

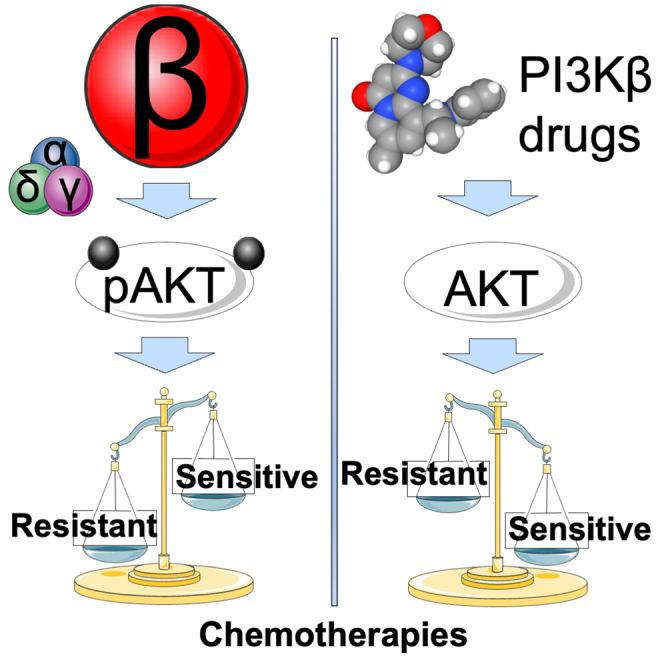
Summary
Resistance to chemotherapies such as temozolomide is a major hurdle to effectively treat therapy-resistant glioblastoma. This challenge arises from the activation of phosphatidylinositol 3-kinase (PI3K), which makes it an appealing therapeutic target. However, non-selectively blocking PI3K kinases PI3Kα/β/δ/γ has yielded undesired clinical outcomes. It is, therefore, imperative to investigate individual kinases in glioblastoma’s chemosensitivity. Here, we report that PI3K kinases were unequally expressed in glioblastoma, with levels of PI3Kβ being the highest. Patients deficient of O6-methylguanine-DNA-methyltransferase (MGMT) and expressing elevated levels of PI3Kβ, defined as MGMT-deficient/PI3Kβ-high, were less responsive to temozolomide and experienced poor prognosis. Consistently, MGMT-deficient/PI3Kβ-high glioblastoma cells were resistant to temozolomide. Perturbation of PI3Kβ, but not other kinases, sensitized MGMT-deficient/PI3Kβ-high glioblastoma cells or tumors to temozolomide. Moreover, PI3Kβ-selective inhibitors and temozolomide synergistically mitigated the growth of glioblastoma stem cells. Our results have demonstrated an essential role of PI3Kβ in chemoresistance, making PI3Kβ-selective blockade an effective chemosensitizer for glioblastoma.
Subject areas: Molecular biology, Cell biology, Cancer
Graphical abstract
Highlights
-
•
Divergent roles of PI3K kinases in glioblastoma’s chemoresistance
-
•
PI3Kβ outcompetes PI3Kα/δ/γ in chemoresistance
-
•
PI3Kβ inhibitors are effective chemosensitizers
-
•
PI3Kβ regulates drug sensitivity in glioblastoma stem cells
Molecular biology; Cell biology; Cancer
Introduction
Glioblastoma (GBM) comprises nearly half of all malignant tumors in the central nervous system, and yet it demonstrates some of the worst survival outcomes. Even with aggressive treatment, median survival is less than 15 months with one-year, five-year, and ten-year survival rates of approximately 42.9%, 6.9%, and 4.3%, respectively.1 The most significant survival benefit is seen with the addition of temozolomide (TMZ), a brain-permeable alkylating agent2 that forms the backbone of GBM treatment.3 Compared to surgical resection with radiation alone, the addition of TMZ increases median overall survival by several months.4 TMZ acts directly on DNA, forming O6-methylguanine lesions that induce DNA breaks and lead to cell death.5 This action can be opposed by endogenous expression of O6-methylguanine-DNA methyltransferase (MGMT), a DNA repair enzyme that removes the O6-methylguanine lesions, making MGMT a key agent in TMZ resistance.6,7 Approximately half of GBM patients demonstrate MGMT promoter hypermethylation, which reduces MGMT protein expression and lesion repair ability, thus significantly increasing GBM’s sensitivity to TMZ and improving patient survival.8 MGMT promoter methylation, which silences the expression of the MGMT gene (MGMT-deficient), is an important predictor of response to TMZ.9,10 However, this response is often not static over the course of treatment, as MGMT-deficient GBMs develop TMZ resistance as well, mechanisms of which are often MGMT-independent, e.g., activation of different DNA-repair pathways or other cell survival pathways.11,12,13,14
The phosphatidylinositol 3-kinase (PI3K) signaling pathway has long been implicated in GBM pathogenesis. PI3K is directly downstream of several frequently mutated or amplified receptor tyrosine kinases (RTKs) in GBM, and dysregulation of the RTKs/PI3K pathway is observed in up to 88% of GBMs.15,16 PI3K converts phosphatidylinositol-4,5-bisphosphate (PIP2) on the inner cell membrane into phosphatidylinositol-3,4,5-trisphosphate (PIP3), which activates AKT serine/threonine kinase (AKT) and exerts widespread effects that support GBM’s proliferation, metabolism, angiogenesis, and survival as well as assist GBM cells to evade from cell death.17,18 This suggests that PI3K inhibition could be a powerful means to combat TMZ resistance. Many preclinical studies have reported increased efficacy of TMZ when combined with various PI3K/molecular targets of rapamycin (mTOR) inhibitors in TMZ-resistant GBM cell lines.19,20,21 Additionally, a phase II clinical study demonstrated a durable response in some participants using the pan-PI3K inhibitor PX-866 as an adjuvant monotherapy in patients who had completed some levels of standard-of-care surgical resection, radiation, and TMZ.22 However, beyond this study, the heavy toxicity and severe side effects often seen with pan-PI3K inhibitors have significantly limited their evaluation for a more robust response.23,24,25 Toxicity associated with pan-PI3K inhibition suggests a more targeted approach is needed. There are four classes of PI3K in mammals (IA, IB, II, III), but classes IA and IB are most relevant to cancer as they directly regulate PI3K activity.17,18 Class IA includes PI3Kα, PI3Kβ, or PI3Kδ, which are encoded by PIK3CA, PIK3CB, and PIK3CD, respectively. Class IB, predominantly expressed in immune cells, includes PI3Kγ encoded by PIK3CG. While it is well documented in in vitro and in vivo preclinical studies that multiple different isoform selective PI3K inhibitors can be effective as monotherapies or synergize with TMZ in GBM,26,27,28,29,30,31 it remains elusive how PI3K kinases regulate the sensitivity of GBM to TMZ.
Recently, our group has identified one of the PI3K kinases, PIK3CB/PI3Kβ, as the dominant isoform that controls GBMs’ survival and growth.30,31 Levels of PIK3CB/PI3Kβ in GBMs are much higher than the other three kinases. Elevated levels of PI3Kβ are associated with poor clinical outcomes in GBMs, particularly recurrent GBMs that are supposed to be therapy-resistant. Knockdown of PI3Kβ, but not PI3Kα/δ/γ, inactivates PI3K signaling and results in a significant growth inhibition in GBM cells expressing prominent levels of PI3Kβ. This pattern was verified by several PI3K isoform-selective inhibitors. It seems that GBMs having more PI3Kβ depend on this kinase to survive. As an added line of evidence to support this idea, our recent investigation on a membrane protein connexin 4329 has revealed that connexin 43 activates PI3K signaling through direct interaction with PI3Kβ and that blocking connexin 43 and PI3Kβ restores chemosensitivity in GBMs. Taken together, research from our groups and others has raised the possibility that the four PI3K kinases have different assignments in GBM’s chemoresistance.
Based on prior results summarized above, we hypothesize that PI3Kβ plays an essential role in MGMT-independent TMZ resistance in GBM. In this report, we have tested this hypothesis by analyzing data from online databases and measuring PI3K activity and responses to TMZ in different GBM cell lines and primary GBM stem cells (GSCs) derived from patient specimens upon CRISPR-Cas9-mediated gene knockout or treatments with isoform-selective inhibitors.
Results
Levels of PIK3CB/PI3Kβ are the highest among the four PI3K kinases in glioma
Previously, we found that PI3K genes are highly enriched in GBM.30 However, it remains elusive if this enrichment is at different degrees in tumors with low or high levels of MGMT (MGMT-deficient vs. MGMT-positive). To address this, we analyzed RNA sequencing (RNA-seq) results from publicly available datasets: The Dependency Map (DepMap,32), The Cancer Genome Atlas (TCGA,33), Genome-Tissue Expression (GTEx,34), and Chinese Glioma Genome Atlas (CGGA,35). To define cell lines based on their MGMT status, we reannotated glioma cell lines (grade II to IV) used in the DepMap based upon results of literature search for MGMT expression, RNA-seq results of MGMT mRNAs (Scores <0.5 were s MGMT-deficient; Scores >0.5 were MGMT-positive), and notes about misidentification and/or contamination in Cellosaurus.36 These analyses (Table S1) yielded 42 GBM cell lines, with 23 being MGMT-deficient and 19 being MGMT-positive, 14 grade II/III glioma cell lines including 11 MGMT-deficient and 3 MGMT-positive, and 8 normal cell lines. Consistent with our earlier findings,30 only PIK3CB was enriched in glioma cell lines, but not in normal cell lines (Figure S1A, p < 0.01), despite that more mRNAs of four PI3K kinases were found in TCGA glioma specimens than in GTEx’s cerebral cortex (Figure S1B, p < 0.0001). Levels of PIK3CB mRNAs were the highest among the four PI3K kinases in both DepMap glioma cell lines and TCGA glioma tumors (Figure S1). We next determined the expression pattern of PI3K genes in gliomas at different grades. Based upon the RNA-seq results from DepMap (Figure 1A), TCGA (Figure 1B), and CGGA (Figure 1C), PIK3CB was consistently expressed at higher levels (p < 0.01 to <0.0001) than other PI3K kinases in grade II and III (GII/III) gliomas or GBM (grade IV) regardless of MGMT levels. While mRNA quantities of PI3K genes were not different in DepMap GII/III and GBM cell lines, TCGA or CGGA GII/III tumors had more PIK3CA and PIK3CB mRNAs than GBM tumors (p < 0.001 or p < 0.0001).
Figure 1.

Levels of PIK3CB/PI3Kβ are the highest among the four PI3K kinases in GBM regardless of MGMT status
RNA-seq data were retrieved from DepMap, TCGA, CGGA, and THPA.
(A) Comparison of PI3K mRNAs in DepMap glioma cell lines. Cell lines from DepMap were reannotated (see Table S1). mRNA levels of PI3K kinases were compared between DepMap grade II/III (GII/III) cell lines and GBM cell lines or between MGMT-deficient and MGMT-positive cells.
(B) Comparison of PI3K mRNAs between TCGA GII/III and GBM tumors or between MGMT-deficient and MGMT-positive tumors.
(C) Comparison of PI3K mRNAs in CGGA gliomas.
(D) Immunohistochemical (IHC) staining of PI3Kα, PI3Kβ, PI3Kδ, PI3Kγ, or MGMT in a THPA MGMT-deficient HGG (Patient ID: 1587). Details of IHC staining are shown in the inset figures. Staining levels determined by the THPA are shown in each image.
(E) Quantification of PI3Kα, PI3Kβ, PI3Kδ, or PI3Kγ based on images shown in D using ImageJ. Four persons performed ImageJ quantification independently. Each data point stands for an intensity reading from one individual.
(F) Quantification of PI3Kα, PI3Kβ, PI3Kδ, or PI3Kγ in THPA HGG or LGG tumors. Four persons performed quantification using ImageJ. Averages of intensity readings from four persons consist of individual data points. Case numbers (N) are shown. Error bars are standard errors. p values were obtained by Student’s t test or One-way ANOVA. ns: not significant; ∗: p < 0.05; ∗∗: p < 0.01; ∗∗∗: p < 0.001; ∗∗∗∗: p < 0.0001.
We next inquired about The Human Protein Atlas (THPA,37), where staining of PI3K proteins using immunohistochemistry (IHC) in gliomas is available (Table S2). In one MGMT-deficient high-grade glioma (HGG) patient (ID: 1587), levels of PI3Kβ proteins were much higher than those of PI3Kα, PI3Kδ, or PI3Kγ (Figure 1D, inset figures), which was verified by ImageJ quantifications (Figure 1E, p < 0.0001). Other HGG tumors also showed a similar expression pattern of PI3K proteins, with PI3Kβ being the highest one and PI3Kγ being undetectable (Figure 1F). Different from this expression pattern in HGGs, PI3K proteins in low-grade gliomas (LGGs) showed similar levels of PI3Kα and PI3Kβ, while PI3Kδ and PI3Kγ were undetected. Because PIK3CB30,38 and MGMT4,39 are prognostic markers for GBMs, we investigated the relationship between PI3K genes and GBM risk factors such as tumor recurrence, mutations in isocitrate dehydrogenase (IDH) genes, molecular subtypes, gender, and age.40,41,42 In all cases tested, levels of PIK3CB were the highest among the four PI3K kinases, irrespective of tumor recurrence, IDH mutations, molecular subtypes, or difference in gender/age (Figures S2–S6). mRNA levels of PI3K genes were not different between primary and recurrent tumors, IDH wild-type (IDH-wt) and mutant (IDH-mut) tumors, male and female, young (age <60 years) and old (age >60 years) patients, or molecular subtypes (classical, mesenchymal, and proneural). Collectively, our results have shown that PI3K kinases are differentially expressed in gliomas and that mRNAs and proteins of PIK3CB/PI3Kβ are highly enriched in GBM regardless of MGMT status.
Levels of PIK3CB/PI3Kβ associate with levels of PI3K activation and chemosensitivity in MGMT-deficient GBMs
We have previously shown that PIK3CB/PI3Kβ activates PI3K in GBM cells,30 suggestive of a correlation between PIK3CB/PI3Kβ levels and PI3K activities. We, therefore, interrogated if levels of PIK3CB/PI3Kβ are functionally relevant to PI3K activities and chemosensitivity in GBMs with various levels of MGMT. As shown in Figures 2A and S7, we retrieved data of phosphorylated proteins, including phosphorylated AKTs (pAKTS473/T308) determined by the reverse phase protein array (RPPA) from DepMap and TCGA. By using the Pearson coefficient, which shows correlations of two subjects, we found that levels of PIK3CB mRNAs positively correlated with levels of pAKTS473/T308 in MGMT-deficient GBM cell lines but not in MGMT-deficient GII/III glioma cell lines and MGMT-positive cell lines, whereas no statistically significant interconnections were detected between pAKTS473/T308 and mRNAs of PIK3CA/D/G. The significant correlation between pAKTS473/T308 and PIK3CB mRNAs (p < 0.05) was recapitulated in MGMT-deficient TCGA GBM tumors (Figures 2B and S8; p < 0.05). However, different from DepMap cell lines, TCGA tumors showed complicated correlations between levels of PI3K genes and pAKTS473 or pAKTT308 with most cases being not statistically significant (p > 0.05). In the cases with p values less than 0.05, levels of PI3K mRNAs were either associated with pAKTS473 only or with pAKTT308 only (e.g., PIK3CA/pAKTS473 in MGMT-deficient GII/III gliomas, PIK3CG/pAKTS473 in MGMT-positive GII/III gliomas, and PIK3CD/pAKTT308 in MGMT-positive GBMs). Unexpected, levels of PIK3CB mRNAs negatively correlated with pAKTS473/T308 in MGMT-positive GII/III gliomas. These random correlations between PI3K genes and pAKTS473 or pAKTT308 in TCGA tumors suggest that, although PI3K genes are enriched, it does not have a definitive impact on PI3K activation in MGMT-deficient or MGMT-positive GII/III gliomas and MGMT-positive GBMs, which have further verified the significance of PIK3CB in activating PI3K selectively in MGMT-deficient GBMs.
Figure 2.

mRNA Levels of PIK3CB associate levels of PI3K activation and chemosensitivity in MGMT-deficient GBMs
RNA-seq and RPPA data were retrieved from DepMap and TCGA. The Pearson correlation assay was used to determine associations between two subjects.
(A) Pearson correlations between pAKTS473/T308 and levels of PIK3CA/B/D/G mRNAs in DepMap MGMT-deficient or MGMT-positive GII/III glioma cell lines or GBM cell lines.
(B) Pearson correlations between pAKTS473/T308 and levels of PIK3CA/B/D/G mRNAs in TCGA MGMT-deficient or MGMT-positive GII/III gliomas or GBMs.
(C) Pearson correlations between TMZ IC50s and levels of PIK3CA/B/D/G mRNAs in DepMap MGMT-deficient or MGMT-positive GII/III glioma cell lines or GBM cell lines.
(D) Pearson correlations between TMZ IC50s and levels of pAKTs or PTEN in DepMap MGMT-deficient or MGMT-positive GII/III glioma cell lines or GBM cell lines. Pearson coefficients (r) with 95% confidence intervals (95% CI) are shown. Representative Pearson correlation results in MGMT-deficient GBMs are also shown. Original results of Pearson correlation in other gliomas are shown in Figures S7, S8, S10, and S11. ∗: p < 0.05; ∗∗: p < 0.01. Statistically not significant Pearson coefficients are not labeled.
Phosphatase and tensin homolog (PTEN), as a PI3K inhibitor, is responsible for the full activation of PI3K.43,44 It is, therefore, expected that PTEN levels inversely correlate with levels of pAKTS473/T308 and PI3K kinases. PTEN protein levels did not show any interconnections with either pAKTS473T308 or mRNA levels of PIK3CA/B/D/G in DepMap GBM cell lines (Figure S9A); however, TCGA gliomas (GII/III and GBM) did exhibit negative Pearson coefficients between PTEN proteins and pAKTS473/T308 or mRNAs of one or more of PI3K kinases (Figure S9B). While these results have demonstrated the negative impact of PTEN on PI3K activation, it seems that PIK3CB does not cooperate with PTEN to activate PI3K signaling, particularly in MGMT-deficient GBMs, suggestive of a dispensable role of PTEN in these tumors.
Next, we determined if levels of PI3K genes had indications in TMZ response, as PI3K is key for TMZ resistance.19,20,21,22,23,24,25,26,27,28,29,30,31 To test this, we collected TMZ’s half maximal inhibitory concentrations (IC50s) in DepMap glioma cell lines or acquired TMZ IC50s by ourselves (Table S3). Pearson correlation analyses detected a positive trend between TMZ IC50s and PIK3CB mRNAs in MGMT-deficient GBMs. (Figures 2C and S10; p < 0.05). In MGMT-deficient GII/III gliomas, PIK3CB mRNA levels negatively, while PIK3CD mRNA levels positively, correlated with TMZ IC50s (Figures 2C and S10; p < 0.05 or p < 0.01). Unexpectedly, levels of PIK3CG positively correlated with TMZ IC50s in MGMT-positive GBMs, which is difficult to be explained by the extremely low levels of PIK3CG in these tumors (Figure 1B). As expected from the results of PIK3CB mRNAs, there was a positive interaction between TMZ IC50s and pAKTS473/T308 (Figures 2D and S11; p < 0.05). Statistically significant correlations between TMZ IC50s and pAKTS473/T308 or PTEN were not found in all other gliomas (Figures 2D and S11; p > 0.05), congruent with no association of PTEN with PI3K activation (Figure S9). Results presented above have demonstrated that levels of PIK3CB, but not those of PTEN or other PI3K kinases, associate with levels of PI3K activation and responses to TMZ in MGMT-deficient GBMs.
PIK3CB predicts clinical outcomes of MGMT-deficient GBMs
We have previously shown that PIK3CB levels are associated with a high incidence of GBM recurrence,30 a clinical condition often resulting from therapy resistance.45 In line with the above results that MGMT-deficient GBMs with high levels of PIK3CB showed resistance to TMZ, we hypothesize that PIK3CB levels would predict clinical outcomes of MGMT-deficient GBMs. To test this hypothesis, we utilized TCGA, CGGA, and 11 more GBM datasets (Table S4) and collected gene expression and clinical data from 114 IDH-wt GII/III gliomas and 377 IDH-wt GBM patients. We focused on IDH-wt primary gliomas in this study because IDH-mut tumors often display increased sensitivity to chemotherapies, thereby displaying better clinical outcomes than IDH-wt patients.46,47 Additionally, PI3K has been identified as a prognostic factor for IDH-wt tumors,48 and clinical relevance of PIK3CB in recurrent GBMs has also been established in our previous work.30 By using the Cox hazard proportional model, we acquired hazard ratios (HRs), which indicate a considerable risk of death and poor prognosis if HR > 1. We also performed the Log Rank analysis to determine the significance of long-term survival and the Wilcoxon analysis to determine short-term survival. We found that HRs of MGMT-deficient/IDH-wt patients with high levels of PIK3CB were 1.891 (GII/III) or 2.186 (GBM), respectively, and that only primary MGMT-deficient/IDH-wt GBMs had statistically significant worse prognosis (Log Rank p = 0.0003/Wilcoxon p = 0.0009, Figures 3A and S12). No significant HRs of PI3K genes were detected in GII/III and MGMT-positive tumors, verifying the importance of PIK3CB in MGMT-deficient primary GBMs. High levels of pAKTS473 (HR = 4.155, Log Rank p = 0.0018/Wilcoxon p = 0.0033) or pAKTT308 (HR = 2.064, Log Rank p = 0.0469/Wilcoxon p = 0.0145) correlated with imminent risk of death in primary MGMT-deficient/IDH-wt GBMs, but not in other gliomas (Figures 3B and S13). Given the strong correlations between PIK3CB and pAKTS437/T308 or between PIK3CB and TMZ IC50s (Figure 2), it is not surprising that GBM patients with prominent levels of PIK3CB will experience poor prognosis.
Figure 3.

PIK3CB predicts clinical outcomes of MGMT-deficient GBMs
RNA-seq/RPPA data and associated clinical data were retrieved from TCGA, CGGA, and other GBM datasets downloaded from the GlioVis online program. Results from primary MGMT-deficient/IDH-wt gliomas were pooled together for Kaplan-Meier survival analyses using JMP and Prism 10. Plotted are the resulting HRs with 95% CIs (HR > 1: more chance of death; HR < 1: more chance of survival).
(A) Comparison of high and low levels of PI3K kinases in primary MGMT-deficient/IDH-wt or MGMT-positive/IDH-wt GII/III or GBM patients.
(B) Comparison of high or low levels of pAKTs in primary MGMT-deficient/IDH-wt or MGMT-positive/IDH-wt GII/III or GBM patients.
(C) Comparison of untreated (Chemo–) and treated (Chemo+) cases in primary MGMT-deficient/IDH-wt or MGMT-positive/IDH-wt GII/III or GBM patients expressing various levels of PI3K kinases. Representative Kaplan-Meir survival curves in MGMT-deficient GBMs are shown. Other Kaplan-Meir survival curves can be found in Figures S12–S14. HRs, Log Rank Ps (long-term survival), and Wilcoxon Ps (short-term survival) with 95% CIs are shown. Only statistically significant HRs are marked with ∗. ∗: p < 0.05.
We next sought to determine if PIK3CB is associated with chemoresistance in GBMs, given its prognostic potential in IDH-wt MGMT-deficient primary GBMs. To test this, we compared untreated patients (Chemo–) with those treated (Chemo+). HR–vs+ shows the risk of death if untreated. We found that primary MGMT-deficient/IDH-wt GBMs responded well to chemo treatments if PIK3CB levels were low because Chemo– patients had much shorter long-term and Chemo+ patients lived longer than those untreated (Figures 3C and S14; HR–vs+ = 2.844, Log Rank p = 0.0414/Wilcoxon p = 0.0988). In contrast, primary MGMT-deficient/IDH-wt GBMs with high levels of PIK3CB exhibited limited benefits from chemotherapies manifested by no difference in prognosis between Chemo+ and Chemo– patients (Figure 3C; HR–vs+ = 1.224, Log Rank p = 0.6777/Wilcoxon p = 0.9761). We next analyzed the effect of PI3K genes on chemotherapies in Chemo+ patients. MGMT-deficient/IDH-wt patients with low levels of PIK3CB lived much longer than patients with high levels of PIK3CB (Figure S15; HRhigh vs low = 2.046, Log Rank p = 0.0447/Wilcoxon p = 0.0305), further confirming chemoresistance in patients with high levels of PIK3CB. Other PI3K kinases did not change the clinical outcomes of Chemo+ patients (Figures 3C, S14, and S15; p > 0.05), except that MGMT-deficient/IDH-wt patients with high levels of PIK3CD responded well to chemotherapy (Figure S14; HR = 2.759, Log Rank p = 0.0183/Wilcoxon p = 0.0706), thereby having a good prognosis (Figure S15; HR = 0.4427, Log Rank p = 0.0213/Wilcoxon p = 0.0632). MGMT-positive GBMs were resistant to chemotherapy regardless of levels of PI3K genes (Figures 3C, S14, and S15). Collectively, PIK3CB, but not other PI3K kinases, accurately predicts the survival and chemosensitivity of primary MGMT-deficient/IDH-wt GBMs, thereby serving as a prognostic biomarker.
PIK3CB/PI3Kβ, but not other PI3K kinases, activates PI3K signaling and regulates TMZ sensitivity in MGMT-deficient GBMs
Results from online datasets have strongly suggested that PI3K kinases play divergent roles in regulating GBM’s chemosensitivity and that PIK3CB/PI3Kβ could be the dominant kinase in this activity. To further clarify the role of PI3K kinases, we knocked down the individual kinases in MGMT-deficient SF295 cells49 or LN229 cells.50 We have previously shown30 that SF295 cells express elevated levels of PI3Kβ (PI3Kβ-high), and LN229 cells have low levels of PI3Kβ (PI3Kβ-low). Because PIK3CG/PI3Kγ was barely detected in GBM,30 we only tested shRNAs against PIK3CA/PI3Kα (shPIK3CA), PIK3CB/PI3Kβ (shPIK3CB), or PIK3CD/PI3Kδ (shPIK3CD). The knockdown efficiency of PI3K genes by these shRNAs has been validated previously.30 We found that shPIK3CA (blue bars), shPIK3CD (green bars), or 200 μM TMZ together with the control non-silencing shRNA (shNS, white bars) significantly reduced the viability of PI3Kβ-low LN229 cells, but not PI3Kβ-high SF295 cells (Figure 4A). Ablation of PIK3CB alone (red bars) induced a modest growth inhibition in both cells despite that more cytotoxic effect was found in SF295 cells. Of note, shPIK3CA (dotted blue bars), shPIK3CB (dotted red bars), or shPIK3CD (dotted green bars) exacerbated growth inhibition in both SF295 and LN229 cells upon a combinational use of TMZ. However, shPIK3CB/TMZ exhibited significantly stronger cytotoxicity to SF295 cells (p < 0.05 or p < 0.01) than shPIK3CA/TMZ and shPIK3CD/TMZ. There was no significance in LN229 cells received these combinational treatments (p > 0.05). Perturbation of PI3K kinases, particularly PIK3CD (Figure 4A, shPIK3CD), ameliorated the survival of LN229 cells, consistent with our previous work in which we have shown that PI-3065, a PI3Kδ inhibitor, induced significant growth inhibition in LN-229 cells.30 However, depleting PIK3CD had no effect on PI3K activation in PI3Kβ-low cells as shown in our earlier report.30 These results together suggest that the cytotoxic effect by inhibiting PIK3CD in PI3Kβ-low cells is independent of PI3K signaling. Hence, we have shown that PI3Kβ-high SF295 cells selectively respond to shPIK3CB/TMZ.
Figure 4.

PIK3CB/PI3Kβ, but not other PI3K kinases, activates PI3K signaling and regulates TMZ sensitivity in MGMT-deficient GBMs
(A) Combinations of TMZ and knockdown of individual PI3K kinases. PI3Kβ-high SF295 or PI3Kβ-low LN229 cells were transfected with viruses harboring non-silencing (NS) shRNA or shRNA of PIK3CA, PIK3CB, or PIK3CD. Cells were then treated with DMSO or 200 μM TMZ. 4 days after treatment, cell viability was measured using the MTS assay. Cells treated with shNS and DMSO serve as controls (100% of viability).
(B) Knockout of PI3Kα or PI3Kβ. PI3Kα/β-high U87MG cells were transduced with viruses harboring gRNA of non-targeting (NT) or PIK3CA. PI3Kβ-high SF295 cells were transduced with viruses harboring gRNA of NT or PIK3CB. Levels of PI3Kα, PI3Kβ, or PI3K signaling were monitored using immunoblotting. β-actin (ACTB) was the loading control.
(C) Combinations of TMZ and knockout of PI3Kα or PI3Kβ. PI3Kα/β-high U87MG or PI3Kβ-high SF295 cells were transduced with viruses having gRNAs of NT, PIK3CA, or PIK3CB. Cells were then treated with DMSO or 200 μM TMZ. 4 days after treatments, cell viability was measured using the MTS viability assay. Cells treated with NT gRNA and DMSO serve as controls (100% of viability).
(D) Overexpression of active AKT1, AKT2, or AKT3. PI3Kα/β-high U87MG cells were transduced with viruses having pBABE (vector control) or plasmids with active myristoylated AKT isoforms (pBABE-Myr-AKT1, pBABE-Myr-AKT2, or pBABE-Myr-AKT3). Cells were then treated with 20 μM of TGX-221 (PI3Kβ-selective inhibitor) and 200 μM TMZ. Cell viability was measured using the MTS assay. Cells treated with DMSO and pBABE serve as controls (100% of viability). Error bars are standard deviations derived from three to four independent replicates. Student’s t test and One-Way ANOVA were used to determine p values. ns: not significant.
To validate the above results, we treated MGMT-deficient U87MG cells that highly express both PI3Kα and PI3Kβ (PI3Kα/β-high) and PI3Kβ-high SF295 cells with a guide RNA (gRNA) of PIK3CA or PIK3CB, respectively. Compared to gRNA of non-targeting (NT), PIK3CA gRNA depleted PI3Kα but did not change levels of PI3Kβ and failed to mitigate PI3K activity in PI3Kα/β-high U87MG cells (Figure 4B, left panel). In contrast, PIK3CB gRNA ablated PI3Kβ without affecting PI3Kα and inactivated PI3K in PI3Kβ-high SF295 cells (Figure 4B, right panel). Given that PIK3CA gRNA had no effect on PI3Kβ expression, PI3Kβ is expected to retain PI3K activity upon PI3Kα-depletion as shown in Figure 4B. In PI3Kβ-depleted SF295 cells, PI3Kα is unable to compensate the loss of PI3Kβ to activate PI3K, however. This finding is consistent with our previous results in which shRNAs of PIK3CB, but not those of PIK3CA or PIK3CD, inactivated PI3K signaling in U87MG cells.30 Hence, PI3Kβ dominates in PI3K activation in PI3Kβ-high GBM cells. This selective activation of PI3K induced by PI3Kβ was further verified by the stronger cytotoxic effect exhibited by PIK3CB gRNA (red dotted bar) than PIK3CA gRNA (blue dotted bar) when combined with TMZ (Figure 4C, p < 0.05). To prove that PI3Kβ acts upstream of AKT in cell survival, we ectopically expressed active (myristoylated) AKT1, AKT2, and AKT3 in U87MG cells. Interestingly, activation of AKT1 (cyan bars) or AKT3 (light green bars), but not AKT2 (yellow bars) rendered GBM cells less responsive to the combination of TGX-221 and TMZ (Figure 4D). TGX-221 is a PI3Kβ-selective inhibitor as its IC50 to PI3Kβ activity is 100-fold lower than those to other PI3K kinases.51 The counteraction of active AKT1 and AKT3 against TGX-221 suggests that they act downstream of PI3Kβ. Indeed, the importance of AKT1 and AKT3 in GBM’s chemoresistance has been previously established.28,52,53,54 Moreover, PI3Kα-E545K, which constitutively activates AKT in U87MG and SF295 cells,29 rescued these cells from growth inhibition induced by TGX-21 at different doses (Figure S16), further verifying the vital role of activated PI3Kβ/AKT in GBM’s survival. Overall, our results have demonstrated that PIK3CB/PI3Kβ is more important than other PI3K kinases in activating PI3K and causing TMZ resistance.
PI3Kβ inhibition synergizes with TMZ to block the growth of PI3Kβ-high MGMT-deficient GBMs
The results described above strongly suggest that targeting PIK3CB/PI3Kβ could significantly impact TMZ resistance. We have previously found that PI3Kβ inhibitors are more selectively toxic to PI3Kβ-high GBMs than other PI3K inhibitors.29,30 We, therefore, hypothesized that the combination of PI3Kβ inhibitors and TMZ would yield synergistic cytotoxicity to PI3Kβ-high GBMs. To test this hypothesis, we treated MGMT-deficient SF295, LN229, and astrocytes with 200 μM TMZ, a lethal dose used in our earlier work.13 Compared to PI3Kβ-low LN229, PI3Kβ-high SF295 cells were resistant to 200 μM TMZ (Figure 5A, white bars). PI3K isoform-selective inhibitors such as PI3Kβ inhibitor TGX-221 (TGX, red bars), PI3Kδ inhibitor CAL101 (CAL; green bars), or PI3Kγ inhibitor CZC24832 (CZC, pink bars) did not induce remarkable growth inhibition at the dose of 20 μM; however, PI3Kα inhibitor BYL-719 (BYL, blue bars) or pan PI3K inhibitor BKM120 (BKM, purple bars) suppressed the viability of three cell lines significantly by itself. When combined with TMZ (dotted bars), TGX/TMZ yielded statistically lower cell viabilities than TGX or TMZ alone (p < 0.0001, 0.01, or 0.05 in SF295, LN229, and astrocyte, respectively). Other PI3K inhibitors either displayed no significant difference between combinations and single agents (e.g., BYL in SF295, CAL in all three cell lines, CZC in SF295, and BKM in astrocytes) or even promoted cell survival when combined with TMZ (e.g., CAL in astrocytes and CZC in LN229 and astrocytes). These diverse responses were more significant when using Excess of Bliss (EOB) to measure drug interaction (EOB >0, synergistic effect; EOB = 0, no effect; EOB <0, antagonistic effect). EOB of TGX/TMZ was 47.1% in SF295 cells, much higher than those of TMZ and other PI3K inhibitors (EOBs = −12.8%–5.2%). PI3K inhibitors, including TGX, did not synergize with TMZ in LN229 cells (EOB = −11.4%–0.6%) and astrocytes (EOB = −21.4% to −3%). This unique drug synergy between TMZ and the PI3Kβ inhibitor in PI3Kβ-high GBM cells has proven the necessity of selectively targeting PI3Kβ in overcoming GBM’s chemoresistance. Because TMZ at 200 μM induced significant cell death in astrocytes (Figure 5A, white bars in astrocytes), suggesting possible harmful effects on normal brain, we reduced TMZ’s dose to 50 μM and tested if 50 μM TMZ would synergize with TGX while having minimal side effects. We found that 20 μM TGX (red bars) or 50 μM TMZ (white bars) alone did not robustly inhibit the proliferation of PI3Kβ-high MGMT-deficient SF295 and U87MG cells; however, TGX/TMZ (dotted red bars) synergistically suppressed cell survival (Figure 5B). This effect was further verified by the EOB of 45.5% or 26.3% in SF295 and U87MG cells, respectively. No drug interactions were detected in PI3Kβ-low MGMT-deficient cells (EOB = −0.02%–2.2%). Whilst the EOB of TGX/TMZ was 6.5% in astrocytes, this combination had almost no effect on the viability of astrocytes, suggesting that TGX/TMZ at these doses has negligible side effects to normal brain. It seems that PI3Kβ is dispensable for astrocytes even though astrocytes express elevated levels of PI3Kβ.30
Figure 5.

PI3Kβ inhibition synergizes with TMZ to block the growth of PI3Kβ-high MGMT-deficient GBMs
(A) Combinations of TMZ and PI3K inhibitors. PI3Kβ-high SF295, PI3Kβ-low LN229, and PI3Kβ-high astrocytes were treated with 200 μM of TMZ and/or 20 μM of different PI3K inhibitors, e.g., PI3Kα inhibitor BYL719 (BYL in blue), PI3Kβ inhibitor TGX-221 (TGX in red), PI3Kδ inhibitor CAL101 (CAL in green), PI3Kγ inhibitor CZC24832 (CZC in pink), and the pan PI3K inhibitor BKM120 (BKM in purple).
(B) Combinations of 50 μM TMZ and 20 μM TGX. PI3Kβ-high SF295/U87MG, PI3Kβ-low LN229/A172, and astrocytes were treated with 50 μM of TMZ and/or 20 μM TGX. Cell viability was measured using the MTS viability assay.
(C) Mice experiments. 6–8 weeks old SCID/Beige mice were injected subcutaneously with SF295 cells. 11 days later, mice were treated with 7.5 mg/kg of TMZ and/or 40 mg/kg of TGX every other day through intraperitoneal injection. DMSO serves as the control. Excess of Bliss (EOB) was calculated based on a Bliss Independence model (See STAR methods for detail). The average of EOB (EOBave) was derived from EOBs of each time point. Error bars are standard deviations derived from three to four independent replicates or standard errors of 5–8 tumors. Student’s t test or One-Way ANOVA was used to determine p values. ns: not significant.
To further corroborate the results from cultured cells, we treated immunodeficient mice harboring human SF295 xenograft tumors with 40 mg/kg of TGX and/or 7.5 mg/kg of TMZ. While TGX-treated (red squares) or TMZ-treated (white squares) tumors grew slower than DMSO-treated tumors (Figure 5C, gray circles), there were no statistically significant differences in tumor volumes (p > 0.05). In stark contrast, the TGX/TMZ combination drastically lowered tumor volumes compared to each treatment alone (p < 0.05). Intriguingly, tumor volumes in mice treated with TGX or TGX/TMZ remained unchanged 11 days after 2-week treatment, indicating no growth of tumors, whereas TMZ-treated tumors continued to grow. This mimics the clinical progression of therapy-resistant GBMs after chemotherapy. Drug interaction analyses showed that TGX/TMZ exhibited a synergistic effect during and after treatments with an average of EOB (EOBave) of 18.5%. Collectively, our results have demonstrated that PI3Kβ inhibition synergizes with TMZ to block the growth of PI3Kβ-high MGMT-deficient GBMs.
PI3Kβ inhibitors overcome TMZ resistance in PI3Kβ-high GSCs
Prior research has found that a small population of GBM cells, named GSCs, is expected to be more resistant to TMZ than differentiated GBM cells.55,56 Our laboratory has isolated some PI3Kβ-high GSC lines from patient specimens.30,57 To test if PI3Kβ inhibition could have the same cytotoxicity as GSCs, we treated PI3Kβ-high VTC-103/GSCs, PI3Kβ-low VTC-056/GSCs, and neural stem cells (NSC) with a combination of TMZ and PI3Kβ inhibitors. Here, we employed the sphere-forming assay to evaluate GSCs’ ability to self-renew. We found that 100 μM TMZ (white bars) had no effect on GSCs’ self-renewal regardless of levels of PI3Kβ (Figures 6A and 6B), proving the TMZ resistance in GSCs. Unexpectedly, 20 μM TGX remarkably suppressed the self-renewal of VTC-103/GSCs (Figure 6A) and NSCs (Figure 6C) with no effect on that of VTC-056/GSCs (Figure 6B) and this cytotoxic effect was exacerbated upon a combination of TMZ (EOBs = 8.3% in VTC-103/GSC and 2.1% in NSCs). Because VTC-103/GSCs and NSCs were overly sensitive to TGX, it would be difficult to measure drug synergy. To resolve this issue, we treated cells with a different PI3Kβ inhibitor, AZD6482 (AZD,58). Unlike TGX, 10 μM AZD by itself had no to limited effect on three cell lines (burnt sienna bars) but yielded a synergistic effect together with TMZ exclusively in VTC103/GSC (EOB = 79.2%) with no drug effect on VTC-056/GSC (EOB = 0%) or NSCs (EOB = −0.08%). These results together have demonstrated a crucial role of PI3Kβ in regulating chemoresistance in GSCs.
Figure 6.

PI3Kβ inhibitors overcome TMZ resistance in PI3Kβ-high GSCs
The sphere-forming assay was used to measure the self-renewal of PI3Kβ-high VTV103/GSC (A), PI3Kβ-low VTC-056/GSC (B), or NSC (C). Cells were treated with 20 μM TGX-221 (TGX), 10 μM AZD6482 (AZD), or 100 μM TMZ, as well as combinations of TGX/TMZ or AZD/TMZ. Cells were imaged using an inverted microscope. Single or sick cells were indicated by red arrows. Wells with healthy spheres were scored, and percentages of wells with spheres (numbers of wells with spheres/numbers of all wells in each treatment group) were plotted. EOBs were calculated using the Bliss Independence model. Error bars stand for standard deviations derived from three to four independent replicates. Student’s t test was used to determine p values between two groups. ns: not significant. Scale bar: 50 μm.
Discussion
GBM accounts for nearly half of all primary brain tumors, but patients continue to exhibit poor clinical outcomes.1 Although many drugs have entered GBM clinical trials, none have shown better antitumoral effects than TMZ, the standard GBM treatment since its approval in March 2005. The combination of TMZ and other treatments, such as radiation, improves median survival and quality of life.4,11,59,60 However, the benefits of TMZ treatment remain palliative due to the inevitable development of TMZ resistance. It is, therefore, imperative to better understand the mechanisms underlying TMZ resistance to maximize TMZ’s efficacy for GBM. In this report, we have found that not only is PIK3CB/PI3Kβ expressed at the highest level among the four PI3K kinases in primary MGMT-deficient GBMs, but its mRNA levels are also associated with levels of PI3K activation, levels of TMZ sensitivity, as well as GBMs’ clinical outcomes. Moreover, PI3Kβ prevails in activating PI3K and inducing chemoresistance over other PI3K kinases in MGMT-deficient GBMs, including GSCs. Our results collectively have demonstrated that selective activation of PIK3CB/PI3Kβ is a dominant mechanism by which MGMT-deficient GBMs develop TMZ resistance; as such, selective blockade of PIK3CB/PI3Kβ is a preferable therapeutic approach to circumventing chemoresistance.
The PI3K signaling appears to be a common denominator in TMZ resistance; however, the most effective strategy for achieving pathway inhibition is crucial but challenging to identify, given the divergent roles of PI3K kinases in cancers.18 Class I PI3K kinases are most implicated in GBM, given their dominant roles in regulating pathway activity. Research presented in this report has focused on class IA subunits because we have previously shown that the class IB subunit is minimally expressed in GBM.30 PIK3CA is a commonly mutated gene across all cancers, but mutation rates are variable and generally low in GBM.61,62,63,64 Genetic ablation of PIK3CA and other PI3K genes does show cytotoxicity to GBM.28,65 However, the high toxicity profile and low efficacy of PIK3CA/PI3Kα inhibitors in the clinic have made this kinase not an appealing drug target for GBM.66 For instance, while PIK3CA/PI3Kα and PIK3CB/PI3Kβ are both widely expressed in almost all tissues including pancreas and PIK3CD/PI3Kδ and PIK3CG/PI3Kγ are almost exclusively expressed in immune cells,18 it is PIK3CA/PI3Kα, but not other PI3K isoforms, activates insulin signaling and controls glucose metabolism67; as such, hyperglycemia is the most common side effect caused by the blockade of PIK3CA/PI3Kα in human patients, which has significantly narrowed the therapeutic window of PIK3CA/PI3Kα inhibitors.66 In line with our observation that astrocytes are sensitive to the PI3Kα inhibitor BYL719, which suggests that inhibiting PIK3CA/PI3Kα could yield significant toxicity to the normal brain, PIK3CA/PI3Kα is not the first therapeutic choice for brain malignancy. The role of PIK3CD/PI3Kδ and PIK3CG/PI3Kγ in GBM has not yet been extensively studied, possibly because they are mainly involved in hematopoietic disorders.68,69 Targeting PIK3CD/PI3Kδ and/or PIK3CG/PI3Kγ is also not good choice for GBM given the potential toxicity to immune systems and our results shown in this study that CAL101 (PI3Kδ inhibitor) or CZC24832 (PI3Kγ inhibitor) exhibited no or limited effect on GBMs. Of the four PI3K kinases, PIK3CB/PI3Kβ is most strongly correlated with GBM survival and recurrence, more specifically regulates AKT activation, proves the most significant and consistent cytotoxicity to GBMs, and reduces cell migration to suppress tumor invasiveness.30,43,44,58,70,71,72,73,74,75,76 Results from these studies are not only congruent with our finding that PIK3CB/PI3Kβ is a selective survival factor for GBM30 but also supply a staunch support to the conclusion derived from this report that PIK3CB/PI3Kβ is a preferred drug target for overcoming GBM’s chemoresistance.
It is still not fully understood why PI3K kinases show divergent functional activities despite significant structural homology. Past research has suggested that the functional divergence among PI3K kinases stems from their different affinities to cell membrane receptors. For instance, PIK3CA/PI3Kα, PIK3CB/PI3Kβ, or PIK3CD/PI3Kδ can interact with RTKs to activate PI3K signaling, while only PIK3CB/PI3Kβ or PIK3CG/PI3Kγ can signal through G-protein coupled receptors.18,73 This explains the unique and dominant activity of PIK3CB/PI3Kβ in GBM and other cancers. Recent research has also revealed that PIK3CB/PI3Kβ is structurally different from other wild-type PI3K kinases, making PIK3CB/PI3Kβ more tumorigenic. For example, wild-type PI3Kβ and mutated PI3Kα (PI3KαE542K, PI3KαE545K, or PI3KαH1047R), but not wild-type PI3Kα, activate PI3K and transform chicken embryo fibroblasts77 or human mammary epithelial cells.78,79 Deletion of PI3Kβ, but not PI3Kα, inhibits the formation of PTEN-deficient prostate cancer,80 colon cancer,44 and brain cancer.30,44,73 Consistently, both wild-type PI3Kβ and oncogenic PI3KαN345K can transform NIH3T3cells.81 However, further studies are needed to characterize structural differences among these PI3K kinases to understand how they function distinctly at atomic levels.
Another gene in close association with the PI3K/AKT pathway that exerts a significant regulatory role is PTEN. PTEN antagonizes the enzymatic reaction of PI3K kinases by dephosphorylating PIP3 into PIP2, thereby inhibiting PI3K/AKT pathway activation. PTEN is the most mutated PI3K gene in GBM,15,16 undergoing inactivating mutations that contribute heavily to aberrant PI3K/AKT activation and TMZ resistance.82,83,84,85 While PTEN inactivation fails to dephosphorylate PIP3, the full activation of PI3K signaling still requires a PI3K kinase. It is therefore possible that PIK3CB/PI3Kβ could synergize with PTEN loss to fully activate PI3K. Consistent with this idea, PIK3CB/PI3Kβ has been found in driving the tumor growth and proliferation in PTEN-deficient GBMs while playing a lesser role when PTEN function is intact.43,44 However, contradictory to this idea, we and others have shown that PTEN levels in GBM patients have no correlations with GBM’s prognosis, particularly in the setting of chemo treatments and tumor recurrence.30,86 We have shown herein that PI3Kβ blockade inactivated PI3K and restored TMZ sensitivity in PTEN-null/PI3Kβ-high SF295 and U87MG cells,87 but failed to do so in both PTEN-positive/PI3Kβ-low LN22988 and PTEN-null A172/PI3Kβ-low A172 cells.30,31,89 Our current study has therefore verified that PTEN or PTEN mutations are dispensable in predicting PI3K activation and TMZ sensitivity at least in a subset of GBMs.
DNA repair mechanisms such as MGMT, mismatch repair, and base excision repair are the most recognized sources of MGMT-dependent or -independent TMZ resistance; however, many molecular pathways also mediate TMZ resistance. One such mediator is the epidermal growth factor receptor (EGFR), which is amplified in a majority of GBM tumors.90,91 Unfortunately, inhibitors of EGFR are highly susceptible to drug resistance because approximately half of amplified EGFR in GBM is of a constitutively activated mutant form EGFRvIII unresponsive to competitive inhibition.92,93 Downstream effectors of EGFR, namely PI3K and its effector AKTs, are thus more efficacious targets as they circumvent such activating mutations. Indeed, PI3K pathway activation is tied to many mediators of TMZ resistance, making it the ideal candidate to mitigate TMZ resistance. For example, PI3K/AKT stabilizes hypoxia-inducible factor 1, which causes increased expression of forkhead box protein M1 that reinforces angiogenesis in TMZ-resistant GBMs.94,95 PI3K/AKT also activates nuclear factor kappa B, leading to increased transcription of MGMT expression and TMZ resistance,96 or forkhead O transcription factors that cause disequilibrium of apoptotic proteins to promote the escape of GBM cells from chemotherapeutics-induced cytotoxicity.97 Moreover, inhibition of PI3K/AKT downstream targets mTOR, JAK/STAT, or B-cell lymphoma 2 activates autophagy and overcomes TMZ resistance.98,99,100,101 In this report, we have found that three AKT isoforms have different activities downstream of PI3Kβ, with AKT1 and AKT3 being the effector kinases involved in TMZ resistance. Our results are consistent with the work from other groups in which they have found that knockdown of AKT1 or AKT3 increases chemosensitivity28,54 and reinforced expression of AKT3 promotes DNA repair and TMZ resistance.52 However, other reports also suggest the involvement of AKT2 in TMZ resistance.102,103 Moving forward, it is essential to further investigate how PIK3CB/PI3Kβ activates its downstream effector proteins to induce chemoresistance in GBM and how to selectively target this PI3K kinase to circumvent GBM’s chemoresistance.
Blocking PI3K signaling by non-selectively inactivating all PI3K kinases has been proven ineffective in the clinic because it yields severe side effects,22,23,24,25 which has substantially hindered the translation of PI3K-based therapies. Such a clinical challenge has prompted an extensive investigation of PI3K isoform-selective functions in various types of cancer including GBM.26,27,28,29,30,31 We have previously shown that certain GBMs depend on PIK3CB/PI3Kβ, but not other isoforms, to survive and grow and that, in a stark contrast, PIK3CB/PI3Kβ is dispensable to normal astrocytes.30,31 Congruent with these results, our work presented in this study has furthered our understanding of PIK3CB/PI3Kβ in TMZ resistance independent of MGMT and added more weight on choosing PIK3CB/PI3Kβ as the prime drug target for difficult-to-treat GBMs. Furthermore, our results have suggested that a subset of MGMT-deficient GBMs expressing high levels of PI3Kβ would benefit from PI3Kβ-based therapies; hence, a preselection of GBMs with methylated MGMT promoters and high levels PIK3CB mRNAs can be a better therapeutic strategy for clinical trials using PI3Kβ drugs. This strategy can also explain the modest effect from the PI3Kβ-selective inhibitor GSK2636771 in PTEN-deficient solid tumors because it is not known of these tumors express high levels of PIK3CB mRNAs.104 Taken together, the research presented herein has demonstrated a strong translational potential of PI3Kβ drugs as clinically applicable treatments for TMZ-resistant GBMs and perhaps other cancers that depend upon PIK3CB/PI3Kβ to survive and grow.
Limitations of the study
In this study, we have identified divergent roles of PI3K catalytic kinases in chemoresistance in GBM and found that PI3Kβ serves as a key signaling point in the response of GBMs to TMZ and a prime target for overcoming TMZ resistance. While we have obtained the results in established GBM cell lines, primary GSCs derived from patients, as well as xenograft animal models, we have not yet been able to verify our key findings in orthotopic GBM mouse models that better mimic human diseases and have challenges in intracranial drug delivery. In fact, the blood-brain barrier is the major hurdle for delivering PI3Kβ inhibitors into the brain and translating our findings into the clinic to help patients. Moreover, we have also not examined the complexity in PI3K activation (e.g., genetic mutations or other survival signaling working in parallel or in concert with PI3K) in the context of drug resistance in GBM patients. We will resolve these issues in our future studies.
Declaration of generative AI and AI-assisted technologies in writing process
Authors declare that no generative AI or AI-assisted technologies in writing process were used in preparing this manuscript. Authors take full responsibility for the content of this publication.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-phospho-AKT-S473 | Cell Signaling Technology | Cat#4051; RRID:AB_331158 |
| Anti-AKT | Cell Signaling Technology | Cat#4685; RRID:AB_2225340 |
| Anti-phospho-AKT-T308 | Cell Signaling Technology | Cat#4056; RRID:AB_331163 |
| Anti-p110α | Cell Signaling Technology | Cat#4249; RRID:AB_2165248 |
| Anti-p110β | Cell Signaling Technology | Cat#3011; RRID:AB_2165246 |
| Anti-β-actin | Millipore-Sigma | Cat#A3854; RRID:AB_262011 |
| Chemicals, peptides, and recombinant proteins | ||
| Human FGF-basic (FGF-2/bFGF) (154 aa) Animal-Free Recombinant Protein PeproTech® | Thermo Fisher Scientific | Cat#AF-100-18B-1MG |
| Human EGF, Animal-Free Recombinant Protein, PeproTech® | Thermo Fisher Scientific | Cat#AF-100-15-1MG |
| AZD6482 | AdooQ Bioscience | Cat#A10112 |
| BKM120 | AdooQ Bioscience | Cat#A11016 |
| CAL101 | AdooQ Bioscience | Cat#A10172 |
| CZC24832 | AdooQ Bioscience | Cat#A11363 |
| TGX-221 | AdooQ Bioscience | Cat#A10923 |
| BYL719 | AdooQ Bioscience | Cat#A11328 |
| Temozolomide | AbMole BioScience | Cat#M2129 |
| Dimethyl sulfoxide | Millipore Sigma | Cat#41639 |
| Puromycin dihydrochloride from Streptomyces alboniger | Millipore Sigma | Cat#P8833 |
| Blasticidin S HCl | Thermo Fisher Scientific | Cat#A1113903 |
| Critical commercial assays | ||
| Effectene Transfection Reagent | QIAGEN | Cat#301425 |
| CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) | Promega | Cat#G3582 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat#34580 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat#34096 |
| High Concentration Corning Matrigel Matrix | Corning | Cat#354248 |
| Experimental models: Cell lines | ||
| U87MG | ATCC | Cat#HTB-14; RRID:CVCL_XZ66 |
| SF295 | Millipore Sigma | Cat#SCC289; RRID:CVCL_1690 |
| LN229 | ATCC | Cat#CRL-2611; RRID:CVCL_0393 |
| A172 | ATCC | Cat#CRL-1620; RRID:CVCL_0131 |
| NHA human astrocytes | Lonza | Cat#cc-2565 |
| HEK293T | ATCC | Cat#CRL-3216; RRID:CVCL_0063 |
| Experimental models: Organisms/strains | ||
| Scid/Beige (C.B-Igh-1b/GbmsTac-Prkdcscid-Lystbg N7) | Taconic Biosciences | Cat#CBSCBG-F; RRID:IMSR_TAC:CBSCBG |
| Software, algorithms, and public datasets | ||
| The Cancer Dependency Map | https://depmap.org | 22Q1; Protein_Array; Proteomics; drug_sensitivity |
| The Cancer Gene Atlas | https://www.cbioportal.org | gbm_tcga |
| The China Glioma Gene Atlas | http://www.cgga.org.cn/download.jsp | mRNAseq_693 |
| The Genotype-Tissue Expression | https://gtexportal.org | gene_tpm_brain_cortex |
| The Human Protein Atlas | https://www.proteinatlas.org | Glioma |
| Plasmids | ||
| pBABE-Myr-AKT1 | Addgene | Cat#15266 |
| pBABE-Myr-AKT2 | Addgene | Cat#9018 |
| pBABE-Myr-AKT3 | Addgene | Cat#9019 |
| PIK3CA gRNA | Addgene | Cat#77415 |
| PIK3CB gRNA | Addgene | Cat#76191 |
| PIK3CB gRNA | Addgene | Cat#76192 |
| pLenti-Cas9-Blast | Addgene | Cat#52962 |
| psPAX2 | Addgene | Cat#12260 |
| pMD2.G | Addgene | Cat#12259 |
| PIK3CA shRNA RHS4844-101656239 | Horizon Discovery | Cat#RHS4012 |
| PIK3CB shRNA RHS4884-10165656350 | Horizon Discovery | Cat#RHS4012 |
| PIK3CD shRNA RHS4884-101655755 | Horizon Discovery | Cat#RHS4012 |
| Other | ||
| Dulbecco’s Modified Eagle Medium | Thermo Fisher Scientific | Cat#12100061 |
| EqualFETALTM bovine serum | Atlas Biologicals | Cat#EF-0500-A |
| Streptomycin & penicillin, Gibco® | Thermo Fisher Scientific | Cat#15140163 |
| Trypsin-EDTA | Santa Cruz Biotechnology | Cat#sc-363354 |
| AGM™ Astrocyte Growth Medium | Lonza | Cat#cc-3138 |
| Fetal Bovine Serum | Atlas Biologicals | Cat#F-0500-A |
| MEM Non-Essential Amino Acids Solution | Thermo Fisher Scientific | Cat#11140050 |
| B-27™ Supplement, Gibco® | Thermo Fisher Scientific | Cat#17504001 |
| TrypLE™ Express Enzyme | Thermo Fisher Scientific | Cat#12605010 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zhi Sheng (zhisheng@vtc.vt.ed).
Materials availability
This study did not generate any new reagents.
Data and code availability
This paper analyzes publicly available data from TCGA or DepMap databases. Information regarding datasets analyzed in this study are listed in the key resources table. This study did not report any original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Cell lines
In this study, four cell lines U87MG, SF295, LN229, and A172 were purchased from and authenticated by ATCC or Millipore Sigma. The authentication essay was used to identify short tandem repeat (STR) markers. Cell lines were also tested for mycoplasma contamination using the ATCC universal mycoplasma detection kit. The U87MG cell line was derived from malignant gliomas of a male patient. The SF295 cell line was derived from GBM of a female patient. The LN229 cell line was derived from GBM of a female patient. The A172 cell line was derived from GBM of a male patient. All cell lines were cultured as described previously.29,30,57,105 Cells were maintained in the Dulbecco’s Modified Eagle Medium supplemented with 10% EqualFETALTM bovine serum, 100 mg/mL streptomycin and 100 IU/mL penicillin. The human astrocyte cell line NHA was purchased from Lonza. NHA cells were cultured in Astrocyte Growth Media having 10% fetal bovine serum, 100 mg/mL streptomycin, 100 IU/mL penicillin, and 0.1 mM MEM nonessential amino acid. When passaging cells, cells were treated with 0.05% Trypsin-EDTA for 2 to 5 min and resuspended in the culture media. After cell counting using a hemacytometer, ∼3-5 × 105 cells were plated in a new 10-cm dish. Detailed information regarding cell lines and culturing reagents can be found in the key resources table.
Primary cultures
VTC-103/GSCs or VTC-056/GSCs were derived from human GBM specimens in the Sheng lab previously.30,57 These human patient specimens were from the Department of Neurosurgery of Carilion Clinic. Because the patient information was recorded by Carilion Clinic and not available to the Sheng lab, sex of these two primary cell lines cannot be reported herein. GBM stem cells were cultured as described previously.30,57 Culture media for GBM stem cells include Dulbecco’s Modified Eagle Medium supplemented with 50X GibcoÒ B-27Ò supplement, 20 ng/mL FGF-2, and 20 ng/mL EGF. GBM stem cells grew as spheres slowly and media were changed weekly. To passage GBM stem cells, spheres were dissociated into single cells by incubating spheres in TrypLE Express Enzyme for 5 to 10 min. After cell counting, ∼3-5 × 105 single stem cells were plated in a T-25 flask.
In vivo animal models
Animal experiments were approved by the Institutional Animal Care and Use Committee of Virginia Tech. This study used 6–8 weeks old female SCID/beige mice purchased from Taconic Bioscience. The nomenclature of this stain is C.B-Igh-1b/GbmsTac-Prkdcscid-Lystbg N7. The genotype is sp/sp; sp/sp. Since our studies did not focus on difference in gender, we used female mice because they were less aggressive when performing cell injections and drug treatments. For drug treatment, mice were randomly allocated into two groups and each group had two to three cages. Detailed information can be found in the key resources table.
Method details
Reagent preparation
Most chemicals listed in the key resources table were reconstituted in dimethyl sulfoxide (DMSO) at a concentration of 50–80 mM. Puromycin was dissolved in sterile water at a concentration of 5 mg/mL. All chemicals were aliquoted (to avoid repeated freeze/thaw cycles that decrease drug activity) and stored at −80° based on our previously published work.29 We previously purchased a TRC kinase shRNA library from Thermo Fisher Scientific, which was discontinued recently. However, the entire library of human shRNAs or individual shRNAs are available at Horizon Discovery. Other plasmids listed in the key resources table were purchased from Addgene. All plasmids were built on lenti- or retro-virus backbone. They were used to generate viruses together with the packaging plasmids psPAX2 and pMD2.G in HEK293T cells. These viruses were aliquoted and stored at −80 freezer and subsequently used to infect cells and transfer DNAs of interest into GBM cells, as described previously.29
Analyses of online databases
Analysis of online databases has been described previously.29,30,106 Datasets of RNA sequencing, immunohistochemical staining, and drug treatments were downloaded from the following databases: (1) The Cancer Dependency Map: https://depmap.org; (2) The Cancer Gene Atlas datasets: https://www.cbioportal.org; (3) The Genotype-Tissue Expression: https://gtexportal.org; (4) The China Glioma Gene Atlas datasets: http://www.cgga.org.cn/download.jsp. Details can be found in the key resources table. The Kaplan-Meier survival analysis and the Cox hazard proportional model were used to acquire hazard ratios, which indicate the risk level of death based on gene expression levels. These analyses were performed using the JMP software. The Pearson correlation coefficient, calculated using Prism 9 software, was used to determine the expression correlation between different genes/proteins and correlations between gene levels and TMZ’s sensitivity quantified as IC50s.
Image analyses
The immunohistochemically stained images were downloaded from The Human Protein Atlas: https://www.proteinatlas.org. Four persons independently quantified immunohistochemically stained images retrieved from The Human Protein Atlas using the ImageJ software. For each image, each person randomly selected 10 different visible cells and measured the intensities of proteins inside cells. 10 intensity readings were averaged to acquire relative protein staining intensities. Measurements from four persons’ experiments were considered as individual data points, where standard errors or standard deviations were derived.
Gene knockout or knockdown
To knock out genes using CRISPR-Cas9 or knock down genes using shRNA, plasmids encoding Cas9, gRNAs, or shRNAs were transfected together with plasmids encoding virus-packaging proteins into HEK293T cells using Effectene. Viral supernatants were collected to infect GBM cells. For CRISPR-Cas9 knockout, GBM cells were selected using blasticidin (5 μg/mL for Cas9 expression) and puromycin (0.5 μg/mL for gRNA expression) for 5 to 7 days. For shRNA knockdown, GBM cells were selected by 0.5 μg/mL puromycin for 7–10 days. Transduced cells were collected for immunoblotting of PI3Kα or PI3Kβ to ensure gene knockout or knockdown or used for drug combination analyses. Details regarding these reagents can be found in the key resources table.
Drug treatment and analyses
To treat cells with single drug or drug combinations, 500 to 2,000 GBM cells were plated into a well of 96-well plates. The next day, media were replaced with drug-containing media, which were prepared from drugs at stock concentrations (usually ∼1,000-fold of working concentrations). For TMZ dose-dependent effect, TMZ at different concentrations (0, 25, 50, 100, 200, 400 μM) was added to cells. DMSO was used as the vehicle control. After 4-day treatment, cell viability was measured using the MTS viability assay as described below. Prism 10 software was used to calculate the half-maximal inhibitory concentration (IC50s) of TMZ. To calculate drug constructive interaction, a Bliss Independence model was used as described previously.29 The predicted cytotoxic effect for dual therapies was calculated using Effect(a+b) = Effecta+Effectb−EffectaEeffectb. The overall cytotoxic synergistic effect was presented as Excess Over Bliss (EOB) scores. EOB >0% shows a synergistic effect, while EOB = 0% shows an additive effect, and EOB <0% shows an antagonistic effect. For animal experiments described below, EOB was calculated based on the results of each time point. EOBave was the average of EOBs from all time points after cell injections.
MTS viability assay
Cell viability was measured using the MTS cell viability assay as described previously.29,30,107 After drug treatment, CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS, Promega) was added to cells and incubated in the dark for 2 h. The absorbance at 490 nm was measured using a FilterMax F3 microplate reader (Molecular Devices). After subtracting 490 nm absorbances of experimental wells with those of blank wells, normalized absorbances of treatment groups were divided by absorbances of respective vehicle control groups, yielding percentages of viability. Error bars are standard deviations from three to four independent replicates.
Sphere-forming assay
A sphere-forming assay was done as previously described.57 ∼ 50–100 GBM stem cells or neural stem cells were plated in a well of 96-well plate. Cells were treated with single agents or combinations, as shown in the figure. Each treatment group had 8 wells. After 4-day treatment, wells with spheres (a spheroid culture that is > 3-fold larger than a single cell) were scored. Images of representative spheres were taken using an inverted microscope. Percentages of wells with spheres in each treatment group were calculated by dividing the number of wells with spheres by the number of wells in each treatment group. Error bars stand for standard deviations from three to four independent replicates.
Immunoblotting
Immunoblotting was done as described previously.29,30,107 In brief, more than 5 × 106 cells were lysed in 50–100 μL of cell lysis buffer including 20 mM HEPES pH 6.8, 140 mM NaCl, 2.5 mM MgCl2, 2.5 mM CaCl2, 1% NP40, 0.5% sodium deoxycholate, protease inhibitor, and phosphatase inhibitors. Cell lysates were collected by centrifugation and concentrations of total proteins were quantified using Coomassie blue staining. 25 to 50 μg total proteins were resolved and separated on SDS-PAGE gels and transferred onto PVDF membranes. Antibodies were purchased from Cell Signaling Technology or from Millipore-Sigma (See the key resources table). Dilution rates of antibodies were 1:1,000 (Anti-PI3Kα, Anti-PI3Kβ, Anti-phospho-AKT-S473, Anti-phospho-AKT-T308, Anti-AKT) or 1:5,000 (Anti-β-actin). Images of chemiluminescence from SuperSignal West Pico or Femto substrates (Thermo Fisher Scientific) were taken using the ChemiDoc MP (BioRad). Images were analyzed using Image Lab (BioRad). Exported images were further cropped in Adobe PhotoShop.
Xenograft mouse models
Animal experiments were done based on previously published work29,30 and were approved by the Institutional Animal Care and Use Committee of Virginia Tech. 2 × 106 cells were premixed with Matrigel Matrix in a cold room and subcutaneously injected into 8 weeks old SCID/beige mice (referring the key resources table for details). 11 days after cell injection, 6 to 8 mice were randomized into different treatment groups. The sample size was determined based on power analysis using G∗Power. Mice were treated with individual drugs (TMZ or TGX-221) or drug combinations (TMZ and TGX-221) at doses shown in the figure. Drugs were delivered using intraperitoneal injection. Tumors were measured every other day using a digital caliper. The endpoints set for animal experiments are, if mice who are significantly ill or with 10% weight loss, they will be excluded from the experiment. In the experiment presented in this study, no mice were excluded based on these endpoints. After 14-day treatment, tumor growth was monitored for 11 more days. On day 35 post-cell infection, mice were euthanized. Tumor volumes (mm3) were acquired using the formula (length x width2)/2.
Quantification and statistical analysis
The distribution and variation of data points in each experiment were analyzed and estimated. Three to four independent experiments were performed in cell-based assays, yielding standard deviations shown as error bars. Drug combinations were repeated in different cell lines. One-way ANOVA with Dunnett test for correction of multiple comparisons, Fisher’s exact test, and Student’s t test were used to determine statistical significance and similarity of variance. All tests were two-sided. The center values shown in each figure were means. Details regarding statistical analyses were also provided in Figure Legends where appropriate.
Acknowledgments
The results published here are in part based upon data generated by The Cancer Genome Atlas Research Network, the Dependency Map, the Genotype-Tissue Expression, or the Chinese Glioma Genome Atlas. This study is supported by National Institutes of Health (NIH) R21 grants R21CA216768 and R21CA245631 to Z.S., VTCSOM medical student research fellowship to K.R.H and E.M., and Translational Neurobiology Summer Undergraduate Research Fellowships from the Fralin Biomedical Research Institute to J.S.
Author contributions
Z.S. and K.J.P. conceptualized this study. Z.S., K.J.P, K.R.H., P.B., M.L., E.X., E.M., V.B., C.P., J.S., and L.H. made contributions to investigation/methodology. Z.S., K.R.H, and P.B. wrote manuscript. Z.S. acquired funds to support this research. Z.S., A.T., and D.F.K. curated/analyzed data and provided resources.
Declaration of interests
All authors declare that they have no conflicts of interest.
Published: May 7, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109921.
Supplemental information
References
- 1.Ostrom Q.T., Price M., Neff C., Cioffi G., Waite K.A., Kruchko C., Barnholtz-Sloan J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016-2020. Neuro Oncol. 2023;25:iv1–iv99. doi: 10.1093/neuonc/noad149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tisdale M.J. Antitumour imidazotetrazines-X. Effect of 8-carbamoyl-3-methylimidazo[5,1-d]-1,2,3,5-tetrazin-4-(3H)-one (CCRG 81045; M & B 39831; NSC 362856) on DNA methylation during induction of haemoglobin synthesis in human leukaemia cell line K562. Biochem. Pharmacol. 1986;35:311–316. doi: 10.1016/0006-2952(86)90531-9. [DOI] [PubMed] [Google Scholar]
- 3.Tan A.C., Ashley D.M., López G.Y., Malinzak M., Friedman H.S., Khasraw M. Management of glioblastoma: State of the art and future directions. CA. Cancer J. Clin. 2020;70:299–312. doi: 10.3322/caac.21613. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J.B., Belanger K., Brandes A.A., Marosi C., Bogdahn U., et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Tisdale M.J. Antitumor imidazotetrazines--XV. Role of guanine O6 alkylation in the mechanism of cytotoxicity of imidazotetrazinones. Biochem. Pharmacol. 1987;36:457–462. doi: 10.1016/0006-2952(87)90351-0. [DOI] [PubMed] [Google Scholar]
- 6.Lee S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016;3:198–210. doi: 10.1016/j.gendis.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butler M., Pongor L., Su Y.T., Xi L., Raffeld M., Quezado M., Trepel J., Aldape K., Pommier Y., Wu J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer. 2020;6:380–391. doi: 10.1016/j.trecan.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegi M.E., Diserens A.C., Gorlia T., Hamou M.F., de Tribolet N., Weller M., Kros J.M., Hainfellner J.A., Mason W., Mariani L., et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 9.Brandes A.A., Franceschi E., Paccapelo A., Tallini G., De Biase D., Ghimenton C., Danieli D., Zunarelli E., Lanza G., Silini E.M., et al. Role of MGMT Methylation Status at Time of Diagnosis and Recurrence for Patients with Glioblastoma: Clinical Implications. Oncologist. 2017;22:432–437. doi: 10.1634/theoncologist.2016-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hegi M.E., Liu L., Herman J.G., Stupp R., Wick W., Weller M., Mehta M.P., Gilbert M.R. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. 2008;26:4189–4199. doi: 10.1200/JCO.2007.11.5964. [DOI] [PubMed] [Google Scholar]
- 11.Stupp R., Hegi M.E., Mason W.P., van den Bent M.J., Taphoorn M.J.B., Janzer R.C., Ludwin S.K., Allgeier A., Fisher B., Belanger K., et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 12.Nakada M., Furuta T., Hayashi Y., Minamoto T., Hamada J.I. The strategy for enhancing temozolomide against malignant glioma. Front. Oncol. 2012;2:98. doi: 10.3389/fonc.2012.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy S.F., Varghese R.T., Lamouille S., Guo S., Pridham K.J., Kanabur P., Osimani A.M., Sharma S., Jourdan J., Rodgers C.M., et al. Connexin 43 Inhibition Sensitizes Chemoresistant Glioblastoma Cells to Temozolomide. Cancer Res. 2016;76:139–149. doi: 10.1158/0008-5472.CAN-15-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh N., Miner A., Hennis L., Mittal S. Mechanisms of temozolomide resistance in glioblastoma - a comprehensive review. Cancer Drug Resist. 2021;4:17–43. doi: 10.20517/cdr.2020.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parsons D.W., Jones S., Zhang X., Lin J.C.H., Leary R.J., Angenendt P., Mankoo P., Carter H., Siu I.M., Gallia G.L., et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoxhaj G., Manning B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer. 2020;20:74–88. doi: 10.1038/s41568-019-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thorpe L.M., Yuzugullu H., Zhao J.J. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi F., Guo H., Zhang R., Liu H., Wu L., Wu Q., Liu J., Liu T., Zhang Q. The PI3K inhibitor GDC-0941 enhances radiosensitization and reduces chemoresistance to temozolomide in GBM cell lines. Neuroscience. 2017;346:298–308. doi: 10.1016/j.neuroscience.2017.01.032. [DOI] [PubMed] [Google Scholar]
- 20.Ji M., Zhang Z., Lin S., Wang C., Jin J., Xue N., Xu H., Chen X. The PI3K Inhibitor XH30 Enhances Response to Temozolomide in Drug-Resistant Glioblastoma via the Noncanonical Hedgehog Signaling Pathway. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.749242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prasad G., Sottero T., Yang X., Mueller S., James C.D., Weiss W.A., Polley M.Y., Ozawa T., Berger M.S., Aftab D.T., et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol. 2011;13:384–392. doi: 10.1093/neuonc/noq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pitz M.W., Eisenhauer E.A., MacNeil M.V., Thiessen B., Easaw J.C., Macdonald D.R., Eisenstat D.D., Kakumanu A.S., Salim M., Chalchal H., et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro Oncol. 2015;17:1270–1274. doi: 10.1093/neuonc/nou365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hainsworth J.D., Becker K.P., Mekhail T., Chowdhary S.A., Eakle J.F., Wright D., Langdon R.M., Yost K.J., Padula G.D.A., West-Osterfield K., et al. Phase I/II study of bevacizumab with BKM120, an oral PI3K inhibitor, in patients with refractory solid tumors (phase I) and relapsed/refractory glioblastoma (phase II) J. Neuro Oncol. 2019;144:303–311. doi: 10.1007/s11060-019-03227-7. [DOI] [PubMed] [Google Scholar]
- 24.Yap T.A., Bjerke L., Clarke P.A., Workman P. Drugging PI3K in cancer: refining targets and therapeutic strategies. Curr. Opin. Pharmacol. 2015;23:98–107. doi: 10.1016/j.coph.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen P.Y., Rodon J.A., Mason W., Beck J.T., DeGroot J., Donnet V., Mills D., El-Hashimy M., Rosenthal M. Phase I, open-label, multicentre study of buparlisib in combination with temozolomide or with concomitant radiation therapy and temozolomide in patients with newly diagnosed glioblastoma. ESMO Open. 2020;5 doi: 10.1136/esmoopen-2020-000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao H.F., Wang J., Shao W., Wu C.P., Chen Z.P., To S.S.T., Li W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: current preclinical and clinical development. Mol. Cancer. 2017;16:100. doi: 10.1186/s12943-017-0670-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J., Kaneda M.M., Ma J., Li M., Shepard R.M., Patel K., Koga T., Sarver A., Furnari F., Xu B., et al. PI3Kgamma inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2009290118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paul-Samojedny M., Pudełko A., Kowalczyk M., Fila-Daniłow A., Suchanek-Raif R., Borkowska P., Kowalski J. Combination Therapy with AKT3 and PI3KCA siRNA Enhances the Antitumor Effect of Temozolomide and Carmustine in T98G Glioblastoma Multiforme Cells. BioDrugs. 2016;30:129–144. doi: 10.1007/s40259-016-0160-y. [DOI] [PubMed] [Google Scholar]
- 29.Pridham K.J., Shah F., Hutchings K.R., Sheng K.L., Guo S., Liu M., Kanabur P., Lamouille S., Lewis G., Morales M., et al. Connexin 43 confers chemoresistance through activating PI3K. Oncogenesis. 2022;11:2. doi: 10.1038/s41389-022-00378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pridham K.J., Le L., Guo S., Varghese R.T., Algino S., Liang Y., Fajardin R., Rodgers C.M., Simonds G.R., Kelly D.F., Sheng Z. PIK3CB/p110beta is a selective survival factor for glioblastoma. Neuro Oncol. 2018;20:494–505. doi: 10.1093/neuonc/nox181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pridham K.J., Varghese R.T., Sheng Z. The Role of Class IA Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunits in Glioblastoma. Front. Oncol. 2017;7:312. doi: 10.3389/fonc.2017.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., 3rd, Barretina J., Gelfand E.T., Bielski C.M., Li H., et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Research Network. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Consortium G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Z., Zhang K.N., Wang Q., Li G., Zeng F., Zhang Y., Wu F., Chai R., Wang Z., Zhang C., et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Dev. Reprod. Biol. 2021;19:1–12. doi: 10.1016/j.gpb.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bairoch A. The Cellosaurus, a Cell-Line Knowledge Resource. J. Biomol. Tech. 2018;29:25–38. doi: 10.7171/jbt.18-2902-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A., et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 38.Varghese R.T., Liang Y., Guan T., Franck C.T., Kelly D.F., Sheng Z. Survival kinase genes present prognostic significance in glioblastoma. Oncotarget. 2016;7:20140–20151. doi: 10.18632/oncotarget.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stupp R., van den Bent M.J., Hegi M.E. Optimal role of temozolomide in the treatment of malignant gliomas. Curr. Neurol. Neurosci. Rep. 2005;5:198–206. doi: 10.1007/s11910-005-0047-7. [DOI] [PubMed] [Google Scholar]
- 40.Alzial G., Renoult O., Paris F., Gratas C., Clavreul A., Pecqueur C. Wild-type isocitrate dehydrogenase under the spotlight in glioblastoma. Oncogene. 2022;41:613–621. doi: 10.1038/s41388-021-02056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruno F., Pellerino A., Palmiero R., Bertero L., Mantovani C., Garbossa D., Soffietti R., Rudà R. Glioblastoma in the Elderly: Review of Molecular and Therapeutic Aspects. Biomedicines. 2022;10 doi: 10.3390/biomedicines10030644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verdugo E., Puerto I., Medina M.A. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun. 2022;42:1083–1111. doi: 10.1002/cac2.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen H., Mei L., Zhou L., Shen X., Guo C., Zheng Y., Zhu H., Zhu Y., Huang L. PTEN restoration and PIK3CB knockdown synergistically suppress glioblastoma growth in vitro and in xenografts. J. Neuro Oncol. 2011;104:155–167. doi: 10.1007/s11060-010-0492-2. [DOI] [PubMed] [Google Scholar]
- 44.Wee S., Wiederschain D., Maira S.M., Loo A., Miller C., deBeaumont R., Stegmeier F., Yao Y.M., Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA. 2008;105:13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Auffinger B., Spencer D., Pytel P., Ahmed A.U., Lesniak M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 2015;15:741–752. doi: 10.1586/14737175.2015.1051968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.SongTao Q., Lei Y., Si G., YanQing D., HuiXia H., XueLin Z., LanXiao W., Fei Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012;103:269–273. doi: 10.1111/j.1349-7006.2011.02134.x. [DOI] [PubMed] [Google Scholar]
- 47.Tran A.N., Lai A., Li S., Pope W.B., Teixeira S., Harris R.J., Woodworth D.C., Nghiemphu P.L., Cloughesy T.F., Ellingson B.M. Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantitative MR volumetry. Neuro Oncol. 2014;16:414–420. doi: 10.1093/neuonc/not198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan Y., Takayasu T., Hines G., Dono A., Hsu S.H., Zhu J.J., Riascos-Castaneda R.F., Kamali A., Bhattacharjee M.B., Blanco A.I., et al. Landscape of Genomic Alterations in IDH Wild-Type Glioblastoma Identifies PI3K as a Favorable Prognostic Factor. JCO Precis. Oncol. 2020;4:575–584. doi: 10.1200/PO.19.00385. [DOI] [PubMed] [Google Scholar]
- 49.Qiu Z.K., Shen D., Chen Y.S., Yang Q.Y., Guo C.C., Feng B.H., Chen Z.P. Enhanced MGMT expression contributes to temozolomide resistance in glioma stem-like cells. Chin. J. Cancer. 2014;33:115–122. doi: 10.5732/cjc.012.10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaspar N., Marshall L., Perryman L., Bax D.A., Little S.E., Viana-Pereira M., Sharp S.Y., Vassal G., Pearson A.D.J., Reis R.M., et al. MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res. 2010;70:9243–9252. doi: 10.1158/0008-5472.CAN-10-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marshall A.J., Lill C.L., Chao M., Kolekar S.V., Lee W.J., Marshall E.S., Baguley B.C., Shepherd P.R., Denny W.A., Flanagan J.U., Rewcastle G.W. Exploring the isoform selectivity of TGX-221 related pyrido[1,2-a]pyrimidinone-based Class IA PI 3-kinase inhibitors: synthesis, biological evaluation and molecular modelling. Bioorg. Med. Chem. 2015;23:3796–3808. doi: 10.1016/j.bmc.2015.03.073. [DOI] [PubMed] [Google Scholar]
- 52.Turner K.M., Sun Y., Ji P., Granberg K.J., Bernard B., Hu L., Cogdell D.E., Zhou X., Yli-Harja O., Nykter M., et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. USA. 2015;112:3421–3426. doi: 10.1073/pnas.1414573112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caporali S., Levati L., Graziani G., Muzi A., Atzori M.G., Bonmassar E., Palmieri G., Ascierto P.A., D'Atri S. NF-kappaB is activated in response to temozolomide in an AKT-dependent manner and confers protection against the growth suppressive effect of the drug. J. Transl. Med. 2012;10:252. doi: 10.1186/1479-5876-10-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caporali S., Levati L., Starace G., Ragone G., Bonmassar E., Alvino E., D'Atri S. AKT is activated in an ataxia-telangiectasia and Rad3-related-dependent manner in response to temozolomide and confers protection against drug-induced cell growth inhibition. Mol. Pharmacol. 2008;74:173–183. doi: 10.1124/mol.107.044743. [DOI] [PubMed] [Google Scholar]
- 55.Beier D., Schulz J.B., Beier C.P. Chemoresistance of glioblastoma cancer stem cells-much more complex than expected. Mol. Cancer. 2011;10:128. doi: 10.1186/1476-4598-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J., Li Y., Yu T.S., McKay R.M., Burns D.K., Kernie S.G., Parada L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanabur P., Guo S., Simonds G.R., Kelly D.F., Gourdie R.G., Verbridge S.S., Sheng Z. Patient-derived glioblastoma stem cells respond differentially to targeted therapies. Oncotarget. 2016;7:86406–86419. doi: 10.18632/oncotarget.13415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu P.F., Yang J.A., Liu J.H., Yang X., Liao J.M., Yuan F.E., Liu B.H., Chen Q.X. PI3Kbeta inhibitor AZD6482 exerts antiproliferative activity and induces apoptosis in human glioblastoma cells. Oncol. Rep. 2019;41:125–132. doi: 10.3892/or.2018.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chakravarti A., Erkkinen M.G., Nestler U., Stupp R., Mehta M., Aldape K., Gilbert M.R., Black P.M., Loeffler J.S. Temozolomide-mediated radiation enhancement in glioblastoma: a report on underlying mechanisms. Clin. Cancer Res. 2006;12:4738–4746. doi: 10.1158/1078-0432.CCR-06-0596. [DOI] [PubMed] [Google Scholar]
- 60.Hegi M.E., Stupp R. Withholding temozolomide in glioblastoma patients with unmethylated MGMT promoter--still a dilemma? Neuro Oncol. 2015;17:1425–1427. doi: 10.1093/neuonc/nov198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hartmann C., Bartels G., Gehlhaar C., Holtkamp N., von Deimling A. PIK3CA mutations in glioblastoma multiforme. Acta Neuropathol. 2005;109:639–642. doi: 10.1007/s00401-005-1000-1. [DOI] [PubMed] [Google Scholar]
- 62.Gallia G.L., Rand V., Siu I.M., Eberhart C.G., James C.D., Marie S.K.N., Oba-Shinjo S.M., Carlotti C.G., Caballero O.L., Simpson A.J.G., et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol. Cancer Res. 2006;4:709–714. doi: 10.1158/1541-7786.MCR-06-0172. [DOI] [PubMed] [Google Scholar]
- 63.Tanaka S., Batchelor T.T., Iafrate A.J., Dias-Santagata D., Borger D.R., Ellisen L.W., Yang D., Louis D.N., Cahill D.P., Chi A.S. PIK3CA activating mutations are associated with more disseminated disease at presentation and earlier recurrence in glioblastoma. Acta Neuropathol. Commun. 2019;7:66. doi: 10.1186/s40478-019-0720-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mueller W., Mizoguchi M., Silen E., D'Amore K., Nutt C.L., Louis D.N. Mutations of the PIK3CA gene are rare in human glioblastoma. Acta Neuropathol. 2005;109:654–655. doi: 10.1007/s00401-005-1001-0. [DOI] [PubMed] [Google Scholar]
- 65.Weber G.L., Parat M.O., Binder Z.A., Gallia G.L., Riggins G.J. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration, and invasion in glioblastoma multiforme cells. Oncotarget. 2011;2:833–849. doi: 10.18632/oncotarget.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nunnery S.E., Mayer I.A. Management of toxicity to isoform alpha-specific PI3K inhibitors. Ann. Oncol. 2019;30:x21–x26. doi: 10.1093/annonc/mdz440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sopasakis V.R., Liu P., Suzuki R., Kondo T., Winnay J., Tran T.T., Asano T., Smyth G., Sajan M.P., Farese R.V., et al. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010;11:220–230. doi: 10.1016/j.cmet.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chandrasekaran S., Funk C.R., Kleber T., Paulos C.M., Shanmugam M., Waller E.K. Strategies to Overcome Failures in T-Cell Immunotherapies by Targeting PI3K-delta and -gamma. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.718621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fruman D.A., Chiu H., Hopkins B.D., Bagrodia S., Cantley L.C., Abraham R.T. The PI3K Pathway in Human Disease. Cell. 2017;170:605–635. doi: 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cen B., Wei Y., Huang W., Teng M., He S., Li J., Wang W., He G., Bai X., Liu X., et al. An Efficient Bivalent Cyclic RGD-PIK3CB siRNA Conjugate for Specific Targeted Therapy against Glioblastoma In Vitro and In Vivo. Mol. Ther. Nucleic Acids. 2018;13:220–232. doi: 10.1016/j.omtn.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen J.S., Zhou L.J., Entin-Meer M., Yang X., Donker M., Knight Z.A., Weiss W., Shokat K.M., Haas-Kogan D., Stokoe D. Characterization of structurally distinct, isoform-selective phosphoinositide 3'-kinase inhibitors in combination with radiation in the treatment of glioblastoma. Mol. Cancer Ther. 2008;7:841–850. doi: 10.1158/1535-7163.MCT-07-0393. [DOI] [PubMed] [Google Scholar]
- 72.Holand K., Boller D., Hagel C., Dolski S., Treszl A., Pardo O.E., Cwiek P., Salm F., Leni Z., Shepherd P.R., et al. Targeting class IA PI3K isoforms selectively impairs cell growth, survival, and migration in glioblastoma. PLoS One. 2014;9 doi: 10.1371/journal.pone.0094132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie S., Ni J., McFaline-Figueroa J.R., Wang Y., Bronson R.T., Ligon K.L., Wen P.Y., Roberts T.M., Zhao J.J. Divergent Roles of PI3K Isoforms in PTEN-Deficient Glioblastomas. Cell Rep. 2020;32 doi: 10.1016/j.celrep.2020.108196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao H.F., Wang J., Jiang H.R., Chen Z.P., To S.S.T. PI3K p110beta isoform synergizes with JNK in the regulation of glioblastoma cell proliferation and migration through Akt and FAK inhibition. J. Exp. Clin. Cancer Res. 2016;35:78. doi: 10.1186/s13046-016-0356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao H.F., Wu C.P., Zhou X.M., Diao P.Y., Xu Y.W., Liu J., Wang J., Huang X.J., Liu W.L., Chen Z.P., et al. Synergism between the phosphatidylinositol 3-kinase p110beta isoform inhibitor AZD6482 and the mixed lineage kinase 3 inhibitor URMC-099 on the blockade of glioblastoma cell motility and focal adhesion formation. Cancer Cell Int. 2021;21:24. doi: 10.1186/s12935-020-01728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pu P., Kang C., Zhang Z., Liu X., Jiang H. Downregulation of PIK3CB by siRNA suppresses malignant glioma cell growth in vitro and in vivo. Technol. Cancer Res. Treat. 2006;5:271–280. doi: 10.1177/153303460600500308. [DOI] [PubMed] [Google Scholar]
- 77.Kang S., Bader A.G., Vogt P.K. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. USA. 2005;102:802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao J.J., Liu Z., Wang L., Shin E., Loda M.F., Roberts T.M. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc. Natl. Acad. Sci. USA. 2005;102:18443–18448. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao J.J., Gjoerup O.V., Subramanian R.R., Cheng Y., Chen W., Roberts T.M., Hahn W.C. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003;3:483–495. doi: 10.1016/s1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]
- 80.Jia S., Liu Z., Zhang S., Liu P., Zhang L., Lee S.H., Zhang J., Signoretti S., Loda M., Roberts T.M., Zhao J.J. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dbouk H.A., Pang H., Fiser A., Backer J.M. A biochemical mechanism for the oncogenic potential of the p110beta catalytic subunit of phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA. 2010;107:19897–19902. doi: 10.1073/pnas.1008739107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benitez J.A., Ma J., D'Antonio M., Boyer A., Camargo M.F., Zanca C., Kelly S., Khodadadi-Jamayran A., Jameson N.M., Andersen M., et al. PTEN regulates glioblastoma oncogenesis through chromatin-associated complexes of DAXX and histone H3.3. Nat. Commun. 2017;8 doi: 10.1038/ncomms15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chuang H.Y., Hsu L.Y., Pan C.M., Pikatan N.W., Yadav V.K., Fong I.H., Chen C.H., Yeh C.T., Chiu S.C. The E3 Ubiquitin Ligase NEDD4-1 Mediates Temozolomide-Resistant Glioblastoma through PTEN Attenuation and Redox Imbalance in Nrf2-HO-1 Axis. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms221910247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dong L., Li Y., Liu L., Meng X., Li S., Han D., Xiao Z., Xia Q. Smurf1 Suppression Enhances Temozolomide Chemosensitivity in Glioblastoma by Facilitating PTEN Nuclear Translocation. Cells. 2022;11 doi: 10.3390/cells11203302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kang Y.J., Balter B., Csizmadia E., Haas B., Sharma H., Bronson R., Yan C.T. Contribution of classical end-joining to PTEN inactivation in p53-mediated glioblastoma formation and drug-resistant survival. Nat. Commun. 2017;8 doi: 10.1038/ncomms14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carico C., Nuño M., Mukherjee D., Elramsisy A., Dantis J., Hu J., Rudnick J., Yu J.S., Black K.L., Bannykh S.I., Patil C.G. Loss of PTEN is not associated with poor survival in newly diagnosed glioblastoma patients of the temozolomide era. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neshat M.S., Mellinghoff I.K., Tran C., Stiles B., Thomas G., Petersen R., Frost P., Gibbons J.J., Wu H., Sawyers C.L. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc. Natl. Acad. Sci. USA. 2001;98:10314–10319. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Furnari F.B., Lin H., Huang H.S., Cavenee W.K. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc. Natl. Acad. Sci. USA. 1997;94:12479–12484. doi: 10.1073/pnas.94.23.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang R., Banik N.L., Ray S.K. Differential sensitivity of human glioblastoma LN18 (PTEN-positive) and A172 (PTEN-negative) cells to Taxol for apoptosis. Brain Res. 2008;1239:216–225. doi: 10.1016/j.brainres.2008.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van den Bent M.J., Gao Y., Kerkhof M., Kros J.M., Gorlia T., van Zwieten K., Prince J., van Duinen S., Sillevis Smitt P.A., Taphoorn M., French P.J. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro Oncol. 2015;17:935–941. doi: 10.1093/neuonc/nov013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mukherjee B., McEllin B., Camacho C.V., Tomimatsu N., Sirasanagandala S., Nannepaga S., Hatanpaa K.J., Mickey B., Madden C., Maher E., et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009;69:4252–4259. doi: 10.1158/0008-5472.CAN-08-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Van Den Bent M., Eoli M., Sepulveda J.M., Smits M., Walenkamp A., Frenel J.S., Franceschi E., Clement P.M., Chinot O., De Vos F., et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro Oncol. 2020;22:684–693. doi: 10.1093/neuonc/noz222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sepulveda-Sanchez J.M., Vaz M.A., Balana C., Gil-Gil M., Reynes G., Gallego O., Martinez-Garcia M., Vicente E., Quindos M., Luque R., et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol. 2017;19:1522–1531. doi: 10.1093/neuonc/nox105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang S., Chen C., Li J., Xu X., Chen W., Li F. The CXCL12/CXCR4 axis confers temozolomide resistance to human glioblastoma cells via up-regulation of FOXM1. J. Neurol. Sci. 2020;414 doi: 10.1016/j.jns.2020.116837. [DOI] [PubMed] [Google Scholar]
- 95.Zhou J., Schmid T., Frank R., Brüne B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1alpha from pVHL-independent degradation. J. Biol. Chem. 2004;279:13506–13513. doi: 10.1074/jbc.M310164200. [DOI] [PubMed] [Google Scholar]
- 96.Kohsaka S., Wang L., Yachi K., Mahabir R., Narita T., Itoh T., Tanino M., Kimura T., Nishihara H., Tanaka S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer Ther. 2012;11:1289–1299. doi: 10.1158/1535-7163.MCT-11-0801. [DOI] [PubMed] [Google Scholar]
- 97.Zhang X., Tang N., Hadden T.J., Rishi A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta. 2011;1813:1978–1986. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 98.Zeng X., Kinsella T.J. A novel role for DNA mismatch repair and the autophagic processing of chemotherapy drugs in human tumor cells. Autophagy. 2007;3:368–370. doi: 10.4161/auto.4205. [DOI] [PubMed] [Google Scholar]
- 99.Chen C.M., Syu J.P., Way T.D., Huang L.J., Kuo S.C., Lin C.T., Lin C.L. BC3EE2,9B, a synthetic carbazole derivative, upregulates autophagy and synergistically sensitizes human GBM8901 glioblastoma cells to temozolomide. Int. J. Mol. Med. 2015;36:1244–1252. doi: 10.3892/ijmm.2015.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu T., Li A., Xu Y., Xin Y. Momelotinib sensitizes glioblastoma cells to temozolomide by enhancement of autophagy via JAK2/STAT3 inhibition. Oncol. Rep. 2019;41:1883–1892. doi: 10.3892/or.2019.6970. [DOI] [PubMed] [Google Scholar]
- 101.Li H., Chen L., Li J.J., Zhou Q., Huang A., Liu W.W., Wang K., Gao L., Qi S.T., Lu Y.T. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J. Hematol. Oncol. 2018;11:70. doi: 10.1186/s13045-018-0618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cui Y., Lin J., Zuo J., Zhang L., Dong Y., Hu G., Luo C., Chen J., Lu Y. AKT2-knockdown suppressed viability with enhanced apoptosis, and attenuated chemoresistance to temozolomide of human glioblastoma cells in vitro and in vivo. OncoTargets Ther. 2015;8:1681–1690. doi: 10.2147/OTT.S83795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang J., Zhang J., Zhang J., Qiu W., Xu S., Yu Q., Liu C., Wang Y., Lu A., Zhang J., Lu X. MicroRNA-625 inhibits the proliferation and increases the chemosensitivity of glioma by directly targeting AKT2. Am. J. Cancer Res. 2017;7:1835–1849. [PMC free article] [PubMed] [Google Scholar]
- 104.Dumbrava E.E., Burton E.M., Subudhi S.K., Milton D.R., Aparicio A., Yap T.A., Naing A., Glen Pilié P., Pilié P.G., Zurita A.J., et al. Phase I/II study of the selective PI3Kβ inhibitor GSK2636771 in combination with pembrolizumab in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and PTEN loss. J. Clin. Oncol. 2022;40:5052. [Google Scholar]
- 105.Varghese R.T., Young S., Pham L., Liang Y., Pridham K.J., Guo S., Murphy S., Kelly D.F., Sheng Z. Casein Kinase 1 Epsilon Regulates Glioblastoma Cell Survival. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-31864-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sheng K.L., Kang L., Pridham K.J., Dunkenberger L.E., Sheng Z., Varghese R.T. An integrated approach to biomarker discovery reveals gene signatures highly predictive of cancer progression. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-78126-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sheng K.L., Pridham K.J., Sheng Z., Lamouille S., Varghese R.T. Functional Blockade of Small GTPase RAN Inhibits Glioblastoma Cell Viability. Front. Oncol. 2018;8:662. doi: 10.3389/fonc.2018.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes publicly available data from TCGA or DepMap databases. Information regarding datasets analyzed in this study are listed in the key resources table. This study did not report any original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.