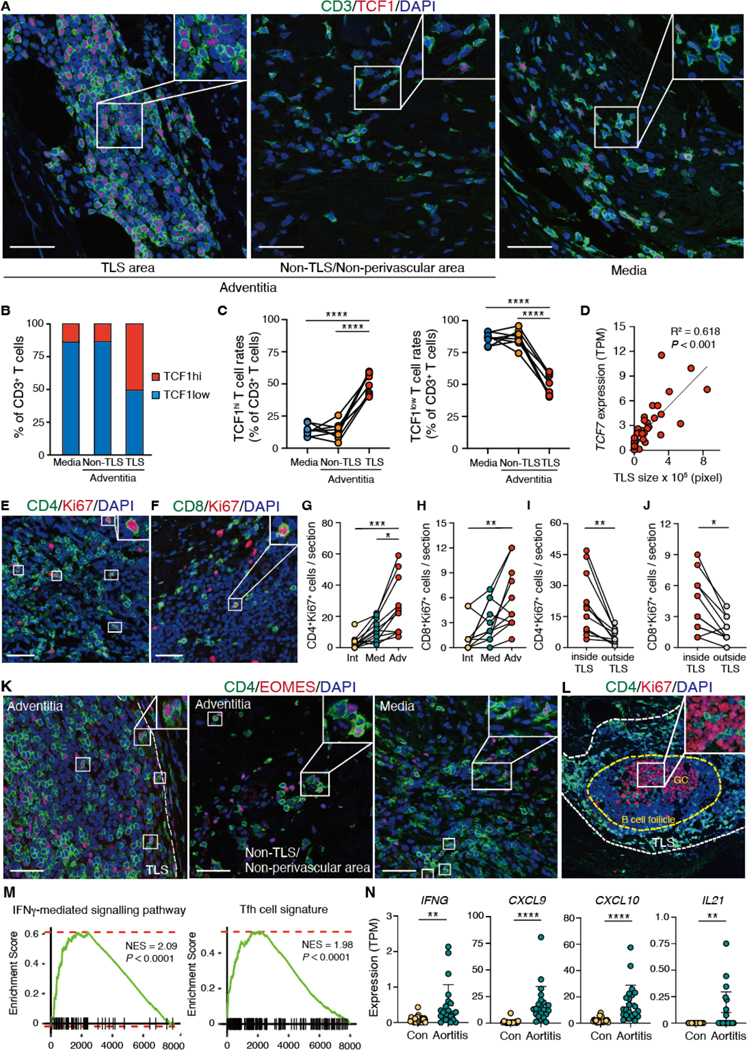
(A) Spatial mapping of TCF1hi T cells in GCA aortitis was visualized by IF staining. Tissues were subdivided into three territories: inside of adventitial TLS, outside of adventitial TLS and medial layer. (B) Proportions of TCF1hi and TCF1lo T cells in the media, inside the TLS, and in non-TLS territory were determined (n=8). (C) Shown is the geographical distribution of TCF1hi and TCF1lo T cells in individual cases of GCA aortitis. Frequencies of TCF1hi and TCF1lo T cells were measured in the medial layer, inside of adventitial TLS, and in the surroundings of the TLS. (D) Shown is the correlation between TCF7 TPM values and TLS sizes in 22 cases of GCA aortitis and 20 cases of disease controls. (E and F) Proliferating CD4+ (E) and CD8+ (F) T cells were mapped. Cycling T cells were detected by Ki67 expression. (G and H) Shown is the geographical distribution of CD4+Ki67+ (G) and CD8+Ki67+ (H) T cells in the intima, the media, and the adventitia of 11 cases of GCA aortitis. (I and J) Shown is the placement of CD4+Ki67+ (I) and CD8+Ki67+ (J) T cells inside or outside of TLS (n=11). (K) CD4 and EOMES expression was analyzed by dual-color IF in different tissue niches: inside TLS, surroundings of TLS, or media. (L) CD4 and Ki67 expression was examined by dual color IF. CD4+ T cells are detected inside of germinal centers (proliferating cell cluster). (M) Shown are GSEAs for the IFN-γ-mediated signaling pathway and for the Tfh cell signature in aortitis (n=22) and in non-inflamed aneurysms (n=20). (N) TPM values for IFNG, CXCL9, CXCL10 and IL21 in aortitis (n=22) and in non-inflamed aneurysms (Con; n=20) are compared. Data are presented as mean ± SD with individual values shown. Data were analyzed by one-way ANOVA with Tukey post hoc test (C, G), Kruskal-Wallis test with post-hoc (H) or Mann-Whitney U test (I, J, N). The correlation in (D) was determined by Pearson’s correlation analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bars, 50 μm.
We investigated whether the high proliferative potential of TCF1hi T cells was maintained when such T cells were placed into the tissue niche. IF for the proliferation marker Ki67 positioned proliferating CD4+ and CD8+ T cells to the adventitial layer (Fig. 6E to H, fig. S8B and C), especially, into the TLS (Fig. 6I and J, fig. S8B and C). Cytotoxic CD4+ T cells, defined as EOMES+CD4+ T cells, were present both inside and outside of the TLS (Fig. 6K). IF identified Tfh-like cells within germinal centers (Fig. 6L). IFNG, IFNGR1 and IFNGR2 transcripts were differentially expressed in the aortitis cases and GSEA provided further evidence for ongoing type II interferon (IFN) signaling in the disease lesions (Fig. 6M and N, fig. S8D), all supporting the presence of IFN-producing cytotoxic CD4+ T cells. GSEA of the bulk RNA-seq dataset confirmed enrichment of a Tfh gene signature and elevated IL21 gene transcripts in aortitis tissues, in support of the presence of Tfh-like cells (Fig. 6M and N). Also, TCF1 and ICOS expression were positively correlated (fig. S8E). Collectively, the data support the concept that TCF1hi T cells give rise to Tfh cells, which reach pathogenic relevance by supporting the assembly and maintenance of TLS.
To further characterize the tissue niche for TCF1hi CD4+ T cells, we determined the geographical distribution of TCF1hi CD4+ T cells within the TLS. TCF1hi CD4+ T cells homed to both T cell zones and T/B mixed zones (Fig. 7A to D). As in lymph nodes, where DC networks are particularly dense at the T cell-B cell zone border, CD11c+ DCs mapped to the T cell zone, often close to the B cell zones (Fig. 7E to G). TLS-residing CD11c+ DCs formed extensive networks, creating a specialized environment for T cell survival (Fig. 7E to G, fig. S9) (34). We also observed Ki67+CD11c+ DCs, suggesting they are capable of replenishing within the TLS (Fig. 7H). Finally, CD11c+ DCs were often wrapped around HEVs, building intercalating cellular networks between the two cell types (Fig. 7I and J).
Fig. 7. TCF1hi T cells survive in defined cellular neighborhoods.
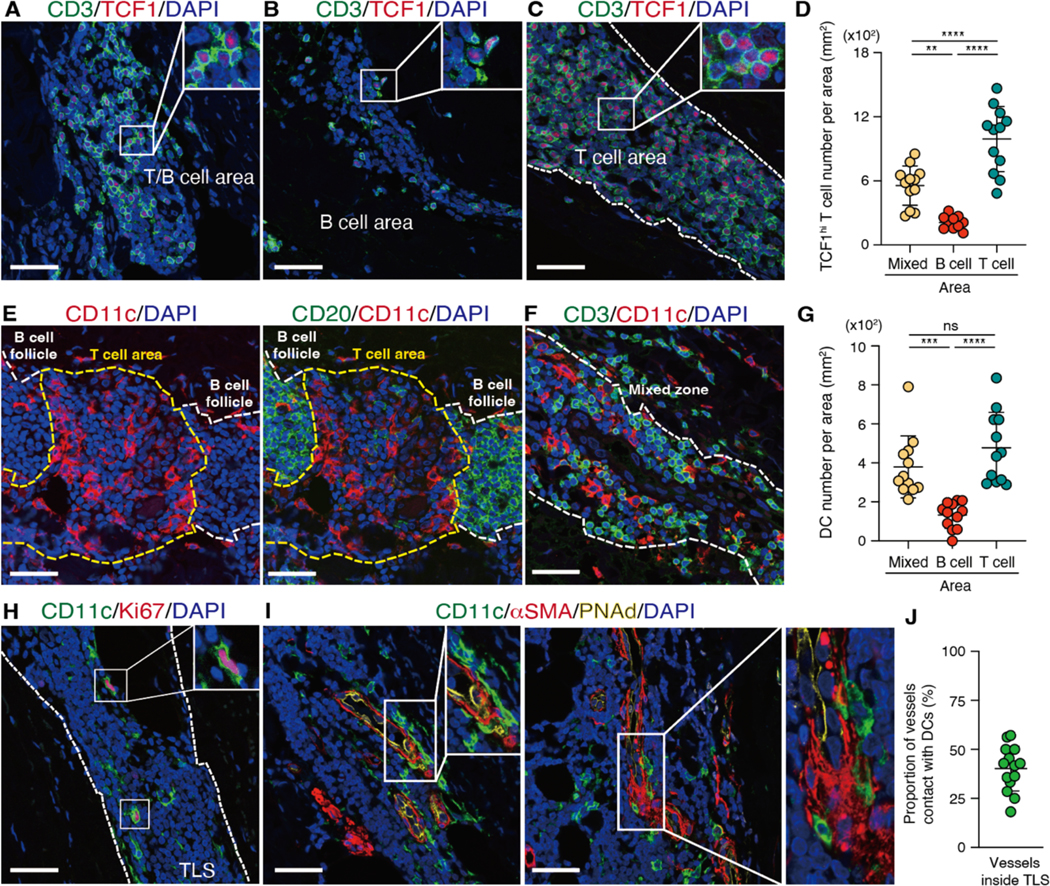
TLS were identified in the aortic wall of GCA-affected tissues and each TLS was subdivided into three zones: T cell zone, B cell zone, and mixed zone. Multi-color IF was used for spatial mapping of all cell types. (A to C) Mapping of the T/B cell area (A), the B cell area (B), and the T cell area (C) as well as quantification of TCF1hi T cells in different zones (n=12) (D) are shown. (E to F) Tissue distribution of CD11c+ dendritic cells (DC) in the T and B cell zone (E) and in the mixed zone (F) are shown and CD11c+ DC numbers in different zones (G) are compared. (H) Proliferating CD11c+ DCs were identified by Ki67 staining. (I) DC-microvessel contact sites are visualized. Microvessels were identified through αSMA and PNAd. (J) Proportions of αSMA+ microvessels inside TLS in direct contact to DC were quantified (n=14). Localization of TLS and T cell zones is indicated by dotted lines. Data are presented as mean ± SD with individual values indicated. Data were analyzed by one-way ANOVA with Tukey post hoc test (D, G). **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bars, 50 μm.
TCF1hiCD4+ T cells function as disease stem cells.
We hypothesized that TCF1hi T cells have a unique pathogenic role due to their ability to replenish the T cell infiltrates by in situ proliferation, give rise to effector T cells and maintain autoimmune vasculitis in an organ-autonomous manner, independent from circulating immune cells. To test this hypothesis, we designed serial transplantation experiments, probing the disease-maintaining capabilities of TCF1hi CD4+ T cells. We first identified IL-7 receptor (IL-7R) as a sortable surface marker correlated with high TCF7 expression (Fig. 8A). IL-7R expression was confined to T cells (Fig. 8B, fig. S10), and high TCF1 expression distinguished IL-7R+ T from IL-7R− T cells (Fig. 8C). Utilizing IL-7R as a surrogate marker for TCF1hi CD4+ T cells, we generated TCF1-low expressing GCA PBMCs by depleting IL-7R+ cells. We then induced vasculitis in humanized NSG mice by adoptive transfer of IL-7R containing (IL-7R+) and IL-7R-depleted (IL-7R−) PBMCs (Fig. 8D). To separate T cells that reside within the inflamed arteries from those living in extravascular lymphoid tissues (mostly the spleen of NSG mice), we serially transplanted the inflamed arteries into “empty” mice free of human cells other than graft-residing cells (Fig. 8D). We monitored numbers of human T cells in the blood and the spleen of the primary and secondary recipients (Fig. 8E). Engraftment of inflamed human arteries into secondary recipients was associated with complete containment of tissue-infiltrating T cells in the vascular grafts. No human cells were detected in the circulation.
Fig. 8. TCF1hiCD4+ T cells function as disease stem cells.

(A) TCF7 and IL-7R gene expression in individual CD4+ T cells isolated from vasculitic lesions are correlated. (B) Representative flow cytometry plot is shown for CD3 and IL-7R in PBMCs from older adults. (C) TCF1 expression in IL-7R positive (IL-7R+) and IL-7R negative (IL-7R−) T cells are compared. Representative histogram and data from 8 individuals are presented. (D) Shown is a scheme of the serial transplantation experiments. Pairs of NSG mice were engrafted with human arteries, reconstituted with IL-7R+ or IL-7R− PBMCs from patients with GCA. IL-7R− PBMCs were prepared by depleting IL-7R+ cells. Vasculitis was induced in engrafted human arteries and inflamed arteries were transplanted into “empty” NSG mice. Finally, arteries were explanted on day 42. (E) Numbers (No.) of human T cells in the blood (100μl) and the spleen of NSG mice reconstituted with IL-7R+ or IL-7R− PBMCs were quantified (n=3). Data of T cell numbers in 100μl blood and spleen 7 days after vasculitis induction were used as positive control (49). (F to M) Tissue sections were prepared from the explanted arteries and the intensity of vasculitis was assessed in arteries. (F) Representative images (H&E) show density of intrawall infiltrates. (G) Shown is a comparison of vascular injury scores in arteries (n=6 each). (H and I) Intralesional microvessels were identified by dual color IF analysis of vWF (green) and αSMA (red). Representative microphotographs (H) and enumeration of lesional microvessels (I) in arteries are shown (n=6 each); HPF, high power field. (J) VEGFA transcripts in arteries were quantified using real-time polymerase chain reaction (RT-PCR); n=6 each. (K and L) Proliferating T cells in the tissue were identified by dual-color IF for CD3 (green) and Ki67 (red). Representative images (K) and total T cell numbers and proportions (L) of Ki67+ T cells in arteries are shown (n=6 each). (M) IFNG and GZMB mRNA was quantified in arteries by RT-PCR; n=6 each. Data are presented as mean ± SD with individual values given. Data were analyzed by paired t-test (C, G, I, L), one-way ANOVA with Tukey post hoc test (E), or Wilcoxon matched-pairs signed rank test (two-sided) (J, M). *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars, 50 μm. IL-7R pos: IL-7R+, IL-7R neg: IL-7R−.
The intensity of vasculitis in explanted arteries was quantified through the histology score, evaluating severity and spread of inflammation as well as extent of vascular damage. Vasculitis induced by IL-7R+ cells depleted PBMCs was significantly less severe (p<0.001) (Fig. 8F and G), demonstrating the pathogenic relevance of TCF1hi T cells. We evaluated the impact of IL-7R+TCF1hi T cells on vascular remodeling. Depletion of IL-7R+ cells from the vasculitis-inducing immune cell populations markedly reduced VEGFA transcription and minimized graft neoangiogenesis (Fig. 8H to J). To mechanistically connect the proliferative capacity of IL-7R+TCF1hi T cells with chronicity and transferability of vasculitis, we compared rates of Ki67+ T cells in arteries from the chimeras reconstituted with IL-7R-containing and IL-7R-depleted grafts. Arterial inflammation induced with IL-7R+ T cell populations was T cell-rich and included high rates of proliferating (Ki67+) T cells (Fig. 8K and L). The depletion of IL-7R+ cells resulted in low-grade wall inflammation that lacked transcripts for the two main effector molecules, IFNG and GZMB (Fig. 8M). Taken together, these data suggest that TCF1hiCD4+ T cells are required to induce transferrable and persistent vasculitis, designating them as disease stem cells.
DISCUSSION
The exclusivity of GCA in medium and large arteries has been attributed to autoantigen availability but current data assign a critical role to structural elements in the tissue microenvironment; specifically, to the vasa vasorum arterioles and venules that serve as an origination point for lymphoid architectures harboring TCF1hiCD4+ T cells that serve as disease stem cells. In vessel-surrounding TLS, TCF1hiCD4+ T cells undergo proliferation and differentiate into pathogenic effector cells that attack the vessel wall. Thus, adventitial TLS function as a hub, steadily supplying T cells to the granulomatous infiltrates and rendering the disease lesions autonomous and self-sustained. The formation of highly sophisticated lymphoid structures seeded with TCF1hi stem-like cells reaches equal importance as autoantigen access. All cases of GCA aortitis analyzed had adventitial TLS, supporting the local renewal of disease stem cells, implicating TLS and local production of stem-like T cells in disease chronicity. These data have implications for the diagnosis and management of autoimmune vasculitis but the principle of local distribution centers for disease-relevant effector cells may be equally important in other autoimmune conditions associated with TLS formation (14). As a minimum, our data suggest that therapies directed only at effector T cells are insufficient to arrest the in situ autoimmune responses and offer strategies for the elimination of disease stem cells.
Structurally, aortic wall TLS are heterogeneous, with only some containing germinal center reactions, whereas 25% were occupied by mostly CD4+ T cells; challenging the concept that TLS are B cell-oriented structures dedicated to autoantibody production (14, 15). GCA patients typically lack autoantibodies; rather macrophages and T cells are the dominant tissue-destructive cell populations (1, 2). Accordingly, GCA TLS engage in the local production and differentiation of CD4+ T cells. Induction or removal of TLS has been considered as a potent strategy to treat cancer, infection, and age-related diseases (18–20, 23, 35–38). In autoimmunity, target organ TLS are generally associated with poor disease prognosis (14, 15, 39). This is also the case in GCA, as the TLS transcriptomic signature is tightly linked to aortic aneurysm formation, but TLS have been noted in temporal artery biopsies (40, 41). Indeed, even early stages of GCA are often resistant to immunosuppressive therapy. In sequential biopsies studies, 50% of patients had persistent vasculitis despite high-dose therapy for 12 months (3). These data emphasize the power of locally produced TCF1hiCD4+ T cells in sustaining autoimmunity.
scRNA-seq studies identified TCF1hiCD4+ T cells as the major subpopulation in the inflamed arteries. Pseudotime analysis suggested that tissue-residing TCF1hiCD4+ T cells decide between self-renewal and differentiation into two major effector populations: EOMES+CD4+ T cells equipped with a cytotoxic machinery and BCL6+ Tfh-like cells, capable of providing help for TLS formation. scTCR-seq analysis confirmed frequent TCR clonotype sharing between TCF1hi CD4+ T cells and all other subpopulations, strongly supporting the hypothesis that TCF1hi CD4+ T cells act as precursor cells for cytotoxic and Tfh-like cells. Stem-cell like CD8+ T cells have been implicated in driving tissue destruction in type I diabetes (42), but notably, these CD8+ Tstem reside in regional lymph nodes. The dimensional step in aortitis lies in the construction of lymph node-like TLS directly in the target organ.
Trm are generated in tissues after site-specific antigen exposure, persist and drive immune responses in an organ-autonomous manner (43). In autoimmunity, Trm confer long-term immunological memory in the target organs, guiding immunologic silence versus tissue inflammation. Here, serial transplantation experiments demonstrated tissue residency of vasculitis-inducing CD4+ T cells that found a survival niche in TLS. Such TLS are prime therapeutic targets for immunotherapy aiming to induce disease remission.
Although TCF1hi CD4+ T cells were shown to be a requirement for autoimmunity in our model, these cells are not directly involved in tissue injury. GCA lesions typically contain a broad spectrum of effector cytokines (IFN-γ, IL-21, IL-9, IL-2, GM-CSF, IL-17, IL-22) but how functional T cell subsets are induced remains unclear (1). EOMES+ cytotoxic CD4+ T cells have high destructive potential, targeting stromal cells, such as endothelial cells and vascular smooth muscle cells (27, 44) and are possibly directly involved in formation of aortic aneurysms. How BCL6+ Tfh-like cells contribute to vasculitis needs to be defined. A major disease pathway lies in macrophage activation, some of which differentiate into multinucleated giant cells. Transcriptomic analysis placed the cytokine GM-CSF into Tfh-like CD4+ T cells, possibly of relevance in vasa vasorum leakiness. Also, better understanding of how the defective PD1/PD-L1 pathway interferes with TLS induction and the turn-over and differentiation of TCF1hi CD4+ T cells requires attention.
NOTCH1+CD4+ T cells interact with vasa vasorum ECs, enabling T cell invasion into the vessel wall (9). Thus, NOTCH signaling may guide early steps of TLS formation, such as the formation of intrawall lymphoid pockets. In aortitis, adventitial vessels acquire a HEV-like phenotype. Loss of Notch signaling selectively in ECs leads to the HEV phenotype in middle-size arteries and to TLS formation (45). Downregulation of Notch signaling, coupled with the loss of EC differentiation markers occurs in high ECs (46). The intimate spatial relationship between HEVs and CD11c+ DCs in aortic TLS suggests a critical position of DCs in HEV development and maintenance, previously proposed for lymph nodes (47). Cancer immunotherapies have been implicated in the EC-to-HEV transformation, promoting TCF1+ T cell niches in the tumor microenvironment (48). Whether pathogenic CD4+ T cells could analogously facilitate the induction of aortic TLS remains to be investigated. Intriguingly, large vessel vasculitis is the predominant type of vasculitis associated with immune checkpoint inhibitors (13).
Our study has limitations. The aortic tissues were resected from patients with advanced disease and it remains to be explore to which extent formation of tertiary lymphoid structures affect early steps in the disease process. Definition of cellular neighborhood by imaging studies is limited to cell types that are present in frequencies >1 to 2% and might therefore miss low-frequency cell types. Also, current technologies do not permit analysis of cells in three-dimensional tissue environments, which may be of particular relevance in the function of TCF1hi CD4+ T cells in the wall of inflamed arteries.
Beneficial TLS in anti-tumor immunity stand in contrast to the detrimental TLS in autoimmune disease. Building complex lymphoid architectures in close vicinity to autoantigen production sites adds a new dimension to the disease process. TLS can capture, enrich, and store autoantigens, maximizing adaptive immune responses toward tissue antigens. Most importantly, TLS function as a site for TCF1hi CD4+ T cells that repopulate in situ and give rise to a diverse set of effector T cells, combining disease maintenance with diversification of tissue damaging effector functions. Understanding the determinants that control TLS development as well as the origin of vasculitis-inducing TCF1hi CD4+ T stem cells has important clinical implications. TLS may provide a focus for biomarker development to surmount the barriers in assessing inflammatory activity in the aortic wall. Equally important, the power of TLS in rendering the disease process autonomous calls for the therapeutic targeting of such structures to overcome the high rate of treatment resistance in this autoimmune disease.
MATERIALS AND METHODS
Study design
The objective of this study was to identify cellular and molecular mechanisms driving persistence of autoimmunity and GCA aortitis was chosen as a model system. Table 1 shows clinical and demographic characteristics of patients with GCA aortitis (n=22) and disease controls (non-inflamed ascending aortic aneurysms, n=20). In case-controlled histological and transcriptomic studies of ascending aortic aneurysm, all cases of aortitis had structured perivascular lymphoid clusters. Next, we dissected the heterogeneity and the function of disease-relevant CD4+ T cells using scRNA-seq and a human artery-NSG mouse chimeric model, in which vasculitis is induced under controlled conditions. Tissue residing CD4+ T cells were composed of 5 subsets. Besides proliferating and effector T cells, the inflamed arteries contained a CD4+ T cell population highly expressing the transcription factor TCF1. We found that TCF1hi CD4+ T cells gave rise to CD4+EOMES+ cytotoxic effector T cells and to CD4+BCL6+ Tfh-like cells in situ, identifying them as disease stem cells. We relied on serial transplantation experiments to examine whether TCF1hi CD4+ T cells can induce durable autoimmunity, even in the absence of cellular influx from outside. Numbers of independent experiments or individuals are given in each figure legend. Sample sizes were determined from pilot experiments. All experiments were approved by the Mayo Clinic Institutional Review Board, and appropriate informed consent was obtained from all participants.
Patients and tissue samples
Patients were recruited in accordance with an approved Mayo Clinic Institutional Review Board protocol, and all patients provided informed consent. Surgically resected ascending aortic aneurysms were obtained from 22 aortitis patients and 20 non-inflammatory disease controls (Table 1). The diagnosis of GCA was established by temporal artery biopsy. Histologic diagnoses were confirmed by at least 2 vascular pathologists. Tissues with severe and diffuse atherosclerosis were excluded from both groups. Normal human arteries were collected from organ donors. Blood samples were donated by patients with GCA, GCA aortitis, or aortic aneurysm as well as by age-matched healthy individuals (Table 2).
Table 2.
Clinical characteristics of patients with GCA.
| Patients with GCA (n = 25) | |
|---|---|
| Age (year, mean ± SD) | 68.8 ± 8.8 |
|
| |
| Male:Female | 4:21 (16.0% : 84.0%) |
|
| |
| Ethnicity | |
| Caucasian | 22 (88.0%) |
| African American | 1 (4.0%) |
| Hispanic | 0 (0%) |
| Asian | 2 (8.0%) |
| Other | 0 (0%) |
|
| |
| Disease Duration (month, mean ± SD) | 4.68 ± 4.43 |
|
| |
| Erythrocyte sedimentation rate (mean ± SD, mm/h) | 33.72 ± 30.38 |
| C-reactive protein (mean ± SD, mg/dL) | 29.19 ± 50.97 |
|
| |
| Clinical symptoms | |
| Headaches | 12 (48.0%) |
| Eye involvement | 7 (28.0%) |
| Claudication | 8 (32.0%) |
| Polymyalgia rheumatica symptoms | 9 (36.0%) |
| Constitutional symptoms | 14 (56.0%) |
| Aortic/large vessel involvement | 19 (76.0%) |
| Treatment of GCA | |
| Untreated | 12 (48.0%) |
| Prednisone | 11 (44.0%) |
| dose (mg/day, mean ± SD) | 25.1 ± 21.8 mg/day |
| Tocilizumab | 4 (16.0%) |
Values are n (%) unless otherwise noted.
Histological and immunohistological analysis
Aortic aneurysms were fixed in formalin, embedded in paraffin, and sectioned (6.0 μm thickness), and then stained with hematoxylin eosin (HE) or Verhoeff-Van Gieson (VVG). The sizes of aortic mononuclear cell aggregates were measured with the HE-stained sections of surgically resected ascending aortic aneurysm samples using Adobe Photoshop software. For immunofluorescence, the paraffin-embedded sections were deparaffinized with xylene, rehydrated, and then steam-heated for 15 min as described previously (38). The sections were incubated with 5% serum appropriate to the secondary antibody for 1 h at room temperature, and then incubated overnight at 4°C with the primary antibodies (table S1). All staining samples were visualized using the appropriate secondary antibodies (at a dilution of 1:300), counterstained with 4’,6-diamidino-2-phenylindole (DAPI), and analyzed using a confocal microscope (LSM780 confocal microscope, Zeiss). TLS were defined as perivascular lymphocyte aggregates with sign of proliferation. Quantification of TLS was performed using immunofluorescence of CD3 and CD20. T and B cell dominated TLS were defined as those composed mainly of T and B cells (> 75%), respectively, and other TLS were defined as mixed type TLS. Ratios were calculated by the total and each type of TLS numbers per one section. Geographical distribution of TCF1hi/low T cells and Ki67+ T cells in aortitis samples were determined by immunofluorescence. The cases with TLS in the media were excluded from the analysis. For immunohistochemistry, the paraffin-embedded tissue sections were rehydrated, and endogenous peroxidase was blocked using 3% H2O2. Antibody labeling was performed with a 3, 3 -diaminobenzidine (DAB) reagent (Vector Laboratories). Sections were counterstained with hematoxylin.
Bulk RNA-Seq data analysis
RNA was extracted from formalin-fixed paraffin-embedded block sections. The tissues were added with buffer PKD and proteinase K and placed in a QIAcube for the RNA purification (extraction) as previously described (21). Libraries were prepared using a TruSeq RNA Exome Capture kit (Illumina) following the manufacturer’s protocol as previously described (21). The raw reads were preprocessed using the nf-core RNA-seq pipeline (v.3.3), aligned to the human reference genome GRCh38. For PCA, the counts were normalized by ‘regularized log’ transformation using DESeq2 (v.1.34.0) (50) followed by removing gender effect using limma (v.3.50.3) (51). To identify the differential gens, the mean-variance trend modelled on logCPM values of each gene along with sample weight adjustment using voomWithQualityWeights (52, 53). Using the normalized weighted matrix linear model is fitted. The differential genes between Aortitis and Control group were calculated by fitting contrast to the model followed by the empirical Bayes moderation. The Gene Set Enrichment Analysis (GSEA) was performed on the differential genes (adjusted pvalue < 0.05) using fgsea (v.1.20.0). The Tfh gene sets were extracted from Meckiff et al. (54) and the Trm gene set was extracted from (55), whereas interferon mediated signaling pathway and positive regulation of T cell proliferation were extracted from Gene Ontology database (56). Pathway enrichment analysis was performed on the 1070 DEGs between aortitis and non-inflamed aortic aneurysm disease controls using enrichR (57). CIBERSORTx analysis were performed on bulk RNAseq data by using the analytical tool (cibersortx.stanford.edu) (58). LM22 was used as a reference signature for analysis. To compare the single cell and bulk RNA-seq datasets, we transformed single cell RNA raw counts to pseudobulk using Seurat’s AggregateExpression function. Genes with raw counts greater than 5 were retained. Subsequently, genes in the bulk RNA were overlapped with the genes in the pseudobulk single cell RNA and percentages identified or missing from single cell to bulk RNA were calculated. The bulkRNAseq data in the current study have been deposited in the NCBI (https://submit.ncbi.nlm.nih.gov/geo/submission/) under accession number GSE224528. The normal aorta sets were extracted from GSE 141032 (59). Raw counts were normalized and compared with the data sets from the aortitis and disease control cases.
Human artery-severe combined immunodeficiency mouse chimeras.
Human artery-mouse chimeras were generated using NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice as previously published (6, 49). In brief, non-inflamed human axillary arteries were engrafted subcutaneously into the back of NSG mice. Six days after the engraftment, 10 μg of lipopolysaccharide (LPS) was injected subcutaneously, and the next day, PBMCs from GCA patients were adoptively transferred into the chimeras.
In scRNA-seq experiments, 10 million cells per mouse were transferred for vasculitis induction. Adventitial tissues from engrafted arteries were harvested 22 days after vasculitis induction. To isolate CD4+ T cells from the tissues, the tissues were physically minced and then shaken at 37 °C for 1 hour with collagenase/DNase I solution (1.5 mg/ml collagenase Ⅰ (#LS004196; Worthington Biochemical Corporation) and 0.1 mg/ml DNase I (#E1011-A; Zymo Research) in Hanks’ balanced salt solution (#14025076, Thermo Fisher Scientific)). After filtration with 100 μm mesh, cells were subject to dead cell removal kits (#17899, STEMCELL Technologies), and filtered again through 35 μm mesh. CD4+ T cells were then isolated using the EasySep Human CD4+ T Cell Isolation Kit (#17952, STEMCELL Technologies).
In serial transplantation experiments, 6 to 10 million cells per mouse were transferred for vasculitis induction. In IL-7R positive cell depleted group, IL-7R positive cells in PBMCs from GCA patients were stained with CD127(IL-7R-α)-phycoerythrin (PE) antibody (clone A019D5, #351304, BioLegend) and removed with anti-PE Microbeads (#130–048-801, Miltenyi Biotec), as commercial protocol indicated. Engrafted arteries were transengrafted into another empty mice 14 days after vasculitis induction and harvested 4 weeks after the transengraftment. For histological analysis, engrafted arteries were fixed in 4% paraformaldehyde, incubated in 20% sucrose for 5 hours and in 30% sucrose in phosphate-buffered saline (PBS) at 4°C overnight, and embedded in OCT compound. OCT-embedded arteries were cryosectioned and used for immunofluorescence and HE stains.
Single cell RNA-Seq
Three humanized mice were used. Beads isolated CD4+ T cells from adventitial lesion of vasculatures were loaded onto a Chromium Single Cell Controller using the Chromium Single Cell 3’ Library and Gel Bead Kit v3 (both from 10x Genomics) following the manufacturer’s instructions. Sample preparation and library preparation were performed following the manufacturer’s instructions. The libraries were sequenced on NextSeq 2000 (Illumina). The scRNA-seq data in the current study have also been deposited in the NCBI (https://submit.ncbi.nlm.nih.gov/geo/submission/) under accession number GSE224528.
Single cell RNA-Seq data analysis
The single cell fastq data is processed using Cell Ranger pipeline version 6.1.2 (10x Genomics) mapped on GRCh38 v.3.0.0 reference. The downstream analysis was performed using R package Seurat v.4.3.0 for quality control (QC), normalization, scaling, dimensionality reduction, clustering, differential expression, and visualization (56). In brief we have retained genes expressed in >3 cells. Cells expressing between 200 and 4000 genes with <10% mitochondrial gene contamination were retained for analysis. We have also removed doublets cells using DoubletFinder (60). Further we have extracted T cells based on the expression of CD3 and CD4 and performed the downstream reanalysis using Seurat. The raw counts were normalized, and variable genes were identified using NormalizeData and FindVariableFeatures function in Seurat, respectively. The variable genes were used for dimensionality reduction followed by clustering using graph-based clustering approach using the Louvian algorithm. We have performed imputation of gene expression using MAGIC (61). The imputed data is being used to perform Pearson correlation between the TCF1 with other genes. The gene module scores were calculated using the “AddModuleScore” function from Seurat applying TCR signaling and Cell cycling gene sets. Pseudotime trajectory analysis was performed using Monocle3 (62).
T cell receptor sequencing from single cell library
To obtain TCR CDR3 sequences from 3’ scRNA sequencing experiments, we enriched and amplified TCR-encoding transcripts from the whole transcriptome starting materials as described previously (63,64). We followed the modified protocol for 10x genomics 3’ starting materials by Millar et al. (63) that was originally developed for Seq-Well by Tu et al. (64). We sequenced TRA- or TRB-enriched libraries on Illumina MiSeq lane (about 1,000 T cells per lane) at a cluster density of about 450,000 per mm2. Twenty-eight cycles were used for read 1 and 100 cycles were used for index 1, which reads the TCR CDR3 region. TCRα- and TCRβ-specific custom sequencing primers were used for index 1 (63). We used WAT3R to analyze the TCR CDR3 sequences (65) and downstream analysis was performed using in-house scripts. For clonal sharing in T cell subsets, we have used TCR repertoire similarity score (TRSS) measured by the Bhattacharyya coefficient as previously described (66).
Flow cytometry antibody staining
PBMCs were isolated by gradient centrifugation with Lymphocyte Separation Medium (Lonza). Single cell suspensions were stained with a cocktail of conjugated antibodies (table S2) at 4 °C for 20 minutes. For intracellular TCF1 and TBX21 staining, cells were surface stained, washed in PBS, permeabilized with Foxp3/Transcription Factor Staining buffer set (eBioscience, catalog number 00–5523-56), and stained with anti-human TCF1 (Cell Signaling Technology, clone C63D9 conjugated with PE, catalog number #14456) and TBX21 (BioLegend, clone 4B10 conjugated with brilliant violet (BV) 421, catalog number 644815). Flow cytometric analyses were performed using a CYTEK NL-1000 or FACSAria II, and analyzed with FlowJo software (Becton, Dickinson Co.).
Tracking proliferation of rested human CD4+ T cells
PBMCs were separated from whole blood using Lymphocyte separation medium (Lonza). Naïve and memory CD4+ T cells were isolated from aged healthy donor PBMCs using the EasySep Human Naive and Memory CD4+ and T Cell Enrichment Kit (STEMCELL Technologies), respectively. CD4+ T cells were labeled with Cell Trace Violet (Thermo Fisher Scientific, C34571). 5×104 cells were plated per well in 96-well plates and stimulated with anti-CD3/anti-CD28-coated Dynabeads (Gibco) at a ratio of 1 bead per 2 cells for 6 days. T cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum, penicillin/streptomycin and 10 ng/mL IL-2 for 6 days and analyzed with flow cytometry. Each peak of the Cell Trace Violet fluorescence signal represents one cell division cycle.
Histological injury score
Histological injury score was scored using HE-stained sections of engrafted arteries in humanized chimeric mice. Inflammation severity, inflammation extent, and vascular damage were graded, same as other humanized disease mice models (67). For each feature, histological scores were graded blindly using a semi-quantitative scale. The histological score was obtained by adding the average of the subscore of each feature.
RNA extraction, reverse transcription and RT-PCR
Total RNA was purified with Direct-zol RNA MiniPrep kits (Genesee Scientific) and was reverse transcribed into complementary DNA with High-Capacity cDNA Reverse Transcription Kits (Thermo Fisher Scientific). Quantitative RT-PCR was performed with PowerUp SYBR Green Master Mix (applied biosystems by Thermo Fisher Scientific) and QuantStudio 6 Pro (applied biosystems by Thermo Fisher Scientific). The primer sequences used in this study are listed in table S3. Expression values were normalized to those of β-actin.
Statistical analysis
All raw, individual-level data for experiments where n<20 are presented in data file S1. Shapiro-Wilk test (Fig. 8E) or Kolmogorov-Smirnov test (others) was performed to compare variance where a parametric test was used (P > 0.05). Statistical analysis was performed by Mann-Whitney U test, Wilcoxon matched-pairs signed rank test (two-sided), paired or unpaired student’s t-test, Kruskal-Wallis test with post-hoc, one-way analysis of variance (ANOVA) with Tukey’s post hoc test, and Pearson’s correlation analysis as indicated in the figure legends. Significance was set at P < 0.05. Statistical analysis was performed using Prism software (version 14, SAS Institute Inc.).
Supplementary Material
Acknowledgments:
Funding:
This work was supported by the National Institutes of Health (R01AR042527, R01AI108906, R01HL142068, and R01HL117913 to CMW and R01AI108891, R01AG045779, U19AI057266, R01AI129191 to JJG). Mayo Foundation for Medical Education and Research contributed funds for the study. YS was supported by a grant from the Astellas Foundation for Research on Metabolic Disorders and IS was supported by the Glenn Foundation Postdoctoral Fellowship.
Footnotes
Competing interests:
HO has received salary support from Shionogi & Co. CMW has provided consulting services to AbbVie Inc., Bristol Myers Squibb and Gilead (Research Scholar Program). GJB has received support from Merck Pharmaceutical and JJG from Retro Biosciences. KJW has participated in clinical trials (Eli Lilly, Bristol Myers Squibb, Kiniksa) and has consulted for Amgen and Sanofi. YS was previously employed by the TMK project, a collaboration between Kyoto University and Mitsubishi Tanabe Pharma.
Data and materials availability:
All data associated with this study are in the paper or supplementary materials. The scRNA-seq and bulk RNA-seq data have been deposited in the NCBI (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE224528. Please direct all other requests for data and further information to C.M.W. at cweyand@stanford.edu.
References and Notes
- 1.Weyand CM, Goronzy JJ, Immunology of Giant Cell Arteritis. Circ Res 132, 238–250 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weyand CM, Goronzy JJ, Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med 371, 1653 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, Weyand CM, Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod Pathol 30, 788–796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weyand CM, Xie C, Goronzy JJ, Homozygosity for the HLA-DRB1 allele selects for extraarticular manifestations in rheumatoid arthritis. J Clin Invest 89, 2033–2039 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weyand CM, Schonberger J, Oppitz U, Hunder NN, Hicok KC, Goronzy JJ, Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes. J Exp Med 179, 951–960 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe R, Maeda T, Zhang H, Berry GJ, Zeisbrich M, Brockett R, Greenstein AE, Tian L, Goronzy JJ, Weyand CM, MMP (Matrix Metalloprotease)-9-Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ Res 123, 700–715 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gloor AD, Berry GJ, Goronzy JJ, Weyand CM, Age as a risk factor in vasculitis. Semin Immunopathol 44, 281–301 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin K, Wen Z, Wu B, Zhang H, Qiu J, Wang Y, Warrington KJ, Berry GJ, Goronzy JJ, Weyand CM, NOTCH-induced rerouting of endosomal trafficking disables regulatory T cells in vasculitis. J Clin Invest 131,(1);e136042 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, Liao YJ, Goronzy JJ, Weyand CM, The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med 9(399);eaal3322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, Goronzy JJ, Weyand CM, Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci U S A 114, E970–E979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohtsuki S, Wang C, Watanabe R, Zhang H, Akiyama M, Bois MC, Maleszewski JJ, Warrington KJ, Berry GJ, Goronzy JJ, Weyand CM, Deficiency of the CD155-CD96 immune checkpoint controls IL-9 production in giant cell arteritis. Cell Rep Med 4, 101012 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe R, Hilhorst M, Zhang H, Zeisbrich M, Berry GJ, Wallis BB, Harrison DG, Giacomini JC, Goronzy JJ, Weyand CM, Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight 3,20:e123047. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daxini A, Cronin K, Sreih AG, Vasculitis associated with immune checkpoint inhibitors-a systematic review. Clin Rheumatol 37, 2579–2584 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Sato Y, Silina K, van den Broek M, Hirahara K, Yanagita M, The roles of tertiary lymphoid structures in chronic diseases. Nat Rev Nephrol, 1–13 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bombardieri M, Lewis M, Pitzalis C, Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat Rev Rheumatol 13, 141–154 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Shinoda K, Hirahara K, Iinuma T, Ichikawa T, Suzuki AS, Sugaya K, Tumes DJ, Yamamoto H, Hara T, Tani-Ichi S, Ikuta K, Okamoto Y, Nakayama T, Thy1+IL-7+ lymphatic endothelial cells in iBALT provide a survival niche for memory T-helper cells in allergic airway inflammation. Proc Natl Acad Sci U S A 113, E2842–2851 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amezcua Vesely MC, Pallis P, Bielecki P, Low JS, Zhao J, Harman CCD, Kroehling L, Jackson R, Bailis W, Licona-Limon P, Xu H, Iijima N, Pillai PS, Kaplan DH, Weaver CT, Kluger Y, Kowalczyk MS, Iwasaki A, Pereira JP, Esplugues E, Gagliani N, Flavell RA, Effector TH17 Cells Give Rise to Long-Lived TRM Cells that Are Essential for an Immediate Response against Bacterial Infection. Cell 178, 1176–1188 e1115 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, Gopalakrishnan V, Xi Y, Zhao H, Amaria RN, Tawbi HA, Cogdill AP, Liu W, LeBleu VS, Kugeratski FG, Patel S, Davies MA, Hwu P, Lee JE, Gershenwald JE, Lucci A, Arora R, Woodman S, Keung EZ, Gaudreau PO, Reuben A, Spencer CN, Burton EM, Haydu LE, Lazar AJ, Zapassodi R, Hudgens CW, Ledesma DA, Ong S, Bailey M, Warren S, Rao D, Krijgsman O, Rozeman EA, Peeper D, Blank CU, Schumacher TN, Butterfield LH, Zelazowska MA, McBride KM, Kalluri R, Allison J, Petitprez F, Fridman WH, Sautes-Fridman C, Hacohen N, Rezvani K, Sharma P, Tetzlaff MT, Wang L, Wargo JA, B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, Jeng YM, Hsiao LP, Lacroix L, Bougouin A, Moreira M, Lacroix G, Natario I, Adam J, Lucchesi C, Laizet YH, Toulmonde M, Burgess MA, Bolejack V, Reinke D, Wani KM, Wang WL, Lazar AJ, Roland CL, Wargo JA, Italiano A, Sautes-Fridman C, Tawbi HA, Fridman WH, B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, van Schoiack A, Lovgren K, Warren S, Jirstrom K, Olsson H, Pietras K, Ingvar C, Isaksson K, Schadendorf D, Schmidt H, Bastholt L, Carneiro A, Wargo JA, Svane IM, Jonsson G, Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Hur B, Koster MJ, Jang JS, Weyand CM, Warrington KJ, Sung J, Global Transcriptomic Profiling Identifies Differential Gene Expression Signatures Between Inflammatory and Noninflammatory Aortic Aneurysms. Arthritis Rheumatol 74, 1376–1386 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, Byrne A, Wein L, Luen SJ, Poliness C, Nightingale SS, Skandarajah AS, Gyorki DE, Thornton CM, Beavis PA, Fox SB, Kathleen C Cuningham Foundation Consortium for Research into Familial Breast, Darcy PK, Speed TP, Mackay LK, Neeson PJ, Loi S, Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med 24, 986–993 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Sato Y, Oguchi A, Fukushima Y, Masuda K, Toriu N, Taniguchi K, Yoshikawa T, Cui X, Kondo M, Hosoi T, Komidori S, Shimizu Y, Fujita H, Jiang L, Kong Y, Yamanashi T, Seita J, Yamamoto T, Toyokuni S, Hamazaki Y, Hattori M, Yoshikai Y, Boor P, Floege J, Kawamoto H, Murakawa Y, Minato N, Yanagita M, CD153/CD30 signaling promotes age-dependent tertiary lymphoid tissue expansion and kidney injury. J Clin Invest 132, e146071 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP, A human memory T cell subset with stem cell-like properties. Nat Med 17, 1290–1297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crotty S, T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 50, 1132–1148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horiuchi S, Wu H, Liu WC, Schmitt N, Provot J, Liu Y, Bentebibel SE, Albrecht RA, Schotsaert M, Forst CV, Zhang B, Ueno H, Tox2 is required for the maintenance of GC T(FH) cells and the generation of memory T(FH) cells. Sci Adv 7, eabj1249 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh DY, Fong L, Cytotoxic CD4(+) T cells in cancer: Expanding the immune effector toolbox. Immunity 54, 2701–2711 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, Wu W, Lang PA, Gattinoni L, McGavern DB, Schwartzberg PL, The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 1,(6);eaai8593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kallies A, Xin A, Belz GT, Nutt SL, Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity 31, 283–295 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, Minnich M, Meckiff BJ, Wei Y, Hou Z, Zotos D, Fenix KA, Atnerkar A, Preston S, Chipman JG, Beilman GJ, Allison CC, Sun L, Wang P, Xu J, Toe JG, Lu HK, Tao Y, Palendira U, Dent AL, Landay AL, Pellegrini M, Comerford I, McColl SR, Schacker TW, Long HM, Estes JD, Busslinger M, Belz GT, Lewin SR, Kallies A, Yu D, CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol 17, 1187–1196 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Gabriel SS, Tsui C, Chisanga D, Weber F, Llano-Leon M, Gubser PM, Bartholin L, Souza-Fonseca-Guimaraes F, Huntington ND, Shi W, Utzschneider DT, Kallies A, Transforming growth factor-beta-regulated mTOR activity preserves cellular metabolism to maintain long-term T cell responses in chronic infection. Immunity 54, 1698–1714 e1695 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Gago da Graca C, van Baarsen LGM, Mebius RE, Tertiary Lymphoid Structures: Diversity in Their Development, Composition, and Role. J Immunol 206, 273–281 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, Clausen BE, Jung S, Greter M, Becher B, The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 43, 502–514 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Takeda S, Rodewald HR, Arakawa H, Bluethmann H, Shimizu T, MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity 5, 217–228 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Sato Y, Mii A, Hamazaki Y, Fujita H, Nakata H, Masuda K, Nishiyama S, Shibuya S, Haga H, Ogawa O, Shimizu A, Narumiya S, Kaisho T, Arita M, Yanagisawa M, Miyasaka M, Sharma K, Minato N, Kawamoto H, Yanagita M, Heterogeneous fibroblasts underlie age-dependent tertiary lymphoid tissues in the kidney. JCI Insight 1, e87680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Camell CD, Gunther P, Lee A, Goldberg EL, Spadaro O, Youm YH, Bartke A, Hubbard GB, Ikeno Y, Ruddle NH, Schultze J, Dixit VD, Aging Induces an Nlrp3 Inflammasome-Dependent Expansion of Adipose B Cells That Impairs Metabolic Homeostasis. Cell Metab 30, 1024–1039 e1026 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, Woodland DL, Lund FE, Randall TD, Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med 10, 927–934 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Sato Y, Boor P, Fukuma S, Klinkhammer BM, Haga H, Ogawa O, Floege J, Yanagita M, Developmental stages of tertiary lymphoid tissue reflect local injury and inflammation in mouse and human kidneys. Kidney Int 98, 448–463 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Jamaly S, Rakaee M, Abdi R, Tsokos GC, Fenton KA, Interplay of immune and kidney resident cells in the formation of tertiary lymphoid structures in lupus nephritis. Autoimmun Rev 20, 102980 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Ciccia F, Rizzo A, Maugeri R, Alessandro R, Croci S, Guggino G, Cavazza A, Raimondo S, Cannizzaro A, Iacopino DG, Salvarani C, Triolo G, Ectopic expression of CXCL13, BAFF, APRIL and LT-beta is associated with artery tertiary lymphoid organs in giant cell arteritis. Ann Rheum Dis 76, 235–243 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Graver JC, Boots AMH, Haacke EA, Diepstra A, Brouwer E, Sandovici M, Massive B-Cell Infiltration and Organization Into Artery Tertiary Lymphoid Organs in the Aorta of Large Vessel Giant Cell Arteritis. Front Immunol 10, 83 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gearty SV, Dundar F, Zumbo P, Espinosa-Carrasco G, Shakiba M, Sanchez-Rivera FJ, Socci ND, Trivedi P, Lowe SW, Lauer P, Mohibullah N, Viale A, DiLorenzo TP, Betel D, Schietinger A, An autoimmune stem-like CD8 T cell population drives type 1 diabetes. Nature 602, 156–161 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mueller SN, Mackay LK, Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol 16, 79–89 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Nakajima T, Schulte S, Warrington KJ, Kopecky SL, Frye RL, Goronzy JJ, Weyand CM, T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation 105, 570–575 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Fleig S, Kapanadze T, Bernier-Latmani J, Lill JK, Wyss T, Gamrekelashvili J, Kijas D, Liu B, Husing AM, Bovay E, Jirmo AC, Halle S, Ricke-Hoch M, Adams RH, Engel DR, von Vietinghoff S, Forster R, Hilfiker-Kleiner D, Haller H, Petrova TV, Limbourg FP, Loss of vascular endothelial notch signaling promotes spontaneous formation of tertiary lymphoid structures. Nat Commun 13, 2022 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee M, Kiefel H, LaJevic MD, Macauley MS, Kawashima H, O’Hara E, Pan J, Paulson JC, Butcher EC, Transcriptional programs of lymphoid tissue capillary and high endothelium reveal control mechanisms for lymphocyte homing. Nat Immunol 15, 982–995 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moussion C, Girard JP, Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature 479, 542–546 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Hua Y, Vella G, Rambow F, Allen E, Antoranz Martinez A, Duhamel M, Takeda A, Jalkanen S, Junius S, Smeets A, Nittner D, Dimmeler S, Hehlgans T, Liston A, Bosisio FM, Floris G, Laoui D, Hollmen M, Lambrechts D, Merchiers P, Marine JC, Schlenner S, Bergers G, Cancer immunotherapies transition endothelial cells into HEVs that generate TCF1(+) T lymphocyte niches through a feed-forward loop. Cancer Cell 40, 1600–1618 e1610 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM, Inhibition of JAK-STAT Signaling Suppresses Pathogenic Immune Responses in Medium and Large Vessel Vasculitis. Circulation 137, 1934–1948 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Law CW, Chen Y, Shi W, Smyth GK, voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin-Labat ML, Smyth GK, Ritchie ME, Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res 43, e97 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meckiff BJ, Ramirez-Suastegui C, Fajardo V, Chee SJ, Kusnadi A, Simon H, Eschweiler S, Grifoni A, Pelosi E, Weiskopf D, Sette A, Ay F, Seumois G, Ottensmeier CH, Vijayanand P, Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4(+) T Cells in COVID-19. Cell 183, 1340–1353 e1316 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, Byrne A, Wein L, Luen SJ, Poliness C, Nightingale SS, Skandarajah AS, Gyorki DE, Thornton CM, Beavis PA, Fox SB, Kathleen C Cuningham Foundation Consortium for Research into Familial Breast, Darcy PK, Speed TP, Mackay LK, Neeson PJ, Loi S, Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med 24, 986–993 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R, Gene Ontology C, The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32, D258–261 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A, Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, Diehn M, Alizadeh AA, Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen PY, Qin L, Li G, Malagon-Lopez J, Wang Z, Bergaya S, Gujja S, Caulk AW, Murtada SI, Zhang X, Zhuang ZW, Rao DA, Wang G, Tobiasova Z, Jiang B, Montgomery RR, Sun L, Sun H, Fisher EA, Gulcher JR, Fernandez-Hernando C, Humphrey JD, Tellides G, Chittenden TW, Simons M, Smooth Muscle Cell Reprogramming in Aortic Aneurysms. Cell Stem Cell 26, 542–557 e511 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McGinnis CS, Murrow LM, Gartner ZJ, DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 e324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, Bierie B, Mazutis L, Wolf G, Krishnaswamy S, Pe’er D, Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 174, 716–729 e727 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL, The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller TE, Lareau CA, Verga JA, DePasquale EAK, Liu V, Ssozi D, Sandor K, Yin Y, Ludwig LS, El Farran CA, Morgan DM, Satpathy AT, Griffin GK, Lane AA, Love JC, Bernstein BE, Sankaran VG, van Galen P, Mitochondrial variant enrichment from high-throughput single-cell RNA sequencing resolves clonal populations. Nat Biotechnol 40, 1030–1034 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tu AA, Gierahn TM, Monian B, Morgan DM, Mehta NK, Ruiter B, Shreffler WG, Shalek AK, Love JC, TCR sequencing paired with massively parallel 3’ RNA-seq reveals clonotypic T cell signatures. Nat Immunol 20, 1692–1699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ainciburu M, Morgan DM, DePasquale EAK, Love JC, Prosper F, van Galen P, WAT3R: recovery of T-cell receptor variable regions from 3’ single-cell RNA-sequencing. Bioinformatics 38, 3645–3647 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schnell A, Huang L, Singer M, Singaraju A, Barilla RM, Regan BML, Bollhagen A, Thakore PI, Dionne D, Delorey TM, Pawlak M, Meyer Zu Horste G, Rozenblatt-Rosen O, Irizarry RA, Regev A, Kuchroo VK, Stem-like intestinal Th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell 184, 6281–6298 e6223 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, Bridonneau C, Jegou S, Hoffmann TW, Natividad JM, Brot L, Taleb S, Couturier-Maillard A, Nion-Larmurier I, Merabtene F, Seksik P, Bourrier A, Cosnes J, Ryffel B, Beaugerie L, Launay JM, Langella P, Xavier RJ, Sokol H, CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med 22, 598–605 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or supplementary materials. The scRNA-seq and bulk RNA-seq data have been deposited in the NCBI (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE224528. Please direct all other requests for data and further information to C.M.W. at cweyand@stanford.edu.