

Abstract
Background:
The composition of the skin microbiome varies from infancy to adulthood and becomes most stable in adulthood. Adult acne patients harbour an ‘acne microbiome’ dominated by specific strains of Cutibacterium acnes. However, the precise timing of skin microbiome evolution, the development of the acne microbiome, and the shift to virulent C. acnes strain composition during puberty is unknown.
Objectives:
We performed a cross-sectional pilot study in a paediatric population to understand how and when the skin microbiome composition transitions during puberty and whether a distinct ‘acne microbiome’ emerges in paediatric subjects.
Methods:
Forty-eight volunteers including males and females, ages 7–17 years, with and without acne were enrolled and evaluated for pubertal development using the Tanner staging criteria. Sebum levels were measured, and skin microbiota were collected by sterile swab on the subject’s forehead. DNA was sequenced by whole genome shotgun sequencing.
Results:
A significant shift in microbial diversity emerged between early (T1-T2) and late (T3-T5) stages of puberty, coinciding with increased sebum production on the face. The overall relative abundance of C. acnes in both normal and acne skin increased during puberty and individual C. acnes strains were uniquely affected by pubertal stage and the presence of acne. Further, an acne microbiome signature associated with unique C. acnes strain composition and metabolic activity emerges in late puberty in those with acne. This unique C. acnes strain composition is predicted to have increased porphyrin production, which may contribute to skin inflammation.
Conclusions:
Our data suggest that the stage of pubertal development influences skin microbiome composition. As children mature, a distinct acne microbiome composition emerges in those with acne. Understanding how both puberty and acne influence the microbiome may support novel therapeutic strategies to combat acne in the paediatric population.
INTRODUCTION
Approximately 85%–90% of individuals develop acne during adolescence, a time period that coincides with the onset and transition through puberty. The aetiology of acne is multifactorial and linked to follicular hyperkeratinization, hormonally triggered sebum production, Cutibacterium acnes (C. acnes) and inflammation.1,2 It is well established that the physical and biochemical properties of the skin are influenced by puberty and that skin colonization by microbes is influenced by moisture and sebum levels.3–6 The composition of the skin microbiome is also linked with acne pathogenesis.7 Thus, dissecting the role of the evolving skin microbiome in response to puberty versus changes related to acne development is challenging.
Our understanding of how the skin microbiota contribute to acne development is incomplete. Adult acne patients harbour an ‘acne microbiome’ dominated by specific strains of C. acnes7 and acne severity is associated with loss of C. acnes diversity.8–10 Compared with strains in healthy individuals, acne-associated strains of C. acnes contain extra virulence related genes,11 produce more porphyrins,12 trigger a pro-inflammatory response13,14 and contain more antibiotic resistance elements.7,15 Yet, when the ‘acne microbiome’ develops and when the shift in C. acnes strain composition occurs during puberty is unclear.
To understand when the skin microbiome composition transitions during puberty and whether a distinct skin microbiome emerges in paediatric acne subjects, we performed a cross-sectional pilot study in children 7–17 years old. We identified key skin microbiome alterations associated with both pubertal transition and acne skin, all of which may support novel therapeutic strategies to combat acne in the paediatric population.
MATERIALS AND METHODS
Human subjects
This study was approved by the Institutional Review Board of the Penn State College of Medicine. Forty-eight volunteers, including males and females, ages 7–17 years, with and without acne were enrolled after providing informed assent with written informed parental consent. At the study visit, dermatologists assessed the pubertal stage according to Tanner stage (T1-T5) criteria, and acne severity was assessed using the 5-point investigator global assessment (IGA) scale (Figure 1a, Supplementary Methods).16–18 All children were healthy with no underlying skin/medical conditions other than acne, if present. All acne subjects had mild-to-moderate acne. The menstrual cycle or use of OCPs was not recorded for females. No subject had a history of systemic retinoid use. A chart review of the 12 months prior to study enrolment did not identify systemic or topical antibiotic exposure for any subject. Exclusion criteria included use of topical antibiotics, benzoyl peroxide or salicylic washes within 2 weeks prior to the start of the study; and use of topical retinoids, oral antibiotics, dermabrasion or facial laser therapy within 4 weeks prior to the start of the study. Demographic and clinical data are provided in Tables S1 and S2.
FIGURE 1.
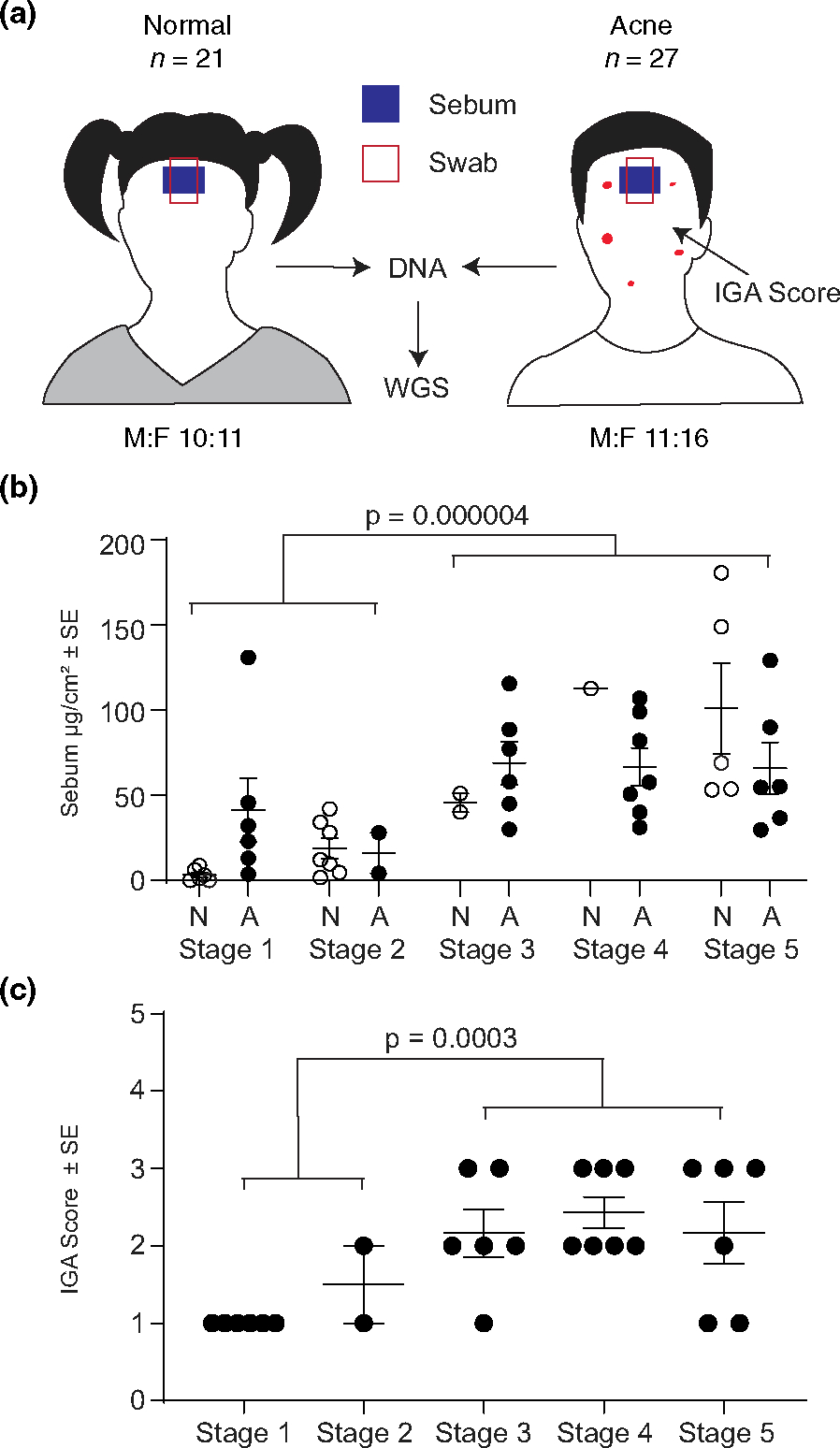
Experimental design and subject demographics. (a) Schematic of cohort characteristics and body site location of sebum and skin microbiota collection. (b) Mean sebumeter reading (μg/cm2) for each subject (dot) stratified by Tanner stage and disease status. (c) IGA score for each acne patient by Tanner stage. M –male, F –female, A –acne, N –normal, IGA –investigator global assessment of acne disease, WGS - whole genome sequencing.
Sebum measurements
Sebum output was measured by Sebumeter 810™ Instrumentation. Measurements were captured from the left, centre and right regions of the forehead prior to skin microbiome sampling and averaged for the overall sebum level (μg/cm2).
Microbiome sampling
A 7 cm2 area of the forehead was scrubbed with a pre-moistened sterile, synthetic fibre swab with moderate pressure for 30 s and stored −80°C until sequencing (Supplemental Methods). Negative sampling control samples were collected throughout the study: swab with air exposure for 30 s (n = 5). Two positive mock community controls were included, a generalized community control (ATCC MSA-1005) and a skin-specific community control (ATCC MSA-1002).
Metagenomic sequencing, processing and taxonomic identification
Frozen swabs were shipped to Microbiome Insights (British Columbia, Canada) for DNA extraction, library preparation and sequencing (Supplemental Methods). Sequencing was performed by Illumina NextSeq sequencing (2 × 150 paired-end reads) and included one negative sequencing control sample. Positive and negative controls were processed in parallel with patient samples (Figure S1). Initial quality evaluation was done using FastQC (v0.11.5).19 High-quality reads were mapped to the human genome (GRCh37) and human sequences were removed. Remaining sequences underwent taxonomic profiling with MetaPhlAn2.20,21 The median number of high-quality filtered reads per sample was 2,840,816 (Table S3). Samples with >50,000 non-human quality-controlled reads were included in analyses.21,22
Diversity analyses and statistics
We identified a total of 321 species. Prior to analyses, we removed species that were present in positive controls (10 species) or those only detected within a single patient sample (101 species; <1% relative abundance, Table S4). Negative sampling controls contained >90% Escherichia; thus, sequences corresponding to Escherichia were removed from all samples before analysis (Figure S1c, Supplemental Methods). All further analyses were done with the remaining 219 species: 201 bacteria, 16 viruses, 1 Eukaryota, and 1 unclassified group. Samples were normalized to sequencing depths (Table S3). Individual taxa, relative abundances and Shannon Diversity indices (α-diversity) of microbial community were compared by the Wilcoxon rank-sum test. Bray–Curtis dissimilarity index (β-diversity) was compared using Permutational Multivariate Analysis of Variance (PERMANOVA).23 p-values were adjusted by the Bonferroni Correction (adj. p-value), where indicated. Viral sequences dominated 8 of 48 samples, but no significant association between viral relative abundance and demographic (age, sex, ethnicity) or clinical data (sebum levels, Tanner stage, IGA score) were found.
Cutibacterium acnes strain identification
Cutibacterium acnes genomes (n = 283 genomes representing 248 unique strains) available in NCBI (https://www.ncbi.nlm.nih.gov/genome/browse/#!/prokaryotes/1140/) on 7 October 2020 were utilized to identify C. acnes strains in our dataset. StrainEst (v1.2.4) was used to build an index of single nucleotide variants (SNV) present in the C. acnes genomes (mapgenomes, map2snp) and to identify strains in our dataset using default parameters except for a 3× coverage threshold.24 Identified C. acnes strains were classified by single locus sequence type (SLST) classification based on homology to the SLST sequence (overlaps PPA2385 and PPA2386 hypothetical proteins) identified by Scholz and colleagues.25
Metabolic profiles
Functional analysis was performed with HUMAnN2 using the default analysis pipeline. UniRef identifiers were converted to KEGG Orthology (KO) terms. 7134 KO terms were used as input into MicrobiomeAnalyst26 on 16 October 2020 using its default filtering parameters. After quality filtering, 4572 KO terms were used in subsequent pathway enrichment analysis. FDR of q = 0.05 (Kruskal–Wallis) identified enzymes and pathways of biological interest.
RESULTS
Sebum production and acne disease severity increase with pubertal development
Normal and acne subjects (7–17 years old) were enrolled after providing written informed assent under an IRB approved protocol (Figure 1a, Supplementary Methods). Consistent with prior studies, sebum production significantly increased throughout puberty.27–29 Mean sebum output was 3.5 times higher in late puberty (T3-T5) compared with early puberty (T1-T2) (Figure 1b). In general, sebum levels were significantly higher (40%) in subjects with acne compared with subjects without acne (p = 0.038).
Acne disease severity also significantly increased with later stages of pubertal development (Figure 1c). Acne patients in early puberty had less severe acne (mean ± SEM; IGA 1.125 ± 0.125, almost clear) than those in late puberty (IGA 2.26 ± 0.168, mild to moderate) (Figure 1c), confirming previous reports that acne severity increases with pubertal development.30,31 Thus, our patient cohort is consistent with published studies on the key metrics of sebum production and acne severity during pubertal development.32–34
Acne status influences the skin microbiome in late puberty
We examined the association of clinical and demographic covariates with microbiome composition. After adjusting for sex and Tanner stage, only acne status was significantly associated with microbiome composition (p = 0.046) (Figure 2a). We divided our cohort into two groups, T1-T2 (early puberty) and T3-T5 (late puberty) based on a principle coordinate analysis showing that T1-T2 samples segregated from T3-T5 samples (Figure S2); this natural separation allowed for a greater number of subjects per group for subsequent comparisons (Figure 2b). Both α-diversity and β-diversity were significantly different (p < 0.05) between early and late puberty regardless of acne status (Figure S2), suggesting that pubertal stage is a strong driver of skin microbiome composition. This transition point coincided with increased sebum production (Figure 1b). After controlling for puberty, there was no difference in α-diversity between normal and acne skin (Figure 2c). Similarly, β-diversity analysis showed no difference in early puberty; however, in late puberty, the presence of acne significantly impacted β-diversity (Figure 2d,e). Collectively, these data indicate that puberty strongly influences the skin microbiome and that a distinct microbiome associated with the presence of acne emerges in late puberty.
FIGURE 2.
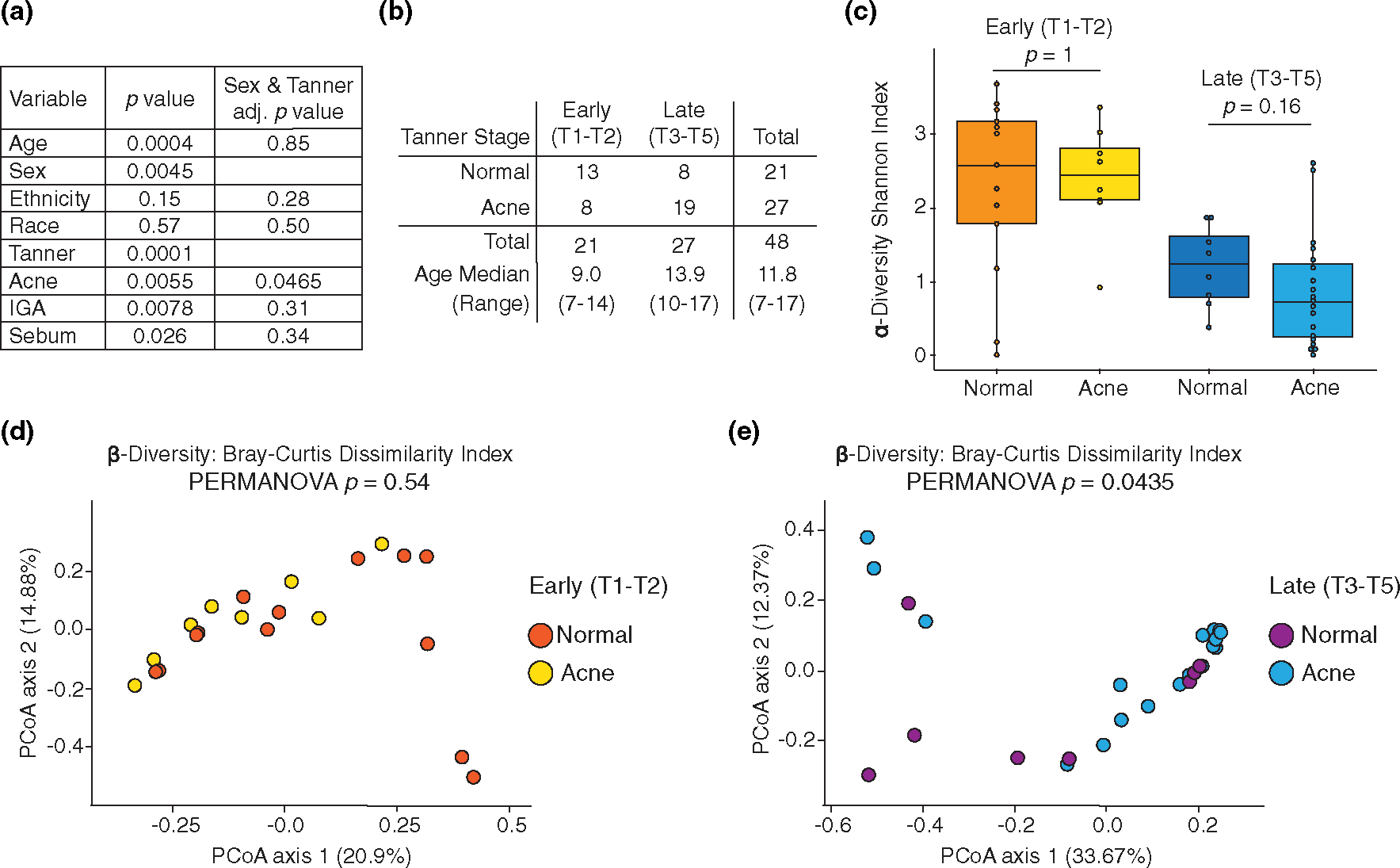
Puberty and acne affect microbial α- and β-diversity. (a) Association between covariates and microbiome composition, before (column 2) and after (column 3) controlling for Tanner stage (T1-T2 vs. T3-T5) and sex. (b) Demographic characteristic of T1-T2 (early puberty) and T3-T5 (late puberty) comparison groups. (c) α-diversity between normal and acne skin; stratified by early (T1-T2) and late (T3-T5) puberty; Wilcoxon rank-sum test. β-diversity between normal and acne skin in (d) early puberty (T1-T2) and (e) late puberty (T3-T5); PERMANOVA
Individual species are influenced by puberty
Next, we asked whether puberty or acne impacted individual species. The relative abundance of the 10 most abundant bacteria is shown for each individual, stratified by Tanner stage and disease state (Figure 3a; Tables S5–S7). As expected, C. acnes was the most abundant species and the relative abundance increased during puberty in both normal and acne skin (Figure 3a,b). In acne skin, C. acnes was significantly higher in late puberty (61.7%) compared with early puberty (28.7%) (p = 0.022) (Figure 3b). However, when controlling for puberty, the relative abundance of C. acnes was not significantly different between normal and acne skin. In all, the relative abundance of C. acnes increased with puberty, but was not significantly different between normal and acne skin.
FIGURE 3.
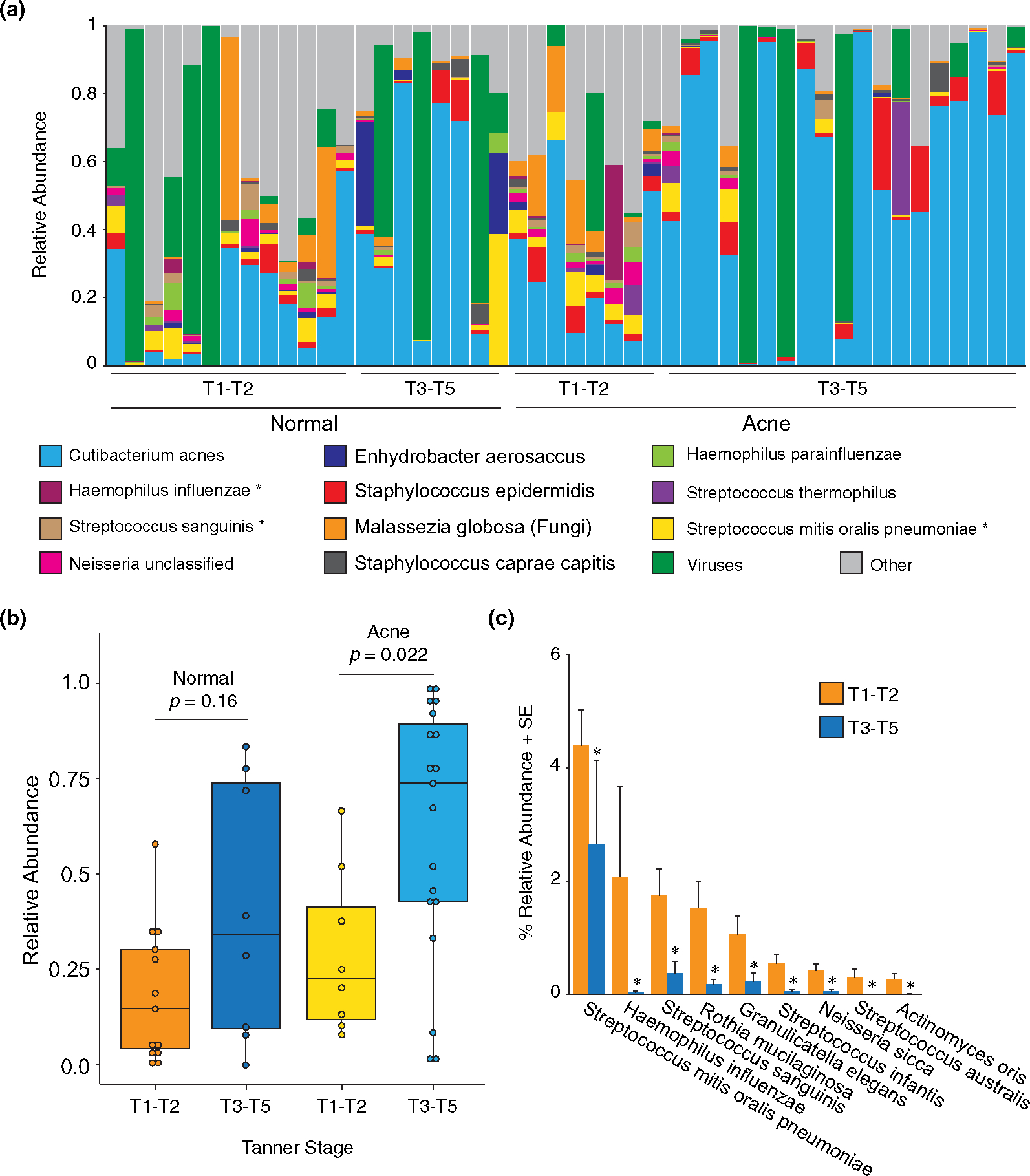
Species relative abundance in normal and acne patients. (a) Relative abundance of the top 10 most abundant bacteria, the single fungal species (Malassezia globosa), and the total viral species detected (16 individual species) for each individual patient (column). *Species that are significantly different between early- and late-stage puberty. (b) Relative abundance of C. acnes by disease state and Tanner stage; Wilcoxon rank-sum test. (c) Differentially abundant taxa between early puberty (T1-T2) and late puberty (T3-T5); Wilcoxon rank-sum test with significance after multiple test adjustment accounting for the 219 species comparisons; p < 0.05
Nine bacterial species were significantly more abundant in early puberty, independent of acne status (Figure 3c), highlighting the loss of microbial diversity that occurs during the transition from childhood to adulthood. The predominance of Streptococcus in younger children is consistent with previous findings.35 Similar to previous findings, the fungal species, Malassezia globosa was detected in 39/48 (81%) samples with an average relative abundance of 4.35% (Figure 3a, Table S5).36 Sixteen viral species were detected in at least one sample (Figure S3). We also identified species that were uniquely present in either normal skin or acne skin: 45 species (37 bacteria, 8 viruses) were only present in normal skin (Table S8) and 16 species (14 bacteria, 2 viruses) were only present in acne skin (Table S9). However, after controlling for puberty, no bacterial, fungal or viral species was significantly different between normal skin and acne skin, suggesting that the entire community composition, rather than a single species, contributes to the acne microbiome.
C. acnes strain composition on skin is influenced by puberty and acne
Adult acne patients harbour specific strains of C. acnes associated with either acne or healthy skin.7,37,38 Given that these prior findings were observed in adults, we investigated whether puberty influenced strain level composition in our paediatric population. We identified a total of 167 unique C. acnes strains. On average, each subject harboured ~34 unique strains (range 9–54) showcasing vast interpersonal variability. We could not identify the specific C. acnes strains in 5 samples, even though C. acnes was identified on the species level in these samples. We next grouped identifiable C. acnes strains by phylogenetic similarity using single locus sequence typing (SLST) classification scheme25 resulting in 11 unique clusters (Figure 4, Tables S10, S11; phylotype designations are noted in brackets).39–41 α- and β-diversity comparisons were assessed as follows: (1) ‘early normal (EN)’ versus ‘early acne (EA)’; (2) ‘early normal (EN)’ vs ‘late normal (LN)’; (3) ‘early acne (EA)’ versus ‘late acne (LA)’ and (4) ‘late normal (LN)’ versus ‘late acne (LA)’ (Tables S12–S15).
FIGURE 4.
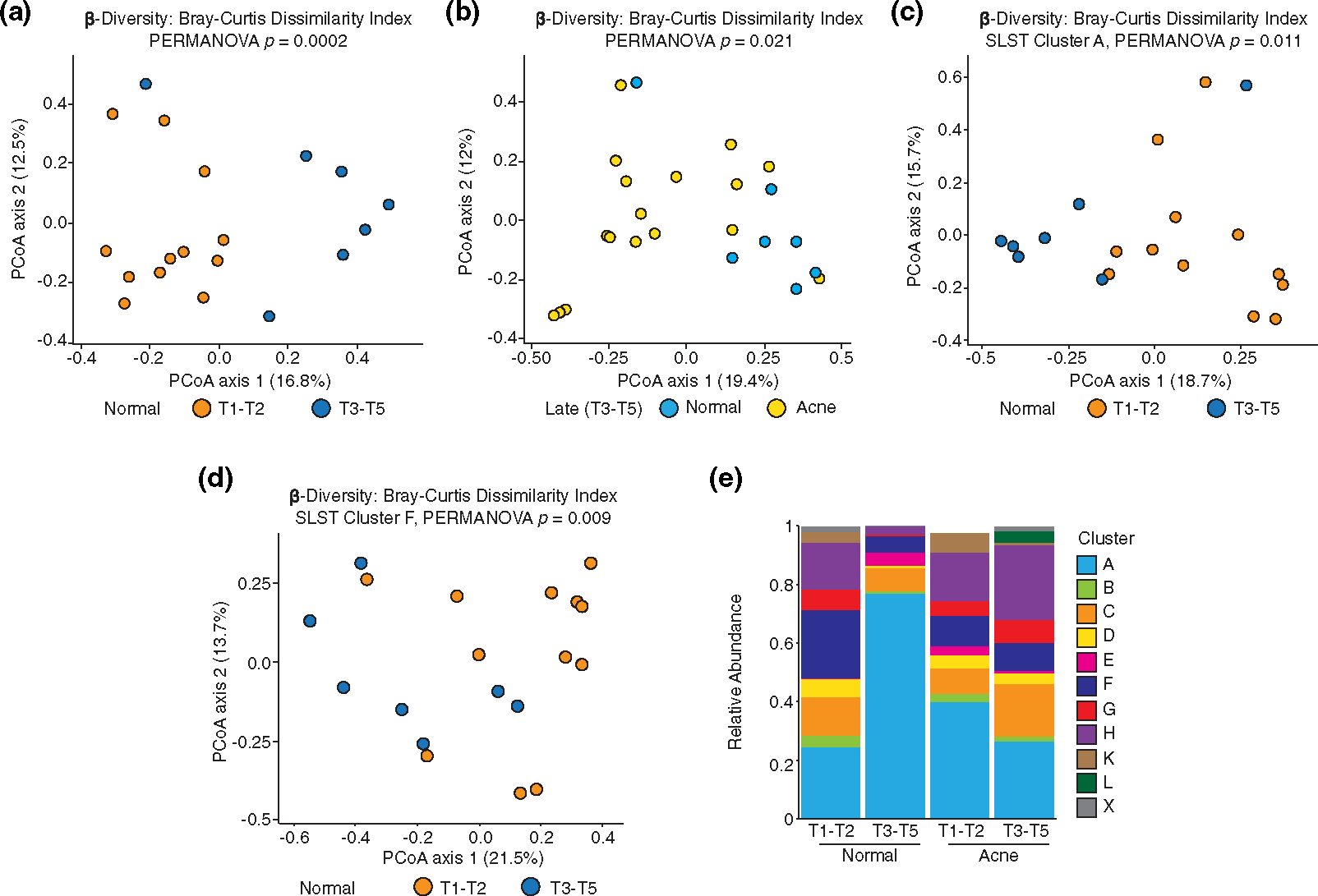
Cutibacterium acnes strain composition is influenced by pubertal development and acne. (a) Principal Coordinates Analysis (PCoA) plot visualizing Bray–Curtis Dissimilarity Index (β-diversity) of SLST clusters in normal patients grouped by Tanner stage; β-diversity statistics with PERMANOVA; T1-T2 versus T3-T5, p = 0.0002. (b) PCoA plot of SLST clusters in T3-T5 stratified by normal and acne skin; PERMANOVA; normal versus acne, p = 0.021. (c) β-diversity of SLST cluster A [IA1] in normal subjects grouped by Tanner stage; PERMANOVA; T1-T2 vs T3-T5, p = 0.011. (d) β-diversity of SLST cluster F [IA2] in normal subjects grouped by Tanner stage; PERMANOVA; T1-T2 versus T3-T5, p = 0.009. (e) Average relative abundance of each SLST cluster and phylotype [brackets] by Tanner stage and disease status. SLST X cluster is unclassified.
There was a significant shift in β-diversity between EN and LN skin (Figure 4a). β-diversity of C. acnes strains was significantly different between LN and LA skin (Figure 4b) but was not significantly different between EN vs EA (p = 0.50) or EA vs. LA (p = 0.87) skin types.
Within specific SLST clusters A [IA1], D [IA1] and F [IA2], significant shifts in α-diversity between EN and LN were observed (Table S12). α-diversity within SLST cluster A significantly increased (p = 0.007) in LN compared with EN, while α-diversity within SLST clusters D and F significantly decreased in LN (p = 0.027 and p = 0.044, respectively). α-diversity for SLST D and H [1B] significantly increased in LA compared with LN (p = 0.047 and p = 0.04, respectively). Similar to α-diversity, significant shifts in β-diversity for each of the SLST A, D and F clusters were found between EN and LN (Figures 4c,d, Table S13). β-diversity was significantly altered between EA and LA for SLST H (p = 0.046). Collectively, C. acnes strains classified within the SLST clusters of A, D and F are most affected by puberty, while SLST cluster H was most affected by the presence of acne. The biological relevance of these findings remains to be determined but may highlight new strategies for acne treatment.
Metabolic pathways are significantly enriched in late acne skin
To understand the functional consequences of these altered microbial communities, sequences were annotated by KEGG Orthology (KO) terms and analysed with MicrobiomeAnalyst.26,42,43 The functional profiles of EN, LN, EA and LA were very similar; yet, variations were evident, suggesting functional differences exist (Figure 5a). 405 KO terms were significantly different between the groups by univariate analysis (Kruskal–Wallis, FDR q = 0.05). Using network mapping to identify pathways associated with these KOs, 10 KEGG metabolic pathways were identified, two of which were significant: histidine metabolism, and porphyrin and chlorophyll metabolism (FDR q = 0.05) (Figure 5b, Table S16, Figure S4). Sixty-nine unique enzymes were associated with the pathways identified in Figure 5b (Table S16).
FIGURE 5.
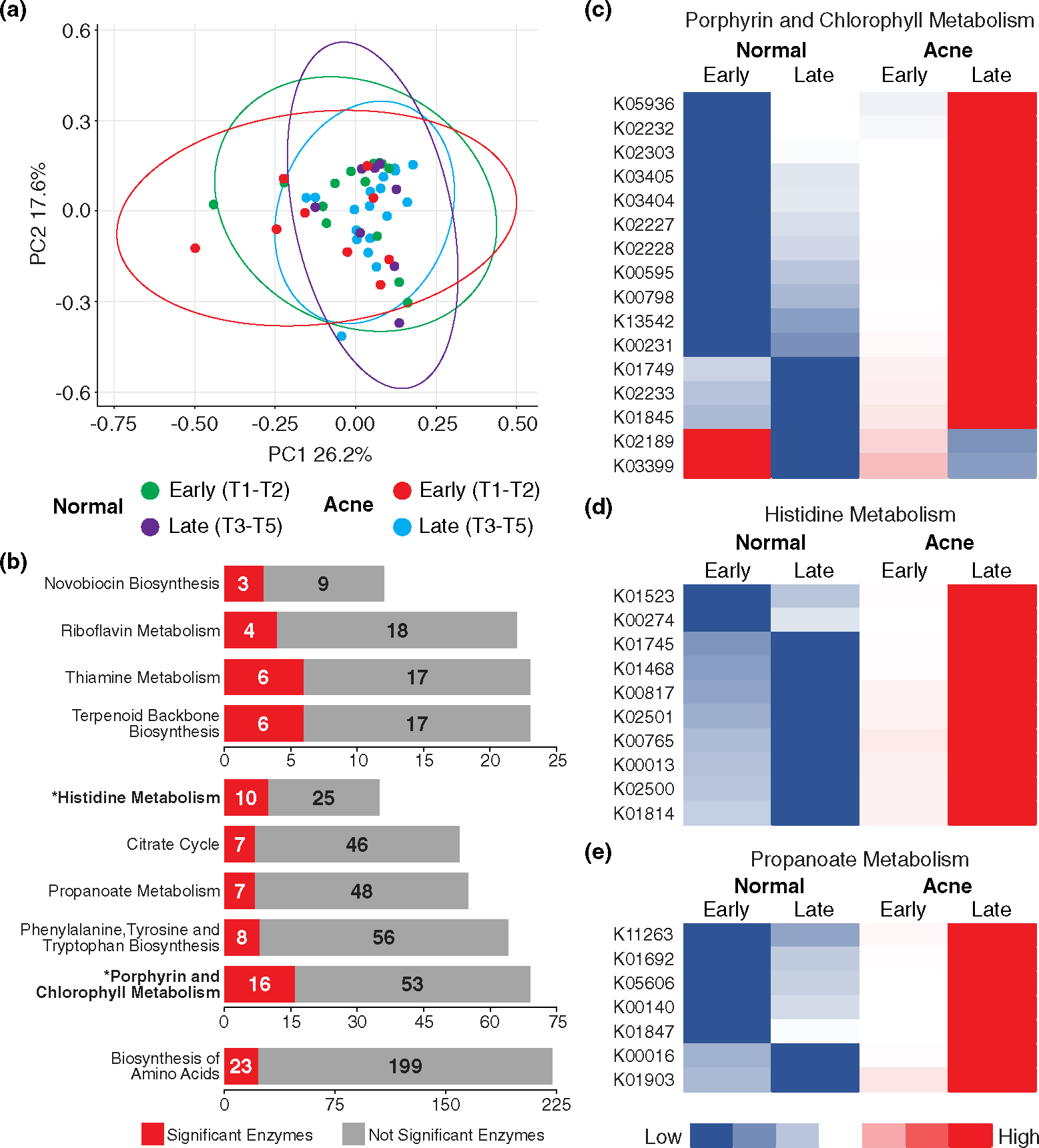
Functional profiles show active metabolic state in late acne skin. (a) PCA plot illustrating functional composition of all four groups; generated by MicrobiomeAnalyst; circles are 95% confidence intervals for respective datasets. (b) Ten metabolic pathways and the number of significant enzymes out of the total number of enzymes affiliated with each pathway. Pathways marked with an * and in bold text are significant by Kruskal-Wallis FDR <0.05. (c) Heatmap showing the 16 significant enzymes associated with KEGG pathway: Porphyrin and chlorophyll metabolism. Heatmap represents high (red) and low (blue) expression of each individual enzyme of the four groups. (d) Heatmap of the 10 significant enzymes associated with histidine metabolism. (e) Heatmap of the seven significant enzymes associated with propanoate metabolism.
The relative abundance of the majority of the KO terms (355/405; 88%) was increased in LA compared with the other groups, and zero features were enriched in LN skin. The functional profiles of LA are clearly distinct and predict a metabolically active acne microbiome with increased porphyrin, histidine and propanoate metabolism (Figure 5c–e; Table S17; Figure S4).
DISCUSSION
In this cross-sectional pilot study, we found that puberty strongly influences the microbiome composition in sebaceous skin. In line with prior studies in adults, we demonstrated that microbial composition is altered in acne skin of children in the later stages of puberty.7,44 A significant shift in both α-diversity and β-diversity occurred between early and late puberty which was characterized by a decrease in bacterial diversity and an increase in the relative abundance of C. acnes. Further, both puberty and acne status impacts C. acnes strain composition in children. As children mature and develop acne, a distinct skin microbiome emerges characterized by decreased bacterial diversity, altered C. acnes strain colonization, and an altered metabolic signature of increased porphyrin, histidine and propanoate metabolism.
Our data highlights a significant transition point for the facial skin microbiome between early and late puberty. This transition coincided with increased sebum production. The Tanner scale for sexual maturity documents changes in secondary sexual characteristics which are responsive to sex hormones.16,17 Increased androgen production and signalling facilitates remodelling and increased activity of the pilosebaceous unit resulting in a new microbial habitat. Thus, enabling C. acnes, a follicle and sebum-loving microbe to dominate the microbial landscape in high sebum areas, while dry-loving microbes decrease in numbers.3,5,7,28 Similarly, a significant transition in the skin microbiome of the nares and forearm occurred between mid and late puberty (T1-T3 vs. T4-T5).45 Shifts in the microbial composition are likely due to the action of sex hormones influencing the skin’s biochemical and physical properties.46 Altogether, these data suggest that different skin microenvironments (dry, moist, sebaceous) may mature asynchronously throughout puberty,4,5,45 but once the skin’s environment has ‘evolved’ enough, a microbial shift to the adult-like state occurs.
Strain diversity exists within the C. acnes species.7,11,13 Nearly 70% (167/248) of sequenced C. acnes strains were identified in this study. Surprisingly, six samples had no identifiable C. acnes strains, even though C. acnes was clearly detected at the species level in 5 of 6 samples. Of these, 3 of 5 were obtained from subjects in pre-puberty (T1) suggesting that the young paediatric population may have unique C. acnes strain composition. Sampling of children is therefore necessary to accurately capture C. acnes diversity in humans.
Our data highlight the impact of puberty and acne on unique SLST clusters, namely that diversity among C. acnes strains within SLST A [IA1], D [IA1] and F [IA2] clusters are more influenced by puberty, while diversity in SLST H [IB] is more influenced by the presence of acne. While this suggests that strains within these clusters are contributing to overall skin health or skin disease, it also suggests that the ability of each C. acnes strain to establish residence on the skin may be influenced by unique factors. We found that puberty and presumably increased sebum production, strongly influences the diversity of SLST A, D and F clusters in our paediatric cohort. Although not observed in our study, an increased abundance of SLST A (specifically A1) or SLST F strain types have been associated with acne skin in adults.47,48 However, SLST H diversity has not previously been associated with acne skin. Changing diversity within these clusters during puberty may influence the development of acne in adults.
The microbial functional profiles of acne skin in late puberty are clearly distinct from early puberty acne skin and healthy skin. This profile predicts a metabolically active community with increased porphyrin metabolism. All 16 significantly enriched KO terms associated with this pathway are directly involved in porphyrin synthesis (Figure S4). Porphyrins are pro-inflammatory metabolites important in vitamin B12 synthesis.49 Vitamin B12 supplementation in humans represses B12 synthesis in C. acnes and increases bacterial porphyrin biosynthesis and the host inflammatory response.50 Acne skin-a ssociated C. acnes strains respond to B12 supplementation by increasing porphyrin production, while healthy skin-associated C. acnes strains do not.12,49 Riboflavin metabolism (vitamin B2) and thiamine metabolism (vitamin B1) pathways were also enriched in late acne skin with riboflavin metabolism feeding directly into the porphyrin and chlorophyll metabolism pathway. Three of the top 10 metabolic pathways enriched in late acne skin involve vitamin B metabolism suggesting that vitamin B metabolism is active and likely contributes to increased porphyrin production and skin inflammation.
We recognize that our sample size in this cross-sectional study is limited and unequal distribution of subjects between Tanner stages could influence results; thus, we analysed our cohort by early (T1-T2) and late puberty (T3-T5) groups based on a natural transition in microbiome diversity to mitigate the influence of this unequal distribution. This study was not powered to detect differences in microbial composition between sexes, mild and moderate acne subjects, the influence of oral contraceptive pills or skin products, like lotion.46,51,52 By study design, absolute abundance data are not available. We cannot exclude the possibility of sampling errors or sequencing errors in our study, but by including negative sampling controls, mock community controls and negative sequencing controls, we used best practices to overcome these inherent limitations and remove contamination before analyses.53
In summary, puberty strongly influences changes in the facial skin microbiome, with a major shift in facial skin microbiome composition that coincides with elevated sebum production. Similar to adult patients, a distinct microbiome signature emerges in children with acne in later stages of puberty, which is characterized by the presence of unique C. acnes strains and increased metabolic activity, which may contribute to skin inflammation.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Quy Pham, M.D. and Chelsey Straight, M.D. for assistance with subject recruitment and Erick Cardenas-Poire, Ph.D. (Microbiome Insights) for initial bioinformatics support. Access to Data and Data Analysis: AMS, ZTN, KB, XZ and AMN had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
FUNDING INFORMATION
Work was funded by the American Acne and Rosacea Society (ARP) and the Penn State Dermatology Research Endowment (AMN). The funding organizations had no role in design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest regarding this work.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
Sequencing data is available at Sequence Read Archive (SRA): PRJNA856701.
REFERENCES
- 1.Scholz CFP, Kilian M. The natural history of cutaneous propionibacteria, and reclassification of selected species within the genus Propionibacterium to the proposed novel genera Acidipropionibacterium gen. nov., Cutibacterium gen. nov. and Pseudopropionibacterium gen. nov. Int J Syst Evol Microbiol. 2016;66:4422–32. [DOI] [PubMed] [Google Scholar]
- 2.Zaenglein AL. Acne vulgaris. N Engl J Med. 2018;379:1343–52. [DOI] [PubMed] [Google Scholar]
- 3.McGinley KJ, Webster GF, Ruggieri MR, Leyden JJ. Regional variations in density of cutaneous propionibacteria: correlation of Propionibacterium acnes populations with sebaceous secretion. J Clin Microbiol. 1980;12:672–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9:244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitz-Gibbon S, Tomida S, Chiu B-H, Nguyen L, du C, Liu M, et al. Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J Invest Dermatol. 2013;133:2152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dréno B, Dagnelie MA, Khammari A, Corvec S. The skin microbiome: a new actor in inflammatory acne. Am J Clin Dermatol. 2020;21:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dagnelie M, Montassier E, Khammari A, Mounier C, Corvec S, Dréno B. Inflammatory skin is associated with changes in the skin microbiota composition on the back of severe acne patients. Exp Dermatol. 2019;28:961–7. [DOI] [PubMed] [Google Scholar]
- 10.Dagnelie M, Corvec S, Saint-Jean M, Nguyen J-M, Khammari A, Dréno B. Cutibacterium acnes phylotypes diversity loss: a trigger for skin inflammatory process. J Eur Acad Dermatol Venereol. 2019;33:2340–8. [DOI] [PubMed] [Google Scholar]
- 11.Tomida S, Nguyen L, Chiu BH, Liu J, Sodergren E, Weinstock GM, et al. Pan-genome and comparative genome analyses of Propionibacterium acnes reveal its genomic diversity in the healthy and diseased human skin microbiome. MBio. 2013;4:e00003–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson T, Kang D, Barnard E, Li H. Strain-level differences in porphyrin production and regulation in Propionibacterium acnes elucidate disease associations. mSphere. 2016;1:e00023–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu Y, Champer J, Agak GW, Kao S, Modlin RL, Kim J. Different Propionibacterium acnes phylotypes induce distinct immune responses and express unique surface and secreted proteomes. J Invest Dermatol. 2016;136:2221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agak GW, Kao S, Ouyang K, Qin M, Moon D, Butt A, et al. Phenotype and antimicrobial activity of Th17 cells induced by Propionibacterium acnes strains associated with healthy and acne skin. J Invest Dermatol. 2018;138:316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Y, Champer J, Garbán H, Kim J. Typing of Propionibacterium acnes: a review of methods and comparative analysis. Br J Dermatol. 2015;172:1204–9. [DOI] [PubMed] [Google Scholar]
- 16.Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child. 1969;44:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child. 1970;45:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(CDER) C for DE and R. Acne vulgaris: developing drugs for treatment. 2005.
- 19.Andrews S (2010). FastQC: a quality control tool for high throughput sequence data [WWW Document]. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 20.Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12:902–3. [DOI] [PubMed] [Google Scholar]
- 21.Oh J, Byrd AL, Deming C, Conlan S, NISC Comparative Sequencing Program, Kong HH, et al. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meisel JS, Hannigan GD, Tyldsley AS, SanMiguel AJ, Hodkinson BP, Zheng Q, et al. Skin microbiome surveys are strongly influenced by experimental design. J Invest Dermatol. 2016;136:947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
- 24.Albanese D, Donati C. Strain profiling and epidemiology of bacterial species from metagenomic sequencing. Nat Commun. 2017;8:2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholz CFP, Jensen A, Lomholt HB, Brüggemann H, Kilian M. A novel high-resolution single locus sequence typing scheme for mixed populations of Propionibacterium acnes in vivo. PLoS One. 2014;9:e104199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dhariwal A, Chong J, Habib S, King IL, Agellon LB, Xia J. MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017;45:W180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramasastry P, Downing DT, Pochi PE, Strauss JS. Chemical composition of human skin surface lipids from birth to puberty. J Invest Dermatol. 1970;54:139–44. [DOI] [PubMed] [Google Scholar]
- 28.Pochi PE, Strauss JS, Downing DT. Age-related changes in sebaceous gland activity. J Invest Dermatol. 1979;73:108–11. [DOI] [PubMed] [Google Scholar]
- 29.Stewart ME, Downing DT. Measurement of sebum secretion rates in young children. J Invest Dermatol. 1985;84:59–61. [DOI] [PubMed] [Google Scholar]
- 30.Lucky AW, Biro FM, Huster GA, Morrison JA, Elder N. Acne vulgaris in early adolescent boys: correlations with pubertal maturation and age. Arch Dermatol. 1991;127:210–6. [PubMed] [Google Scholar]
- 31.Lucky AW, Biro FM, Huster GA, Leach AD, Morrison JA, Ratterman J. Acne vulgaris in premenarchal girls: an early sign of puberty associated with rising levels of dehydroepiandrosterone. Arch Dermatol. 1994;130:308–14. [DOI] [PubMed] [Google Scholar]
- 32.Zouboulis CC, Jourdan E, Picardo M. Acne is an inflammatory disease and alterations of sebum composition initiate acne lesions. J Eur Acad Dermatol Venereol. 2014;28:527–32. [DOI] [PubMed] [Google Scholar]
- 33.Okoro OE, Adenle A, Ludovici M, Truglio M, Marini F, Camera E. Lipidomics of facial sebum in the comparison between acne and non-acne adolescents with dark skin. Sci Rep. 2021;11:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Youn SW. The role of facial sebum secretion in acne pathogenesis: facts and controversies. Clin Dermatol. 2010;28:8–11. [DOI] [PubMed] [Google Scholar]
- 35.Coughlin CC, Swink SM, Horwinski J, Sfyroera G, Bugayev J, Grice EA, et al. The preadolescent acne microbiome: a prospective, randomized, pilot study investigating characterization and effects of acne therapy. Pediatr Dermatol. 2017;34:661–4. [DOI] [PubMed] [Google Scholar]
- 36.Jo J-H, Deming C, Kennedy EA, Conlan S, Polley EC, Ng WI, et al. Diverse human skin fungal communities in children converge in adulthood. J Invest Dermatol. 2016;136:2356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lomholt HB, Kilian M. Population genetic analysis of Propionibacterium acnes identifies a subpopulation and epidemic clones associated with acne. PLoS One. 2010;5:e12277. 10.1371/journal.pone.0012277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDowell A, Barnard E, Nagy I, Gao A, Tomida S, Li H, et al. An expanded multilocus sequence typing scheme for Propionibacterium acnes: investigation of ‘pathogenic’, ‘commensal’ and antibiotic resistant strains. PLoS One. 2012;7:e41480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brüggemann H, Salar-Vidal L, Gollnick HPM, Lood R. A Janus-faced bacterium: host-beneficial and - detrimental roles of Cutibacterium acnes. Front Microbiol. 2021;12:673845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scholz CFP, Brüggemann H, Lomholt HB, Tettelin H, Kilian M. Genome stability of Propionibacterium acnes: a comprehensive study of indels and homopolymeric tracts. Sci Rep. 2016;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDowell A. Over a decade of recA and tly gene sequence typing of the skin bacterium Propionibacterium acnes: what have we learnt? Microorganisms. 2017;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15:962–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chong J, Liu P, Zhou G, Xia J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat Protoc. 2020;15:799–821. [DOI] [PubMed] [Google Scholar]
- 44.Li C, You Z, Lin Y, Liu HY, Su J. Skin microbiome differences relate to the grade of acne vulgaris. J Dermatol. 2019;46:787–90. [DOI] [PubMed] [Google Scholar]
- 45.Oh J, Conlan S, Polley EC, Segre JA, Kong HH. Shifts in human skin and nares microbiota of healthy children and adults. Genome Med. 2012;4:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park J, Schwardt NH, Jo JH, Zhang Z, Pillai V, Phang S, et al. Shifts in the skin bacterial and fungal communities of healthy children transitioning through puberty. J Invest Dermatol. 2022;142:212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dagnelie M, Corvec S, Saint-Jean M, Bourdès V, Nguyen J, Khammari A, et al. Decrease in diversity of Propionibacterium acnes phylotypes in patients with severe acne on the Back. Acta Derm Venereol. 2018;98:262–7. [DOI] [PubMed] [Google Scholar]
- 48.Nakase K, Hayashi N, Akiyama Y, Aoki S, Noguchi N. Antimicrobial susceptibility and phylogenetic analysis of Propionibacterium acnes isolated from acne patients in Japan between 2013 and 2015. J Dermatol. 2017;44:1248–54. [DOI] [PubMed] [Google Scholar]
- 49.Barnard E, Johnson T, Ngo T, Arora U, Leuterio G, McDowell A, et al. Porphyrin production and regulation in cutaneous propionibacteria. mSphere. 2020;5:e00793–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang D, Shi B, Erfe MC, Craft N, Li H. Vitamin B12 modulates the transcriptome of the skin microbiota in acne pathogenesis. Sci Transl Med. 2015;7:293ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bouslimani A, da Silva R, Kosciolek T, Janssen S, Callewaert C, Amir A, et al. The impact of skin care products on skin chemistry and microbiome dynamics. BMC Biol. 2019;17:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy B, Grimshaw S, Hoptroff M, Paterson S, Arnold D, Cawley A, et al. Alteration of barrier properties, stratum corneum ceramides and microbiome composition in response to lotion application on cosmetic dry skin. Sci Rep. 2022;12:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kong HH. Details matter: designing skin microbiome studies. J Invest Dermatol. 2016;136:900–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data is available at Sequence Read Archive (SRA): PRJNA856701.