

Abstract

We introduce the boryloxide ligand {(HCNDipp)2BO}− (NBODipp, Dipp = 2,6-di-isopropylphenyl) to actinide chemistry. Protonolysis of [U{N(SiMe3)2}3] with 3 equiv of NBODippH produced the uranium(III) tris(boryloxide) complex [U(NBODipp)3] (1). In contrast, treatment of UCl4 with 3 equiv of NBODippK in THF at room temperature or reflux conditions produced only [U(NBODipp)2(Cl)2(THF)2] (2) with 1 equiv of NBODippK remaining unreacted. However, refluxing the mixture of 2 and unreacted NBODippK in toluene instead of THF afforded the target complex [U(NBODipp)3(Cl)(THF)] (3). Two-electron oxidation of 1 with AdN3 (Ad = 1-adamantyl) afforded the uranium(V)–imido complex [U(NBODipp)3(NAd)] (4). The solid-state structure of 1 reveals a uranium–arene bonding motif, and structural, spectroscopic, and DFT calculations all suggest modest uranium–arene δ-back-bonding with approximately equal donation into the arene π4 and π5 δ-symmetry π* molecular orbitals. Complex 4 exhibits a short uranium(V)–imido distance, and computational modeling enabled its electronic structure to be compared to related uranium–imido and uranium–oxo complexes, revealing a substantial 5f-orbital crystal field splitting and extensive mixing of 5f |ml,ms⟩ states and mj projections. Complexes 1–4 have been variously characterized by single-crystal X-ray diffraction, 1H NMR, IR, UV/vis/NIR, and EPR spectroscopies, SQUID magnetometry, elemental analysis, and CONDON, F-shell, DFT, NLMO, and QTAIM crystal field and quantum chemical calculations.
Short abstract
Through the synthesis of four uranium−boryloxide complexes, we introduce the boryloxide ligand {(HCNDipp)2BO}− (NBODipp, Dipp = 2,6-di-isopropylphenyl) to actinide chemistry. The uranium(III)−arene derivative exhibits two modest, approximately equal uranium−arene δ-back-bonding interactions. The imido complex exhibits a short uranium(V)−imido distance, and computational modeling enabled its electronic structure to be compared to related uranium−imido and uranium−oxo complexes, revealing a substantial total 5f-orbital crystal field splitting and extensive mixing of 5f |ml,ms⟩ states and mj projections.
Introduction
It is often stated that ligand–metal complementarity is an essential component of the recipe for controlling the behavior of metal ions in coordination and organometallic complexes. This is certainly the case in synthetic actinide chemistry, where the nature of the ancillary ligands plays a decisive role in stabilizing or destabilizing actinide oxidation states, bonding motifs, reactivity, magnetism, and optical properties.1−14 In recent years, we have made extensive use of triamidoamine (Tren) ligands to stabilize a wide range of novel An–ligand multiple bonds,15−28 An–metal bonds,15,29−35 novel main group moieties,36−43 uranyl activation,44 small molecule activation,45−48 single-molecule magnets (SMMs),49 and novel photochemical rearrangements,50 and provide insight into fundamental f-block phenomena such as disproportionation, the inverse trans influence, pushing from below,29,49,51−54 and NMR covalency studies.55,56 However, there are situations where Tren ligands reach their limitations. For example, while high oxidation state uranium(V) and uranium(VI) oxos, imidos, and nitridos and even a neptunium(V)–oxo are now straightforwardly accessible with Tren ancillary ligands,16,19,22,26−28,44−47,49,51,52,56 to date, uranium(IV) has proven to be the highest accessible oxidation state when paired with softer elements such as phosphorus, arsenic, antimony, and bismuth, with all attempts to oxidize resulting in uranium(IV) products exclusively being isolated.17,24,36,40 Although alkoxides and aryloxides are logical alternatives on hard–soft acid–base (HSAB) grounds, they do not always possess the ideal steric profiles for specific applications.
We have previously utilized the boryloxide ligand {(HCNDipp)2BO}− (NBODipp, Dipp = 2,6-di-isopropylphenyl)57,58 to prepare rare earth complexes59 including the dysprosium complex [Dy{OB(NDippCH)2}2(THF)4][BPh4] that exhibits a notably high SMM relaxation energy barrier.59 Having considered the structure of that dysprosium complex, we surmised that assembling three of those NBODipp ligands at actinide ions might produce sterically encumbered complexes with one or at most two well-protected reaction pockets at the metal, resulting from a “picket-fence” arrangement of Dipp groups, while also being capable of stabilizing high oxidation state actinide ions on HSAB principles. We also considered that the boryl center could provide an electronic “buffer”, where the oxide can modulate its donor strength to a metal and transfer any excess electron density into the vacant boryl p orbital. Alternatively, if the boryl center is still electron deficient, it could make up the difference with π donation from the two α-nitrogen atoms in the NBO scaffold or not accept any N atom π donation if already electronically saturated. Further motivation stemmed from the fact that there are relatively few f-element boryloxide complexes,60,61 which as R2BO– species are distinct from the far more prevalent borate derivatives,62−78 and they have very different steric profiles from NBODipp. This is because R2BO– ligands tend to have the ligand sterics pointing away from a coordinated metal, whereas with NBODipp, they point toward the metal. Thus, we considered the NBODipp ligand to have significant potential for metal–ligand complementarity.
Here, we report the introduction of the NBODipp ligand into actinide chemistry through the synthesis of uranium(III), uranium(IV), and uranium(V) derivatives which showcases the flexibility of the NBODipp ligand to stabilize a range of actinide oxidation states. The uranium(III) derivative exhibits a uranium–arene interaction, where structural, spectroscopic, and computational methods consistently suggest uranium–arene δ-back-bonding with approximately equal donation into the π4 and π5 δ-symmetry π* molecular orbital combinations of the arene. However, the uranium–arene interaction is evidently weak, as demonstrated by straightforward two-electron oxidation to provide an imido complex. NIR spectroscopic and computational studies on the imido enable its contextualization with existing related uranium–imido, uranium–nitrido, and uranium–oxo derivatives. These new complexes thus represent a new family of synthetic precursors for further elaboration.
Results and Discussion
Synthesis
Treatment of [U{N(SiMe3)2}3]79,80 with 3 equiv of NBODippH57,58 results, after a 2 day stir and workup, in the uranium(III)–tris(boryloxide) complex [U(NBODipp)3] (1) as an analytically pure dark purple solid in 51% yield, Scheme 1a; the HN(SiMe3)2 byproduct is conveniently removed, either when the reaction solvent is removed in vacuo or when the resulting solid is washed with pentane. Although the reaction is evidently driven thermodynamically by the formation of U–O bonds and the respective pKa values of the protic components of the reaction, it is clearly kinetically sluggish. This likely reflects the kinetic barrier to installing three sterically demanding NBODipp ligands at a single metal center, even one as large as uranium(III) (Shannon uranium(III) six-coordinate ionic radius = 1.025 Å).81 Support for this suggestion can be found in the issues encountered in the synthesis of the chlorido analogs (see below).
Scheme 1. Synthesis of Complexes 1–4.

(a) Reaction of [U{N(SiMe3)2}3] with 3 equiv of NBODippH affords 1 with elimination of 3 equiv of amine, and subsequent treatment of 1 with AdN3 produces 4 with elimination of N2. (b) Reaction of UCl4 with 3 equiv of NBODippK in THF produces 2, 2 equiv of KCl, and 1 equiv of unreacted NBODippK, and reflux in toluene after THF affords 3 and 3 equiv of KCl.
Compound 1 constitutes a potentially useful starting material, where the redox chemistry of uranium(III) may be utilized (see below for an example), and seeking to extend the range of uranium–NBODipp synthetic precursors, we targeted a uranium(IV)–chlorido derivative for applications in salt elimination chemistry, Scheme 1b. Accordingly, we treated UCl4 with 3 equiv of NBODippK in THF, and the green solution turned brown during a 16 h stir. After workup, we isolated yellow [U(NBODipp)2(Cl)2(THF)2] (2) in 80% crystalline yield. The third equivalent of unreacted NBODippK was found to be in the mother liquor by 1H NMR spectroscopy. The fact that only 2 equiv of NBODipp substituted onto uranium by salt elimination is consistent with the slow substitution by protonolysis that produces 1. Repeating the reaction but refluxing in THF made no difference to the reaction outcome. However, when first stirring the 3:1 NBODippK:UCl4 mixture in THF, then replacing the THF with toluene, and refluxing, a dark green solid was isolated after workup. Recrystallization from toluene afforded [U(NBODipp)3(Cl)(THF)] (3) as yellow-green crystals in 71% yield. We suggest that in THF the coordination sphere of uranium is permanently saturated with THF molecules, blocking installation of the third NBODipp ligand but that in toluene any loss of THF is not compensated by the bulk solvent, thus opening up the coordination sphere at uranium to enable salt elimination and installation of the third NBODipp ligand at uranium to occur. Alternatively, or in addition to, the partial solubility of KCl in THF may establish an equilibrium that is driven to products by the insolubility of KCl in toluene.
Experiments to derivatize 2 and 3 are ongoing and will be reported in due course. However, in a preliminary demonstration of the utility of 1 as a useful synthetic precursor the brown uranium(V)–imido complex [U(NBODipp)3(NAd)] (4, Ad = 1-adamantyl) was prepared in a straightforward and essentially quantitative two-electron oxidation of 1 with AdN3 with concomitant elimination of N2 (65% crystalline yield), Scheme 1a. Thus, although the coordinated arene in 1 may be regarded as providing steric protection of the uranium ion, it is labile enough to not impede further reactivity.
Solid-State Structures
To confirm the formulations of 1–4, their solid-state structures were determined by single-crystal X-ray diffraction, Figures 1–4 and Table S1.
Figure 1.

Molecular structure of 1 at 100 K with selected atom labels. Displacement ellipsoids are set at 40%, and hydrogen atoms and disordered components are omitted for clarity.
Figure 4.

Molecular structure of 4 at 120 K with selected atom labels. Displacement ellipsoids are set at 30%, and hydrogen atoms and disordered components are omitted for clarity.
Complex 1, Figure 1, crystallizes with two of NBODipp ligands coordinated in terminal monodentate modes with the third NBODipp ligand coordinated as an η6-κ1-chelate, coordinated through one of the Dipp rings as well as the boryloxide O atom. Thus, counting the centroid of the arene ring as a coordination point, 1 adopts a distorted tetrahedral coordination geometry at uranium with a piano-stool geometry defined by the η6-arene and the three boryloxide O-donor centers. The three U–O distances are 2.217(3), 2.183(3), and 2.173(3) Å, similar to other uranium–boryloxide distances,60,61 with the longer U–O distance being associated with the chelating NBODipp ligand, reflecting the bent U–O–B angle of 127.6(3)° in the former compared to the latter two U–O–B angles of 162.1(3)° and 162.4(3)° which are much closer to being linear. Although care must be taken when invoking π donation as a function of bond angles,48,82,83 the more linear U–O–B coordination linkages do, in principle, offer the possibility of stronger donation to uranium and hence shorter U–O bonds compared to the bent U–O–B linkage. The U–C distances span the range 2.913(4)–2.999(4) Å (average 2.970 Å) with a U–Ccentroid distance of 2.616 Å, which falls in the middle to upper range of uranium(III)–arene distances,84 likely due to the strain of the chelate ring, which is evident in the slightly bent Ccentroid–Cipso–NNBO angle of 168.2°. The coordinated arene ring is essentially planar (maximum rms deviations from the arene plane from −0.016 to 0.026 Å), but its C–C distances span the range 1.382(6)–1.421(6) Å (average 1.406 Å), which suggests some transfer of electron density into its π* orbitals because the average C–C distance in the other Dipp rings in 1 is ∼1.396 Å, though the difference is marginal.
Complex 2, Figure 2, exhibits a pseudo-octahedral uranium(IV) ion situated on a crystallographic center of inversion, coordinated to two NBODipp, two chloride, and two THF molecules in an all-trans arrangement. The U–ONBO distances are 2.127(2) Å, which is ∼0.05 Å shorter than the terminal U–O distances in 1; this is around one-half the difference in the ionic radii of six-coordinate uranium(III) and uranium(IV) (1.025 vs 0.89 Å, respectively),81 but this likely reflects the very different formal coordination numbers and geometries of 1 vs 2. The U–Cl and U–OTHF distances are unremarkable.
Figure 2.

Molecular structure of 2 at 120 K with selected atom labels. Displacement ellipsoids are set at 40%, and hydrogen atoms and disordered components are omitted for clarity.
The structure of 3, Figure 3, reveals a pseudo-trigonal bipyramidal geometry at uranium with the three NBODipp ligands positioned in the equatorial plane and the chloride and THF ligands occupying the axial sites. The U–ONBO distances span the range 2.124(2)–2.134(2) Å (average 2.130 Å) and by the 3σ criterion are statistically indistinguishable from the corresponding U–ONBO distances in 2. The U–Cl and U–OTHF distances in 3 (2.568(7) and 2.442(2) Å) are unremarkable, though we note that they are shorter and longer, respectively, than the corresponding distances in 2 (U–Cl = 2.648(2) Å; U–OTHF = 2.424(2) Å); acknowledging the different uranium coordination numbers of 2 and 3, this most likely reflects the trans-influence effects of Cl vs THF in 3 and Cl vs Cl and THF vs THF ligand arrangements in 2.
Figure 3.

Molecular structure of 3 at 150 K with selected atom labels. Displacement ellipsoids are set at 40%, and hydrogen atoms and disordered components are omitted for clarity.
Complex 4, Figure 4, contains a uranium(V) ion that is four coordinate, adopting a distorted tetrahedral geometry. The U–O distances span the range 2.129(5)–2.153(5) Å (average 2.140 Å) and by the 3σ criterion are indistinguishable from the corresponding U–ONBO distances in 2 and 3. This is likely due to the size decrease on going from uranium(IV) to uranium(V) being offset by the sterically demanding NBODipp not being able to approach any closer due to interligand clashing, and also the presence of the strongly donating imido ligand. The U–N distance in 4 is short at 1.911(7) Å, which can be compared to U–Nimido distances of 1.945(2), 1.910(6), and 1.967(12) Å in [U(NAd){N(SiMe3)2}3],85 [U(NSiMe3){N(SiMe3)2}3],85,86 and [U(NAd)(TrenTIPS)] (TrenTIPS = {N(CH2CH2NSiiPr3)3}3–),27 respectively. The N–U–O angles in 4 span the range 99.7(2)–104.1(2)° (average 102.07°), between the ideal angles for trigonal monopyramidal (90°) and tetrahedral (109.5°), closer to the latter as found in [U(NAd){N(SiMe3)2}3]85 and [U(NSiMe3){N(SiMe3)2}3],86 but in contrast to [U(O){N(SiMe3)2}3]87 which is closer to the former.
NMR and IR Spectroscopies
1H NMR and IR spectroscopic data for 1–4 can be found in the Supporting Information (Figures S1–S8). Despite the different coordination modes of the NBODipp ligands in the solid-state structure of 1, the 1H NMR spectrum of 1 spans the range from −2 to 10 ppm and indicates a symmetrical species in solution on the NMR time scale with 3-fold symmetry; this was not probed by variable-temperature experiments due to precipitation of 1. However, two iPr-Me resonances and hence environments are observed, reflecting 12 Me groups that point toward the uranium ion and 12 that point away due to restricted rotation of the Dipp-iPr groups. The ATR-IR spectrum of 1 is unremarkable, as expected.
The 1H NMR spectra of 2 and 3 span the ranges from −72 to 46 and from −20 to 13 ppm, which for the former is qualitatively an unusually large range for uranium(IV), but this likely reflects the varied coordination environment in that complex; the range for 3 is unexceptional for uranium(IV). The IR data for 2 and 3 are unremarkable but are consistent with their formulations.
The 1H NMR spectrum of 4 spans the range from −1 to 17 ppm, which is qualitatively as expected for a uranium(V) complex. The IR spectrum of 4 is as expected, and we note that there are absorptions around 1056–972 and 707–648 cm–1 consistent with the prediction of U=N stretches at 1000 and 663 cm–1 from an analytical frequencies calculation; those vibrations are best described as AdN=U bond stretches where the entire AdN unit moves together and at lower energy stretches where the U and Ad groups stretch out and in together about the N center.
SQUID Magnetometry
Variable-temperature SQUID magnetometry data for 1–4 can be found in the Supporting Information (Figures S9–S16) The uranium(III) formulation of 1 is confirmed by variable-temperature SQUID magnetometry of powdered 1, Figure 5, where the effective magnetic moment is 2.50 μB at 300 K, and this decreases smoothly to ∼30 K, at which point there is a more rapid decrease, reaching 1.59 μB at 1.8 K; the high-temperature effective magnetic moment of 1 is low for uranium(III), but this has been observed for uranium(III)–aryloxides,88,89 though otherwise this behavior is typical of uranium(III).9,88,90 Magnetization vs field experiments at 2 K are approaching but do not reach saturation at the highest available field (∼0.9 μB, 7 T), which is consistent with a Kramers uranium(III) ion.91
Figure 5.

SQUID magnetometry data for 1–4. (a) Variable-temperature μeff vs T data: 1 (black circles), 2 (red circles), 3 (green circles), and 4 (blue circles). (b) M vs H data at 2 K: 1 (purple trace), 2 (orange trace), 3 (green trace), and 4 (red trace). Note: the data for 2 and 3 are virtually identical. Lines are a guide to the eye only.
The variable-temperature SQUID magnetometry results of 2 and 3, Figure 5, reveal effective magnetic moments of 2.31 and 2.31 μB at 300 K, respectively, and these values decrease smoothly over the temperature range reaching 0.12 and 0.25 μB at 1.8 K and tending to zero, which is typical of magnetic singlet uranium(IV) ions subject to modest temperature-independent paramagnetism.9,88 However, we do note that the 300 K μeff values for 2 and 3 are lower than the theoretical Russell–Saunders magnetic moment of 3.58 μB for 3H4 ions, but this is known for uranium–aryloxides.88,92 Nevertheless, at 2 K the magnetization vs field data for 2 and 3 are both still rising with no sign of approaching saturation up to the highest available field (∼0.09 μB for 2 and 3, 7 T), which confirms the uranium(IV) formulations of 2 and 3.93
The variable-temperature SQUID magnetometry data on 4, Figure 5, reveal an almost constant effective magnetic moment of 1.69 μB across the temperature range measured, except for a small, decisive drop below 10 K to a final value of 1.60 μB at 1.8 K, likely resulting from depopulation of some low-lying states at very low temperature. This is similar to [U(NAd)(TrenTIPS)]27 and [U(O)(TrenTIPS)],49 but distinct from [U(NAd){N(SiMe3)2}3]85 and [U(NSiMe3){N(SiMe3)2}3]85,86 which exhibit effective magnetic moments of 2.47 and 2.12 μB. The flat μeff vs T plot for 4 suggests that at high temperature either only the ground state is thermally populated or any excited states have similar effective magnetic moments. The magnetization vs field data for 4 at 2 K are approaching saturation, suggesting a well-isolated electronic ground state, though saturation is not quite achieved up to the highest available field strength available (∼0.8 μB, 7 T). Based on our prior analysis of the electronic structure of uranium nitrido complexes as a guide22 and noting that precise values of magnetization data will vary depending on the ligand field and the g values, magnetization data of ∼0.9 μB imply a mj ground state of 5/2 for 4 rather than 3/2, which would have a lower magnetization value of ∼0.5 μB.
EPR Spectroscopy
The X-band EPR spectrum of powdered 1, Figure 6a, is consistent with the rhombic formulation anticipated from arene coordination to uranium(III) without a strong axial ligand,89 exhibiting anisotropic g values of gx = 2.680, gy = 1.776, and gz = 1.039. In all samples examined there is also an isotropic feature at 2.002. We note that the g value for benzenide radical anions is also 2.002,94 and given the relative absorption intensities, the feature at g = 2.002 is assigned as a minor radical impurity. From μeff = 1/2[(gx2 + gy2 + gz2)1/2], the anisotropic EPR g values suggest that the magnetic moment of 1 at 5 K should be 1.68 μeff, which is in good agreement with the measured effective magnetic moment at 5 K (1.73 μB).
Figure 6.

EPR X-band (∼9.4 GHz) spectra of powdered samples of 1 and 4 at 5 K. Black lines are experimental data, and red lines represent simulated data. (a) 1: rhombic-type spectrum with g values of gx = 2.680, gy = 1.776, and gz = 1.039 and a radical feature at g = 2.002 attributed to a minor organic radical impurity. (b) 4: axial spectrum with gx = gy = 0.564 and gz = 3.045. The sharp feature at g = 2.002 (marked with an asterisk (*)) is associated with a small portion of an organic radical impurity in the sample of 4.
The X-band EPR spectrum of powdered 4, Figure 6b, presents an axial-type spectrum with g values of gx = gy = 0.564 and gz = 3.045. A g = 2.002 feature is also observed but is assigned as a minor organic radical impurity, likely of benzenide origin.94 Using μeff = 1/2[(gx2 + gy2 + gz2)1/2], the anisotropic EPR g values suggest that the magnetic moment of 4 at 5 K should be 1.57 μeff, which is in good agreement with the measured effective magnetic moment at 5 K (1.63 μB). In C3v symmetry, a pure mj = 3/2 state would be EPR silent (gx = gy = 0), as found for [U(O)(TrenTIPS)],49 because the J = ±1 EPR selection rule cannot be met; however, with changes to the geometry and ligand field and with mj mixing, that selection rule can be met and g values of gx = gy = 0.1–0.5 and gz = ∼2.4 would be anticipated. For mj = 5/2, that is typically mixed with mj = 7/2, g values of gx = gy = ∼0.3 and gz = ∼3.7 are usually found. Thus, the observed gz values for 4 can be compared to those of [U(NAd){N(SiMe3)2}3]85 (gz = 3.60), [U(NSiMe3){N(SiMe3)2}3]85 (gz = 2.50), and [U(O){N(SiMe3)2}3]85,87 (gz = 2.17), suggesting that the ground state of 4 is mj = 5/2, which is also consistent with the magnetometry data.
UV/vis/NIR Spectroscopy
UV/vis/NIR spectra for 1–4 were collected (see Supporting Information, Figures S17–S20). All complexes exhibit absorptions in the range from 25 000 to 30 000 cm–1, which correspond to π–π* transitions of the NBODipp ligands. The UV/vis/NIR spectrum of 1, Figure 7, exhibits absorptions in the range from 5500 to ∼26 000 cm–1, which are assigned as f–f transitions due to their approximate intensities (ε = ∼250–800 M–1 cm–1, these values should be regarded as upper bounds rather than intrinsic molar absorptivities).9 Above 26 000 cm–1, the spectrum is dominated by charge transfer bands. It is notable that there are not any absorptions in the region from 15 000 to 22 000 cm–1 with large enough intensities (∼1500 M–1 cm–1) to be assigned as f–d transitions. This suggests little stabilization of the 6d orbitals by the ligand field of 1 and that the f–d absorptions of 1 reside under the charge transfer bands at higher energy.
Figure 7.
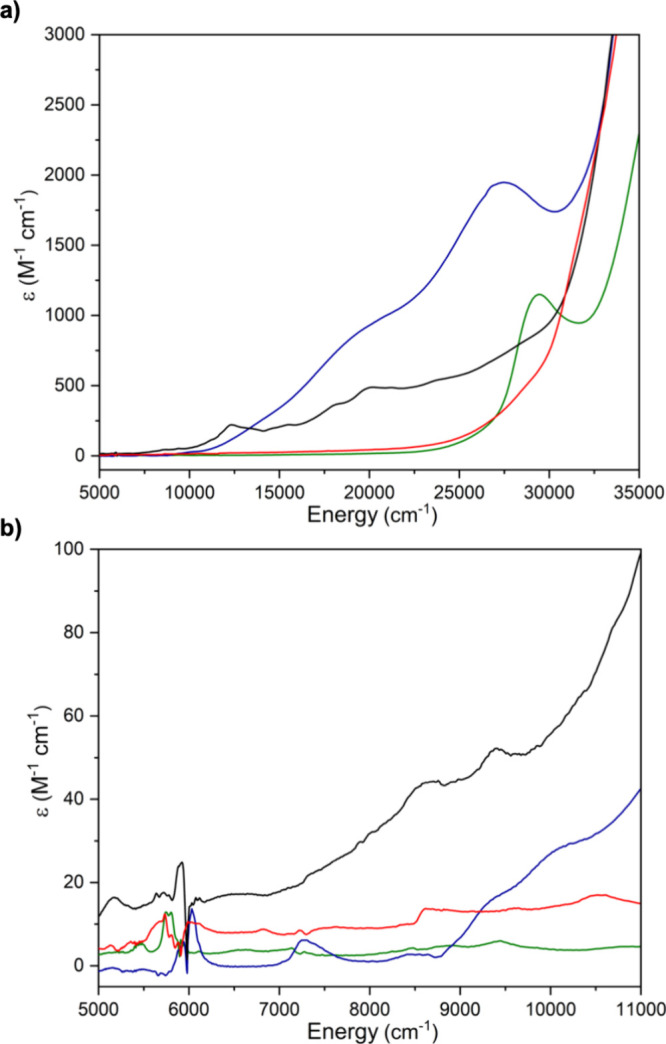
UV/vis/NIR spectra of 1 (black line; 1 mM in toluene), 2 (red line; 1 mM in THF), 3 (green line; 1 mM in THF), and 4 (blue line; 1 mM in toluene). (a) Spectra in the range from 5000 to 35000 cm–1. (b) Spectra in the range from 5000 to 11 000 cm–1.
The UV/vis/NIR spectra of 2 and 3, Figure 7, exhibit weak absorptions in the NIR region that are characteristic of uranium(IV) ions,9 and then, charge transfer bands tail into the visible region from the UV zone.
The UV/vis/NIR spectrum of 4, Figure 7, exhibits absorptions in the NIR region that are characteristic of 2F5/2 to 2F7/2 intraconfigurational transitions of the uranium(V) ion in pseudo C3v symmetry.9 To provide a framework for visualizing the following discussion on the characteristic uranium(V) absorptions observed in the NIR region of 4, Figure 7, we use the approach of Eisenstein and Pryce,95,96 previously used by us to describe the electronic structure of uranium nitrido complexes,22 and Hayton and Lukens to describe the electronic structure of nitrido and imido complexes.85,97 In brief, using |ml,ms⟩ notation, the U–Nimido σ bond will derive from a N sp orbital combined with |0,±1/2⟩ (5fσ, two electrons) and the two π bonds from N p orbitals with |1, ±1/2⟩ (5fπ, four electrons). The |2,±1/2⟩ (5fδ) would be nonbonding, and then, the |3,±1/2⟩ (5fϕ) pair of orbitals will contain the 5f electron of uranium(V).22 There will then be the π* (ml = ±1) and σ* (ml = 0) orbital combinations.22 Using a zeroth-order 5f-splitting approach, where the energies of states vary but mixing of states is neglected, energetically these would be ordered ml = ±3 < ml = ±2 < ml = ±1 < ml = 0 for the (U=NAd)3+ fragment, Figure 8 (note only ml ≥ 0 are shown for clarity). The crystal field (CF) and spin–orbit coupling (SOC) will modify the energies of the resulting individual Kramers doublet states, where it is assumed that the magnitudes of splitting are ordered imido-CF > SOC > NBODipp-CF.22,85,93 Now considering the NBODipp-CF, the presence of equatorial or near-equatorial ligands will modulate the energies of the ml = ±3 and ±2 orbitals, producing an ordering of ml = ±2 < ml = ±3 < ml = ±1 < ml = 0, Figure 8. This is because the NBODipp ligands will variously engage in σ- and π-antibonding interactions with the ml = ±3 and ±2 orbitals, where the precise strength of those interactions will depend on how equatorial or pseudotetrahedral the position of those ligands is. The action of SOC will then further split the states as shown in Figure 8. The result is that the ground state will be |3,–1/2⟩ (mj = 5/2) in pseudo-tetrahedral complexes or |2,–1/2⟩ (mj = 3/2) in trigonal monopyramidal complexes. The baseline g values for |3,–1/2⟩ and |2,±1/2⟩ are 4 and 2, and then, they will vary depending on the exact CF and SOC. Hence, the EPR data for 4 are consistent with a |3,–1/2⟩ (mj = 5/2) 5fϕ ground state, which is the same as that of [U(NAd){N(SiMe3)2}3]85 but in contrast to the |2,–1/2⟩ (mj = 3/2) 5fδ ground state of [U(O){N(SiMe3)2}3].85,87
Figure 8.

Qualitative zeroth-order splitting of the 5f1 states of 4. Horizontal lines correspond to Kramers doublets, where only ml ≥ 0 is shown and the states are shown as |ml,ms⟩ (ms = +1/2 (+) and −1/2 (−) for brevity.
The NIR region of 4 exhibits absorptions approximately at 5945, 6031, 7042, 7267, 8666, 9328, 10 121, and ∼17 500 cm–1, and these data are similar to those obtained for [U(NAd){N(SiMe3)2}3] (6245, 7202, 7568, 9144, 9973, and 15 434 cm–1)85 and [U(NSiMe3){N(SiMe3)2}3] (6097, 7258, 7467, 8734, 9414, and 16 955 cm–1).85 We note, as was also found for [U(NR){N(SiMe3)2}3] (R = Ad, SiMe3)85 and the CF computational analysis that follows, that there are too many NIR absorptions for 4 on the basis of the zeroth-order model. Since there would be predicted to be only four transitions above 5000 cm–1,22,85 a likely explanation is vibronic coupling. Certainly, the absorption at ∼17 500 cm–1 is very broad, likely from unresolved vibronic coupling—recall the U=NAd group stretching frequencies of ∼970 and ∼650 cm–1—and that this absorption is a shoulder on the side of a more intense feature. That this feature is so intense likely stems from intensity stealing (5fσ and N sp orbital mixing)98 of this U–N σ* orbital. Similar features were also found with [U(NAd){N(SiMe3)2}3], [U(NSiMe3){N(SiMe3)2}3], and [U(O){N(SiMe3)2}3].85 The absorptions in the approximate range 7042–9328 cm–1 also appear to exhibit vibronic coupling, though this is not obviously resolved, but we note that the approximate separations of the three lowest lying absorptions are similar to the most energetic U–NAd stretching mode. Assuming that the peaks at ∼7042 and ∼7267 cm–1 are a split feature and likewise for the absorptions at ∼8666 and ∼9328 cm–1, then they are averaged (to 7155 and 8997 cm–1, respectively). Using the zeroth-order splitting, Figure 8, and discarding the lowest two absorptions from the analysis, the absorptions can be initially assigned as 7155 (5fϕ, |3,+1/2⟩), 8997 (5fπ, |1,–1/2⟩), 10 121 (5fπ, |1,+1/2⟩), and 17 500 (5fσ, |0,+1/2⟩) cm–1, Table 1. The |2,+1/2⟩ (5fδ) and |2,–1/2⟩ (5fδ) states fall below the measurement window, and the |3,–1/2⟩ (5fϕ) ground state is known from the EPR and magnetic data.
Table 1. Calculated Energies and Principal Components of the 5f States for Complex 4 Derived from CONDON 3.0 and F Shella.
| absorption
energy (cm–1) |
||||||
|---|---|---|---|---|---|---|
| zeroth-order assignment | observed | averaged | calculated | |ml,ms⟩ composition (%) | parent orbital | Russell–Saunders state and mj projection |
| |0,+1/2⟩ | 17 500 | 17 500 | 17 293 | 35 |0,±1/2⟩ | 5fσ | 55 2F7/2 |±1/2⟩ |
| 29 |±1,∓1/2⟩ | 5fπ | 22 2F7/2 |±7/2⟩ | ||||
| 22 |±3,±1/2⟩ | 5fϕ | 9 2F5/2 |±1/2⟩ | ||||
| 7 |∓3,±1/2⟩ | 5fϕ | 7 2F7/2 |∓5/2⟩ | ||||
| 7 |∓2,∓1/2⟩ | 5fδ | 7 2F5/2 |∓5/2⟩ | ||||
| |1,+1/2⟩ | 10 121 | 10 121 | 10 489 | 61 |±1,±1/2⟩ | 5fπ | 86 2F7/2 |±3/2⟩ |
| 25 |±2,∓1/2⟩ | 5fδ | 12 2F5/2 |∓3/2⟩ | ||||
| 9 |∓2,±1/2⟩ | 5fδ | |||||
| 3 |∓1,∓1/2⟩ | 5fπ | |||||
| |1,–1/2⟩ | 9328 | 8997 | 9392 | 34 |±1,∓1/2⟩ | 5fπ | 44 2F5/2 |±1/2⟩ |
| 8666 | 30 |0,±1/2⟩ | 5fσ | 20 2F7/2 |∓5/2⟩ | |||
| 17 |∓2,∓1/2⟩ | 5fδ | 20 2F7/2 |±1/2⟩ | ||||
| 10 |±3,±1/2⟩ | 5fϕ | 10 2F7/2 |±7/2⟩ | ||||
| 3 |∓3,±1/2⟩ | 5fϕ | |||||
| |3,+1/2⟩ | 7267 | 7155 | 7525 | 33 |±2,±1/2⟩ | 5fδ | 39 2F7/2 |±5/2⟩ |
| 7042 | 27 |∓3,∓1/2⟩ | 5fϕ | 27 2F7/2 |∓7/2⟩ | |||
| 17 |∓1,±1/2⟩ | 5fπ | 17 2F5/2 |∓1/2⟩ | ||||
| 16 |0,∓1/2⟩ | 5fσ | 16 2F7/2 |∓1/2⟩ | ||||
| 6 |±3,∓1/2⟩ | 5fϕ | |||||
| |2,+1/2⟩ | b | 3273 | 27 |±2,±1/2⟩ | 5fδ | 36 2F5/2 |∓1/2⟩ | |
| 24 |∓1,±1/2⟩ | 5fπ | 30 2F7/2 |±5/2⟩ | ||||
| 19 |0,∓1/2⟩ | 5fσ | 19 2F7/2 |∓7/2⟩ | ||||
| 19 |∓3,∓1/2⟩ | 5fϕ | 8 2F5/2 |±5/2⟩ | ||||
| 11 |±3,∓1/2⟩ | 5fϕ | 7 2F7/2 |∓1/2⟩ | ||||
| |2,–1/2⟩ | b | 69 | 52 |±2,∓1/2⟩ | 5fδ | 73 2F5/2 |±3/2⟩ | |
| 21 |±1,±1/2⟩ | 5fπ | 14 2F7/2 |∓3/2⟩ | ||||
| 14 |∓1,∓1/2⟩ | 5fπ | 13 2F5/2 |∓3/2⟩ | ||||
| 13 |∓2,±1/2⟩ | 5fδ | |||||
| |3,–1/2⟩ | b | 0 | 64 |±3,∓1/2⟩ | 5fϕ | 75 2F5/2 |±5/2⟩ | |
| 11 |±2,±1/2⟩ | 5fδ | 17 2F5/2 |∓1/2⟩ | ||||
| 10 |∓1,±1/2⟩ | 5fπ | 6 2F7/2 |∓7/2⟩ | ||||
| 7 |0,∓1/2⟩ | 5fσ | |||||
| 6 |∓3,∓1/2⟩ | 5fϕ | |||||
Magnetic data and energies were fitted simultaneously using CONDON 3.0 in C3v symmetry, and F-shell was used to determine the composition of the states. F-shell outputs the composition as |L, S, J, mj⟩, and these were converted into the |ml, ms⟩ basis using Clebsch–Gordan coefficients.
Outside the measurement window.
While the zeroth-order analysis is intuitive and provides a useful initial electronic structure description, it neglects symmetry-allowed mixing of the states. In order to probe this aspect, magnetic data and energies were fitted simultaneously using CONDON 3.099,100 in C3v symmetry (see Figure S21 for magnetic data fits), and F-shell101 was used to determine the full composition of the states (see the SI for details about the modeling procedure). F-shell outputs the compositions in the |L, S, J, mj⟩ basis (coupled basis); therefore, these were converted into the |ml, ms⟩ basis (uncoupled basis) using Clebsch–Gordan coefficients102 as reported in Table 1. The fit produced the crystal field parameters (expressed in Wybourne notation) B02 = 5655, B04 = 14 932, B06 = 6791, B34 = −16 885, B36 = −6148, and B66 = 10 887 cm–1 and a spin–orbit coupling value of ζ = 1761 cm–1. In general, the experimental magnetic data and NIR energies are modeled well, especially considering that a single ζ value is used in the calculations, but in reality, this and the associated orbital angular momentum would vary across different orbitals depending on their nature; for example, orbital mixing and hence reduced ζ and orbital angular momentum would be ordered 5fσ > 5fπ ≫ 5fϕ ≈ 5fδ.
Since in 4 the SOC and CF parameters are comparable in strength, it would be predicted that there would be substantial mixing of the J = 7/2 and J = 5/2 manifolds. Inspection of Table 1 reveals that when mixing of states is modeled, the initial zeroth-order model is largely maintained. Indeed, the largest component of all states is the one predicted from the zeroth order except for the doublet at 7525 cm–1, which is composed by similar amounts of |2,+1/2⟩ and |3,–1/2⟩. It is noticeable that the calculated extent of mixing of states in 4 appears to be significantly more than that reported for [U(NR){N(SiMe3)2}3] (R = Ad, SiMe3),85 which we suggest stems from a larger crystal field for 4 resulting in more mixing overall. Hence, it would seem that the NBODipp ligand brings more crystal field splitting than the bis(trimethylsilyl)amide, consistent with their O vs N donor natures. Therefore, Table 1 also lists each component as its Russell–Saunders state and mj projection, showing that they are indeed significantly mixed, and largely the calculated mj ordering (|±1/2⟩ > |±3/2⟩ > |∓1/2⟩ > |±5/2⟩ > |∓1/2⟩ > |±3/2⟩ > |±5/2⟩) for 4 is essentially the same as that calculated for [U(NAd){N(SiMe3)2}3] and [U(N)(TrenTIPS)]− (|±1/2⟩ > |±3/2⟩ > |±5/2⟩ > |±7/2⟩ > |±1/2⟩ > |±3/2⟩ > |±5/2⟩);22,85 again, where there are differences (the third and fourth absorptions), the second largest component of each mj projection is the same as that for [U(NAd){N(SiMe3)2}3] and [U(N)(TrenTIPS)]−. This emphasizes that although 4 has pseudo-C3v symmetry (and is in reality lower symmetry), the strength of the imido ligand confers substantial effective axial symmetry, as found for [U(E){N(SiMe3)2}3]− (E = NSiMe3, O).93
Although it should be acknowledged that there is some uncertainty in the precise CF splittings of 4 (∼17 500 cm–1), [U(NAd){N(SiMe3)2}3] (15 434 cm–1), [U(NSiMe3){N(SiMe3)2}3] (16 955 cm–1), and [U(O){N(SiMe3)2}3] (20 262 cm–1),22,85 due to the broadness and shoulder character of their U–Eσ* (E = NR, O) absorptions, it is instructive to consider them in qualitative terms. The larger CF splitting of 4 vs [U(NAd){N(SiMe3)2}3] can be primarily rationalized on the basis that the U=NAd distance in 4 is ∼0.04 Å shorter than that in the latter,85 and then, secondarily, the NBODipp ligands also contribute to a larger CF. For 4 vs [U(NSiMe3){N(SiMe3)2}3], the U=NR distances are essentially the same,86 so even though the U=NSiMe3 σ* antibonding state should be higher in energy than U=NAd on a like for like basis (because the silylimido is less strongly donating than the alkylimido so will have slightly more stabilized bonding orbitals), the similar U=NR distances coupled to the NBODipp ligands in 4 seemingly result in a slightly larger net CF for 4 compared to [U(NSiMe3){N(SiMe3)2}3]. Since the U=E (E = O, NR) distance is the principal driver for the energy of the UEσ* state, 4 has a smaller CF splitting than [U(O){N(SiMe3)2}3] because the latter has a U=E distance that is ∼0.09 Å shorter87 than that in 4, and this seems to outweigh any secondary NBODipp vs amide CF effects.
Computational Characterization
In order to further understand complexes 1–4, we performed GGA LDA BP86 DFT calculations on the whole structures (Tables S2–S5). The gas-phase geometry-optimized structures (Table S6) match the experimental solid structures well overall, with U–ONBO, U–OTHF, U–Cl, B–O, and B–N distances computed to within 0.03 Å and 5°. Computed bond orders, charges, spin densities, natural localized molecular orbital (NLMO), and quantum theory of atoms in molecules (QTAIM) data are compiled in Tables S6–S8. We note a discrepancy between experimental and computed U–Carene distances in 1 (Δ = 0.13 Å), which likely arises from this being a relatively “soft” interaction and that the calculations are free of solid-state effects in the experimental structure of 1. Geometry optimization using the hybrid PBE0 functional only marginally improved the discrepancy of the U–Carene distance (Δ = 0.09 Å), so we examined the crystallographic coordinates with heavy atoms frozen and H atom positions geometry optimized and found only minor changes to the electronic structure of 1. Thus, to provide an internally consistent set of data, we focus on the geometry-optimized BP86 data here. We conclude that the BP86 data provide qualitatively reliable models of the electronic structures of these two complexes since prior work has shown good bond order and NBO/NLMO analysis agreement between BP86 and experimentally (NMR) benchmarked hybrid B3LYP chemical shift anisotropy calculations.55,56,103
The computed uranium MDCq charges for 1–4, Table S6, are 2.52, 2.24, 2.40, and 3.01 and are as anticipated for uranium ions in +3, +4, +4, and +5 oxidation states, respectively. The corresponding MDCm spin densities of −2.51, −2.24, −2.25, and −1.31 are consistent for 2, 3, and 4 in terms of their formal fn counts (n = 2, 2, and 1, respectively) augmented by net donation of electron density from the ligands. However, the spin density for 1 is less than the anticipated value of 3 for a 5f3 ion, suggesting that the uranium ion in 1 is a net exporter of electron density. The arene bonded to uranium has a computed net MDCq charge of −0.94 (C6 ring, 3 × H, 2 × C, 1 × N atoms), and the C6 unit has a total net MDCm spin density of 0.73. Confirmation of uranium–arene interactions is obtained by visualization of the α-spin frontier Kohn–Sham molecular orbitals (KSMOs) of 1, Figure 9. Specifically, although the HOMO is a singly occupied electron of largely 5f character (5f/6d/7s, 83.93/1.40/9.67%), HOMOs−1 and −2 while both being principally of 5f character evidence modest carbon 2p-orbital contributions from δ-bonding interactions (U 5f/6d:C 2p HOMO−1, 70.08/5.04:18.06%; HOMO−2, 64.76/5.39:20.35%). The presence of two distinct but similarly composed and quasi-degenerate δ bonds is significant because it suggests almost equal population of both the π4 and the π5 δ-symmetry π* molecular orbitals of the arene; hence, no Jahn–Teller distortion of the arene,104−106 which would induce ring-puckering,107,108 occurs (recall the planar nature of the coordinated arene in the solid-state structure above). The presence of a U–arene interaction in 1 likely reflects its low-coordinate and electron-rich 5f3 formulation compared to 2–4, which have higher coordination numbers and are 5f2 or 5f1. The KSMOs of 2 and 3 are unremarkable (Figures S22 and S23). Visualization of the α-spin KSMOs of 4 (Figure S24) reveals that the HOMO is an essentially pure 5f orbital (5f/6d, 93.66/5.48), HOMOs−4 and −5 are the U=N π bonds, and HOMO−21 is the U=N σ bond. However, to obtain a more intuitive bonding description, we focus on NLMO analysis (see later).
Figure 9.

Selected frontier Kohn–Sham molecular orbitals of 1: (a) HOMO (5f, 376a, −2.942 eV), (b) HOMO–1 (δ bond, 375a, −3.226 eV), and (c) HOMO–2 (δ bond, 374a, −3.257 eV). Hydrogen atoms are omitted for clarity.
The U–ONBO Nalewajski–Mrozek bond orders average 1.46, 1.52, 1.49, and 1.38 for 1, 2, 3, and 4, Table S6. This shows that the NBODipp ligands are strong donors and reflects the increasing formal uranium charges on moving from trivalent 1 to tetravalent 2 and 3. The fall in U–ONBO bond order in 4 is likely due to the presence of the strong imido donor ligand. Reflecting that the U–arene δ-symmetry bonding interactions exhibit only modest carbon contributions, the U–C Nalewajski–Mrozek bond orders average 0.31, which is certainly consistent with those interactions being weak given the apparent loss of the uranium–arene interaction in solution as evidenced by 1H NMR spectroscopy. Although the changes are small, Table S6, the variation of the B–O and B–N Nalewajski–Mrozek bond orders generally confirms that when the U–ONBO bond order increases/decreases, the B–O and B–N bond orders decrease/increase and increase/decrease, respectively. This reflects the aforementioned electronic buffering role of the boryl as the oxide donates less and more to electron-rich and -poor metals, respectively. The U–Nimido Nalewajski–Mrozek bond order is 2.80, reflecting that the imido ligand is a triple-bond donor in the KSMO scheme.
Complexes 1–4 provide an opportunity to examine the variation of the U–ONBO bond as a function of the uranium oxidation state, and NLMO data are compiled in Table S7. The NLMO analysis reveals rather polar U–ONBO bonds, and although the entire range of uranium contributions is only 3–8%, it can be generally noted that the uranium contributions increase with increasing oxidation state and the uranium 5f character tends to dominate over 6d contributions (5f:6d ≈ 2:1 for σ bonds and ∼3:1 for π bonds). Complex 2 exhibits the largest uranium contributions to the U–ONBO bonds, likely reflecting the presence of two neutral THF donors. This can also be seen in the U–Cl bonds, where the uranium contributes more to the U–Cl bonds in 2 than 3. NLMO analysis of 4, Figure 10, reveals a U=N σ bond that is 11% uranium and 88% N character. The uranium component is composed of 1/3/38/59% 7s/7p/6d/5f character, and the nitrogen is 65:35 2s:2p character. The U=N π bonds are essentially the same and are composed of 22% and 77% uranium and nitrogen contributions, respectively. The uranium component is 0/0/13/87% 7s/7p/6d/5f character, and the nitrogen components are 100% 2p character.
Figure 10.

Selected natural localized molecular orbitals of 4: (a) U=NAd σ bond, (b) U=NAd π bond, and (c) U=NAd π bond. Hydrogen atoms are omitted for clarity.
To gain a topological- rather than orbital-based perspective, we examined the 3,–1 bond critical point QTAIM data for 1–4, Table S8. As expected, the data reveal slightly polar–covalent B–O and B–N bonds (ρav = 0.21 and 0.19 e per Bohr3; Hav = −0.15 and −0.16 hartree per Bohr3, respectively), whereas the U–ONBO bonds are borderline ionic (ρav = 0.10 e per Bohr3) and much weaker in terms of energy (Hav = −0.04 hartree per Bohr3). In contrast, the U–Nimido bond is a much stronger interaction, with ρ and H values of 0.19 e per Bohr3 and −0.14 hartree per Bohr3, respectively, and being close to zero, the ε value of 0.05 confirms a U–N triple bond.109 Overall, these values are similar to data previously calculated for [U(NSiMe3){N(SiMe3)2}3]27 but indicate a slightly stronger U=N bond than that calculated for [U(NR)(TrenTIPS)] (R = SiMe3, Ad)27 but, as expected, a weaker U=N bond than the oxo and nitrido bonds in [U(O){N(SiMe3)2}3],87 [U(O)(TrenTIPS)],49 [U(N)(TrenTIPS)]n (n = 0, −1),27,28 and [Np(O)(TrenTIPS)].16 Lastly, the QTAIM data for the U–arene interactions in 1 reveal ρav and Hav values of 0.03 e per Bohr3 and −0.01 hartree per Bohr3, respectively, and, hence, in agreement with the analysis earlier, rather weak uranium–arene interactions. The uranium–arene εav value of 0.49 reflects the asymmetry of the individual orbital coefficient bonding interactions that construct the uranium–arene δ bonds.
Conclusions
In conclusion, furthering the application of the boryloxide NBODipp ligand in f-element chemistry, we have introduced this ligand to uranium chemistry. This has furnished a uranium(III)–tris(boryloxide) complex that features a uranium–arene interaction, two chlorido derivatives, and a uranium(V)–imido complex. Although the preparation of these complexes is thermodynamically favorable, it is notable that there is kinetic hindrance installing three sterically demanding NBODipp ligands at a single metal center, even one as large as uranium(III). The coordinated arene may provide a useful protecting-group role but is evidently flexible enough to not impede reactivity as evidenced by preliminary work that has accessed a uranium(V)–imido complex by a two-electron oxidation strategy.
DFT studies qualitatively evidence modest uranium(III)–arene δ bonding, which appears to be approximately equal donation into the π4 and π5 δ-symmetry π* orbitals of the arene; this accounts for an apparent lack of Jahn–Teller distortion of the arene despite the fact it is shown experimentally and computationally to be carrying excess open-shell spin density. NIR and computational analysis of the imido complex shows that it has a similar CF to [U(NSiMe3){N(SiMe3)2}3], a larger CF than [U(NAd){N(SiMe3)2}3], but, as expected, a weaker CF than [U(O){N(SiMe3)2}3]. However, replacing amides with the NBODipp ligand results in much more mixing of the |ml,ms⟩ states and Russell–Saunders mj projections, but overall, it is the case that their orderings approximate well to a basic zeroth-order model that neglects mixing of states.
The synthesis of uranium(III), uranium(IV), and uranium(V) complexes supported by a common NBODipp ligand demonstrates the versatility of this boryloxide ligand over several oxidation states of uranium and has provided an opportunity to examine the U–NBODipp interactions over three uranium oxidation states. This versatility can in part be ascribed to the “buffer” nature of the boryl enabling the NBODipp ligand to vary its electron donation as required. Hence, the NBODipp ligand promises to stabilize complementary or new bonding motifs in actinide chemistry compared to existing Tren, amide, and aryloxide ligand classes and also suggests potential for further elaboration in lanthanide chemistry. Certainly, the NBO ligand class demonstrates the importance of ligand–metal cooperativity to bring new vistas to f-element chemistry, and work in that regard is underway in our laboratory.
Acknowledgments
We gratefully acknowledge funding and support from the UK Engineering and Physical Sciences Research Council (EP/T011289/1, EP/S033181/1, EP/P001386/1, EP/M027015/1), the European Research Council (CoG612724), and the University of Manchester including computational resources and associated support services of the Computational Shared Facility. We thank the EPSRC UK National Electron Paramagnetic Resonance Service (EP/W014521/1 and EP/V035231/1). S.T.L. thanks the Alexander von Humboldt Foundation for a Friedrich Wilhelm Bessel Research Award. Financial support provided by the MUR, Dipartimenti di Eccellenza 2023-2027 (DICUS 2.0) to the Department of Chemistry “Ugo Schiff” of the University of Florence is acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.3c04275.
Experimental and computational procedures; crystallographic details for 1–4; NMR, IR, and UV/vis/NIR spectroscopic details for 1–4; SQUID magnetometric characterization data for 1–4; computational data for 1–4 (PDF)
Author Present Address
§ College of Chemistry, Zhengzhou University, Zhengzhou 450001, China
Author Contributions
‡ X.D., J.D., S.Z.: These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Ephritikhine M. The vitality of uranium molecular chemistry at the dawn of the XXIst century. Dalton Trans. 2006, 2501–2516. 10.1039/b603463b. [DOI] [PubMed] [Google Scholar]
- Fox A. R.; Bart S. C.; Meyer K.; Cummins C. C. Towards uranium catalysts. Nature 2008, 455, 341–349. 10.1038/nature07372. [DOI] [PubMed] [Google Scholar]
- Hayton T. W. Metal-Ligand Multiple Bonding in Uranium: Structure and Reactivity. Dalton Trans. 2010, 39, 1145–1158. 10.1039/B909238B. [DOI] [PubMed] [Google Scholar]
- Jones M. B.; Gaunt A. J. Recent Developments in Synthesis and Structural Chemistry of Nonaqueous Actinide Complexes. Chem. Rev. 2013, 113, 1137–1198. 10.1021/cr300198m. [DOI] [PubMed] [Google Scholar]
- Hayton T. W. Recent Developments in Actinide-Ligand Multiple Bonding. Chem. Commun. 2013, 49, 2956–2973. 10.1039/c3cc39053e. [DOI] [PubMed] [Google Scholar]
- La Pierre H. S.; Meyer K. Activation of Small Molecules by Molecular Uranium Complexes. Prog. Inorg. Chem. 2014, 58, 303–415. 10.1002/9781118792797.ch05. [DOI] [Google Scholar]
- Gregson M.; Wooles A. J.; Cooper O. J.; Liddle S. T. Covalent uranium carbene chemistry. Comments on Inorganic Chemistry 2015, 35, 262–294. 10.1080/02603594.2015.1020154. [DOI] [Google Scholar]
- Johnson S. A.; Bart S. C. Achievements in uranium alkyl chemistry: celebrating sixty years of synthetic pursuits. Dalton Trans. 2015, 44, 7710–7726. 10.1039/C4DT01621A. [DOI] [PubMed] [Google Scholar]
- Liddle S. T. The Renaissance of Non-Aqueous Uranium Chemistry. Angew. Chem., Int. Ed. 2015, 54, 8604–8641. 10.1002/anie.201412168. [DOI] [PubMed] [Google Scholar]
- Gardner B. M.; Liddle S. T. Uranium triamidoamine chemistry. Chem. Commun. 2015, 51, 10589–10607. 10.1039/C5CC01360G. [DOI] [PubMed] [Google Scholar]
- Schädle D.; Anwander R. Rare-Earth Metal and Actinide Organoimide Chemistry. Chem. Soc. Rev. 2019, 48, 5752–5805. 10.1039/C8CS00932E. [DOI] [PubMed] [Google Scholar]
- Boreen M. A.; Arnold J. The synthesis and versatile reducing power of low-valent uranium complexes. Dalton Trans. 2020, 49, 15124–15138. 10.1039/D0DT03151H. [DOI] [PubMed] [Google Scholar]
- Hartline D. R.; Meyer K. From chemical curiosities and trophy molecules to uranium-based catalysis: developments for uranium catalysis as a new facet in molecular uranium chemistry. JACS Au 2021, 1, 698–709. 10.1021/jacsau.1c00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keener M.; Maria L.; Mazzanti M. Progress in the chemistry of molecular actinide-nitride compounds. Chem. Sci. 2023, 14, 6493–6521. 10.1039/D3SC01435E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Dollberg K.; Seed J. A.; Wooles A. J.; Von Hänisch C.; Liddle S. T.. Thorium(IV)-antimony complexes exhibiting single, double, and triple polar covalent metal-metal bonds. Nat. Chem. 2024, 16, 10.1038/s41557-024-01448-6. [DOI] [PubMed] [Google Scholar]
- Dutkiewicz M. S.; Goodwin C. A. P.; Perfetti M.; Gaunt A. J.; Griveau J.-C.; Colineau E.; Kovács A.; Wooles A. J.; Caciuffo R.; Walter O.; Liddle S. T. A Terminal Neptunium(V)-Mono(Oxo) Complex. Nat. Chem. 2022, 14, 342–349. 10.1038/s41557-021-00858-0. [DOI] [PubMed] [Google Scholar]
- Du J.; Balazs G.; Seed J. A.; Cryer J. D.; Wooles A. J.; Scheer M.; Liddle S. T. Actinide Pnictinidene Chemistry: A Terminal Thorium Parent-Arsinidene Complex Stabilised by a Super-Bulky Triamidoamine Ligand. Angew. Chem., Int. Ed. 2022, 61, e202211627 10.1002/anie.202211627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seed J. A.; Sharpe H. R.; Futcher H. J.; Wooles A. J.; Liddle S. T. Nature of the Arsonium-Ylide Ph3As = CH2 and a Uranium(IV) Arsonium-Carbene Complex. Angew. Chem., Int. Ed. 2020, 59, 15870–15874. 10.1002/anie.202004983. [DOI] [PubMed] [Google Scholar]
- Du J.; King D. M.; Chatelain L.; Lu E.; Tuna F.; McInnes E. J. L.; Wooles A. J.; Maron L.; Liddle S. T. Thorium- and Uranium-Azide Reductions: A Transient Dithorium-Nitride Versus Isolable Diuranium-Nitrides. Chem. Sci. 2019, 10, 3738–3745. 10.1039/C8SC05473H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rookes T. M.; Gardner B. M.; Balázs G.; Gregson M.; Tuna F.; Wooles A. J.; Scheer M.; Liddle S. T. Crystalline Diuranium-Phosphinidiide and -μ-Phosphido Complexes with Symmetric and Asymmetric UPU Cores. Angew. Chem., Int. Ed. 2017, 56, 10495–10500. 10.1002/anie.201706002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildman E. P.; Balázs G.; Wooles A. J.; Scheer M.; Liddle S. T. Triamidoamine Thorium-Arsenic Complexes with Parent Arsenide, Arsinidiide and Arsenido Structural Motifs. Nat. Commun. 2017, 8, 14769. 10.1038/ncomms14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King D. M.; Cleaves P. A.; Wooles A. J.; Gardner B. M.; Chilton N. F.; Tuna F.; Lewis W.; McInnes E. J. L.; Liddle S. T. Molecular and Electronic Structure of Terminal and Alkali Metal-Capped Uranium(V)-Nitride Complexes. Nat. Commun. 2016, 7, 13773. 10.1038/ncomms13773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildman E. P.; Balázs G.; Wooles A. J.; Scheer M.; Liddle S. T. Thorium-Phosphorus Triamidoamine Complexes Containing Th-P Single- and Multiple-Bond Interactions. Nat. Commun. 2016, 7, 12884. 10.1038/ncomms12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner B. M.; Balázs G.; Scheer M.; Tuna F.; McInnes E. J. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Triamidoamine Uranium(IV)-Arsenic Complexes Containing One-, Two-, and Three-fold U-As Bonding Interactions. Nat. Chem. 2015, 7, 582–590. 10.1038/nchem.2279. [DOI] [PubMed] [Google Scholar]
- Gardner B. M.; Balázs G.; Scheer M.; Tuna F.; McInnes E. J. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Triamidoamine-Uranium(IV)-Stabilized Terminal Parent Phosphide and Phosphinidene Complexes. Angew. Chem., Int. Ed. 2014, 53, 4484–4488. 10.1002/anie.201400798. [DOI] [PubMed] [Google Scholar]
- King D. M.; McMaster J.; Tuna F.; McInnes E. J. L.; Lewis W.; Blake A. J.; Liddle S. T. Synthesis and Characterization of an f-Block Terminal Parent Imido [U=NH] Complex: A Masked Uranium(IV)-Nitride. J. Am. Chem. Soc. 2014, 136, 5619–5622. 10.1021/ja502405e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King D. M.; Tuna F.; McInnes E. J. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Isolation and characterisation of a uranium(VI)-nitride triple bond. Nat. Chem. 2013, 5, 482–488. 10.1038/nchem.1642. [DOI] [PubMed] [Google Scholar]
- King D. M.; Tuna F.; McInnes E. J. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Synthesis and Structure of a Terminal Uranium Nitride Complex. Science 2012, 337, 717–720. 10.1126/science.1223488. [DOI] [PubMed] [Google Scholar]
- Rookes T. M.; Balázs G.; Gardner B. M.; Wooles A. J.; Scheer M.; Liddle S. T. Actinide-Pnictide Chemistry: A Uranium Primary Alkyl Stibinide and a Diuranium Hexaantimonide-Tetralithium Zintl Cluster. ChemistryEurope 2023, 1, e202300067 10.1002/ceur.202300067. [DOI] [Google Scholar]
- Gardner B. M.; Patel D.; Cornish A. D.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. The Nature of Unsupported Uranium-Ruthenium Bonds: A Combined Experimental and Theoretical Analysis. Chem. Eur. J. 2011, 17, 11266–11273. 10.1002/chem.201101394. [DOI] [PubMed] [Google Scholar]
- Vlaisavljevich B.; Miró P.; Cramer C. J.; Gagliardi L.; Infante I.; Liddle S. T. On the Nature of Actinide- and Lanthanide-Metal Bonds in Heterobimetallic Compounds. Chem. Eur. J. 2011, 17, 8424–8433. 10.1002/chem.201100774. [DOI] [PubMed] [Google Scholar]
- Gardner B. M.; McMaster J.; Moro F.; Lewis W.; Blake A. J.; Liddle S. T. An Unsupported Uranium-Rhenium Complex Prepared by Alkane Elimination. Chem. Eur. J. 2011, 17, 6909–6912. 10.1002/chem.201100682. [DOI] [PubMed] [Google Scholar]
- Patel D.; King D. M.; Gardner B. M.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Structural and theoretical insights into the perturbation of uranium-rhenium bonds by dative Lewis base ancillary ligands. Chem. Commun. 2011, 47, 295–297. 10.1039/C0CC01387K. [DOI] [PubMed] [Google Scholar]
- Gardner B. M.; McMaster J.; Lewis W.; Liddle S. T. Synthesis and Structure of [{N(CH2CH2NSiMe3)3}URe(η5-C5H5)2]: A Heterobimetallic Complex with an Unsupported Uranium-Rhenium Bond. Chem. Commun. 2009, 2851–2853. 10.1039/b906554g. [DOI] [PubMed] [Google Scholar]
- Liddle S. T.; McMaster J.; Mills D. P.; Blake A. J.; Jones C.; Woodul W. D. σ and π donation in an unsupported uranium-gallium bond. Angew. Chem., Int. Ed. 2009, 48, 1077–1080. 10.1002/anie.200805481. [DOI] [PubMed] [Google Scholar]
- Du J.; Hunger D.; Seed J. A.; Cryer J. D.; King D. M.; Wooles A. J.; Van Slageren J.; Liddle S. T. Dipnictogen f-Element Chemistry: A Diphosphorus Uranium Complex. J. Am. Chem. Soc. 2021, 143, 5343–5348. 10.1021/jacs.1c02482. [DOI] [PubMed] [Google Scholar]
- Du J.; Balázs G.; Wooles A. J.; Scheer M.; Liddle S. T. The “Hidden” Reductive [2 + 2+1]-Cycloaddition Chemistry of 2-Phosphaethynolate Revealed by Reduction of a Th-OCP Linkage. Angew. Chem., Int. Ed. 2021, 60, 1197–1202. 10.1002/anie.202012506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnall R.; Balázs G.; Lu E.; Kern M.; Van Slageren J.; Tuna F.; Wooles A. J.; Scheer M.; Liddle S. T. Photolytic and Reductive Activations of 2-Arsaethynolate in a Uranium-Triamidoamine Complex: Decarbonylation Arsenic Group-Transfer and Trapping of a Highly Bent and Reduced Form. Chem. Eur. J. 2019, 25, 14246–14252. 10.1002/chem.201903973. [DOI] [PubMed] [Google Scholar]
- Magnall R.; Balázs G.; Lu E.; Tuna F.; Wooles A. J.; Scheer M.; Liddle S. T. Trapping of a Highly Bent and Reduced Form of 2-Phosphaethynolate in a Mixed-Valence Diuranium-Triamidoamine Complex. Angew. Chem., Int. Ed. 2019, 58, 10215–10219. 10.1002/anie.201904676. [DOI] [PubMed] [Google Scholar]
- Rookes T. M.; Wildman E. P.; Balázs G.; Gardner B. M.; Wooles A. J.; Gregson M.; Tuna F.; Scheer M.; Liddle S. T. Actinide-Pnictide (An-Pn) Bonds Spanning Non-Metal, Metalloid, and Metal Combinations (An = U, Th; Pn = P, As, Sb, Bi). Angew. Chem., Int. Ed. 2018, 57, 1332–1336. 10.1002/anie.201711824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner B. M.; Balázs G.; Scheer M.; Wooles A. J.; Tuna F.; McInnes E. J. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Isolation of Elusive HAsAsH in a Crystalline Diuranium(IV) Complex. Angew. Chem., Int. Ed. 2015, 54, 15250–15254. 10.1002/anie.201508600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner B. M.; Tuna F.; McInnes E. J. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. An Inverted Sandwich Diuranium μ-η5:η5-Cyclo-P5 Complex Supported by U-P5 δ-Bonding. Angew. Chem., Int. Ed. 2015, 54, 7068–7072. 10.1002/anie.201501728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner B. M.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. A Crystallizable Dinuclear Tuck-In-Tuck-Over Tuck-Over Dialkyl Tren Uranium Complex and Double Dearylation of BPh4– to give the BPh2-Functionalized Metallocycle [U{N(CH2CH2NSiMe3)2(CH2CH2NSiMe2CHBPh2)}(THF)]. J. Am. Chem. Soc. 2009, 131, 10388–10389. 10.1021/ja904459q. [DOI] [PubMed] [Google Scholar]
- Cobb P. J.; Wooles A. J.; Liddle S. T. A Uranium(VI)-Oxo-Imido Dimer Complex Derived from a Sterically Demanding Triamidoamine. Inorg. Chem. 2020, 59, 10034–10041. 10.1021/acs.inorgchem.0c01207. [DOI] [PubMed] [Google Scholar]
- Chatelain L.; Louyriac E.; Douair I.; Lu E.; Tuna F.; Wooles A. J.; Gardner B. M.; Maron L.; Liddle S. T. Terminal Uranium(V)-Nitride Hydrogenations Involving Direct Addition or Frustrated Lewis Pair Mechanisms. Nat. Commun. 2020, 11, 337. 10.1038/s41467-019-14221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaves P. A.; Kefalidis C. E.; Gardner B. M.; Tuna F.; McInnes E. J. L.; Lewis W.; Maron L.; Liddle S. T. Terminal Uranium(V/VI)-Nitride Activation of Carbon Dioxide and Carbon Disulfide: Factors Governing Diverse and Well-Defined Cleavage and Redox Reactions. Chem. Eur. J. 2017, 23, 2950–2959. 10.1002/chem.201605620. [DOI] [PubMed] [Google Scholar]
- Cleaves P. A.; King D. M.; Kefalidis C. E.; Maron L.; Tuna F.; McInnes E. J. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Two-Electron Reductive Carbonylation of Terminal Uranium(V) and Uranium(VI) Nitrides to Cyanate by Carbon Monoxide. Angew. Chem., Int. Ed. 2014, 53, 10412–10415. 10.1002/anie.201406203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner B. M.; Stewart J. C.; Davis A. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Homologation and Functionalization of Carbon Monoxide by a Recyclable Uranium Complex. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9265–9270. 10.1073/pnas.1203417109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King D. M.; Tuna F.; McMaster J.; Lewis W.; Blake A. J.; McInnes E. J. L.; Liddle S. T. Single-Molecule Magnetism in a Single-Ion Triamidoamine Uranium(V) Terminal Mono-Oxo Complex. Angew. Chem., Int. Ed. 2013, 52, 4921–4924. 10.1002/anie.201301007. [DOI] [PubMed] [Google Scholar]
- Gardner B. M.; Patel D.; Lewis W.; Blake A. J.; Liddle S. T. Photochemically Promoted Bond-Cleavage and -Capture in a Diazomethane Derivative of a Triamidoamine Uranium(IV) Complex. Angew. Chem., Int. Ed. 2011, 50, 10440–10443. 10.1002/anie.201105098. [DOI] [PubMed] [Google Scholar]
- Du J.; Douair I.; Lu E.; Seed J. A.; Tuna F.; Wooles A. J.; Maron L.; Liddle S. T. Evidence for ligand- and solvent-induced disproportionation of uranium(IV). Nat. Commun. 2021, 12, 4832. 10.1038/s41467-021-25151-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Alvarez-Lamsfus C.; Wildman E. P.; Wooles A. J.; Maron L.; Liddle S. T. Thorium-Nitrogen Multiple Bonds Provide Evidence for Pushing-from-Below for Early Actinides. Nat. Commun. 2019, 10, 4203. 10.1038/s41467-019-12206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner B. M.; King D. M.; Tuna F.; Wooles A. J.; Chilton N. F.; Liddle S. T. Assessing Crystal Field and Magnetic Interactions in Diuranium-μ-Chalcogenide Triamidoamine Complexes With UIV-E-UIV Cores (E = S, Se, Te): Implications for Determining the Presence or Absence of Actinide-Actinide Magnetic Exchange. Chem. Sci. 2017, 8, 6207–6217. 10.1039/C7SC01998J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner B. M.; Cleaves P. A.; Kefalidis C. E.; Fang J.; Maron L.; Lewis W.; Blake A. J.; Liddle S. T. The Role of 5f-Orbital Participation in Unexpected Inversion of the σ-Bond Metathesis Reactivity Trend of Triamidoamine Thorium(IV) and Uranium(IV) Alkyls. Chem. Sci. 2014, 5, 2489–2497. 10.1039/C4SC00182F. [DOI] [Google Scholar]
- Du J.; Hurd J.; Seed J. A.; Balazs G.; Scheer M.; Adams R. W.; Lee D.; Liddle S. T. 31P Nuclear Magnetic Resonance Spectroscopy as a Probe of Thorium-Phosphorus Bond Covalency: Correlating Phosphorus Chemical Shift to Metal-Phosphorus Bond Order. J. Am. Chem. Soc. 2023, 145, 21766–21784. 10.1021/jacs.3c02775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Seed J. A.; Berryman V. E. J.; Kaltsoyannis N.; Adams R. W.; Lee D.; Liddle S. T. Exceptional Uranium(VI)-Nitride Triple Bond Covalency from 15N Nuclear Magnetic Resonance Spectroscopy and Quantum Chemical Analysis. Nat. Commun. 2021, 12, 5649. 10.1038/s41467-021-25863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa Y.; Suzuki Y.; Yamashita M.; Nozaki K. Chemistry of Boryllithium: Synthesis, Structure, and Reactivity. J. Am. Chem. Soc. 2008, 130, 16069–16079. 10.1021/ja8057919. [DOI] [PubMed] [Google Scholar]
- Loh Y. K.; Ying L.; Angeles Fuentes M.; Do D. C. H.; Aldridge S. An N-Heterocyclic Boryloxy Ligand Isoelectronic with N-Heterocyclic Imines: Access to an Acyclic Dioxysilylene and its Heavier Congeners. Angew. Chem., Int. Ed. 2019, 58, 4847–4851. 10.1002/anie.201812058. [DOI] [PubMed] [Google Scholar]
- Thomas-Hargreaves L. R.; Hunger D.; Kern M.; Wooles A. J.; Van Slageren J.; Chilton N. F.; Liddle S. T. Insights into D4h@Metal-Symmetry Single-Molecule Magnetism: The Case of a Dysprosium-bis(Boryloxide) Complex. Chem. Commun. 2021, 57, 733–736. 10.1039/D0CC07446B. [DOI] [PubMed] [Google Scholar]
- Arnold P. L.; Halliday C. J. V.; Puig-Urrea L.; Nichol G. S. Instantaneous and phosphine-catalyzed arene binding and reduction by U(III) complexes. Inorg. Chem. 2021, 60, 4162–4170. 10.1021/acs.inorgchem.1c00327. [DOI] [PubMed] [Google Scholar]
- Arnold P. L.; Puig-Urrea L.; Wells J. A. L.; Yuan D.; Cruickshank F. L.; Young R. D. Applications of boroxide ligands in supporting small molecule activation by U(III) and U(IV) complexes. Dalton Trans. 2019, 48, 4894–4905. 10.1039/C8DT05051A. [DOI] [PubMed] [Google Scholar]
- Faizova R.; Fadaei-Tirani F.; Bernier-Latmani R.; Mazzanti M. Ligand-supported facile conversion of uranyl(VI) into uranium(IV) in organic and aqueous media. Angew. Chem., Int. Ed. 2020, 59, 6756–6759. 10.1002/anie.201916334. [DOI] [PubMed] [Google Scholar]
- Cowie B. E.; Douair I.; Maron L.; Love J. B.; Arnold P. L. Selective oxo ligand functionalisation and substitution reactivity in an oxo/catecholate-bridged UIV/UIV Pacman complex. Chem. Sci. 2020, 11, 7144–7157. 10.1039/D0SC02297G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Song E.; Cheng L.; Song L.; Xie J.; Li G.; Zhang Y.; Wang Y.; Wang Y.; Xia Z.; Chai Z.; Wang S. Introducing uranium as the activator toward highly stable narrow-band green emitters with near-unity quantum efficiency. Chem. Mater. 2019, 31, 9684–9690. 10.1021/acs.chemmater.9b03130. [DOI] [Google Scholar]
- Cowie B. E.; Nichol G. S.; Love J. B.; Arnold P. L. Double uranium oxo cations derived from uranyl by borane or silane reduction. Chem. Commun. 2018, 54, 3839–3842. 10.1039/C8CC00341F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrick E. A.; Wu G.; Kaltsoyannis N.; Hayton T. W. Reductive silylation of a uranyl dibenzoylmethanate complex: an example of controlled uranyl oxo ligand cleavage. Chem. Sci. 2014, 5, 3204–3213. 10.1039/C4SC00996G. [DOI] [Google Scholar]
- Wang S.; Parker T. G.; Grant D. J.; Diwu J.; Alekseev E. V.; Depmeier W.; Gagliardi L.; Albrecht-Schmitt T. E. Elucidation of tetraboric acid with a new borate fundamental building block in a chiral uranyl fluoroborate. Inorg. Chem. 2012, 51, 11211–11213. 10.1021/ic300741s. [DOI] [PubMed] [Google Scholar]
- Wang S.; Alekseev E. V.; Diwu J.; Miller H. M.; Oliver A. G.; Liu G.; Depmeier W.; Albrecht-Schmitt T. E. Functionalization of borate networks by the incorporation of fluoride: syntheses, crystal structures, and nonlinear optical properties of novel actinide fluoroborates. Chem. Mater. 2011, 23, 2931–2939. 10.1021/cm2004984. [DOI] [Google Scholar]
- Wang S.; Alekseev E. V.; Ling J.; Liu G.; Depmeier W.; Albrecht-Schmitt T. E. Polarity and chirality in uranyl borates: insights into understanding the vitrification of nuclear waste and the development of nonlinear optical materials. Chem. Mater. 2010, 22, 2155–2163. 10.1021/cm9037796. [DOI] [Google Scholar]
- Schnaars D. W.; Wu G.; Hayton T. W. Silylation of the uranyl ion using B(C6F5)3-activated Et3SiH. Inorg. Chem. 2011, 50, 9642–9649. 10.1021/ic201385h. [DOI] [PubMed] [Google Scholar]
- Schnaars D. D.; Wu G.; Hayton T. W. Borane-mediated silylation of a metal-oxo ligand. Inorg. Chem. 2011, 50, 4695–4697. 10.1021/ic2008649. [DOI] [PubMed] [Google Scholar]
- Wang S.; Alekseev E. V.; Miller H. M.; Depmeier W.; Albrecht-Schmitt T. E. Boronic acid flux synthesis and crystal growth of uranium and neptunium boronates and borates: a low-temperature route to the first neptunium(V) borate. Inorg. Chem. 2010, 49, 9755–9757. 10.1021/ic101678d. [DOI] [PubMed] [Google Scholar]
- Wang S.; Alekseev E. V.; Stritzinger J. T.; Depmeier W.; Albrecht-Schmitt T. E. Crystal chemistry of the potassium and rubidium uranyl borate families derived from boric acid fluxes. Inorg. Chem. 2010, 49, 6690–6696. 10.1021/ic100728s. [DOI] [PubMed] [Google Scholar]
- Das R. K.; Barnea E.; Andrea T.; Kapon M.; Fridman N.; Botoshansky M.; Eisen M. S. Group 4 lanthanide and actinide organometallic inclusion complexes. Organometallics 2015, 34, 742–752. 10.1021/om501103v. [DOI] [Google Scholar]
- Schnaars D. D.; Wu G.; Hayton T. W. Reduction of pentavalent uranyl to U(IV) facilitated by oxo functionalization. J. Am. Chem. Soc. 2009, 131, 17532–17533. 10.1021/ja906880d. [DOI] [PubMed] [Google Scholar]
- Barnea E.; Andrea T.; Kapon M.; Eisen M. S. Formation of inclusion organoactinide complexes with boron-containing macrocycles. J. Am. Chem. Soc. 2004, 126, 5066–5067. 10.1021/ja0498843. [DOI] [PubMed] [Google Scholar]
- Campello M. P. C.; Calhorda M. J.; Domingos A.; Galvao A.; Leal J. P.; Pires de Matos A.; Santos I. Hydrocarbyl derivatives of [UCl2HB(pz)32]: synthesis, characterization and reactivity studies towards protic substrates and ketones. J. Organomet. Chem. 1997, 538, 223–239. 10.1016/S0022-328X(97)00078-8. [DOI] [Google Scholar]
- Carvalho A.; Domingos A.; Gaspar P.; Marques N.; de Matos A. P.; Santos I. Dihydrobis(3,5-dimethylpyrazolyl)borate derivatives of f elements. Polyhedron 1992, 11, 1481–1488. 10.1016/S0277-5387(00)83141-1. [DOI] [Google Scholar]
- Avens L. R.; Bott S. G.; Clark D. L.; Sattelberger A. P.; Watkin J. G.; Zwick B. D. A convenient entry into trivalent actinide chemistry: synthesis and characterization of AnI3(THF)4 and An[N(SiMe3)2]3 (An = U, Np, Pu). Inorg. Chem. 1994, 33, 2248–2256. 10.1021/ic00088a030. [DOI] [Google Scholar]
- Stewart J. L.; Andersen R. A. Trivalent uranium chemistry: molecular structure of [(Me3Si)2N]3U. Polyhedron 1998, 17, 953–958. 10.1016/S0277-5387(97)00244-1. [DOI] [Google Scholar]
- Shannon R. D. Revised effective ionic radii and systematic studies of interactomic distances in halides and chalcogenides. Acta Cryst. A 1976, 32, 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Russo M. R.; Kaltsoyannis N.; Sella A. Are Metal Alkoxides Linear Owing to Electrostatic Repulsion?. Chem. Commun. 2002, 2458–2459. 10.1039/b207435d. [DOI] [PubMed] [Google Scholar]
- Steffey B. D.; Fanwick P. E.; Rothwell I. P. Solid state structure of the tantalum bis-aryl compounds Ta(OAr-2,6R2)3(C6H5)2 (R = CH3, Pri; OAr-2,6R2 = 2,6-dialkylphenoxide): Observation of a lack of correlation of M-OR distances and M-O-Ar angles for aryloxide derivatives of niobium(V) and tantalum(V). Polyhedron 1990, 9, 963–968. 10.1016/S0277-5387(00)84298-9. [DOI] [Google Scholar]
- Cryer J. D.; Liddle S. T.. Arene complexes of the actinides. In Comprehensive Organometallic Chemistry IV; Parkin G., Meyer K., O’Hare D., Eds.; Elsevier, 2022; Vol. 4, pp 460–501. [Google Scholar]
- Nguyen T. H.; Paul E. L.; Lukens W. W.; Hayton T. W. Evaluating f-orbital participation in the UV=E multiple bonds of [U(E)(NR2)3] (E = O, NSiMe3, NAd; R = SiMe3). Inorg. Chem. 2023, 62, 6447–6457. 10.1021/acs.inorgchem.3c00455. [DOI] [PubMed] [Google Scholar]
- Zalkin A.; Brennan J. G.; Andersen R. A. Tris[bis-trimethylsilyl)amido](trimethylsilylimido)uranium(V). Acta Cryst. C 1988, 44, 1553–1554. 10.1107/S0108270188005116. [DOI] [Google Scholar]
- Fortier S.; Brown J. L.; Kaltsoyannis N.; Wu G.; Hayton T. W. Synthesis, Molecular and Electronic Structure of UV(O)[N(SiMe3)2]3. Inorg. Chem. 2012, 51, 1625–1633. 10.1021/ic201936j. [DOI] [PubMed] [Google Scholar]
- Kindra D. R.; Evans W. J. Magnetic susceptibility of uranium complexes. Chem. Rev. 2014, 114, 8865–8882. 10.1021/cr500242w. [DOI] [PubMed] [Google Scholar]
- Bart S. C.; Heinemann F. W.; Anthon C.; Hauser C.; Meyer K. A new tripodal ligand system with steric and electronic modularity for uranium coordination chemistry. Inorg. Chem. 2009, 48, 9419–9426. 10.1021/ic9012697. [DOI] [PubMed] [Google Scholar]
- Pividori D.; Miehlich M. E.; Kestel B.; Heinemann F. W.; Scheurer A.; Patzschke M.; Meyer K. Uranium going the soft way: low-valent uranium(III) coordinated to an arene-anchored tris-thiophenolate ligand. Inorg. Chem. 2021, 60, 16455–16465. 10.1021/acs.inorgchem.1c02310. [DOI] [PubMed] [Google Scholar]
- Moro F.; Mills D. P.; Liddle S. T.; Van Slageren J. The Inherent Single Molecule Magnet Character of Trivalent Uranium. Angew. Chem., Int. Ed. 2013, 52, 3430–3433. 10.1002/anie.201208015. [DOI] [PubMed] [Google Scholar]
- Halter D. P.; La Pierre H. S.; Heinemann F. W.; Meyer K. Uranium(IV) halide (F–, Cl–, Br–, and I–) monoarene complexes. Inorg. Chem. 2014, 53, 8418–8424. 10.1021/ic501011p. [DOI] [PubMed] [Google Scholar]
- Seed J. A.; Birnoschi L.; Lu E.; Tuna F.; Wooles A. J.; Chilton N. F.; Liddle S. T. Anomalous Magnetism of Uranium(IV)-Oxo and -Imido Complexes Reveals Unusual Doubly Degenerate Electronic Ground States. Chem. 2021, 7, 1666–1680. 10.1016/j.chempr.2021.05.001. [DOI] [Google Scholar]
- Jones M. T.; Kuechler T. C. An electron spin resonance study of the benzene anion radical. A model of its ion pair with alkali metal ions. J. Phys. Chem. 1977, 81, 360–367. 10.1021/j100519a016. [DOI] [Google Scholar]
- Eisenstein J. C.; Pryce M. H. L. The electronic structure and magnetic properties of uranyl-like ions. I. Uranyl and neptunyl. Proc. R. Soc. London Math. Phys. Eng. Sci. 1955, 229, 20–38. 10.1098/rspa.1955.0071. [DOI] [Google Scholar]
- Eisenstein J. C.; Pryce M. H. L. Electronic structure and magnetic properties of the neptunyl ion. J. Res. Natl. Bur. Std. 1965, 69A, 217–235. 10.6028/jres.069A.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staun S. L.; Wu G.; Lukens W. W.; Hayton T. W. Synthesis of a heterobimetallic actinide nitride and an analysis of its bonding. Chem. Sci. 2021, 12, 15519–15527. 10.1039/D1SC05072A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever A. B. P.Inorganic Electronic Spectroscopy, 2nd ed.; Elsevier: New York, 1984. [Google Scholar]
- Van Leusen J.; Speldrich M.; Schilder H.; Koegerler P. Comprehensive insight into molecular magnetism via CONDON: Full vs. effective models. Coord. Chem. Rev. 2015, 289, 137–148. 10.1016/j.ccr.2014.10.011. [DOI] [Google Scholar]
- Speldrich M.; Van Leusen J.; Koegerler P. CONDON 3.0: An Updated Software Package for Magnetochemical Analysis-All the Way to Polynuclear Actinide Complexes. J. Comput. Chem. 2018, 39, 2133–2145. 10.1002/jcc.25389. [DOI] [PubMed] [Google Scholar]
- Reid M.F-shell program; University of Canterbury, New Zealand, 1984. [Google Scholar]
- Edmonds A. R.Angular Momentum in Quantum Mechanics; Princeton University Press: Princeton, NJ, 1957. [Google Scholar]
- Baker C. F.; Seed J. A.; Adams R. W.; Lee D.; Liddle S. T. 13Ccarbene nuclear magnetic resonance chemical shift analysis confirms CeIV=C double bonding in cerium(IV)-diphosphonioalkylidene complexes. Chem. Sci. 2024, 15, 238–249. 10.1039/D3SC04449A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brezina K.; Kostal V.; Jungwirth P.; Marsalek O. Electronic structure of the solvated benzene radical anion. J. Chem. Phys. 2022, 156, 014501. 10.1063/5.0076115. [DOI] [PubMed] [Google Scholar]
- Brezina K.; Jungwirth P.; Marsalek O. Benzene radical anion in the context of the Birch reduction: when solvation is the key. J. Phys. Chem. Lett. 2020, 11, 6032–6038. 10.1021/acs.jpclett.0c01505. [DOI] [PubMed] [Google Scholar]
- Bazante A. P.; Davidson E. R.; Bartlett R. J. The benzene radical anion: a computationally demanding prototype for aromatic anions. J. Chem. Phys. 2015, 142, 204304. 10.1063/1.4921261. [DOI] [PubMed] [Google Scholar]
- Hitchcock P. B.; Lappert M. F.; Protchenko A. V. Synthesis and structure of the silylated benzene radical anion salts [K([18]crown-6){C6H4(SiMe3)2-1,4}] and [K([18]crown-6)(THF)2][C6H2(SiMe3)4-1,2,4,5]. J. Organomet. Chem. 2011, 696, 2161–2164. 10.1016/j.jorganchem.2010.11.040. [DOI] [Google Scholar]
- Hitchcock P. B.; Lappert M. F.; Protchenko A. V. The First Crystalline Alkali Metal Salt of a Benzenoid Radical Anion without a Stabilizing Substituent and of a Related Dimer: X-ray Structures of the Toluene Radical Anion and of the Benzene Radical Anion Dimer Potassium-Crown Ether Salts. J. Am. Chem. Soc. 2001, 123, 189–190. 10.1021/ja005580e. [DOI] [PubMed] [Google Scholar]
- Bader R. F. W.; Slee T. S.; Cremer D.; Kraka E. Description of conjugation and hyperconjugation in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5061–5068. 10.1021/ja00353a035. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.