

Abstract
There is long-standing interest in nonaqueous uranium chemistry because of fundamental questions about uranium’s variable chemical bonding and the similarities of this pseudo-Group 6 element to its congener d-block elements molybdenum and tungsten. To provide historical context, with reference to a conference presentation slide presented around 1988 that advanced a defining collection of top targets, and the challenge, for synthetic actinide chemistry to realize in isolable complexes under normal experimental conditions, this Viewpoint surveys progress against those targets, including (i) CO and related π-acid ligand complexes, (ii) alkylidenes, carbynes, and carbidos, (iii) imidos and terminal nitrides, (iv) homoleptic polyalkyls, -alkoxides, and -aryloxides, (v) uranium–uranium bonds, and (vi) examples of topics that can be regarded as branching out in parallel from the leading targets. Having summarized advances from the past four decades, opportunities to build on that progress, and hence possible future directions for the field, are highlighted. The wealth and diversity of uranium chemistry that is described emphasizes the importance of ligand–metal complementarity in developing exciting new chemistry that builds our knowledge and understanding of elements in a relativistic regime.
Short abstract
There is long-standing interest in nonaqueous uranium chemistry because of fundamental questions about its basic chemistry. With reference to a “to-do” list from around 1988 that advanced a defining collection of top targets, this Viewpoint surveys progress against those targets. Opportunities to build on that progress and hence possible future directions are highlighted. The wealth and diversity of uranium chemistry that is described emphasizes the importance of ligand−metal complementarity.
Introduction
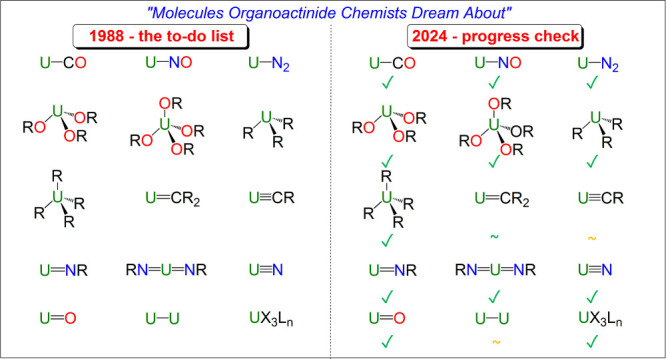
Being subject to a rich interplay of relativistic, interelectronic repulsion, spin–orbit coupling, and crystal field effects, the chemistry of actinides is complex and fascinating, and there remains much to learn about these still somewhat enigmatic elements at a basic level.1 From a molecular perspective, uranium, in depleted or natural forms, is one of the more intensively investigated actinides. This is not only because of its prominent role in nuclear technologies—with associated extraction, recycling, and cleanup legacy challenges—and relative ease to work with as a weak α-emitter but also because of fundamental questions over the nature of its chemical bonding. With variable oxidation states and a large range of valence orbitals available for hydridization with ligand frontier orbitals, uranium can behave like a covalent transition metal through to being rather ionic like trivalent lanthanides.2 Indeed, the fact that uranium was originally classified as a Group 6 transition metal until its rightful place in the 5f actinide series was recognized underlines just how variable the chemical bonding of uranium can be.1 Given the need for new knowledge and understanding in nuclear research, for many years the molecular chemistry of uranium was dominated by aqueous studies of the uranyl dication (UO2)2+.1,2 However, seeking to answer the question of how transition-metal-like uranium can be and the role of 5f, 6p, 6d, 7s, and 7p orbitals in its chemical bonding, a debate sparked by the revolutionary molecule uranocene [U(η8-C8H8)2] (1; Figure 1) from Streitwieser and Raymond,3,4 nonaqueous uranium chemistry has flourished over the past four decades.1,2 Underpinning all of the advances that have been made in nonaqueous uranium chemistry, and indeed more widely in aqueous studies, is the concept of ligand–metal complementarity because variation of the steric and electronic properties of ancillary ligands is key to enabling and developing new uranium structural motifs, reactivity, and physicochemical properties.
Figure 1.
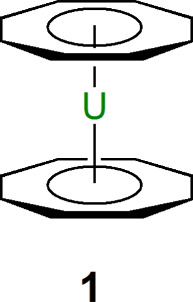
Reflecting the aforementioned motivation to understand how transition-metal-like uranium is and given an appreciation of uranium’s similarities to molybdenum and tungsten—and hence the likely ability of the former to engage in equivalent bonding motifs to the latter pair—around 1988 there was “that slide” on Molecules Organoactinide Chemists Dream About(5) presented by Sattelberger at the Third Chemical Congress of North America (including the 195th American Chemical Society National Meeting) in Toronto that year, an adapted version of which is illustrated in Figure 2.6 The slide has since assumed a somewhat legendary status in actinide “folk lore” because it was presented in a conference talk rather than becoming fixed in a journal publication. However, it was an important call-to-arms to the synthetic actinide community to advance the nonaqueous chemistry of uranium in terms of structural linkages that could be isolated under normal experimental conditions. It is intended that, by providing some historical context, viz., Figure 2, and its role in inspiring the progress that followed, the journey and status of the field can be more fully appreciated than by simply presenting advances in isolation.
Figure 2.

Adapted version of “that slide” on Molecules Organoactinide Chemists Dream About from the Los Alamos National Laboratory archive.5,6
It is a widely held view that the chemistry of the early actinides lags behind that of the transition metals. However, the astonishing aspect of Figure 2 is just how much was still waiting to be realized ca. 1988 compared to the d block that had undergone major advances in the 1960–1980s. Much has been accomplished in the intervening decades, and so this Viewpoint aims to provide an overview of how the principal themes of Figure 2 developed, and indeed expanded, but will make the occasional detour into motifs or notable analogues with other f elements that assist in contextualizing the area. Hence, the discussion will focus principally on advances directly related to Figure 2 and will then summarize other advances that developed in parallel. The interested reader is referred to several excellent recent reviews and books on the subject, and the cited references herein, for further detailed insight.1,2,7−21
CO and Related π–Acid Ligand Complexes
There are numerous transition-metal carbonyls; indeed, this is a fundamental class of organometallic complex, so the absence of uranium analogues for many years stood in stark contrast. When Figure 2 was presented, a structurally authenticated uranium carbonyl remained elusive. However, uranium carbonyl had been identified in matrix isolation experiments in 1975 by Sheline and Slater,22 and in 1986 spectroscopic evidence by Andersen showed that placing [U(η5-C5H4SiMe3)3] under an atmosphere of CO produced [U(η5-C5H4SiMe3)3(CO)] (2; Figure 3), but the CO coordination was reversible.23 Nevertheless, 1995 marked the first structurally authenticated uranium carbonyl, [U(η5-C5Me4H)3(CO)] (3; Figure 3),24 reported by Parry, Carmona, and Hursthouse. Since then, only a few uranium carbonyl complexes have been reported (Figure 3): [U(η5-C5Me5)3(CO)] (4) by Evans in 2003;25 [{U(tacn[CH2C6H2-2-O-3,5-tBu2]3)}2(μ-CO)] (5) by Meyer in 2005;26 [U{η8-C8H6-1,4-(SiiPr3)2}(η5-C5Me5)(CO)] (6) by Cloke in 2008;27 [U(η5-C5Me5)(As2Mes2)(CO)] (7; Mes = 2,4,6-trimethylphenyl) by Walensky in 2021;28 [U(η5-C5Me5)2(O-2,6-tBu2-4-Me-C6H2)(CO)] (8) by Walensky in 2023.29 Evidently, U–CO bonds are not as strong as d-block metal–CO bonds and are hence more difficult to stabilize and isolate.
Figure 3.
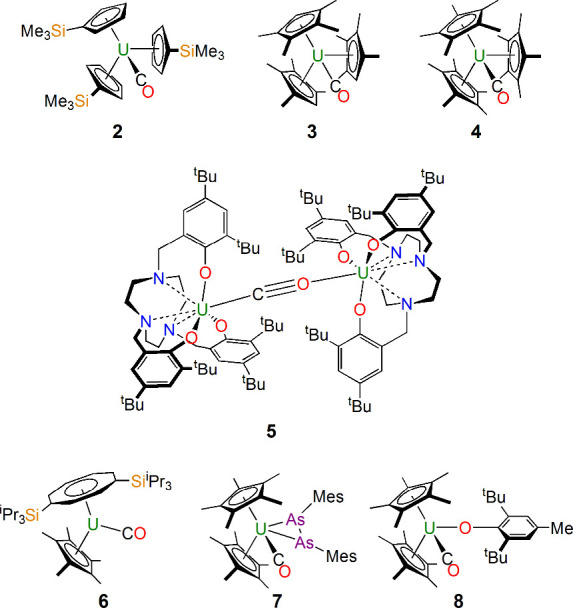
Interestingly, the IR spectra of 2–6 reveal that while the CO stretching frequencies are in the range 1880–1976 cm–1, indicating back-bonding into the CO π* orbitals, individual CO stretching frequencies do not correlate with their corresponding Cp–U distances but instead vary with the Cp substituents. In 2009, Eisenstein rationalized this on the basis of U–CO back-bonding from Cp–U bonding molecular orbitals of mainly Cp-ligand character.30 Thus, in contrast to the conventional metal-to-ligand back-bonding model for transition-metal π-acid complexes, the back-bonding in tris(cyclopentadienyl)uranium complexes has been classed as ligand-to-ligand back-bonding. Weak ligand-to-ligand back-bonding was also found by Evans and Furche for the cationic thorium complex [Th(η5-C5Me5)3(CO)][BPh4] reported in 2017,31 which, formally, as a 5f06d0 metal has no metal-based electrons with which to back-bond. Complex 7 was found to engage in Th–As σ to CO π* back-bonding, and hence that system also engages in ligand-to-ligand back-bonding to stabilize the U–CO linkage.28 However, quantum-chemical calculations on 8 suggested that the U–CO back-bonding is from a U 5f/6d hybrid orbital29 and hence of metal-to-ligand back-bonding character. The exciting implication is that uranium can switch between ligand-to-ligand and metal-to-ligand back-bonding modes as a function of the ancillary ligands because the only difference between 4 and 8 is the replacement of one pentamethylcyclopentadienyl ligand with an aryloxide. This touches on the variable, responsive bonding nature of uranium, vide supra, exemplified by the parallel notion that uranium tends to π-bond to small ligands with mainly 5f character but often bonds to more expansive ligands through δ-bonding with increasing 6d character.32
Complexes 1–8 set the scene for reductive homologation of CO at uranium (Figure 4), which contrasts to the more traditional 1,1-migratory insertion chemistry of CO at transition-metal centers. In 2006, Cloke reported the remarkable cyclotrimerization of CO using [U{η8-C8H6-1,4-(SiiPr3)2}(η5-C5Me5)],33 a structurally more sterically demanding analogue of [U(η8-C8H8)(η5-C5Me5)] reported in 1993 by Burns,34 to produce the deltate complex [U{η8-C8H6-1,4-(SiiPr3)2}(η5-C5Me5)]2(μ-η1:η2-C3O3) (9) and then through variation of the Cp substituents or reaction conditions could isolate the cyclotetramerized squarate and dimerized ethynediolate forms of CO in [U{η8-C8H6-1,4-(SiiPr3)2}(η5-C5Me4H)]2(μ-η2:η2-C4O4) (10)35 and [U{η8-C8H6-1,4-(SiiPr3)2}(η5-C5Me5)]2(μ-η1:η1-C2O2) (11),27 in 2006 and 2008, respectively. The formation of ethynediolate at uranium was also accomplished by P. Arnold in 2011 and Liddle in 2012 in [U{N(SiMe3)2}3]2(μ-η1:η1-C2O2) (12)36 and [U(TrenDMBS)]2(μ-η1:η1-C2O2) (13; TrenDMBS = {N(CH2CH2NSiMe2tBu)3}3–),37 respectively. A synthetic cycle could be closed for the latter where a substituted furanone was liberated,37 hinting at a possible catalytic process where uranium meditates the conversion of CO and silyl iodides into a functionalized furnanone. More recently, in 2023 Walensky demonstrated that [U(η5-C5Me5)2(O-2,4,6-Me3-4-Me-C6H2)] also reacts with CO to make the ethynediolate complex [U(η5-C5Me5)2(O-2,4,6-Me3-4-Me-C6H2)]2(μ-η1:η1-C2O2) (14),29 from which a range of complexes featuring further C–C bond-functionalized products could be accessed. A particularly notable result in this arena was the finding by Cloke in 2011 that the complex [U{η8-C8H6-1,4-(SiiPr3)2}(η5-C5Me5)] reacts with CO and H2 to form the methoxide complex [U{η8-C8H6-1,4-(SiiPr3)2}(η5-C5Me5)(OCH3)] (15).38 The methoxide in 15 could be released as a methanol equivalent in Me3SiOMe to, in principle, close a synthetic cycle, and this essentially corresponds to a selective molecular version of Fischer–Tropsch chemistry. Overall, complexes 9–15 demonstrate the highly reducing power of low-valent uranium, but thus far this has not gone beyond closed synthetic cycles to true catalysis. This likely reflects unbalanced cycles when factoring in returning uranium to the initial reactive trivalent state.
Figure 4.
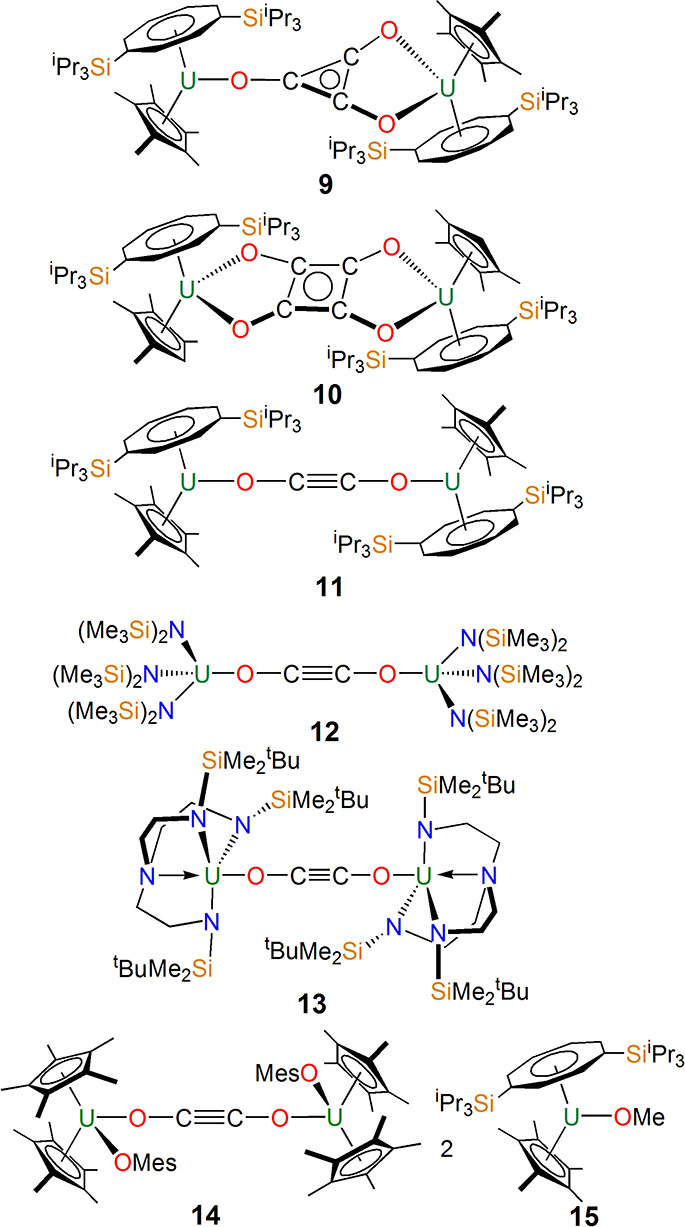
Reductively homologized CO complexes of uranium 9–15.29,33,35−38
In parallel to uranium–CO chemistry has been the development of uranium–CO2 chemistry. In contrast to the classical 1,2-migratory insertion chemistry of CO2, uranium–CO2 chemistry took a different turn when Meyer reported the synthesis of the terminal uranium–CO2 radical-anion adduct [U{tacn(CH2C6H2-2-O-3-Ad-5-tBu)3}(η1-OCO)] (16; Figure 5) in 2004.39 No further reactivity has been reported for that complex, likely because the very steric profile required to stabilize the U–CO2 linkage inhibits subsequent reactivity. However, it presented a basis for subsequent studies by Meyer and Mazzanti reporting reductive CO2-to-carbonate reactivities including closed synthetic cycles and heteroleptic heavy carbonate analogues.40−42
Figure 5.
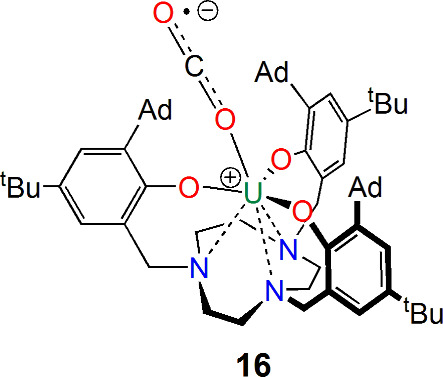
End-on bound uranium–CO2 complex 16.39
Closely related to CO is isoelectronic (NO)+, which has an extensive array of coordination chemistry with transition metals. In 1989, Bursten predicted that a [U(η5-C5H5)3(NO)] complex would curiously feature a linear U–N–O linkage that could be rationalized as a combination of uranium(IV) Cp3U+ and not (NO)+ but (NO)− fragments, with a further notable prediction of that complex being diamagnetic.43 However, experimental validation of those predictions would take 23 years to emerge. In 2012, Evans, Furche, and Long reported the synthesis of [U(η5-C5Me4H)3(NO)] (17),44Figure 6, and it was found to have an essentially linear U–N–O bond angle. Furthermore, quantum-chemical calculations44 revealed that the ground state is a diamagnetic singlet, which can be represented as (C5Me4H)3U≡N+—O–, with a low-lying triplet state corresponding to the UIV/(NO)− structure (C5Me4H)3U—N=O, which nicely accounted for the experimentally determined temperature-independent paramagnetism of 17. Complex 17 remains the sole example of a uranium nitrosyl complex to date.
Figure 6.
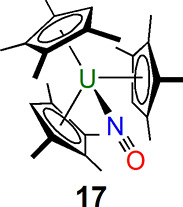
Uranium–NO complex 17.44
With U–CO and U–NO complexes structurally verified and predicted, respectively, by the mid-1990s, attention focused on the essential isoelectronic diatomic N2. In 1998, 3 years after 3, Scott reported the first actinide–N2 complex [U(TrenDMBS)]2(μ-η2:η2-N2) (18; Figure 7).45 The side-on bridging coordination of N2 in that complex was reversible, which led to the initial belief that the uranium ions were trivalent, but it now recognized that N2 is reduced to its dianionic form by back-bonding into a π* orbital of N2 but reversibly so.
Figure 7.

Building on 18(45) and recognizing the relevance of uranium–N2 complexes to Haber–Bosch fixation of N2,46 in the intervening years to the present day, a range of uranium–N2 complexes have been isolated, with most adopting side-on (μ-η2:η2-N2) binding modes that are not reversible.47 However, a few of the more unusual end-on or labile side-on-bound derivatives have been reported (Figure 7), including the end-on bridging heterobimetallic complex [{R(R′)N}3Mo(μ-η1:η1-N2)U{N(tBu)Xy}3] (19, R = tBu, R′ = Ph; 20, R = adamantyl, R′ = Xy, where Xy = 3,5-Me2C6H3) reported by Cummins in 1998,48 [{U(η8-C8H4[SiiPr3-1,4]2)(η5-C5Me5)}2(μ-η2:η2-N2)] (21) by Cloke in 2002,49 the terminal end-on N2 complex [U(η5-C5Me5)3(η1-N2)] (22) reported by Evans in 2003,50 and the end-on bridging complex [(BIPMTMS)U(NAd)2(μ-η1:η1-N2)Li(2.2.2-crypt)] (23; BIPMTMS = {C(PPh2NSiMe3)2}2–) reported by Liddle in 2019.51 Complexes 21 and 22 are notable for the facile reversibility of N2 coordination, whereas 23 features a high-oxidation-state complex that goes against traditional the donor–acceptor requirements of low-oxidation-state, electron-rich metals.
Other notable achievements in this area (Figure 7) include the splitting of N2 into a bis(nitride) in the complex [K(DME)4][{K(DME)(Et8-calix[4]tetrapyrrole)U}2(μ-NK)2] (24) by Gambarotta in 2002,52 hydrogenation to afford ammonia by [{U(OSi[OtBu]3)}2(μ-N)(μ-η2:η2-N2)K3] (25) by Mazzanti in 2017,53 and recently the formation of N23– at uranium in [K(L)n][{U(OC6H2-2,4,6-tBu3)3}2(μ-η2:η2-N2)] (26, L = 2.2.2-cryptand, n = 1; 27, L = THF, n = 6) and subsequent N–N cleavage to afford polynitrides by Mazzanti in 2023.54 Collectively, these advances highlight the ability of uranium to activate N2, confirming the observation that uranium is a highly effective promoter for the formation of NH3 from N2 and H2, as stated in the original Haber–Bosch patent from over a century ago.46
Alkylidenes, Carbynes, and Carbidos
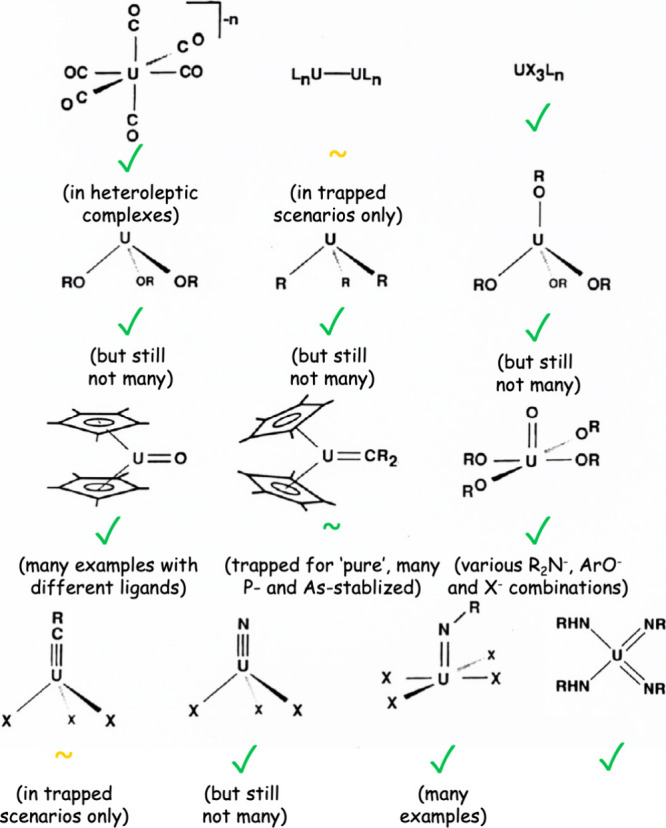
Because the M = CR2 (R = H, alkyl, silyl) motif is a fundamental structural class in transition-metal chemistry, there has long been an interest in realizing uranium alkylidenes. However, outside of matrix isolation—where species such as H2C=U(X)(Y) (X, Y = F, Cl, Br, I), H2C=U(H)X (X = F, Cl, Br), and H2C=UH2 have been reported by Andrews and Li in the period 2006–200855−58—it is a target that has remained elusive in “pure” M = CR2 (R = H, alkyl, silyl) form outside of matrix isolation experiments and so is one of the targets in Figure 2 that remains unmet to this day in isolable molecules made under normal conditions.
In 1981, 7 years before Figure 2, Gilje reported the first U=C double bond in [U(η5-C5H5)3(CHPMe2Ph)] (28) by utilizing a phosphonioalkylidene ligand (Figure 8).59 The complex undoubtedly contains a U=C multiple bond, albeit polarized, but two competing resonance forms can be drawn [U–C(H)=PMe3Ph and U–=C(H)-P+Me2Ph] due to phosphonium-substituent stabilization, which renders the double bond not as clear-cut as that in a “pure” alkylidene. However, a range of reactivity studies were all consistent with UC double-bond character.12
Figure 8.

The area then became dormant for the best part of three decades before Ephritikhine, Mézailles, and Le Floch revived it in 2009 with the synthesis of U=C double bonds using the diphosphoniomethanediide {C(PPh2S)2}2– (Figure 8), as exemplified by the uranium(IV) complex [U{C(PPh2S)2}(BH4)2(THF)2] (29),60 and then in 2011 the uranyl complex [U(O)2{C(PPh2S)2}(py)2] (30),61 a rare example of a uranyl organometallic. In parallel, with the related diphosphoniomethanediide BIPMTMS (Figure 8), in 2011 and 2012 Liddle reported the uranium(IV), -(V), and -(VI) complexes [U(BIPMTMS)(Cl)3Li(THF)2] (31),62,63 [U(BIPMTMS)(Cl)2(I)] (32),63 and [U(BIPMTMS)(O)(Cl)2] (33),64 respectively, allowing comparisons of the U=C bond over three oxidation states of uranium, with the majority of the ligand field conserved. This series was then completed by Liddle and Vlaisavljevich in 2018 with the synthesis of [{U(BIPMTMS)}6(μ-I)3(μ-η6:η6-C7H8)3] (34), which formally contains uranium(III) U=C double bonds65 (Figure 8). In 2014, Liddle reported the uranium(VI) derivatives [U(BIPMTMS)(O)(NMes)(dmap)2] [35; dmap = 4-(dimethylamino)pyridine]66 and [U(BIPMTMS)(O)2(dmap)2] (36),66 providing complexes with up to three different multiply bonded ligands at uranium and another rare example of an organouranyl complex (Figure 8). Further prominent examples from the period 2013–2020 of uranium phosphonioalkylidenes (Figure 8) include [U(CHPPh3){N(SiMe3)2}3] (37) by Hayton and Walensky,67 [U(CHPPh3)(η5-C5Me5)2(X)] (38–40; X = Cl, Br, I) by Walensky and Maron,68 and [U(CHPPh3)(TrenTIPS)] (41; TrenTIPS = {N(CH2CH2NSiiPr3)3}3–) by Liddle.69 The latter provided impetus to prepare the arsonioalkylidene analogue [U(CHAsPh3)(TrenTIPS)] (42),69 which was the first arsonioalkylidene complex of any metal and which displays a more well-developed U=C double bond compared to the phosphonioalkylidene analogue, consistent with diminished As versus P stabilization of the alkylidene center. The assertion of the presence of U=C double bonds in these complexes has proven controversial at times, but the weight of reactivity and computational analysis combined with a 13C NMR chemical shift anisotropy study in 2024 supporting the Ce=C double-bond formulations in related Ce(IV) complexes70 all point to these complexes possessing polarized U=C double bonds.
The years 2018 and then 2021 marked two milestones in U=C double-bond chemistry (Figure 8) with reports of the phosphinosilylalkylidene complexes exemplified by [U{C(PPh2)SiMe3}(BIPMTMS)(dmap)2] (43) by Liddle71 and the allenylidene complex [Li(2.2.2-cryptand)][U(CCCPh2){N(SiMe3)2}3] (44) by Hayton and Autschbach,72 respectively. Both complexes are notable for exhibiting U=C double-bond interactions that depart from the use of pentavalent pnictonium alkylidene stabilization.
Compared to alkylidenes, the corresponding chemistry of uranium carbyne and carbido complexes is sparsely developed. Matrix isolation studies have led the way, with reports of fundamental, elegant species such as CUO, CUO–, UC, CUC, UCH, U(CC)2, X3U≡CH (X = F, Cl, Br), F2ClU≡CH, and F3U≡CF first being reported around the years 1999–2012 by Andrews, Bursten, and Li.73−78 More recently, in recent years (2019–2023), work led by Chen has exploited the unique confinement effects of endohedral fullerenes to isolate a range of carbide compounds, including U(μ-η1:η1-C)U@C80,79 U(μ-η2:η2-C2)U@C78,80 U(μ-η2:η2-C2)U@C80,80 U(μ3-η1:η1-C)Sc2@C80,81 U(μ-η1:η1-C)Ce@C72,82 and U(μ-η1:η1-C)Ce@C80.82 Akin to the eventually successful quest for terminal nitrides in isolable molecular species (see below), the prevalence of these species in confined trapping scenarios suggests that, with suitable ancillary ligands, isolable terminal molecular uranium alkylidenes, carbynes, and carbidos under normal experimental conditions should eventually be secured.
Imidos and Terminal Nitride Complexes
By the time the contents of Figure 2 emerged as a presentation slide, uranium mono(imido) complexes had already been realized, and some subsequent key complexes are illustrated in Figure 9.15 Initially, in 1984 Gilje isolated the uranium(IV) imido complex [U(η5-C5H5)3{NC(Me)C(H)PMePh2}] (45) from the insertion of CH3CN into the U=C bond of the PMePh2 analogue of 28, although this complex is not a “pure” imido linkage.83 Soon after, in 1985 Andersen reported two-electron oxidation of [U(η5-C5H4Me)3(THF)] by azides to produce the first clear-cut uranium(V) imidos [U(η5-C5H4Me)3(NPh)] (46) and [U(η5-C5H4Me)3(NSiMe3)] (47),84 and the same approach with [U{N(SiMe3)2}3] yielded [U{N(SiMe3)2}3(NPh)] (48) and [U{N(SiMe3)2}3(NSiMe3)] (49) in 1988.85 Apart from accessing imido functionalities, these were important reactions because they developed two-electron-oxidation chemistry, in contrast to the reputation that the f block has for one-electron-redox couples. In 1990 Sattelberger reported that 48 and 49 could be oxidized to produce the uranium(VI) imido complexes [U{N(SiMe3)2}3(NPh)(F)] (50) and [U{N(SiMe3)2}3(NSiMe3)(F)] (51),86 which were the first uranium(VI) complexes to have multiple bonds to nitrogen.
Figure 9.
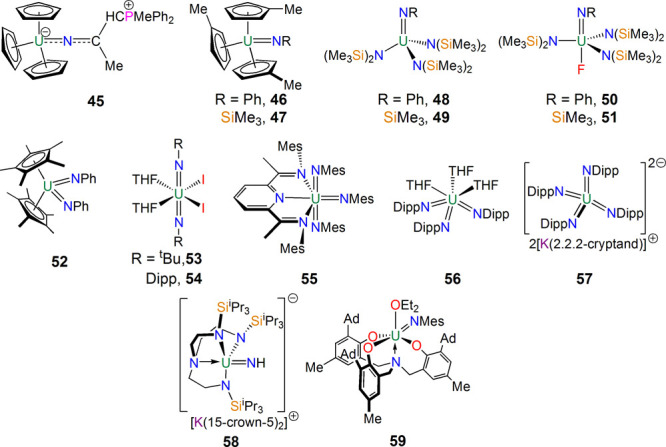
With mono(imido) uranium complexes established, attention turned to polyimidos, and relatively quickly in 1992 Burns showed that the treatment of [U(η5-C5Me5)2(Cl)(Me)] with LiN(H)Ph and Me2NCH2CH2NMe2(tmeda) (tmeda = tetramethylethylenediamine) afforded [U(η5-C5Me5)2(μ-NPh)(μ-Cl)Li(tmeda)], which was oxidized by N3Ph to afford the first uranium bis(imido) complex [U(η5-C5Me5)2(NPh)2] (52),87 which was also the first organouranium(VI) complex. Notably, due to the presence of the two Cp* rings, the N–U–N linkage is bent [98.7(4)°], raising interesting questions about its relationship to uranyl and, in particular, the still yet to be routinely isolated cis-uranyl. As an aside, noting that 52 was prepared by a two-electron oxidation, in 1993 Burns also found that the oxidation of [U(η5-C5Me5)2(ODipp)] (Dipp = 2,6-diisopropylphenyl) and [U(η5-C5Me5)2(NDipp)] with pyridine N-oxide afforded the first uranium(V) and -(VI) complexes to contain mono(oxo) linkages, namely, [U(η5-C5Me5)2(O)(ODipp)] and [U(η5-C5Me5)2(O)(NDipp)].88 Again, this demonstrated that the uranium(III/V) two-electron-redox couple is a powerful vehicle for installing multiply bonded ligands at uranium.
Complex 52 remained the only class of uranium bis(imido) complexes for 13 years (the N-adamantyl version of 52 was reported in 1998)89 until in 2005–2006 Boncella reported the synthesis of linear uranium bis(imido)uranyl analogues.90,91 Oxidation of uranium metal or [U(I)3(THF)4] with I2 in the presence of amines produced alkyl and arylbis(imido) complexes of the form [U(NR)2(I)2(THF)2] (53, R = tBu; 54, R = Dipp) with the elimination of ammonium iodide salts. The linear formulation of these bis(imido) complexes suggests that an inverse trans influence operates as it does in isoelectronic uranyl. A tris(imido)uranium complex, isoelectronic to UO3, was introduced by Bart in 2014.92 The complex mer-[U{C5H3N-2,6-(C[Me]NMes)2}(NMes)3] (55) was obtained by the reaction of a highly reduced, i.e., noninnocent, pyridylbis(imino)uranium complex with MesN3, where the U(NMe)3 component is T-shaped. This was followed soon after in 2015 by another tris(imido) by Bart in a reaction that is elegant by virtue of its simplicity, where the reduction of [U(I)3(THF)4] by KC8 in the presence of DippN3 produced fac-[U(NDipp)3(THF)3] (56).93
Remarkably, in 2017 Bart reported that the polyimido motif could be extended to a range of tetrakis(imido)uranate(VI) complexes exemplified by [K(2.2.2-crypt)]2[U(NDipp)4] (57).94 Quantum-chemical calculations showed that the significant amount of charge loading resulted in more activated U=NR bonds than in tris(imido) and bis(imido) analogues. It will be interesting to see if a pentakis(imido)uranium complex can be realized, given the range and number of vacant valence orbitals that uranium possesses.
There are now many uranium imido complexes, but two merit specific mention. The first is the parent imido complex [K(15-crown-5)2][U(NH)(TrenTIPS)] (58) reported by Liddle in 2014.95 Complex 58 is stable despite lacking any sterically demanding substituent protection at the imido, although the anion formulation of the imido component of 58 evidently plays a role because oxidation of 58 results in disproportionation. The imido complex [U{N(CH2C6H2-2-O-3-Ad-5-Me)3}NMes] (59) was reported by Meyer in 2012.96 Notably, the imido resides trans to one of the aryloxides, where it would be more intuitive to predict the imido residing in the axial site trans to the tertiary amine. This implies the presence of an inverse trans influence in 59.
The search for terminal uranium nitrides can trace its origins back to 1976, when Green and Reedy identified UN in a frozen argon matrix.97 Then, in the period 1993–2016, fundamental species such as NUN, NUO, NUO+, F3UN, NUN-H, and U2N2 were variously reported or studied in matrix isolation or as spectroscopic transients by Andrews, Bursten, Gagliardi, Pyykkö, Roos, Schwarz, and Vlaisavljevich,98−102 and UN was reported in the C82 endohedral fullerene by Chen and Autschbach in 2022.103 Nevertheless, when Figure 2 was making its debut in 1988, placing an emphasis on a molecular terminal uranium nitride as a key synthetic target and bonding benchmark, there were no molecular uranium nitrides at all.
Several polymetallic nitrides of uranium were reported in the 2000s21,104 before (Figure 10) Cummins reported the borane-capped nitride complexes [NBun4][U{NB(C6F5)3}{N(tBu)C6H3-3,5-Me2}3] (60) and [U{NB(C6F5)3}{N(tBu)C6H3-3,5-Me2}3] (61) in 2009.105 Complexes 60 and 61 can alternatively be formulated as imidoborates, but computational analysis reveals significant U≡N triple bonds. In 2010 Kiplinger provided evidence of a transient terminal uranium nitride through isolation of the C–H activated complex [U(η5-C5Me4CH2NH)(η5-C5Me5){N(SiMe3)2}] (62) resulting from photolysis of the azide precursor [U(η5-C5Me5)2(N3){N(SiMe3)2}].106
Figure 10.
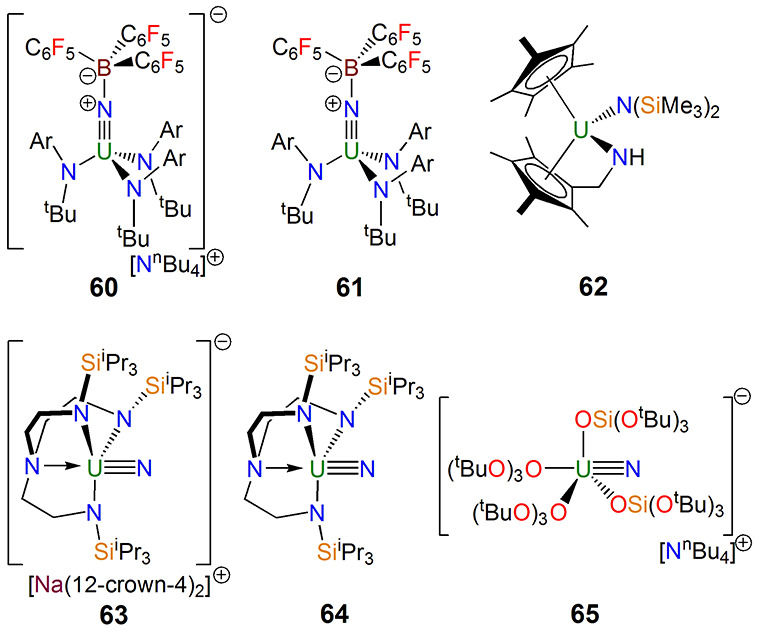
The terminal uranium nitride was finally reported in 2012 by Liddle in the uranium(V) nitride complex [Na(12-crown-4)2][U(N)(TrenTIPS)] (63),107Figure 10, prepared by [U(TrenTIPS)]-mediated two-electron azide reduction and subsequent sodium sequestration with 12-crown-4 ether. Success hinged on TrenTIPS providing exactly the right size and shape pocket for the nitride, combined with azide activation but stabilization by the sodium cation and then its gentle subsequent removal. In 2013, the uranium(VI) nitride [U(N)(TrenTIPS)] (64) was prepared by oxidation of 63(108) (Figure 10), concluding the search for terminal uranium(VI) nitrides previously restricted to spectroscopic experiments as well as confirming the presence of intermediate nitrides in C–H activation such as 62–64. A range of derivatives of 63 proved to be fertile ground for detailed electronic structure investigations.109 Complex 64 was computationally predicted108 and experimentally confirmed by 15N NMR spectroscopy110 to contain a highly covalent U≡N triple bond, and more so than Group 6 terminal nitrides, which is an astonishing result that goes to the heart of one of the original motivations behind Figure 2 to elucidate the bonding relationship of uranium to Group 6 elements like molybdenum and tungsten. Only one other class of terminal uranium nitride has since been reported, where photolysis of [NBun4][U(N3){OSi(OtBu)3}4] was reported to produce [NBun4][U(N){OSi(OtBu)3}4] (65) by Mazzanti in 2020 (Figure 10).111
This area has now expanded to include many examples of astonishing small-molecule activations and structural motifs,21,104 with notable examples including hydrogenation of 25 to produce ammonia by Mazzanti in 201753,112 and elegant preparations from UX5 (X = Cl, Br) and NH3 of bis(nitride) complexes containing the cations [(H3N)8UNUN(NH3)5U(NH3)8]8+, [(H3N)8UNUN(NH3)4(Br)U(NH3)8]7+, and [(H3N)8UNUN(NH3)3(Cl)2U(NH3)8]6+ reported by Kraus in 2020.113
Homoleptic Polyalkyl, -alkoxides, and -aryloxides
As a fundamental ligand type in organometallic chemistry, there has always been interest since the 1940s in uranium alkyl complexes particularly because at one stage volatile uranium alkyls were candidates for isotope enrichment work in the Manhattan Project.114 In the 1980s, Marks pioneered the study of heteroleptic uranium bis(cyclopentadienyl)alkyls, having reported in 1974 that attempts to prepare tetrakis(alkyl) compounds resulted in decomposition.115 Likewise, in 1982 Evans concluded that hydride species formed,116 although in 1984 Andersen subsequently found that tetrakis(alkyl) complexes could be stabilized as heteroleptic derivatives by the addition of chelating diphosphine ligands to saturate the coordination sphere of uranium, for example, in [U(CH2Ph)3(Me)(Me2PCH2CH2PMe2)] (66).117 Thus, Figure 2 focused attention on homoleptic polyalkyl complexes of uranium.
As it turned out, a homoleptic polyalkyl was delivered rapidly (Figure 11), and in 1988 Sattelberger reported the first example of a neutral homoleptic uranium alkyl with the synthesis of the tris(alkyl) complex [U{CH(SiMe3)2}3] (67).118 Like lanthanide analogues, 67 had to be prepared by the reaction of LiCH(SiMe3)2 with a uranium tris(aryloxide) because the more conventional route of reacting UCl3(THF)n resulted in formation of the “ate” complex [U{CH(SiMe3)2}3(Cl)Li(THF)3].118 Complex 67 is isolable because of the sterically demanding alkyls, but it is not coordinatively saturated, so it decomposes in solution, underscoring the inherent reactivity of uranium alkyls.
Figure 11.
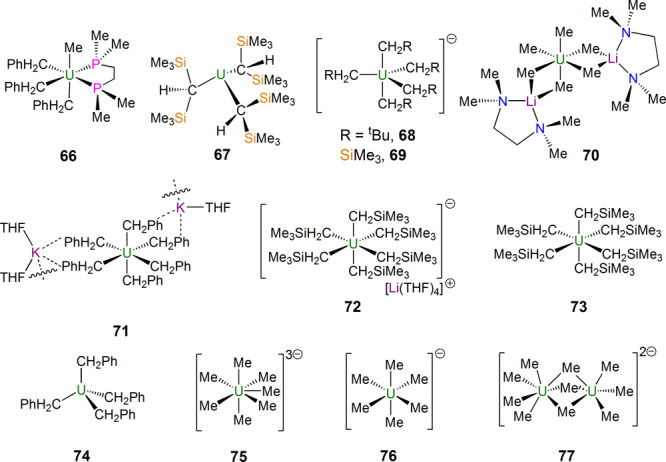
Homoleptic uranium alkyl complexes 66–77. Only the anionic components of 68, 69, and 75–77 are shown for clarity.117−123
The year 2009 marked a fresh impetus in the area (Figure 11) when Hayton reported the synthesis of several homoleptic uranium(IV) complexes, specifically separated ion-pair “ate” complexes of the anions [U(CH2But)5]− (68) and [U(CH2SiMe3)5]− (69) and contact ion triple assemblies of [U(Me)2(μ-Me)4{μ-Li(tmeda)}2] (70) and {[K(THF)][K(THF)2][U(CH2Ph)6]}∞ (71).119 Shortly after, in 2011 Hayton went on to report [Li(THF)4][U(CH2SiMe3)6] (72) and its oxidation to the remarkable hexakis(alkyl) [U(CH2SiMe3)6] (73),120 although the latter was found to be thermally unstable and decompose above −25 °C. Soon after 72 and 73, in 2012 Bart reported the synthesis and isolation of [U(η2-CH2Ph)4] (74),121 where the η2-coordination mode of the four benzyls evidently contributes to the stability of this tetrakis(alkyl) complex, and this led to a wide range of [U(η2-CH2R)4] (R = substituted aryls) complexes being reported by Bart in 2015.122
As mentioned above, homoleptic polyalkyl complexes of uranium often undergo facile decomposition and can be thermally unstable. This prompted Neidig to undertake low-temperature studies (Figure 11), where compounds were prepared and crystallized at −70 to −80 °C. The resulting range of compounds reported in 2020 underscored the complexity of uranium polyalkyl chemistry because [Li(THF)4][U(Me)4(μ-Me)2{μ-Li(THF)2}], [U(Me)(μ-Me)6{μ-Li(THF)2}{μ3-Li(THF)}(μ3-Li)] (75), [Li(18-crown-6(THF)2][U(Me)6] (76), and [Li(THF)4]2[Me4U(μ-Me)3UMe3] (77), built around hexakis- or septakis(methyl) motifs, could all be isolated under those conditions.123
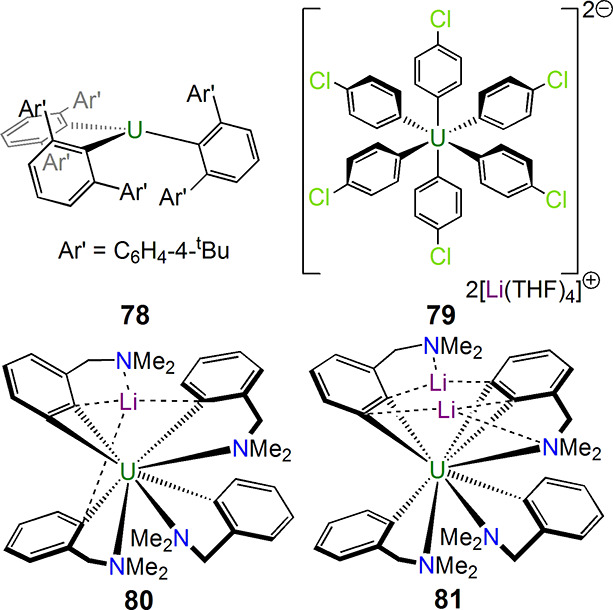
The above activity in homoleptic polyalkyluranium chemistry has spurred renewed interest in related homoleptic polyaryluranium chemistry (Figure 12), with notable examples including the uranium(III) tris(terphenyl) complex [U{C6H3-2,6-(C6H4-4-tBu)2}3] (78) by J. Arnold in 2016124 and uranium(IV) hexakis(aryls) exemplified by [Li(THF)4][U(C6H5)6Li(THF)] and [Li(THF)4]2[U(C6H4-4-Cl)6] (79) by Neidig in 2019;125 like Neidig’s alkyl work, the latter pair of aryls were synthesized and crystallized at low (−80 °C) temperature. Last, Hayton isolated exceedingly rare examples of uranium benzyne complexes, namely, [U(η2-C6H3-2-CH2NMe2)(C6H4-2-CH2NMe2)3Li] (80) in 2013126 and [U(η2-C6H3-2-CH2NMe2)2(C6H4-2-CH2NMe2)2Li2] (81) and the THF-solvate congener [U(η2-C6H3-2-CH2NMe2)2(C6H4-2-CH2NMe2)2[Li(THF)2}(Li)] in 2016.127
Figure 12.
Although uranium alkoxides had been known since the 1950s, rather than being straightforward homoleptic formulations, they were often polymetallic aggregates with “ate” character, mixed uranium oxidation states, or were constructed around oxide dianions (Figure 13). Prominent examples include [U2(μ-OtBu)3(μ3-OtBu)2(OtBu)4K] (82), [U2(μ-OtBu)3(OtBu)6] (83), and [U3(μ3-O)(μ3-OtBu)(μ-OtBu)3(OtBu)6] (84) reported in 1984 by Cotton,128−130 and even the “pure” homoleptic [U2(μ-OtBu)2(OtBu)8] (85) reported by Eller in 1983 is dimeric.131 Furthermore, aryloxides were relatively scarce, and so Figure 2 sought to prompt an expansion of mononuclear homoleptic polyalkoxides and -aryloxides.
Figure 13.
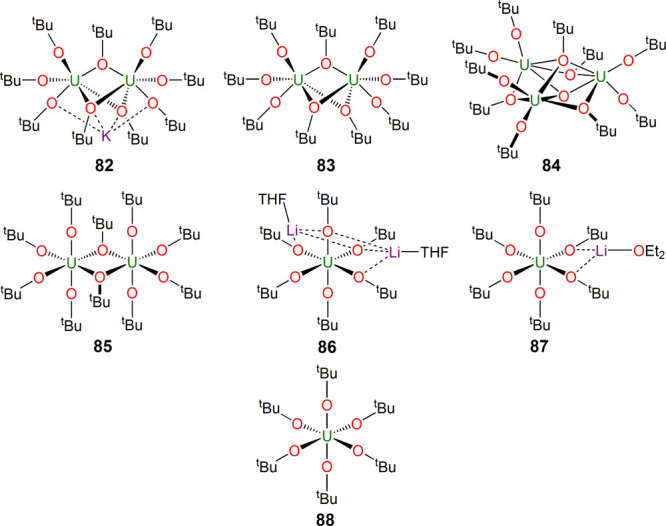
Some of the basic uranium alkoxide chemistry was reinvestigated in 2008 by Hayton,132 who found that the tendency of alkoxides to form “ate” complexes could be synthetically exploited. Hence, the preparation of [U(OtBu)2(μ-OtBu)4{(μ3-Li(THF)}2] (86) was performed, and then stepwise oxidations with iodine first secured [U(OtBu)4(μ-OtBu)2{μ-Li(OEt2)}] (87) and then [U(OtBu)6] (86); it is notable that this chemistry works when utilizing lithium to stabilize the aggregates rather than potassium, which tends to produce clusters such as 82.128 Electrochemical studies suggested significant stabilization of the uranium(VI) ion in 88 compared to the uranium(VI) hexakis(halide) series, which are generally considered to be quite oxidizing.
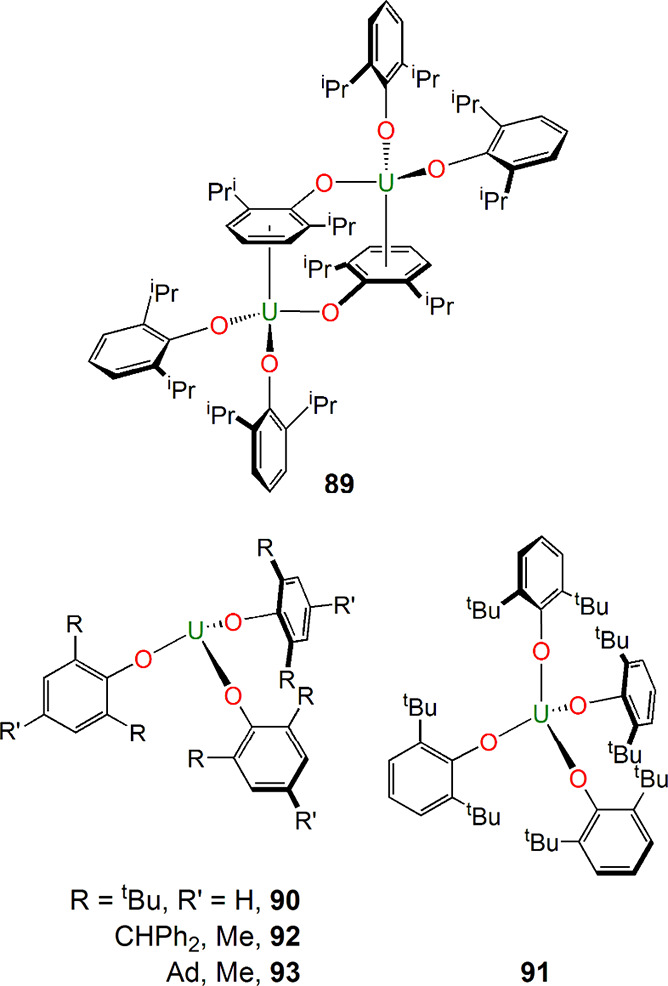
Where aryloxides are concerned, there are still relatively few homoleptic variants (Figure 14), with reports by Sattelberger in 1988 of dimeric [{U(μ-η1:η6-ODipp)(ODipp)2}2] (89)133 and the monomers [U(O-C6H3-2,6-tBu2)3] (90, suggested to be monomeric from IR data in the initial report133 but only structurally confirmed as such in 2011 by P. Arnold134) and [U(O-C6H3-2,6-tBu2)4] (91).135,136 The more sterically demanding [U{OC6H2[2,6-CHPh2]2-4-Me}3] (92) reported by Meyer and Mindiola137 and [U(OC6H2-2,6-Ad2-4-Me)3] (93) disclosed by Meyer appeared in 2013 and 2016, respectively.138 It is worth noting that some homoleptic uranium aryloxides exist but have not been structurally authenticated; however, they have been used to make N22–, N23–, and CO-coupled ethynediolate derivatives.54,134
Figure 14.
U–U Bonds
Given the prevalence of Mo–Mo and W–W bonding in transition-metal chemistry, the absence of U–U bonds led to the latter being a natural target in Figure 2 in 1988. This was not for a lack of attempts to prepare U–U bonds by 1988, where one study by Cotton in 1984130 investigating the possibility of accessing U–U bonding supported by alkoxides, given the tendency of alkoxides to support Mo–Mo and W–W bonding, stated that, “While we are not suggesting that on the basis of these two structural results all hope of observing U–U bonds is futile, we do feel that such hopes are rather dim.” Indeed, in 2006 energy decomposition analysis calculations carried out on hypothetical U–U bonds in classical [U2X8]2– (X = Cl, Br) dianions by Kaltsoyannis139 consistently found weak metal–metal bonds. Hence, this suggested that U–U bonds, at least in the [U2X8]2– formulation, would be unlikely to be formed or be isolable experimentally, in contrast to the large range of heterobimetallic uranium–metal bonds that have been reported.20 However, like terminal uranium nitrides, the quest for isolable U–U bonds under normal experimental conditions has been stoked by advances in spectroscopic and trapped-species scenarios.
The U2 and OUUO dimers were observed as spectroscopic transients as long ago as 1974 by Khodeev,140 and in a theoretical study of actinide dimers by Roos in 2006, there is mention of U2 and U2+ as spectroscopic transients from a private communication from Heaven,141 but the nature of the bonding in U2 has proven to be a challenge to definitively model due to the relativistic regime.142,143 In 1996 and 1997, Andrews showed that HUUH and H2UUH2 form in cryogenic matrix isolation experiments.144,145 It took until 2018 in a report by Chen, Feng, Echegoyen, and Poblet for U2 to be formed and isolated in U2@C80,146 although extensive disorder of the U2 unit has made analysis of the U2 unit challenging. Computational studies suggest a complicated bonding picture that is highly dependent on the U–U distance,146−148 but the consensus appears to be that two uranium(III) ions are present with an overall septet spin state but with two ferromagnetic two-center one-electron bonds that correspond to a single bond. Unfortunately, it has not been possible to verify this experimentally due to the lack of magnetic data, which likely reflects the extremely challenging nature of the synthesis and which in itself underscores the achievement of preparing U2 at all. The U–U bond in U2@C80 was described in 2015 by Straka and Foroutan-Nejad as attractive but “unwilling”,147 which was debated by Rodríguez-Fortea, Graaf, and Poblet,148 but if correct would be in line with prior work suggesting the weak nature of 5f–5f bonding.130,139
Interestingly, more recently, in 2021 Th2@Ih(7)-C80 was reported by Chen and Poblet149 and the trimer [{Th(η8-C8H8)(μ3-Cl)2}3{K(THF)2}2]∞,150 accessible under normal experimental conditions and on multigram scale, containing three-center two-electron σ-aromatic bonding,151 was reported by Liddle and Kaltsoyannis, also in 2021. These advances in thorium chemistry, together with the matrix isolation and endohedral fullerene advances with uranium, suggest that U–U bonding in a complex made under normal experimental conditions may eventually be realizable.
Topics That Developed in Parallel to “That Slide”
Figure 2 aimed to capture the spirit of high-value targets to primarily focus efforts on securing. However, of course, it could not envisage every subarea to target or predict what new lines of enquiry those primary endeavors might eventually branch out into, and indeed in many ways, that was also a motivation of Figure 2. This section will briefly summarize other key advances that have branched out in parallel.
One necessary spin-off has been the development of uranium halide starting materials, the importance of which can easily be overlooked when targeting high-value structural motifs, but, of course, the successful isolation of new compounds depends on having suitable starting materials to begin with. There are now many uranium halide starting materials, with UCl4 playing a prominent role,2,15 but perhaps the one that has had the most obvious sustained impact in terms of uplifting research outputs is that of [U(I)3(THF)4], reported in publications in 1989 and 1994 by Clark, Sattelberger, and Zwick,152,153 for example, already being mentioned above as a key starting point to accessing 53–57.90,93
Many of the linkages in Figure 2 are organometallic, and, of course, organouranium chemistry has a rich heritage spanning back to the 1940s, but definitive compounds began emerging around 1956 and onward, with examples (Figure 15) including [U(η5-C5H5)3Cl] (94) by Wilkinson in 1956,154 [U(η5-C5H5)4] (95) by Fischer in 1962155 [K(18-crown-6)][U(η7-C7H7)2] (96) in 1995 and [U(BH4)2(THF)5][{U(BH4)3}2(μ-η7:η7-C7H7)] (97) in 1994 by Ephritikhine,156,157 and the aforementioned 1 in 1968/1969.3,4 Arene complexes, for example, [U(η6-C6H6)(AlCl4)3] (98) reported by Marconi,158 started appearing in the literature around 1971 and onward, although against the backdrop of Figure 2, a notable advance was the report of the inverse-sandwich complexes [{U(N[Xy]R)2}2(μ-η6:η6-C6H5Me)] (R = Ad; 99, R = tBu, 100) in 2000 by Cummins.159 There are now numerous inverse-sandwich complexes of uranium, most of which are best regarded as diuranium(III) with arene dianions,160 although there are a few notable exceptions of diuranium(V) arene tetraanions, such as [{U(TsR}2(μ-η6:η6C6H5Me)] [TsR = {HC(SiMe2NR)3}3–; R = C6H3-3-5-Me2 (Xy), 101; R = C6H4-4-Me (Tol), 102;161Figure 15], as unequivocally confirmed by spectroscopic and magnetic studies. The synthetic credentials of 101 and 102 were confirmed by their use as precursors to the first f-element diuranium cyclobutadienyl and diphosphacyclobutadienyl complexes [{U(TsXy)}2(μ-η5:η5-C4Ph4)] (103) and [{U(TsTol)}2(μ-η4:η4-C2P2tBu2)] (104) (Figure 15) reported by Liddle in 2013.162 Continuing the small-ring theme, Walter, Ding, and Zi reported uranium metallacyclopropene complexes such as [U(η5-C5Me5)2(η2-Me3SiCCSiMe3)] (105).163 A recurring theme of 94–105 is significant 5f-orbital contributions to the bonding, including π- and δ-bonding motifs, again emphasizing once again how uranium, like transition metals, can engage in different bonding depending on the nature of the coordinated ligands.
Figure 15.
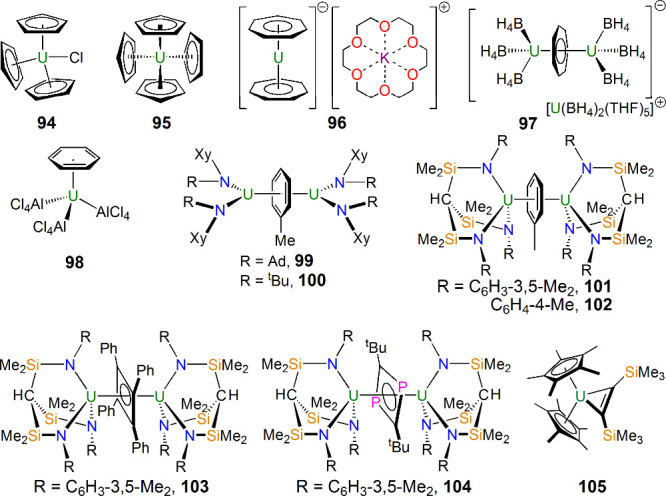
Uranium complexes 94–105 with Cn-type ligands (n = 2, 4–8).154−159,161−163
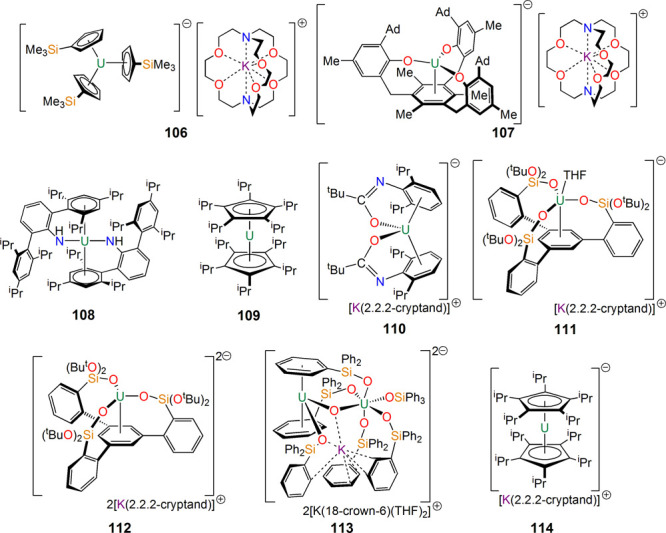
Although not directly a result of inverse sandwich arene complexes, the oxidation state ambiguity of inverse sandwich arene complexes certainly prompted thoughts of uranium complexes with oxidation states below 3+. Thus, related to inverse sandwich uranium arene complexes, the chemistry of uranium in 2+ and 1+ oxidation states was developed (Figure 16). The first isolable uranium(II) complex was [K(2.2.2-cryptand)][U(η5-C5H4SiMe3)3] (106) reported by Evans in 2013,164 and then in 2014, Meyer reported [K(2.2.2-crypt)][U{η6-C6Me3(CH2C6H2-2-O-3-Ad-5-Me)3}] (107).165 These compounds were both important in terms of formally containing uranium(II) but also because the former was found to be 5f36d1 and the latter 5f46d0. That is a clear demonstration of how the ligand field at uranium can determine the electronic ground-state structure, which is very transition-metal-like behavior. This subarea has expanded significantly, with several ligand classes supporting uranium(II), including the terphenylamide [U{N(H)C6H3-2,6-[C6H2-2,4,6-iPr3]2}2] (108) by Odom Boncella, and Shores in 2018,166 the parallel metallocene [U(η5-C5iPr5)2] (109) by Layfield in 2020,167 the amidate [K(2.2.2-cryptand)][U{OC(tBu)N-η6-Dipp}2] (110) by J. Arnold in 2021,168 and the arene-tris(siloxide) [K(2.2.2-cryptand)][U{C6H3-1,3,5-(C6H4Si[OtBu]2O)3}(THF)] (111) by Mazzanti in 2023.169 Several uranium(I) synthons have now been isolated, including 110 by J. Arnold in 2021,168 the arene-tris(siloxide) [K(2.2.2-cryptand)]2[U{C6H3-1,3,5-(C6H4Si[OtBu]2O)3}] (112) by Mazzanti in 2023,169 and [K(THF)2(18-crown-6)]2[K{(Ph3SiO)U}(μ-O)(μ-κ2:η6-Ph,O-PhSiPh2O)(μ-κ2:η4-Ph,O-PhSiPh2O){U-(Ph3SiO)3}] (113) also by Mazzanti in 2023.170 Uranium(I) has been identified in disordered [K(2.2.2-cryptand)][U(η5-C5iPr5)2] (114) by Layfield.171 These results have paralleled advances isolating thorium(III) and, remarkably, thorium(II) in molecular tris(cyclopentadienyl) complexes.172−174 This has even been extended to include neptunium(II)175 and plutonium(II),176 showing the impact that studying uranium can have on neighboring actinide elements.
Figure 16.
As indicated above, there are now many amides,15 imidos,16 nitrides,21,104 and oxos,9−12,15 so attention naturally turned to developing to accessing multiply bonded heavier group 15 and 16 derivatives of uranium by way of phosphinidene, phosphido, diphosphorus, arsinidene, arsenido, sulfido, selenido, tellurido, and Zintl cluster complexes.177−183 The result is that there is now a significant range of U=PR (R = H, aryl), U=P(R)K, U=P=U, U–P(H)–U, U(P2)U, U(P3)U, U=AsR, U=As(R)K, U=As=U, U≡AsK2, U(As2)U, U(As2H2)U, U=S, U=Se, and U=Te bonds reported with a range of supporting ligands. A selection of representative complexes reported by Burns, Liddle, Ephritikhine, Hayton, Mazzanti, Meyer, Kiplinger, and Walter can be found in Figure 17 (115–134), and the reader is directed to recent reviews177,178 and subsequent publications.179−184 Overall, the range of heavier Group 15 and 16 derivatives emphasizes how multiple bond linkages more often associated with the d block can be stabilized and isolated at uranium through appropriate synthetic approaches coupled to ligand–metal complementarity.
Figure 17.

Selected examples of heavier Group 15 and 16 multiple bonds to uranium including 115–134.177−184 Mes* = 2,4,6-tBu3C6H2. Dipp = 2,6-iPr2C6H2. Cation components of 121−130 are omitted for clarity.
In addition to all of the above fascinating chemistry, the long-known uranyl dication has continued to produce new chemistry time and time again. Although the uranyl dication is often referred to as inert, Clark showed in 1999 that, under highly alkaline conditions, oxo–ligand exchange can occur in uranyl hydroxides.185 In the years that followed, uranyl activation developed into two distinct but interrelated areas, that of pentavalent uranyl and its disproportionation chemistry, and functionalization of uranyl producing O-element bonds from the “yl” oxos, which often involved reduction and hence pentavalent uranyl-type intermediates.186−193 Through a range of silylation and borane-silylation chemistry, activation of uranyl and reduction to uranium(IV) species is now well-established, which when taken together with the facile oxo exchange by Clark renders the classical textbook description of the inert nature of the uranyl dication, except for in acidic media, somewhat in need of revision. Another textbook description of uranyl is that it is rigorously linear, but several studies have now reported uranyl O–U–O angles of ∼162–168°.66,194,195 Furthermore, cis-uranyl was proposed by Meyer in 2023 as a credible reaction intermediate,196 suggesting that with suitable trapping a cis-uranyl may be within reach, which would also contribute to a need to rewrite textbook descriptions of uranyl. Last, there is continued interest in the extraction of uranyl, with a recent highlight being redox-switchable carboranes for uranium capture and release reported by Ménard and Hayton in 2020.197,198 Again, all of these advances rely on ligand–metal complementarity to be successful.
Earlier, this Viewpoint touched on small-molecule activation and catalysis by uranium, mainly with CO, CO2, and N2, but uranium has a rich chemistry in this area with a range of small molecules and substrates,8,12,19 even, in nonaqueous media, remarkably including water splitting reported by Meyer.199,200 This is just one subarea of several novel physicochemical properties that uranium exhibits by virtue of its position in the Periodic Table, with others including studies encompassing single-molecule magnetism,201 the inverse trans influence,202 6p-orbital pushing from below,203 sterically induced reduction chemistry,204 and even noble gas adducts under matrix isolation conditions.205
With such a rich range of new molecular complexes to study and with characterization techniques and methods becoming ever more capable and widely available, there has been growing interest in probing the covalency of uranium complexes; after all, this goes to the very heart of one of the prime motivations for pursuing molecular nonaqueous uranium chemistry, and methodological advances mean that studies that would have been unimaginable in 1988 are now verging on becoming relatively routine. From 2009 and onward, ligand K-edge X-ray absorption spectroscopy (XAS) has enabled uranium-ligand covalency to be probed from the perspective of the ligand,206−212 and increasingly resonant inelastic X-ray spectroscopy (RIXS)213−215 is providing a complementary perspective from the metal side. However, given that covalency can be understood and defined216,217 as the spatial overlap of parent atomic orbitals or near-energy matching of parent atomic orbitals, or simply the net amalgamated result of both, precisely what XAS and RIXS data are reporting is an interesting debate.218 Pulsed electron paramagnetic resonance spectroscopy has now been used to probe unpaired spin density,219 although again how that relates exactly to describing covalency is an interesting question. Optical spectroscopy has been used to quantify 5f-orbital covalency and can be the basis of a quite detailed dissection of uranium bonding, but so far this has been limited to probing only the 5f-orbital contributions.110,220−222 Last, NMR spectroscopy has emerged as a powerful way to probe the covalency of molecular actinide–ligand linkages, where a detailed interrogation of the shielding parameters can quantify the bonding. However, this approach is currently restricted to diamagnetic complexes and so has focused on uranium(VI) and thorium(IV) complexes.111,223−231
Conclusions and Outlook
Some 36 years after the vision of Figure 2 first emerged, this Viewpoint has sought to highlight the broad range of resulting advances that have directly, or in parallel, been delivered. An updated version of Figure 2 is presented in Figure 18, showing that most of the major targets have been secured or have close approximations. Many advances have resulted, and in particular an ever better understanding of chemical bonding in a relativistic regime has been developed, and the redox chemistry of uranium has proven to be exploitable in numerous scenarios to secure new bonding motifs, reactivity, and physical properties. Perhaps one of the most important advances is the knowledge that even targets likely initially thought to be more aspirational than actually achievable were eventually secured—persistence is the victor.
Figure 18.
Updated version of Figure 2, the result of ∼36 years of progress.
What started as a presentation slide now requires this Viewpoint to barely scratch the surface of all of the advances that have occurred. That underscores just how much has been achieved in the intervening four decades, and those advances have undoubtedly prompted the community to reevaluate the nature of actinides. This naturally leads to the question, “Where to next?” While not claiming to be a definitive and exclusive list, the following emerge as obvious areas of focus:
A “pure” alkylidene linkage of the form M=CR2 (R = H, alkyl, silyl) is yet to be secured in an isolable molecular actinide complex under normal experimental conditions.
Actinide carbyne and carbido complexes, in particular terminal variants, are yet to be secured in an isolable molecular actinide complex under normal experimental conditions.
Heavier Group 14 and 15 element bonding to uranium requires further development.
U–U bonding in an isolable molecular complex under normal experimental conditions is yet to be secured.
A clear-cut cis-uranyl in an isolable molecular complex under normal experimental conditions is yet to be secured.
The above all emphasize a need to develop the molecular chemistry of transuranium elements. Noting recent reports on a neptunium(V) bis(imido) in 2015,232 a neptunium(V) mono(oxo) in 2022,233 and neptunium(III) and plutonium(III) diphosphonioalkylidenes in 2022 and 2024,234,235 respectively, and early reports of alkyls and alkoxides that lack definitive structural authentication, many of the bonding motifs from Figure 2 that have been delivered with uranium demand realization in transuranium chemistry. This applies to thorium as well, although to a lesser extent given recent advances in its chemistry. It is also worth noting that protactinium chemistry is arguably the “sleeping beauty” of the actinides whose development is long overdue.
All of the areas listed under parallel topics above would also certainly benefit from being translated to transuranium analogues in order to truly build a rigorous picture of actinide periodic trends.
The prior discussion above is not exhaustive by any means but aims to provide context, highlight what has been done and why, and perhaps provide inspiration to focus attention onto the possible opportunities and directions of future travel that researchers in the area might pursue. Finally, the above also serves as a powerful example of the importance of ligand–metal complementarity in developing exciting new chemistry to build our knowledge and understanding of the f elements, especially in a relativistic regime.
Acknowledgments
The author is grateful for support over the years from the Royal Society, Engineering and Physical Sciences Research Council, European Research Council, Marie Curie Fellowship Scheme, and Alexander von Humboldt Foundation that has enabled some of the science featured in this Viewpoint. Previous co-workers of the author whose efforts have realized some of the compounds covered in this Viewpoint are thanked, and their names can be found in the reference list. The author sincerely thanks David L. Clark (Los Alamos National Laboratory, LANL) and Alfred P. Sattelberger (University of Central Florida) for delving into the LANL archive and for discussions about historical and scientific aspects of this Viewpoint, thus helping to hand over the “knowledge-baton” for posterity. The LANL archive is thanked for providing access to its predigital records.
Biography

Steve Liddle is Professor and Head of Inorganic Chemistry and Co-Director of the Centre for Radiochemistry Research at The University of Manchester. His research interest spans experimental and computational investigations of metal–ligand multiple bonding, metal–metal bonding, small-molecule activation and catalysis, and magnetism, with a focus on early-transition and f-block metals but in particular the wonders of researching thorium, uranium, neptunium, and plutonium.
Author Contributions
The manuscript was written by S.T.L., who has given approval for the final version of the manuscript.
The author declares no competing financial interest.
References
- The Chemistry of the Actinide and Transactinide Elements, 3rd ed.; Morss L. R., Edelstein N. M., Fuger J., Katz J. J., Eds.; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- The Lanthanides and Actinides, Synthesis, Reactivity, Properties and Applications; Liddle S. T., Mills D. P., Natrajan L. S., Eds.; World Scientific: Covent Garden, London, 2022. [Google Scholar]
- Streitwieser A. Jr.; Müller-Westerhoff U. Bis(cyclooctatetraenyl)uranium (uranocene). A new class of sandwich complexes that utilize atomic f orbitals. J. Am. Chem. Soc. 1968, 90, 7364–7364. 10.1021/ja01028a044. [DOI] [Google Scholar]
- Zalkin A.; Raymond K. N. The Structure of Di-π-cyclooctatetraeneuranium (Uranocene). J. Am. Chem. Soc. 1969, 91, 5667–5668. 10.1021/ja01048a055. [DOI] [Google Scholar]
- Sattelberger A. P.Organo-f-Element Chemistry—Balancing Theory and Experiment; Report LA-UR-88-0594; Los Alamos National Laboratory: Los Alamos, NM, 1988.
- Burns C. J.; Smith W. H.; Ryan R. R.; Sattelberger A. P.. High Valent Organouranium Complexes—Synthesis, Characterization and Reactivity; Report LA-UR-88-0593, presented at the Third Chemical Congress of North America (including the 195th American Chemical Society National Meeting in Toronto); Los Alamos National Laboratory: Los Alamos, NM, 1988.
- Ephritikhine M. The vitality of uranium molecular chemistry at the dawn of the XXIst century. Dalton Trans. 2006, 2501–2516. 10.1039/b603463b. [DOI] [PubMed] [Google Scholar]
- Fox A. R.; Bart S. C.; Meyer K.; Cummins C. C. Towards uranium catalysts. Nature 2008, 455, 341–349. 10.1038/nature07372. [DOI] [PubMed] [Google Scholar]
- Hayton T. W. Metal-Ligand Multiple Bonding in Uranium: Structure and Reactivity. Dalton Trans. 2010, 39, 1145–1158. 10.1039/B909238B. [DOI] [PubMed] [Google Scholar]
- Jones M. B.; Gaunt A. J. Recent Developments in Synthesis and Structural Chemistry of Nonaqueous Actinide Complexes. Chem. Rev. 2013, 113, 1137–1198. 10.1021/cr300198m. [DOI] [PubMed] [Google Scholar]
- Hayton T. W. Recent Developments in Actinide-Ligand Multiple Bonding. Chem. Commun. 2013, 49, 2956–2973. 10.1039/c3cc39053e. [DOI] [PubMed] [Google Scholar]
- La Pierre H. S.; Meyer K. Activation of Small Molecules by Molecular Uranium Complexes. Prog. Inorg. Chem. 2014, 58, 303–415. 10.1002/9781118792797.ch05. [DOI] [Google Scholar]
- Gregson M.; Wooles A. J.; Cooper O. J.; Liddle S. T. Covalent uranium carbene chemistry. Comments on Inorganic Chemistry 2015, 35, 262–294. 10.1080/02603594.2015.1020154. [DOI] [Google Scholar]
- Johnson S. A.; Bart S. C. Achievements in uranium alkyl chemistry: celebrating sixty years of synthetic pursuits. Dalton Trans. 2015, 44, 7710–7726. 10.1039/C4DT01621A. [DOI] [PubMed] [Google Scholar]
- Liddle S. T. The Renaissance of Non-Aqueous Uranium Chemistry. Angew. Chem., Int. Ed. 2015, 54, 8604–8641. 10.1002/anie.201412168. [DOI] [PubMed] [Google Scholar]
- Schädle D.; Anwander R. Rare-Earth Metal and Actinide Organoimide Chemistry. Chem. Soc. Rev. 2019, 48, 5752–5805. 10.1039/C8CS00932E. [DOI] [PubMed] [Google Scholar]
- Boronski J. T.; Liddle S. T. The emergence of actinide cyclobutadienyl chemistry. Eur. J. Inorg. Chem. 2020, 2020, 2851–2861. 10.1002/ejic.202000383. [DOI] [Google Scholar]
- Boreen M. A.; Arnold J. The synthesis and versatile reducing power of low-valent uranium complexes. Dalton Trans. 2020, 49, 15124–15138. 10.1039/D0DT03151H. [DOI] [PubMed] [Google Scholar]
- Hartline D. R.; Meyer K. From chemical curiosities and trophy molecules to uranium-based catalysis: developments for uranium catalysis as a new facet in molecular uranium chemistry. JACS Au 2021, 1, 698–709. 10.1021/jacsau.1c00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W.; Maron L.; Zhu C. Chapter 327 - Recent advances in f-block metal-metal bonds. Handbook on the Physics and Chemistry of Rare Earths 2023, 63, 1–54. 10.1016/bs.hpcre.2023.01.001. [DOI] [Google Scholar]
- Keener M.; Maria L.; Mazzanti M. Progress in the chemistry of molecular actinide-nitride compounds. Chem. Sci. 2023, 14, 6493–6521. 10.1039/D3SC01435E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline R. K.; Slater J. L. Spectral evidence for lanthanoid and actinoid carbonyl compounds. Angew. Chem., Int. Ed. 1975, 14, 309–313. 10.1002/anie.197503091. [DOI] [Google Scholar]
- Brennan J. G.; Andersen R. A.; Robbins J. L. Prepararion of the first molecular carbon monoxide complex of uranium, (Me3SiC5H4)3UCO. J. Am. Chem. Soc. 1986, 108, 335–336. 10.1021/ja00262a046. [DOI] [Google Scholar]
- Parry J.; Carmona E.; Coles S.; Hursthouse M. Synthesis and single crystal X-ray diffraction study on the first isolable carbonyl complex of an actinide, (C5Me4H)3U(CO). J. Am. Chem. Soc. 1995, 117, 2649–2650. 10.1021/ja00114a030. [DOI] [Google Scholar]
- Evans W. J.; Kozimor S. A.; Nyce G. W.; Ziller J. W. Comparative Reactivity of Sterically Crowded nf3 (C5Me5)3Nd and (C5Me5)3U Complexes with CO: Formation of a Nonclassical Carbonium Ion versus an f Element Metal Carbonyl Complex. J. Am. Chem. Soc. 2003, 125, 13831–13835. 10.1021/ja036631l. [DOI] [PubMed] [Google Scholar]
- Castro-Rodriguez I.; Meyer K. Carbon dioxide reduction and carbon monoxide activation employing a reactive uranium(III) complex. J. Am. Chem. Soc. 2005, 127, 11242–11243. 10.1021/ja053497r. [DOI] [PubMed] [Google Scholar]
- Frey A. S. P.; Cloke F. G. N.; Hitchcock P. B.; Day I. J.; Green J. C.; Aitken G. Mechanistic Studies on the Reductive Cyclooligomerisation of CO by U(III) Mixed Sandwich Complexes; the Molecular Structure of [(U(η-C8H6{SiiPr3-1,4}2)(η-Cp*)]2(μ-η1:η1-C2O2). J. Am. Chem. Soc. 2008, 130, 13816–13817. 10.1021/ja8059792. [DOI] [PubMed] [Google Scholar]
- Tarlton M. L.; Yu X.; Ward R. J.; Kelley S. P.; Autschbach J.; Walensky J. R. Backbonding in thorium(IV) and uranium(IV) diarsenido complexes with tBuNC and CO. Chem. Eur. J. 2021, 27, 14396–14400. 10.1002/chem.202102670. [DOI] [PubMed] [Google Scholar]
- Ward R. J.; Del Rosal I.; Kelley S. P.; Maron L.; Walensky J. R. isolation of C1 through C4 derivatives from CO using heteroleptic uranium(III) metallocene aryloxide complexes. Chem. Sci. 2023, 14, 2024–2032. 10.1039/D2SC06375A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron L.; Eisenstein O.; Andersen R. A. The Bond between CO and Cp′3U in Cp′3U(CO) Involves Back-bonding from the Cp′3U Ligand-Based Orbitals of π-Symmetry, where Cp′ Represents a Substituted Cyclopentadienyl Ligand. Organometallics 2009, 28, 3629–3635. 10.1021/om801098b. [DOI] [Google Scholar]
- Langeslay R. R.; Chen G. P.; Windorff C. J.; Chan A. K.; Ziller J. W.; Furche F.; Evans W. J. Synthesis, Structure, and Reactivity of the Sterically Crowded Th3+ Complex (C5Me5)3Th Including Formation of the Thorium Carbonyl, [(C5Me5)3Th(CO)][BPh4]. J. Am. Chem. Soc. 2017, 139, 3387–3398. 10.1021/jacs.6b10826. [DOI] [PubMed] [Google Scholar]
- Lu E.; Sajjad S.; Berryman V. E. J.; Wooles A. J.; Kaltsoyannis N.; Liddle S. T. Emergence of the structure dictating role of f-orbital overlap-driven covalency. Nat. Commun. 2019, 10, 634. 10.1038/s41467-019-08553-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerscales O. T.; Cloke F. G. N.; Hitchcock P. B.; Green J. C.; Hazari N. Reductive cyclotrimerization of carbon monoxide to the deltate dianion by an organometallic uranium complex. Science 2006, 311, 829–831. 10.1126/science.1121784. [DOI] [PubMed] [Google Scholar]
- Schake A. R.; Avens L. R.; Burns C. J.; Clark D. L.; Sattelberger A. P.; Smith W. H. Synthesis of mixed-ring organoactinide complexes. 2. (Cyclooctatetraene)(pentamethylcyclopentadienyl)(THF)uranium [(C8H8)(C5Me5)U(THF)] and its 4,4’-dimethyl-2,2’-bipyridine derivative. Organometallics 1993, 12, 1497–1498. 10.1021/om00029a004. [DOI] [Google Scholar]
- Summerscales O. T.; Cloke F. G. N.; Hitchcock P. B.; Green J. C.; Hazari N. Reductive Cyclotetramerization of CO to Squarate by a U(III) Complex: The X-ray Crystal Structure of [(U(η-C8H6{SiiPr3-1,4}2)(η-C5Me4H)]2(μ-η2:η2-C4O4). J. Am. Chem. Soc. 2006, 128, 9602–9603. 10.1021/ja063222r. [DOI] [PubMed] [Google Scholar]
- Arnold P. L.; Turner Z. R.; Bellabarba R. M.; Tooze R. P. Carbon monoxide coupling and functionalisation at a simple uranium coordination complex. Chem. Sci. 2011, 2, 77–79. 10.1039/C0SC00452A. [DOI] [Google Scholar]
- Gardner B. M.; Stewart J. C.; Davis A. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Homologation and Functionalization of Carbon Monoxide by a Recyclable Uranium Complex. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9265–9270. 10.1073/pnas.1203417109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey A. S. P.; Cloke F. G. N.; Coles M. P.; Maron L.; Davin T. Facile conversion of CO/H2 into methoxide at a uranium(III) center. Angew. Chem., Int. Ed. 2011, 50, 6881–6883. 10.1002/anie.201101509. [DOI] [PubMed] [Google Scholar]
- Castro-Rodriguez I.; Nakai H.; Zakharov L. N.; Rheingold A. L.; Meyer K. A linear, O-coordinated η1-CO2 bound to uranium. Science 2004, 305, 1757–1759. 10.1126/science.1102602. [DOI] [PubMed] [Google Scholar]
- Schmidt A.-C.; Nizovtsev A. V.; Scheurer A.; Heinemann F. W.; Meyer K. Uranium-mediated reductive conversión of CO2 to CO and carbonate in a single-vessel, closed synthetic cycle. Chem. Commun. 2012, 48, 8634–8636. 10.1039/c2cc34150f. [DOI] [PubMed] [Google Scholar]
- Cooper O.; Camp C.; Pécaut J.; Kefalidis C. E.; Maron L.; Gambarelli S.; Mazzanti M. Multimetallic cooperativity in uranium-mediated CO2 activation. J. Am. Chem. Soc. 2014, 136, 6716–6723. 10.1021/ja5017624. [DOI] [PubMed] [Google Scholar]
- Lam O. P.; Franke S. M.; Heinemann F.; Meyer K. Reactivity of U-E-U (E = S, Se) Toward CO2, CS2, and COS: New Mixed-Carbonate Complexes of the Types U-CO2E-U (E = S, Se), U-CS2E-U (E = O, Se), and U-COSSe-U. J. Am. Chem. Soc. 2012, 134, 16877–16881. 10.1021/ja307539w. [DOI] [PubMed] [Google Scholar]
- Bursten B. E.; Rhodes L. F.; Strittmatter R. J. Bonding of tris(η5-cyclopentadienyl)-actinide complexes. 3. Interaction of π-neutral, π-acidic, and π-basic ligands with (η5-C5H5)3U. J. Am. Chem. Soc. 1989, 111, 2758–2766. 10.1021/ja00190a003. [DOI] [Google Scholar]
- Siladke N. A.; Meihaus K. R.; Ziller J. W.; Fang M.; Furche F.; Long J. R.; Evans W. J. Synthesis, Structure, and Magnetism of an f Element Nitrosyl Complex, (C5Me4H)3UNO. J. Am. Chem. Soc. 2012, 134, 1243–1249. 10.1021/ja2096128. [DOI] [PubMed] [Google Scholar]
- Roussel P.; Scott P. Complex of dinitrogen with trivalent uranium. J. Am. Chem. Soc. 1998, 120, 1070–1071. 10.1021/ja972933+. [DOI] [Google Scholar]
- Haber F.Verfahrenzur Herstellung von Ammoniak durch katalytische Vereinigung von Stickstoff und Wasserstoff, zweckmäßig unter hohem Druch. German Patent DE 229126, 1909.
- Mansell S. M.; Farnaby J. H.; Germeroth A. I.; Arnold P. L. Thermally stable uranium dinitrogen complex with siloxide supporting ligands. Organometallics 2013, 32, 4214–4222. 10.1021/om4003957. [DOI] [Google Scholar]
- Odom A. L.; Arnold P. L.; Cummins C. C. Heterodinuclear uranium/molybdenum dinitrogen complexes. J. Am. Chem. Soc. 1998, 120, 5836–5837. 10.1021/ja980095t. [DOI] [Google Scholar]
- Cloke F. G. N.; Hitchcock P. B. Reversible binding and reduction of dinitrogen by a uranium(III) pentalene complex. J. Am. Chem. Soc. 2002, 124, 9352–9353. 10.1021/ja027000e. [DOI] [PubMed] [Google Scholar]
- Evans W. J.; Kozimor S. A.; Ziller J. W. A monometallic f element complex of dinitrogen: (C5Me5)3U(η1-N2). J. Am. Chem. Soc. 2003, 125, 14264–14265. 10.1021/ja037647e. [DOI] [PubMed] [Google Scholar]
- Lu E.; Atkinson B. E.; Wooles A. J.; Boronski J. T.; Doyle L. R.; Tuna F.; Cryer J. D.; Cobb P. J.; Vitorica-Yrezabal I. J.; Whitehead G. F. S.; Kaltsoyannis N.; Liddle S. T. Back-bonding between an electron-poor, high-oxidation-state metal and poor π-acceptor ligand in a uranium(V)-dinitrogen complex. Nat. Chem. 2019, 11, 806–811. 10.1038/s41557-019-0306-x. [DOI] [PubMed] [Google Scholar]
- Korobkov I.; Gambarotta S.; Yap G. P. A. A highly reactive uranium complex supported by the calix[4]tetrapyrrole tetraanion affording dinitrogen cleavage, solvent deoxygenation, and polysilanol depolymerization. Angew. Chem., Int. Ed. 2002, 41, 3433–3436. . [DOI] [PubMed] [Google Scholar]
- Falcone M.; Chatelain L.; Scopelliti R.; Živković I.; Mazzanti M. Nitrogen reduction and functionalization by a multimetallic uranium nitride complex. Nature 2017, 547, 332–335. 10.1038/nature23279. [DOI] [PubMed] [Google Scholar]
- Batov M. S.; Del Rosal I.; Scopelliti R.; Fadaei-Tirani F.; Živković I.; Maron L.; Mazzanti M. Multimetallic uranium nitride cubane clusters from dinitrogen cleavage. J. Am. Chem. Soc. 2023, 145, 26435–26443. 10.1021/jacs.3c10617. [DOI] [PubMed] [Google Scholar]
- Lyon J. T.; Andrews L. Formation and characterization of the uranium methylidene complexes CH2=UHX (X = F, Cl, Br). Inorg. Chem. 2006, 45, 1847–1852. 10.1021/ic051785i. [DOI] [PubMed] [Google Scholar]
- Lyon J. T.; Andrews L.; Malmqvist P. -Å.; Roos B. O.; Yang T.; Bursten B. E. Infrared spectrum and bonding in uranium methylidene dihydride, CH2=UH2. Inorg. Chem. 2007, 46, 4917–4925. 10.1021/ic062407w. [DOI] [PubMed] [Google Scholar]
- Li J.; Hu H.-S.; Lyon J. T.; Andrews L. Chirality, agostic interactions, and pyramidality in actinide methylidene complexes. Angew. Chem., Int. Ed. 2007, 46, 9045–9049. 10.1002/anie.200702771. [DOI] [PubMed] [Google Scholar]
- Lyon J. T.; Andrews L.; Hu H.-S.; Li J. Infrared spectra and electronic structures of agostic uranium methylidene molecules. Inorg. Chem. 2008, 47, 1435–1442. 10.1021/ic701786h. [DOI] [PubMed] [Google Scholar]
- Cramer R. E.; Maynard R. B.; Paw J. C.; Gilje J. W. A uranium-carbon multiple bond. Crystal and molecular structure of (η5-C5H5)3UCHP(CH3)2(C6H5). J. Am. Chem. Soc. 1981, 103, 3589–3590. 10.1021/ja00402a065. [DOI] [Google Scholar]
- Cantat T.; Arliguie T.; Noël A.; Thuéry P.; Ephritikhine M.; Le Floch P.; Mézailles N. The U=C double bond: synthesis and study of uranium nucleophilic carbene complexes. J. Am. Chem. Soc. 2009, 131, 963–972. 10.1021/ja807282s. [DOI] [PubMed] [Google Scholar]
- Tourneux J.-C.; Berthet J.-C.; Cantat T.; Thuéry P.; Mézailles N.; Ephritikhine M. Exploring the uranyl organometallic chemistry: from single to double uranium-carbon bonds. J. Am. Chem. Soc. 2011, 133, 6162–6165. 10.1021/ja201276h. [DOI] [PubMed] [Google Scholar]
- Mills D. P.; Moro F.; McMaster J.; Van Slageren J.; Lewis W.; Blake A. J.; Liddle S. T. A delocalized arene-bridged diuranium single molecule magnet. Nat. Chem. 2011, 3, 454–460. 10.1038/nchem.1028. [DOI] [PubMed] [Google Scholar]
- Cooper O. J.; Mills D. P.; McMaster J.; Moro F.; Davies E. S.; Lewis W.; Blake A. J.; Liddle S. T. Uranium-Carbon Multiple Bonding: Facile Access to the Pentavalent Uranium Carbene [U{C(PPh2NSiMe3)2}(Cl)2(I)] and Comparison of UV=C and UIV=C Double Bonds. Angew. Chem., Int. Ed. 2011, 50, 2383–2386. 10.1002/anie.201007675. [DOI] [PubMed] [Google Scholar]
- Mills D. P.; Cooper O. J.; Tuna F.; McInnes E. J. L.; Davies E. S.; McMaster J.; Moro F.; Lewis W.; Blake A. J.; Liddle S. T. Synthesis of a Uranium(VI)-Carbene: Reductive Formation of Uranyl(V)-Methanides, Oxidative Preparation of a [R2C = U=O]2+ Analogue of the [O = U=O]2+ Uranyl Ion (R = Ph2PNSiMe3), and Comparison of the Nature of UIV=C, UV=C and UVI=C Double Bonds. J. Am. Chem. Soc. 2012, 134, 10047–10054. 10.1021/ja301333f. [DOI] [PubMed] [Google Scholar]
- Wooles A. J.; Mills D. P.; Tuna F.; McInnes E. J. L.; Law G. T. W.; Fuller A. J.; Kremer F.; Ridgway M.; Lewis W.; Gagliardi L.; Vlaisavljevich B.; Liddle S. T. Uranium(III)-Carbon Multiple Bonding Supported by Arene δ-Bonding in Mixed-Valence Hexauranium Nanometre-Scale Rings. Nat. Commun. 2018, 9, 2097. 10.1038/s41467-018-04560-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu E.; Cooper O. J.; McMaster J.; Tuna F.; McInnes E. J. L.; Lewis W.; Blake A. J.; Liddle S. T. Synthesis, Characterization, and Reactivity of a Uranium(VI) Carbene Imido Oxo Complex. Angew. Chem., Int. Ed. 2014, 53, 6696–6700. 10.1002/anie.201403892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortier S.; Walensky J. R.; Wu G.; Hayton T. W. Synthesis of a Phosphorano-Stabilized U(IV)-Carbene via One-Electron Oxidation of a U(III)-Ylide Adduct. J. Am. Chem. Soc. 2011, 133, 6894–6897. 10.1021/ja2001133. [DOI] [PubMed] [Google Scholar]
- Rungthanaphatsophon P.; Bathelier A.; Castro L.; Behrle A. C.; Barnes C. L.; Maron L.; Walensky J. R. Formation of Methane versus Benzene in the Reactions of (C5Me5)2Th(CH3)2 with [CH3PPh3]X (X = Cl, Br, I) Yielding Thorium-Carbene or Thorium-Ylide Complexes. Angew. Chem., Int. Ed. 2017, 56, 12925–12929. 10.1002/anie.201706496. [DOI] [PubMed] [Google Scholar]
- Seed J. A.; Sharpe H. R.; Futcher H. J.; Wooles A. J.; Liddle S. T. Nature of the Arsonium-Ylide Ph3As = CH2 and a Uranium(IV) Arsonium-Carbene Complex. Angew. Chem., Int. Ed. 2020, 59, 15870–15874. 10.1002/anie.202004983. [DOI] [PubMed] [Google Scholar]
- Baker C. F.; Seed J. A.; Adams R. W.; Lee D.; Liddle S. T. 13Ccarbene nuclear magnetic resonance chemical shift analysis confirms CeIVC double bonding in cerium(iv)-diphosphonioalkylidene complexes. Chem. Sci. 2024, 15, 238–249. 10.1039/D3SC04449A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu E.; Boronski J. T.; Gregson M.; Wooles A. J.; Liddle S. T. Silyl-phosphino-carbene complexes of uranium(IV). Angew. Chem., Int. Ed. 2018, 57, 5506–5511. 10.1002/anie.201802080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent G. T.; Yu X.; Wu G.; Autschbach J.; Hayton T. W. Synthesis and electronic structure analysis of the actinide allenylidenes, [{(NR2)3}An(CCCPh2)]− (An = U, Th; R = SiMe3). Chem. Sci. 2021, 12, 14383–14388. 10.1039/D1SC04666G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Andrews L.; Li J.; Bursten B. E. Reaction of Laser-Ablated Uranium Atoms with CO: Infrared Spectra of the CUO, CUO–, OUCCO, (η2-C2)UO2, and U(CO)x (x = 1–6) Molecules in Solid Neon. J. Am. Chem. Soc. 1999, 121, 9712–9721. 10.1021/ja9921322. [DOI] [Google Scholar]
- Andrews L.; Liang B.; Li J.; Bursten B. E. Ground-state reversal by matrix interaction: electronic states and vibrational frequencies of CUO in solid argon and neon. Angew. Chem., Int. Ed. 2000, 39, 4565–4567. . [DOI] [PubMed] [Google Scholar]
- Lyon J. T.; Hu H.-S.; Andrews L.; Li J. Formation of unprecedented actinide≡carbon triple bonds in uranium methylidyne molecules. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18919–18924. 10.1073/pnas.0707035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Andrews L.; Malmqvist P. -Å.; Roos B. O.; Gonçalves A. P.; Pereira C. C. L.; Marçalo J.; Godart C.; Villeroy B. Infrared spectra and quantum chemical calculations of the uranium carbide molecules UC and CUC with triple bonds. J. Am. Chem. Soc. 2010, 132, 8484–8488. 10.1021/ja102475t. [DOI] [PubMed] [Google Scholar]
- Wang X.; Andrews L.; Ma D.; Gagliardi L.; Gonçalves A. P.; Pereira C. C. L.; Marçalo J.; Godart C.; Villeroy B. Infrared spectra and quantum chemical calculations on the uranium-carbon molecules UC, CUC, UCH, and U(CC)2. J. Chem. Phys. 2011, 134, 244313. 10.1063/1.3602325. [DOI] [PubMed] [Google Scholar]
- Hu H.-S.; Qiu Y.-H.; Xiong X.-G.; Schwarz W. H. E.; Li J. On the maximum bond multiplicity of carbon: unusual C≣U quadruple bonding in molecular CUO. Chem. Sci. 2012, 3, 2786–2796. 10.1039/c2sc20329d. [DOI] [Google Scholar]
- Zhang X.; Li W.; Feng L.; Chen X.; Hansen A.; Grimme S.; Fortier S.; Sergentu D.-C.; Duignan T. J.; Autschbach J.; Wang S.; Wang Y.; Velkos G.; Popov A. A.; Aghdassi N.; Duhm S.; Li X.; Li J.; Echegoyen L.; Schwarz W. H. E.; Chen N. A diuranium carbide cluster stabilized inside a C80 fullerene cage. Nat. Commun. 2018, 9, 2753. 10.1038/s41467-018-05210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J.; Abella L.; Sergentu D.-C.; Yao Y.-R.; Jin M.; Yang W.; Zhang X.; Li X.; Zhang D.; Zhao Y.; Li X.; Wang S.; Echegoyen L.; Autschbach J.; Chen N. Diuranium(IV) carbide cluster U2C2 stabilized inside fullerene cages. J. Am. Chem. Soc. 2019, 141, 20249–20260. 10.1021/jacs.9b10247. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Yu X.; Guo M.; Yao Y.-R.; Meng Q.; Echegoyen L.; Autschbach J.; Chen N. USc2C2 and USc2NC clusters with U-C triple bond character stabilized inside fullerene cages. J. Am. Chem. Soc. 2023, 145, 5645–5654. 10.1021/jacs.2c10231. [DOI] [PubMed] [Google Scholar]
- Yao Y.-R.; Zhao J.; Meng Q.; Hu H.-S.; Guo M.; Yan Y.; Zhuang J.; Yang S.; Fortier S.; Echegoyen L.; Schwarz W. H. E.; Li J.; Chen N. Synthesis and characterization of U≡C triple bonds in fullerene compounds. J. Am. Chem. Soc. 2023, 145, 25440–25449. 10.1021/jacs.3c10042. [DOI] [PubMed] [Google Scholar]
- Cramer R. E.; Panchanatheswaran K.; Gilje J. W. Uranium carbon multiple-bond chemistry. 3. Insertion of acetonitrile and the formation of a uranium nitrogen multiple bond. J. Am. Chem. Soc. 1984, 106, 1853–1854. 10.1021/ja00318a059. [DOI] [Google Scholar]
- Brennan J. G.; Andersen R. A. Electron-transfer reactions of trivalent uranium. Preparation and structure of the uranium metallocene compounds (MeC5H4)3U = NPh and [(MeC5H4)3U]2[μ-η1,η2-PhNCO]. J. Am. Chem. Soc. 1985, 107, 514–516. 10.1021/ja00288a047. [DOI] [Google Scholar]
- Zalkin A.; Brennan J. G.; Andersen R. A. Tris[bis(trimethylsilyl)amido](trimethylsilylimido)uranium(V). Acta Cryst. C 1988, 44, 1553–1554. 10.1107/S0108270188005116. [DOI] [Google Scholar]
- Burns C. J.; Smith W. H.; Huffman J. C.; Sattelberger A. P. Uranium(VI) organoimido complexes. J. Am. Chem. Soc. 1990, 112, 3237–3239. 10.1021/ja00164a069. [DOI] [Google Scholar]
- Arney D. S.; Burns C. J.; Smith D. C. Synthesis and structure of the first uranium(VI) organometallic complex. J. Am. Chem. Soc. 1992, 114, 10068–10069. 10.1021/ja00051a053. [DOI] [Google Scholar]
- Arney D. S.; Burns C. J. Synthesis and structure of high-valent organouranium complexes containing terminal monooxo functional groups. J. Am. Chem. Soc. 1993, 115, 9840–9841. 10.1021/ja00074a077. [DOI] [Google Scholar]
- Warner B. P.; Scott B. L.; Burns C. J. A simple preparative route to bis(imido)uranium(VI) complexes by the direct reductions of diazenes and azides. Angew. Chem., Int. Ed. 1998, 37, 959–960. . [DOI] [PubMed] [Google Scholar]
- Hayton T. W.; Boncella J. M.; Scott B. L.; Palmer P. D.; Batista E. R.; Hay P. J. Synthesis of imido analogs of the uranyl ion. Science 2005, 310, 1941–1943. 10.1126/science.1120069. [DOI] [PubMed] [Google Scholar]
- Hayton T. W.; Boncella J. M.; Scott B. L.; Batista E. R.; Hay P. J. Synthesis and reactivity of the imido analogues of the uranyl ion. J. Am. Chem. Soc. 2006, 128, 10549–10559. 10.1021/ja0629155. [DOI] [PubMed] [Google Scholar]
- Anderson N. H.; Odoh S. O.; Yao Y.; Williams U. J.; Schaefer B. A.; Kiernicki J. J.; Lewis A. J.; Goshert M. D.; Fanwick P. E.; Schelter E. J.; Walensky J. R.; Gagliardi L.; Bart S. C. Harnessing redox activity for the formation of uranium tris(imido) compounds. Nat. Chem. 2014, 6, 919–926. 10.1038/nchem.2009. [DOI] [PubMed] [Google Scholar]
- Anderson N. H.; Yin H.; Kiernicki J. J.; Fanwick P. E.; Schelter E. J.; Bart S. C. Investigation of uranium tris(imido) complexes: synthesis, characterization, and reduction chemistry of [U(NDIPP)3(thf)3]. Angew. Chem., Int. Ed. 2015, 54, 9386–9389. 10.1002/anie.201503771. [DOI] [PubMed] [Google Scholar]
- Anderson N. H.; Xie J.; Ray D.; Zeller M.; Gagliardi L.; Bart S. C. Elucidating bonding preferences in tetrakis(imido)uranate(VI) dianions. Nat. Chem. 2017, 9, 850–855. 10.1038/nchem.2767. [DOI] [PubMed] [Google Scholar]
- King D.; McMaster J.; Tuna F.; McInnes E. J. L.; Lewis W.; Blake A. J.; Liddle S. T. Synthesis and Characterization of an f-Block Terminal Parent Imido [U = NH] Complex: A Masked Uranium(IV)-Nitride. J. Am. Chem. Soc. 2014, 136, 5619–5622. 10.1021/ja502405e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam O. P.; Franke S. M.; Nakai H.; Heinemann F. W.; Hieringer W.; Meyer K. Observation of the Inverse Trans Influence (ITI) in a Uranium(V) Imide Coordination Complex: An Experimental Study and Theoretical Evaluation. Inorg. Chem. 2012, 51, 6190–6199. 10.1021/ic300273d. [DOI] [PubMed] [Google Scholar]
- Green D. W.; Reedy G. T. The identification of UN in Ar matrices. J. Chem. Phys. 1976, 65, 2921–2922. 10.1063/1.433406. [DOI] [Google Scholar]
- Hunt R. D.; Yustein J. T.; Andrews L. Matrix infrared spectra of NUN formed by the insertion of uranium atoms into molecular nitrogen. J. Chem. Phys. 1993, 98, 6070–6074. 10.1063/1.464845. [DOI] [Google Scholar]
- Zhou M.; Andrews L. Infrared spectra and pseudopotential calculations for NUO+, NUO, and NThO in solid neon. J. Chem. Phys. 1999, 111, 11044–11049. 10.1063/1.480463. [DOI] [Google Scholar]
- Andrews L.; Wang X.; Lindh R.; Roos B. O.; Marsden C. J. Simple N≡UF3 and P≡UF3 molecules with triple bonds to uranium. Angew. Chem., Int. Ed. 2008, 47, 5366–5370. 10.1002/anie.200801120. [DOI] [PubMed] [Google Scholar]
- Wang X.; Andrews L.; Vlaisavljevich B.; Gagliardi L. Combined triple and double bonds to uranium: the N≡U = N-H uranimine nitride molecule prepared in solid argon. Inorg. Chem. 2011, 50, 3826–3831. 10.1021/ic2003244. [DOI] [PubMed] [Google Scholar]
- Vlaisavljevich B.; Andrews L.; Wang X.; Gong Y.; Kushto G. P.; Bursten B. E. Detection and electronic structure of naked actinide complexes: rhombic-ring (AnN)2 molecules stabilized by delocalized π-bonding. J. Am. Chem. Soc. 2016, 138, 893–905. 10.1021/jacs.5b10458. [DOI] [PubMed] [Google Scholar]
- Meng Q.; Abella L.; Yao Y.-R.; Sergentu D.-C.; Yang W.; Liu X.; Zhuang J.; Echegoyen L.; Autschbach J.; Chen N. A charged diatomic triple-bonded U≡N species trapped in C82 fullerene cages. Nat. Commun. 2022, 13, 7192. 10.1038/s41467-022-34651-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King D. M.; Liddle S. T. Progress in molecular uranium-nitride chemistry. Coord. Chem. Rev. 2014, 266–267, 2–15. 10.1016/j.ccr.2013.06.013. [DOI] [Google Scholar]
- Fox A. R.; Cummins C. C. Uranium-nitrogen multiple bonding: the case of a four-coordinate uranium(VI) nitridoborate complex. J. Am. Chem. Soc. 2009, 131, 5716–5717. 10.1021/ja8095812. [DOI] [PubMed] [Google Scholar]
- Thomson R. K.; Cantat T.; Scott B. L.; Morris D. E.; Batista E. R.; Kiplinger J. L. Uranium azide photolysis results in C-H bond activation and provides evidence for a terminal uranium nitride. Nat. Chem. 2010, 2, 723–729. 10.1038/nchem.705. [DOI] [PubMed] [Google Scholar]
- King D. M.; Tuna F.; McInnes E. J. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Synthesis and Structure of a Terminal Uranium Nitride Complex. Science 2012, 337, 717–720. 10.1126/science.1223488. [DOI] [PubMed] [Google Scholar]
- King D. M.; Tuna F.; McInnes E. J. L.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Isolation and characterisation of a uranium(VI)-nitride triple bond. Nat. Chem. 2013, 5, 482–488. 10.1038/nchem.1642. [DOI] [PubMed] [Google Scholar]
- King D. M.; Cleaves P. A.; Wooles A. J.; Gardner B. M.; Chilton N. F.; Tuna F.; Lewis W.; McInnes E. J. L.; Liddle S. T. Molecular and Electronic Structure of Terminal and Alkali Metal-Capped Uranium(V)-Nitride Complexes. Nat. Commun. 2016, 7, 13773. 10.1038/ncomms13773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Seed J. A.; Berryman V. E. J.; Kaltsoyannis N.; Adams R. W.; Lee D.; Liddle S. T. Exceptional Uranium(VI)-Nitride Triple Bond Covalency from 15N Nuclear Magnetic Resonance Spectroscopy and Quantum Chemical Analysis. Nat. Commun. 2021, 12, 5649. 10.1038/s41467-021-25863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barluzzi L.; Scopelliti R.; Mazzanti M. Photochemical synthesis of a stable terminal uranium(VI) nitride. J. Am. Chem. Soc. 2020, 142, 19047–19051. 10.1021/jacs.0c09814. [DOI] [PubMed] [Google Scholar]
- Chatelain L.; Louyriac E.; Douair I.; Lu E.; Tuna F.; Wooles A. J.; Gardner B. M.; Maron L.; Liddle S. T. Terminal Uranium(V)-Nitride Hydrogenations Involving Direct Addition or Frustrated Lewis Pair Mechanisms. Nat. Commun. 2020, 11, 337. 10.1038/s41467-019-14221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel S. S.; Deubner H. L.; Müller M.; Karttunen A. J.; Kraus F. Complexes featuring a linear [N≡U≡N] core isoelectronic to the uranyl cation. Nat. Chem. 2020, 12, 962–967. 10.1038/s41557-020-0505-5. [DOI] [PubMed] [Google Scholar]
- Seaman L. A.; Walensky J. R.; Wu G.; Hayton T. W. In pursuit of homoleptic actinide alkyl complexes. Inorg. Chem. 2013, 52, 3556–3564. 10.1021/ic300867m. [DOI] [PubMed] [Google Scholar]
- Marks T. J.; Seyam A. M. Observations on the thermal decomposition of some uranium(IV) tetraalkyls. J. Organomet. Chem. 1974, 67, 61–66. 10.1016/S0022-328X(00)93684-2. [DOI] [Google Scholar]
- Evans W. J.; Wink D. J.; Stanley D. R. Reinvestigation of the reaction of tert-butyllithium with uranium tetrachloride: formation of catalytically active uranium(III) hydride complexes. Inorg. Chem. 1982, 21, 2565–2573. 10.1021/ic00137a008. [DOI] [Google Scholar]
- Edwards P. G.; Andersen R. A.; Zalkin A. Preparation of tetraalkyl phosphine complexes of the f-block metals. Crystal structure of Th(CH2Ph)4(Me2PCH2CH2PMe2) and U(CH2Ph)3Me(Me2PCH2CH2PMe2). Organometallics 1984, 3, 293–398. 10.1021/om00080a023. [DOI] [Google Scholar]
- Van der Sluys W. G.; Burns C. J.; Sattelberger A. P. First example of a neutral homoleptic uranium alkyl. Synthesis, properties, and structure of U[CH(SiMe3)2]3. Organometallics 1989, 8, 855–857. 10.1021/om00105a051. [DOI] [Google Scholar]
- Fortier S.; Melot B. C.; Wu G.; Hayton T. W. Homoleptic uranium(IV) alkyl complexes: synthesis and characterization. J. Am. Chem. Soc. 2009, 131, 15512–15521. 10.1021/ja906516e. [DOI] [PubMed] [Google Scholar]
- Fortier S.; Walensky J. R.; Wu G.; Hayton T. W. High-Valent Uranium Alkyls: Evidence for the Formation of UVI(CH2SiMe3)6. J. Am. Chem. Soc. 2011, 133, 11732–11743. 10.1021/ja204151v. [DOI] [PubMed] [Google Scholar]
- Kraft S. J.; Fanwick P. E.; Bart S. C. Carbon-Carbon Reductive Elimination from Homoleptic Uranium(IV) Alkyls Induced by Redox-Active Ligands. J. Am. Chem. Soc. 2012, 134, 6160–6168. 10.1021/ja209524u. [DOI] [PubMed] [Google Scholar]
- Johnson S. A.; Kiernicki J. J.; Fanwick P. E.; Bart S. C. New Benzylpotassium Reagents and Their Utility for the Synthesis of Homoleptic Uranium(IV) Benzyl Derivatives. Organometallics 2015, 34, 2889–2895. 10.1021/acs.organomet.5b00212. [DOI] [Google Scholar]
- Sears J. D.; Sergentu D.-C.; Baker T. M.; Brennessel W. W.; Autschbach J.; Neidig M. L. The exceptional diversity of homoleptic uranium-methyl complexes. Angew. Chem., Int. Ed. 2020, 59, 13586–13590. 10.1002/anie.202005138. [DOI] [PubMed] [Google Scholar]
- Boreen M. A.; Parker B. F.; Lohrey T. D.; Arnold J. A homoleptic uranium(III) tris(aryl) complex. J. Am. Chem. Soc. 2016, 138, 15865–15868. 10.1021/jacs.6b11182. [DOI] [PubMed] [Google Scholar]
- Wolford N. J.; Sergentu D.-C.; Brennessel W. W.; Autschbach J.; Neidig M. L. Homoleptic aryl complexes of uranium(IV). Angew. Chem., Int. Ed. 2019, 58, 10266–10270. 10.1002/anie.201905423. [DOI] [PubMed] [Google Scholar]
- Seaman L. A.; Pedrick E. A.; Tsuchiya T.; Wu G.; Jakubikova E.; Hayton T. W. Comparison of the Reactivity of 2-Li-C6H4CH2NMe2 with MCl4 (M = Th, U): Isolation of a Thorium Aryl Complex or a Uranium Benzyne Complex. Angew. Chem., Int. Ed. 2013, 52, 10589–10592. 10.1002/anie.201303992. [DOI] [PubMed] [Google Scholar]
- Pedrick E. A.; Seaman L. A.; Scott J. C.; Griego L.; Wu G.; Hayton T. W. Synthesis and reactivity of a U(IV) dibenzyne complex. Organometallics 2016, 35, 494–502. 10.1021/acs.organomet.5b00929. [DOI] [Google Scholar]
- Cotton F. A.; Marler D. O.; Schwotzer W. Dinuclear uranium alkoxides. Preparation and structures of KU2(OCMe3)9, U2(OCMe3)9, and U2(OCHMe2)10, containing [uranium(IV), uranium(IV)], [uranium(IV), uranium(V)], and [uranium(V), uranium(V)], respectively. Inorg. Chem. 1984, 23, 4211–4215. 10.1021/ic00193a023. [DOI] [Google Scholar]
- Cotton F. A.; Marler D. O.; Schwotzer W. The structure of U3(O)(OCMe3)10, an unusual trinuclear U(IV) oxo-alkoxide. Inorg. Chim. Acta 1984, 95, 207–209. 10.1016/S0020-1693(00)87468-X. [DOI] [Google Scholar]
- Cotton F. A.; Marler D. O.; Schwotzer W. Uranium-to-uranium bonds: do they exist?. Inorg. Chim. Acta 1984, 85, L31–L32. 10.1016/S0020-1693(00)81015-4. [DOI] [Google Scholar]
- Eller P. G.; Vergamini P. J. Nuclear magnetic resonance and chemical studies of uranium(V) alkoxides. Inorg. Chem. 1983, 22, 3184–3189. 10.1021/ic00164a005. [DOI] [Google Scholar]
- Fortier S.; Wu G.; Hayton T. W. Synthesis and Characterization of Three Homoleptic Alkoxides of Uranium: [Li(THF)]2[UIV(OtBu)6], [Li(Et2O)][UV(OtBu)6], and UVI(OtBu)6. Inorg. Chem. 2008, 47, 4752–4761. 10.1021/ic800067k. [DOI] [PubMed] [Google Scholar]
- Van der Sluys W. G.; Burns C. J.; Huffman J. C.; Sattelberger A. P. Uranium alkoxide chemistry. 1. Synthesis and the novel dimeric structure of the first homoleptic uranium(III) aryloxide complex. J. Am. Chem. Soc. 1988, 110, 5924–5925. 10.1021/ja00225a067. [DOI] [Google Scholar]
- Mansell S. M.; Kaltsoyannis N.; Arnold P. L. Small Molecule Activation by Uranium Tris(aryloxides): Experimental and Computational Studies of Binding of N2, Coupling of CO, and Deoxygenation Insertion of CO2 under Ambient Conditions. J. Am. Chem. Soc. 2011, 133, 9036–9051. 10.1021/ja2019492. [DOI] [PubMed] [Google Scholar]
- Van der Sluys W. G.; Sattelberger A. P.; Streib W. E.; Huffman J. C. Tetrakis(2,6-di-t-butylphenoxy)uranium(IV): The first structurally characterized neutral homoleptic aryloxide complex of uranium(IV). Polyhedron 1989, 8, 1247–1249. 10.1016/S0277-5387(00)81149-3. [DOI] [Google Scholar]
- Berg J. M.; Clark D. L.; Huffman J. C.; Morris D. E.; Sattelberger A. P.; Streib W. E.; Van der Sluys W. G.; Watkin J. G. Early actinide alkoxide chemistry. Synthesis, characterization, and molecular structures of Th(IV) and U(IV) aryloxide complexes. J. Am. Chem. Soc. 1992, 114, 10811–10821. 10.1021/ja00053a017. [DOI] [Google Scholar]
- Franke S. M.; Tran B. L.; Heinemann F. W.; Hieringer W.; Mindiola D. J.; Meyer K. Uranium(III) Complexes with Bulky Aryloxide Ligands Featuring Metal-Arene Interactions and Their Reactivity Toward Nitrous Oxide. Inorg. Chem. 2013, 52, 10552–10558. 10.1021/ic401532j. [DOI] [PubMed] [Google Scholar]
- Hoerger C. J.; La Pierre H. S.; Maron L.; Scheurer A.; Heinemann F. W.; Meyer K. Reductive disproportionation of nitric oxide mediated by low-valent uranium. Chem. Commun. 2016, 52, 10854–10857. 10.1039/C6CC06095A. [DOI] [PubMed] [Google Scholar]
- Cavigliasso G.; Kaltsoyannis N. On the Paucity of Molecular Actinide Complexes with Unsupported Metal-Metal Bonds: A Comparative Investigation of the Electronic Structure and Metal-Metal Bonding in U2X6 (X = Cl, F, OH, NH2, CH3) Complexes and d-Block Analogues. Inorg. Chem. 2006, 45, 6828–6839. 10.1021/ic060777e. [DOI] [PubMed] [Google Scholar]
- Gorokhov L. N.; Emelyanov A. M.; Khodeev Y. S. Mass-spectroscopic investigation of stability of gaseous U2O2 and U2. High Temp. 1974, 12, 1307–1309. [Google Scholar]
- Roos B. O.; Malmqvist P. -Å.; Gagliardi L. Exploring the actinide-actinide bond: theoretical studies of the chemical bond in Ac2, Th2, Pa2, and U2. J. Am. Chem. Soc. 2006, 128, 17000–17006. 10.1021/ja066615z. [DOI] [PubMed] [Google Scholar]
- Gagliardi L.; Roos B. O. Quantum chemical calculations show that the uranium molecule U2 has a quintuple bond. Nature 2005, 433, 848–851. 10.1038/nature03249. [DOI] [PubMed] [Google Scholar]
- Knecht S.; Jensen H. J. A.; Saue T. Relativistic quantum chemical calculations show that the uranium molecule U2 has a quadruple bond. Nat. Chem. 2019, 11, 40–44. 10.1038/s41557-018-0158-9. [DOI] [PubMed] [Google Scholar]
- Souter P. F.; Kushto G. P.; Andrews L. IR spectra of uranium hydride molecules isolated in solid argon. Chem. Commun. 1996, 2401–2402. 10.1039/cc9960002401. [DOI] [Google Scholar]
- Souter P. F.; Kushto G. P.; Andrews L.; Neurock M. Experimental and Theoretical Evidence for the Formation of Several Uranium Hydride Molecules. J. Am. Chem. Soc. 1997, 119, 1682–1687. 10.1021/ja9630809. [DOI] [Google Scholar]
- Zhang X.; Wang Y.; Morales-Martínez R.; Zhong J.; De Graaf C.; Rodríguez-Fortea A.; Poblet J. M.; Echegoyen L.; Feng L.; Chen N. U2@Ih(7)-C80: crystallographic characterization of a long-sought dimetallic actinide endohedral fullerene. J. Am. Chem. Soc. 2018, 140, 3907–3915. 10.1021/jacs.7b10865. [DOI] [PubMed] [Google Scholar]
- Foroutan-Nejad C.; Vícha J.; Marek R.; Patzschke M.; Straka M. Unwilling U-U bonding in U2@C80: cage-driven metal-metal bonds in di-uranium fullerenes. Phys. Chem. Chem. Phys. 2015, 17, 24182–24192. 10.1039/C5CP04280A. [DOI] [PubMed] [Google Scholar]
- Moreno-Vicente A.; Roselló Y.; Chen N.; Echegoyen L.; Dunk P. W.; Rodríguez-Fortea A.; De Graaf C.; Poblet J. M. Are U–U bonds inside fullerenes really unwilling bonds?. J. Am. Chem. Soc. 2023, 145, 6710–6718. 10.1021/jacs.2c12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J.; Morales-Martínez R.; Zhang J.; Wang Y.; Yao Y.-R.; Pei C.; Rodríguez-Fortea A.; Wang S.; Echegoyen L.; De Graaf C.; Poblet J. M.; Chen N. Characterization of a strong covalent Th3+-Th3+ bond inside an Ih(7)-C80 fullerene cage. Nat. Commun. 2021, 12, 2372. 10.1038/s41467-021-22659-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boronski J. T.; Seed J. A.; Hunger D.; Woodward A. W.; Van Slageren J.; Wooles A. J.; Natrajan L. S.; Kaltsoyannis N.; Liddle S. T. A crystalline tri-thorium cluster with σ-aromatic metal-metal bonding. Nature 2021, 598, 72–75. 10.1038/s41586-021-03888-3. [DOI] [PubMed] [Google Scholar]
- Tomeček J.; Liddle S. T.; Kaltsoyannis N. Actinide-Actinide Bonding: Electron Delocalisation and σ-Aromaticity in the Tri-Thorium Cluster [{Th(η8-C8H8)(μ-Cl)2}3K2]. ChemPhysChem 2023, 24, e202300366 10.1002/cphc.202300366. [DOI] [PubMed] [Google Scholar]
- Clark D. L.; Sattelberger A. P.; Bott S. G.; Vrtis R. N. Lewis base adducts of uranium triiodide: a new class of synthetically useful precursors for trivalent uranium chemistry. Inorg. Chem. 1989, 28, 1771–1773. 10.1021/ic00309a004. [DOI] [Google Scholar]
- Avens L. R.; Bott S. G.; Clark D. L.; Sattelberger A. P.; Watkin J. G.; Zwick B. D. A convenient entry into trivalent actinide chemistry: synthesis and characterization of AnI3(THF)4 and An[N(SiMe3)2]3 (An = U, Np, Pu). Inorg. Chem. 1994, 33, 2248–2256. 10.1021/ic00088a030. [DOI] [Google Scholar]
- Reynolds L. T.; Wilkinson G. π-Cylopentadienyl compounds of uranium-IV and thorium-IV. J. Inorg. Nucl. Chem. 1956, 2, 246–253. 10.1016/0022-1902(56)80051-1. [DOI] [Google Scholar]
- Fischer E. O.; Hristidu Y. Uber aromatenkomplexe von metallen. 57. Uran-tetracyclopentadienyl. Z. Naturforsch. 1962, 17, 275–275. 10.1515/znb-1962-0410. [DOI] [Google Scholar]
- Arliguie T.; Lance M.; Nierlich M.; Vigner J.; Ephritikhine M. Synthesis and crystal structure of [K(C12H24O6)][U(η-C7H7)2], the first cycloheptatrienyl sandwich compound. J. Chem. Soc. Chem. Commun. 1995, 183–184. 10.1039/C39950000183. [DOI] [Google Scholar]
- Arliguie T.; Lance M.; Nierlich M.; Vigner J.; Ephritikhine M. Inverse cycloheptatrienyl sandwich complexes. Crystal structure of [U(BH4)2(OC4H8)5][(BH4)3U(μ-η7,η7-C7H7)U(BH4)3]. J. Chem. Soc. Chem. Commun. 1994, 847–848. 10.1039/C39940000847. [DOI] [Google Scholar]
- Cesari M.; Pedretti U.; Zazzetta A.; Lugli G.; Marconi W. Synthesis and structure of a π-arene complex of uranium(III) - aluminum chloride. Inorg. Chim. Acta 1971, 5, 439–444. 10.1016/S0020-1693(00)95960-7. [DOI] [Google Scholar]
- Diaconescu P. L.; Arnold P. L.; Baker T. A.; Mindiola D. J.; Cummins C. C. Arene-Bridged Diuranium Complexes: Inverted Sandwiches Supported by δ Backbonding. J. Am. Chem. Soc. 2000, 122, 6108–6109. 10.1021/ja994484e. [DOI] [Google Scholar]
- Cryer J. D.; Liddle S. T.. Arene complexes of the actinides. In Comprehensive Organometallic Chemistry IV; Parkin G.; Meyer K.; O’Hare D., Eds.; Elsevier, 2022; Vol. 4, pp 460–501. [Google Scholar]
- Patel D.; Moro F.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. A Formal High Oxidation State Inverse-Sandwich Diuranium Complex: A New Route to f-Block-Metal Bonds. Angew. Chem., Int. Ed. 2011, 50, 10388–10392. 10.1002/anie.201104110. [DOI] [PubMed] [Google Scholar]
- Patel D.; McMaster J.; Lewis W.; Blake A. J.; Liddle S. T. Reductive assembly of cyclobutadienyl and diphosphacyclobutadienyl rings at uranium. Nat. Commun. 2013, 4, 2323. 10.1038/ncomms3323. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Hou G.; Zi G.; Ding W.; Walter M. D. Influence of the 5f orbitals on the bonding and reactivity in organoactinides: experimental and computational studies on a uranium metallacyclopropene. J. Am. Chem. Soc. 2016, 138, 5130–5142. 10.1021/jacs.6b01391. [DOI] [PubMed] [Google Scholar]
- MacDonald M. R.; Fieser M. E.; Bates J. E.; Ziller J. W.; Furche F.; Evans W. J. Identification of the + 2 Oxidation State for Uranium in a Crystalline Molecular Complex, [K(2.2.2-Cryptand)][(C5H4SiMe3)3U]. J. Am. Chem. Soc. 2013, 135, 13310–133113. 10.1021/ja406791t. [DOI] [PubMed] [Google Scholar]
- La Pierre H. S.; Scheurer A.; Heinemann F. W.; Hieringer W.; Meyer K. Synthesis and Characterization of a Uranium(II) Monoarene Complex Supported by δ Backbonding. Angew. Chem., Int. Ed. 2014, 53, 7158–7162. 10.1002/anie.201402050. [DOI] [PubMed] [Google Scholar]
- Billow B. S.; Livesay B. N.; Mokhtarzadeh C. C.; McCracken J.; Shores M. P.; Boncella J. M.; Odom A. L. Synthesis and characterization of a neutral U(II) arene sandwich complex. J. Am. Chem. Soc. 2018, 140, 17369–17373. 10.1021/jacs.8b10888. [DOI] [PubMed] [Google Scholar]
- Guo F.-S.; Tsoureas N.; Huang G.-Z.; Tong M.-L.; Mansikkamäki A.; Layfield R. A. Isolation of a perfectly linear uranium(II) metallocene. Angew. Chem., Int. Ed. 2020, 59, 2299–2303. 10.1002/anie.201912663. [DOI] [PubMed] [Google Scholar]
- Straub M. D.; Ouellette E. T.; Boreen M. A.; Britt R. D.; Chakarawet K.; Douair I.; Gould C. A.; Maron L.; Del Rosal I.; Villarreal D.; Minasian S. G.; Arnold J. A Uranium(II) Arene Complex That Acts as a Uranium(I) Synthon. J. Am. Chem. Soc. 2021, 143, 19748–19760. 10.1021/jacs.1c07854. [DOI] [PubMed] [Google Scholar]
- Keener M.; Shivaraam R. A.; Rajeshkumar T.; Tricoire M.; Scopelliti R.; Živković I.; Chauvin A.-S.; Maron L.; Mazzanti M. Multielectron Redox Chemistry of Uranium by Accessing the + II Oxidation State and Enabling Reduction to a U(I) Synthon. J. Am. Chem. Soc. 2023, 145, 16271–16283. 10.1021/jacs.3c05626. [DOI] [PubMed] [Google Scholar]
- Shivaraam R. A. K.; Keener M.; Modder D. K.; Rajeshkumar T.; Živković I.; Scopelliti R.; Maron L.; Mazzanti M. A Route to Stabilize Uranium(II) and Uranium(I) Synthons in Multimetallic Complexes. Angew. Chem., Int. Ed. 2023, 62, e202304051 10.1002/anie.202304051. [DOI] [PubMed] [Google Scholar]
- Barluzzi L.; Giblin S. R.; Mansikkamäki A.; Layfield R. A. Identification of oxidation state + 1 in a molecular uranium complex. J. Am. Chem. Soc. 2022, 144, 18229–18233. 10.1021/jacs.2c06519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langeslay R. R.; Fieser M. E.; Ziller J. W.; Furche F.; Evans W. J. Synthesis, structure, and reactivity of crystalline molecular complexes of the {[C5H3(SiMe3)2]3Th}1- anion containing thorium in the formal + 2 oxidation state. Chem. Sci. 2015, 6, 517–521. 10.1039/C4SC03033H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortu F.; Formanuik A.; Innes J. R.; Mills D. P. New vistas in the molecular chemistry of thorium. Dalton Trans. 2016, 45, 7537–7549. 10.1039/C6DT01111J. [DOI] [PubMed] [Google Scholar]
- Huh D. N.; Roy S.; Ziller J. W.; Furche F.; Evans W. J. Isolation of a Square-Planar Th(III) Complex: Synthesis and Structure of [Th(OC6H2tBu2-2,6-Me-4)4]1--. J. Am. Chem. Soc. 2019, 141, 12458–12463. 10.1021/jacs.9b04399. [DOI] [PubMed] [Google Scholar]
- Su J.; Windorff C. J.; Batista E. R.; Evans W. J.; Gaunt A. J.; Janicke M. T.; Kozimor S. A.; Scott B. L.; Woen D. H.; Yang P. Identification of the Formal + 2 Oxidation State of Neptunium: Synthesis and Structural Characterization of {NpII[C5H3(SiMe3)2]3}1--. J. Am. Chem. Soc. 2018, 140, 7425–7428. 10.1021/jacs.8b03907. [DOI] [PubMed] [Google Scholar]
- Windorff C. J.; Chen G. P.; Cross J. N.; Evans W. J.; Furche F.; Gaunt A. J.; Janicke M. T.; Kozimor S. A.; Scott B. L. Identification of the Formal + 2 Oxidation State of Plutonium: Synthesis and Characterization of {PuII[C5H3(SiMe3)2]3}−. J. Am. Chem. Soc. 2017, 139, 3970–3973. 10.1021/jacs.7b00706. [DOI] [PubMed] [Google Scholar]
- Du J.; Cobb P. J.; Ding J.; Mills D. P.; Liddle S. T. f-Element heavy pnictogen chemistry. Chem. Sci. 2023, 15, 13–45. 10.1039/D3SC05056D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ephritikhine M. Molecular actinide compounds with soft chalcogen ligands. Coord. Chem. Rev. 2016, 319, 35–62. 10.1016/j.ccr.2016.04.020. [DOI] [Google Scholar]
- Kelly R. P.; Falcone M.; Lamsfus C. A.; Scopelliti R.; Maron L.; Meyer K.; Mazzanti M. Metathesis of a UV imido complex: a route to a terminal UV sulfide. Chem. Sci. 2017, 8, 5319–5328. 10.1039/C7SC01111C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrez J.; Pécaut J.; Scopelliti R.; Kefalidis C. E.; Maron L.; Rosenzweig M. W.; Meyer K.; Mazzanti M. Synthesis and reactivity of a terminal uranium(IV) sulfide supported by siloxide ligands. Chem. Sci. 2016, 7, 5846–5856. 10.1039/C6SC00675B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig M. W.; Scheurer A.; Lamsfus C. A.; Heinemann F. W.; Maron L.; Andrez J.; Mazzanti M.; Meyer K. Uranium(IV) terminal hydrosulfido and sulfido complexes: insights into the nature of the uranium-sulfur bond. Chem. Sci. 2016, 7, 5857–5866. 10.1039/C6SC00677A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano J. K.; Arney D. S. J.; Scott B. L.; Morris D. E.; Kiplinger J. L.; Burns C. J. A sulphur and uranium fiesta! Synthesis, structure, and characterization of neutral terminal uranium(VI) monosulphide, uranium(VI) η2-disulphide, and uranium(IV) phosphine sulphide complexes. Dalton Trans. 2019, 48, 50–57. 10.1039/C8DT02932F. [DOI] [PubMed] [Google Scholar]
- Rosenzweig M. W.; Hümmer J.; Scheurer A.; Lamsfus C. A.; Heinemann F. W.; Maron L.; Mazzanti M.; Meyer K. A complete series of uranium(IV) complexes with terminal hydrochalcogenido (EH) and chalcogenido (E) ligands E = O, S, Se, Te. Dalton Trans. 2019, 48, 10853–10864. 10.1039/C9DT00530G. [DOI] [PubMed] [Google Scholar]
- Li T.; Wang D.; Heng Y.; Hou G.; Zi G.; Ding W.; Walter M. D. A Comprehensive Study Concerning the Synthesis, Structure, and Reactivity of Terminal Uranium Oxido, Sulfido, and Selenido Metallocenes. J. Am. Chem. Soc. 2023, 145, 14839–14855. 10.1021/jacs.3c03753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D. L.; Conradson S. D.; Donohoe R. J.; Keogh D. W.; Morris D. E.; Palmer P. D.; Rogers R. D.; Tait C. D. Chemical Speciation of the Uranyl Ion under Highly Alkaline Conditions. Synthesis, Structures, and Oxo Ligand Exchange Dynamics. Inorg. Chem. 1999, 38, 1456–1466. 10.1021/ic981137h. [DOI] [Google Scholar]
- Graves C. R.; Kiplinger J. L. Pentavalent uranium chemistry - synthetic pursuit of a rare oxidation state. Chem. Commun. 2009, 3831–3853. 10.1039/b902969a. [DOI] [PubMed] [Google Scholar]
- Arnold P. L.; Love J. B.; Patel D. Pentavalent uranyl complexes. Coord. Chem. Rev. 2009, 253, 1973–1978. 10.1016/j.ccr.2009.03.014. [DOI] [Google Scholar]
- Fortier S.; Hayton T. W. Oxo ligand functionalization in the uranyl ion (UO22+). Coord. Chem. Rev. 2010, 254, 197–214. 10.1016/j.ccr.2009.06.003. [DOI] [Google Scholar]
- Pedrick E. A.; Wu G.; Kaltsoyannis N.; Hayton T. W. Reductive silylation of a uranyl dibenzoylmethanate complex: an example of controlled uranyl oxo ligand cleavage. Chem. Sci. 2014, 5, 3204–3213. 10.1039/C4SC00996G. [DOI] [Google Scholar]
- Pankhurst J. R.; Bell N. L.; Zegke M.; Platts L. N.; Lamfsus C. A.; Maron L.; Natrajan L. S.; Sproules S.; Arnold P. L.; Love J. B. Inner-sphere vs. outer-sphere reduction of uranyl supported by a redox-active, donor-expanded dipyrrin. Chem. Sci. 2017, 8, 108–116. 10.1039/C6SC02912D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin E. J.; Qiao Y.; Lapsheva E.; Zeller M.; Schelter E. J.; Bart S. C. Uranyl Functionalization Mediated by Redox-Active Ligands: Generation of O-C Bonds via Acylation. J. Am. Chem. Soc. 2019, 141, 1016–1026. 10.1021/jacs.8b11302. [DOI] [PubMed] [Google Scholar]
- Assefa M. K.; Wu G.; Hayton T. W. Uranyl oxo silylation promoted by silsesquioxane coordination. J. Am. Chem. Soc. 2020, 142, 8738–8747. 10.1021/jacs.0c00990. [DOI] [PubMed] [Google Scholar]
- Faizova R.; Fadaei-Tirani F.; Chauvin A.-S.; Mazzanti M. Synthesis and characterization of water stable uranyl(V) complexes. Angew. Chem., Int. Ed. 2021, 60, 8227–8235. 10.1002/anie.202016123. [DOI] [PubMed] [Google Scholar]
- Hayton T. W. Understanding the origins of Oyl-U-Oyl bending in the uranyl (UO22+) ion. Dalton Trans. 2018, 47, 1003–1009. 10.1039/C7DT04123C. [DOI] [PubMed] [Google Scholar]
- Kent G. T.; Murillo J.; Wu G.; Fortier S.; Hayton T. W. Coordination of uranyl to the redox-active calix[4]pyrrole ligand. Inorg. Chem. 2020, 59, 8629–8634. 10.1021/acs.inorgchem.0c01224. [DOI] [PubMed] [Google Scholar]
- Hartline D. R.; Löffler S. T.; Fehn D.; Kasper J. M.; Heinemann F. W.; Yang P.; Batista E. R.; Meyer K. Uranium-Mediated Peroxide Activation and a Precursor toward an Elusive Uranium cis-Dioxo Fleeting Intermediate. J. Am. Chem. Soc. 2023, 145, 8927–8938. 10.1021/jacs.2c12868. [DOI] [PubMed] [Google Scholar]
- Keener M.; Hunt C.; Carroll T. G.; Kampel V.; Dobrovetsky R.; Hayton T. W.; Ménard G. Redox-switchable carboranes for uranium capture and release. Nature 2020, 577, 652–655. 10.1038/s41586-019-1926-4. [DOI] [PubMed] [Google Scholar]
- Keener M.; Mattejat M.; Zheng S.-L.; Wu G.; Hayton T. W.; Ménard G. Selective electrochemical capture and release of uranyl from aqueous alkali, lanthanide, and actinide mixtures using redox-switchable carboranes. Chem. Sci. 2022, 13, 3369–3374. 10.1039/D1SC07070C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halter D. P.; Heinemann F. W.; Bachmann J.; Meyer K. Uranium-mediated electrocatalytic dihydrogen production from water. Nature 2016, 530, 317–321. 10.1038/nature16530. [DOI] [PubMed] [Google Scholar]
- Halter D. P.; Heinemann F. W.; Maron L.; Meyer K. The role of uranium-arene bonding in H2O reduction catalysis. Nat. Chem. 2018, 10, 259–267. 10.1038/nchem.2899. [DOI] [PubMed] [Google Scholar]
- Magnani N.; Caciuffo R. Future directions for transuranic single molecule magnets. Inorganics 2018, 6, 26. 10.3390/inorganics6010026. [DOI] [Google Scholar]
- La Pierre H. S.; Meyer K. Uranium-Ligand Multiple Bonding in Uranyl Analogues, [L = U=L]n+, and the Inverse Trans Influence. Inorg. Chem. 2013, 52, 529–539. 10.1021/ic302412j. [DOI] [PubMed] [Google Scholar]
- Motta L. C.; Autschbach J. Actinide inverse trans influence versus cooperative pushing from below and multi-center bonding. Nat. Commun. 2023, 14, 4307. 10.1038/s41467-023-39626-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans W. J. Advances in f element reductive reactivity as a paradigm for expanding lanthanide and actinide science and technology. J. Alloys Compd. 2009, 488, 493–510. 10.1016/j.jallcom.2009.02.018. [DOI] [Google Scholar]
- Li J.; Bursten B. E.; Liang B.; Andrews L. Noble gas-actinide compounds: complexation of the CUO molecule by Ar, Kr, and Xe atoms in Noble gas matrices. Science 2002, 295, 2242–2245. 10.1126/science.1069342. [DOI] [PubMed] [Google Scholar]
- Kozimor S. A.; Yang P.; Batista E. R.; Boland K. S.; Burns C. J.; Clark D. L.; Conradson S. D.; Martin R. L.; Wilkerson M. P.; Wolfsberg L. E. Trends in Covalency for d- and f-Element Metallocene Dichlorides Identified Using Chlorine K-Edge X-ray Absorption Spectroscopy and Time-Dependent Density Functional Theory. J. Am. Chem. Soc. 2009, 131, 12125–12136. 10.1021/ja9015759. [DOI] [PubMed] [Google Scholar]
- Minasian S. G.; Keith J. M.; Batista E. R.; Boland K. S.; Clark D. L.; Conradson S. D.; Kozimor S. A.; Martin R. L.; Schwarz D. E.; Shuh D. K.; Wagner G. L.; Wilkerson M. P.; Wolfsberg L. E.; Yang P. Determining Relative f and d Orbital Contributions to M-Cl Covalency in MCl62-- (M = Ti, Zr, Hf, U) and UOCl5– Using Cl K-Edge X-ray Absorption Spectroscopy and Time-Dependent Density Functional Theory. J. Am. Chem. Soc. 2012, 134, 5586–5597. 10.1021/ja2105015. [DOI] [PubMed] [Google Scholar]
- Spencer L. P.; Yang P.; Minasian S. G.; Jilek R. E.; Batista E. R.; Boland K. S.; Boncella J. M.; Conradson S. D.; Clark D. L.; Hayton T. W.; Kozimor S. A.; Martin R. L.; MacInnes M. M.; Olson A. C.; Scott B. L.; Shuh D. K.; Wilkerson M. P. Tetrahalide Complexes of the [U(NR)2]2+ Ion: Synthesis, Theory, and Chlorine K-Edge X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2013, 135, 2279–2290. 10.1021/ja310575j. [DOI] [PubMed] [Google Scholar]
- Minasian S. G.; Keith J. M.; Batista E. R.; Boland K. S.; Clark D. L.; Kozimor S. A.; Martin R. L.; Shuh D. K.; Tyliszczak T. New evidence for 5f covalency in actinocenes determined from carbon K-edge XAS and electronic structure theory. Chem. Sci. 2014, 5, 351–359. 10.1039/C3SC52030G. [DOI] [Google Scholar]
- Su J.; Batista E. R.; Boland K. S.; Bone S. E.; Bradley J. A.; Cary S. K.; Clark D. L.; Conradson S. D.; Ditter A. S.; Kaltsoyannis N.; Keith J. M.; Kerridge A.; Kozimor S. A.; Löble M. W.; Martin R. L.; Minasian S. G.; Mocko V.; La Pierre H. S.; Seidler G. T.; Shuh D. K.; Wilkerson M. P.; Wolfsberg L. E.; Yang P. Energy-Degeneracy-Driven Covalency in Actinide Bonding. J. Am. Chem. Soc. 2018, 140, 17977–17984. 10.1021/jacs.8b09436. [DOI] [PubMed] [Google Scholar]
- Smiles D. E.; Batista E. R.; Booth C. H.; Clark D. L.; Keith J. M.; Kozimor S. A.; Martin R. L.; Minasian S. G.; Shuh D. K.; Stieber S. C.E.; Tyliszczak T. The duality of electron localization and covalency in lanthanide and actinide metallocenes. Chem. Sci. 2020, 11, 2796–2809. 10.1039/C9SC06114B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y.; Ganguly G.; Booth C. H.; Branson J. A.; Ditter A. S.; Lussier D. J.; Moreau L. M.; Russo D. R.; Sergentu D.-C.; Shuh D. K.; Sun T.; Autschbach J.; Minasian S. G. Enhanced 5f-δ bonding in [U(C7H7)2]−: C K-edge XAS, magnetism, and ab initio calculations. Chem. Commun. 2021, 57, 9562–9565. 10.1039/D1CC03414F. [DOI] [PubMed] [Google Scholar]
- Vitova T.; Pidchenko I.; Fellhauer D.; Bagus P. S.; Joly Y.; Pruessmann T.; Bahl S.; Gonzalez-Robles E.; Rothe J.; Altmaier M.; Denecke M. A.; Geckeis H. The role of the 5f valence orbitals of early actinides in chemical bonding. Nat. Commun. 2017, 8, 16053. 10.1038/ncomms16053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegke M.; Zhang X.; Pidchenko I.; Hlina J. A.; Lord R. M.; Purkis J.; Nichol G. S.; Magnani N.; Schreckenbach G.; Vitova T.; Love J. B.; Arnold P. L. Differential uranylVv) oxo-group bonding between the uranium and metal cations from groups 1, 2, 4, and 12; a high energy resolution X-ray absorption, computational, and synthetic study. Chem. Sci. 2019, 10, 9740–9751. 10.1039/C8SC05717F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvashnina K. O.; Butorin S. M. High-energy resolution X-ray spectroscopy at actinide M4,5 and ligand K edges: what we know, what we want to know, and what we can know. Chem. Commun. 2022, 58, 327–342. 10.1039/D1CC04851A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltsoyannis N. Does Covalency Increase or Decrease across the Actinide Series? Implications for Minor Actinide Partitioning. Inorg. Chem. 2013, 52, 3407–3413. 10.1021/ic3006025. [DOI] [PubMed] [Google Scholar]
- Neidig M. L.; Clark D. L.; Martin R. L. Covalency in f-Element Complexes. Coord. Chem. Rev. 2013, 257, 394–406. 10.1016/j.ccr.2012.04.029. [DOI] [Google Scholar]
- Sergentu D.-C.; Autschbach J. Covalency in actinide(IV) hexachlorides in relation to the chlorine K-edge X-ray absorption structure. Chem. Sci. 2022, 13, 3194–3207. 10.1039/D1SC06454A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formanuik A.; Ariciu A.-M.; Ortu F.; Beekmeyer R.; Kerridge A.; Tuna A.; McInnes E. J. L.; Mills D. P. Actinide covalency measured by pulsed electron paramagnetic resonance spectroscopy. Nat. Chem. 2017, 9, 578–583. 10.1038/nchem.2692. [DOI] [PubMed] [Google Scholar]
- Lukens W. W.; Edelstein N. M.; Magnani N.; Hayton T. W.; Fortier S.; Seaman L. A. Quantifying the σ and π Interactions between U(V) f Orbitals and Halide, Alkyl, Alkoxide, Amide and Ketimide Ligands. J. Am. Chem. Soc. 2013, 135, 10742–10754. 10.1021/ja403815h. [DOI] [PubMed] [Google Scholar]
- Staun S. L.; Wu G.; Lukens W. W.; Hayton T. W. Synthesis of a heterobimetallic actinide nitride and an analysis of its bonding. Chem. Sci. 2021, 12, 15519–15527. 10.1039/D1SC05072A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. H.; Paul E. L.; Lukens W. W.; Hayton T. W. Evaluating f-Orbital Participation in the UV=E Multiple Bonds of [U(E)(NR2)3] (E = O, NSiMe3, NAd; R = SiMe3). Inorg. Chem. 2023, 62, 6447–6457. 10.1021/acs.inorgchem.3c00455. [DOI] [PubMed] [Google Scholar]
- Hrobárik P.; Hrobáriková V.; Greif A. H.; Kaupp M. Giant Spin-Orbit Effects on NMR Shifts in Diamagnetic Actinide Complexes: Guiding the Search of Uranium(VI) Hydride Complexes in the Correct Spectral Range. Angew. Chem., Int. Ed. 2012, 51, 10884–10888. 10.1002/anie.201204634. [DOI] [PubMed] [Google Scholar]
- Seaman L. A.; Hrobárik P.; Schettini M. F.; Fortier S.; Kaupp M.; Hayton T. W. A Rare Uranyl(VI)-Alkyl Ate Complex [Li(DME)1.5]2[UO2(CH2SiMe3)4] and Its Comparison with a Homoleptic Uranium(VI)-Hexaalkyl. Angew. Chem., Int. Ed. 2013, 52, 3259–3263. 10.1002/anie.201209611. [DOI] [PubMed] [Google Scholar]
- Smiles D. E.; Wu G.; Hrobárik P.; Hayton T. W. Use of 77Se and 125Te NMR Spectroscopy to Probe Covalency of the Actinide-Chalcogen Bonding in [Th(En){N(SiMe3)2}3]− (E = Se, Te; N = 1, 2) and Their Oxo-Uranium(VI) Congeners. J. Am. Chem. Soc. 2016, 138, 814–825. 10.1021/jacs.5b07767. [DOI] [PubMed] [Google Scholar]
- Smiles D. E.; Wu G.; Hrobárik P.; Hayton T. W. Synthesis, Thermochemistry, Bonding, and 13C NMR Chemical Shift Analysis of a Phosphorano-Stabilized Carbene of Thorium. Organometallics 2017, 36, 4519–4524. 10.1021/acs.organomet.7b00202. [DOI] [Google Scholar]
- Rungthanaphatsophon P.; Huang P.; Walensky J. R. Phosphorano-Stabilized Carbene Complexes with Short Thorium(IV)- and Uranium(IV)-Carbon Bonds. Organometallics 2018, 37, 1884–1891. 10.1021/acs.organomet.8b00137. [DOI] [Google Scholar]
- Mullane K. C.; Hrobárik P.; Cheisson T.; Manor B. C.; Carroll P. J.; Schelter E. J. 13C NMR Shifts as an Indicator of U-C Bond Covalency in Uranium(VI) Acetylide Complexes: An Experimental and Computational Study. Inorg. Chem. 2019, 58, 4152–4163. 10.1021/acs.inorgchem.8b03175. [DOI] [PubMed] [Google Scholar]
- Staun S. L.; Sergentu D. C.; Wu G.; Autschbach J.; Hayton T. W. Use of 15N NMR Spectroscopy to Probe Covalency in a Thorium Nitride. Chem. Sci. 2019, 10, 6431–6436. 10.1039/C9SC01960J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergentu D. C.; Kent G. T.; Staun S. L.; Yu X.; Cho H.; Autschbach J.; Hayton T. W. Probing the Electronic Structure of a Thorium Nitride Complex by Solid-State 15N NMR Spectroscopy. Inorg. Chem. 2020, 59, 10138–10145. 10.1021/acs.inorgchem.0c01263. [DOI] [PubMed] [Google Scholar]
- Du J.; Hurd J.; Seed J. A.; Balázs G.; Scheer M.; Adams R. W.; Lee D.; Liddle S. T. 31P Nuclear Magnetic Resonance Spectroscopy as a Probe of Thorium-Phosphorus Bond Covalency: Correlating Phosphorus Chemical Shift to Metal-Phosphorus Bond Order. J. Am. Chem. Soc. 2023, 145, 21766–21784. 10.1021/jacs.3c02775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. L.; Batista E. R.; Boncella J. M.; Gaunt A. J.; Reilly S. D.; Scott B. L.; Tomson N. C. A Linear trans-Bis(imido) Neptunium(V) Actinyl Analog: NpV(NDipp)2(tBu2bipy)2Cl (Dipp = 2,6-iPr2C6H3). J. Am. Chem. Soc. 2015, 137, 9583–9586. 10.1021/jacs.5b06667. [DOI] [PubMed] [Google Scholar]
- Dutkiewicz M. S.; Goodwin C. A. P.; Perfetti M.; Gaunt A. J.; Griveau J.-C.; Colineau E.; Kovács A.; Wooles A. J.; Caciuffo R.; Walter O.; Liddle S. T. A Terminal Neptunium(V)-Mono(Oxo) Complex. Nat. Chem. 2022, 14, 342–349. 10.1038/s41557-021-00858-0. [DOI] [PubMed] [Google Scholar]
- Goodwin C. A. P.; Wooles A. J.; Murillo J.; Lu E.; Boronski J. T.; Scott B. L.; Gaunt A. J.; Liddle S. T. Carbene complexes of neptunium. J. Am. Chem. Soc. 2022, 144, 9764–9774. 10.1021/jacs.2c02152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murillo J.; Seed J. A.; Wooles A. J.; Oakley M. S.; Goodwin C. A. P.; Gregson M.; Dan D.; Chilton N. F.; Gaunt A. J.; Kozimor S. A.; Liddle S. T.; Scott B. L. Carbene complexes of plutonium: structure, bonding, and divergent reactivity to lanthanide analogs. J. Am. Chem. Soc. 2024, 146, 4098–4111. 10.1021/jacs.3c12719. [DOI] [PMC free article] [PubMed] [Google Scholar]