

Abstract
The tumor microenvironment is a complex and dynamic ecosystem composed of various physical cues and biochemical signals that facilitate cancer progression, and tumor-associated macrophages are especially of interest as a treatable target due to their diverse pro-tumorigenic functions. Engineered three-dimensional models of tumors more effectively mimic the tumor microenvironment than monolayer cultures and can serve as a platform for investigating specific aspects of tumor biology within a controlled setting. To study the combinatorial effects of tumor-associated macrophages and microenvironment mechanical properties on osteosarcoma, we co-cultured human osteosarcoma cells with macrophages within biomaterials-based bone tumor niches with tunable stiffness. In the first 24 hours of direct interaction between the two cell types, macrophages induced an inflammatory environment consisting of high concentrations of tumor necrosis factor alpha (TNFα) and interleukin (IL)-6 within moderately stiff scaffolds. Expression of Yes-associated protein (YAP), but not its homolog, transcriptional activator with PDZ-binding motif (TAZ), in osteosarcoma cells was significantly higher than in macrophages, and co-culture of the two cells slightly upregulated YAP in both cells, although not to a significant degree. Resistance to doxorubicin treatment in osteosarcoma cells was correlated with inflammation in the microenvironment, and signal transducer and activator of transcription 3 (STAT3) inhibition diminished the inflammation-related differences in drug resistance but ultimately did not improve the efficacy of doxorubicin. This work highlights that the biochemical cues conferred by tumor-associated macrophages in osteosarcoma are highly variable, and signals derived from the immune system should be considered in the development and testing of novel drugs for cancer.
Keywords: osteosarcoma, tumor-associated macrophages, tissue engineered tumor model, tumor microenvironment mechanical properties, YAP/TAZ, IL-6/STAT3
Graphical Abstract
Introduction
Osteosarcoma is the most common primary tumor of the bone with peak incidence occurring in pediatric patients and a second peak occurring in patients over the age of 50 [1]. Osteosarcoma is characterized by the production of osteoid tissue or immature bone by malignant osteoblastic cells [2, 3]. Although osteosarcoma is a rare disease, comprising 2% of all childhood cancers, bone and joint neoplasms account for nearly 9% of pediatric and adolescent cancer deaths [4]. No specific genetic mutation is associated with osteosarcoma, although children with inherited germline mutations such as Li-Fraumeni syndrome or retinoblastoma syndrome are more predisposed to developing it, especially during growth spurts [5]. Massive genomic rearrangements, known as chromothripsis, are also frequently observed in osteosarcoma [6–8]. Taken together, osteosarcoma exhibits a high degree of intra- and inter-tumor heterogeneity. The current standard of care for osteosarcoma treatment, which consists of neoadjuvant chemotherapy, surgical resection, followed by adjuvant chemotherapy, has remained largely unchanged since it was established in the 1980s [6, 9]. This treatment results in 65–70% overall 5-year survival in patients with localized disease but only 20% overall 5-year survival in patients with metastasis on presentation [1, 10, 11]. Bringing new therapies to treat osteosarcoma to the clinic has remained a challenge due to its rarity, high tumor heterogeneity, and poor preclinical in vitro and in vivo models in which to test new therapeutic agents [12].
Typical in vitro studies conducted in tissue culture flasks lack crucial elements of the tumor microenvironment, a complex three-dimensional network composed of stroma, vasculature, and immune cells within a dynamic extracellular matrix that hosts a diverse array of signals including architecture, heterotypic cell interactions, biochemical cues, and mechanical cues [13, 14]. In addition to osteosarcoma cells, the osteosarcoma tumor microenvironment consists of bone cells, stromal cells, vascular cells, immune cells, and extracellular matrix with calcification [5]. This variety of signals combined with genomic instability enables to disease progression by facilitating the generation of diverse and adaptable cell populations within the tumor that contribute to resistance to conventional therapies [15].
Of growing interest is the role of the mechanical microenvironment on tumor progression. The mechanical microenvironment in porous trabecular bone where osteosarcoma frequently occurs is highly heterogeneous; the elastic moduli of trabecular bone sections can vary over an order of magnitude within a single long bone of the human skeleton [16, 17]. As such, that the stiffness of the surrounding tissue has been shown to direct lineage commitment in stem cells, including mesenchymal stem cells (MSCs) and cancer stem cells, is relevant to osteosarcoma tumor biology as the former are the supposed cell of origin of osteosarcoma and the latter make up a fraction of osteosarcoma cell populations [18, 19].
In addition to its unique mechanical properties, bone tissue is comprised of multiple cell types, which interact with the immune system in several ways [20]. Of the immune cells in the bone tumor microenvironment, tumor-associated macrophages (TAMs) possess the highest rate of infiltration in osteosarcoma and many other solid tumors, and they can make up 50% of the tumor mass [21–23]. Macrophages are antigen-presenting cells that are activated or polarized to effect different functions in homeostasis and disease; those that exhibit inflammatory functions are referred to as M1 or classically activated macrophages while those that exhibit anti-inflammatory functions are referred to as M2 or alternatively activated macrophages [24]. However, macrophage activation is more complex than the simple M1/M2 binary would suggest, and the polarization states of macrophage subpopulations are heterogeneous. Tumor-associated macrophages are highly plastic and have both tumor-promoting and anti-tumoral functions. On one hand, TAMs can exhibit M2-like functions and promote tumor cell proliferation, tumor angiogenesis and vascularization, metastatic potential, and immunosuppression. On the other, they can have M1-like functions and kill tumor cells directly or indirectly by mediating the response of T cells and natural killer cells [25, 26]. Findings about the roles of TAMs in osteosarcoma have been mixed. Some reports linked macrophage infiltration to improved overall survival and reduced metastasis in osteosarcoma, while others have suggested that macrophages facilitate epithelial-to-mesenchymal transition and promote osteosarcoma cell migration and metastatic potential [27–29].
Tissue-engineered models of tumors serve as platforms to address the inadequacies of monolayer culture and avoid the confounding factors associated with xenograft models in animals. Tissue architecture and mechanical properties can be tuned by the choice of scaffold fabrication method and material, and specific heterotypic interactions can be introduced through co-culture with different types of cells [13]. We have previously developed a three-dimensional osteosarcoma tumor model with tunable mechanical properties and found that the three-dimensional architecture led to a downregulation of the IGF-1R/mTOR axis and an upregulation of Sox2 compared to osteosarcoma cells grown in monolayers. We also found that osteosarcoma cells are sensitive to the matrix stiffness; decreasing the substrate stiffness led to increased nuclear localization of the mechanoresponsive Hippo pathway effectors Yes-associated protein 1 (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) in osteosarcoma cells [30]. Moreover, the nuclear-to-cytoplasmic (N:C) ratio of YAP and TAZ in cells cultured in these scaffolds was comparable to N:C ratio measurements in osteosarcoma patient tumor biopsies, whereas the N:C ratio of YAP and TAZ in cells grown in monolayers was above the patient range [30, 31].
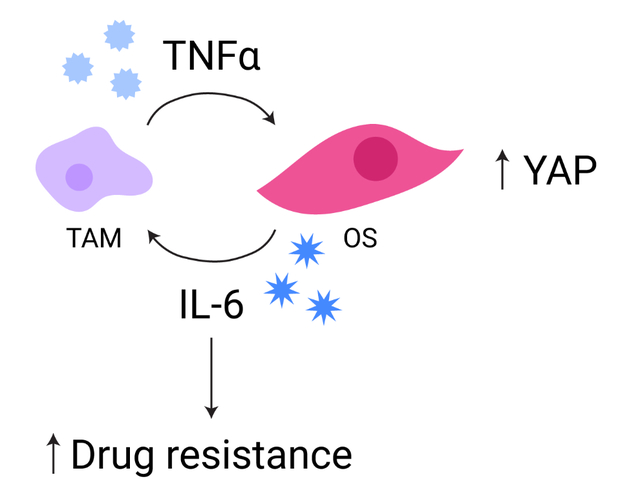
Our goal for this study was to introduce tumor-associated macrophages into the mechanically tunable model of osteosarcoma to investigate how scaffold mechanical properties and macrophages synergistically affect osteosarcoma cell response. MG-63 human osteosarcoma cells were directly co-cultured in electrospun scaffolds with macrophages derived from THP-1 cells in four ratios to mimic different levels of immune infiltration into the tumor. We characterized the immune microenvironment induced by the interaction of macrophages with osteosarcoma cells by probing the released cytokines. Recent work from others suggests that YAP is a key molecule in sensing stiffness and modulating inflammation in macrophages [32]. Having established that macrophages significantly affect the biochemical signals present in the tumor, we then assessed changes in the mechanoresponse of cells by evaluating the expression of YAP and TAZ in osteosarcoma cells and macrophages. We additionally characterized the differential response of osteosarcoma cells to doxorubicin and combination treatment with STAT3 inhibition in the presence of macrophages. Our models underscore that osteosarcoma cell phenotype and behavior in part dictated to signals conferred by the immune system, and we discuss the implications for response to therapy.
Materials and Methods
Experimental design
In this study we sought to investigate the combined effects of tumor-associated macrophages (TAMs) and the mechanical properties of a three-dimensional tumor microenvironment on the progression, phenotype, and resistance to therapy in osteosarcoma (OS) cells. MG-63 human osteosarcoma cells and TAMs derived from THP-1 cells were co-cultured in four ratios (OS only, OS/TAM 3:1, OS/TAM 1:1, and TAM only) within polymeric electrospun scaffolds with a range of mechanical properties.
Electrospinning
Meshes of randomly oriented microfibers were electrospun using a YFlow Professional Device V2.0 as previously described [30]. Briefly, Poly(ε-caprolactone) (PCL, average Mn 80,000, Sigma-Aldrich, CAS RN 24980-41-4) and gelatin type B (Nitta Gelatin, CAS RN 9000-70-8) were each dissolved at 15% w/v in 2,2,2-trifluoroethanol (TFE, Sigma-Aldrich, CAS RN 75-89-8) overnight in sealed round bottom flasks. The dissolved polymer solutions were loaded into syringes (BD) and loaded into the electrospinner syringe pump pointed downwards towards a grounded aluminum collecting plate, which was covered in non-stick aluminum foil (Reynolds) to prevent fibers from sticking to the collecting surface.
For pure PCL or pure gelatin (referred to as 100% gelatin) fibers, polymer was extruded through a 16 G blunt tip needle at 5 mL/h at a distance of 10–12 cm from the collecting plate. Meshes were spun for 45 min with an applied voltage of 6–8 kV. To produce coaxial PCL-gelatin core-shell fibers, PCL was extruded through a 20 G inner needle and gelatin was extruded through a 16 G outer needle of a blunt tip coaxial needle (ramé-hart instrument co.). The two polymers were extruded from each syringe at variable flow rates totaling 10 mL/h (2 mL/h gelatin and 8 mL/h PCL, 5 mL/h gelatin and 5 mL/h PCL, and 8 mL/h gelatin and 2 mL/h PCL to achieve 20% gelatin, 50% gelatin, and 80% gelatin, respectively), and meshes were spun for 25–30 min with an applied voltage of 9–11 kV at a distance of 15–16 cm from the collecting plate.
Scaffold preparation from electrospun meshes
Electrospun meshes were removed from the non-stick foil. Scaffolds for 96-well plates were cut from the meshes using a 6 mm diameter dermal biopsy punch (Integra Miltex). Similarly, scaffolds for 48-well plates were cut using a 10 mm diameter dermal biopsy punch (Accu Punch), and scaffolds for 12-well plates were prepared by cutting circles of 20 mm diameter from the meshes. Gelatin in 20% gelatin, 50% gelatin, 80% gelatin, and 100% gelatin scaffolds was crosslinked in glutaraldehyde (Sigma-Aldrich, CAS RN 111-30-8) vapor for 24 h in a sealed desiccator.
Scaffolds were placed into 50 mL conical tubes (BD Falcon) or well plates (BD Falcon) and sterilized for 12 h in ethylene oxide (AN73, Andersen Products, CAS RN 75-21-8) and then vented for 24 h. Gelatin-containing scaffolds were quenched in sterile filtered 0.1 M aqueous solution of glycine (Sigma, CAS RN 56-40-6) overnight to block excess aldehyde residues from the glutaraldehyde crosslinking. PCL scaffolds were wet in an ethanol dilution gradient as previously described [33–35]. All scaffolds were then washed three to five times in Dulbecco’s phosphate-buffered saline (PBS; Gibco) and placed in RPMI 1640 cell culture medium (Gibco) overnight in a humidified incubator at 37 °C with 5% CO2.
Cell culture
MG-63 osteosarcoma cells and THP-1 monocyte cells were purchased from ATCC and used in all experiments. Cells were cultured in RPMI 1640 with 10% fetal bovine serum (GeminiBio) and 1% Penicillin-Streptomycin or Antibiotic-Antimycotic (Gibco).
Cell seeding and OS/TAM co-culture
MG-63 and THP-1 cells were seeded upon electrospun scaffolds and cultured alone (OS only and TAM only) or co-cultured in two different ratios to mimic different levels of immune cell infiltration in the tumor microenvironment (OS/TAM 3:1 and OS/TAM 1:1). For all experiments, the total number of cells seeded on the scaffolds was kept constant between the different cell combinations. Cells were seeded at 10,000 cells per 96-well plate sized (6 mm diameter) electrospun scaffold. Seeding density was scaled to the area of the scaffold; 48-well plate sized (10 mm diameter) and 12-well plate sized (20 mm diameter) scaffolds were seeded with 27,000 cells per scaffold and 110,000 cells per scaffold, respectively. Cell proliferation and cytokine secretion assays were conducted with all four culture conditions. Flow cytometry studies were conducted with OS only, OS/TAM 1:1 co-culture, TAM only groups. Drug response studies were conducted with OS only and OS/TAM 1:1 co-culture.
Pre-wet scaffolds were placed into the corresponding size of well plate with half of the working volume of media using sterile technique and then cells were seeded upon the scaffolds in the remaining half of the media. The total volume per well was 200 μL in 96-well plates, 500 μL in 48-well plates, and 2 mL in 12-well plates.
For the OS only culture condition, MG-63 cells were seeded on scaffolds in RPMI 1640 cell culture media. Cells were allowed to adhere for 24 h in a humidified incubator at 37 °C. After 24 h, the cell-laden scaffolds were transferred to new well plates with fresh cell culture media, and the cells were cultured for up to five days.
For TAM monoculture, THP-1 cells were seeded on scaffolds in RPMI 1640 containing 25 nM phorbol 12-myristate 13-acetate (PMA; Sigma, CAS RN 16561-29-8). Cells were allowed to differentiate into macrophages and adhere to scaffolds for 48 h. After differentiation, the cells were washed in PBS, transferred to new well plates with PMA-free media, and cultured for up to five days.
For OS/TAM co-cultures, THP-1 cells were first seeded onto scaffolds in RPMI 1640 with 25 nM PMA and then allowed to differentiate into macrophages and adhere to scaffolds for 48 h. In OS/TAM 3:1 co-culture, the number of macrophages seeded was 25% of the seeding density for a given scaffold size. For OS/TAM 1:1 co-culture, the number of macrophages seeded was 50% of the seeding density for a given scaffold size. After 48 h, the scaffolds were washed in excess PBS and placed into a new well plate with PMA-free RPMI 1640. MG-63 cells were then seeded upon the TAM-containing scaffolds to bring the total number of cells up to 100% of the seeding density for the scaffold and allowed to adhere for 24 h. After 24 h, the scaffolds were transferred to new well plates with RPMI 1640 and cultured for up to five days.
M1 and M2 macrophage polarization
THP-1 cells differentiated into macrophages (M0) and polarized towards M1 or M2 phenotypes were used as comparisons for cell morphology. For all conditions, THP-1 cells were seeded at 250,000 cells/mL in tissue culture treated 24-well plates in RPMI 1640 with 25 nM PMA. Cells were polarized towards M2 with the addition of 20 ng/mL recombinant human interleukin (IL)-4 (PeproTech) and 20 ng/mL recombinant human IL-13 (PeproTech) in the PMA media for 48 h. Cells were polarized towards M1 with the addition of 20 ng/mL recombinant human IFNγ (PeproTech) and 10 ng/mL lipopolysaccharides (Santa Cruz Biotechnology, CAS RN 93572-42-0) in the PMA media for 24 h.
Immunofluorescent staining, confocal microscopy, and epifluorescent microscopy
OS, OS/TAM 1:1, and TAM cultures were seeded in 10 mm diameter PCL scaffolds in 48-well plates as described in Cell seeding and OS/TAM co-culture. TAM cultures were also seeded in 10 mm diameter 50% gelatin and 100% gelatin scaffolds. Scaffolds were collected after 24 h of culture or co-culture to confirm the presence of cells within the scaffolds. All samples were washed 3× with PBS, fixed for 15 min in 4% w/v methanol-free formaldehyde (Pierce, CAS RN 50-00-0) at room temperature, and washed 3× with PBS for 5 min each to remove excess fixative. Fixed cells on PCL scaffolds were blocked in blocking buffer (1× PBS with 5% normal goat serum [Cell Signaling Technology] and 0.3% Triton™ X-100 [CAS RN 9036-19-5]) for 1 h. Primary antibodies for CD68 (Cell Signaling Technology, diluted 1:100) and Sox2 (Cell Signaling Technology, diluted 1:100) were diluted in antibody dilution buffer (1× PBS with 1% bovine serum albumin [BSA, Cell Signaling Technology, CAS RN 9048-46-8] and 0.3% Triton™ X-100), and samples were incubated in the primary antibody solution overnight at 4 °C. Samples were washed 3× with PBS and then incubated with fluorochrome-conjugated secondary antibodies (Cell Signaling Technology, goat anti-rabbit IgG Alexa Fluor 594 conjugate, diluted 1:500; Cell Signaling Technology, goat anti-mouse IgG Alexa Fluor 647 conjugate, diluted 1:500), iFluor 488-conjugated phalloidin (Abcam, diluted 1:1000), and Hoechst 33342 (ThermoFisher, diluted 1:2000, CAS RN 875756-97-1) for 1 h in the dark at room temperature. TAM only samples on 50% gelatin and 100% gelatin scaffolds were not blocked; after fixing, antibody dilution buffer with phalloidin-iFluor 488 (Abcam, diluted 1:1000) and Hoechst (diluted 1:2000) was applied to the cells for 1 h in the dark at room temperature. All samples were washed 3× in PBS and scaffolds were placed in 1 mL PBS and imaged on 35 mm diameter 1.5 glass bottom dishes (Ibidi). Images were collected using a Nikon A1-RSi confocal microscope using the appropriate laser wavelengths and filters for each stain.
After differentiation and polarization, M0, M1, and M2 macrophages in 24-well plates were washed with 3× with PBS and fixed for 15 min in 4% w/v methanol-free paraformaldehyde at room temperature. Cells were then washed 3× with PBS. Antibody dilution buffer with phalloidin-iFluor 594 (Abcam, diluted 1:1000) and Hoechst (diluted 1:2000) was applied to the cells for 1 h in the dark at room temperature. Samples were washed 3× in PBS, covered with 0.5 mL PBS, and imaged with a Nikon Eclipse Ti2-U microscope using the TRITC and DAPI filters.
DNA quantitation cell proliferation assays
OS, OS/TAM 3:1, OS/TAM 1:1, and TAM cultures (n = 6) were seeded in 6 mm diameter scaffolds in 96-well plates as described in Cell seeding and OS/TAM co-culture and grown for up to five days. Scaffolds were collected after cell adhesion to the scaffold (day 0), and then following 1, 3, and 5 days of culture. Collected scaffolds were washed in PBS and the placed into 1.7 mL microcentrifuge tubes (VWR) containing 200 μL sterile MilliQ water. Cells were lysed and DNA was extracted from samples through three freeze-thaw cycles in liquid nitrogen and 37 °C water. Lysed samples were stored at −20 °C until use.
Double stranded DNA (dsDNA) was quantified using a Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen). Cell lysate was combined with TE buffer and PicoGreen dye solution and placed in opaque black 96-well plates. Fluorescence (485 nm excitation, 538 nm emission) was measured on a SpectraMax M2 microplate reader (Molecular Devices) and compared with a standard λ DNA curve prepared according to the manufacturer’s instructions.
Cytokine secretion assays and bootstrapping analysis
OS, OS/TAM 3:1, OS/TAM 1:1, and TAM cultures (n = 3 or n = 4) were seeded in 10 mm diameter scaffolds in 48-well plates as described in Cell seeding and OS/TAM co-culture. After cell adhesion, scaffolds were washed in PBS, transferred to new 48-well plates with serum-free RPMI 1640, and cultured for an additional 24 h. After 24 h, the cell culture supernatant was collected, centrifuged at 500 g for 5 min, and aliquoted into sterile microcentrifuge tubes. Supernatant samples were stored at −80 °C until use.
Secreted human IL-10 (n = 3; R&D Systems DuoSet), CCL18/PARC (n = 3; R&D Systems Quantikine), TNFα (n = 3; R&D Systems DuoSet), and IL-6 (n = 4; R&D Systems DuoSet) were measured via ELISA according to the manufacturer’s instructions. Optical absorbance was measured at 450 nm wavelength with 540 nm correction on a PowerWave X 340 plate reader (Bio-Tek Instruments). Cytokine adsorption to the scaffolds was assumed to be negligible. Sample absorbances were compared to a four-parameter logistical standard curve prepared according to the manufacturer’s instruction.
The assays for TNFα and IL-6 resulted in secreted cytokine concentrations both within and below the assay range, so samples with measurements below the calculated standard curve were simulated with parametric bootstrapping to facilitate statistical analysis. Sample values were assumed to follow a Normal distribution on a log scale, and the pooled variance and standard deviation were calculated from the samples with values within the assay range. An R script was written to simulate values below the assay range on a log Normal scale using the pooled standard deviation and was run 1000 times. In the log transformation, the mean was assumed to be log(5) for TNFα and log(0.5) for IL-6; these selected because they were an order of magnitude below the lower limit of the provided standards for the respective assays. Simulated values were transformed back to a linear scale, and all values were compared using two-way ANOVA with post-hoc Tukey’s HSD.
Cell collection and fixing for flow cytometry
OS, OS/TAM 1:1, and TAM cultures were seeded in 20 mm diameter scaffolds in 12-well plates as described Cell seeding and OS/TAM co-culture. Cells from TAM groups were collected after 48 h PMA treatment and adhesion to scaffolds. OS and OS/TAM 1:1 groups were cultured for an additional 24 h after cell adhesion before cell collection.
To collect sufficient cells from electrospun scaffolds for analysis, 3 or 4 scaffolds were pooled into a single sample. Using aseptic technique, scaffolds were washed twice in excess PBS and then placed into a 50 mL conical tube with 4 mL TrypLE Express Enzyme (Gibco) that had been pre-warmed to 37 °C. The conical tubes were placed on an orbital shaker table at 100 rpm at 37 °C and shaken for 20 min. The tubes were then tapped lightly, 6 mL RPMI 1640 cell culture media was added to quench the TrypLE, and the scaffolds were removed from the conical tubes. Samples were centrifuged for 5 min at 1000 rpm to pellet the cells, and supernatant was removed. The cells were resuspended in 1 mL FACS buffer (1% BSA in 1× PBS) to wash, transferred to a 1.7 mL microcentrifuge tube, and centrifuged for 5 min at 2000 rpm to pellet the cells.
The supernatant was removed from the pellet and the cells were resuspended in 200 μL BD CytoFix/Cytoperm Fixation/Permeabilization Solution and fixed for 15 min. The cells were washed twice by centrifugation with 1× BD CytoFix/Cytoperm perm/wash buffer at 2500 rpm for 5 min, resuspended in 500 μL FACS buffer, and stored at 4 °C until staining.
Cell staining and flow cytometry
Fixed samples were pelleted and the supernatant was aspirated. Cells were resuspended in 50 μL 1× Human Fc Block (BD) and blocked for 20 min at room temperature. Fluorophore-conjugated primary antibodies for CD90 (BD Pharmingen, PE-Cy7 conjugate, diluted 1:40), CD11b (BD Horizon, Brilliant Blue 515 conjugate, diluted 1:20), YAP (Cell Signaling Technology, Alexa Fluor 647 conjugate, diluted 1:40), and TAZ (Santa Cruz Biotechnology, Alexa Fluor 594 conjugate, diluted 1:40) were added to the samples to a final volume of 100 μL. Cells were stained for 30 min in the dark at 4 °C. After staining, cells were washed twice by centrifugation with BD perm/wash buffer and resuspended in 150 μL FACS buffer. Flow cytometry was run on stained and unstained samples with a Sony SA3800 Spectral Analyzer. Fluorescence data were analyzed with the Sony SA3800 software, Community Cytobank (Beckman Coulter Life Sciences), and FlowJo (BD). The geometric mean fluorescence of the unstained sample for the respective cell population and scaffold type was subtracted from that of the stained samples to get the expression of a given marker of interest.
Dose-response and drug testing
OS and OS/TAM 1:1 cultures were seeded on 6 mm diameter electrospun scaffolds in 96-well plates as described in Cell seeding and OS/TAM co-culture. After 24 h of allowing MG-63 cells to adhere to the scaffolds in 200 μL RPMI 1640, the scaffolds were transferred sterilely to a new 96-well plate with 200 μL of therapeutic agent at the appropriate concentration in RPMI 1640 medium. To generate dose-response curves for doxorubicin (DOX; Selleck, CAS RN 25316-40-9), stock solution was diluted to 10 μM and then serially diluted 1:2 over nine DOX concentrations (0.039 μM – 10 μM) with a no-drug control. For combination drug experiments, the STAT3 inhibitor Stattic (Selleck, CAS RN 19983-44-9) was added at constant concentration of 5.1 μM in the same serial dilution of DOX. The DOX-free control for the combination drug study contained only 5.1 μM Stattic. All drug concentrations were applied to n = 4 wells in the OS and OS/TAM 1:1 cultures in separate 96-well plates for each group to prevent cross-contamination.
After three days of drug treatment, 100 μL of cell culture medium was removed from each well and replaced with WST-1 solution (Abcam) in RPMI 1640 to a final working concentration specified by the manufacturer in 200 μL cell culture medium per well. Cells were incubated at 37 °C for 1 h. After incubating, 100 μL was removed from the sample and placed into a new clear 96-well plate. Absorbance was read at 450 nm with a reference wavelength of 650 nm in a PowerWave X 340 plate reader (Bio-Tek Instruments). Baseline absorbance from cell culture medium alone was subtracted from all sample values. Absorbance values that were negative after baseline subtraction were taken to be zero viability. Sample values for the respective DOX-free groups were averaged and used as live cell controls for each condition.
Statistical analyses
All studies were performed with at least n = 3 for all groups as indicated below and in figure captions. All statistics were performed using Microsoft Excel, GraphPad Prism 9, or JMP. Data are displayed as mean with standard deviation or standard error as indicated in figure captions. All values were compared between groups using two-way analysis of variance (ANOVA) with post-hoc Tukey’s honestly significant difference (HSD) or multiple paired t test with Holm-Šídák multiple comparisons as indicated with p < 0.05.
Results
OS cells can be co-cultured with TAMs in electrospun scaffolds
Our objective for this work was to investigate the combined effects of tumor-associated macrophages (TAMs) and the mechanical properties of a three-dimensional substrate on the progression, phenotype, and resistance to therapy in osteosarcoma (OS) cells, which we aimed to do by co-culturing MG-63 human osteosarcoma cells and TAMs derived from THP-1 cells in four ratios (OS only, OS/TAM 3:1, OS/TAM 1:1, and TAM only) within PCL/gelatin electrospun scaffolds with stiffnesses ranging over several orders of magnitude (Figure 1A). We have previously characterized the mechanical properties of the scaffolds and found that substrate stiffness decreased as gelatin percentage increased. Specifically, the elastic modulus of the fibers as measured by AFM indentation testing was 71 ± 10 MPa, 10 ± 2 MPa, 4 ± 3 MPa, 2.3 ± 0.7 MPa, and 0.004 ± 0.004 MPa for PCL, 20% gelatin, 50% gelatin, 80% gelatin, and 100% gelatin, respectively [30]. We first used epifluorescent and confocal microscopy to visually confirm that THP-1 tumor-associated macrophages (TAMs) can be cultured upon and co-cultured with MG-63 osteosarcoma cells in the electrospun scaffolds. OS cultures and OS/TAM 1:1 co-cultures were seeded on PCL scaffolds (Figure 1B). Homotypic TAM cultures were seeded on PCL, coaxial 50% gelatin, and 100% gelatin scaffolds and compared to THP-1-derived M0, M1, and M2 macrophages in well plates (Figure 1B, Supplemental Figure 1).
Figure 1: Study groups and confirmation of cell attachment.
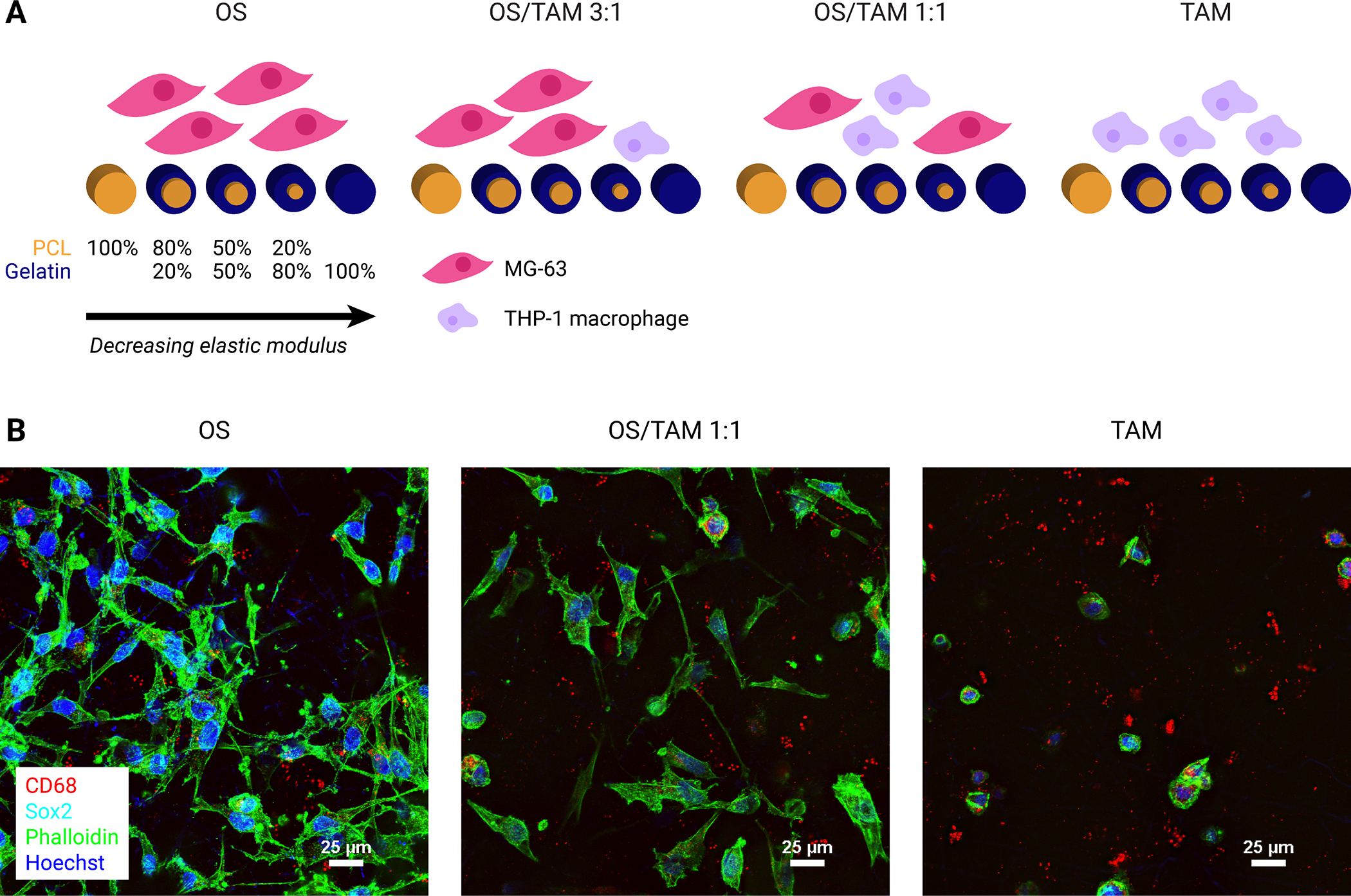
(A) Schematic representation of experimental groups. Four combinations of OS cells and TAMs were cultured on five sets of electrospun scaffolds ranging from 100% PCL with the highest stiffness to 100% gelatin with the lowest stiffness. (B) Representative confocal microscopy images demonstrating the morphology of MG-63 osteosarcoma and THP-1 macrophage cells on electrospun PCL scaffolds after 24 h culture or co-culture. Cells were co-stained for CD68 (red), Sox2 (cyan), phalloidin (actin, green), and Hoechst (nuclei, blue). Focused projection images of 20 μm z-stacks. Scale = 25 μm.
Upon PMA treatment to differentiate THP-1 cells from monocytes to macrophages, TAMs in scaffolds adhere to fibers, generally exhibit a round morphology similar to M0 macrophages in monolayers, and express CD68, which is seen in cytoplasmic granules (Figure 1B, Supplemental Figure 1A). TAMs in scaffolds do not exhibit the thin, sharp-looking cytoplasmic extensions seen in classically activated M1 macrophages, nor do they exhibit the elongation seen in alternatively activated M2 macrophages (Supplemental Figure 1A), which suggests that the substrate stiffness alone does not strongly polarize the macrophages like biochemical factors do. In contrast to TAMs, MG-63 cells have spindle-like morphology and spread out along microfibers (Figure 1B). Together, these results confirmed that PMA treatment to differentiate THP-1 into macrophages was compatible with electrospun scaffolds across a variety of compositions, and the scaffolds were a suitable three-dimensional platform for studying the interactions between OS cells and TAMs.
Depending on their polarization, TAMs recruited to the tumor microenvironment can be either cytotoxic to or promote the proliferation of cancer cells [36]. The growth of MG-63 osteosarcoma cells in the presence of TAMs over five days was examined by dsDNA quantification. In previous work, we found that homotypic MG-63 cultures in the electrospun scaffolds become confluent in five days [30]. Consistent with the literature, we observed that TAMs lose proliferative capacity after differentiation with PMA [37]. We can therefore reasonably attribute the change in DNA content observed over the five days in the OS/TAM co-culture conditions to the proliferation of the MG-63 cells, not a combination of OS cell and macrophage growth (Supplemental Figure 2A). In the first 24 h of co-culture, OS cells grew more slowly compared to OS cells grown on their own, but by the end of five days, OS cell proliferation in both OS/TAM 3:1 and OS/TAM 1:1 co-cultures met or exceeded that of homotypic OS cultures (Figure 2). In 100% gelatin scaffolds, cell growth in OS/TAM 3:1 co-culture was significantly higher than in OS only culture and OS/TAM 1:1 co-culture (Supplemental Figure 2B).
Figure 2: Proliferation of MG-63 osteosarcoma cells in PCL/gelatin scaffolds co-cultured with THP-1 TAMs.
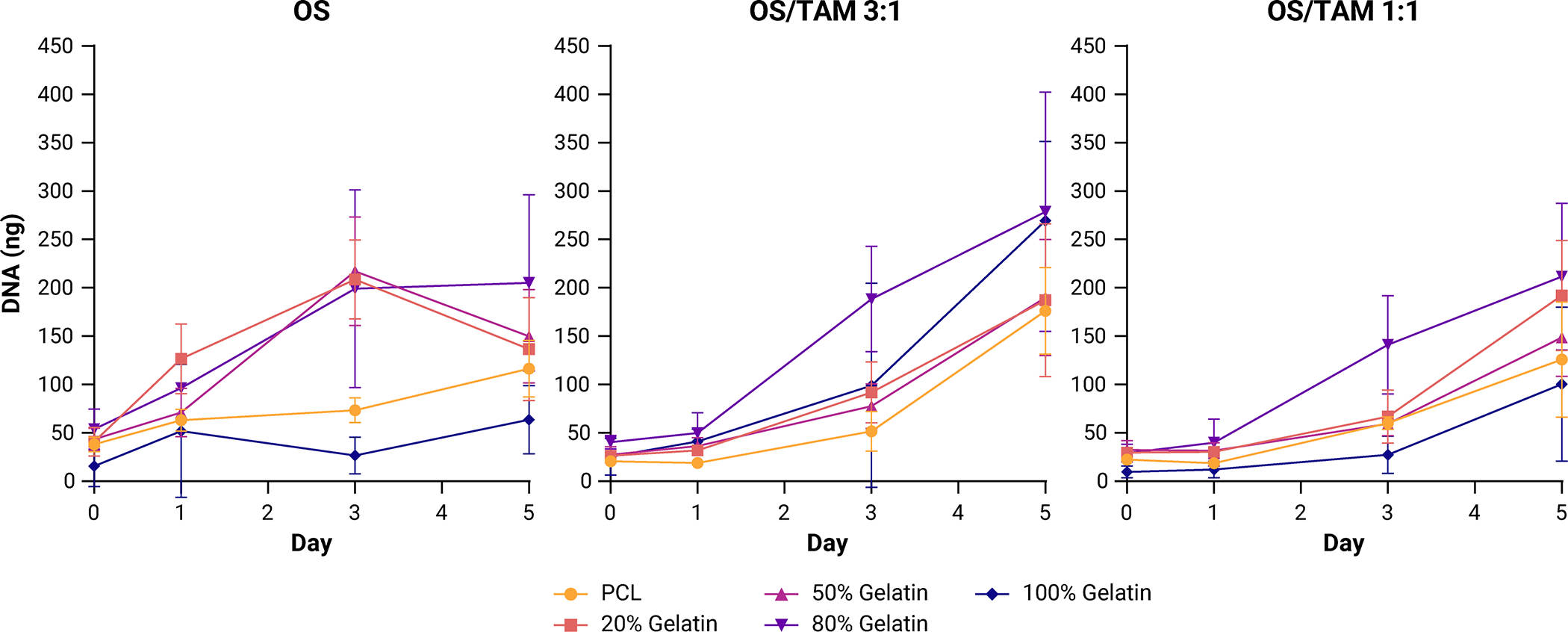
Data shown as mean ± SD of the mass of dsDNA per 6 mm scaffold (n = 6).
OS microenvironment is highly inflammatory in the initial period of tumor development
Given that the OS cell growth lagged within the first 24 h of co-culture with the TAMs, we wanted to further probe the phenotype of the cells at this timepoint. To determine the polarization state of macrophages in osteosarcoma tumor microenvironments with different mechanical properties, cell culture supernatant collected after 24 h of culture or co-culture was analyzed for anti-inflammatory cytokines IL-10 (n = 3) and CCL18 (n = 3) and pro-inflammatory cytokines tumor necrosis factor alpha (TNFα; n = 3) and IL-6 (n = 4). We hypothesized that all cytokine levels would be elevated in OS/TAM co-cultures compared to homotypic OS and TAM cultures and that the relative quantities of anti-inflammatory and pro-inflammatory cytokines would vary with scaffold stiffness, with softer scaffolds generating a more pro-inflammatory microenvironment.
ELISA showed that across the secretion of anti-inflammatory cytokines was negligible in all cell combinations across all scaffolds; specifically, the concentration of IL-10 was below 31.2 pg/mL and CC18 was below 18.8 pg/mL, the lower ends of the respective assay range, in all samples. However, cytokines associated with classical macrophage activation, TNFα and IL-6, were secreted in measurable quantities, with levels varying across different culture conditions.
TNFα was not detected in homotypic OS culture but was detected in 50% gelatin and 80% gelatin scaffolds in OS/TAM 3:1 co-culture, in all scaffolds in OS/TAM 1:1 co-culture, and 80% gelatin scaffolds in homotypic TAM culture, with the highest quantities on average in the OS/TAM 1:1 co-culture condition (Supplemental Figure 3A, Supplemental Table 1). For a given scaffold, the production of TNFα did not vary linearly with the proportion of macrophages present in the co-culture. In 50% gelatin scaffolds, the concentration of TNFα was 6.9-fold higher in OS/TAM 1:1 than OS/TAM 3:1 co-culture; in 80% gelatin scaffolds, it was 12.9-fold higher in OS/TAM 1:1 co-culture compared to OS/TAM 3:1 co-culture. There was also no clear correlation between substrate stiffness and the amount of cytokine released, but interestingly, TNFα secretion exhibited a non-monotonic relationship with decreasing scaffold stiffness in the OS/TAM 1:1 co-culture condition. Specifically, the average concentration of TNFα increased as the substrate stiffness decreased from PCL to 80% gelatin, but then was lowest in the softest 100% gelatin scaffolds.
We employed parametric bootstrapping to simulate TNFα values in the samples where the cytokine was not measurable (below the assay range) to better evaluate the role of the cell combination and scaffolds on TNFα production (Supplemental Figure 3C). Using the median bootstrapped values along with the measured values, it was found that the scaffold type (p < 0.05), culture condition (p < 0.0001), and their interaction (p < 0.05) were significant sources of variation in the quantity of TNFα produced by the cells (Figure 3A). Indeed, the amount of TNFα is significantly higher in OS/TAM 1:1 co-culture in the 80% scaffold than all other conditions except for OS/TAM 1:1 co-culture in 20% gelatin and 50% gelatin scaffolds.
Figure 3: Secreted pro-inflammatory cytokines in OS and TAM cultures.
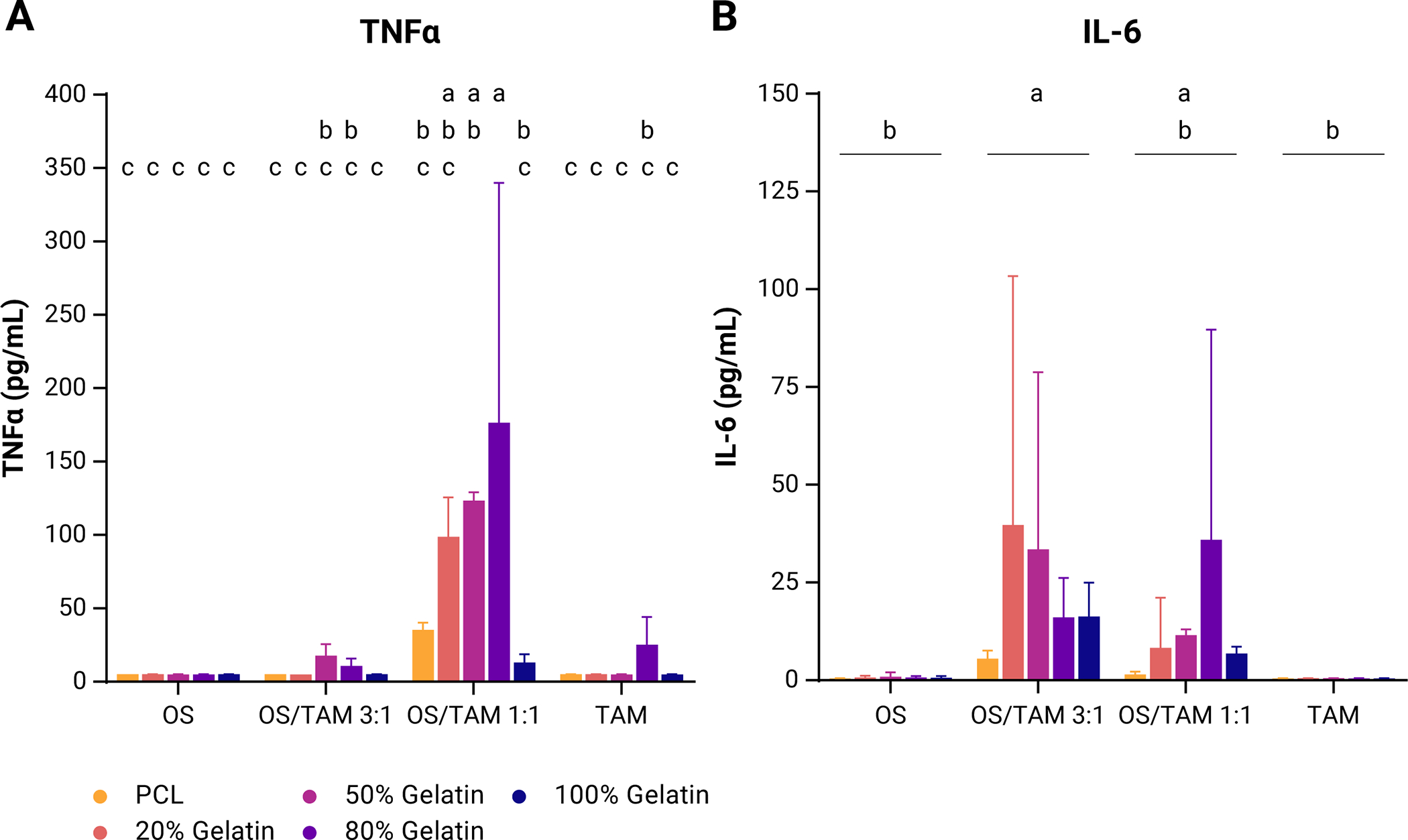
Data shown as mean + SD from measured values and 1000 bootstrapped simulations for (A) TNFα (n = 3) and (B) IL-6 (n = 4). Two-way ANOVA with post-hoc Tukey’s HSD, p < 0.05. Groups that do not share the same letter are statistically different.
Secreted IL-6 was measured from all scaffolds except PCL in OS culture and across all scaffolds in both OS/TAM 3:1 and OS/TAM 1:1 co-culture, but not at all in TAM only culture, and on average was highest in OS/TAM 3:1 co-culture (Supplemental Figure 3B, Supplemental Table 1). There was no clear correlation between scaffold stiffness and the amount of IL-6 secreted within a given culture condition, but notably, in the OS/TAM 1:1 co-culture, like what was observed with TNFα release, there was a non-monotonic relationship between IL-6 release and decreasing scaffold stiffness. The average concentration of IL-6 increased as the substrate stiffness decreased from PCL scaffolds to 80% gelatin scaffolds, but cytokine levels dropped in 100% gelatin scaffolds. Applying the median bootstrap values in the analysis confirms that the culture condition (p < 0.01) is the only significant source of variation in released IL-6 (Figure 3B, Supplemental Figure 3D). Secretion is significantly higher in the OS/TAM 3:1 co-culture group than both homotypic OS and homotypic TAM cultures; IL-6 production in the OS/TAM 1:1 co-culture is statistically similar to all groups.
Taken together, these results suggest that early on in tumor development, the interaction between TAMs and OS cells plays a significant role in generating a strong inflammatory response in the osteosarcoma tumor microenvironment that is not observed in the absence of macrophages.
Macrophages induce increased YAP expression in OS cells
In previous work using PCL and gelatin scaffolds to model the osteosarcoma tumor microenvironment, we found that the mechanical properties of the microenvironment alter the expression of the Hippo pathway effectors YAP and TAZ. Specifically, total YAP expression was downregulated and the nuclear-to-cytoplasmic ratio of YAP and TAZ increased in MG-63 osteosarcoma cells as scaffold stiffness decreased [30]. Given the growing body of work demonstrating the crosstalk between the YAP/TAZ and STAT3 pathways in other cancers and the elevated production of TNFα and IL-6 observed in our OS/TAM co-culture, we wanted to investigate how the presence of TAMs within the osteosarcoma microenvironment may be affecting the expression of YAP and TAZ [38, 39].
Cells collected from OS, OS/TAM 1:1, and TAM samples cultured for 24 h within the electrospun scaffolds were stained for CD90, CD11b, YAP, and TAZ. We found OS cells exhibited a bimodal distribution of CD90+ across all scaffolds after paraformaldehyde fixing (Supplemental Figure 4A). In OS samples, CD90+ OS cells were analyzed for the expression of total YAP (n = 4) and TAZ (n = 3) as measured by the geometric mean of the fluorescence intensity (MFI) of Alexa Fluor 647 and Alexa Fluor 594, respectively. Similarly, singles cells from TAM monoculture samples were analyzed for the expression of total YAP and TAZ (n = 3).
When comparing within OS cell populations, there was no significant difference in YAP or TAZ expression across OS cells cultured on the different electrospun scaffolds but YAP expression between individual samples was relatively consistent, whereas TAZ expression was more variable (Figure 4A). YAP expression in TAMs was significantly lower than in OS cells and was found to be downregulated as the substrate stiffness decreased (Figure 4B). TAZ expression in TAMs was also lower compared to OS cells but only significantly lower in the 80% gelatin scaffolds (Figure 4B).
Figure 4: Comparison of Hippo pathway effector expression in OS/TAM cultures.
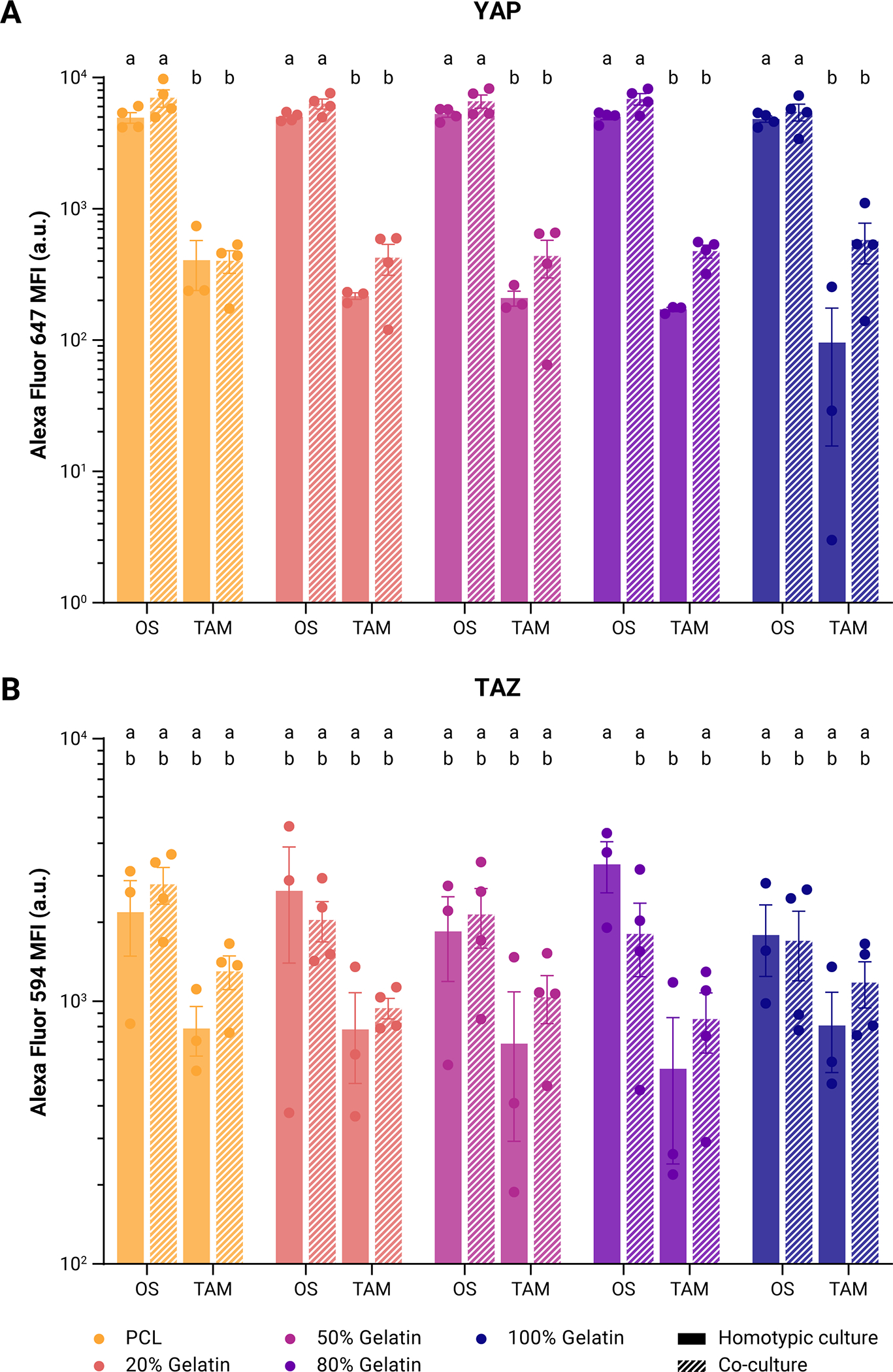
Expression of (A) YAP as measured by mean fluorescence of Alexa Fluor 647 and (B) TAZ as measured by mean fluorescence of Alexa Fluor 594 in OS cells and TAMs in homotypic culture or co-culture. Points represent individual measurements of the geometric mean. Bars represent mean ± SE. Two-way ANOVA with post-hoc Tukey’s HSD, p < 0.05. Groups that do not share the same letter are statistically different.
In co-cultures, OS cells could not be clearly distinguished from TAMs on the basis of CD90 and CD11b expression, so analysis was performed on a representative population of single cells (>90%) expressing CD90 and CD11b (Supplemental Figure 4B). Within this population of cells, there was a bimodal distribution of YAP. The population of cells with high YAP also expressed the bimodal distribution of CD90+ seen in OS monocultures, so these were taken to be the OS cells; the low YAP cells had similar YAP levels seen in macrophage monocultures and were taken to be the TAMs (Supplemental Figure 4C). The expression of total YAP and TAZ were analyzed in these two subpopulations and compared with their respective counterparts in the homotypic culture (Supplemental Figure 5, Figure 4).
Notably, while differences were not statistically significant, YAP expression was upregulated in co-cultured OS cells compared to OS cells alone in the corresponding scaffold, ranging from a 13% increase in MFI in 100% gelatin to a 41% increase in MFI in PCL; YAP was also upregulated in co-cultured TAMs compared to TAMs alone, except in PCL scaffolds (Figure 4A). Comparing across all cell combinations and scaffolds, YAP expression is significantly higher in OS cells than in TAMs, with MFI higher in the OS cells by one order of magnitude. Interestingly, TAZ expression is also slightly upregulated in TAMs in co-culture compared to homotypic TAM culture across all scaffolds, but this was not observed in OS cells (Figure 4B). Apart from that seen between homotypic OS and homotypic TAM cultures in 80% gelatin scaffolds, there are no significant differences in TAZ expression across the different groups.
Taken together, the increase in total YAP across co-cultured cells and the high variability of TAZ expression between individual samples suggests that YAP expression is more specifically sensitive than TAZ to changes in the immune microenvironment.
Inflammation confers resistance to doxorubicin treatment
Signal transducer and activator of transcription 3 (STAT3) is overexpressed and constitutively activated in osteosarcoma, leading to the upregulation of expression of target oncogenes that contribute to disease progression [40]. IL-6 is one of the main cytokines that regulates and activates the expression STAT3. Having demonstrated IL-6 is elevated in the osteosarcoma microenvironment with the presence of macrophages, we wanted to investigate the effect how treatment with doxorubicin (DOX), one of the common chemotherapeutic agents used for osteosarcoma, is affected and whether specific inhibition of STAT3 would alter the effectiveness of doxorubicin treatment.
We generated dose-response curves from serial dilutions of DOX to calculate the 50% inhibitory concentration (IC50) in OS cultures and OS/TAM 1:1 co-cultures (Supplemental Figure 6A, Figure 5A). We previously found that scaffold architecture and substrate stiffness have little effect on the response to doxorubicin in MG-63 cells [30]. Here we found that OS cells alone on 20% gelatin are more susceptible to doxorubicin treatment, but there is no difference between the other scaffolds (Supplemental Figure 6B). In OS/TAM co-cultures, the IC50 increased as substrate stiffness decreased except for in the 100% gelatin scaffolds, which had the lowest IC50 (Figure 5B). The 95% confidence intervals (95% CI) between different scaffolds do not overlap completely, but each scaffold overlaps with at least two other scaffolds, so it appears that in the presence of the immune compartment the substrate stiffness affects the response of OS cells to doxorubicin at least to some degree. Interestingly, within the OS/TAM co-culture, the IC50 correlates with TNFα secretion and IL-6 secretion such that as more cytokine is released, the IC50 increases. Thus, the non-monotonic relationship between cell response and substrate stiffness observed for cytokine release is also observed in the calculated IC50. Pro-inflammatory environments are frequently considered cytotoxic to cancer cells, but in this case, it appears that inflammation makes the OS cells less susceptible to doxorubicin treatment [41, 42].
Figure 5: Drug response in OS/TAM co-cultures.
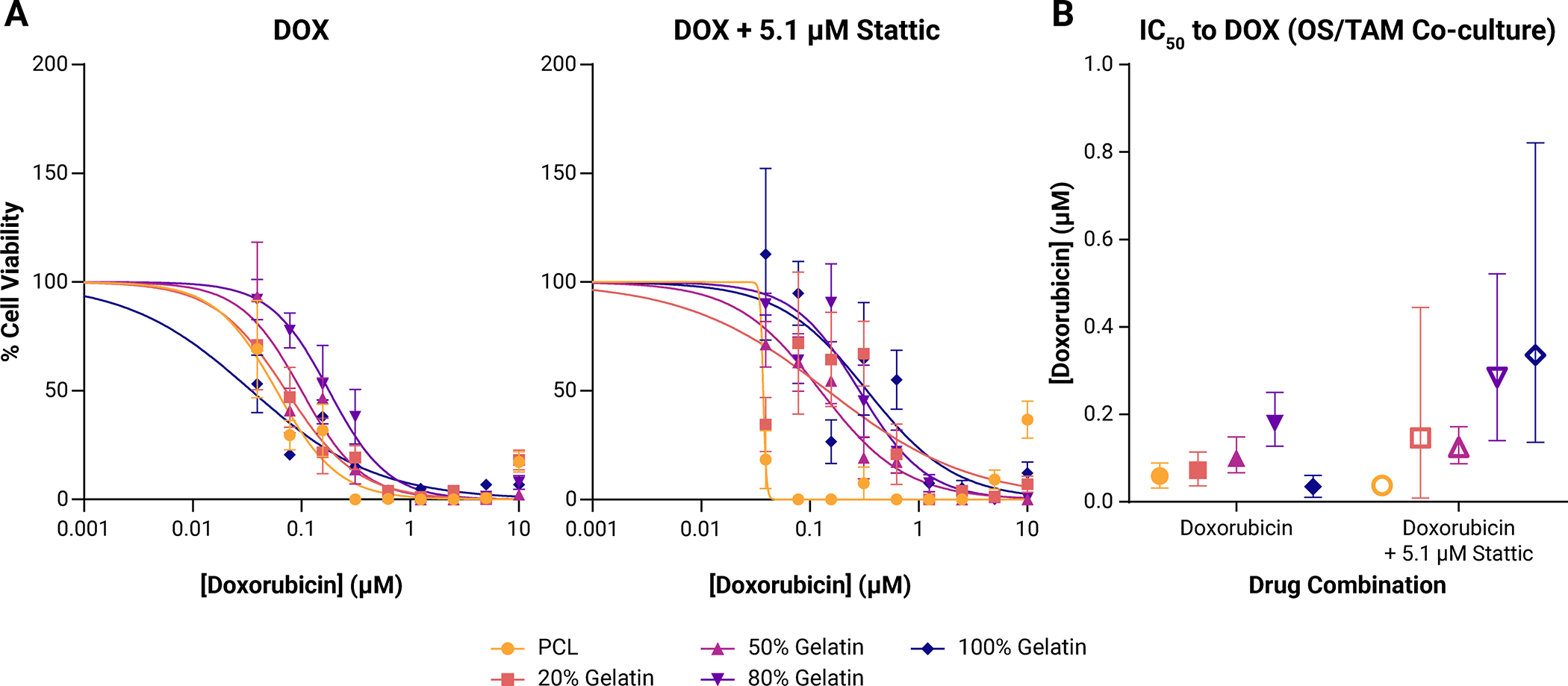
(A) Dose-response curves to doxorubicin (DOX; left) or doxorubicin with 5.1 μM Stattic (right) in OS/TAM co-cultures. Points shown represent mean ± SD (n = 4 for each drug concentration for each scaffold). (B) Calculated IC50 (points) with 95% confidence interval (95% CI; bars) to doxorubicin. Doxorubicin only treatment is shown in filled points. Combination treatment of doxorubicin with 5.1 μM Stattic is shown in open points.
We hypothesized that STAT3 inhibition would make osteosarcoma cells in pro-inflammatory environments more sensitive to doxorubicin treatment. When a single concentration (5.1 μM; acellular IC50) of the small molecule STAT3 inhibitory compound (Stattic) was added to the doxorubicin, there was no synergistic effect in improving the treatment against osteosarcoma cells. When OS cells on their own were exposed to 5.1 μM Stattic for 72 h, we observed either no change or an insignificant decrease in the overall viability compared to live cells exposed to no drug (Supplemental Figure 6). In doxorubicin/Stattic combination therapy, the 95% CI of the IC50 calculated for a given scaffold overlapped with the IC50 and 95% CI of the respective doxorubicin only group, indicating that STAT3 inhibition did not increase the efficacy of doxorubicin treatment (Supplemental Figure 6). One notable exception is that OS cells grown on PCL scaffolds showed a 9.3-fold increase in the calculated IC50.
In the OS/TAM co-culture, Stattic treatment led to significant decreases in cell viability in 20% gelatin and 100% gelatin scaffolds suggesting that cells in environments with altered immune signals are more sensitive to STAT3 inhibition (Supplemental Figure 7). In combination therapy of doxorubicin and a single concentration of Stattic, the inflammation-correlated differences in calculated IC50 observed in the doxorubicin only treatment were abrogated by the addition of Stattic, as indicated by the overlap in the 95% CI between the 20% gelatin, 50% gelatin, 80% gelatin, and 100% gelatin scaffolds (Figure 5B). Cells cultured upon PCL exhibited low viability regardless of doxorubicin concentration when exposed to Stattic, thus the 95% CI for the IC50 could not be calculated for this condition. Interestingly, STAT3 inhibition decreased the efficacy of doxorubicin treatment on cells in the 100% gelatin scaffolds (IC50 without Stattic: 0.035, 95% CI: 0.010 μM – 0.060 μM; IC50 with Stattic: 0.336 μM, 95% CI: 0.136 μM – 0.821 μM), suggesting that even though the addition of Stattic decreased the overall viability of cells in 100% gelatin scaffolds, a higher concentration of doxorubicin was required to kill the remaining cells. In the remaining groups except PCL, the IC50 increased slightly as indicated by the right shift of the dose-response curves (Figure 5B), but as the 95% CIs overlapped with the respective doxorubicin only condition, this increase is not meaningful. All calculated IC50 and 95% CIs are shown in Table 1.
Table 1.
IC50 to doxorubicin.
| Scaffold | OS | OS | OS/TAM | OS/TAM |
|---|---|---|---|---|
| DOX | DOX + 5.1 μM Stattic | DOX | DOX + 5.1 μM Stattic | |
| PCL | 0.086 (0.041 – 0.133) | 0.802 (0.394 – 2.595) | 0.059 (0.032 – 0.089) | 0.037 |
| 20% Gelatin | 0.011 (1.69 × 10−5 – 0.031) | 0.037 (0.014 – 0.065) | 0.072 (0.037 – 0.114) | 0.146 (0.008 – 0.444) |
| 50% Gelatin | 0.117 (0.093 – 0.145) | 0.193 (0.115 – 0.308) | 0.100 (0.067 – 0.148) | 0.125 (0.087 – 0.172) |
| 80% Gelatin | 0.149 (0.118 – 0.187) | 0.176 (0.102 – 0.284) | 0.179 (0.127 – 0.250) | 0.285 (0.140 – 0.522) |
| 100% Gelatin | 0.113 (0.078 – 0.159) | 0.093 (0.053 – 0.142) | 0.035 (0.010 – 0.060) | 0.336 (0.136 – 0.821) |
IC50 is given in μM of doxorubicin (DOX) with the 95% confidence interval shown in parentheses.
Discussion
Tumor-associated macrophages account for the largest fraction of the infiltrating immune cells in many solid tumors and are of growing interest as a target for anti-cancer agents, including for osteosarcoma [22, 23, 42, 43]. In many solid tumors, macrophages play several roles in tumor progression, including modulation of the inflammatory response; facilitation of migration, invasion, and metastatic processes; and resistance to therapy [42, 44]. Most studies indicate that macrophages in the osteosarcoma microenvironment exhibit M2 polarization, which is usually associated with poor survival outcome in patients. However, the effect of macrophage polarization on the prognosis of osteosarcoma remains unclear [22, 44].
In this work we introduced tumor-associated macrophages to a mechanically tunable model of the osteosarcoma tumor niche and found that macrophages and osteosarcoma cells together produce biochemical signals that significantly alter the immune landscape of the tumor in a manner that is not observed when either cell type is cultured on its own. The presence of macrophages early in tumor development led to an elevation in molecules associated with classical or pro-inflammatory activation, TNFα and IL-6, but not those associated with alternative or anti-inflammatory activation, IL-10 and CCL18. Although the surface properties differ between the PCL scaffolds and the remaining four scaffold types, which all have crosslinked gelatin on the surface, the absence of a significant immune response in the OS only and TAM only conditions in all of the scaffolds suggests that the change in inflammation observed in the OS/TAM co-cultures is due to the co-culture condition and the underlying substrate mechanical properties rather than incompatibility with the scaffold material.
For both TNFα and IL-6 we observed differences in cytokine levels as the mechanical properties of the underlying scaffolds changed, but this was only significant in the case of TNFα. Even so, there was no clear correlation between the scaffold stiffness and the amount of cytokine released. Scaffolds with moderate stiffness, specifically 20% gelatin, 50% gelatin, and 80% gelatin, had similarly elevated concentrations of TNFα, with the 80% gelatin scaffolds releasing the highest concentrations of both TNFα and IL-6 in the OS/TAM 1:1 co-culture [30]. Indeed, under this co-culture condition, we observed a non-monotonic relationship of TNFα and IL-6 concentrations with scaffold stiffness: cytokine production increased as the scaffold stiffness decreased from stiffest (PCL, 71 ± 70 MPa) to the second softest (80% gelatin), but then decreased in the softest scaffolds (100% gelatin, 0.004 ± 0.004 MPa) [30]. Previous studies have reported conflicting results about the effect of stiffness on macrophage phenotype; some indicate that softer substrates lead to more pro-inflammatory polarization and stiffer substrates induce a more anti-inflammatory phenotype in bone marrow-derived macrophages, while others report the opposite [32, 45–47]. Indeed, in our system, we found that in the absence of stimulation by exogenous factors such as LPS or cytokines, polarization towards M1 or M2 phenotypes in response to the scaffold elastic modulus was negligible, which suggests that substrate stiffness influences, but is not the most important factor affecting macrophage activation. Instead, it appears that the direct cell-cell contact between the osteosarcoma cells and macrophages plays a greater role in tuning the immune response. Since macrophages are present in nearly all tissues in the body and their transcription profiles vary depending on the tissue of origin, taken together, our results and others’ underscore the plasticity and sensitivity of macrophages to the surrounding environment [48, 49]. It is imperative that in future work, macrophages are investigated within models that mimic the specific contexts in which they would be found to fully understand their phenotype and function.
One major limitation of our system is that it is difficult to study phenomena between osteosarcoma cells and macrophages beyond short time scales. It is generally thought that tumor-associated macrophages exhibit M1-like polarization at the beginning of tumor formation and shift towards the M2-like polarization typically associated with tumor-associated macrophages as the disease progresses. While our model is suitable for examining osteosarcoma and macrophage interactions at 24 h and we do observe that pro-inflammatory activation of the macrophages, it lacks the capacity to recruit additional immune cells, such as T cells, into the tumor microenvironment that would promote the shift towards the anti-inflammatory phenotype and drive tumor-promoting functions [23, 41, 50]. The ability to investigate the roles of tumor-associated macrophages in osteosarcoma at a later stage of disease progression, for instance when the tumor has metastasized, would be incredibly impactful since it has been reported that approximately 18% to 25% of osteosarcoma patients present with metastasis at the time of diagnosis [51, 52]. However, the role of inflammation in the tumor microenvironment cannot be discounted. In fact, in a mouse model of osteosarcoma, TNFα produced by macrophages maintained osteosarcoma cells in a stem-like state, which is required for tumorigenesis, and a growing body of work implicates IL-6 as a mediator of multi-drug resistance in cancer [53–55].
Our model suggests that macrophage-induced inflammation by way of TNFα and IL-6 contributes to the resistance of osteosarcoma cells to doxorubicin treatment. In OS/TAM co-cultures, the 50% inhibitory concentration (IC50) for doxorubicin on each scaffold correlated with the concentrations of secreted TNFα and IL-6 such that scaffolds upon which higher concentrations of cytokine were released required more doxorubicin to be effective. This is corroborated by work demonstrating that IL-6 produced by cancer-associated fibroblasts reduces the efficacy of doxorubicin in prostate cancer [56]. The role of TNFα to drug resistance is more complex because TNFα can serve both pro-tumoral and anti-tumoral functions and the debate about whether it should be targeted in treatment or used as a therapeutic agent continues [57, 58]. TNFα expression has been linked to doxorubicin resistance in lung cancer, breast cancer, and leukemia [59–62]. A growing body of work points to the role of TNFα in regulating cancer stem cell populations in multiple cancers including osteosarcoma, breast cancer, and colon cancer, and that cancer stem cells are responsible for resistance to therapy and metastatic potential [53, 63–68]. We have previously found that the stem cell marker Sox2 is elevated in MG-63 osteosarcoma cells cultured within three-dimensional scaffolds compared to cells grown in monolayers, and it is possible that TNFα leads to doxorubicin resistance by supporting this stem-like nature in osteosarcoma cells, although further investigation is required to identify the mechanisms by which this occurs [30]. The role of TNFα in drug resistance is further complicated by the fact that the administration of doxorubicin increases system inflammation, including the induction of TNFα, in cancer patients [69–71]. TNFα has been shown to increase the apoptotic effect of doxorubicin on cardiomyocytes but not U2OS osteosarcoma cells, indicating that doxorubicin-mediated changes to TNFα signaling are dependent on cell type [72]. Taken together, the correlation between inflammatory cytokine content and the calculated IC50 that we observed suggests that the TNFα and IL-6 present within the microenvironment from co-culture with macrophages limits the initial efficacy of doxorubicin treatment on osteosarcoma cells, potentially by mediating cancer stemness, and any TNFα stimulated in response to the drug exposure either sustains this drug resistance or has no effect in improving treatment.
The downstream effectors of the Hippo pathway YAP and TAZ are dysregulated in many cancers, and their hyperactivation contributes to disease progression, including tumor initiation, proliferation, and metastasis [73, 74]. Critically, YAP and TAZ are sensitive to mechanical cues and mediate mechanotransduction largely independently of the canonical Hippo pathway [73, 75, 76]. Studies have confirmed that YAP is upregulated in osteosarcoma, and knockdown of YAP1 inhibits proliferation in both MG-63 and HOS osteosarcoma cells [77, 78]. Interestingly, the resistance to therapy observed in our system may in part be regulated by YAP since YAP has emerged as a mediator of inflammatory signals in crucial processes like tumor initiation. It has been demonstrated that TNFα activates YAP in multiple cell types including breast cancer cells and endothelial cells [79–81]. Additionally, YAP induces IL-6 and promotes stemness in basal-like breast cancer, and IL-6 induced by YAP promotes tumor-associated macrophage recruitment in hepatocellular carcinoma [82–84]. These results from others, in combination with our previous finding that osteosarcoma cells upregulate Sox2 expression in three-dimensional architectures and our observation in the present study that YAP is slightly elevated in OS/TAM co-culture conditions, suggest that inflammation by way of TNFα induces YAP activation, which in turn induces IL-6 to further drive activation of macrophages and maintain a stem-like phenotype in the osteosarcoma cells that is resistant to therapy [30]. However, further mechanistic studies investigating the crosstalk between YAP and TAZ signaling, pathways mediating the maintenance of cancer stem cell phenotypes, and inflammatory pathways within osteosarcoma are required to fully elucidate how these phenomena mediate the efficacy of treatment.
Because STAT3 is overexpressed and associated with poor prognosis in osteosarcoma and is a key molecule in IL-6 signal transduction, we were interested in investigating whether STAT3 inhibition by the small molecule STAT3 inhibitory compound (Stattic) would improve the efficacy of treatment by doxorubicin in tumor microenvironments with elevated IL-6 [40]. We found that Stattic can decrease the baseline viability of osteosarcoma cells in inflammatory microenvironments and reduces the inflammation-mediated differences in doxorubicin efficacy. Since the IC50 of doxorubicin with the addition of Stattic was comparable regardless of the underlying scaffold, it appears that the phenomena are not specific to the underlying substrates and STAT3 inhibition does not alter mechanoresponse. Instead, it seems that STAT3 inhibition reduces inflammation-mediated differences in therapy resistance by disrupting cell-cell communication between the osteosarcoma cells and macrophages. Even though inflammation is reduced, the osteosarcoma cells that remain after STAT3 inhibition are more resistant to therapy such that the survival of the remaining osteosarcoma cells is independent of STAT3, which contrasts with previous studies indicating that direct or indirect inhibition of STAT3 improves the sensitivity of osteosarcoma cells to doxorubicin [85, 86]. Interestingly, at least one study has found that JAK/STAT pathway inhibition has competing effects on cancer cells and other cells within the tumor microenvironment. For instance, JAK/STAT signal inhibition is anti-tumoral to breast cancer cells, but deletion of STAT3 and inhibition of JAK/STAT in macrophages leads to the production of pro-tumorigenic cyclooxygenase-2, which leads to therapeutic resistance in the cancer cells [87]. Future studies are required to identify whether our results are indicative of similar competing effects of STAT3 inhibition on the osteosarcoma cells and macrophages or whether the three-dimensional architecture and mechanical microenvironment are activating a different set of pathways that confer resistance to therapy.
This study has potential limitations. We developed our model using the MG-63 osteosarcoma cell line and the THP-1 cell line, the latter of which was selected due to its accessibility compared to human peripheral blood monocytes and establishment as a cell line for macrophage studies [88, 89]. Yet, THP-1s are not a perfect cell line and their utility as a model cell line for macrophages has come under scrutiny in recent years [90]. Thus, it may be the case that the results we have observed are specific to the cell lines utilized; given the inter- and intra-tumor heterogeneity of osteosarcoma, we would likely observe different results using patient-derived cancer and peripheral blood cells. We have also not compared the results to an animal model; using a tissue-engineered model affords us a greater degree of control in investigating the functions of specific elements within the tumor microenvironment. One of the long-term objectives for engineering tumor models is to develop a tunable platform in which a cancer patient’s cells can be tested for targetable pathways and personalized treatment in an environment that is more physiologically relevant than traditional in vitro monolayers and minimizes the cost and ethical concerns associated with animal models. Additional studies are required to compare the clinical utility of these novel systems with the current standards used in the drug development pipeline, including patient-derived xenografts in animals. Despite these constraints, our study adds to the body of work that demonstrates the critical role that supporting cells within the tumor microenvironment and tumor heterogeneity play in osteosarcoma disease progression and response to therapies. Continuing to engineer materials-based tumor models that aid in identifying patient-specific targets for therapy may ultimately improve treatment outcomes [31, 91, 92].
Conclusion
In this study we wanted to investigate the combined roles of tumor-associated macrophages and substrate mechanical properties on osteosarcoma progression by characterizing the immune environment in a three-dimensional model of osteosarcoma with tunable substrate stiffness. We have demonstrated that including an immune compartment within relevant trabecular bone-like architecture may prove useful for identifying and developing targeted therapies at different stages of osteosarcoma progression. We found that early in tumor development, macrophages confer a pro-inflammatory environment with elevated TNFα and IL-6 in scaffolds of moderate stiffness. Moreover, macrophage polarization towards M1 or M2 phenotypes is not strictly dictated by the mechanical properties of the substrate, but instead is both affected by the mechanical properties and heterotypic signaling with cancer cells. In our model, inflammation in the tumor microenvironment decreased the sensitivity of osteosarcoma cells to doxorubicin, and while STAT3 inhibition led to the loss of those inflammation-mediated differences, it ultimately did not increase the efficacy of doxorubicin. Altogether, we have highlighted that utilizing scaffolds that recapitulate the structure and mechanical properties of the microenvironment and including relevant non-cancer cells in tumor models helps elucidate pathways that are potentially targetable and mechanisms by which cells are resistant to treatment.
Supplementary Material
Supplemental Figure 1: Morphological comparison of THP-1-derived macrophages. (A) Representative epifluorescent microscopy images of M0, M1, and M2 macrophages derived from THP-1 in monolayers co-stained with phalloidin (actin, red) and Hoechst (nuclei, blue). M1 macrophages were polarized with INFγ and LPS. M2 macrophages were polarized with IL-4 and IL-13. Scale = 25 μm. (B) Representative confocal microscopy images of THP-1 cells after 48 h PMA treatment on electrospun scaffolds co-stained with phalloidin (actin, green) and Hoechst (nuclei, blue). Scale = 50 μm. Images are taken from a single plane on the surface of the scaffold.
Supplemental Figure 2: Cell proliferation over five days. (A) Proliferation of THP-1 macrophages in electrospun scaffolds. Data shown as mean ± SD of mass of dsDNA per 6 mm diameter scaffold (n = 6 per scaffold type at each time point). (B) Cell growth as measured by dsDNA content in OS, OS/TAM 3:1, and OS/TAM 1:1 co-cultures in different electrospun scaffolds after 5 days. Data shown as mean + SD (n = 6). Two-way ANOVA with post-hoc Tukey’s HSD, ***p < 0.001, ****p < 0.0001.
Supplemental Figure 3: Raw (non-bootstrapped) and simulated (bootstrapped) measurements of secreted cytokines from OS and TAM cultures in electrospun scaffolds. Top: Individual measurements within the standard curve range for (A) TNFα (n = 3) and (B) IL-6 (n = 4); bars represent mean ± SD. Measurements below the standard curve are not displayed. Bottom: An example of bootstrapped cytokine secretion data. Each point represents an individual value; filled points represent measured data (also shown in A and B) and outlined points represent bootstrapped values for (C) TNFα (n = 3) and (D) IL-6 (n = 4); bars represent mean + SD. Two-way ANOVA with post-hoc Tukey’s HSD, p < 0.05. Groups that do not share the same letter are statistically different.
Supplemental Figure 4: Population selection for flow cytometry analysis. (A) Representative histograms of CD90 expression in MG-63 OS cells across different scaffolds. (B) Example of gating for high YAP and low YAP cells in OS/TAM co-cultures. Data shown are from cells grown on PCL scaffolds. (1) Analysis was conducted on single cells expressing CD90 and CD11b (Q2). (2) Cells were found to be in two distinct populations expressing either high YAP (Q2) or low YAP (Q3). (3) The geometric means of Alexa Fluor 647-conjugated anti-YAP and Alexa Fluor 594-conjugated anti-TAZ were measured in the two quadrants of interest. High YAP cells were taken to be OS cells and low YAP cells were taken to be TAMs. (C) Representative scatter plots (top) and histograms (bottom) of different culture conditions showing that high YAP cells in OS/TAM co-culture (purple) align with OS only cultures (green) and low YAP cells in OS/TAM co-culture align with TAM only cultures (yellow). Data shown are from cells grown on PCL scaffolds.
Supplemental Figure 5: YAP and TAZ expression by flow cytometry. Representative scatter plots showing expression of (A) YAP and (B) TAZ across different culture conditions. Osteosarcoma cells and macrophages were identified by the differential expression of YAP. Osteosarcoma cells were observed to have bimodal CD90 and high YAP (upper right quadrant in OS and OS/TAM 1:1 rows) and macrophages were observed to have low YAP (lower right gate in OS/TAM 1:1 and TAM rows). Measurements of TAZ in OS/TAM co-culture were taken after applying the gates for YAP as described in Supplemental Figure 4B.
Supplemental Figure 6: Response of homotypic osteosarcoma cell culture to doxorubicin (DOX) and combination therapy with Stattic. (A) Fitted dose-response curves of osteosarcoma cells to DOX (left) and DOX with 5.1 μM Stattic (right). Data shown as mean ± SD (n = 4 for each scaffold and drug combination). (B) Calculated IC50 (points) and 95% CI (bars) of osteosarcoma cells to doxorubicin (left) and doxorubicin with Stattic (right).
Supplemental Figure 7: Effect of STAT3 inhibitory compound (Stattic) on overall cell viability. Cell viability in OS (left) and OS/TAM (right) cultures without drug or with 5.1 μM Stattic was measured by WST-1 after 72 h of culture on electrospun scaffolds. Cell viability is shown normalized to the no drug condition. Data shown as mean + SD (n = 4). Multiple paired t tests with Holm-Šídák multiple comparisons, *p < 0.05. Comparisons not shown are not statistically significant.
Acknowledgements:
We would like to thank Dr. Eric R. Molina for his discussions on analyzing dose-response curves for combination therapy. We would also like to thank Dr. David W. Scott for his expertise and guidance on statistical analyses and parametric bootstrapping.
Funding:
This work was supported by the National Institutes of Health [grant numbers R01CA180279, P41EB023833].
Data Availability:
The raw and processed data required to reproduce these findings are available upon request.
References
- [1].Luetke A, Meyers PA, Lewis I, Juergens H, Osteosarcoma treatment - where do we stand? A state of the art review, Cancer Treat Rev 40(4) (2014) 523–32. [DOI] [PubMed] [Google Scholar]
- [2].Gianferante DM, Mirabello L, Savage SA, Germline and somatic genetics of osteosarcoma - connecting aetiology, biology and therapy, Nat Rev Endocrinol 13(8) (2017) 480–491. [DOI] [PubMed] [Google Scholar]
- [3].Kansara M, Teng MW, Smyth MJ, Thomas DM, Translational biology of osteosarcoma, Nat Rev Cancer 14(11) (2014) 722–35. [DOI] [PubMed] [Google Scholar]
- [4].Ottaviani G, Jaffe N, The epidemiology of osteosarcoma, Cancer Treat Res 152 (2009) 3–13. [DOI] [PubMed] [Google Scholar]
- [5].Czarnecka AM, Synoradzki K, Firlej W, Bartnik E, Sobczuk P, Fiedorowicz M, Grieb P, Rutkowski P, Molecular Biology of Osteosarcoma, Cancers (Basel) 12(8) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gill J, Gorlick R, Advancing therapy for osteosarcoma, Nat Rev Clin Oncol 18(10) (2021) 609–624. [DOI] [PubMed] [Google Scholar]
- [7].Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ, Massive genomic rearrangement acquired in a single catastrophic event during cancer development, Cell 144(1) (2011) 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cortes-Ciriano I, Lee JJ, Xi R, Jain D, Jung YL, Yang L, Gordenin D, Klimczak LJ, Zhang CZ, Pellman DS, Group PSVW, Park PJ, Consortium P, Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing, Nat Genet 52(3) (2020) 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Misaghi A, Goldin A, Awad M, Kulidjian AA, Osteosarcoma: a comprehensive review, SICOT J 4 (2018) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Allison DC, Carney SC, Ahlmann ER, Hendifar A, Chawla S, Fedenko A, Angeles C, Menendez LR, A meta-analysis of osteosarcoma outcomes in the modern medical era, Sarcoma 2012 (2012) 704872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Saraf AJ, Fenger JM, Roberts RD, Osteosarcoma: Accelerating Progress Makes for a Hopeful Future, Front Oncol 8 (2018) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Botter SM, Neri D, Fuchs B, Recent advances in osteosarcoma, Curr Opin Pharmacol 16 (2014) 15–23. [DOI] [PubMed] [Google Scholar]
- [13].Chim LK, Mikos AG, Biomechanical forces in tissue engineered tumor models, Curr Opin Biomed Eng 6 (2018) 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Molina ER, Chim LK, Barrios S, Ludwig JA, Mikos AG, Modeling the Tumor Microenvironment and Pathogenic Signaling in Bone Sarcoma, Tissue Eng Part B Rev 26(3) (2020) 249–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marusyk A, Almendro V, Polyak K, Intra-tumour heterogeneity: a looking glass for cancer?, Nat Rev Cancer 12(5) (2012) 323–34. [DOI] [PubMed] [Google Scholar]
- [16].Keaveny TM, Morgan EF, Niebur GL, Yeh OC, Biomechanics of trabecular bone, Annu Rev Biomed Eng 3 (2001) 307–33. [DOI] [PubMed] [Google Scholar]
- [17].Oftadeh R, Perez-Viloria M, Villa-Camacho JC, Vaziri A, Nazarian A, Biomechanics and mechanobiology of trabecular bone: a review, J Biomech Eng 137(1) (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mutsaers AJ, Walkley CR, Cells of origin in osteosarcoma: mesenchymal stem cells or osteoblast committed cells?, Bone 62 (2014) 56–63. [DOI] [PubMed] [Google Scholar]
- [19].Maurizi G, Verma N, Gadi A, Mansukhani A, Basilico C, Sox2 is required for tumor development and cancer cell proliferation in osteosarcoma, Oncogene 37(33) (2018) 4626–4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Deng Z, Zhang Q, Zhao Z, Li Y, Chen X, Lin Z, Deng Z, Liu J, Duan L, Wang D, Li W, Crosstalk between immune cells and bone cells or chondrocytes, Int Immunopharmacol 101(Pt A) (2021) 108179. [DOI] [PubMed] [Google Scholar]
- [21].Endo-Munoz L, Evdokiou A, Saunders NA, The role of osteoclasts and tumour-associated macrophages in osteosarcoma metastasis, Biochim Biophys Acta 1826(2) (2012) 434–42. [DOI] [PubMed] [Google Scholar]
- [22].Wang Z, Wang Z, Li B, Wang S, Chen T, Ye Z, Innate Immune Cells: A Potential and Promising Cell Population for Treating Osteosarcoma, Front Immunol 10 (2019) 1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Solinas G, Germano G, Mantovani A, Allavena P, Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation, J Leukoc Biol 86(5) (2009) 1065–73. [DOI] [PubMed] [Google Scholar]
- [24].Komohara Y, Fujiwara Y, Ohnishi K, Takeya M, Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy, Adv Drug Deliv Rev 99(Pt B) (2016) 180–185. [DOI] [PubMed] [Google Scholar]
- [25].Engblom C, Pfirschke C, Pittet MJ, The role of myeloid cells in cancer therapies, Nat Rev Cancer 16(7) (2016) 447–62. [DOI] [PubMed] [Google Scholar]
- [26].Qian BZ, Pollard JW, Macrophage diversity enhances tumor progression and metastasis, Cell 141(1) (2010) 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Buddingh EP, Kuijjer ML, Duim RA, Burger H, Agelopoulos K, Myklebost O, Serra M, Mertens F, Hogendoorn PC, Lankester AC, Cleton-Jansen AM, Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: a rationale for treatment with macrophage activating agents, Clin Cancer Res 17(8) (2011) 2110–9. [DOI] [PubMed] [Google Scholar]
- [28].Dumars C, Ngyuen JM, Gaultier A, Lanel R, Corradini N, Gouin F, Heymann D, Heymann MF, Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma, Oncotarget 7(48) (2016) 78343–78354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Han Y, Guo W, Ren T, Huang Y, Wang S, Liu K, Zheng B, Yang K, Zhang H, Liang X, Tumor-associated macrophages promote lung metastasis and induce epithelial-mesenchymal transition in osteosarcoma by activating the COX-2/STAT3 axis, Cancer Lett 440–441 (2019) 116–125. [DOI] [PubMed] [Google Scholar]
- [30].Molina ER, Chim LK, Salazar MC, Mehta SM, Menegaz BA, Lamhamedi-Cherradi SE, Satish T, Mohiuddin S, McCall D, Zaske AM, Cuglievan B, Lazar AJ, Scott DW, Grande-Allen JK, Ludwig JA, Mikos AG, Mechanically tunable coaxial electrospun models of YAP/TAZ mechanoresponse and IGF-1R activation in osteosarcoma, Acta Biomater 100 (2019) 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Molina ER, Chim LK, Lamhamedi-Cherradi SE, Mohiuddin S, McCall D, Cuglievan B, Krishnan S, Porter RW, Ingram DR, Wang WL, Lazar AJ, Scott DW, Truong DD, Daw NC, Ludwig JA, Mikos AG, Correlation of nuclear pIGF-1R/IGF-1R and YAP/TAZ in a tissue microarray with outcomes in osteosarcoma patients, Oncotarget 13 (2022) 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Meli VS, Atcha H, Veerasubramanian PK, Nagalla RR, Luu TU, Chen EY, Guerrero-Juarez CF, Yamaga K, Pandori W, Hsieh JY, Downing TL, Fruman DA, Lodoen MB, Plikus MV, Wang W, Liu WF, YAP-mediated mechanotransduction tunes the macrophage inflammatory response, Sci Adv 6(49) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fong EL, Lamhamedi-Cherradi SE, Burdett E, Ramamoorthy V, Lazar AJ, Kasper FK, Farach-Carson MC, Vishwamitra D, Demicco EG, Menegaz BA, Amin HM, Mikos AG, Ludwig JA, Modeling Ewing sarcoma tumors in vitro with 3D scaffolds, Proc Natl Acad Sci U S A 110(16) (2013) 6500–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Santoro M, Lamhamedi-Cherradi SE, Menegaz BA, Ludwig JA, Mikos AG, Flow perfusion effects on three-dimensional culture and drug sensitivity of Ewing sarcoma, Proc Natl Acad Sci U S A 112(33) (2015) 10304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Santoro M, Menegaz BA, Lamhamedi-Cherradi SE, Molina ER, Wu D, Priebe W, Ludwig JA, Mikos AG, Modeling Stroma-Induced Drug Resistance in a Tissue-Engineered Tumor Model of Ewing Sarcoma, Tissue Eng Part A 23(1–2) (2017) 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang J, Li D, Cang H, Guo B, Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment, Cancer Med 8(10) (2019) 4709–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Schwende H, Fitzke E, Ambs P, Dieter P, Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3, J Leukoc Biol 59(4) (1996) 555–61. [PubMed] [Google Scholar]
- [38].Hsu PC, Li JM, Yang CT, Forced Overexpression of Signal Transducer and Activator of Transcription 3 (STAT3) Activates Yes-Associated Protein (YAP) Expression and Increases the Invasion and Proliferation Abilities of Small Cell Lung Cancer (SCLC) Cells, Biomedicines 10(7) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].He L, Pratt H, Gao M, Wei F, Weng Z, Struhl K, YAP and TAZ are transcriptional co-activators of AP-1 proteins and STAT3 during breast cellular transformation, Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu Y, Liao S, Bennett S, Tang H, Song D, Wood D, Zhan X, Xu J, STAT3 and its targeting inhibitors in osteosarcoma, Cell Prolif 54(2) (2021) e12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Malfitano AM, Pisanti S, Napolitano F, Di Somma S, Martinelli R, Portella G, Tumor-Associated Macrophage Status in Cancer Treatment, Cancers (Basel) 12(7) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Luo ZW, Liu PP, Wang ZX, Chen CY, Xie H, Macrophages in Osteosarcoma Immune Microenvironment: Implications for Immunotherapy, Front Oncol 10 (2020) 586580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P, Tumour-associated macrophages as treatment targets in oncology, Nat Rev Clin Oncol 14(7) (2017) 399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fujiwara T, Healey J, Ogura K, Yoshida A, Kondo H, Hata T, Kure M, Tazawa H, Nakata E, Kunisada T, Fujiwara T, Ozaki T, Role of Tumor-Associated Macrophages in Sarcomas, Cancers (Basel) 13(5) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen M, Zhang Y, Zhou P, Liu X, Zhao H, Zhou X, Gu Q, Li B, Zhu X, Shi Q, Substrate stiffness modulates bone marrow-derived macrophage polarization through NF-kappaB signaling pathway, Bioact Mater 5(4) (2020) 880–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Liu Y, Segura T, Biomaterials-Mediated Regulation of Macrophage Cell Fate, Front Bioeng Biotechnol 8 (2020) 609297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sridharan R, Cavanagh B, Cameron AR, Kelly DJ, O’Brien FJ, Material stiffness influences the polarization state, function and migration mode of macrophages, Acta Biomater 89 (2019) 47–59. [DOI] [PubMed] [Google Scholar]
- [48].Epelman S, Lavine KJ, Randolph GJ, Origin and functions of tissue macrophages, Immunity 41(1) (2014) 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ, Immunological Genome C, Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages, Nat Immunol 13(11) (2012) 1118–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pathria P, Louis TL, Varner JA, Targeting Tumor-Associated Macrophages in Cancer, Trends Immunol 40(4) (2019) 310–327. [DOI] [PubMed] [Google Scholar]
- [51].Tsukamoto S, Errani C, Angelini A, Mavrogenis AF, Current Treatment Considerations for Osteosarcoma Metastatic at Presentation, Orthopedics 43(5) (2020) e345–e358. [DOI] [PubMed] [Google Scholar]
- [52].Marko TA, Diessner BJ, Spector LG, Prevalence of Metastasis at Diagnosis of Osteosarcoma: An International Comparison, Pediatr Blood Cancer 63(6) (2016) 1006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mori T, Sato Y, Miyamoto K, Kobayashi T, Shimizu T, Kanagawa H, Katsuyama E, Fujie A, Hao W, Tando T, Iwasaki R, Kawana H, Morioka H, Matsumoto M, Saya H, Toyama Y, Miyamoto T, TNFalpha promotes osteosarcoma progression by maintaining tumor cells in an undifferentiated state, Oncogene 33(33) (2014) 4236–41. [DOI] [PubMed] [Google Scholar]
- [54].Ghandadi M, Sahebkar A, Interleukin-6: A Critical Cytokine in Cancer Multidrug Resistance, Curr Pharm Des 22(5) (2016) 518–26. [DOI] [PubMed] [Google Scholar]
- [55].Wang K, Zhu X, Zhang K, Yin Y, Chen Y, Zhang T, Interleukin-6 contributes to chemoresistance in MDA-MB-231 cells via targeting HIF-1alpha, J Biochem Mol Toxicol 32(3) (2018) e22039. [DOI] [PubMed] [Google Scholar]
- [56].Cheteh EH, Sarne V, Ceder S, Bianchi J, Augsten M, Rundqvist H, Egevad L, Ostman A, Wiman KG, Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells, Cell Death Discov 6 (2020) 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Montfort A, Colacios C, Levade T, Andrieu-Abadie N, Meyer N, Segui B, The TNF Paradox in Cancer Progression and Immunotherapy, Front Immunol 10 (2019) 1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Balkwill F, Tumour necrosis factor and cancer, Nat Rev Cancer 9(5) (2009) 361–71. [DOI] [PubMed] [Google Scholar]
- [59].Prewitt TW, Matthews W, Chaudhri G, Pogrebniak HW, Pass HI, Tumor necrosis factor induces doxorubicin resistance to lung cancer cells in vitro, The Journal of Thoracic and Cardiovascular Surgery 107(1) (1994) 43–49. [PubMed] [Google Scholar]
- [60].Zhang Z, Lin G, Yan Y, Li X, Hu Y, Wang J, Yin B, Wu Y, Li Z, Yang XP, Transmembrane TNF-alpha promotes chemoresistance in breast cancer cells, Oncogene 37(25) (2018) 3456–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mercogliano MF, Bruni S, Elizalde PV, Schillaci R, Tumor Necrosis Factor alpha Blockade: An Opportunity to Tackle Breast Cancer, Front Oncol 10 (2020) 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kobayashi D, Watanabe N, Yamauchi N, Tsuji N, Sato T, Niitsu Y, Endogenous Tumor Necrosis Factor as a Predictor of Doxorubicin Sensitivity in Leukemic Patients, Blood 89(7) (1997) 2472–2479. [PubMed] [Google Scholar]
- [63].Yao J, Lin J, He L, Huang J, Liu Q, TNF-alpha/miR-155 axis induces the transformation of osteosarcoma cancer stem cells independent of TP53INP1, Gene 726 (2020) 144224. [DOI] [PubMed] [Google Scholar]
- [64].Storci G, Sansone P, Mari S, D’Uva G, Tavolari S, Guarnieri T, Taffurelli M, Ceccarelli C, Santini D, Chieco P, Marcu KB, Bonafe M, TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype, J Cell Physiol 225(3) (2010) 682–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhao X, Ma L, Dai L, Zuo D, Li X, Zhu H, Xu F, TNF‑alpha promotes the malignant transformation of intestinal stem cells through the NF‑kappaB and Wnt/beta‑catenin signaling pathways, Oncol Rep 44(2) (2020) 577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zheng Q, Zhang M, Zhou F, Zhang L, Meng X, The Breast Cancer Stem Cells Traits and Drug Resistance, Front Pharmacol 11 (2020) 599965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Paramanantham A, Jung EJ, Kim HJ, Jeong BK, Jung JM, Kim GS, Chan HS, Lee WS, Doxorubicin-Resistant TNBC Cells Exhibit Rapid Growth with Cancer Stem Cell-like Properties and EMT Phenotype, Which Can Be Transferred to Parental Cells through Autocrine Signaling, Int J Mol Sci 22(22) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Meirelles K, Benedict LA, Dombkowski D, Pepin D, Preffer FI, Teixeira J, Tanwar PS, Young RH, MacLaughlin DT, Donahoe PK, Wei X, Human ovarian cancer stem/progenitor cells are stimulated by doxorubicin but inhibited by Mullerian inhibiting substance, Proc Natl Acad Sci U S A 109(7) (2012) 2358–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Niiya M, Niiya K, Kiguchi T, Shibakura M, Asaumi N, Shinagawa K, Ishimaru F, Kiura K, Ikeda K, Ueoka H, Tanimoto M, Induction of TNF-alpha, uPA, IL-8 and MCP-1 by doxorubicin in human lung carcinoma cells, Cancer Chemother Pharmacol 52(5) (2003) 391–8. [DOI] [PubMed] [Google Scholar]
- [70].Wang L, Chen Q, Qi H, Wang C, Wang C, Zhang J, Dong L, Doxorubicin-Induced Systemic Inflammation Is Driven by Upregulation of Toll-Like Receptor TLR4 and Endotoxin Leakage, Cancer Res 76(22) (2016) 6631–6642. [DOI] [PubMed] [Google Scholar]
- [71].Gilliam LA, Ferreira LF, Bruton JD, Moylan JS, Westerblad H, St Clair DK, Reid MB, Doxorubicin acts through tumor necrosis factor receptor subtype 1 to cause dysfunction of murine skeletal muscle, J Appl Physiol (1985) 107(6) (2009) 1935–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chiosi E, Spina A, Sorrentino A, Romano M, Sorvillo L, Senatore G, D’Auria R, Abbruzzese A, Caraglia M, Naviglio S, Illiano G, Change in TNF-alpha receptor expression is a relevant event in doxorubicin-induced H9c2 cardiomyocyte cell death, J Interferon Cytokine Res 27(7) (2007) 589–97. [DOI] [PubMed] [Google Scholar]
- [73].Zanconato F, Cordenonsi M, Piccolo S, YAP/TAZ at the Roots of Cancer, Cancer Cell 29(6) (2016) 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Moroishi T, Hansen CG, Guan KL, The emerging roles of YAP and TAZ in cancer, Nat Rev Cancer 15(2) (2015) 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S, Role of YAP/TAZ in mechanotransduction, Nature 474(7350) (2011) 179–83. [DOI] [PubMed] [Google Scholar]
- [76].Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S, A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors, Cell 154(5) (2013) 1047–1059. [DOI] [PubMed] [Google Scholar]
- [77].Zhang YH, Li B, Shen L, Shen Y, Chen XD, The role and clinical significance of YES-associated protein 1 in human osteosarcoma, Int J Immunopathol Pharmacol 26(1) (2013) 157–67. [DOI] [PubMed] [Google Scholar]
- [78].Yang Z, Zhang M, Xu K, Liu L, Hou WK, Cai YZ, Xu P, Yao JF, Knockdown of YAP1 inhibits the proliferation of osteosarcoma cells in vitro and in vivo, Oncol Rep 32(3) (2014) 1265–72. [DOI] [PubMed] [Google Scholar]
- [79].Gao Y, Yang Y, Yuan F, Huang J, Xu W, Mao B, Yuan Z, Bi W, TNFalpha-YAP/p65-HK2 axis mediates breast cancer cell migration, Oncogenesis 6(9) (2017) e383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Shen Y, Wang X, Liu Y, Singhal M, Gurkaslar C, Valls AF, Lei Y, Hu W, Schermann G, Adler H, Yu FX, Fischer T, Zhu Y, Augustin HG, Schmidt T, de Almodovar CR, STAT3-YAP/TAZ signaling in endothelial cells promotes tumor angiogenesis, Sci Signal 14(712) (2021) eabj8393. [DOI] [PubMed] [Google Scholar]
- [81].Choi HJ, Kim NE, Kim BM, Seo M, Heo JH, TNF-alpha-Induced YAP/TAZ Activity Mediates Leukocyte-Endothelial Adhesion by Regulating VCAM1 Expression in Endothelial Cells, Int J Mol Sci 19(11) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kim T, Yang SJ, Hwang D, Song J, Kim M, Kyum Kim S, Kang K, Ahn J, Lee D, Kim MY, Kim S, Seung Koo J, Seok Koh S, Kim SY, Lim DS, A basal-like breast cancer-specific role for SRF-IL6 in YAP-induced cancer stemness, Nat Commun 6 (2015) 10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kim T, Lim DS, The SRF-YAP-IL6 axis promotes breast cancer stemness, Cell Cycle 15(10) (2016) 1311–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Zhou TY, Zhou YL, Qian MJ, Fang YZ, Ye S, Xin WX, Yang XC, Wu HH, Interleukin-6 induced by YAP in hepatocellular carcinoma cells recruits tumor-associated macrophages, J Pharmacol Sci 138(2) (2018) 89–95. [DOI] [PubMed] [Google Scholar]
- [85].Tian ZC, Wang JQ, Ge H, Apatinib ameliorates doxorubicin-induced migration and cancer stemness of osteosarcoma cells by inhibiting Sox2 via STAT3 signalling, J Orthop Translat 22 (2020) 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Tu B, Zhu J, Liu S, Wang L, Fan Q, Hao Y, Fan C, Tang TT, Mesenchymal stem cells promote osteosarcoma cell survival and drug resistance through activation of STAT3, Oncotarget 7(30) (2016) 48296–48308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Irey EA, Lassiter CM, Brady NJ, Chuntova P, Wang Y, Knutson TP, Henzler C, Chaffee TS, Vogel RI, Nelson AC, Farrar MA, Schwertfeger KL, JAK/STAT inhibition in macrophages promotes therapeutic resistance by inducing expression of protumorigenic factors, Proc Natl Acad Sci U S A 116(25) (2019) 12442–12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Auwerx J, The human leukemia cell line, THP-1: a multifacetted model for the study of monocyte-macrophage differentiation, Experientia 47(1) (1991) 22–31. [DOI] [PubMed] [Google Scholar]
- [89].Lund ME, To J, O’Brien BA, Donnelly S, The choice of phorbol 12-myristate 13-acetate differentiation protocol influences the response of THP-1 macrophages to a pro-inflammatory stimulus, J Immunol Methods 430 (2016) 64–70. [DOI] [PubMed] [Google Scholar]
- [90].Tedesco S, De Majo F, Kim J, Trenti A, Trevisi L, Fadini GP, Bolego C, Zandstra PW, Cignarella A, Vitiello L, Convenience versus Biological Significance: Are PMA-Differentiated THP-1 Cells a Reliable Substitute for Blood-Derived Macrophages When Studying in Vitro Polarization?, Front Pharmacol 9 (2018) 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lilienthal I, Herold N, Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in Osteosarcoma: A Review of Current and Future Strategies, Int J Mol Sci 21(18) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Smrke A, Anderson PM, Gulia A, Gennatas S, Huang PH, Jones RL, Future Directions in the Treatment of Osteosarcoma, Cells 10(1) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Morphological comparison of THP-1-derived macrophages. (A) Representative epifluorescent microscopy images of M0, M1, and M2 macrophages derived from THP-1 in monolayers co-stained with phalloidin (actin, red) and Hoechst (nuclei, blue). M1 macrophages were polarized with INFγ and LPS. M2 macrophages were polarized with IL-4 and IL-13. Scale = 25 μm. (B) Representative confocal microscopy images of THP-1 cells after 48 h PMA treatment on electrospun scaffolds co-stained with phalloidin (actin, green) and Hoechst (nuclei, blue). Scale = 50 μm. Images are taken from a single plane on the surface of the scaffold.
Supplemental Figure 2: Cell proliferation over five days. (A) Proliferation of THP-1 macrophages in electrospun scaffolds. Data shown as mean ± SD of mass of dsDNA per 6 mm diameter scaffold (n = 6 per scaffold type at each time point). (B) Cell growth as measured by dsDNA content in OS, OS/TAM 3:1, and OS/TAM 1:1 co-cultures in different electrospun scaffolds after 5 days. Data shown as mean + SD (n = 6). Two-way ANOVA with post-hoc Tukey’s HSD, ***p < 0.001, ****p < 0.0001.
Supplemental Figure 3: Raw (non-bootstrapped) and simulated (bootstrapped) measurements of secreted cytokines from OS and TAM cultures in electrospun scaffolds. Top: Individual measurements within the standard curve range for (A) TNFα (n = 3) and (B) IL-6 (n = 4); bars represent mean ± SD. Measurements below the standard curve are not displayed. Bottom: An example of bootstrapped cytokine secretion data. Each point represents an individual value; filled points represent measured data (also shown in A and B) and outlined points represent bootstrapped values for (C) TNFα (n = 3) and (D) IL-6 (n = 4); bars represent mean + SD. Two-way ANOVA with post-hoc Tukey’s HSD, p < 0.05. Groups that do not share the same letter are statistically different.
Supplemental Figure 4: Population selection for flow cytometry analysis. (A) Representative histograms of CD90 expression in MG-63 OS cells across different scaffolds. (B) Example of gating for high YAP and low YAP cells in OS/TAM co-cultures. Data shown are from cells grown on PCL scaffolds. (1) Analysis was conducted on single cells expressing CD90 and CD11b (Q2). (2) Cells were found to be in two distinct populations expressing either high YAP (Q2) or low YAP (Q3). (3) The geometric means of Alexa Fluor 647-conjugated anti-YAP and Alexa Fluor 594-conjugated anti-TAZ were measured in the two quadrants of interest. High YAP cells were taken to be OS cells and low YAP cells were taken to be TAMs. (C) Representative scatter plots (top) and histograms (bottom) of different culture conditions showing that high YAP cells in OS/TAM co-culture (purple) align with OS only cultures (green) and low YAP cells in OS/TAM co-culture align with TAM only cultures (yellow). Data shown are from cells grown on PCL scaffolds.
Supplemental Figure 5: YAP and TAZ expression by flow cytometry. Representative scatter plots showing expression of (A) YAP and (B) TAZ across different culture conditions. Osteosarcoma cells and macrophages were identified by the differential expression of YAP. Osteosarcoma cells were observed to have bimodal CD90 and high YAP (upper right quadrant in OS and OS/TAM 1:1 rows) and macrophages were observed to have low YAP (lower right gate in OS/TAM 1:1 and TAM rows). Measurements of TAZ in OS/TAM co-culture were taken after applying the gates for YAP as described in Supplemental Figure 4B.
Supplemental Figure 6: Response of homotypic osteosarcoma cell culture to doxorubicin (DOX) and combination therapy with Stattic. (A) Fitted dose-response curves of osteosarcoma cells to DOX (left) and DOX with 5.1 μM Stattic (right). Data shown as mean ± SD (n = 4 for each scaffold and drug combination). (B) Calculated IC50 (points) and 95% CI (bars) of osteosarcoma cells to doxorubicin (left) and doxorubicin with Stattic (right).
Supplemental Figure 7: Effect of STAT3 inhibitory compound (Stattic) on overall cell viability. Cell viability in OS (left) and OS/TAM (right) cultures without drug or with 5.1 μM Stattic was measured by WST-1 after 72 h of culture on electrospun scaffolds. Cell viability is shown normalized to the no drug condition. Data shown as mean + SD (n = 4). Multiple paired t tests with Holm-Šídák multiple comparisons, *p < 0.05. Comparisons not shown are not statistically significant.
Data Availability Statement
The raw and processed data required to reproduce these findings are available upon request.