

Abstract
Replication stress is a major cause of genomic instability and a crucial vulnerability of cancer cells. This vulnerability can be therapeutically targeted by inhibiting kinases that coordinate the DNA damage response with cell cycle control, including ATR, CHK1, WEE1 and MYT1 checkpoint kinases. In addition, inhibiting the DNA damage response releases DNA fragments into the cytoplasm, eliciting an innate immune response. Therefore, several ATR, CHK1, WEE1 and MYT1 inhibitors are undergoing clinical evaluation as monotherapies or in combination with chemotherapy, poly[ADP-ribose]polymerase (PARP) inhibitors, or immune checkpoint inhibitors to capitalize on high replication stress, overcome therapeutic resistance and promote effective antitumour immunity. Here, we review current and emerging approaches for targeting replication stress in cancer, from preclinical and biomarker development to clinical trial evaluation.
One of the most striking characteristics of cancer cells is their ability to sustain chronic proliferation1. For these cells, maintaining DNA integrity during DNA replication is a highly complex task that requires the coordination of DNA replication fork progression, which in turn requires an adequate supply of deoxyribonucleotides, functional DNA repair mechanisms and intact cell cycle checkpoints2. Cancer cells have alterations in many of these processes, which results in obstacles to replication fork progression, slowing down of the replication fork and impaired DNA synthesis. If left unresolved, these obstacles result in detachment of the DNA replication machinery from the DNA fibre, a result known as replication fork collapse3. Replication stress is traditionally defined as the slowing or stalling of replication fork progression2, but this is an evolving concept and other definitions are present in the literature4.
As replication stress is higher in cancer cells, these cells depend on replication stress response pathways for survival3. If the cellular response to replication stress is ineffective, cells may enter mitosis with an excess of DNA error, leading to genomic instability or even cell death owing to mitotic catastrophe2,3,5. Indeed, replication stress is one of the leading causes of genomic instability in cancer cells2,3,5, and targeting replication stress has enabled the identification of novel cancer vulnerabilities6.
Cellular response to replication stress comprises the activation of DNA damage tolerance pathways and the activation of the ataxia telangiectasia mutated (ATM)- and Rad3-related (ATR) pathway. Since the description of ATR in 1996 (REFS.7,8), several protein interactions responsible for the cellular response to replication stress have been elucidated9. Similarly, new techniques that enable the direct measurement of nucleotide incorporation into the DNA, such as the DNA fibre assay, have unravelled replication fork dynamics under replication stress. Moreover, large-scale genomic studies have highlighted the genomic disorganization of tumour cells and described potential molecular mechanisms of replication stress9. In this regard, many types of cancer commonly harbour genomic alterations associated with replication stress (such as amplification of CCNE1, which encodes G1/S-specific cyclin E1), including high-grade serous ovarian carcinoma (HGSOC), high-grade serous endometrial carcinoma (HGSEC), non-small-cell lung cancer (NSCLC), triple-negative breast cancer (TNBC), pancreatic cancer and gastric cancer. HGSOC is an excellent tumour model of high replication stress because almost all cases exhibit loss of cell cycle regulation, oncogene activation and/or DNA repair defects10.
In this Review, we discuss the intrinsic and extrinsic causes of replication stress, outline the biological mechanisms involved in the replication stress response, and review current and emerging approaches for targeting replication stress to induce cancer cell lethality, overcome drug resistance and facilitate effective antitumour immunity.
Response to replication stress
Any obstacle that causes fork slowing or fork stalling can lead to replication stress. In normal cells, the most frequent causes of replication stress include endogenous replicative errors caused by reactive oxygen species (ROS), misincorporation of ribonucleotides, and secondary DNA structures such as hairpins, DNA triplexes and quadruplexes formed in DNA regions with specific base repeats. In addition, replication stress can result from collisions between the replication and transcription machineries and from rehybridization of nascent mRNA to DNA, forming R-loops2 (FIG. 1).
Fig. 1 |. Illustration of causes of replication stress.
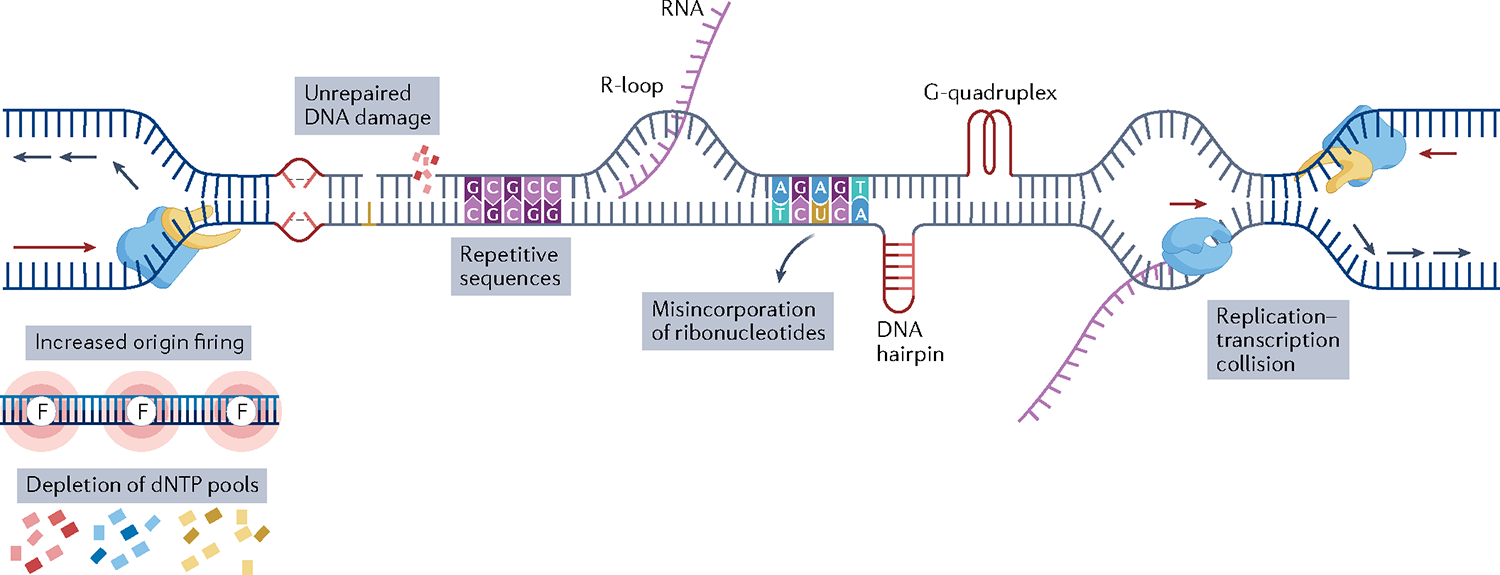
A replication fork consists of an enzyme with helicase activity (shown in yellow) and an enzyme with polymerase activity (shown in blue). Replicative stress results from endogenous or exogenous obstacles to DNA replication. The red arrows indicate the direction of the continuous DNA synthesis on the leading strand and the blue arrows indicate the direction of the non-continuous DNA synthesis on the lagging strand. Oncogenic activation and some chemotherapeutic agents such as gemcitabine cause depletion of deoxynucleotides (dNTPs), which impairs the progression of ongoing DNA replication. Increased origin firing (shown by pink circles labelled with the letter F) may be caused by oncogenic activation or loss of tumour suppressor genes. Different types of DNA damage caused by endogenous and exogenous agents lead to replication stress if left unrepaired. Repetitive DNA sequence, rehybridization of the nascent RNA to DNA, forming R-loops, misincorporation of ribonucleotides and secondary DNA structures such as hairpins and quadruplexes pose a challenge to DNA replication and are endogenous causes of replication stress even in healthy cells. Finally, collisions between replication and transcription machinery result in replication stress. RNA polymerase is shown as the blue protein and the newly synthesized RNA molecule is shown in purple.
In cancer cells, unrepaired DNA lesions are among the most frequent causes of replication stress and pose a physical barrier to fork progression. Depletion of nucleotide pools, caused by chemotherapeutic agents such as gemcitabine and 5-fluorouracil, can also limit DNA replication and fork progression. Additionally, activation of oncogenes can lead to depletion of nucleotide pools and collisions between the replication fork and transcription complexes. Finally, loss of the G1/S checkpoint leads to the accumulation of DNA errors and early entry into S phase2,5.
ATR kinase occupies a central role in the cellular response to replication stress. Replication protein A (RPA) is the first sensor of this pathway. RPA binds to single-stranded DNA (ssDNA) at the stalled fork and recruits ATR through the partner ATR-interacting protein (ATRIP). ATR is then activated by DNA topoisomerase 2-binding protein 1 (TOPBP1), activating CHK1 through phosphorylation, thus triggering the ATR–CHK1 pathway. ATR–CHK1 pathway activation leads to cell cycle arrest in S–G2, and to activation of the homologous recombination repair (HRR), inter-strand crosslink and nucleotide excision repair (NER) DNA repair pathways. Importantly, ATR–CHK1 pathway also results in replication fork stabilization and suppression of dormant replication origin firing, ensuring enough supply of deoxynucleotides (dNTPs)11,12 (FIG. 2).
Fig. 2 |. Schematics of the ATR pathway.
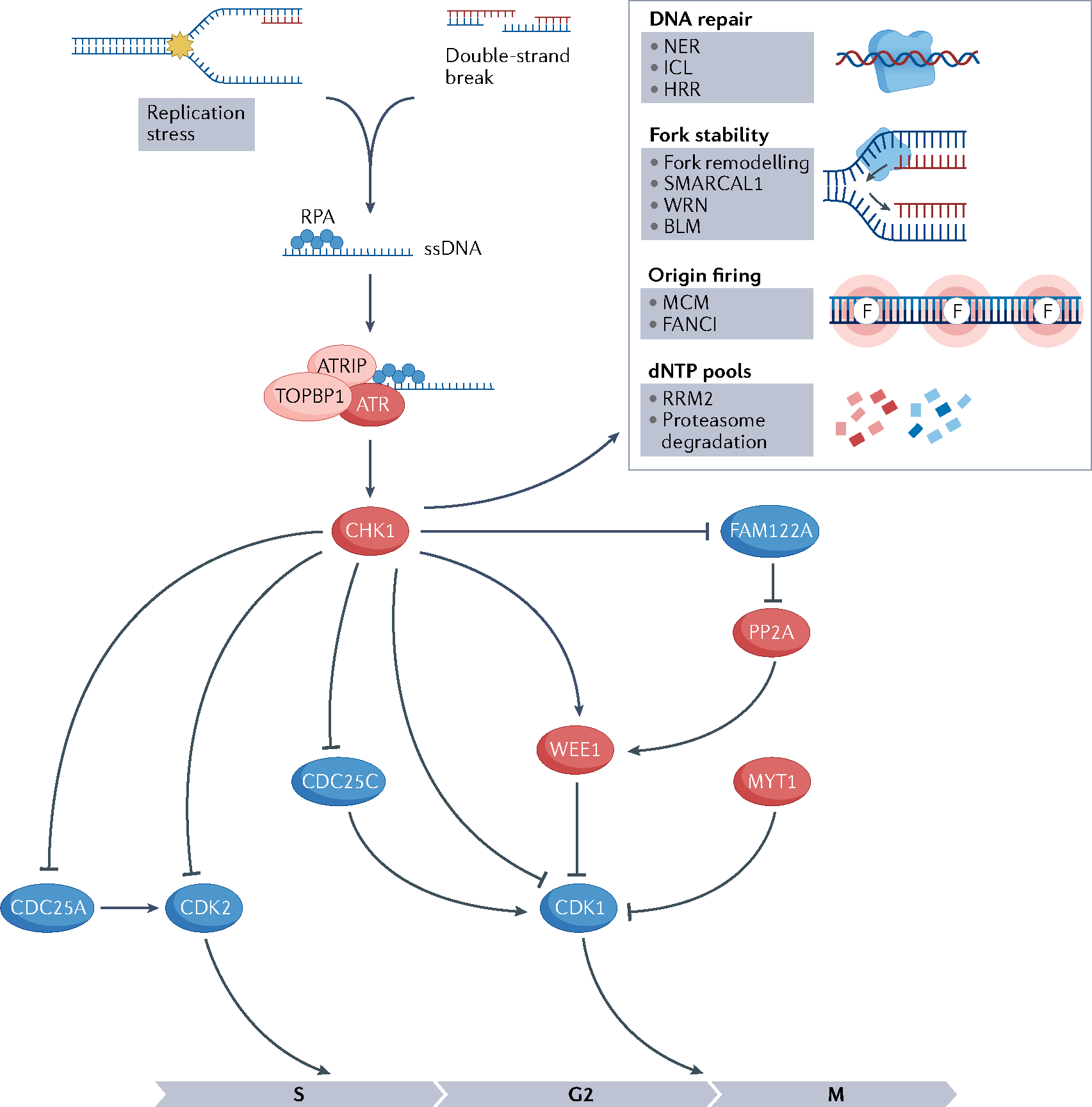
Activation of ataxia telangiectasia mutated (ATM) and Rad3-related (ATR) pathway because of replication stress or double-strand breaks results in cell cycle arrest, activation of DNA repair pathways, fork stabilization, inhibition of origin firing, decrease in deoxynucleotide (dNTP) degradation and increase in dNTP synthesis. Barriers to replication fork progression (shown by the yellow star) lead to the uncoupling of helicases and polymerases, generating single-stranded DNA (ssDNA) at the stalled fork. Replication protein A (RPA) binds to ssDNA at the stalled fork and recruits ATR through the partner ATR-interacting protein (ATRIP). ATR is then activated by DNA topoisomerase 2-binding protein 1 (TOPBP1), activates CHK1 through phosphorylation and triggers the ATR–CHK1 pathway. CHK1 mediates cell cycle arrest in the S–G2 phase through phosphorylation and reduction of cyclin-dependent kinase 2 (CDK2) activation. CDK1 is negatively regulated via phosphorylation by WEE1 and MYT1 kinases and positively regulated by CDC25 phosphatases. CHK1 phosphorylates and inactivates CDC25C, activates WEE1 and promotes the degradation of CDC25A11,13. FAM122A is a negative regulator of the G2/M checkpoint and forms a complex with the phosphatase PP2A. When CHK1 phosphorylates FAM122A, PP2A is disinhibited and dephosphorylates WEE1, preventing its degradation and activating the G2/M checkpoint by WEE1. Proteins whose activity leads to cell cycle arrest are shown in red and proteins whose activity leads to cell cycle progression are shown in blue. Red DNA strands indicate newly synthesized DNA. Pointed arrows indicate action on that protein (phosphorylation) and blunted arrows indicate inhibition. BLM, Bloom syndrome protein; HRR, homologous recombination repair pathway; ICL, inter-strand crosslink pathway; MCM, minichromosome maintenance; NER, nucleotide excision repair pathway; RRM2, ribonucleoside-diphosphate reductase subunit M2; WRN, Werner syndrome ATP-dependent helicase.
CHK1 mediates cell cycle arrest in the S–G2 phase through reduction of cyclin-dependent kinase 2 (CDK2) activation, thus slowing down DNA replication in S phase. CHK1 also limits mitotic entry through phosphorylation of cyclin-dependent kinase 1 (CDK1). CDK1 is negatively regulated via phosphorylation by WEE1 and membrane-associated tyrosine- and threonine-specific cdc2-inhibitory kinase (PKMYT1, also known as MYT1) kinases and positively regulated by CDC25 phosphatases. CHK1 activates WEE1 and promotes the degradation of CDC25A11,13.
Replication fork plasticity is a fundamental feature of the replication stress response. To ensure that obstacles in DNA replication do not result in loss of genetic information, cells develop DNA damage repair and tolerance mechanisms to ensure fork progression. Thus, the stalled fork can reverse its direction (fork reversal) (FIG. 3) or replicate over lesions. Fork reversal slows down the replication fork, shifts the position of the replication fork backwards, thus repositioning the DNA damage in a double-stranded DNA segment, and enables an error-free DNA repair, whereas DNA damage tolerance pathways, such as translesion synthesis or repriming, lead to replication fork progression through the DNA lesion in an error-prone manner. In the case of translesion synthesis, specialized translesion synthesis polymerases can synthesize the daughter strand through the lesion owing to a more flexible active polymerase site and the lack of exonucleolytic proofreading, whereas in the case of repriming, DNA-directed primase/polymerase protein (PRIMPOL) can restart DNA synthesis immediately after the lesion without removing it, leaving a ssDNA gap behind it14. The mechanisms involved in the choice of pathway of fork recovery are not completely understood, but ATR orchestrates many of them, leading to the final result of fork stability11. On the one hand, ATR activation causes a global response to fork reversal through RAD51–ZRANB3 activity to slow DNA replication in response to inter-strand crosslinks in BRCA1-proficient cells14. On the other hand, ATR limits fork reversal promoted by the chromatin remodeller SMARCAL1 and modulates the fork remodelling proteins Bloom syndrome protein (BLM) and Werner syndrome ATP-dependent helicase (WRN), thereby limiting excessive fork cleavage and facilitating fork restart11. ATR also has a role in promoting PRIMPOL repriming of stalled forks14. These seemingly contradictory effects of ATR in replication fork reversal may be related to different types of DNA damage eliciting ATR activation in the context of specific genomic backgrounds and constitute an area of active research.
Fig. 3 |. Fork dynamics regulation by the ATR pathway.
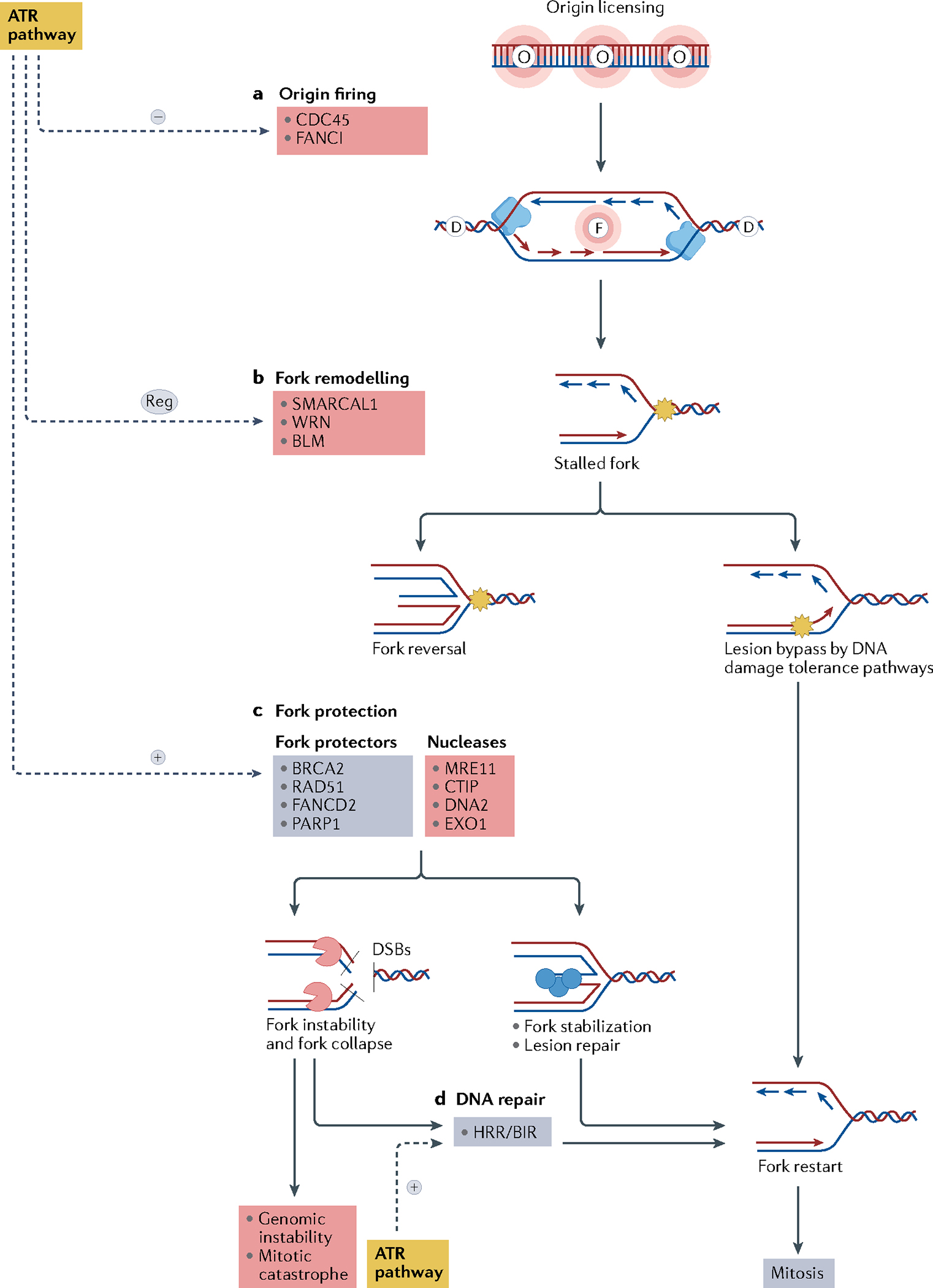
a | The ataxia telangiectasia mutated (ATM) and Rad3-related (ATR) pathway inhibits replication fork origin firing through inhibition of the minichromosome maintenance 2–7 complex (MCM2–7) and Fanconi anaemia group I protein (FANCI). Replication origin firing is indicated by the letter F, and dormant replication origins — licensed but not activated — are indicated by the letter D. Replisome (blue), is a protein complex with helicase and polymerase activities that progresses bidirectionally, starting from the origin firing and forming the replication forks. b | Stalled forks owing to an obstacle to fork progression (shown by the yellow star) may be rescued directly by DNA damage tolerance pathways or may be reversed by fork remodelling proteins. Fork reversal may result in fork protection or fork degradation depending on the balance of activity of various remodelling factors and on the DNA repair proficiency of the cell. c | The ATR pathway modulates fork reversal and activates fork protectors, promoting fork stabilization. Reversed forks are substrates for nucleases that cleave the DNA to form double-strand breaks (DSBs). Fork protectors such as BRCA2, RAD51 and poly[ADP-ribose]polymerase 1 (PARP1) prevent fork degradation by nucleases, allowing lesion repair and fork restart. d | Collapsed replication forks may be repaired by the homologous recombination repair (HRR) and the break-induced replication (BIR) pathways, allowing fork restart and normal mitosis. If DNA repair is not effective, fork collapse may lead to genomic instability and mitotic catastrophe. Mechanisms that lead to fork stabilization and restart and normal mitosis are shown in blue, mechanisms that lead to fork instability, fork collapse and genomic instability are shown in red. BLM, Bloom syndrome protein; CDC45, cell division cycle 45; WRN, Werner syndrome ATP-dependent helicase.
Prevention of the depletion of dNTP supply is another feature of the replication stress response. ATR–CHK1 pathway activation ensures dNTPs supply by suppressing global dormant replication origin firing through phosphorylation of FANCI and the inhibition of cell division cycle 45 (CDC45) recruitment to the minichromosome maintenance 2–7 complex (MCM2–7)13 (FIG. 3). Replication origins close to the stalled forks are subsequently activated to rescue the fork. In addition, ATR pathway activation leads to upregulation of the cellular dNTP supply by decreasing degradation of dNTPs through proteasome inhibition and increasing dNTP synthesis through upregulation of transcription of ribonucleoside-diphosphate reductase subunit M2 (RRM2), which is primarily mediated by WEE1 activity11,13,15. Finally, WEE1 activation also contributes to fork stability by inhibiting nucleases such as MUS81, preventing fork degradation16.
The essential function of the ATR–CHK1–WEE1 pathway in replication stress response is highlighted by the extremely low rates of loss-of-function genomic alterations involving this pathway in various cancer types (<3% for ATR and <1% for CHK1 and WEE1)17. Of note, mouse models with severe reduction of ATR function are resistant to development of tumours11.
Replication stress in cancer
Sources of replication stress in cancer cells include loss of G1/S checkpoint (for example, owing to deleterious TP53 mutations), premature entry into S phase (due to RB1 loss, CCNE1 amplification or FBXW7 loss), oncogenic drive (oncogene-related replication stress such as that due to KRAS activating mutations or MYC amplification) and DNA repair deficiencies (such as HRR or NER pathway deficiency)9. It is important to underscore that different mechanisms may lead to varying levels of replication stress, also depending on the specific genomic context. For example, oncogenic activation or CCNE1 amplification results in a higher level of replication stress compared with loss of function of p53 or RB1. Moreover, some of these mechanisms, such as RB1 loss, may lead to genomic instability by means other than replication stress18.
Oncogene activation from KRAS, CCNE1 or MYC can lead to replication stress through the increase in replication origin firing, thus resulting in more topological stress and depletion of nucleotide pools. Oncogene activation also increases transcription, leading to more transcription–replication collisions, and generates ROS through changes in cell metabolism19,20. Cyclin E1 regulates the transition from G1 to S phase through binding and activation of CDK2. It also increases replication origin firing while also causing deficiency in pre-replication complex formation. For these reasons, cancers with cyclin E1 overexpression are among those with the highest level of replication stress19,21.
HGSOC is an excellent tumour model of high replication stress (Supplementary Fig. 1). Almost all HGSOCs bear TP53 mutations, whereas CCNE1 amplification occurs in 20% of cases, biallelic RB1 loss in 10% and CDKN2A mRNA downregulation in 32%, all leading to premature entry into S phase. Of note, low CDKN2A mRNA expression is mutually exclusive with CCNE1 amplification and RB1 deletion and/or mutation events, reflecting an extreme selection pressure for loss of RB pathway regulation and rapid G1 to S phase entry in HGSOC10. MYC amplification is present in 40% of cases of HGSOC, NF1 loss in 10% and KRAS amplification and/or mutation in 11%, all leading to oncogene-driven replication stress. Finally, HRR alterations are present in about 50% of cases and NER alterations in 4–8%, leading to accumulation of DNA lesions10. Most chemotherapeutic agents used to treat HGSOC, such as gemcitabine, topotecan and platinum salts, are effective, at least in part, because of their ability to further aggravate pre-existing replication stress in these tumours22 (Supplementary Table 1).
Several other cancers bear different genomic alterations that can lead to a high degree of replication stress. HGSEC shares many molecular features with HGSOC, although it is associated with oncogene-induced replication stress (due to ERBB2 amplification and FBXW7 mutations) even more frequently than HGSOC but is less commonly associated with HRR deficiency (Supplementary Fig. 1). In addition, NSCLC, TNBC, pancreatic and gastric cancers commonly exhibit genomic alterations associated with a high degree of replication stress (Supplementary Fig. 1). Human papillomavirus (HPV)-related cancers such as head and neck squamous cell (HNSCC) and cervical cancers also exhibit high levels of replication stress due to loss of RB1 and p53 control of G1/S checkpoint caused by HPV E6 and E7 proteins as well as other mechanisms23.
Targeting replication stress
Cytotoxic agents and replication stress
Most chemotherapeutic agents that are used to treat cancer patients induce replication stress through various mechanisms (FIG. 4). Platinum salts and alkylating agents generate intra-strand and inter-strand crosslinks. Intra-strand crosslinks are bypassed by translesion synthesis polymerases or repaired via NER, whereas inter-strand crosslinks cause fork stalling and require a synchronized response of several pathways, including NER, base excision repair (BER), HRR and Fanconi anaemia (FA) pathways24 (BOX 1). Topoisomerase 1 inhibitors, such as topotecan and irinotecan generate topoisomerase 1–DNA cleavage complexes that block replication by forming a barrier to fork progression9. Nucleoside analogues, such as gemcitabine, promote replication stress through two different mechanisms. First, they are directly incorporated into DNA during replication, leading to fork stalling. Secondly, they reduce intracellular nucleotide pools through inhibition of the enzymes responsible for dNTP synthesis (gemcitabine inhibits ribonucleotide reductase, whereas 5-fluorouracil inhibits thymidylate synthetase)25.
Fig. 4 |. Drug combinations with ATR–CHK1–WEE1 inhibitors.
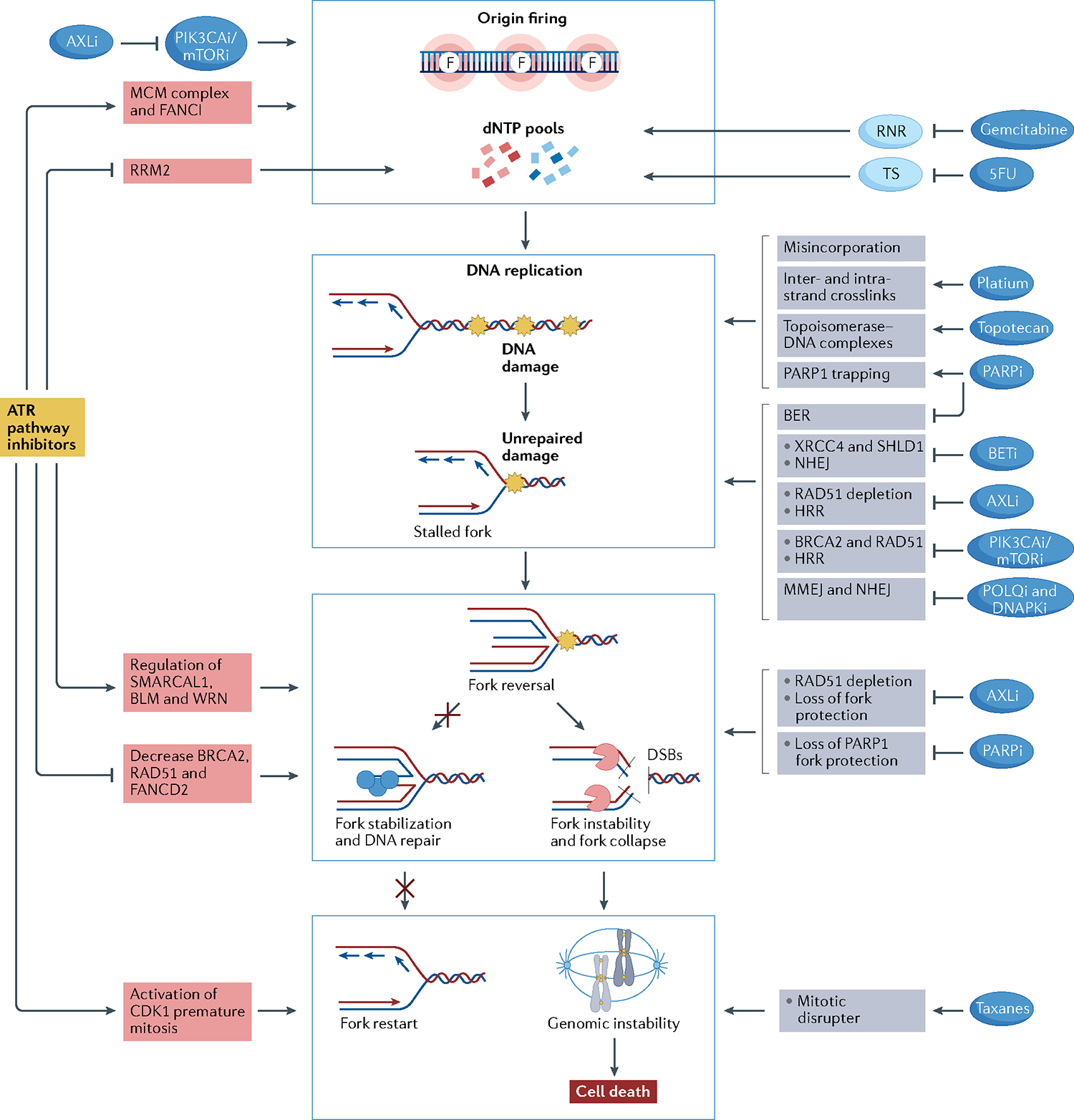
Rationale for drug combinations that promote replication stress, potentiating the effect of ATR–CHK1–WEE1 inhibitors. Red boxes indicate mechanisms of action related to ataxia telangiectasia mutated (ATM) and Rad3-related (ATR) pathway inhibitors; grey boxes indicate mechanisms of action of the other drugs. Central blue squares group the main mechanisms through which drugs may activate replication stress. Drugs may activate replication stress by decreasing deoxynucleotide (dNTP) pools directly or through increasing origin firing (indicated by the letter F, top blue box), increasing DNA damage (second blue box), promoting fork instability (third blue box) and disrupting mitosis (bottom blue box). ATR inhibitors (ATRi), CHK1 inhibitors (CHK1i) and WEE1 inhibitors (WEE1i) decrease dNTP pools by activating origin firing through loss of Fanconi anaemia group I protein (FANCI) phosphorylation and minichromosome maintenance 2–7 complex (MCM2–7) inhibition and by decreasing ribonucleoside-diphosphate reductase subunit M2 (RRM2) expression. PIK3CA/mTOR inhibitors synergize with ATRi, CHK1i or WEE1i by promoting origin firing through increasing cell division cycle 45 (CDC45) expression, which leads to activation of the MCM2–7 complex. Moreover, AXL inhibitors (AXLi) decrease dNTP pools by inhibiting the PI3K–mTOR pathway. Nucleoside analogues reduce intracellular dNTP pools by inhibiting the enzymes responsible for dNTP synthesis. Gemcitabine inhibits ribonucleotide reductase (RNR), and 5-fluorouracil (5FU) inhibits thymidylate synthetase (TS). Drugs may increase DNA damage by directly damaging the DNA or by inhibiting DNA repair mechanisms. Gemcitabine and 5FU lead to DNA damage by their misincorporation into DNA. Platinum salts cause inter- and intra-strand crosslinks, and topotecan generates topoisomerase–DNA complexes, which become obstacles to fork progression. Poly[ADP-ribose]polymerase (PARP) inhibitors (PARPi) create obstacles to fork progression through PARP1 trapping and inhibition of the base excision repair (BER) pathway. Bromodomain and extra-terminal domain (BET) inhibitors (BETi) lead to non-homologous end-joining (NHEJ) inhibition through downregulation of XRCC4 and SHLD1. AXL inhibition results in inhibition of homologous recombination repair (HRR) through RAD51 depletion. PIK3CA/mTOR inhibition leads to HRR inhibition through downregulation of BRCA2 and RAD51. POLQ inhibitors and DNAPK inhibitors directly inhibit microhomology-mediated end-joining (MMEJ) and NHEJ pathways, respectively. ATRi, CHK1i and WEE1i cause fork instability by regulating fork remodellers such as SMARCAL1, Bloom syndrome protein (BLM) and Werner syndrome ATP-dependent helicase (WRN) and by decreasing the activity of fork protectors such as BRCA2, RAD51 and FANCD2. AXLi and PARPi also lead to fork instability by depletion of RAD51 and inhibition of PARP1 fork protection, respectively. Finally, ATRi, CHK1i and WEE1i lead cells to premature mitosis and may synergize with mitotic disrupter agents such as taxanes.
Box 1 |. Summary of DNA repair pathways.
Base excision repair (BER):
The BER pathway is responsible for the repair of single-nucleotide lesions that usually do not lead to distortion in the double helix of DNA.
Nucleotide excision repair (NER):
NER is responsible for the repair of bulky adducts that cause helix distortions, and intra- and inter-strand DNA crosslinks.
Homologous recombination repair (HRR):
HRR is a high-fidelity repair pathway that has an essential role in double-strand break repair, replication fork stabilization and recovery of stalled or collapsed replication forks during DNA replication. It is active during S and G2 cell cycle phases and uses the sister chromatid as a template to rebuilt lost sequences in double-strand breaks.
Non-homologous end-joining (NHEJ):
NHEJ is a simpler, faster and more versatile response to double-strand breaks. It detects double-strand breaks, protects them from nuclease activity that would otherwise lead to other repair pathways, binds the double-strand break ends together and processes them to seal the break.
Fanconi anaemia (FA) pathway:
The FA pathway repairs inter-strand crosslinks in the genome. FA proteins work as sensors of inter-strand crosslinks, recruit the FA core complex proteins (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL), which activate a second FA complex (FANCI–FANCD2). Once activated, the FA complex cleaves the DNA damage site and generates two sister chromatids, one with a double-strand break that is repaired by HRR and one with an inter-strand crosslink that will be repaired by translesion DNA synthesis.
Translesion synthesis (TLS):
TLS is a DNA damage tolerance pathway. It rescues stalled forks, allowing fork progression using special error-prone polymerases that are able to synthesize DNA across a DNA damaged template.
Among targeted agents, poly[ADP-ribose]polymerase (PARP) inhibitors (PARPi) are potent inducers of replication stress via multiple mechanisms including inhibition of BER, which leads to impairment of single-strand break repair, PARP entrapment on the DNA, causing replication forks to stall, and inhibition of PARP activities in fork protection and restart26,27. PI3K–mTOR pathway inhibitors also induce replication stress by promoting new origin firing, leading to decelerated fork speed; they also attenuate DNA HRR activity by decreasing trans-nuclear localization of RAD51 and downregulating BRCA2 and RAD51 expression28. AXL is a receptor tyrosine kinase that promotes cell proliferation and survival via the RAS–MEK–ERK and the PI3K–AKT–mTOR pathways and drives epithelial to mesenchymal transition. Inhibition of AXL promotes DNA damage and replication stress, leading to activation and dependence on the ATR–CHK1 pathway29,30. AXL promotes resistance to chemotherapy and to many targeted therapies such as EGFR, ALK and BRAF inhibitors in lung cancer and HNSCC31,32. Bromodomain and extra-terminal domain (BET) inhibitors, which have been used to target MYC-driven cancers, also induce replication stress. Besides its role as transcription factor, BRD4, one of the BET family proteins, plays a part in the DNA damage response. BRD4 interacts with the pre-replication factor CDC6, regulating the activation of CHK1–WEE1 signalling and preventing the formation of R-loops. BET inhibitors (BETi) also promote double-strand breaks through inhibition of the non-homologous end-joining repair (NHEJ) pathway33–37.
More recently, the implication of DNA damage tolerance pathways such as translesion synthesis and repriming by PRIMPOL as alternative responses to replication stress has led to the development of inhibitors of these pathways14,38. Furthermore, the improved characterization of specific post-translational modifications of replication stress response factors through ubiquitylation and deubiquitylation has renewed the interest in drugs that target these processes, such as inhibitors of the deubiquitylating enzymes USP13 and USP1, which take part in ATR activation39 and translesion synthesis39 regulation, respectively, and inhibitors of the ubiquitylating enzyme TRAF4, which is involved in proper CHK1 activation40.
Given their ability to induce replication stress, all aforementioned agents represent ideal partners for synergistic combinatorial strategies with agents that target the replication stress response such as ATR inhibitors (ATRi), CHK1 inhibitors (CHK1i) and WEE1 inhibitors (WEE1i).
Replication stress and drug resistance.
Although tumours with HRR deficiency are highly sensitive to platinum salts and PARPi, acquired resistance commonly occurs via distinct mechanisms. First, tumours may restore HRR function through secondary somatic reversion mutations or re-expression of HRR-related genes that had been silenced through promoter hypermethylation. Second, HRR-deficient tumour cells may restore double-strand break end resection and HRR through loss of checkpoint proteins that inhibit end resection and promote NHEJ such as 53BP1, REV7 (also known as MAD2L2) or dynein light chain 1 (DYNLL1), or by upregulation of proteins that promote end resection and activate HRR such as TRIP13 and ubiquitin carboxyl-terminal hydrolase 15 (USP15)41–43. Third, HRR-deficient tumour cells can stabilize their replication forks through loss of PAX-interacting protein 1 (PTIP), which recruits end resection proteins, loss of SMARCAL1 or loss of Schlafen family member 11 (SLFN11)41,42. Fourth, mutations or post-translational modifications of PARP1 itself can lead to PARPi resistance41,42. Fifth, loss of expression of poly(ADP-ribose) (PAR) glycohydrolase (PARG) can lead to upregulation of PAR despite PARP1 inhibition44. Finally, the overexpression of the PARPi efflux pump MDR1 (REFS.41,42,44) can cause PARPi resistance (FIG. 5). Restoration of HRR function has a central role in PARPi resistance, whereas stabilization of the replication fork is important for platinum resistance45.
Fig. 5 |. Mechanisms of ATR–CHK1–WEE1 inhibitors to overcome PARP inhibitor resistance.
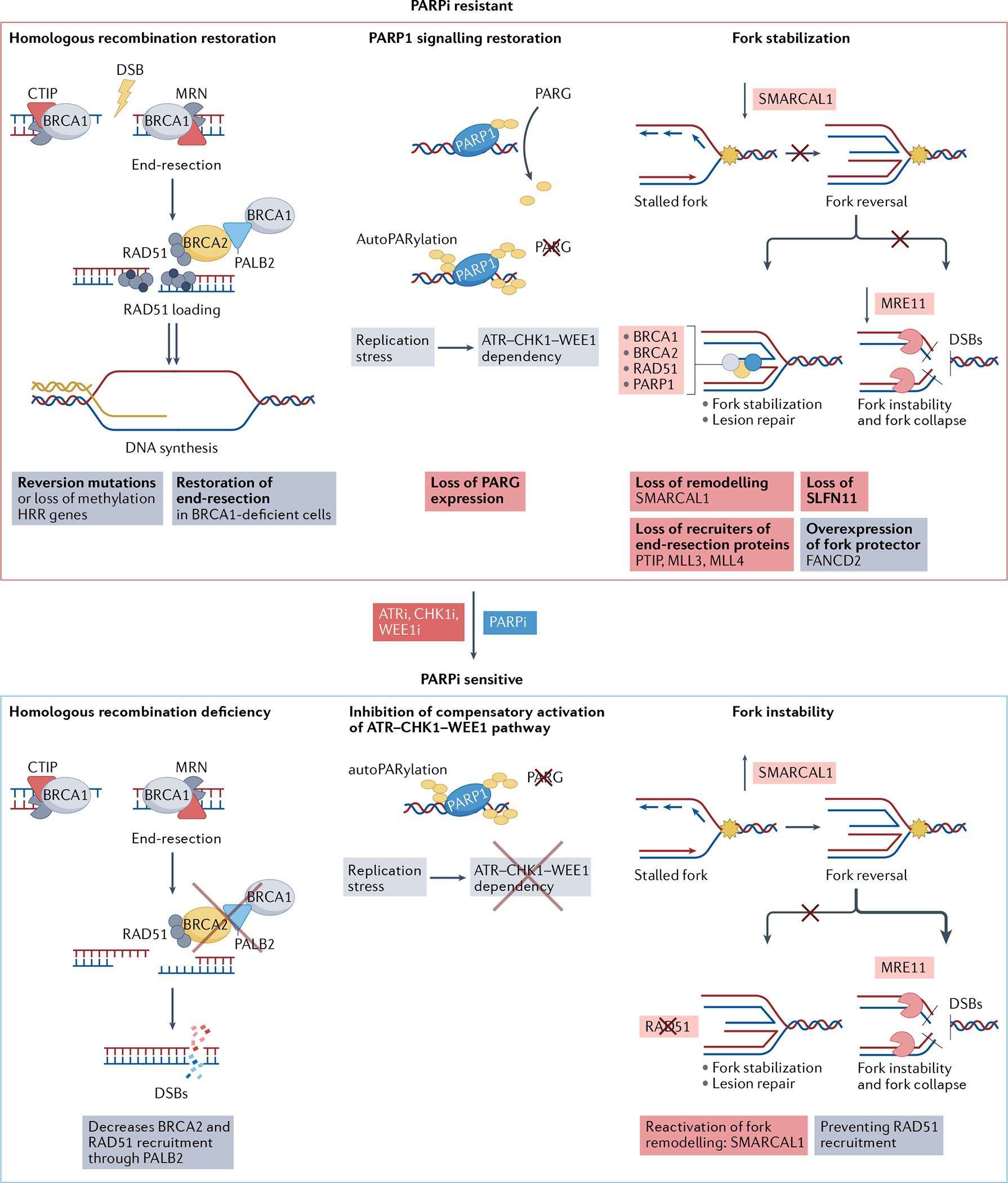
The top panel shows mechanisms of poly[ADP-ribose]polymerase (PARP) inhibitor (PARPi) resistance. The main steps of the homologous recombination repair (HRR) pathway are shown at the left: DNA end resection on double-strand breaks (DSBs), RAD51 loading, homologous strand invasion and DNA synthesis. HRR deficiency makes cells sensitive to PARPi. Reversion mutations in BRCA1 and BRCA2, and RAD51 paralogues can restore HRR. CtIP and MRN are nucleases responsible for initial DNA end resection. MRN is a nuclease complex formed by MRE11-RAD50-NBS1. Loss of proteins that inhibit DNA end resection, such as 53BP1 and shieldin complex proteins, or upregulation of proteins that promote end resection, such as TRIP13 and USP15, can restore end resection in BRCA1-deficient cells, promoting PARPi resistance. The central panel shows how poly(ADP-ribose) glycohydrolase (PARG) counteracts PARP1-mediated PARylation (shown in yellow). Accordingly, loss of expression of PARG can lead to upregulation of PAR despite PARP1 inhibition, increasing replication stress and activating the ATR–CHK1–WEE1 pathway. The top right panel shows a stalled fork undergoing fork reversal. Fork protectors (shown by the grey, yellow and blue circles) such as BRCA1, BRCA2, RAD51 and PARP1 stabilize the fork, allowing DNA repair and fork restart. In the absence of fork protectors, nucleases (shown in red) such as MRE11 can degrade the reversed forks. Accordingly, loss of fork remodelling factors such as SMARCAL1, loss of recruiters of end resection such as PTIP, MLL3 and MLL4, and overexpression of fork protectors such as Fanconi anaemia group D2 protein (FANCD2), can lead to fork stabilization and PARPi resistance. The bottom panels show how ataxia telangiectasia mutated (ATM) and Rad3-related inhibitors (ATRi), CHK1 inhibitors (CHK1i) and WEE1 inhibitors (WEE1i) can overcome PARPi resistance. ATRi, CHK1i and WEE1i can cause HRR deficiency by decreasing BRCA2 and RAD51 recruitment (bottom left panel). They can suppress activation of the ATR pathway, which may be overactivated by PARG loss (bottom centre panel), and promote fork instability by reactivating SMARCAL1 and preventing RAD51 recruitment to the reversed fork (bottom right panel). SLFN11, Schlafen family member 11.
Preclinical studies have demonstrated that ATRi, CHK1i and WEE1i may overcome PARPi resistance and platinum resistance, addressing different resistance mechanisms46–55. ATRi, CHK1i and WEE1i can promote HRR deficiency in cases in which HRR function is restored. In the cases in which stable replication forks drive PARPi resistance, ATRi, CHK1i and WEE1i can cause replication stress and replication fork instability as measured by the DNA fibre assay46,48–50. Interestingly, ATRi, CHK1i and WEE1i may also overcome primary PARPi resistance in BRCA1/2 wild-type tumour models, such as CCNE1-amplified cancers46,51. Finally, cells that become PARPi resistant through PARG downregulation exhibit high replication stress and dependence on the ATR–CHK1–WEE1 pathway for survival. Accordingly, PARG downregulation confers sensitivity to CHK1 and WEE1 inhibition56,57.
Loss of SLFN11 expression is a unique mechanism of PARPi and platinum resistance. SLFN11 is an executioner of replication stress, as it binds to RPA-coated ssDNA and permanently blocks replication independently of ATR, leading to cell death58. SLFN11 expression changes are dynamic during tumour evolution and loss of SLFN11 expression, mainly through methylation and silencing, has been associated with resistance to DNA-damaging agents such as PARPi, cisplatin, gemcitabine, topotecan, etoposide and doxorubicin58. Tumours with low expression of SLFN11 depend on the ATR–CHK1–WEE1 axis to tolerate replication stress and inhibition of this pathway resensitizes tumour cells, in vitro and in vivo, to both platinum and PARPi59,60. Taken together, ATR–CHK1–WEE1 inhibitors may overcome multiple mechanisms of de novo and acquired PARPi and platinum resistance.
Replication stress and immunotherapy
Combination of DNA-damaging agents with immunotherapy has recently yielded promising results. Indeed, PARPi can elicit an innate immune response by releasing DNA particles into the cytoplasm, activating the cGAS–stimulator of interferon genes protein (STING) pathway, and upregulating the type I interferon-γ response and, subsequently, the nuclear factor-κB (NF-κB) pathway61,62. Besides this pro-inflammatory signal, cGAS–STING can also upregulate programmed cell death 1 ligand 1 (PDL1) expression. Hence, there is a strong rationale for combining monoclonal antibodies against PD1 or PDL1 with PARPi61,62. However, although DNA damage checkpoint inhibitors may contribute to cGAS–STING activation through the induction of double-strand breaks63, some studies indicate that the ATR–CHK1 pathway also has a fundamental role in the PDL1 upregulation caused by DNA damage64,65. Indeed, at least one preclinical study demonstrated that ATR inhibition leads to PDL1 downregulation66. In that setting, although PDL1 downregulation could sensitize tumour cells to T cell-mediated killing, there may not be a synergism between DNA damage checkpoint inhibitors and PD1/PDL1 blockade. Moreover, at least one study using different cell lines demonstrated that inhibition of ATR, CHK1 or WEE1 increases cytoplasmic double-stranded DNA and activates the STING pathway but does not result in activation of a type I interferon response67.
However, other studies have demonstrated synergistic antitumour activity for combinations of DNA damage checkpoint inhibitors with PD1/PDL1 blockade68–71. In models of NSCLC, the addition of a CHK1i to PD1 inhibition was synergistic owing to activation of the cGAS–STING pathway and upregulation of PDL1 expression69. Remarkably, two other studies using syngeneic mouse and organoid models of HGSOC with CCNE1 amplification and KRAS mutation demonstrated a significant benefit from the combination of immunotherapy agents (anti-CTLA4 or anti-PDL1) with replication stress-inducing agents (the CHK1i prexasertib or gemcitabine), compared with the immunotherapy doublet or combinations of immunotherapy and other chemotherapies72,73.
In one study that addressed the various effects of PARPi and ATRi in activating the immune response and regulating PDL1 expression, both PARPi and ATRi activated the cGAS–STING pathway; however, ATR inhibition led to PDL1 protein degradation by activating the CDK1–speckle-type POZ protein (SPOP) pathway. Conversely, PARPi led to CDK1 inactivation and did not activate the CDK1–SPOP pathway, therefore having no effect on PDL1 degradation. By downregulating PDL1, ATR inhibition derepressed an IFNβ-mediated pro-apoptotic pathway, the cGAS–STING–IFNα/β receptor (IFNAR1) pathway, resulting in autocrine cytotoxic signalling. Whether these results are tumour specific or restricted to ATRi, compared with CHK1i or WEE1i, remains to be clarified74 (FIG. 6).
Fig. 6 |. Mechanism of immune response sensitization.
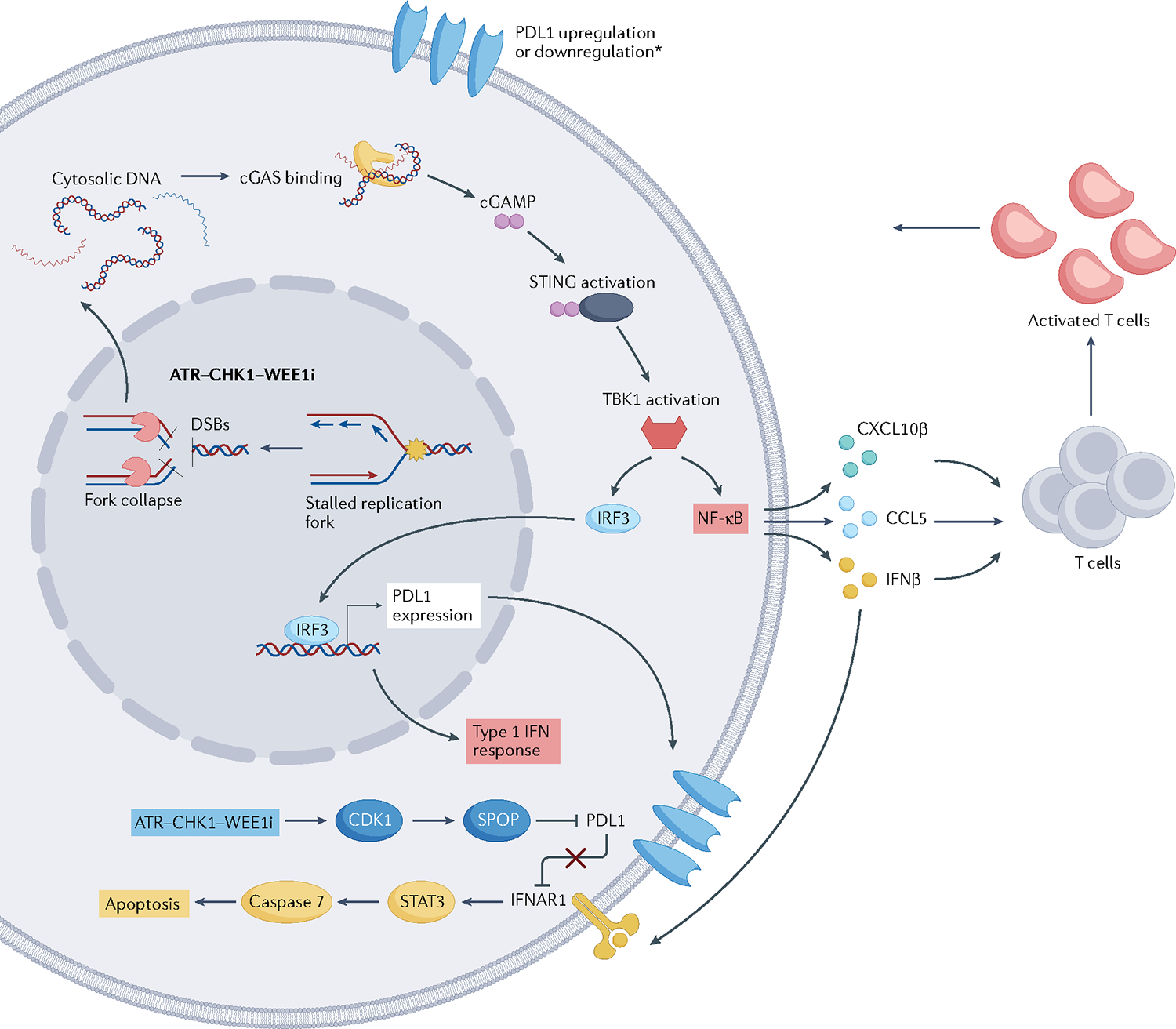
Replication stress can elicit an innate immune response by releasing DNA particles into the cytoplasm, activating the cGAS–stimulator of interferon genes (STING) pathway, and upregulating the type I interferon-γ (IFNγ) response and, subsequently, the nuclear factor-κB (NF-κB) pathway. Besides this pro-inflammatory signal, cGAS–STING can also upregulate PDL1 expression. In addition, the ATR–CHK1 pathway can also lead to PDL1 protein degradation by activating the CDK1–speckle-type POZ protein (SPOP) pathway. Downregulation of PDL1 can inhibit an IFNβ-mediated pro-apoptotic pathway, resulting in autocrine cytotoxic signalling. IFNAR1, interferon-α/β receptor 1. *The resulting effect of ATR-CHK1-WEE1 inhibition on PD-L1 is disputable with studies showing both upregulation or downregulation upon blockage of the ATR-CHK1-WEE1 pathway.
A different strategy is to combine ATR, CHK1 and WEE1 inhibition with radioimmunotherapy. Although radiotherapy stimulates an immune response against tumour cells, it also upregulates PDL1 expression and increases tumour infiltration of regulatory T (Treg) cells through the activation of DNA damage repair pathways75. In a hepatocellular carcinoma model, the addition of ATRi promoted CD8+ T cell infiltration and activity, reduced infiltration of Treg cells, and increased memory T cell infiltration76. Thus, triple therapy with radiotherapy, immune checkpoint inhibitors and ATRi prolonged survival and delayed recurrence in tumour-bearing mice.
Taken together, these studies highlight the active role of replication stress and DNA damage response in immunomodulation and suggest that there may be synergism between DNA damage checkpoint and immune checkpoint blockade, although this synergism seems to be context specific.
ATRi, CHK1i, WEE1i and MYT1i
ATR–CHK1–WEE1 inhibitors are potent and selective agents when used as single agents. Their primary mechanism of action is to increase origin firing and to inhibit the S phase and G2/M cell cycle checkpoints, which leads to enhanced replication stress and premature entry to mitosis77–83. It also leads to impairment of HRR during the S and G2 phases and induction of double-strand breaks83–91.
Early studies hypothesized that cells that bear TP53 mutations would be more sensitive to ATR–CHK1–WEE1 inhibition. ATRi, CHK1i and WEE1i have shown selective efficacy in p53-deficient cells92–95, but this effectiveness is irrespective of p53 status90,91,96–98. The development of these drugs then followed two paths, the first searching for predictive biomarkers to identify tumour cells that are particularly sensitive to monotherapy and the second testing various drug combinations. Several compounds that target ATR, CHK1 and WEE1 are currently in preclinical and clinical development (TABLE 1 and Supplementary Tables 2, 3 and 4).
Table 1 |.
Selected ongoing clinical trials with ATR, CHK1 and WEE1 inhibitors according to strategy
| Design | Target | Agents | Indication | Status | Study Identifier |
|---|---|---|---|---|---|
| Biomarker driven monotherapy trials | |||||
| Phase II | CHK1 | Prexasertib | Advanced solid tumours with replication stress or homologous recombination deficiency | Active, not recruiting | NCT02873975 |
| Phase II | WEE1 | Adavosertib | Advanced refractory solid tumours | Suspended | NCT03253679 |
| Combination with chemotherapy | |||||
| Phase I | ATR | Gemcitabine, carboplatin and berzosertib | Platinum-sensitive ovarian cancer | Recruiting | NCT02627443 |
| Phase I | ATR | Gemcitabine and BAY1895344 | Incurable solid tumours; expansion cohort platinum-resistant ovarian cancer | Not yet recruiting | NCT04616534 |
| Phase I | WEE1 | Gemcitabine, nab-paclitaxel and adavosertib | Unresectable pancreatic adenocarcinoma | Active, not recruiting | NCT02194829 |
| PARPi synergism | |||||
| Phase II | ATR | Ceralasertib and olaparib | Recurrent HGSOC irrespective of platinum sensitivity | Recruiting | NCT03462342 (CAPRI trial) |
| Phase II | ATR | Ceralasertib, olaparib | Recurrent gynaecological cancers, including CCC and rare gynaecological cancer subtypes with frequent ARID1A loss | Recruiting | NCT04065269 (ATARI trial) |
| Phase II | ATR; WEE1 | Olaparib alone; olaparib and capivasertib; olaparib and ceralasertib; olaparib and adavosertib | Advanced solid tumours with positive predictive biomarkers | Not yet recruiting | NCT02576444 (OLPACO trial) |
| Randomized phase II | ATR | Ceralasertib, olaparib; placebo, olaparib | HGSOC relapsed after at least 6 months maintenance with PARPi | Recruiting | NCT04239014 (DUETTE trial) |
| Randomized phase II | ATR; WEE1 | Olaparib; ceralasertib and olaparib; adavosertib and olaparib | Metastatic triple-negative breast cancer | Active, not recruiting | NCT03330847 (VIOLETTE trial) |
| Phase I | ATR | BAY1895344 and niraparib | Advanced solid tumours and ovarian cancer; two expansion ovarian cancer cohorts: PARPi naive, PARPi resistant | Recruiting | NCT04267939 |
| Phase I | WEE1 | Olaparib, adavosertib | Advanced refractory solid tumour with mutations in DDR genes | Recruiting | NCT04197713 (STAR trial) |
| Phase I/II | ATR | Ceralasertib; ceralasertib and carboplatin; ceralasertib and olaparib; ceralasertib and durvalumab | Head and neck squamous cell carcinoma, non-small-cell lung carcinoma, gastric, breast and ovarian cancer | Recruiting | NCT02264678 |
| Overcoming PARPi resistance | |||||
| Phase I/II | ATR | Olaparib and ceralasertib | Platinum-sensitive ovarian cancer after PARPi progression | Recruiting | NCT02264678 |
| Phase I | ATR | Niraparib once daily plus M4344 100–200 mg daily | Patients with ovarian cancer with disease progression while on PARPi | Not yet recruiting | NCT04149145 |
| Phase I | CHK1 | Olaparib and prexasertib | Advanced solid tumours with prior PARPi treatment | Active, not recruiting | NCT03057145 |
| Phase II | WEE1 | Adavosertib; adavosertib and olaparib | Patients with ovarian cancer with disease progression while on PARPi | Recruiting | NCT03579316 |
| Immunotherapy combinations | |||||
| Phase I/II | ATR | Berzosertib and avelumab | Advanced solid tumours | Recruiting | NCT04266912 |
| Phase I | ATR | BAY1895344 and pembrolizumab | Advanced solid tumours | Recruiting | NCT04095273 |
ATR, ataxia telangiectasia mutated (ATM)- and Rad3-related; CCC, clear cell cancer; DDR, DNA damage response; HGSOC, high-grade serous ovarian cancer; PARPi, poly[ADP-ribose]polymerase 1 inhibitor.
ATRi monotherapy.
ATRi have shown promising efficacy and are currently being tested in randomized, late-phase clinical trials. At least five compounds have reached clinical trial evaluation: berzosertib (M6620/VE822), ceralasertib (AZD6738), elimusertib (BAY1895344), M1774 and RP-3500.
A phase I trial evaluated ceralasertib, an oral ATRi, as monotherapy in advanced-stage solid tumours that were refractory to conventional treatment. Ceralasertib was well tolerated at 160 mg delivered twice daily on a 2-week-on, 2-week-off schedule and showed an overall response rate (ORR) of 7%99. Furthermore, preliminary clinical data from a phase II study indicated promising antitumour activity of ceralasertib given as a single agent (at a dose of 160 mg twice daily for days 1–14 of a 28-day cycle) in patients with ARID1A-deficient solid tumours, including two of ten patients (both with endometrial cancer) with durable and ongoing complete responses100.
Another phase I trial evaluated another oral ATRi, elimusertib, in 21 patients with advanced-stage solid tumours. The ORR was 19% (four of 21 patients), and all four responding tumours harboured loss or alterations in ATM. Grade 3 anaemia, a class effect of ATRi, occurred in 81.8% of patients. Notably, one long-term responder patient carried a BRCA1 pathogenic mutation and was resistant to previous treatment with PARPi101. Phase I evaluation of berzosertib, an intravenous ATRi administered as monotherapy in 17 patients, showed a complete response in one patient with colorectal cancer bearing ATM loss and an ARID1A mutation102. Finally, RP-3500 was tested in a phase I/II clinical trial in patients with solid tumours selected by genomic alterations in DNA damage repair genes. Interestingly, the ORR was 25% (5/20) among patients with ovarian cancer, 17 of whom presented with platinum-resistant disease and 18 of whom had prior PARPi treatment103.
CHK1i monotherapy.
Several CHK1i have reached early-phase clinical trials, including prexasertib, rabusertib, MK-877, SRA737, GDC-0575, PF-00477736 and AZD7762. In many cases, such as with rabusertib, MK-877, GDC-0575, PF-0047773 and AZD7762, toxicity or low efficacy led to termination of further drug development104–111.
Two phase I trials tested prexasertib, an intravenous agent, as monotherapy. The first trial included 45 patients with advanced-stage solid tumours; one patient with HNSCC and one patient with squamous cell carcinoma of the anus had a partial response112. This result led to an expansion cohort, including patients with squamous cell carcinoma of various origins; 15% of patients with anal cancer and 5% of patients with HNSCC had partial responses. Genomic alterations in BRCA1, BRCA2, ATR, MRE11A, PIK3CA and the E3 ubiquitin ligase genes FBXW7 and PARK2 were present in patients with clinical benefit. HPV positivity correlated with higher clinical benefit for patients with HNSCC113. Prexasertib was also tested in patients with ovarian cancer in a single-arm phase II trial that included high-grade serous or endometrioid histologies, regardless of platinum sensitivity. Among 24 patients without mutations in BRCA1 or BRCA2, eight patients had partial responses (ORR 33%). Interestingly, four of the eight responses occurred in patients with tumours bearing CCNE1 amplification114. Among 18 patients with a mutation in BRCA1 or BRCA2, only two responses were observed (ORR 11%), suggesting that BRCA1 and BRCA2 wild-type tumours may be enriched for other replication stress mechanisms115. Unlike ovarian cancer, prexasertib treatment resulted in lower response rates in other tumour types.
In these monotherapy studies, a high incidence of myelosuppression was observed, including grade ≥3 neutropenia in 70–90% of patients, febrile neutropenia in 6–12% of patients and grade ≥3 thrombocytopenia in 20% of patients112–115. This degree of haematological toxicity with CHK1i seems higher than that of other DNA damage checkpoint inhibitors, possibly reflecting concurrent inhibition of CHK2 or the presence of independent crosstalk mechanisms in haematopoietic cells. Other toxicities associated with earlier CHK1i such as hyperglycaemia and cardiotoxicity are generally considered off-target effects. In this regard, prexasertib is considered a more specific CHK1i116.
SRA737 is an orally bioavailable CHK1i that was first tested as monotherapy in a phase I/II trial with an expansion cohort of HGSOC enriched for CCNE1 amplification. Intriguingly, despite a disease control rate of 54%, there were no partial or complete responses, and there was no association of CCNE1 with clinical benefit117. LY2880070 is an oral CHK1i tested in an initial phase I trial as monotherapy with a maximal tolerated dose of 200 mg delivered twice daily118.
WEE1i monotherapy.
The WEE1i adavosertib was initially tested in two phase I trials, with partial responses observed in two of eight patients with mutations in BRCA1 or BRCA2 (REF.119), four of ten patients with HGSOC and two of three patients with endometrial cancer, both of whom had TP53 mutations120. Frequent adverse events included gastrointestinal effects (nausea, vomiting, diarrhoea), fatigue and haematological toxicity. Similarly, in a phase I trial with another WEE1i, ZN-c3, the most frequent adverse events were nausea, vomiting, diarrhoea and fatigue. In that study, two of 16 patients had a partial response121. ZN-c3 is also being tested in combination with standard-of-care chemotherapy in platinum-resistant HGSOC. Preliminary results showed an ORR of 30.2% among 43 patients122.
In a phase II single-arm study of adavosertib in HGSEC, 34 patients who had received at least one prior chemotherapy line were treated with adavosertib as a single treatment 300 mg orally, delivered once daily on days 1–5 and 8–12 of a 21-day cycle. In that study, adavosertib monotherapy demonstrated very promising and durable evidence of activity in women with HGSEC, with an ORR of 29.4%, median progression-free survival (PFS) of 6.1 months and median duration of response of 9 months123. Of note, one patient with two-copy loss of FBXW7, which is expected to lead to upregulation of both MYC and cyclin E, experienced a significant and prolonged response of more than a year. In a phase I study, ZN-c3 treatment resulted in one complete response and two partial responses among 11 patients with metastatic HGSEC, further suggesting the sensitivity of HGSEC to WEE1i124.
In another phase II trial in patients with CCNE1 amplification (defined as ≥7 copy numbers) using the same dosing schedule, ORR was 27%, with five objective responses among 13 patients with ovarian cancer (ORR 38.5%)125.
MYT1i monotherapy.
MYT1 (FIG. 2) is another target for inhibition of the cellular response to replication stress. MYT1 targets both Tyr14 and Tyr15 residues of CDK1, and MYT1 inhibition causes unscheduled activation of CDK1, leading to early mitosis. MYT1 is more CDK1-selective than WEE1, which regulates both CDK1 and CDK2. Unlike WEE1, which is localized primarily in the nucleus, MYT1 is associated with the endoplasmic reticulum and Golgi apparatus and can promote CDK1 cytosolic segregation126. Preclinical studies have demonstrated that the MYT1i RP-6306 has higher selectivity for cyclin E1-overexpressing cells both in vitro and in vivo and that synergizes with gemcitabine127.
Taken together, ATRi, CHK1i or WEEi have only modest activity as monotherapy in biomarker-unselected patient populations. Certain genomic alterations such as CCNE1 amplification, ATM loss and mutations in BRCA1 or BRCA2 as well as specific tumour subtypes such as HGSOC and HGSEC, which are characterized by high prevalence of alterations associated with a high degree of replication stress, may predict response to these agents, but more studies are needed to confirm these observations. All these agents are characterized by dose-limiting haematological toxicity, with anaemia more prominent with ATRi and neutropenia more prominent with CHK1 and WEE1i, although individual differences do exist.
Combining ATRi, CHK1i, WEE1i and MYT1i
Cytotoxic and targeted agents that induce replication stress are attractive partners for combinatorial strategies with ATRi, CHK1i and WEE1i, and many combinations are currently under development (FIG. 4).
Gemcitabine combinations.
Gemcitabine induces a high level of replication stress and has been effectively combined with ATRi, CHK1i and WEE1i in preclinical studies128–139. Interestingly, CHK1i are less effective than ATRi and WEE1i in sensitizing tumour cells to gemcitabine129,132.
A phase I trial tested adavosertib alone or in combination with carboplatin, cisplatin or gemcitabine in advanced solid tumours that were refractory to standard treatments. Among 81 patients treated with the gemcitabine combination, four (4.9%) achieved a partial response140. In pancreatic cancer, adavosertib was combined with gemcitabine and radiotherapy. Carbohydrate antigen 19–9 (CA19.9) levels were downregulated in the 26 patients for whom measurements of this biomarker were available141.
CHK1i SRA737 and LY2880070 were also tested in combination with gemcitabine. In a phase I study in 81 patients with tumours from various primary sites (28 patients with HGSOC), ORR was 11.7%, including four of 16 patients (25%) who had FA pathway/BRCA mutations and who showed an objective response. Grade 3, or higher, diarrhoea occurred in 20% of patients, and nausea and vomiting occurred in 50% of patients142. Another phase I trial tested LY2880070 plus low-dose gemcitabine in patients with advanced-stage solid tumours; one partial response was observed among 12 patients with ovarian cancer143.
Two randomized phase II trials compared gemcitabine monotherapy with gemcitabine plus ATRi or WEE1i. The first trial compared berzosertib plus gemcitabine with gemcitabine alone in 70 patients with HGSOC and platinum-resistant relapse stratified by platinum-free interval (PFI) (PFI <3 months against PFI 3–6 months). The median PFS was 22.9 weeks in the gemcitabine plus berzosertib group versus 14.7 weeks in the gemcitabine-alone group (HR 0.57). Interestingly, benefit was observed primarily in the PFI <3 months subgroup, in which median PFS was 27.7 weeks with gemcitabine plus berzosertib compared with 9.0 weeks with gemcitabine alone144.
The other randomized phase II study compared gemcitabine plus adavosertib with gemcitabine alone in 124 patients with platinum-resistant ovarian cancer. Median PFS was 4.6 months versus 3.0 months, ORR was 23% compared with 6% and median overall survival was 11.4 versus 7.2 months, all in favour of the adavosertib plus gemcitabine combination. Patients with tumours that had CCNE1 amplification showed a higher response rate with the combination regimen, whereas SLFN11 expression was not associated with clinical benefit123.
The combination of adavosertib with various chemotherapies resulted in significant bone marrow toxicity. Grade 3 or higher thrombocytopenia was observed in 31–48% of patients, grade 3 or higher neutropenia in 37–46% of patients, and dose reductions were necessary in 38–80% of patients123,145–147 (TABLE 1).
Platinum combinations.
As discussed above, platinum compounds induce replication stress and dependency on ATR–CHK1–WEE1 through formation of intra- and inter-strand crosslinks leading to slowing/stalling of the replication fork. Synergism of ATRi, WEE1i or CHK1i with platinum is further mediated by the additional role of these kinases in the activation of the FA–inter-strand crosslink DNA repair pathway that is responsible for repair of the DNA inter-strand crosslinks induced by platinum55,133,148–151.
Platinum agents were among the first agents to be tested in phase I trials in combination with ATRi and WEE1i. In a phase I trial, among 120 patients with various solid tumours treated with carboplatin or cisplatin in combination with adavosertib, 13 (10.8%) achieved a partial response140. Combination of adavosertib with docetaxel and cisplatin in patients with HNSCC was well tolerated, and five of ten patients had a partial response152. The ATRi berzosertib in combination with carboplatin102 produced a complete response in one patient with platinum-refractory and PARPi-resistant HGSOC102. Finally, in a phase I trial of ceralasertib plus carboplatin, two of 36 patients with absent or low expression of ATM or SLFN11 had a partial response153.
Two phase II studies evaluated carboplatin plus adavosertib in platinum-resistant ovarian cancer. The first trial included patients with TP53-mutated ovarian cancer refractory or resistant (PFI <3 months) to first-line platinum and showed an ORR in 43% of patients significantly higher than expected in that setting145. The other phase II trial was a non-randomized study testing four different combinations of adavosertib with gemcitabine, paclitaxel, carboplatin or liposomal doxorubicin in six different schedules. Of 94 patients who entered the study, 31.9% had an ORR. Among the various combinations, carboplatin and adavosertib resulted in an ORR of 66.7% (eight of 12) patients. Among 13 patients whose tumours exhibited CCNE1 amplification, six patients showed partial responses, for an ORR of 46.1% of patients. The small number of patients in each group limits any definitive conclusions about the best drug to combine with adavosertib and these data require confirmation in larger cohorts of patients147. Besides ovarian cancer, a phase II trial of cisplatin plus adavosertib in 34 patients with TNBC demonstrated an ORR in 26% of patients, with median PFS of 4.9 months123.
Of note, two randomized phase II studies compared ATRi or WEE1i plus chemotherapy versus chemotherapy alone. The first trial randomly assigned patients with TP53-mutated ovarian cancer to receive carboplatin and paclitaxel together with adavosertib or placebo. The study met its primary end point of an enhanced response evaluation criteria in solid tumours (RECIST) PFS criterion (ePFS), showing an ePFS of 7.9 months versus 7.3 months (HR 0.63, 95% CI 0.38–1.06, P = 0.080)146. The other study was conducted in patients with urothelial carcinoma who were randomly assigned to receive cisplatin and gemcitabine or cisplatin, gemcitabine and berzosertib. There was no difference in ORR or PFS, and patients in the experimental arm had higher rates of grade 3 or 4 thrombocytopenia (59% versus 39%), neutropenia (37% versus 27%), dis continuation of treatment (24% versus 15%), and lower cumulative cisplatin dose, likely explaining why the addition of berzosertib did not provide benefit125.
Topotecan and taxane combinations.
Combinations with topoisomerase I inhibitors such as topotecan were also synergistic in preclinical studies133,148. A phase I trial evaluated berzosertib plus topotecan154; toxicity was mild, with no dose-limiting toxic events in the monotherapy group and only one patient in the combination treatment group reporting toxicity. Three of five patients with platinum-refractory small-cell lung cancer (SCLC) achieved partial response or prolonged stable disease for 10 months, more than 6 months and more than 7 months, suggesting that this may be a promising regimen for this challenging tumour type154.
Combinatorial strategies with taxanes are supported by various mechanisms. The most obvious of these mechanisms is to further enhance the activity of taxanes by bypassing the G2/M checkpoint and forcing cells with DNA damage and replication stress into mitosis. However, an additional mechanism may be relevant to the emerging role of ATR in controlling chromosome instability during mitosis155. Specifically, during mitosis, Aurora A promotes the localization of ATR to centromeres by enabling its binding to centromere protein F. Centromere protein F recruits ATR to RPA-coated centromeric R-loops, where ATR stimulates Aurora B and prevents formation of lagging chromosomes and ensures accurate chromosomal segregation during mitosis. Accordingly, ATRi, CHK1i and WEE1i may synergize with anti-mitotic agents such as taxanes5,155. Indeed, a phase I study tested ceralasertib plus paclitaxel. The response rate was 33% in patients with melanoma post-immunotherapy. Interestingly, although not clearly associated with response, patients with melanoma exhibited frequent somatic NF1 or NRAS activating mutations156.
In conclusion, combinations of ATRi and WEE1i with chemotherapy have shown promising preliminary results. Gemcitabine and platinum have been the agents most successfully used in these combinations so far, especially in platinum-resistant HGSOC. Conversely, chemotherapy combinations with CHK1i were limited by toxicity and demonstrated only modest activity.
PARPi combinations.
Several preclinical studies demonstrated synergism of PARPi with ATRi, CHK1i and WEE1i in ovarian cancer models148,157–160, models of TNBC51,161,162 and other primary tumours163–165. The synergism was present in both tumours with wild-type and those with mutated BRCA1 and BRCA2, although the effect was more evident in wild-type tumours159,166. As discussed above, ATR–CHK1–WEE1 inhibition may overcome key mechanisms of PARPi resistance including restoration of HRR deficiency, replication fork stabilization, SLFN11 inactivation and loss of PARG expression. Mechanistically, preclinical studies have demonstrated decreased levels of pATR and pCHK1, decreased RAD51 recruitment and increased pH2AX accumulation51,63,148,157,158,161,166.
Concomitant or sequential administration of PARPi with WEE1i in ovarian cancer xenografts had similar effects in cells with high degree of replication stress, but the sequential regimen led to a smaller effect in cells with a low degree of replication stress and was less toxic. Simultaneous administration led to higher levels of replication stress and more DNA damage than sequential administration of the drugs, which may explain the different cell sensitivity according to basal levels of replication stress158. This suggests that sequential administration may be an equally effective, less toxic dosing strategy for PARPi combinations with cell cycle checkpoint inhibitors.
A proof-of-mechanism phase I trial tested prexasertib in combination with the PARPi olaparib in an expansion cohort of patients with HGSOC who had disease progression after PARPi treatment. Four of 18 patients with PARPi-resistant disease had partial responses143. In a phase I study of ceralasertib plus olaparib, the recommended phase II dose was 160 mg delivered once daily from day 1 to day 7 plus olaparib 300 mg delivered twice daily on days 1–28. Thrombocytopenia and neutropenia were the dose-limiting toxic effects, and one complete response and five partial responses were seen among 45 patients167.
PARPi in combination with the WEE1i adavosertib and the ATRi ceralasertib was evaluated in two phase II studies in patients with PARPi-resistant ovarian cancer. The CAPRI trial assessed ceralasertib plus olaparib in patients with platinum-sensitive ovarian carcinoma who had progressed through PARPi therapy after at least 6 months of treatment. Among 13 patients, six patients had a partial response for an ORR of 46%168. The olaparib plus ceralasertib combination resulted in mostly haematological toxicity with grade 3 or 4 thrombocytopenia in 23.1% of patients, and anaemia and neutropenia in 8% of patients, each. In the EFFORT study, patients who showed disease progression while taking PARPi were randomly assigned to treatment with adavosertib alone or in combination with olaparib. The ORR was 23% and 29% of patients, and median PFS was 5.5 months and 6.8 months, respectively. Among patients with wild-type BRCA1 and BRCA2, ORR was 31% in the single treatment arm and 39% in the combination treatment arm169. The combination treatment resulted in grade 3 or 4 thrombocytopenia and anaemia in 10% of patients, each, and in grade 3 or 4 diarrhoea and fatigue in 12% of patients, each168,169. The results of CAPRI and EFFORT studies are clearly promising, with objective responses higher than expected for PARPi monotherapy in these settings.
Immunotherapy combinations.
The combination of the CHK1i prexasertib with the anti-PDL1 antibody LY3300054 in a phase I clinical trial was safe, without unexpected toxicity. Three of six patients with CCNE1-amplified HGSOC had partial responses and one patient had stable disease for more than 12 months170. A phase I study tested the ATRi ceralasertib plus the anti-PDL1 antibody durvalumab in 21 patients, leading to one complete response and two partial responses in patients with NSCLC and HNSCC167.
A phase II trial tested ceralasertib plus durvalumab in patients with melanoma who had progressed after treatment with anti-PD1 or anti-PDL1 therapy immediately before enrolment in the trial. The ORR was 30% and the median PFS was 7.1 months among 30 patients. Grade 3 or 4 toxicity was mainly haematological, with anaemia in 33.3% and thrombocytopenia in 16.7% of patients171.
Combinations with other targeted drugs.
Other targeted drugs are synergistic in combination with ATRi, CHK1i or WEE1i. PIK3CA inhibitors and mTOR inhibitors have shown synergism with CHK1i and WEE1i, leading to increased DNA damage, chromosomal breaks and mitotic catastrophe in preclinical models of HGSOC, NSCLC, TNBC and glioblastoma172–177. A phase I trial tested prexasertib plus the PI3K/mTOR inhibitor samotolosib. The addition of samotolosib resulted in a higher rate of haematological toxicity and nausea; ORR was 13.3% among patients with PIK3CA-mutated tumours and 25% among patients with TNBC in the expansion cohorts143.
Besides PI3K pathway inhibitors, AXL inhibitors plus ATRi were synergistic in melanoma and NSLC cells, notably in cells with low expression of SLFN11, a marker of platinum and PARPi resistance29,30. BETi synergized with ATRi, CHK1i or WEE1i in various preclinical models of lung cancer, ovarian cancer, melanoma and lymphoma. In these studies, inhibition of BRD4 led to increased replication stress and activation of pCHK1 (REFS.34–37). Translesion synthesis inhibitors may also be attractive partners for ATRi, CHK1i and WEE1i, given the role of translesion synthesis as an alternative to the ATR–CHK1–WEE1 pathway in overcoming replication stress178. Other drugs tested in combination with ATRi, CHK1i or WEE1i in preclinical studies have been Aurora kinase inhibitors179, histone deacetylase inhibitors180–182 and BCL2 inhibitors183, but more data are needed179.
Dual blockage of ATR–CHK1–WEE1 pathway.
ATR, CHK1 and WEE1 function at different points of the replication stress response and have distinct roles. Therefore, their inhibition leads to varied cellular toxic effects and elicits different compensatory mechanisms. Because WEE1 is the downstream effector of the pathway, WEE1i are more potent than ATRi or CHK1i, inducing cell cycle arrest and death in the S phase of the same cell cycle owing to replication catastrophe77,131,184. Moreover, WEE1 upregulation by various mechanisms has been implicated as an adaptive mechanism of resistance to ATRi and CHK1i185,186. Therefore, combinations of WEE1i with ATRi and CHK1i are mechanistically relevant, and preclinical models have already demonstrated the synergistic potential of such combinations131,187–192.
Emerging replication stress biomarkers
Functional assays combined with genomic analyses may be useful to identify tumours with high levels of replication stress, but these assays are far from being incorporated into routine clinical practice. In preclinical models, the DNA fibre assay can directly measure the speed and symmetry of replication fork progression. Hill et al.45 described the generation of short-term patient-derived organoids from patients with ovarian cancer that are suitable for evaluation by functional assays such as the DNA fibre assay. In these organoid models, replication fork instability was associated with response to ATRi and CHK1i and replication stress-inducing chemotherapies.
Biomarkers of response to ATRi, CHK1i or WEE1i may be grouped according to the following principles: first, genomic alterations that are potential causes of replication stress; second, biomarkers of the presence of an active cellular response to replication stress; third, biomarkers that present a synthetic lethal relationship with the ATR–CHK1–WEE1 pathway; and fourth, biomarkers of treatment resistance to ATRi, CHK1i and WEE1i (Supplementary Tables 5 and 6). Biomarkers in the first and second groups can also be used as surrogate markers of replication stress.
Among genomic alterations, tumour cells with amplification of CCNE1 overexpress biomarkers of replication stress such as pRPA32, pKAP1, pATR, pCHK1 and pCDK1 and are more sensitive to ATRi, CHK1i or WEE1i in preclinical models47,48. Moreover, patients with CCNE1-amplified tumours had higher response rates to WEE1i and CHK1i in early-phase clinical trials114,119,123,125,145.
MYC-amplified tumours show high levels of replication stress. MYC induces CDK4 and cyclin D expression and inhibits transcription of the CDK inhibitors CDKN2B (also known as p15) and CDKN1A (also known as p21). These alterations lead to increased replication origin firing and increased cell cycle progression from G1 to S phase19,193. In this regard, MYC-amplified multiple myeloma and mantle-cell lymphoma models are sensitive to ATR and CHK1 inhibition77,194,195. FBXW7 and PPP2R1A mutations, which are both prevalent (each in 22% of patients) and mutually exclusive with CCNE1 amplification in HGSEC (Supplementary Fig. 1) may also be biomarkers of response to WEE1i and MYT1i and may explain the excellent response of adavosertib in this setting196.
DNA repair defects also promote replication stress through the accumulation of DNA errors and the disturbance of replication fork dynamics. In the first phase I study of the WEE1i adavosertib, two patients carrying mutations in BRCA1 and BRCA2 had partial responses119 consistent with preclinical studies demonstrating an association between loss of FA and HRR pathway components with response to WEE1 inhibition89.
NF1 is a negative regulator of the RAS pathway, and deletions in NF1 lead to activation of the RAS pathway, resulting in replication stress19,193,197. In addition, RB1 loss and CDKN2A mRNA downregulation lead to early S phase entry. These alterations, together with KRAS, CCNE1 and MYC amplification, were incorporated as candidate biomarkers of replication stress, which was associated with response to gemcitabine compared with combined gemcitabine and berzosertib in the randomized phase II study in HGSOC discussed previously198. This study suggested that the degree of replication stress in a tumour is probably determined by more than one genomic alteration.
The phosphorylated forms of ATR, CHK1 and RPA32 are markers of ATR pathway activation and their expression is widely used as a marker of replication stress in preclinical studies using protein detection methods such as western blot45,48,196. KAP1 and H2AX are primary substrates of the ATM kinase. Because replication stress generates secondary double-strand breaks and subsequently activates ATM, the expression of pKAP1 and pH2AX is also used as a marker of cellular response to replication stress45,48,196. The clinical utility of these biomarkers depends on the development of reproducible immunohistochemistry methods to enable the evaluation of archival tumour samples and is currently under investigation101. Gene expression profiling is another alternative to identify a cellular state of high replication stress; a recent study identified a six-gene signature of differentially expressed genes in cells harbouring genomic alterations related to replication stress199.
Besides their utility as candidate predictive biomarkers of response to ATRi, CHK1i or WEE1i, changes in expression and phosphorylation of proteins related to the cellular response to replication stress may also be used as pharmacodynamic biomarkers of target engagement. Although all DNA damage inhibitors lead to increased expression of pH2AX and fork instability as determined by the DNA fibre assay, the effects of different ATRi, CHK1i or WEE1i on pATR and pCHK1 depend on the point of the pathway that is blocked. Supplementary Table 6 presents various pharmacodynamic biomarkers for ATRi, CHK1i or WEE1i200.
Inhibition of ATR in cells with ATM loss is synthetically lethal in preclinical models of various primary tumours197,201–204. ATM and ATR are the primary sensors of the DNA damage response. Cells with ATM loss rely on ATR response to orchestrate DNA repair and cell cycle checkpoints. Indeed, in early-phase clinical studies of ATRi monotherapy, patients with ATM-deficient tumours responded to treatment101.
Another synthetic lethal interaction to highlight is that between WEE1 inhibition and deficiency of histone methyltransferase SETD2 (REF.15). RRM2 is the target of this synthetic lethal interaction. Specifically, RRM2 is regulated by two pathways: first, trimethylation of the histone 3 protein (H3K36me3) by SETD2 upregulates transcription of RRM2; and second, WEE1 increases RRM2 protein levels by inhibiting CDK1 and preventing RRM2 degradation. When SETD2 is lost (SETD2 mutations occur in about 15% of endometrial cancers) or the demethylase gene KDM4A is amplified (as is in ~6% of ovarian cancers), then there is a decrease in histone trimethylation, decrease in RRM2 levels and decrease in dNTP pools. In that setting, WEE1 inhibition, which also leads to aberrant origin firing, induces even further, critical, reduction in RRM2, critical dNTP depletion and subsequent lethality15.
Mutations in ARID1A and SMARCA4 (encoding transcription activator BRG1) are also synthetic lethal with ATR inhibition. ARID1A and BRG1 are subunits of the BAF complex, one of the SWI/SNF complexes. These are multisubunit protein complexes that interact with histones and transcription factors and modulate DNA accessibility for cellular processes such as transcription, DNA replication and DNA repair205. Mutations in the genes encoding SWI/SNF complex components are present in 20% of all cancer types, mutations in ARID1A are present in up to 50% of ovarian clear cell carcinomas and 30% of ovarian endometrioid carcinomas, and mutations in SMARCA4 are almost universally present in the rare small-cell hypercalcaemic ovarian tumours205. Loss of ARID1A leads to a reduced localization of topoisomerase 2A (TOP2A) to DNA, resulting in a defect in chromosome decatenation. This is relevant to the role of ATR in regulating the G2 phase decatenation checkpoint, which delays entry into mitosis until chromosomes have been decatenated (disentangled) by TOP2A206. ATR mediates the delay of entry to mitosis by inhibition of Polo-like kinase 1, which leads to inhibition of cyclin B1. Interestingly, the SWI/SNF or BAF complexes have a very important function in this process by recruiting TOP2 to the chromatin207. ARID1A interacts with TOP2A, and BRG1 is required for TOP2A binding to chromatin. So, when there is loss of ARID1A function or loss of SMARCA4, this results in a deficiency in decatenation and dependence on the ATR decatenation checkpoint. As a result, ARID1A-deficient cells have a reduced rate of S phase progression and an increased dependency on the G2/M checkpoint208. Moreover, loss of ARID1A increases the abundance of R-loops and transcription–replication transcripts, thus increasing replication stress197. Therefore, in the setting of ARID1A deficiency, ATR inhibition results in increased S phase DNA damage and premature mitotic entry, leading to chromosomal instability and apoptosis208. Consistent with these preclinical data, preliminary clinical data from a phase II study presented at the European Society of Medical Oncology (ESMO) annual meeting in 2021 indicated promising antitumour activity of the ATRi ceralasertib given as a single agent in patients with ARID1A-deficient solid tumours, including two patients (both with endometrial cancer) with durable and ongoing complete responses100.
Finally, certain studies have suggested that the alternative lengthening of telomeres (ALT) pathway used by some cancer cells to overcome replicative senescence is synthetically lethal with ATR inhibition. ALT depends on stabilizing the RPA coating of telomeres during the G2 phase to start the HRR pathway to elongate telomeres. RPA activates the HRR pathway through the ATR pathway. As a consequence, cancer cells that rely on ALT are particularly sensitive to ATR inhibition209. Although ALT is uncommon in HGSOC, it has been found in 4% of clear cell ovarian carcinomas and 1% of ovarian endometrioid cancers210. However, other studies did not find an association of ATR sensitivity with ALT, and this synthetic lethal relationship remains to be better defined211,212.
ATM and ARID1A mutations can be detected by gene sequencing, but as the functional impact of ATM mutations is occasionally difficult to determine, ATM protein levels have been evaluated by immunohistochemistry as a complementary method in clinical trials101. Although loss of H3K36me3 may be caused by mutations in SETD2 or amplification of KDM4A, both detectable by genomic evaluation, immunohistochemistry may interrogate the expression of H3K36me3 or its direct target, RRM2 (REF.15). Clinical studies are currently using these methods to confirm the preclinical findings of the synthetic lethal relationship between them and ATR, CHK1 and WEE1 inhibition.
More recent studies have focused on potential biomarkers of resistance to ATRi, CHK1i or WEE1i. Most of these biomarkers are downstream factors of the ATR pathway. Change in expression of these proteins may develop as compensatory mechanisms during treatment or may be present as a constitutive survival mechanism of the cell and result in the bypass of the upstream inhibition of the pathway by ATRi, CHK1i and WEE1i. FAM122A is a negative regulator of the G2/M checkpoint and forms a complex with the phosphatase PP2A. When CHK1 phosphorylates FAM122A, PP2A is disinhibited and dephosphorylates WEE1, preventing its degradation and activating the G2/M checkpoint by WEE1 (FIG. 2). Cells with low expression of FAM122A exhibit constitutional activation of the G2/M checkpoint independently of ATR–CHK1 activity, becoming more tolerant to replication stress and resistant to treatment with CHK1i. Interestingly, the addition of WEE1i to CHK1i overcomes this resistance mechanism185,186.
As described above, MYT1 is a kinase related to WEE1, with redundant roles in the phosphorylation and inhibition of CDK1. Cells treated with WEE1i upregulate MYT1 as a compensatory mechanism, leading to reactivation of the G2/M checkpoint. Moreover, patients with breast cancer with high levels of MYT1 mRNA have worse prognosis, underscoring that MYT1 mRNA expression could be both a prognostic biomarker and a predictive biomarker of WEE1i response213. Another downstream complex in the ATR–CHK1–WEE1 pathway is the MMB–FOXM1 complex, the presence of which is required to activate premature mitosis triggered by CDK1 activity induced by CHK1 inhibition, resulting in replication catastrophe214. Other potential resistance biomarkers for CHK1i and WEE1i, such as p21 and YAP, are also under investigation213,215.
Conclusions
Thus far, clinical evaluation of ATRi, CHK1i and WEE1i in early-phase clinical trials has demonstrated significant and durable monotherapy activity only in a subset of cancer patients. Monotherapy activity has been promising in very specific settings, such as WEE1i in HGSEC and CCNE1-amplified HGSOC, and ATRi in tumours with ATM loss and ARID1A mutations. Combinatorial strategies have shown considerable promise, with gemcitabine combinations with WEE1i and ATRi showing superior activity to gemcitabine alone in randomized phase II trials. The combination of gemcitabine and berzosertib is planned to undergo randomized phase III clinical trial evaluation in platinum-resistant ovarian cancer, and adavosertib monotherapy is being evaluated with a registrational intent in HGSEC in the ADAGIO trial. Significant interest exists for combination of ATRi, CHK1i or WEE1i with PARPi to overcome or prevent PARPi resistance as well as combination with radiotherapy and immune checkpoint inhibitors; a summary of ongoing clinical trials evaluating ATRi, CHK1i and WEE1i is provided in Supplementary Table 7. In addition, several new compounds are under preclinical development and/or undergoing first-in-human clinical trials, such as the ATRi M1774, RP-350 and ART0380, and the PKMYT1 inhibitor RP-6306.
Nonetheless, a major challenge for the clinical development of these agents remains toxicity, especially bone marrow toxicity. To address this, alternative dosing schedules and sequencing of administration of drug combinations are being explored and this is an active area of clinical investigation. For instance, the STAR trial is designed to test sequential therapy of WEE1i plus PARPi in an attempt to limit the toxicity of this promising combination158.
Another challenge is that there is currently no single, clinically validated biomarker of replication stress or response to ATRi, CHK1i and WEE1i. Also, there is no accurate way to measure replication stress in cancer cells, nor is it known what threshold of baseline replication stress is necessary to confer response to these agents. CCNE1 amplification represents a biomarker of particular interest, given its high prevalence among some types of cancer such as HGSOC and HGSEC, its homogeneity within tumour samples, its mutual exclusivity with BRCA mutations and its association with platinum-resistant or refractory disease. CCNE1 amplification confers enhanced replication stress and possibly response to cell cycle checkpoint inhibitors, based on emerging, early-phase, clinical data. Other genomic alterations such as FBXW7 and PPP2R1A mutations, which are prevalent and mutually exclusive in HGSEC, may also represent genomic biomarkers of response to these agents, particularly WEE1i and MYT1i. Preclinical studies and early clinical data suggest that mutation status of BRCA1 and BRCA2 may be associated with response to ATRi, CHK1i and WEE1i, including in the setting of PARPi resistance. This is consistent with preclinical studies showing that ATR–CHK1–WEE1 inhibition can overcome multiple mechanisms of PARPi resistance in tumours that are deficient in BRCA and HRR. These studies suggest that BRCA-mutated, PARPi-resistant tumours may be a particularly attractive group to consider for cell cycle checkpoint inhibition (preferably combinatorial strategies, as monotherapy seems to have low activity based on early clinical data). Furthermore, the synthetic lethality between ATM deficiency and ATR inhibition, which has been demonstrated preclinically, seems to be supported by early-phase clinical data (mostly in non-ovarian cancers), although more data are needed. Taken together, the development of biomarkers of response to ATR–CHK1–WEE1 inhibition is an evolving field with a rapidly growing body of preclinical data. Clinical data are immature as they include only small, early-phase trials with results that can only be considered hypothesis-generating at this juncture.
In conclusion, targeting replication stress has emerged as a very promising anticancer strategy over the past decade. Outstanding challenges include identification of predictive biomarkers to optimize selection of patients and addressing toxicity, which may be overcome by using alternative dosing and administration schedules based on preclinical, pharmacokinetic and pharmacodynamic data.
Supplementary Material
Acknowledgements
This work was funded by the Dana-Farber/Harvard Cancer Center Specialized Program of Research Excellence (SPORE) in Ovarian Cancer (NIH/NCI 2P50CA240243) and in Gastrointestinal (GI) Cancer (P50 CA127003). The work was also supported by a Lustgarten Foundation/Stand Up To Cancer Pancreatic Cancer Challenge grant, the Breast Cancer Research Foundation, the Gray Foundation, and the Ludwig Center at Harvard.
Glossary
- DNA replication fork
A structure that is formed during DNA replication by the unwinding of the DNA double helix, so that the fork has a DNA double helix downstream, towards which the fork progresses, and two single-stranded DNA strands upstream. The replisome, a multiprotein complex present at the DNA fork, is responsible for DNA unwinding and synthesizes new DNA strands.
- Mitotic catastrophe
A mechanism of cell death that is the consequence of the appearance of numerous DNA breaks during DNA replication owing to obstructions of the replication fork progression that the cell is not able to overcome.
- Replication origin firing
Replication origins are the genomic regions in which DNA replication starts in a two-step process. First, origin licensing occurs during G1 phase when the pre-replication complex assembles to the DNA and identifies the origin sites. Then, origin firing happens in the G1–s transition and s phase by the formation and activation of the replisome, starting DNA replication at the origin site.
- Repriming
A DNA damage tolerance pathway through which the replication fork bypasses DNA damage sites. The enzyme primase–polymerase (PRIMPOL) inserts a new primer immediately after the damage site and allows polymerases to restart DNA synthesis.
- DNA fibre assay
An in vitro image technique to visualize single DNA molecules and DNA replication forks. Replicating DNA is labelled with two thymidine analogues such as 5-iodo-2′-deoxyuridine (IdU) and 5-chloro-2′-deoxyuridine (CldU), cells are lysed and DNA fibre stretched on glass coverslips. Fibres are then visualized under a microscope by immunofluorescence, allowing the study of replication fork dynamics.
- Enhanced response evaluation criteria in solid tumours
Response evaluation criteria in solid tumours (RECIsT) sets the criteria to define tumour response or progression to treatments. Enhanced RECIsT are modified RECIsT criteria that assess changes in the longest diameter of the lesion, allowing earlier detection of response or progression.
- Decatenation
The disentanglement of chromosomes during mitosis to allow proper cell division. Sister chromatids entangle as a consequence of DNA replication, but non-replicative entanglements may also occur during interphase.
Footnotes
Competing interests
G.I.S. is a consultant/advisory board member for Lilly, Sierra Oncology, Merck-EMD Serono, Pfizer, Astex, Almac, Roche, Bicycle Therapeutics, Fusion Pharmaceuticals, G1 Therapeutics, Bayer, Ip-sen, Cybrexa Therapeutics, Angiex, Daiichi Sankyo and Seattle Genetics, and reports receipt of commercial research grants from Lilly, Sierra Oncology, Merck-EMD Serono and Merck & Co. P.A.K. reports participation in advisory boards from GlaxoSmithKline/Tesaro, Merck, AstraZeneca and Bayer. A.D.D. is a consultant and/or advisory board member for AstraZeneca, Bayer AG, Cedilla Therapeutics, Celgene, Cyteir Therapeutics, Epizyme, GalaxoSmithKline, Ideaya, Impact Therapeutics, LAV Global Management Company Limited. D.C. and A.A.B.A.C. declare no competing interests.
Supplementary information
The online version contains supplementary material available at https://doi.org/10.1038/s41573-022-00558-5.
References
- 1.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Zeman MK & Cimprich KA Causes and consequences of replication stress. Nat. Cell Biol. 16, 2–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaillard H, García-Muse T & Aguilera A Replication stress and cancer. Nat. Rev. Cancer 15, 276–280 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Halazonetis TD, Gorgoulis VG & Bartek J An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Wilhelm T, Said M & Naim V DNA replication stress and chromosomal instability: dangerous liaisons. Genes 11, 1–35 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Connor MJ Targeting the DNA damage response in cancer. Mol. Cell 60, 547–560 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Cimprich KA, Shin TB, Keith CT & Schreiber SL cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc. Natl Acad. Sci. USA 93, 2850–2855 (1996). This paper is the first to describe the protein encoded by the ATR gene, then called FRAP-related protein, and considered the human counterpart of known essential proteins for S and G2/M checkpoints in other species.
- 8.Bentley NJ et al. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBOJ 15, 6641–6651 (1996). [PMC free article] [PubMed] [Google Scholar]
- 9.Dobbelstein M & Sørensen CS Exploiting replicative stress to treat cancer. Nat. Rev. Drug. Discov. 14, 405–423 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Bell D et al. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lecona E & Fernandez-Capetillo O Targeting ATR in cancer. Nat. Rev. Cancer 18, 586–595 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Karnitz LM & Zou L Molecular pathways: targeting ATR in cancer therapy. Clin. Cancer Res. 21, 4780–4785 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saldivar JC, Cortez D & Cimprich KA The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 18, 622–636 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quinet A, Tirman S, Cybulla E, Meroni A & Vindigni A To skip or not to skip: choosing repriming to tolerate DNA damage. Mol. Cell 81, 649–658 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfister SX et al. Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell 28, 557–568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domínguez-Kelly R et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 194, 567–579 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin JC et al. Exploiting replication stress as a novel therapeutic intervention. Mol. Cancer Res. 19, 192–206 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coschi CH et al. Haploinsufficiency of an RB-E2F1-condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov. 4, 840–853 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Schoonen PM, Guerrero Llobet S & van Vugt MATM Replication stress: driver and therapeutic target in genomically instable cancers. Adv. Protein Chem. Struct. Biol. 115, 157–201 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Macheret M & Halazonetis TD Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555, 112–116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartkova J et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Nazareth D, Jones MJK & Gabrielli B Everything in moderation: lessons learned by exploiting moderate replication stress in cancer. Cancers 11, 1320 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medda A, Duca D & Chiocca S Human papillomavirus and cellular pathways: hits and targets. Pathogens 10, 262 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rottenberg S, Disler C & Perego P The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 21, 37–50 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Zhu H, Swami U, Preet R & Zhang J Harnessing DNA replication stress for novel cancer therapy. Genes 11, 990 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gralewska P et al. PARP inhibition increases the reliance on ATR/CHK1 checkpoint signaling leading to synthetic lethality — an alternative treatment strategy for epithelial ovarian cancer cells independent from HR effectiveness. Int. J. Mol. Sci. 21, 9715 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao H, Ji F, Helleday T & Ying S Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 19, e46263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang TT et al. Targeting the PI3K/mTOR pathway augments CHK1 inhibitor-induced replication stress and antitumor activity in high-grade serous ovarian cancer. Cancer Res. 80, 5380–5392 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flem-Karlsen K et al. Targeting AXL and the DNA damage response pathway as a novel therapeutic strategy in melanoma. Mol. Cancer Ther. 19, 895–905 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Ramkumar K et al. AXL inhibition induces DNA damage and replication stress in non-small cell lung cancer cells and promotes sensitivity to ATR inhibitors. Mol. Cancer Res. 19, 485–497 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan D, Shelton Earp H, DeRyckere D & Graham DK Targeting MERTK and AXL in EGFR mutant non-small cell lung cancer. Cancers 13, 5639 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDaniel NK et al. AXL mediates cetuximab and radiation resistance through tyrosine 821 and the c-abl kinase pathway in head and neck cancer. Clin. Cancer Res. 26, 4349–4359 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bowry A & Kelly RDW Hypertranscription and replication stress in cancer. Trends Cancer 7, 863–877 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Zhang J et al. BRD4 facilitates replication stress-induced DNA damage response. Oncogene 37, 3763–3777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang P et al. BRD4 inhibitor AZD5153 suppresses the proliferation of colorectal cancer cells and sensitizes the anticancer effect of PARP inhibitor. Int. J. Biol. Sci. 15, 1942–1954 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takashima Y et al. Bromodomain and extraterminal domain inhibition synergizes with WEE1-inhibitor AZD1775 effect by impairing nonhomologous end joining and enhancing DNA damage in nonsmall cell lung cancer. Int. J. Cancer 146, 1114–1124 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Muralidharan SV et al. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene 35, 4689–4697 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Nayak S et al. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv. 6, eaaz7808 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim W et al. USP13 regulates the replication stress response by deubiquitinating TopBP1. DNA Repair 100, 103063 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu X et al. Ubiquitination of the DNA-damage checkpoint kinase CHK1 by TRAF4 is required for CHK1 activation. J. Hematol. Oncol. 13, 1–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.D’Andrea AD Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 71, 172–176 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Lee EK & Matulonis UA Parp inhibitor resistance mechanisms and implications for post-progression combination therapies. Cancers 12, 1–25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng Y et al. The deubiquitylating enzyme USP15 regulates homologous recombination repair and cancer cell response to PARP inhibitors. Nat. Commun. 10, 1224 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gogola E et al. Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell 33, 1078–1093.e12 (2018). [DOI] [PubMed] [Google Scholar]
- 45. Hill SJ et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 8, 1404–1421 (2018). This seminal paper on ovarian cancer organoids shows the utility of these preclinical models to assess drug sensitivity and perform functional assays such as the DNA combining assay to assess the level and response to replication stress in patient tumour samples.
- 46.Kim H et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 11, 3726 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dréan A et al. Modeling therapy resistance in BRCA1/2-mutant cancers. Mol. Cancer Ther. 16, 2022–2034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Parmar K et al. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin. Cancer Res. 25, 6127–6140 (2019). This paper shows the monotherapy activity of the CHK1 inhibitor prexasertib in in vitro and in vivo preclinical models of PARPi-resistant ovarian cancer by promoting homologous recombination deficiency and replication fork instability.
- 49. Yazinski SA et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 31, 318–332 (2017). This is a comprehensive preclinical study evaluating the role of ATR inhibition in overcoming PARPi resistance in BRCA1-deficient cells by promoting homologous recombination deficiency and replication fork instability.
- 50.Murai J et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 7, 76534–76550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ha D-H et al. Antitumor effect of a WEE1 inhibitor and potentiation of olaparib sensitivity by DNA damage response modulation in triple-negative breast cancer. Sci. Rep. 10, 9930 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moens S et al. The mitotic checkpoint is a targetable vulnerability of carboplatin-resistant triple negative breast cancers. Sci. Rep. 11, 3176 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi Q et al. The identification of the ATR inhibitor VE-822 as a therapeutic strategy for enhancing cisplatin chemosensitivity in esophageal squamous cell carcinoma. Cancer Lett. 432, 56–68 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Hall AB et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 5, 5674–5685 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leonard BC et al. ATR inhibition sensitizes HPV− and HPV+ head and neck squamous cell carcinoma to cisplatin. Oral. Oncol. 95, 35–42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pillay N et al. DNA replication vulnerabilities render ovarian cancer cells sensitive to poly(ADP-ribose) glycohydrolase inhibitors. Cancer Cell 35, 519–533. e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Min W et al. Poly(ADP-ribose) binding to CHK1 at stalled replication forks is required for S-phase checkpoint activation. Nat. Commun. 4, 2993 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Murai J, Thomas A, Miettinen M & Pommier Y Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anticancer therapies. Pharmacol. Ther. 201, 94–102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mao S et al. Resistance to pyrrolobenzodiazepine dimers is associated with SLFN11 downregulation and can be reversed through inhibition of ATR. Mol. Cancer Ther. 20, 541–552 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Winkler C et al. SLFN11 informs on standard of care and novel treatments in a wide range of cancer models. Br. J. Cancer 124, 951–962 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen J et al. PARPI triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCANEss. Cancer Res. 79, 311–319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ding L et al. PARP inhibition elicits STING-dependent antitumor immunity in BRCA1-deficient ovarian cancer. Cell Rep. 25, 2972–2980.e5 (2018). This is one of the first studies to show that PARPi activate the cGAS–STING pathway to promote antitumour immunity.
- 63.Schoonen PM et al. Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol. Oncol. 13, 2422–2440 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mouw KW & Konstantinopoulos PA From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation. Br. J. Cancer 118, 933–935 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sato H et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 8, 1751 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun L-L et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am. J. Cancer Res. 8, 1307–1316 (2018). [PMC free article] [PubMed] [Google Scholar]
- 67.Wayne J, Brooks T, Landras A & Massey AJ Targeting DNA damage response pathways to activate the STING innate immune signaling pathway in human cancer cells. FEBS J. 288, 4507–4540 (2021). [DOI] [PubMed] [Google Scholar]
- 68.Patel P et al. Enhancing direct cytotoxicity and response to immune checkpoint blockade following ionizing radiation with Wee1 kinase inhibition. Oncoimmunology 8, e1638207 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sen T et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 9, 646–661 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sen T et al. Combination treatment of the oral CHK1 inhibitor, SRA737, and low-dose gemcitabine enhances the effect of programmed death ligand 1 blockade by modulating the immune microenvironment in SCLC. J. Thorac. Oncol. 14, 2152–2163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alimzhanov M et al. Abstract 2269: ATR inhibitor M6620 enhances anti-tumor efficacy of the combination of the anti-PD-L1 antibody avelumab with platinum-based chemotherapy. Cancer Res. 79 (Suppl. 13), Abstr. 2269 (2019). [Google Scholar]
- 72.Zhang S et al. Genetically defined, syngeneic organoid platform for developing combination therapies for ovarian cancer. Cancer Discov. 11, 362–383 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Iyer S et al. Genetically defined syngeneic mouse models of ovarian cancer as tools for the discovery of combination immunotherapy. Cancer Discov. 11, 384–407 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang Z et al. ATR inhibition induces CDK1–SPOP signaling and enhances anti-PD-L1 cytotoxicity in prostate cancer. Clin. Cancer Res. 27, 4898–4909 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pilger D, Seymour LW & Jackson SP Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes. Dev. 35, 602–618 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sheng H et al. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J. Immunother. Cancer 8, e000340 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Young LA et al. Differential activity of ATR and Wee1 inhibitors in a highly sensitive subpopulation of DLBCL linked to replication stress. Cancer Res. 79, 3762–3775 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Aarts M et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2, 524–539 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Moiseeva TN, Qian C, Sugitani N, Osmanbeyoglu HU & Bakkenist CJ WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and S-phase cells. Proc. Natl Acad. Sci. USA 116, 23891–23893 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beck H et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell Biol. 32, 4226–4236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cuneo KC et al. Wee1 kinase inhibitor AZD1775 radiosensitizes hepatocellular carcinoma regardless of TP53 mutational status through induction of replication stress. Int. J. Radiat. Oncol. Biol. Phys. 95, 782–790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.King C et al. LY2606368 causes replication catastrophe and antitumor effects through CHK1-dependent mechanisms. Mol. Cancer Ther. 14, 2004–2013 (2015). [DOI] [PubMed] [Google Scholar]
- 83.Morgan MA et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 70, 4972–4981 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dibitetto D et al. Intrinsic ATR signaling shapes DNA end resection and suppresses toxic DNA-PKcs signaling. Nar. Cancer 2, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sørensen CS et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 7, 195–201 (2005). This is the first study to propose that CHK1 is not only crucial for cell-cycle regulation but also has a fundamental role in DNA repair.
- 86.Pefani DE et al. RASSF1A-LATS1 signalling stabilizes replication forks by restricting CDK2-mediated phosphorylation of BRCA2. Nat. Cell Biol. 16, 962–971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parsels LA et al. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol. Cancer Ther. 8, 45–54 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kausar T et al. Sensitization of pancreatic cancers to gemcitabine chemoradiation by WEE1 kinase inhibition depends on homologous recombination repair. Neoplasia 17, 757–766 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aarts M et al. Functional genetic screen identifies increased sensitivity to WEE1 inhibition in cells with defects in Fanconi anemia and HR pathways. Mol. Cancer Ther. 14, 865–876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guertin AD et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol. Cancer Ther. 12, 1442–1452 (2013). [DOI] [PubMed] [Google Scholar]
- 91.Kreahling JM et al. MK1775, a selective WEE1 inhibitor, shows single-agent antitumor activity against sarcoma cells. Mol. Cancer Ther. 11, 174–182 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reaper PM et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 7, 428–430 (2011). [DOI] [PubMed] [Google Scholar]
- 93.Hirai H et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 8, 2992–3000 (2009). [DOI] [PubMed] [Google Scholar]
- 94.Bridges KA et al. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin. Cancer Res. 17, 5638–5648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pappano WN, Zhang Q, Tucker LA, Tse C & Wang J Genetic inhibition of the atypical kinase Wee1 selectively drives apoptosis of p53 inactive tumor cells. BMC Cancer 14, 430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dillon MT et al. Radiosensitization by the ATR inhibitor AZD6738 through generation of acentric micronuclei. Mol. Cancer Ther. 16, 25–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Middleton FK, Pollard JR & Curtin NJ The impact of p53 dysfunction in ATR inhibitor cytotoxicity and chemo- and radiosensitisation. Cancers 10, 275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Van Linden AA et al. Inhibition of wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol. Cancer Ther. 12, 2675–2684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dillon M et al. A phase I study of ATR inhibitor, AZD6738, as monotherapy in advanced solid tumours (PATRIOT part A, B). Ann. Oncol. 30, v165–v166 (2019). [Google Scholar]
- 100.Aggarwal R et al. 512O. Interim results from a phase II study of the ATR inhibitor ceralasertib in ARID1A-deficient and ARID1A-intact advanced solid tumor malignancies. Ann. Oncol. 32, S583 (2021). [Google Scholar]
- 101.Yap TA et al. First-in-human trial of the oral ataxia telangiectasia and Rad3-related inhibitor BAY 1895344 in patients with advanced solid tumors. Cancer Discov. 11, 80–91 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yap TA et al. Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J. Clin. Oncol. 38, 3195–3204 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yap TA et al. Genomic and pathologic determinants of response to RP-3500, an ataxia telangiectasia and Rad3-related inhibitor (ATRi), in patients (pts) with DNA damage repair (DDR) loss-of-function (LOF) mutant tumors in the phase 1/2 TRESR trial. Cancer Res. 82 (Suppl. 12), Abstr. CT030 (2022). [Google Scholar]
- 104.Daud AI et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 33, 1060–1066 (2015). [DOI] [PubMed] [Google Scholar]
- 105.Italiano A et al. Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Ann. Oncol. 29, 1304–1311 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Laquente B et al. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer 17, 137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Scagliotti G et al. Phase II evaluation of LY2603618, a first-generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non-small cell lung cancer. Invest. New Drugs 34, 625–635 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Doi T et al. Phase I study of LY2603618, a CHK1 inhibitor, in combination with gemcitabine in Japanese patients with solid tumors. Anticancer Drugs 26, 1043–1053 (2015). [DOI] [PubMed] [Google Scholar]
- 109.Calvo E et al. Phase I study of CHK1 inhibitor LY2603618 in combination with gemcitabine in patients with solid tumors. Oncology 91, 251–260 (2016). [DOI] [PubMed] [Google Scholar]
- 110.Seto T et al. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer Chemother. Pharmacol. 72, 619–627 (2013). [DOI] [PubMed] [Google Scholar]
- 111.Sausville E et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 73, 539–549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hong D et al. Phase i study of LY2606368, a checkpoint kinase 1 inhibitor, in patients with advanced cancer. J. Clin. Oncol. 34, 1764–1771 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hong DS et al. Evaluation of prexasertib, a checkpoint kinase 1 inhibitor, in a phase Ib study of patients with squamous cell carcinoma. Clin. Cancer Res. 24, 3263–3272 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lee JM et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol. 19, 207–215 (2018). This clinical study is the first to show the activity of a CHK1 inhibitor to treat HGSOC.
- 115.Lampert EJ et al. Prexasertib, a cell cycle checkpoint kinase 1 inhibitor, in BRCA mutant recurrent high-grade serous ovarian cancer (HGSOC): a proof-of-concept single arm phase II study. J. Clin. Oncol. 38, 6038 (2020). [Google Scholar]
- 116.Ditano JP & Eastman A comparative activity and off-target effects in cells of the CHK1 inhibitors MK-8776, SRA737, and LY2606368. ACS Pharmacol. Transl. Sci. 4, 730–743 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Plummer ER et al. A first-in-human phase I/II trial of SRA737 (a Chk1 Inhibitor) in subjects with advanced cancer. J. Clin. Oncol. 37, 3094 (2019). [Google Scholar]
- 118.Miller WH et al. A phase Ib study of oral Chk1 inhibitor LY2880070 as monotherapy in patients with advanced or metastatic cancer. J. Clin. Oncol. 38, 3579 (2020). [Google Scholar]
- 119.Do K et al. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J. Clin. Oncol. 33, 3409–3415 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Takebe N et al. Safety, antitumor activity, and biomarker analysis in a phase I trial of the once-daily wee1 inhibitor adavosertib (AZD1775) in patients with advanced solid tumors. Clin. Cancer Res. 27, 3834–3844 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tolcher A et al. Clinical activity of single-agent ZN-c3, an oral WEE1 inhibitor, in a phase 1 dose-escalation trial in patients with advanced solid tumors. Cancer Res. 81 (Suppl. 13), Abstr. CT016 (2021). [Google Scholar]
- 122.Pasic A et al. A phase 1b dose-escalation study of ZN-c3, a WEE1 inhibitor, in combination with chemotherapy (CT) in subjects with platinum-resistant or refractory ovarian, peritoneal, or fallopian tube cancer. Cancer Res. 82 (Suppl. 12), Abstr. CT148 (2022). [Google Scholar]
- 123. Lheureux S et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 397, 281–292 (2021). This is one of the two main randomized trials showing activity of the combination of a WEE1i with gemcitabine in platinum-resistant ovarian cancers.
- 124.Meric-Bernstam F et al. Safety and clinical activity of single-agent ZN-c3, an oral WEE1 inhibitor, in a phase 1 trial in subjects with recurrent or advanced uterine serous carcinoma (USC). Cancer Res. 82 (Suppl. 12), Abstr. CT029 (2022). [Google Scholar]
- 125.Fu S et al. Phase II trial of the Wee1 inhibitor adavosertib in advanced refractory solid tumors with CCNE1 amplification. Cancer Res. 81 (Suppl. 13), Abstr. 974 (2021). [Google Scholar]
- 126.Ghelli Luserna Di Rorà A, Cerchione C, Martinelli G & Simonetti G A WEE1 family business: regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 13, 126 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Gallo D et al. CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition. Nature 604, 749–756 (2022). This is the first preclinical study to show the synthetic lethality of PKMYT1 inhibition with CCNE1 amplification.
- 128.Meng X et al. AZD1775 increases sensitivity to olaparib and gemcitabine in cancer cells with p53 mutations. Cancers 10, 149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xiao Y et al. Identification of preferred chemotherapeutics for combining with a CHK1 Inhibitor. Mol. Cancer Ther. 12, 2285–2295 (2013). [DOI] [PubMed] [Google Scholar]
- 130.Kreahling JM et al. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PLoS ONE 8, e57523 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Koh SB et al. Mechanistic distinctions between CHK1 and WEE1 inhibition guide the scheduling of triple therapy with gemcitabine. Cancer Res. 78, 3054–3066 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Montano R, Chung I, Garner KM, Parry D & Eastman A Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol. Cancer Ther. 11, 427–438 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hirai H et al. MK-1775, a small molecule Wee1 inhibitor, enhances antitumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther. 9, 514–522 (2010). [DOI] [PubMed] [Google Scholar]
- 134.Fordham SE et al. Inhibition of ATR acutely sensitizes acute myeloid leukemia cells to nucleoside analogs that target ribonucleotide reductase. Blood Adv. 2, 1157–1169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu S et al. Inhibition of ATR potentiates the cytotoxic effect of gemcitabine on pancreatic cancer cells through enhancement of DNA damage and abrogation of ribonucleotide reductase induction by gemcitabine. Oncol. Rep. 37, 3377–3386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wallez Y et al. The ATR inhibitor AZD6738 synergizes with gemcitabine in vitro and in vivo to induce pancreatic ductal adenocarcinoma regression. Mol. Cancer Ther. 17, 1670–1682 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Venkatesha VA et al. Sensitization of pancreatic cancer stem cells to gemcitabine by Chk1 inhibition. Neoplasia 14, 519–525 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Warren NJH & Eastman A Inhibition of checkpoint kinase 1 following gemcitabine-mediated S phase arrest results in CDC7- and CDK2-dependent replication catastrophe. J. Biol. Chem. 294, 1763–1778 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Montano R et al. Sensitization of human cancer cells to gemcitabine by the Chk1 inhibitor MK-8776: cell cycle perturbation and impact of administration schedule in vitro and in vivo. BMC Cancer 13, 604 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Leijen S et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J. Clin. Oncol. 34, 4371–4380 (2016). This was one of the first clinical studies showing the safety and activity of WEE inhibition in patients with solid tumors.
- 141.Cuneo KC et al. Dose escalation trial of the WEE1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J. Clin. Oncol. 37, 2643–2650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Banerji U et al. A phase I/II first-in-human trial of oral SRA737 (a Chk1 inhibitor) given in combination with low-dose gemcitabine in subjects with advanced cancer. J. Clin. Oncol. 37, 3095 (2019). [Google Scholar]
- 143.Hong DS et al. Preclinical evaluation and phase Ib study of prexasertib, a CHK1 inhibitor, and samotolisib (LY3023414), a dual PI3K/mTOR inhibitor. Clin. Cancer Res. 27, 1864–1874 (2021). [DOI] [PubMed] [Google Scholar]
- 144. Konstantinopoulos PA et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 21, 957–968 (2020). This is the first randomized study to show the activity of the ATRi berzosertib in patients with platinum-resistant HGSOC.
- 145.Leijen S et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J. Clin. Oncol. 34, 4354–4361 (2016). [DOI] [PubMed] [Google Scholar]
- 146.Oza AM et al. A biomarker-enriched, randomized phase II trial of adavosertib (AZD1775) plus paclitaxel and carboplatin for women with platinum-sensitive TP53-mutant ovarian cancer. Clin. Cancer Res. 26, 4767–4776 (2020). [DOI] [PubMed] [Google Scholar]
- 147.Moore KN et al. Adavosertib with chemotherapy in patients with primary platinum-resistant ovarian, fallopian tube, or peritoneal cancer: an open-label, four-arm, phase II study. Clin. Cancer Res. 28, 36–44 (2022). [DOI] [PubMed] [Google Scholar]
- 148.Huntoon CJ et al. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 73, 3683–3691 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zheng H, Shao F, Martin S, Xu X & Deng CX WEE1 inhibition targets cell cycle checkpoints for triple negative breast cancers to overcome cisplatin resistance. Sci. Rep. 7, 43517 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jhuraney A et al. PAXIP1 potentiates the combination of WEE1 inhibitor AZD1775 and platinum agents in lung cancer. Mol. Cancer Ther. 15, 1669–1681 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Osman AA et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol. Cancer Ther. 14, 608–619 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mendez E et al. A phase I clinical trial of AZD1775 in combination with neoadjuvant weekly docetaxel and cisplatin before definitive therapy in head and neck squamous cell carcinoma. Clin. Cancer Res. 24, 2740–2748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yap TA et al. Ceralasertib (AZD6738), an oral ATR kinase inhibitor, in combination with carboplatin in patients with advanced solid tumors: a phase I study. Clin. Cancer Res. 27, 5213–5224 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Thomas A et al. Phase I study of ATR inhibitor M6620 in combination with topotecan in patients with advanced solid tumors. J. Clin. Oncol. 36, 1594–1602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kabeche L, Nguyen HD, Buisson R & Zou L A mitosis-specific and R loop–driven ATR pathway promotes faithful chromosome segregation. Science 359, 108–114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kim ST et al. Phase I study of ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer. Clin. Cancer Res. 27, 4700–4709 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim H et al. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin. Cancer Res. 23, 3097–3108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Fang Y et al. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell 35, 851–867.e7 (2019). This study proposes an innovative schedule for the combination of PARPi and WEEi to overcome the toxicity of the combination.
- 159.Burgess BT et al. Olaparib combined with an ATR or Chk1 inhibitor as a treatment strategy for acquired olaparib-resistant BRCA1 mutant ovarian cells. Diagnostics 10, 121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Brill E et al. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 8, 111026–111040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mani C et al. Prexasertib treatment induces homologous recombination deficiency and synergizes with olaparib in triple-negative breast cancer cells. Breast Cancer Res. 21, 104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen X et al. Targeting replicative stress and DNA repair by combining PARP and Wee1 kinase inhibitors is synergistic in triple negative breast cancers with cyclin E or BRCA1 alteration. Cancers 13, 1656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Heidler CL et al. Prexasertib (LY2606368) reduces clonogenic survival by inducing apoptosis in primary patient-derived osteosarcoma cells and synergizes with cisplatin and talazoparib. Int. J. Cancer 147, 1059–1070 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Garcia TB et al. A small-molecule inhibitor of WEE1, AZD1775, synergizes with olaparib by impairing homologous recombination and enhancing DNA damage and apoptosis in acute leukemia. Mol. Cancer Ther. 16, 2058–2068 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Karnak D et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin. Cancer Res. 20, 5085–5096 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Smith HL, Prendergast L & Curtin NJ Exploring the synergy between PARP and CHK1 inhibition in matched BRCA2 mutant and corrected cells. Cancers 12, 878 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Krebs MG et al. Abstract CT026: Phase I study of AZD6738, an inhibitor of ataxia telangiectasia Rad3-related (ATR), in combination with olaparib or durvalumab in patients (pts) with advanced solid cancers. Cancer Res. 78 (Suppl. 13), CT026 (2018). [Google Scholar]
- 168.Wethington SL et al. Combination of PARP and ATR inhibitors (olaparib and ceralasertib) shows clinical activity in acquired PARP inhibitor-resistant recurrent ovarian cancer. J. Clin. Oncol. 39, 5516 (2021). [Google Scholar]
- 169. Westin SN et al. EFFORT: EFFicacy Of adavosertib in parp ResisTance: a randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J. Clin. Oncol. 39, 5505 (2021). This is the first clinical study to show the activity of the WEE1 inhibitor adavosertib in which all patients presented PARPi-resistant ovarian cancer.
- 170.Do KT et al. Immune modulating activity of the CHK1 inhibitor prexasertib and anti-PD-L1 antibody LY3300054 in patients with high-grade serous ovarian cancer and other solid tumors. Cancer Immunol. Immunother. 70, 2991–3000 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Kwon M et al. Phase II study of ceralasertib (AZD6738), in combination with durvalumab in patients with metastatic melanoma who have failed prior anti-PD-1 therapy. J. Clin. Oncol. 39, 9514 (2021). This study shows the most promising clinical result of the combination of an ATRi with anti-PD1/PDL1 therapy.
- 172.Li F et al. mTOR inhibition overcomes primary and acquired resistance to Wee1 inhibition by augmenting replication stress in epithelial ovarian cancers. Am. J. Cancer Res. 10, 908–924 (2020). [PMC free article] [PubMed] [Google Scholar]
- 173.Hai J et al. Synergy of WEE1 and mTOR inhibition in mutant KRAS-driven lung cancers. Clin. Cancer Res. 23, 6993–7005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wu S et al. Activation of WEE1 confers resistance to PI3K inhibition in glioblastoma. NeuroOncol. 20, 78–91 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wu W et al. Combination of the Chk1 inhibitor (prexasertib) with a PI3K/mTOR inhibitor (LY3023414) induces synergistic anti-tumor activity in triple negative breast cancer (TNBC) models. Cancer Res. 79 (Suppl. 13), Abstr. 3508 (2019). [Google Scholar]
- 176.Ding Y et al. PI3K/AKT signaling pathway is transcriptionally elevated in prexasertib-resistant TNBC PDX models. Cancer Res. 78 (Suppl. 13), Abstr. 2586 (2018). [Google Scholar]
- 177.Song X et al. Synergistic targeting of CHK1 and mTOR in MYC-driven tumors. Carcinogenesis 42, 448–460 (2021). [DOI] [PubMed] [Google Scholar]
- 178.Nayak S, Calvo JA & Cantor SB Targeting translesion synthesis (TLS) to expose replication gaps, a unique cancer vulnerability. Expert Opin. Ther. Targets 25, 27–36 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lee JW et al. Combined Aurora kinase A (AURKA) and WEE1 inhibition demonstrates synergistic antitumor effect in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 25, 3430–3442 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhou L et al. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia 29, 807–818 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Schwartz J et al. Synergistic anti-leukemic interactions between ABT-199 and panobinostat in acute myeloid leukemia ex vivo. Am. J. Transl. Res. 8, 3893–3902 (2016). [PMC free article] [PubMed] [Google Scholar]
- 182.Wang G et al. Synergistic antitumor interactions between MK-1775 and panobinostat in preclinical models of pancreatic cancer. Cancer Lett. 356, 656–668 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.de Jong MRW et al. WEE1 inhibition enhances anti-apoptotic dependency as a reult of premature mitotic entry and DNA damage. Cancers 11, 1743 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Nojima H et al. Differential properties of mitosis-associated events following CHK1 and WEE1 inhibitor treatments in human tongue carcinoma cells. Exp. Cell Res. 386, 111720 (2020). [DOI] [PubMed] [Google Scholar]
- 185.Caeser R & Sen T Should WEE(1) CHK(1) in on the FAM(122A)ily? Mol. Cell 80, 377–378 (2020). [DOI] [PubMed] [Google Scholar]
- 186.Li F et al. CHK1 inhibitor blocks phosphorylation of FAM122A and promotes replication stress. Mol. Cell 80, 410–422.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nam AR et al. Inhibition of ATR increases the sensitivity to WEE1 inhibitor in biliary tract cancer. Cancer Res. Treat. 52, 945–956 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bukhari AB et al. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Invest. 129, 1329–1344 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Restelli V et al. DNA damage response inhibitor combinations exert synergistic antitumor activity in aggressive B-cell lymphomas. Mol. Cancer Ther. 18, 1255–1264 (2019). [DOI] [PubMed] [Google Scholar]
- 190.Qi W et al. Inhibition of Wee1 sensitizes AML cells to ATR inhibitor VE-822-induced DNA damage and apoptosis. Biochem. Pharmacol. 164, 273–282 (2019). [DOI] [PubMed] [Google Scholar]
- 191.Deneka AY et al. Synthetic lethal targeting of mitotic checkpoints in HPV-negative head and neck cancer. Cancers 12, 306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Di Rorá AGL et al. Synergism through WEE1 and CHK1 inhibition in acute lymphoblastic leukemia. Cancers 11, 1654 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Maya-Mendoza A et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 9, 601–616 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cottini F et al. Synthetic lethal approaches exploiting DNA damage in aggressive myeloma. Cancer Discov. 5, 972–987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Murga M et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 18, 1331–1335 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Liu JF et al. Phase II study of the WEE1 inhibitor adavosertib in recurrent uterine serous carcinoma. J. Clin. Oncol. 39, 1531–1539 (2021). [DOI] [PubMed] [Google Scholar]
- 197.Al Zubaidi T et al. Targeting the DNA replication stress phenotype of KRAS mutant cancer cells. Sci. Rep. 11, 3656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Konstantinopoulos PA et al. A replication stress biomarker is associated with response to gemcitabine versus combined gemcitabine and ATR inhibitor therapy in ovarian cancer. Nat. Commun. 12, 5574 (2021). This study tests several biomarkers of replication stress in patient tumour samples from a randomized clinical trial. It proposes a score to identify tumours that are more sensitive to replication stress-inducing agents.
- 199.Guerrero Llobet S et al. An mRNA expression-based signature for oncogene-induced replication-stress. Oncogene 41, 1216–1224 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Cleary JM, Aguirre AJ, Shapiro GI & D’Andrea AD Biomarker-guided development of DNA repair inhibitors. Mol. Cell 78, 1070–1085 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dunlop CR et al. Complete loss of ATM function augments replication catastrophe induced by ATR inhibition and gemcitabine in pancreatic cancer models. Br. J. Cancer 123, 1424–1436 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Perkhofer L et al. ATM deficiency generating genomic instability sensitizes pancreatic ductal adenocarcinoma cells to therapy-induced DNA damage. Cancer Res. 77, 5576–5590 (2017). [DOI] [PubMed] [Google Scholar]
- 203.Schmitt A et al. ATM deficiency is associated with sensitivity to PARP1- and ATR inhibitors in lung adenocarcinoma. Cancer Res. 77, 3040–3056 (2017). [DOI] [PubMed] [Google Scholar]
- 204.Min A et al. AZD6738, A novel oral inhibitor of ATR, induces synthetic lethality with ATM deficiency in gastric cancer cells. Mol. Cancer Ther. 16, 566–577 (2017). [DOI] [PubMed] [Google Scholar]
- 205.Wang Y, Hoang L, Ji JX & Huntsman DG SWI/SNF complex mutations in gynecologic cancers: molecular mechanisms and models. Annu. Rev. Pathol. Mech. Dis. 15, 467–492 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Damelin M & Bestor TH The decatenation checkpoint. Br. J. Cancer 96, 201–205 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Dykhuizen EC et al. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature 497, 624–627 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Williamson CT et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 7, 13837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Flynn RL et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347, 273–277 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Heaphy CM et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 179, 1608–1615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Laroche-Clary A et al. ATR inhibition broadly sensitizes soft-tissue sarcoma cells to chemotherapy independent of alternative lengthening telomere (ALT) status. Sci. Rep. 10, 7488 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Deeg KI, Chung I, Bauer C & Rippe K Cancer cells with alternative lengthening of telomeres do not display a general hypersensitivity to ATR inhibition. Front. Oncol. 6, 186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lewis CW et al. Upregulation of MyT1 promotes acquired resistance of cancer cells to WEE1 inhibition. Cancer Res. 79, 5971–5985 (2019). [DOI] [PubMed] [Google Scholar]
- 214.Tsai S et al. ARID1A regulates R-loop associated DNA replication stress. PLoS Genet. 17, e1009238 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Oku Y et al. Augmentation of the therapeutic efficacy of WEE1 kinase inhibitor AZD1775 by inhibiting the YAP-E2F1-DNA damage response pathway axis. FEBS Open. Bio 8, 1001–1012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.