

Abstract
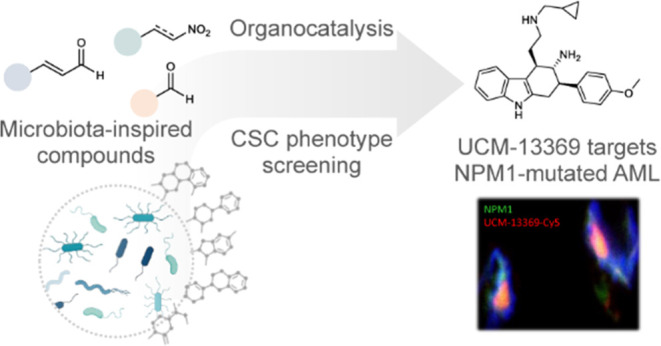
The human microbiota plays an important role in human health and disease, through the secretion of metabolites that regulate key biological functions. We propose that microbiota metabolites represent an unexplored chemical space of small drug-like molecules in the search of new hits for drug discovery. Here, we describe the generation of a set of complex chemotypes inspired on selected microbiota metabolites, which have been synthesized using asymmetric organocatalytic reactions. Following a primary screening in CSC models, we identified the novel compound UCM-13369 (4b) whose cytotoxicity was mediated by NPM1. This protein is one of the most frequent mutations of AML, and NPM1-mutated AML is recognized by the WHO as a distinct hematopoietic malignancy. UCM-13369 inhibits NPM1 expression, downregulates the pathway associated with mutant NPM1 C+, and specifically recognizes the C-end DNA-binding domain of NPM1 C+, avoiding the nucleus-cytoplasm translocation involved in the AML tumorological process. The new NPM1 inhibitor triggers apoptosis in AML cell lines and primary cells from AML patients and reduces tumor infiltration in a mouse model of AML with NPM1 C+ mutation. The disclosed phenotype-guided discovery of UCM-13369, a novel small molecule inspired on microbiota metabolites, confirms that CSC death induced by NPM1 inhibition represents a promising therapeutic opportunity for NPM1-mutated AML, a high-mortality disease.
Keywords: drug discovery, small molecule, microbiota, organocatalysis, stem cells, NPM1, leukemia
Introduction
The human microbiota is a complex ecosystem of symbiotic microorganisms that play an important role in human health and disease, in part through the secretion of metabolites that can regulate host proteins.1,2 Microbiota can synthesize, transform, and degrade a large repertoire of organic molecules that are key for maintaining homeostasis providing beneficial functions to the host. Consequently, microbiota dysbiosis, i.e., alterations in its composition that result from exposure to environmental factors, such as diet, xenobiotics, drugs, and pathogens, eventually contributes to the pathogenesis of various diseases, such as obesity, type 2 diabetes, cardiovascular disease, cancer, and neurological disorders.3−6 In this context, several metabolites produced by the microbiota have been shown to confer protection against disease due to their antioxidant and anti-inflammatory properties.7−9 For example, equol exhibits strong antiproliferative effects on breast neoplasm and hepatocellular carcinoma;10,11 urolithins have been suggested to target specific tumorigenesis pathways in prostate cancer;12,13 niacin (vitamin B3) has been shown to suppress colitis and colon cancer in mice;14,15 indole appears to modulate the important antihyperglycemic hormone glucagon-like peptide (GLP1), with a substantial impact on pancreatic function, insulin release, and food intake regulation;16 protocatechiuc acid prevents esophageal cancer;17 and butyrate has been reported to modulate the activity of the nervous system in mouse models.18 Hence, microbiota metabolism produces a wide and diverse range of bioactive small molecules that are available to the host to regulate key biological functions. The compatibility of the microbiome with healthy human systems suggests that microbiota metabolites represent drug-like molecules that are highly specific modulators with no risk of adverse effects.8 This supports the hypothesis that the metabolites can be considered an unexplored chemical space of small organic molecules in the search of initial hits toward the identification of novel drug candidates for the treatment of a wide range of pathological conditions. Focused on cancer, in the present work, we have designed and synthesized a set of new small molecules inspired on human microbiota metabolites and explored their potential in cancer cellular models with the ultimate goal to identify novel antitumor drug candidates. Following a primary screening in cancer stem cells (CSCs), we have identified the synthetic compound UCM-13369 that has been subsequently characterized as a nucleophosmin 1 (NPM1) inhibitor that specifically recognizes the C-end DNA-binding domain of the protein. The capacity of the new NPM1 inhibitor to trigger stem cell apoptosis has been translated to ex vivo and in vivo efficacy in models of acute myeloid leukemia (AML).19 Our results support phenotypic drug discovery as a valuable approach to develop active molecules in a pathologically relevant model, which together with the validation of the target protein involved in the disease, contributes to enhance innovation and deliver truly novel therapeutics for unmet medical needs.20,21
Results
Design and Synthesis of New Microbiota-Inspired Compounds
We propose that microbiota metabolites represent an interesting starting point for the design of new compounds for drug discovery. Toward this end, we have analyzed the structures of identified small-molecule metabolites22 and explored the repertoire of organocatalytic reactions, to generate a set of microbiota-inspired compounds that combine metabolite structural diversity with synthetic accessibility using a key methodology that allows to obtain chemotypes with high structural complexity in a straightforward and stereocontrolled manner. This strategy offers the advantage of combining two or more reagents in a one-pot fashion to effectively yield a target chemotype. Thus, based on privileged scaffolds (PrSc) found in the metabolites selected in Figure 1A and amino catalytic reactions starting with a small group of aldehydes and nitro derivatives (Figure 1B), we designed new chemotypes that contain a PrSc-based central core linked to the common PrSc p-methoxyphenyl as well as to a drug-like moiety that could favor good pharmacokinetic properties. In the designed molecules 1–13 (Figure 1C), the central cores are piperidin-2-one, piperidine, tetrahydro-1H-carbazole, chromane, and dihydropyrido[2,3-b]pyrazine. These systems are inspired on metabolites including 3-amino-2-piperidone, pipecolic acid, methyl 3-carbazolecarboxylate, equol, γ-carboxyethylhydroxychroman (γ-CEHC) related to vitamin E, folic acid, and trimethylpyrazine shown in Figure 1A. The central cores are constructed in a single step and with high diastereoselectivities by reaction between an α,β-unsaturated aldehyde and a nitro compound or a second aldehyde promoted by diphenylprolinol trimethylsilyl ether catalyst (Figure 1C). Further transformation of aldehyde and/or nitro groups following a general sequence allows us to obtain the desired compounds 1–13 that contain the drug-like moiety (cyclopropylmethyl)amine and an amino group. For each chemotype, we have synthesized both enantiomers—designated as compounds a and b—with high enantioselectivity by using the corresponding catalyst.
Figure 1.
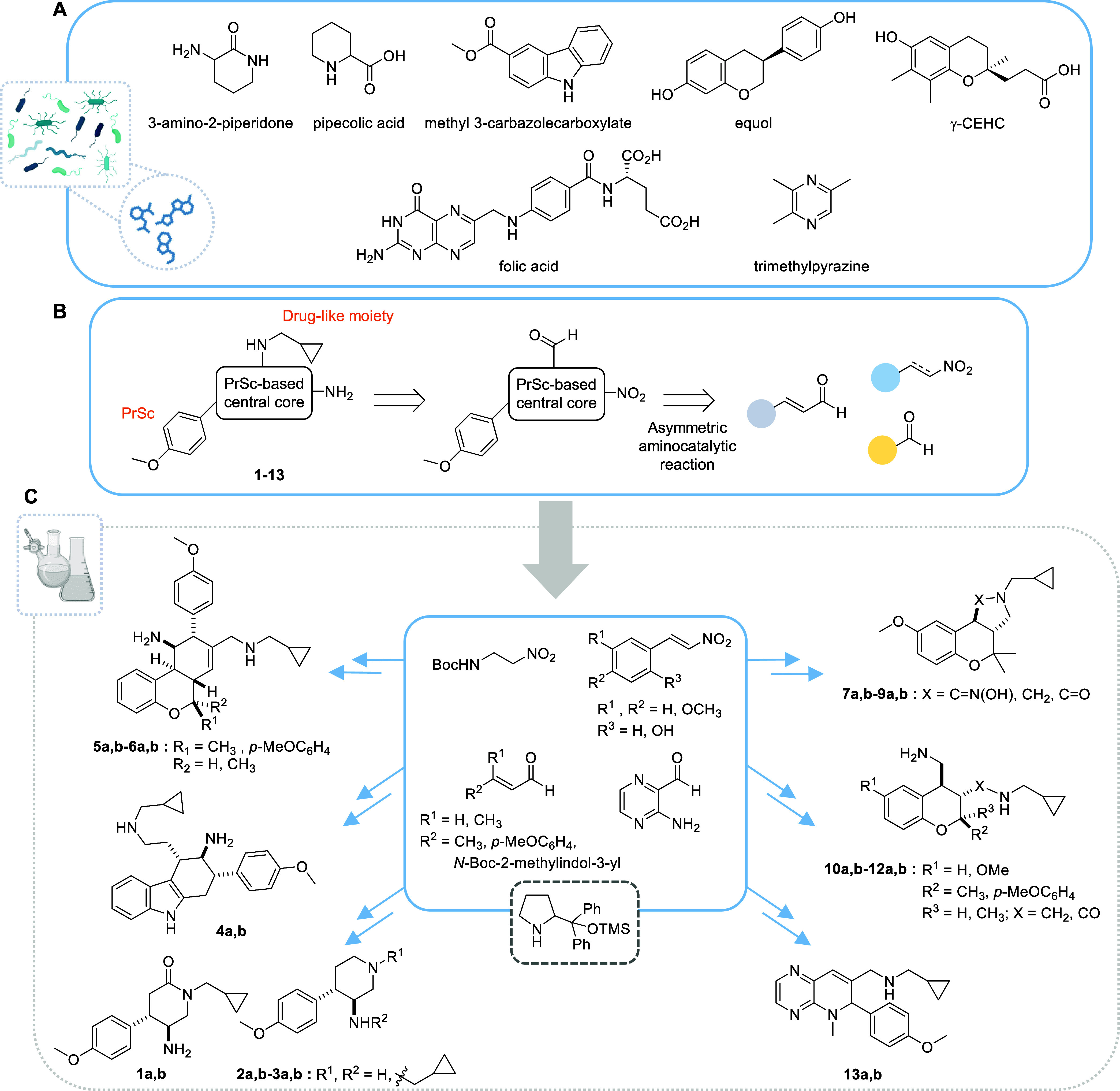
Design of new chemotypes inspired on human microbiota metabolites. (A) Selected small-molecule metabolites produced by the microbiota. (B) Synthetic approach via asymmetric aminocatalysis to generate microbiota-inspired molecules 1–13. (C) Overview of the synthetic strategy based on a reaction between an α,β-unsaturated aldehyde and a nitro compound or a second aldehyde. PrSc: privileged scaffold. Untapered lines represent the relative stereochemistry; a and b are used to designate the two enantiomers of each compound.
The synthesis of final compounds 1–3 (Figure 2A) containing a disubstituted piperidin-2-one or a piperidine scaffold started with the reaction between N-Boc-2-amino-1-nitroethane and p-methoxycinnamaldehyde to obtain intermediates 14 and 15.23 Michael addition catalyzed by the S or R enantiomer of 2-{diphenyl[(trimethylsilyl)oxy]methyl}pyrrolidine in the presence of benzoic acid, and subsequent in situ oxidation of the resulting hemiaminal afforded piperidin-2-one scaffold 14 (diastereoisomeric ratio (dr) = 9:1, determined by 1H-nuclear magnetic resonance (NMR) analysis of the crude). Intermediate 14 was then deprotected before one-pot reduction of the nitro group and protection of the resulting amine to give N-Boc-5-aminopiperidin-2-one (22), which was alkylated at the amide nitrogen with (bromomethyl)cyclopropane and treated with trifluoroacetic acid (TFA) to obtain compound 1a or 1b (enantiomeric ratio (er) = 97:3 for 1a, 96:4 for 1b). Reduction of the carbonyl group of 1 yielded target compound 2a or 2b (er = 99:1). On the other hand, piperidine scaffold 15 (Figure 2A) was prepared via the same asymmetric organocatalytic Michael addition to p-methoxycinnamaldehyde, but this time the resulting cyclic hemiaminal was in situ dehydrated (dr = 8:2, determined by high-performance liquid chromatography–mass spectrometry (HPLC-MS) analysis of the crude). Subsequent reduction of both the nitro group and the double bond yielded 5-aminopiperidine 23, which afforded final compound 3a or 3b via reductive amination and deprotection (er = 98:2).
Figure 2.
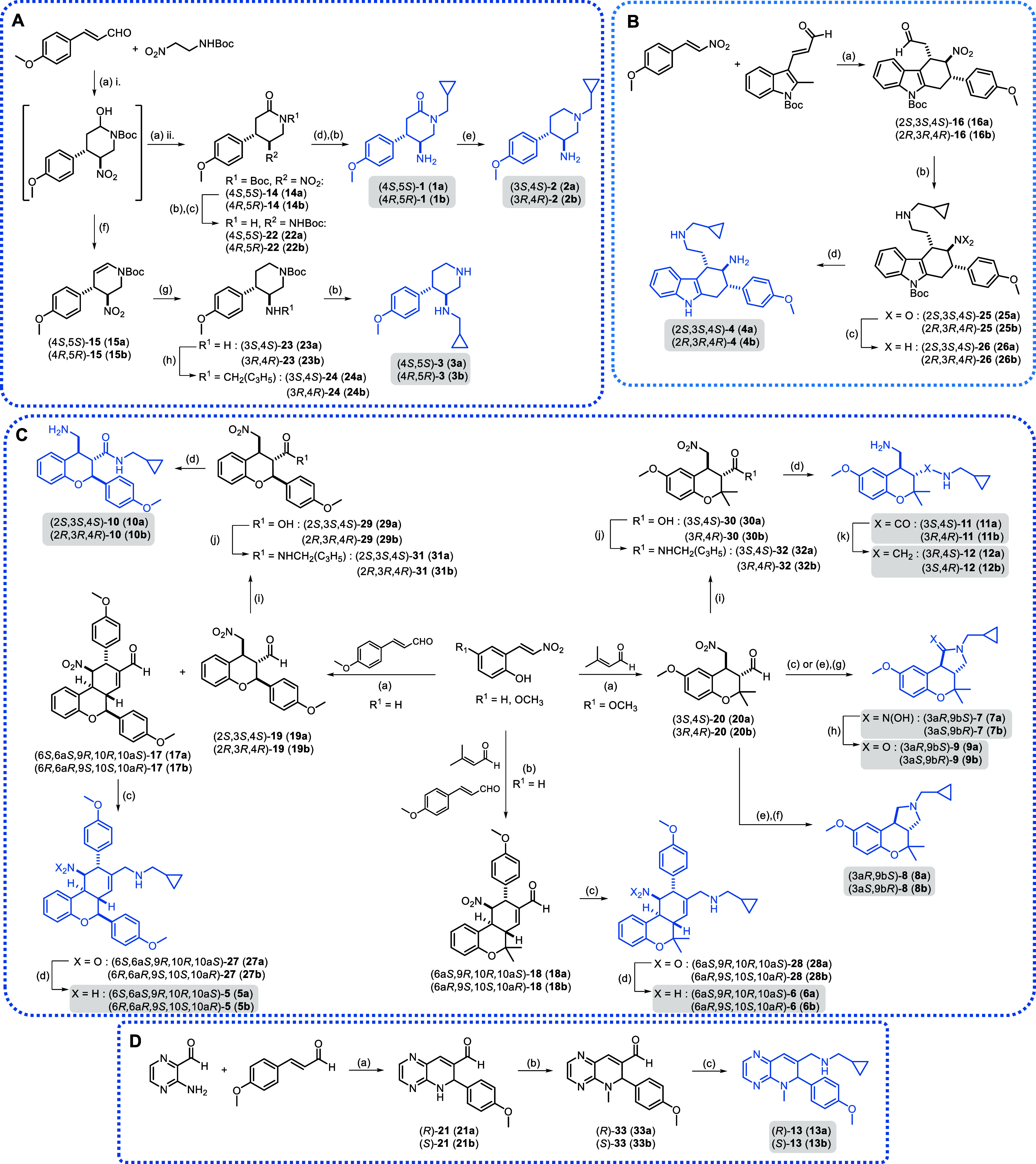
(A) Synthesis of compounds 1–3 containing 2-piperidinone or piperidine scaffolds. Reagents and conditions: (a) (i) 10 mol % chiral cat., 20 mol % benzoic acid, DCM, 0 °C to rt, 5 h; (ii) PDC, rt, on, 45–58%; (b) TFA, DCM, rt, 45 min-4 h, 73–93%; (c) (i) NiCl2·6H2O, NaBH4, MeOH, 0 °C, 1 h; (ii) Boc2O, rt, on, quantitative; (d) (i) NaH, DMF, 0 °C, 1 h; (ii) (bromomethyl)cyclopropane, NaI, rt, on, 36–42%; (e) LiAlH4, THF, rt, on, 77–85%; (f) TFA, rt, 2 h, 84–87%; (g) NiCl2·6H2O, NaBH4, MeOH, 0 °C to rt, on, 53–60%; (h) (i) cyclopropranecarbaldehyde, MeOH, rt, 4 h; (ii) NaBH4, rt, 2 h, 63–64%. (B) Synthesis of compound 4 containing a tetrahydrocarbazole scaffold. Reagents and conditions: (a) 20 mol % chiral cat., 20 mol % benzoic acid, toluene, MW, 110 °C, 5.5 h, 78%; (b) (i) (cyclopropylmethyl)amine, MeOH/DCM, rt, 3 h; (ii) NaBH4, rt, 2 h, 83–86%; (c) Zn, AcOH/MeOH, 2 h, quantitative; (d) HCl, MeOH, rt, 4 h, 40–50%. (C) Synthesis of compounds 5–12 containing chromane and tetrahydrobenzo[c]chromene scaffolds. Reagents and conditions: (a) 20 mol % chiral cat., 20 mol % AcOH, CHCl3, rt, on, 21–26% for 17, 22–31% for 19, 52–61% for 20; (b) 20 mol % chiral cat., 20 mol % AcOH, CHCl3, rt, 1 h then 24 h, 43–48%; (c) (i) (cyclopropylmethyl)amine, MeOH/DCM, rt, 3 h; (ii) NaBH4, rt, 2 h, 17%-quantitative; (d) Zn, AcOH/MeOH, rt, 2 h, 41–93%; (e) (cyclopropylmethyl)amine, DCM, rt, 3 h, quantitative; (f) NaBH4, MeOH, rt, 3 h, 30–33%; (g) HSiCl3, DCM, DMF, rt, on, 80–89%; (h) NaNO2, AcOH, MeOH/H2O, rt, on, 30–40%; (i) NaClO2, KH2PO4, 2-methylbut-2-ene, t-BuOH/THF, 30 °C, on, 79%-quantitative; (j) (i) (COCl)2 (2 M in DCM), cat. DMF, DCM, rt, 1 h; (ii) (cyclopropylmethyl)amine, 0 °C to rt, 3–18 h, 55%-quantitative; (k) LiAlH4, THF, reflux, 24 h, 20–22%. (D) Synthesis of compound 13 containing a dihydropyrido[2,3-b]pyrazine scaffold. Reagents and conditions: (a) 20 mol % chiral cat., 20% mol AcOH, CHCl3, rt, 7 days, 40–45%; (b) CH3I, Cs2CO3, DMF, rt, 1 h, 95%-quantitative; (c) (i) (cyclopropylmethyl)amine, MeOH/DCM, rt, 3 h; (ii) NaBH4, 0 °C to rt, 1–2 h, 49–50%.
For the synthesis of the tetrahydrocarbazole core present in compound 4 (Figure 2B), we applied an asymmetric Diels–Alder reaction between a β-indolyl α,β-unsaturated aldehyde and trans-p-methoxy-β-nitrostyrene catalyzed by the S or R form of the diphenylprolinol silyl ether.24 In this case, we optimized the reported thermal Diels–Alder reaction by using microwave (MW) conditions (irradiation at 110 °C for 5.5 h) to obtain cycloadduct 16 (dr = 98:2, 1H NMR; er = 96:4 for 16a, 98:2 for 16b). These new conditions increased the yield and reduced considerably the time with respect to the thermal reaction without affecting the stereoselectivity outcome. Compound 16 was further derivatized via reductive amination to introduce a (cyclopropylmethyl)amino moiety, subsequent reduction of the nitro group and Boc deprotection, to afford final compound 4a or 4b (er = 94:6 for 4a, 95:5 for 4b).
The chromane and tetrahydrobenzo[c]chromene scaffolds 17–20 (Figure 2C), precursors of compounds 5–12, were synthesized via an asymmetric organocatalytic domino reaction between an o-nitrovinylphenol and a properly substituted α,β-unsaturated aldehyde.25,26 Thus, intermediates 17 and 19 were both obtained using p-methoxycinnamaldehyde and o-nitrovinylphenol through a domino oxa-Michael–Michael reaction and a quadruple cascade oxa-Michael–Michael–Michael-aldol condensation reaction, respectively. Upon optimization of the reported conditions, tetrahydrobenzo[c]chromene 17 (single diastereoisomer) and chromane 19 (dr 8:2, 1H NMR) were obtained in a 1:1 ratio. Regarding tetrahydrobenzo[c]chromene and chromane intermediates 18 and 20 (Figure 2C), bearing a geminal dimethyl group, the organocatalytic cascade between 3,3-dimethylacrolein and trans-2-hydroxy-5-methoxy-β-nitrostyrene afforded only the chromane derivative 20 (dr = 8:2, 1H NMR), through a domino oxa-Michael–Michael reaction. In this case, the corresponding tetrahydrobenzo[c]chromene scaffold was not formed, probably due to the steric hindrance of the geminal dimethyl group, which may prevent the second Michael addition to a third molecule of 3,3-dimethylacrolein. Tetrahydrobenzo[c]chromene 18 was then obtained following a two-step procedure. Initial reaction between trans-2-hydroxy-β-nitrostyrene and 3,3-dimethylacrolein resulted in a chromane intermediate, which was treated in situ with less hindered Michael acceptor p-methoxycinnamaldehyde to induce a subsequent Michael-aldol condensation that provided 18 as a single diastereoisomer. Next, reductive amination with (cyclopropylmethyl)amine followed by nitro reduction of tetrahydrobenzo[c]chromenes 17 and 18 provided final compounds 5a or 5b and 6a or 6b, respectively (er = 99:1 for 5a, 98:2 for 5b, 99:1 for 6a,b) (Figure 2C). However, when the one-pot reductive amination was applied to chromane intermediate 20, the expected amine was not formed, but oxime 7 was obtained instead. The procedure in two steps did not yield the expected product either but the cyclic amine 8 resulting from the reduction of the corresponding imine. Alternative reduction of the imine with trichlorosilane combined with N,N-dimethylformamide (DMF) as a soft Lewis base afforded again compound 7, which was hydrolyzed to 9. Unexpected chemotypes 7a,b-9a,b (Figure 2C, er = 99:1 for 7a,b, 96:4 for 8a,b, 98:2 for 9a,b) were considered as additional final compounds to be tested in the phenotypic assays. The synthesis of desired compounds 10–12 was achieved following an alternative synthetic route starting from chromanes 19 and 20 (Figure 2C). The aldehyde group was oxidized to the corresponding carboxylic acid through the Pinnick reaction, and the (cyclopropylmethyl)amine moiety was then incorporated by coupling via the acid chloride. Next, reduction of the nitro group under standard conditions afforded compounds 10a or 10b and 11a or 11b, respectively (er = 99:1 for 10a,b and 11a,b). Finally, the amide group in the latter was reduced to obtain analogue 12a or 12b (er = 99:1).
The synthesis of the dihydropyrido[2,3-b]pyrazine scaffold 21 (Figure 2D) was performed via an asymmetric domino aza-Michael-aldol reaction between 3-aminopyrazine-2-carbaldehyde and p-methoxycinnamaldehyde. N-Methylation followed by reductive amination with (cyclopropylmethyl)amine led to the final compound 13a or 13b (er = 98:2).
Cancer-Stem-Cell (CSC) Phenotype Screening: Identification of UCM-13369
Once microbiota-inspired compounds 1a–13a and 1b–13b were synthesized, we carried out a cellular phenotypic screening in cancer and CSC models. The cytotoxic effect was determined in breast (MCF-7) and colon (HCT-116) tumor cell lines using MTT assays. Then, the formation of MCF-7-derived mamospheres and HCT-116-derived colonospheres was evaluated in the presence of a noncytotoxic concentration of the compounds, using bright-field microscopy. As shown in Table S1, compounds 4b, 5a, 5b, 6a, and 6b were able to completely inhibit the formation of tumorspheres, a characteristic growth in CSCs, without affecting the viability of differentiated tumor cells. To select those compounds endowed with a good balance between efficacy and safety, we subsequently determined if the compounds that are able to act on CSCs do not affect the viability of IMR90 fibroblasts, which are commonly used as control normal cells. From the data in Table S1, tetrahydrocarbazole 4b was not toxic in nontumor cells at 5 μM, while tetrahydrobenzo[c]chromenes 5a, 5b, 6a, and 6b exhibited toxicity (20–57% cell viability @5 μM). Among them, compound 4b (UCM-13369) induced low levels of cytotoxicity in tumor cells (96 and 70% cell viability), complete inhibition of tumorsphere formation, and no toxicity in fibroblasts (100% cell viability) (Table S1 and Figure S1A), and was selected for further studies.
Compound UCM-13369 Targets Nucleophosmin 1 (NPM1) Protein
One of the most paradigmatic diseases with critical involvement of CSCs is AML. Among the AML stem cell population, CD34+/CD38- are the only ones capable of inducing leukemia in vivo.27 Hence, with the aim to decipher the molecular targets of UCM-13369, 1D-nano LC ESI-MSMS proteomic analysis was performed in CD34+ cells before and after 24 h of treatment with 5 μM UCM-13369. The results showed multiple differentially expressed proteins. We focused on proteins that disappeared completely after treatment with the compound and whose concentration could be quantified in untreated samples. According to this profile, 340 proteins, including targets related with stemness and cancer, were identified (see Supplementary Excel file). Among them, nucleophosmin 1 (NPM1) especially attracted our attention, as it is one of the most common mutated proteins in AML.28 To support that the inhibitory capacity of UCM-13369 observed in CSC cells (see Table S1) is mediated by NPM1, the compound was tested on OCI-AML3 and MOLM13 cell lines, as in vitro AML models bearing mutant (NPM1 C+) and wild-type (WT) protein, respectively. As shown in Figure 3A, the compound exhibits cytotoxicity against both cell lines in the μM range. In addition, different efficacies of UCM-13369 were observed between both AML models, being higher in NPM1 C+ expressing cells (OCI-AML3: IC50 = 7.26 μM, MOLM13: IC50 = 10.89 μM, p-value ≤0.01, Figure 3A).
Figure 3.

Compound UCM-13369 targets NPM1 protein, binding mainly through the C-end domain of the C+ mutant form. (A) Dose–response curves of UCM-13369 in AML cell lines MOLM13 (NPM1 WT) and OCI-AML3 (NPM1 C+). (B) Confocal microscopy images with staining of NPM1 (Alexa Fluor-488, green), UCM-13369-Cy5 (red), and nuclei (DAPI, blue) in OCI-AML3 cells. (C,D) 1D 1H NMR spectra of the C-end domain of NPM1 WT (C) and C+ (D). (E) ITC thermograms and binding isotherms of the interaction of UCM-13369 with the WT (blue) and C+ mutant (red) forms of NPM1. ITC-derived thermodynamic parameters of UCM-13369–NPM1 interaction. Gibbs free energy (ΔG), enthalpy (ΔH), entropic term (-TΔS), equilibrium dissociation constant (KD), and binding stoichiometry (n). (F) Visualization of UCM-13369 binding interfaces to NPM1 C-end domain. The centroids of UCM-13369 in the four lowest-energy structures of each 10 clusters are plotted as balls colored according to the HADDOCK score, as indicated in the color legend. The structure of UCM-13369 of the top-ranked structure of each cluster is represented in the insets (i-v).
To further support NPM1 as a molecular target of UCM-13369, we synthesized UCM-13369-Cy5 (Figure S2), a derivative of UCM-13369 containing the fluorescent tag Cy5, as a chemical probe for use in confocal microscopy. As shown in Figure 3B, we visualized the overlapping of UCM-13369-Cy5 (in red) with an Alexa Fluor-488-conjugated NPM1 primary antibody (in green), indicating colocalization of the compound with the protein in OCI-AML3 cells (see video in the Supporting Information).
To confirm a direct interaction of UCM-13369 with NPM1 and to address which domain of the protein would be responsible for binding the compound, pure protein samples of the pentameric N-end domain of NPM1, along with the C-terminal DNA-binding domains of WT and mutant NPM1 C+, were used in calorimetry assays. As a confirmation of its native state, analysis of the 1D 1H NMR spectrum of NPM1 C-end WT showed well-dispersed sharp peaks corresponding to proper protein folding (Figure 3C). In contrast, the spectrum obtained for the C+ mutant variant exhibited drastically less dispersed and broader signals, particularly in the amide region (6–10 ppm) (Figure 3D). This is indicative of a lower diversity in the chemical environment of amide protons consistent with the previously reported protein unfolding.29 The lower signal intensities in this region also suggest more pronounced relaxation effects, which might be related to the higher aggregation tendency of the mutant.30 Isothermal titration calorimetry (ITC) experiments revealed that the interaction between the NPM1 protein and UCM-13369 occurs mainly through the DNA-binding domain C-end of NPM1, with the C-end of the NPM1 C+ variant showing a 2-fold higher affinity toward UCM-13369 than the WT domain (Figure 3D). On the other hand, the pentameric construct of the NPM1 N-end, which lacks the AML-related region, yielded a much lower binding affinity (KD ca. 100 μM range, Figure 3D). Interestingly, the interaction is entropically driven in all cases. Altogether, these data suggest that the new NPM1 inhibitor UCM-13369 binds specifically to the C-end domain of the C+ mutant protein.
To identify putative binding sites of UCM-13369 in this region of NPM1, ab initio docking predictions were performed using HADDOCK software.31,32 Docking solutions were clustered in 10 groups distributed across the surface of this domain, suggesting that UCM-13369 explores the surface of NPM1 C-end domain stochastically in the WT construct (Figure 3E, Figure S4, and Table S2). Notably among these, cluster 3 matched with a previously reported NPM1 binding site of a small molecule that would allow the conformation stabilization of the C+ mutant.33 In line with this finding, this cluster exhibited the lowest desolvation energy and harbored the second largest buried surface area of all analyzed docking clusters (Table S2). Further work should aim to confirm whether enhanced disorder in the NPM1 C+ species results in the unraveling of cryptic binding sites that lead to (i) a higher binding affinity to UCM-13369 and (ii) the stabilization of NPM1 conformational states.
Unraveling the Molecular Mechanism of the New NPM1 Inhibitor UCM-13369
We next studied the impact of UCM-13369 in the genes and/or expression levels of NPM1 and NPM1 C+ pathway components (Figure 4A), including the positive regulation of c-Myc34 and the negative regulation of FBXW7 and the pro-apoptotic CASP8.35 Upon treatment with the compound in OCI-AML3 cell line, qPCR and Western blot revealed reduced levels for NPM1 and c-Myc, while FBXW7 was found to be upregulated (NPM1 p-value ≤0.0001, Figure 4B,C). The inhibition of NPM1 was also observed in MOLM13 cells expressing WT NPM1 (Figure S1B).
Figure 4.

UCM-13369 inhibits gene and mutant NPM1 C+ protein expression and restores nucleolar localization. (A) Schematic representation of the molecular signaling pathways of NPM1 mutation in AML. (B) Dot plot showing decreased NPM1 expression with UCM-13369 treatment (10 μM) in OCI-AML3 cells. (C) Western blot showing decreased NPM1 and c-Myc expression and increased FBXW7 expression with UCM-13369 treatment (10 μM) in OCI-AML3 cells at 24 and 48 h. Densitometry values for NPM1: reduction of 50% and 70%, respectively. (D) Top: Confocal microscopy images with NPM1 (Alexa Fluor-647, red) and nuclei (DAPI, blue) staining in MOLM13, OCI-AML3, and UCM-13369-treated OCI-AML3 cells (10 μM). Bottom: Violin plot of NPM1 nucleus/cytoplasm ratio in MOLM13 and OCI-AML3 cell lines, and in untreated and UCM-13369-treated OCI-AML3 cells.
To further elucidate the mechanism of action of UCM-13369, we studied the influence of the compound on the subcellular localization of the protein by confocal microscopy, using an Alexa Fluor-647-conjugated NPM1 primary antibody (Figure 4D). In OCI-AML3 cells, we visualized that UCM-13369 induced the reduction of the cytoplasmic form of the protein, typically associated with the NPM1 mutation (NPM1 C+),36 restoring the nucleolar localization phenotype of the protein, more similar to the WT state of NPM1 in MOLM13 cells.
Validation of the Therapeutic Potential of UCM-13369 in AML Patient Cells
Considering the inhibitory capacity of UCM-13369 on AML cell lines (shown in Figure 3A), the therapeutic potential was assessed in primary CD34+ cells from AML patients. Two ex vivo models were used from AML patients and healthy donors: CD34+ liquid HSC (hematopoietic stem cell) culture as a short-term model and CFU-E (colony-forming unit–erythroid) as a midterm model. Primary CD34+ cells were sensitive to UCM-13369, exhibiting decreased viability, as shown in Figure 5A for CFU-E (IC50 = 0.30 μM for AML patients, IC50 = 1.73 μM for healthy donors, p-value ≤0.0001) and in Figure 5B for liquid HSC culture. Interestingly, the compound displayed higher efficacy against primary cells in AML patients than in healthy donors (IC50 = 1.877 and 6.399 μM, respectively, p-value ≤0.0001). Moreover, the compound showed higher efficacy in patients with NPM1 C+ AML (IC50 = 1.725 μM, n = 4) than in WT NPM1 AML patients (IC50 = 3.774 μM, n = 3, p-value ≤0.007) (Figure 5B).
Figure 5.

UCM-13369 has efficacy against AML cell lines and primary cells. (A) Dose–response curve of UCM-13369 in colony-forming unit (CFU) assays performed with primary HSCs bone marrow mononuclear fraction from healthy donors and AML patients. (B) Left: Dose–response curve of UCM-13369 in short-term cultures of HSCs performed with primary HSCs from bone marrow mononuclear fraction from healthy donors and AML patients. Right: Dose–response curve of the same assay with samples from AML patients with NPM1 WT vs NPM1 C+. (C) Dot plot of a representative sample from flow cytometry analysis using CD34-PE/Annexin V-FITC staining of short-term cultures of primary HSCs from AML patients performed with primary HSCs from bone marrow mononuclear fraction with and without treatment.
The inhibition of NPM1, especially NPM1 C+ pathway (as shown in Figure 4), by UCM-13369 correlates with the increase of apoptosis observed in treated primary CD34+ cells upon staining with Annexin V (Figure 5C).
Efficacy in an AML Animal Model
Based on the encouraging results obtained in ex vivo models of AML, we further assessed the therapeutic potential of the new NPM1 inhibitor in vivo. Prior to the evaluation of the compound in an animal model of AML, in vivo pharmacokinetic studies were addressed. Following a single intraperitoneal administration of UCM-13369 at a dose of 25 mg/kg, levels of 200–600 μg/L were detected in mouse peripheral blood up to 6 h (Figure 6A). Plasma protein binding of the compound in mouse serum was measured using an equilibrium dialysis assay, indicating a bound fraction of 83 ± 1%. Next, the efficacy of the compound was evaluated in immunodeficient mice after injection of OCI-AML3-ffLuc-GFP cells as an AML animal model. Notably, as shown in Figure 6B, the UCM-13369-treated group (50 mg/kg, tail vein injection) showed a reduction of tumor infiltration compared to untreated group at end point (the reduction in tumor mass was negligible in 25-mg/kg-treated animals, data not shown). However, this effect did not result in an increase in survival as no difference was observed between treated and untreated animals (Figure S1C). Hematoxylin-eosin (H&E) staining of the bone marrow revealed less tumor cell infiltration in treated mice, supporting the efficacy of the compound against malignant cells (Figure 6C). Caspase-3 pathology analysis of treated mice revealed the absence of toxicity in bone marrow and liver (data not shown), while some caspase-3 cleavage was observed in the kidney, pointing to some toxicity in this organ, although most part of this staining was unspecific (Figure S1D). Indeed, the dose–response viability assay in human renal proximal tubule epithelial cells (hRPT Epi Cells) revealed toxicity (Figure S1E), suggesting that renal toxicity is one of the probable causes of lower-than-expected survival of treated AML animals.
Figure 6.

UCM-13369 shows efficacy against AML cells in vivo. (A) Values of UCM-13369 concentration in vivo at different time points after injection (25 mg/kg) analyzed by HPLC-MS. (B) Bioluminescence images acquired using an in vivo imaging system of NSG mice after injection of OCI-AML3 cell line with and without UCM-13369 treatment (50 mg/kg). (C) Microscopy analysis of the OCI-AML3 infiltration by H&E staining slides of bone marrow samples from NSG mice with paired time points after engraftment with and without UCM-13369 treatment (50 mg/kg).
Discussion
In the past decade, the human microbiota has emerged as one of the most important topics in cancer research, as multiple associations with cancer development, progression, and response to treatment have been reported.4,37,38 Considering the cancer protective effect of several microbiota metabolites,39−41 in the present work, we describe the design, screening, and validation of novel small molecules inspired on microbiota metabolites in order to identify new drug candidates for cancer treatment. We designed a set of new chemotypes containing privileged scaffolds present in selected metabolites22 (Figure 1) and obtained them following synthetic routes based on asymmetric organocatalytic reactions (Figure 2). This valuable methodology provides complex chemotypes characterized by the presence of chiral carbon centers, which also address the demand for compounds with an increased number of sp3 carbon atoms, a parameter that has achieved great significance in successfully guiding drug research in recent years.42 Following a phenotypic drug discovery approach, our synthetic microbiota-inspired molecules were screened in CSC models. Among the different cells that contribute to tumor growth, CSCs have been described as a subpopulation of cells that potentially originate the cancer process and contribute to expansion, resistance, recurrence, and metastasis after therapy.43−45 We selected the compound UCM-13369 (4b) that showed capacity to inhibit tumorsphere growth of CSCs and confirmed its safety against healthy cells (Table S1). Differential proteomic profiling in CD34+ stem cells was analyzed to unravel the molecular target(s) associated with the efficacy of UCM-13369 against CSCs. Among the different stem- and cancer-related proteins that were ablated by the compound (see Supplementary Excel file), NPM1 stands out as a particularly relevant protein in AML, a hematological disease with a high mortality rate.28 AML affects the hematopoietic system through abnormal proliferation of myeloid cells in the bone marrow and ultimately suppresses the production of healthy blood cells. The standard treatment for AML for approximately 50 years has consisted of a combination of cytarabine (an antimetabolic agent that generates DNA damage in S-phase cells) and an anthracycline (an intercalating agent) in a program known as the “7 + 3” regimen. This intensive treatment is only effective in a small subset of AML patients and the therapeutic landscape has broadened in the last five years, driven by the development of targeted therapies focused on genetic mutations present in the disease.46,47 NPM1 and fms-like tyrosine kinase 3 (FLT3) are the most frequent mutations in AML, with an incidence of more than 50%,48 and therapies targeting specific mutations appear to have great potential to improve prognosis and treatment. Thus, the combination of the 7 + 3 regimen with mutant FLT3 inhibitors, such as midostaurin (Rydapt), improves response rates and outcomes in 20% of diagnosed patients whose leukemia has this mutation.49,50 Therefore, primary and secondary resistance remains a critical problem for most patients and treatment failure remains common. Although the NPM1 mutation is considered a good prognostic marker, even in this scenario, the 5-year rate of NPM1-mutated cases is below 50% overall survival and the crude numbers of deceased AML patients harbor NPM1 mutation, mainly due to the high incidence of this genetic alteration.51 Due to its biological and clinical characteristics, AML with NPM1 mutation was recognized in 2017 as a distinct entity in the WHO classification of hematopoietic malignancies. Unfortunately, there are no NPM1 inhibitors currently used in clinical practice, and a few candidate drugs are in clinical trials, such as NucAnt 6L and CIGB-300, but only ATRA is being studied as an adjuvant for the treatment of AML.52,53 Therefore, the development of new targeted therapy treatments is critical for the future of patients, to improve the dramatically low survival rate of AML.
An optimal therapeutic strategy against AML is expected to be a targeted therapy that abrogates the CSC population. Interestingly, the cytotoxicity against CSCs observed for UCM-13369 (Table S1) was mediated by NPM1, as the compound exhibited efficacy in the micromolar range in AML cell lines expressing WT and mutant NPM1 C+, especially in mutant cells (Figure 3A). Our results were validated in AML patient cells, in which UCM-13369 induced decreased viability while we observed lower cytotoxicity in healthy donor cells (Figure 5A,B), confirming the therapeutic window opportunity of the compound. In addition, greater efficacy was observed in AML patients harboring the NPM1 C+ mutation than in those lacking it (Figure 5B). This therapeutic potential was translated to a mouse model of AML with NPM1 C+ mutation, as intraperitoneal administration of UCM-13369 resulted in a reduction of tumor infiltration compared to the untreated group, although no increase in mice survival was observed (Figure 6B,C).
NPM1 is a nucleolar phosphoprotein, whereas the mutant form designated as NPM1 C+ protein translocates to the cytoplasm and confers undifferentiating properties to myeloid stem cells that transform into CSCs, being responsible for the stem cell signature characteristic of AML.54−62 Notably, the new compound, by interacting specifically with the C-end monomeric DNA-binding domain of NPM1 C+ form, was able to restore the nucleolar localization phenotype of the protein, reducing the presence of NPM1 in the cytoplasm of OCI-AML3 cells (Figure 4D), which is characteristic of the mutation and critical for the maintenance of the disease.36 In addition, upon UCM-13369 treatment, we observed downregulation of the pathway associated with the NPM1 C+ form34,35 (Figure 4B,C) and activation of stem cell apoptosis (Figure 5C). These results further support the mechanism of action of the compound through the mutant NPM1 pathway, which explains why the inhibitor has a higher efficacy in NPM1-mutated cells compared to NPM1-expressing cells. Moreover, cellular colocalization and ITC measurements of the compound with the protein (Figure 3B,C) confirmed the formation of the NPM1-UCM-13369 complex, validating the NPM1 protein as a molecular target of the new small-molecule targeting AML.
In summary, we disclose the phenotype-guided discovery of UCM-13369, a novel small molecule inspired on microbiota metabolites. Together, our results revealed that the activity of UCM-13369 in CSCs is linked to the regulation of the NPM1 protein and support the potential of the new synthetic inhibitor especially for the treatment of NPM1-mutated AML. The efficacy exhibited by UCM-13369 in ex vivo and in vivo AML models establishes tetrahydrocarbazole chemotype as worthy of further advancement as a promising therapeutic opportunity for the targeted treatment of leukemia.
Methods
Synthesis of tert-Butyl-2-(4-methoxyphenyl)-3-nitro-4-(2-oxoethyl)-1,2,3,4-tetrahydro-9H-carbazole-9-carboxylate, 16
To a solution of (S)- or (R)-2-{diphenyl[(trimethylsilyl)oxy]methyl}pyrrolidine (0.20 equiv) in toluene (2 mL/mmol), benzoic acid (0.20 equiv) was added and the mixture was stirred at rt for 10 min under air. Then, tert-butyl-2-methyl-3-[(1E)-3-oxoprop-1-en-1-yl]-1H-indole-1-carboxylate (1.50 equiv) and trans-4-methoxy-β-nitrostyrene (1.00 equiv) were added and the reaction was heated at 110 °C under MW irradiation for 5.5 h. Afterward, the crude was flushed through a short plug of silica, using a 1:1 mixture of DCM/diethyl ether, and the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography (toluene/diethyl ether, 98:2) to afford the title compound (2S,3S,4S)- or (2R,3R,4R)-16.
(2S,3S,4S)-16 (16a) was obtained from trans-4-methoxy-β-nitrostyrene (213 mg, 1.18 mmol), using the S enantiomer of the catalyst (77 mg, 0.24 mmol), as a yellow solid (294 mg, 53%). The spectroscopic data were consistent with those previously reported.24Rf: 0.30 (hexane/EtOAc 8:2). [α]20D = −15.9 (c = 0.97, CHCl3) lit.24 [α]20D = −12.01 (c = 0.85, CHCl3). 1H NMR (CDCl3): δ 1.64 (s, 9H, 3CH3), 2.97 (dd, J = 18.7, 2.2, 1H, 1/2CH2CHO), 3.25–3.38 (m, 2H, H1, 1/2CH2CHO), 3.52–3.62 (m, 2H, H1, H2), 3.79 (s, 3H, OCH3), 4.13–4.21 (m, 1H, H4), 5.21 (dd, J = 10.7, 9.1, 1H, H3), 6.87 (d, J = 8.7, 2H, H3′, H5′), 7.22 (d, J = 8.8, 2H, H2′, H6′), 7.21–7.33 (m, 2H, H6, H7), 7.39 (d, J = 7.8, 1H, H5), 8.14 (d, J = 7.8, 1H, H8), 9.71 (s, 1H, CHO). 1D 1H NMR NOE: irradiation of the signal at δ 5.21 ppm (dd, H3) yielded NOE on 2.97(dd, 1/2CH2CHO), and 7.22 (d, H2′, H6′).
(2R,3R,4R)-16 (16b) was obtained from trans-4-methoxy-β-nitrostyrene (168 mg, 0.93 mmol), using the R enantiomer of the catalyst (61 mg, 0.19 mmol), as a yellow solid (190 mg, 45%, er = 96:4). [α]20D = +11.8 (c = 1.31, CHCl3). Spectroscopic data were in agreement with those described for enantiomer 16a. The er was determined after reduction of 16b to the corresponding alcohol (2.5 equiv NaBH4, methanol, rt, 3 h, 92%) and chiral HPLC analysis (Chiralpak IC, 5 μm, 4.6 mm × 250 mm; 80:20 hexane/i-PrOH, flow rate 1.00 mL/min, λ = 254 nm, tR(major) = 13.22 min, tR(minor) = 18.42 min).
Synthesis of tert-Butyl-4-{2-[(cyclopropylmethyl)amino]ethyl}-2-(4-methoxyphenyl)-3-nitro-1,2,3,4-tetrahydro-9H-carbazole-9-carboxylate, 25
(2S,3S,4S)-25 (25a)
Following general procedure B for reductive amination (see the Supporting Information) using 16a (225 mg, 0.48 mmol) and (cyclopropylmethyl)amine (80 μL, 0.97 mmol), compound 25a was obtained as an oil (218 mg, 86%), which was used in the next step without further purification. Rf: 0.48 (DCM/ethanol 9:1). [α]20D = −13.8 (c = 1.00, CHCl3). IR (ATR): ν 1730 (C=O), 1550 (NO2), 1251 (COC). 1H NMR (CDCl3): δ 0.02–0.07 (m, 2H, CH2cpr), 0.39–0.45 (m, 2H, CH2cpr), 0.80–0.93 (CHcpr), 1.64 (s, 9H, 3CH3), 1.96–2.07 (m, 1H, 1/2NHCH2CH2), 2.29–2.43 (m, 3H, 1/2NHCH2CH2, NHCH2CH), 2.46–2.55 (m, 1H, 1/2NHCH2CH2), 2.61–2.70 (m, 1H, 1/2NHCH2CH2), 3.18–3.28 (m, 1H, H1), 3.42–3.56 (m, 2H, H1, H2), 3.79 (s, 3H, OCH3), 3.88–3.93 (m, 1H, H4), 5.25–5.32 (m, 1H, H3), 6.87 (d, J = 8.7, H3′, H5′), 7.20–7.33 (m, 2H, H6, H7), 7.21 (d, J = 8.7, 2H, H2′, H6′), 7.59 (d, J = 7.1, 1H, H5), 8.12 (d, J = 7.7, 1H, H8). 13C NMR (CDCl3): δ 3.4 (CH2cpr), 11.2 (CHcpr), 28.4 (3CH3), 30.6 (NHCH2CH2), 32.9 (C1), 38.5 (C4), 45.4 (NHCH2CH2), 45.5 (C2), 54.9 (NHCH2CH), 55.4 (OCH3), 84.4 (C(CH3)3), 93.6 (C3), 114.4 (C3′, C5′), 115.3 (C4a), 115.8 (C8), 119.1 (C5), 123.1, 124.2 (C6, C7), 127.9 (C8a), 128.8 (C2′, C6′), 131.1 (C1′), 134.3 (C9a), 136.3 (C4b), 150.4 (NCOO), 159.3 (C4′). HPLC (tR, min): 19.58. MS (ESI, m/z, %): 520.0 ([M + H]+, 100).
(2R,3R,4R)-25 (25b)
Following general procedure B for reductive amination (see the Supporting Information) using 16b (200 mg, 0.43 mmol) and (cyclopropylmethyl)amine (75 μL, 0.86 mmol), compound 25b was obtained as an oil (186 mg, 83%), which was used in the next step without further purification. [α]20D = +12.13 (c = 0.75, CHCl3). Spectroscopic data were in agreement with those described for enantiomer 25a.
Synthesis of tert-Butyl-3-amino-4-{2-[(cyclopropylmethyl)amino]ethyl}-2-(4-methoxy-phenyl)-1,2,3,4-tetrahydro-9H-carbazole-9-carboxylate, 26
(2S,3S,4S)-26 (26a)
Following general procedure C for nitro group reduction (see the Supporting Information) using 25a (218 mg, 0.42 mmol), compound 26a was obtained as an oil (220 mg, quantitative), which was used in the next step without further purification. Rf: 0.34 (DCM/methanol/NH3 9:1:0.1). [α]20D = −15.1 (c = 1.00, CHCl3). IR (ATR): ν 1727 (C=O), 1363 (C–N), 1249 (COC). 1H NMR (CDCl3): δ 0.03–0.08 (m, 2H, CH2cpr), 0.40–0.46 (m, 2H, CH2cpr), 0.84–0.98 (m, 1H, CHcpr), 1.62 (s, 9H, 3CH3), 2.17–2.27 (m, 1H, 1/2NHCH2CH2), 2.31–2.36 (m, 1H, 1/2NHCH2CH2), 2.41 (d, J = 6.9, 2H, NHCH2CH), 2.53–2.62 (m, 1H, 1/2NHCH2CH2), 2.64–2.82 (m, 2H, H2, 1/2NHCH2CH2), 2.92–2.97 (m, 1H, H4), 3.09 (ddd, J = 18.0, 11.3, 2.6, 1H, H1), 3.19 (dd, J = 10.5, 8.5, 1H, H3), 3.35 (dd, J = 17.9, 4.3, 1H, H1), 3.82 (s, 3H, OCH3) 6.91 (d, J = 8.7, 2H, H3′, H5′), 7.19–7.28 (m, 2H, H6, H7), 7.22 (d, J = 8.4, 2H, H2′, H6′), 7.57 (dd, J = 6.8, 2.1, 1H, H5), 8.11 (dd, J = 7.1, 1.9, 1H, H8). 13C NMR (CDCl3): δ 3.5 (2CH2cpr), 11.1 (CHcpr), 28.4 (3CH3), 32.1 (NHCH2CH2), 33.7 (C1), 41.4 (C4), 46.9 (NHCH2CH2), 49.7 (C2), 55.1 (NHCH2CH), 55.4 (OCH3), 56.5 (C3), 83.8 (C(CH3)3), 114.4 (C3′, C5′), 115.6 (C8), 118.4 (C4a), 119.0 (C5), 122.7 (C6), 123.5 (C7), 128.9 (C4b), 129.1 (C2′, C6′), 135.2 (C9a), 135.4 (C1′), 136.4 (C8a), 150.6 (NCOO), 158.7 (C4′). HPLC (tR, min): 13.78. MS (ESI, m/z, %): 490.1 ([M + H]+, 100).
(2R,3R,4R)-26 (26b)
Following general procedure C for nitro group reduction (see the Supporting Information) using 25b (160 mg, 0.31 mmol), compound 26b was obtained as an oil (152 mg, quantitative), which was used in the next step without further purification. [α]20D= +17.8 (c = 1.03, CHCl3). Spectroscopic data were in agreement with those described for enantiomer 26a.
Synthesis of 4-{2-[(Cyclopropylmethyl)amino]ethyl}-2-(4-methoxyphenyl)-2,3,4,9-tetrahydro-1H-carbazol-3-amine, 4
To a solution of enantiomer 26a or 26b (1.00 equiv) in methanol (2.5 mL/mmol), HCl (35.0 equiv, 37% aq. solution) was added and the reaction mixture was stirred at rt for 4 h. Then, solvents were evaporated under reduced pressure and the resulting solid was washed with anhydrous diethyl ether and dried under vacuum to afford the corresponding hydrochloride salt of (2S,3S,4S) or (2R,3R,4R)-4.
(2S,3S,4S)-4 (4a) was obtained from 26a (85 mg, 0.17 mmol) as hydrochloride salt (42 mg, 50%, er = 96:4). Rf: 0.30 (DCM/methanol/NH3 9:1:0.1). [α]20D = −5.4 (c = 0.24, methanol). Chiral HPLC (method J, tR, min): 4.74. IR (ATR): ν 3375 (NH), 1458 (C–N). 1H NMR (500 MHz, methanol-d4): δ 0.26–0.36 (m, 2H, CH2cpr), 0.55–0.65 (m, 2H, 2CH2cpr), 0.93–1.02 (m, 1H, CHcpr), 2.21–2.43 (m, 2H, NHCH2CH2), 2.64–2.82 (m, 3H, NHCH2CH, 1/2NHCH2CH2), 3.01–3.24 (m, 3H, 2H1, 1/2NHCH2CH2), 3.29–3.38 (m, 1H, H2), 3.48–3.55 (m, H4), 3.79 (s, 3H, CH3), 3.92–3.97 (m, 1H, H3), 6.96 (d, J = 8.3, 2H, H3′, H5′), 7.01–7.11 (m, 2H, H6, H7), 7.32 (d, J = 7.7, 1H, H8), 7.37 (d, J = 8.5, 2H, H2′, H6′), 7.56 (d, J = 7.7, 1H, H5). 13C NMR (125 MHz, methanol-d4): δ 4.4 (CH2cpr), 4.6 (CH2cpr), 8.13 (CHcpr), 27.8 (NHCH2CH2), 29.4 (C1), 37.6 (C4), 44.4 (C2), 45.1 (NHCH2CH2), 53.7 (NHCH2CH), 55.8 (CH3), 56.1 (C3), 106.7 (C4a), 112.3 (C5), 115.8 (C3′, C5′), 118.8 (C8), 120.4 (C6), 122.5 (C7), 127.4 (C4b), 130.3 (C2′, C6′), 132.7 (C1′), 134.8 (C9a), 138.3 (C8a), 161.0 (C4′). HPLC (tR, min): 11.45. MS (ESI, m/z, %): 390.1 ([M + H]+, 100). Elemental analysis calculated for C25H31N3O·2HCl·2H2O: %C 60.24, %H 7.98, %N 8.43; experimental: %C 60.98, %H 7.74, %N 8.11.
(2R,3R,4R)-4 (4b, UCM-13369) was obtained from 26b (100 mg, 0,2 mmol) as hydrochloride salt (40 mg, 40%, er = 95:5). [α]20D= +6.8 (c = 0.30, methanol). Chiral HPLC (method J, tR, min): 6.42. HRMS (ESI, m/z): calculated for C25H32N3O [M + H]+: 390.2540, found: 390.2526. Elemental analysis calculated for C25H31N3O·2HCl·2H2O: %C 60.24, %H 7.98, %N 8.43; experimental: %C 60.63, %H 7.68, %N 8.15. Spectroscopic data were in agreement with those described for enantiomer 4a.
Cell Viability Assays
The impact of the novel compound UCM-13369 on cell viability was assessed in cell lines, MCF-7, HCT-116, IMR90, OCI-AML3, and MOLM13; mamospheres and colonospheres; bone marrow mononuclear primary cells; and colony-forming unit assays. Cells were seeded at 3 × 104 cells/mL in 96-well plates. The test compound was diluted in PBS. Cell lines were treated for 48 h with 4–6 different concentrations of UCM-13369 (10 μM, 5 μM, 1 μM, 0,5 μM, 0,1 μM) or vehicle (PBS). Viability was evaluated by MTT or WST-1 and dose/response curves were performed.
Flow Cytometry
CFU-E and CD34+ primary cells with and without UCM-13369 treatment were analyzed. The following monoclonal antibodies were used: CD34-PE, Annexin V-FITC (BD Biosciences). The cells were incubated with antibodies in PBS on ice for 15 min in the dark. The analysis of CD34+ and Annexin V+ subpopulation was carried out in a flow cytometer FACSCalibur (Becton Dickinson, Citometría y Microscopía de Fluorescencia core facility, UCM) and the data were processed using FlowJo_V10 software.
Proteomic Analysis
CD34+ from healthy donors (n = 3) were cultured for 24 h with and without 5 μM UCM-13369. Control and treated samples were harvested and pooled and protein extracts were obtained after cell lysis with urea-containing CHAPS buffer and sonication at 4 °C. Proteins were precipitated with methanol/chloroform and protein concentration was measured using Pierce 660 nm Protein Assay Reagent. For tryptic digestion, 10 μg of each depleted protein sample was individually dissolved in 8 M urea and 25 mM ammonium bicarbonate, reduced, and alkylated with iodoacetamide, according to a method previously described.63 Urea concentration was reduced to 2 M with 25 mM ammonium bicarbonate and the samples were digested overnight at 37 °C with trypsin (Sigma-Aldrich), with a 25:1 ratio. After digestion, the samples were desalted using ZipTip (Merck) as described. For liquid chromatography and mass spectrometer analysis, a 2 μg aliquot of each digested sample was subjected to 1D-nano LC ESI-MSMS analysis using a nano liquid chromatography system (Eksigent Technologies nano LC Ultra 1D plus, SCIEX, Foster City, CA) coupled to a high-speed Triple TOF 5600 mass spectrometer (SCIEX, Foster City, CA) with a Nanospray III source. The analytical column used was a silica-based reversed-phase Acquity UPLC M-Class Peptide BEH C18 Column, 75 μm × 150 mm, 1.7 μm particle size and 130 Å pore size (Waters). The trap column was a C18 Acclaim PepMap TM 100 (Thermo Scientific), 100 μm × 2 cm, 5 μm particle diameter, 100 Å pore size, switched online with the analytical column. The loading pump delivered a solution of 0.1% formic acid in water at 2 μL/min. The nanopump provided a flow rate of 250 nL/min and was operated under gradient elution conditions. Peptides were separated using a 250 min gradient ranging from 2% to 90% mobile phase B (mobile phase A: 2% ACN, 0.1% formic acid; mobile phase B: 100% ACN, 0.1% formic acid). The injection volume was 5 μL. The data acquisition was performed using a Triple TOF 5600 System (SCIEX, Foster City, CA). Data were acquired using an ionspray voltage floating (ISVF) 2300 V, curtain gas (CUR) 35, interface heater temperature (IHT) 150, ion source gas 1 (GS1) 25, declustering potential (DP) 100 V. All data were acquired using information-dependent acquisition (IDA) mode with Analyst TF 1.7 software (SCIEX). For IDA parameters, 0.25 s MS survey scans (mass range of 350–1250 Da) were followed by 15 MS/MS scans of 100 ms (mass range of 100–1800, total cycle time was 2 s). Switching criteria were: ion m/z greater than 350 and smaller than 1250, with a charge state of 2–5 and an abundance threshold of more than 90 counts (cps). Former target ions were excluded for 15 s. IDA rolling collision energy (CE) parameters script was used for automatically controlling the CE. The mass spectrometry data analysis and quantification were processed using PeakView 2.2 Software (SCIEX, Foster City, CA) and exported as mgf files. Protein data searches were performed either using Mascot Server v.2.6.1 or Peaks Studio 7.5 search engines against a target/decoy database built from sequences in the Homo sapiens reference proteome at Uniprot Knowledgebase. Search parameters were set as follows: trypsin enzyme; 2 missed cleavages were allowed; carbamidomethyl (C) as fixed modification and acetyl (Protein N-term) and oxidation (M) as variable modifications. Peptide mass tolerance was set to ±25 ppm for precursors and 0.02 Da for fragment masses. Spectral counting was used to perform a semiquantitative analysis (see Venn diagram in the Supporting Information and Supplementary Excel file).
Confocal Microscopy
Cells were plated onto glass coverslips or Greiner Bio-One μClear black 96-well plate (7.5 × 104 cells per well) and treated with compound UCM-13369 for 48 h. Then, cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 30 min, and blocked in 5% bovine serum albumin for 1 h at rt. Incubation with primary antibodies against NPM1 (7H10B9, NOVUS Biologicals) was carried out overnight at 4 °C. Alexa Fluor-488-conjugated antimouse or antirabbit antibodies (Molecular Probes) were used for detection of the reported proteins by confocal microscopy. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) bath. A total of 35 fields per well were acquired with ×40 magnification lens for those samples run in the Opera HCS system (PerkinElmer). Images were segmented using the DAPI staining and the coefficient of variation of NPM1 intensity per nucleus/cytoplasm was calculated using Definiens Developer XD software.
Isothermal Titration Calorimetry
ITC experiments were performed in a Nano ITC Low Volume (TA Instruments) at 25 °C. The experiments consisted of 17 injections of 2.9 μL of 1 mM UCM-13369 in 20 mM sodium phosphate buffer (pH 7.2) with 150 mM NaCl and 1 mM TCEP into the sample cell, initially filled with 161 μL of 20 μM pentameric WT NPM1 N-end or 100 μM monomeric WT or C+ NPM1 C-end proteins. The injection interval was set to 180 s, allowing the thermal power signal to return to the baseline before the next injection. Homogeneous mixing was achieved by maintaining stirring speed at 300 rpm. The reference cell was filled with degassed ultrapure water. All samples were degassed prior to the experiments. Injection heats normalized per mole of injectant vs molar ratio data were fitted to an independent binding site model using Origin 7.0 (OriginLab) for the analysis.
In Vivo Experiments
The animal studies were performed according to the guidelines of the Institutional Animal Care and Use Committees of the Comunidad de Madrid, Spain, under approved protocol (PROEX 023/17).
For the pharmacokinetic study of UCM-13369, after injecting 25 mg/kg dose of the compound, samples of blood were withdrawn at different times (30, 60, 120, 240, 360 min), and the presence of tested compound was analyzed by HPLC-MS. The area of the molecular peak allowed us to calculate the concentration of the compound present in mice serum at each time.
An AML xenograft mouse model was used for the in vivo study of efficacy. Female 6- to 8-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Vivotecnia, Madrid, Spain) were inoculated intravenously by tail vein injection with OCI-AML3-ffLuc-GFP cells (1 × 106). Four days later, the mice were randomized into two groups of 10 animals: the first group received intraperitoneal injections of vehicle (0.9% saline solution) 5 days a week; the second group received intraperitoneal injections of 25 mg/kg or 50 mg/kg UCM-13369 five times weekly for 3 weeks. The mice were monitored for tumor burden, distribution, and engraftment every week by IVIS whole-body bioluminescence imaging. Bioimaging of MM burden in vivo was performed by IVIS Imaging system (Caliper Life Science). A solution of luciferin (D-luciferin monosodium salt, Thermo Fisher) in PBS was injected (150 mg/kg, ip), and luminescence signal was determined after 10 min. The data were analyzed using the Living Image 4.2 (Caliper Life Science) software.
Statistical analysis was performed using GraphPad Software, Inc., CA (version 6.0). All data are presented as mean ± SEM and are representative of three independent experimental replicates (n = 3). Normally distributed data were analyzed by unpaired Student’s t test or one-way ANOVA. For nonparametric data, Mann–Whitney test was used. Overall survival (OS) and PFS were analyzed by Kaplan–Meier curves. p-Values < .05 were considered statistically significant.
Acknowledgments
This work was supported by grants PID2022-138797OB-I00, PGC2018-096049-B-I00 and PID2021-126663NB-I00 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”; grant PID2019-106279RB-I00 funded by MCIN/AEI/10.13039/501100011033; grant PDC2022-133488-I00 funded by MCIN/AEI/10.13039/501100011033 and by the “European Union NextGenerationEU/PRTR”; grants PI21/00191 and CP19/00140 funded by Instituto de Salud Carlos III; CNIO agreements 2017-2020, 2020-2023 funded by Foundation CRIS contra el Cancer; grants BIO-198 and P18-FR-3487 funded by Junta de Andalucía; VI PPIT program funded by Universidad de Sevilla; and by Ramón Areces Foundation. The authors acknowledge technological support from NMR, mass spectrometry and elemental analysis CAIs (Complutense University of Madrid), Biointeractomicts Platform (cicCartuja, Seville), and the Services at CITIUS (University of Seville). S.A., A.S.-M., I.A.-A. and R.L.G.-A. are grateful to Ministerio de Ciencia e Innovación and Complutense University of Madrid for predoctoral fellowships; M.V.-E. to European Union’s Horizon 2020 for Marie Sklodowska-Curie grant; and P.A.-G. to Fundación Española de Hematología y Hemoterapia for grant. The authors thank Dr. Adrián Velázquez-Campoy at the University of Saragossa for helping in fitting ITC analysis and Prof. Miguel A. De la Rosa at the University of Seville for critical reading of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00682.
Additional results of in vitro and in vivo experiments (Figure S1), synthesis of probe UCM-13369-Cy5 (Figure S2), purification of NPM1 species and validation of the peptide sequence of NPM1 C-end C+ (Figure S3), statistical analysis of ab initio UCM-13369 binding site prediction in WT NPM1 C-end domain (Figure S4), phenotypic screening data of compounds 1–13 (Table S1), statistical analysis of HADDOCK data after clustering the solutions for the NPM1 C-end WT and UCM-13369 (Table S2); detailed description of the methods of all of the experiments performed: synthesis and characterization data for all compounds, cellular assays, NPM1 protein studies, and plasma protein binding (PPB) assay; NMR spectra for final compounds 1–13 and probe UCM-13369-Cy5 and HPLC traces (PDF)
Proteomic data (XLSX)
Colocalization of compound UCM-13369 with NPM1 protein (AVI)
Author Contributions
# S.A. and H.V.-V. contributed equally. CRediT: Sergio Algar methodology; Henar Vázquez-Villa conceptualization, investigation, supervision, writing-original draft; Pedro Aguilar-Garrido methodology; Miguel Angel Navarro-Aguadero methodology; Maria Velasco-Estevez methodology; Anabel Sanchez-Merino methodology; Ivan Arribas-Alvarez methodology; Alberto Paradela methodology; Rafael L Giner-Arroyo methodology; Joaquin Tamargo-Azpilicueta methodology; Irene Díaz-Moreno funding acquisition, investigation, supervision, writing-original draft; Joaquin Martinez-Lopez investigation; Miguel Gallardo conceptualization, funding acquisition, investigation, supervision, writing-original draft; María L. López-Rodríguez conceptualization, funding acquisition, investigation, supervision, writing-original draft; Bellinda Benhamú conceptualization, investigation, supervision, writing-original draft.
The authors declare no competing financial interest.
Supplementary Material
References
- McCarville J. L.; Chen G. Y.; Cuevas V. D.; Troha K.; Ayres J. S. Microbiota Metabolites in Health and Disease. Annu. Rev. Immunol. 2020, 38, 147–170. 10.1146/annurev-immunol-071219-125715. [DOI] [PubMed] [Google Scholar]
- Aggarwal N.; Kitano S.; Puah G. R. Y.; Kittelmann S.; Hwang I. Y.; Chang M. W. Microbiome and Human Health: Current Understanding, Engineering, and Enabling Technologies. Chem. Rev. 2023, 123, 31–72. 10.1021/acs.chemrev.2c00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari S. N.; McCurry M. D.; Devlin A. S. Chains of Evidence from Correlations to Causal Molecules in Microbiome-Linked Diseases. Nat. Chem. Biol. 2021, 17, 1046–1056. 10.1038/s41589-021-00861-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink B. A.; Khan M. A. W.; Hermann A.; Gopalakrishnan V.; Wargo J. A. The Microbiome, Cancer, and Cancer Therapy. Nat. Med. 2019, 25, 377–388. 10.1038/s41591-019-0377-7. [DOI] [PubMed] [Google Scholar]
- Papadopoulos P. D.; Tsigalou C.; Valsamaki P. N.; Konstantinidis T. G.; Voidarou C.; Bezirtzoglou E. The Emerging Role of the Gut Microbiome in Cardiovascular Disease: Current Knowledge and Perspectives. Biomedicines 2022, 10, 948. 10.3390/biomedicines10050948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasso J. M.; Ammar R. M.; Tenchov R.; Lemmel S.; Kelber O.; Grieswelle M.; Zhou Q. A. Gut Microbiome-Brain Alliance: A Landscape View into Mental and Gastrointestinal Health and Disorders. ACS Chem. Neurosci. 2023, 14, 1717–1763. 10.1021/acschemneuro.3c00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. J.; Hase K. Gut Microbiota-Generated Metabolites in Animal Health and Disease. Nat. Chem. Biol. 2014, 10, 416–424. 10.1038/nchembio.1535. [DOI] [PubMed] [Google Scholar]
- Saha S.; Rajpal D. K.; Brown J. R. Human Microbial Metabolites as a Source of New Drugs. Drug discovery today 2016, 21, 692–698. 10.1016/j.drudis.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Turroni S.; Brigidi P.; Cavalli A.; Candela M. Microbiota-Host Transgenomic Metabolism, Bioactive Molecules from the Inside. J. Med. Chem. 2018, 61, 47–61. 10.1021/acs.jmedchem.7b00244. [DOI] [PubMed] [Google Scholar]
- Cho K. W.; Lee O. H.; Banz W. J.; Moustaid-Moussa N.; Shay N. F.; Kim Y. C. Daidzein and the Daidzein Metabolite, Equol, Enhance Adipocyte Differentiation and Ppargamma Transcriptional Activity. J. Nutr. Biochem. 2010, 21, 841–847. 10.1016/j.jnutbio.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Horiuchi H.; Harada N.; Adachi T.; Nakano Y.; Inui H.; Yamaji R. S-Equol Enantioselectively Activates Camp-Protein Kinase a Signaling and Reduces Alloxan-Induced Cell Death in Ins-1 Pancreatic Beta-Cells. J. Nutr. Sci. Vitaminol. 2014, 60, 291–296. 10.3177/jnsv.60.291. [DOI] [PubMed] [Google Scholar]
- Kasimsetty S. G.; Bialonska D.; Reddy M. K.; Thornton C.; Willett K. L.; Ferreira D. Effects of Pomegranate Chemical Constituents/Intestinal Microbial Metabolites on Cyp1b1 in 22rv1 Prostate Cancer Cells. J. Agric. Food Chem. 2009, 57, 10636–10644. 10.1021/jf902716r. [DOI] [PubMed] [Google Scholar]
- Piwowarski J. P.; Kiss A. K.; Granica S.; Moeslinger T. Urolithins, Gut Microbiota-Derived Metabolites of Ellagitannins, Inhibit Lps-Induced Inflammation in Raw 264.7 Murine Macrophages. Mol. Nutr. Food Res. 2015, 59, 2168–2177. 10.1002/mnfr.201500264. [DOI] [PubMed] [Google Scholar]
- Digby J. E.; Martinez F.; Jefferson A.; Ruparelia N.; Chai J.; Wamil M.; Greaves D. R.; Choudhury R. P. Anti-Inflammatory Effects of Nicotinic Acid in Human Monocytes Are Mediated by Gpr109a Dependent Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 669–676. 10.1161/ATVBAHA.111.241836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N.; Gurav A.; Sivaprakasam S.; Brady E.; Padia R.; Shi H.; Thangaraju M.; Prasad P. D.; Manicassamy S.; Munn D. H.; Lee J. R.; Offermanns S.; Ganapathy V. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 2014, 40, 128–139. 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal T.; Alaniz R. C.; Wood T. K.; Jayaraman A. The Bacterial Signal Indole Increases Epithelial-Cell Tight-Junction Resistance and Attenuates Indicators of Inflammation. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 228–233. 10.1073/pnas.0906112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer D. S.; Zimmerman N. P.; Wang L. S.; Ransom B. W. S.; Carmella S. G.; Kuo C. T.; Siddiqui J.; Chen J. H.; Oshima K.; Huang Y. W.; Hecht S. S.; Stoner G. D. Chemoprevention of Esophageal Cancer with Black Raspberries, Their Component Anthocyanins, and a Major Anthocyanin Metabolite, Protocatechuic Acid. Cancer Prev. Res. 2014, 7, 574–584. 10.1158/1940-6207.CAPR-14-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y.; Obata Y.; Fukuda S.; Endo T. A.; Nakato G.; Takahashi D.; Nakanishi Y.; Uetake C.; Kato K.; Kato T.; Takahashi M.; Fukuda N. N.; Murakami S.; Miyauchi E.; Hino S.; Atarashi K.; Onawa S.; Fujimura Y.; Lockett T.; Clarke J. M.; Topping D. L.; Tomita M.; Hori S.; Ohara O.; Morita T.; Koseki H.; Kikuchi J.; Honda K.; Hase K.; Ohno H. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature 2013, 504, 446–450. 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- López-Rodríguez M. L.; Benhamú B.; Vázquez-Villa H.; Algar S.; Sánchez-Merino A.; Gallardo M.. Npm1-Depending Leukemia Agents. Patent PCT/EP2022/076831, WO2023/0523542021.
- Moffat J. G.; Vincent F.; Lee J. A.; Eder J.; Prunotto M. Opportunities and Challenges in Phenotypic Drug Discovery: An Industry Perspective. Nat. Rev. Drug Discovery 2017, 16, 531–543. 10.1038/nrd.2017.111. [DOI] [PubMed] [Google Scholar]
- Vincent F.; Nueda A.; Lee J.; Schenone M.; Prunotto M.; Mercola M. Phenotypic Drug Discovery: Recent Successes, Lessons Learned and New Directions. Nat. Rev. Drug Discovery 2022, 21, 899–914. 10.1038/s41573-022-00472-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MiMeDB. Microbiota Metabolites Database. https://mimedb.org/ (accessed 07 Dec, 2023).
- Huo L. L.; Ma A. Q.; Zhang Y. H.; Ma D. W. Assembly of 4-Substituted 3-Nitro-1,2,3,4-Tetrahydropyridines Via Organocatalytic Michael Addition. Adv. Synth. Catal. 2012, 354, 991–994. 10.1002/adsc.201100903. [DOI] [Google Scholar]
- Liu Y.; Nappi M.; Arceo E.; Vera S.; Melchiorre P. Asymmetric Catalysis of Diels-Alder Reactions with in Situ Generated Heterocyclic Ortho-Quinodimethanes. J. Am. Chem. Soc. 2011, 133, 15212–15218. 10.1021/ja206517s. [DOI] [PubMed] [Google Scholar]
- Kotame P.; Hong B. C.; Liao J. H. Enantioselective Synthesis of the Tetrahydro-6h-Benzo[C]Chromenes Via Domino Michael-Aldol Condensation: Control of Five Stereocenters in a Quadruple-Cascade Organocatalytic Multi-Component Reaction. Tetrahedron Lett. 2009, 50, 704–707. 10.1016/j.tetlet.2008.11.106. [DOI] [Google Scholar]
- Hong B. C.; Kotame P.; Tsai C. W.; Liao J. H. Enantioselective Total Synthesis of (+)-Conicol Via Cascade Three-Component Organocatalysis. Org. Lett. 2010, 12, 776–779. 10.1021/ol902840x. [DOI] [PubMed] [Google Scholar]
- Bonnet D.; Dick J. E. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates from a Primitive Hematopoietic Cell. Nat. Med. 1997, 3, 730–737. 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Sharma N.; Liesveld J. L. Npm 1 Mutations in Aml-the Landscape in 2023. Cancers 2023, 15, 1177. 10.3390/cancers15041177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grummitt C. G.; Townsley F. M.; Johnson C. M.; Warren A. J.; Bycroft M. Structural Consequences of Nucleophosmin Mutations in Acute Myeloid Leukemia. J. Biol. Chem. 2008, 283, 23326–23332. 10.1074/jbc.M801706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manna S.; Florio D.; Di Natale C.; Lagreca E.; Sibillano T.; Giannini C.; Marasco D. Type C Mutation of Nucleophosmin 1 Acute Myeloid Leukemia: Consequences of Intrinsic Disorder. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130173 10.1016/j.bbagen.2022.130173. [DOI] [PubMed] [Google Scholar]
- Honorato R. V.; Koukos P. I.; Jimenez-Garcia B.; Tsaregorodtsev A.; Verlato M.; Giachetti A.; Rosato A.; Bonvin A. Structural Biology in the Clouds: The Wenmr-Eosc Ecosystem. Front. Mol. Biosci. 2021, 8, 729513 10.3389/fmolb.2021.729513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert G. C. P.; Rodrigues J.; Trellet M.; Schmitz C.; Kastritis P. L.; Karaca E.; Melquiond A. S. J.; van Dijk M.; de Vries S. J.; Bonvin A. The Haddock2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Urbaneja M. A.; Skjaerven L.; Aubi O.; Underhaug J.; Lopez D. J.; Arregi I.; Alonso-Marino M.; Cuevas A.; Rodriguez J. A.; Martinez A.; Banuelos S. Conformational Stabilization as a Strategy to Prevent Nucleophosmin Mislocalization in Leukemia. Sci. Rep. 2017, 7, 13959 10.1038/s41598-017-14497-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti P.; Davoli T.; Sironi C.; Amati B.; Pelicci P. G.; Colombo E. Nucleophosmin and Its Aml-Associated Mutant Regulate C-Myc Turnover through Fbw7 Gamma. J. Cell Biol. 2008, 182, 19–26. 10.1083/jcb.200711040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong S. M.; Tan B. X.; Ahmad B. B.; Yan T.; Chee L. Y.; Ang S. T.; Tay K. G.; Koh L. P.; Yeoh A. E. J.; Koay E. S. C.; Mok Y. K.; Lim T. M. Mutant Nucleophosmin Deregulates Cell Death and Myeloid Differentiation through Excessive Caspase-6 and-8 Inhibition. Blood 2010, 116, 3286–3296. 10.1182/blood-2009-12-256149. [DOI] [PubMed] [Google Scholar]
- Falini B.; Mecucci C.; Tiacci E.; Alcalay M.; Rosati R.; Pasqualucci L.; La Starza R.; Diverio D.; Colombo E.; Santucci A.; Bigerna B.; Pacini R.; Pucciarini A.; Liso A.; Vignetti M.; Fazi P.; Meani N.; Pettirossi V.; Saglio G.; Mandelli F.; Lo-Coco F.; Pelicci P. G.; Martelli M. F.; Party G. A. L. W. Cytoplasmic Nucleophosmin in Acute Myelogenous Leukemia with a Normal Karyotype. N. Engl. J. Med. 2005, 352, 254–266. 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- Perez-Chanona E.; Trinchieri G. The Role of Microbiota in Cancer Therapy. Curr. Opin. Immunol. 2016, 39, 75–81. 10.1016/j.coi.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarnoutse R.; Ziemons J.; Penders J.; Rensen S. S.; de Vos-Geelen J.; Smidt M. L. The Clinical Link between Human Intestinal Microbiota and Systemic Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 4145. 10.3390/ijms20174145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis P.; Hold G. L.; Flint H. J. The Gut Microbiota, Bacterial Metabolites and Colorectal Cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- Ma C.; Han M.; Heinrich B.; Fu Q.; Zhang Q.; Sandhu M.; Agdashian D.; Terabe M.; Berzofsky J. A.; Fako V.; Ritz T.; Longerich T.; Theriot C. M.; McCulloch J. A.; Roy S.; Yuan W.; Thovarai V.; Sen S. K.; Ruchirawat M.; Korangy F.; Wang X. W.; Trinchieri G.; Greten T. F. Gut Microbiome-Mediated Bile Acid Metabolism Regulates Liver Cancer Via Nkt Cells. Science 2018, 360, eaan5931 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintelnot J.; Xu Y.; Lesker T. R.; Schonlein M.; Konczalla L.; Giannou A. D.; Pelczar P.; Kylies D.; Puelles V. G.; Bielecka A. A.; Peschka M.; Cortesi F.; Riecken K.; Jung M.; Amend L.; Broring T. S.; Trajkovic-Arsic M.; Siveke J. T.; Renne T.; Zhang D.; Boeck S.; Strowig T.; Uzunoglu F. G.; Gungor C.; Stein A.; Izbicki J. R.; Bokemeyer C.; Sinn M.; Kimmelman A. C.; Huber S.; Gagliani N. Microbiota-Derived 3-Iaa Influences Chemotherapy Efficacy in Pancreatic Cancer. Nature 2023, 615, 168–174. 10.1038/s41586-023-05728-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buskes M. J.; Coffin A.; Troast D. M.; Stein R.; Blanco M. J. Accelerating Drug Discovery: Synthesis of Complex Chemotypes Via Multicomponent Reactions. ACS Med. Chem. Lett. 2023, 14, 376–385. 10.1021/acsmedchemlett.3c00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Guzmán M. E.; Bigoni-Ordonez G. D.; Ibanez Hernandez M.; Ortiz-Sanchez E. Cancer Stem Cell Impact on Clinical Oncology. World J. Stem Cells 2018, 10, 183–195. 10.4252/wjsc.v10.i12.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader J. E.; Lindeman G. J. Cancer Stem Cells in Solid Tumours: Accumulating Evidence and Unresolved Questions. Nat. Rev. Cancer 2008, 8, 755–768. 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- Hernández-Camarero P.; Jimenez G.; Lopez-Ruiz E.; Barungi S.; Marchal J. A.; Peran M. Revisiting the Dynamic Cancer Stem Cell Model: Importance of Tumour Edges. Crit. Rev. Oncol. Hematol. 2018, 131, 35–45. 10.1016/j.critrevonc.2018.08.004. [DOI] [PubMed] [Google Scholar]
- Bohl S. R.; Bullinger L.; Rucker F. G. New Targeted Agents in Acute Myeloid Leukemia: New Hope on the Rise. Int. J. Mol. Sci. 2019, 20, 1983. 10.3390/ijms20081983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short N. J.; Konopleva M.; Kadia T. M.; Borthakur G.; Ravandi F.; DiNardo C. D.; Daver N. Advances in the Treatment of Acute Myeloid Leukemia: New Drugs and New Challenges. Cancer Discovery 2020, 10, 506–525. 10.1158/2159-8290.CD-19-1011. [DOI] [PubMed] [Google Scholar]
- Ley T. J.; Miller C.; Ding L.; Raphael B. J.; Mungall A. J.; Robertson A.; Hoadley K.; Triche T. J. Jr.; Laird P. W.; Baty J. D.; Fulton L. L.; Fulton R.; Heath S. E.; Kalicki-Veizer J.; Kandoth C.; Klco J. M.; Koboldt D. C.; Kanchi K. L.; Kulkarni S.; Lamprecht T. L.; Larson D. E.; Lin L.; Lu C.; McLellan M. D.; McMichael J. F.; Payton J.; Schmidt H.; Spencer D. H.; Tomasson M. H.; Wallis J. W.; Wartman L. D.; Watson M. A.; Welch J.; Wendl M. C.; Ally A.; Balasundaram M.; Birol I.; Butterfield Y.; Chiu R.; Chu A.; Chuah E.; Chun H. J.; Corbett R.; Dhalla N.; Guin R.; He A.; Hirst C.; Hirst M.; Holt R. A.; Jones S.; Karsan A.; Lee D.; Li H. I.; Marra M. A.; Mayo M.; Moore R. A.; Mungall K.; Parker J.; Pleasance E.; Plettner P.; Schein J.; Stoll D.; Swanson L.; Tam A.; Thiessen N.; Varhol R.; Wye N.; Zhao Y.; Gabriel S.; Getz G.; Sougnez C.; Zou L.; Leiserson M. D.; Vandin F.; Wu H. T.; Applebaum F.; Baylin S. B.; Akbani R.; Broom B. M.; Chen K.; Motter T. C.; Nguyen K.; Weinstein J. N.; Zhang N.; Ferguson M. L.; Adams C.; Black A.; Bowen J.; Gastier-Foster J.; Grossman T.; Lichtenberg T.; Wise L.; Davidsen T.; Demchok J. A.; Shaw K. R.; Sheth M.; Sofia H. J.; Yang L.; Downing J. R.; Eley G. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamyan G.; Kadia T.; Ravandi F.; Borthakur G.; Cortes J.; Jabbour E.; Daver N.; Ohanian M.; Kantarjian H.; Konopleva M. Frontline Treatment of Acute Myeloid Leukemia in Adults. Crit. Rev. Oncol. Hematol. 2017, 110, 20–34. 10.1016/j.critrevonc.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solana-Altabella A.; Ballesta-Lopez O.; Megias-Vericat J. E.; Martinez-Cuadron D.; Montesinos P. Emerging Flt3 Inhibitors for the Treatment of Acute Myeloid Leukemia. Exp. Opin. Emerg. Drugs 2022, 27, 1–18. 10.1080/14728214.2021.2009800. [DOI] [PubMed] [Google Scholar]
- Straube J.; Ling V. Y.; Hill G. R.; Lane S. W. The Impact of Age, Npm1(Mut), and Flt3(Itd) Allelic Ratio in Patients with Acute Myeloid Leukemia. Blood 2018, 131, 1148–1153. 10.1182/blood-2017-09-807438. [DOI] [PubMed] [Google Scholar]
- Di Matteo A.; Franceschini M.; Chiarella S.; Rocchio S.; Travaglini-Allocatelli C.; Federici L. Molecules That Target Nucleophosmin for Cancer Treatment: An Update. Oncotarget 2016, 7, 44821–44840. 10.18632/oncotarget.8599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranieri R.; Pianigiani G.; Sciabolacci S.; Perriello V. M.; Marra A.; Cardinali V.; Pierangeli S.; Milano F.; Gionfriddo I.; Brunetti L.; Martelli M. P.; Falini B. Current Status and Future Perspectives in Targeted Therapy of Npm1-Mutated Aml. Leukemia 2022, 36, 2351–2367. 10.1038/s41375-022-01666-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murano K.; Okuwaki M.; Hisaoka M.; Nagata K. Transcription Regulation of the Rrna Gene by a Multifunctional Nucleolar Protein, B23/Nucleophosmin, through Its Histone Chaperone Activity. Mol. Cell. Biol. 2008, 28, 3114–3126. 10.1128/MCB.02078-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falini B.; Gionfriddo I.; Cecchetti F.; Ballanti S.; Pettirossi V.; Martelli M. P. Acute Myeloid Leukemia with Mutated Nucleophosmin (Npm1): Any Hope for a Targeted Therapy?. Blood Rev. 2011, 25, 247–254. 10.1016/j.blre.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Maggi L. B.; Brady S. N.; Apicelli A. J.; Dai M. S.; Lu H.; Weber J. D. Nucleophosmin Is Essential for Ribosomal Protein L5 Nuclear Export. Mol. Cell. Biol. 2006, 26, 3798–3809. 10.1128/MCB.26.10.3798-3809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan V.; Kishore A. H.; Febitha K. K.; Kundu T. K. Human Histone Chaperone Nucleophosmin Enhances Acetylation-Dependent Chromatin Transcription. Mol. Cell. Biol. 2005, 25, 7534–7545. 10.1128/MCB.25.17.7534-7545.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda M. The Role of Nucleophosmin in Centrosome Duplication. Oncogene 2002, 21, 6170–6174. 10.1038/sj.onc.1205708. [DOI] [PubMed] [Google Scholar]
- Kang Y. J.; Olson M. O.; Jones C.; Busch H. Nucleolar Phosphoproteins of Normal Rat Liver and Novikoff Hepatoma Ascites Cells. Cancer Res. 1975, 35, 1470–1475. [PubMed] [Google Scholar]
- Kang Y. J.; Olson M. O.; Busch H. Phosphorylation of Acid-Soluble Proteins in Isolated Nucleoli of Novikoff Hepatoma Ascites Cells. Effects of Divalent Cations. J. Biol. Chem. 1974, 249, 5580–5585. 10.1016/S0021-9258(20)79767-X. [DOI] [PubMed] [Google Scholar]
- Grisendi S.; Mecucci C.; Falini B.; Pandolfi P. P. Nucleophosmin and Cancer. Nat. Rev. Cancer 2006, 6, 493–505. 10.1038/nrc1885. [DOI] [PubMed] [Google Scholar]
- López D. J.; Rodriguez J. A.; Banuelos S. Nucleophosmin, a Multifunctional Nucleolar Organizer with a Role in DNA Repair. Biochim. Biophys. Acta, Proteins Proteomics 2020, 1868, 140532 10.1016/j.bbapap.2020.140532. [DOI] [PubMed] [Google Scholar]
- López-Ferrer D.; Martinez-Bartolome S.; Villar M.; Campillos M.; Martin-Maroto F.; Vazquez J. Statistical Model for Large-Scale Peptide Identification in Databases from Tandem Mass Spectra Using Sequest. Anal. Chem. 2004, 76, 6853–6860. 10.1021/ac049305c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.