

Summary
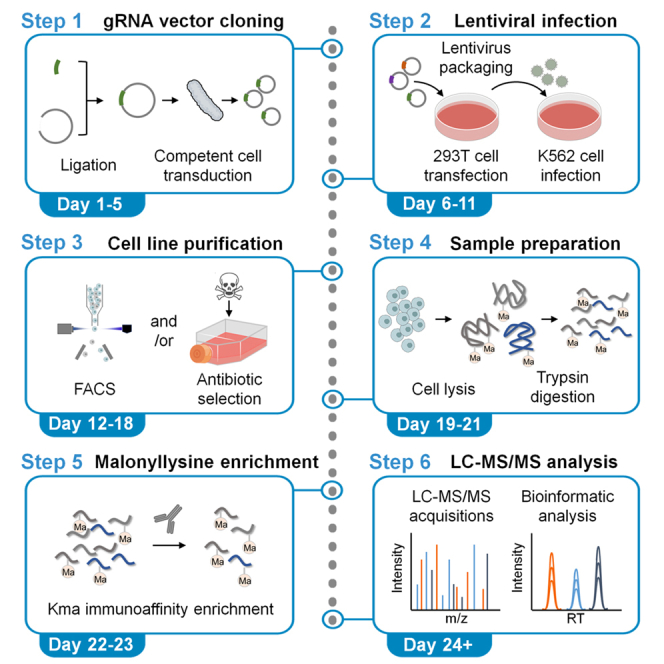
Lysine malonylation is a protein posttranslational modification. We present a protocol to generate stable gene-knockdown K562 cell lines through lentiviral infection of a CRISPR interference (CRISPRi) system followed by lysine malonylation measurement using mass spectrometry (MS). We detail guide RNA (gRNA) vector cloning, lentiviral infection, cell line purification, protein digestion, malonyl-lysine enrichment, desalting, and MS acquisition and analysis.
For complete details on the use and execution of this protocol, please refer to Zhang et al.1 and Bons et al.2
Subject areas: Molecular Biology, protein Biochemistry, Proteomics
Graphical abstract
Highlights
-
•
The CRISPRi system enables efficient and stable gene knockdown in K562 cells
-
•
CRISPRi K562 cell lines can be purified through FACS or antibiotic selection
-
•
Malonyl-lysine-specific antibody beads allow the enrichment of malonylated peptides
-
•
Malonylated peptides can be measured by data-independent acquisition mass spectrometry
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Lysine malonylation is a protein posttranslational modification. We present a protocol to generate stable gene-knockdown K562 cell lines through lentiviral infection of a CRISPR interference (CRISPRi) system followed by lysine malonylation measurement using mass spectrometry (MS). We detail guide RNA (gRNA) vector cloning, lentiviral infection, cell line purification, protein digestion, malonyl-lysine enrichment, desalting, and MS acquisition and analysis.
Before you begin
The protocol below describes the specific steps for creating a stable gene knockdown K562 cell line and subsequently identifying malonylated proteins in the cells. However, we have also used this protocol to knock down genes in Jurkat and HEK293 cell lines, and to measure lysine malonylation in mouse tissues.
Obtaining your choice of dCas9-KRAB-expressing cell line
-
1.
Generate dCas9-KRAB (or dCas9-KARB-MeCP2)-expressing K562 cell line in house according to Gilbert et al.3
Note: Ours were a gift from the Jonathan Weissman Lab. At the time of preparation of this protocol, dCas9-KRAB-expressing HEK-293, MCF7, and U-87 MG cell lines were available from ATCC (ATCC.org) if needed.
Implementation of BSL2 containment
-
2.
Make sure that BSL2 containment is implemented since lentiviruses have the capability of transducing human cells.
Oligonucleotide synthesis
Timing: 3 days
-
3.Choose the top ranked (two or more) sgRNA protospacer sequences targeting your gene of interest scored by CRISPRi-v2.1 algorithm.4 Order synthesized DNA oligonucleotide pairs (both sense and anti-sense) that contain the protospacer sequence from the service provider of your choice (e.g., Elim Biopharm, CA).Note: Gel- or HPLC-purification is not necessary. Standard non-phosphorylated oligos would suffice. From 5′ to 3′ end, the paired oligos should contain a BstXI site (sticky end), ‘G’, the protospacer (19 bp), the protospacer adjacent motif (PAM), and a BlpI site (sticky end). The sequences are as shown in the following format:
-
a.Sense (33 nt): 5′-TTGGNNNNNNNNNNNNNNNNNNNGTTTAAGAGC-3′.
-
b.Anti-sense (40 nt): 5′- TTAGCTCTTAAACNNNNNNNNNNNNNNNNNNNCCAACAAG-3′.Note: Remember to include scramble negative control sgRNA sequences in your study.
-
a.
-
4.
Design primer pairs for qPCR examination of the expression level of your gene of interest using your favorite software (e.g., Primer Premier). Order synthesized DNA oligonucleotide pairs (both sense and anti-sense) from the service provider of your choice (e.g., Elim Biopharm, CA).
Note: Gel- or HPLC-purification is not necessary. Standard non-phosphorylated oligos would suffice.
-
5.
Reconstitute lyophilized oligonucleotides with nuclease-free water to make 100 μM stock solutions, and store at −20°C.
Culture cell lines
-
6.
Recover a frozen aliquot of 293T cells, and culture cells in complete high glucose DMEM (containing 10% FBS) used for lentivirus production. 1:4 split cells once the cells reach 80% confluence.
Note: Make sure that 293T cells are in log phase for lentivirus packaging.
-
7.
Recover a frozen aliquot of K562 dCas9-KRAB cells, and culture cells in complete RPMI 1640 medium (containing 10% FBS) used for making CRISPRi K562 cell lines via lentiviral infection. Make sure that K562 dCas9-KRAB cells are in log phase for lentivirus infection.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-β-tubulin (dilution 1:1,000 for western blotting) | Cell Signaling Technology | Cat. #2128, RRID:AB_823664 |
| Rabbit monoclonal anti-KAT2A (dilution 1:1,000 for western blotting) | Cell Signaling Technology | Cat. #3305, RRID:AB_2128281 |
| Rabbit monoclonal anti-KAT2B (dilution 1:1,000 for western blotting) | Cell Signaling Technology | Cat. #3378 RRID:AB_2128409 |
| Rabbit monoclonal anti-SIRT5 (dilution 1:1,000 for western blotting) | Cell Signaling Technology | Cat. #8782, RRID:AB_2716763 |
| Chemicals, peptides, and recombinant proteins | ||
| Acetonitrile (Burdick & Jackson; LC-MS grade) | Honeywell | Cat. #LC015 |
| BlpI | New England Biolabs | Cat. #R0585 |
| BstXI | New England Biolabs | Cat. #R0113 |
| DL-dithiothreitol (DTT) | Sigma-Aldrich | Cat. #D9779 |
| Formic acid (FA; Fluka; Puriss.-grade) | Honeywell | Cat. #27001 |
| Halt protease/phosphatase single-use inhibitor cocktail (100x) | Thermo Fisher Scientific | Cat. #78442 |
| Iodoacetamide (IAA) | Sigma-Aldrich | Cat. #I1149 |
| Nicotinamide | Sigma-Aldrich | Cat. #72340 |
| Quick Ligation kit | New England Biolabs | Cat. #M2200 |
| Phosphate-buffered saline (PBS; Gibson), pH 7.4 (1x) | Thermo Fisher Scientific | Cat. #10010-023 |
| Puromycin | Santa Cruz Biotechnology | Cat. #sc-108071 |
| Sodium chloride | VWR | Cat. #BDH9286 |
| Trichostatin A | Cayman Chemical | Cat. #89730 |
| Triethylammonium bicarbonate (TEAB), pH8 (1 M) | Sigma-Aldrich | Cat. #7408 |
| Trifluoroacetic acid (TFA; HPLC-grade) | Sigma-Aldrich | Cat. #302031 |
| Trypsin (sequencing-grade modified) | Promega | Cat. #V5111 |
| Urea | Thermo Fisher Scientific | Cat. #29700 |
| Water (Burdick & Jackson; LC-MS grade) | Honeywell | Cat. #LC365 |
| Critical commercial assays | ||
| Lipofectamine 3000 | Invitrogen | Cat. #L3000 |
| Quick-DNA microprep kit | Zymo Research | Cat. #3020 |
| ZymoPURE plasmid miniprep kit | Zymo Research | Cat. #4210 |
| iRT kit | Biognosys | https://biognosys.com/product/irt-kit/ |
| Pierce BCA protein assay | Thermo Fisher Scientific | Cat. #23225 |
| PTMScan malonyl-lysine [Mal-K] kit | Cell Signaling Technology | Cat. #93872 |
| Deposited data | ||
| Raw data and complete CRISPRi K562 cell malonylome and proteome MS datasets | N/A | MassIVE ID number: MSV000093564; ProteomeXchange ID: PXD047514 ftp://massive.ucsd.edu/v06/MSV000093564/ |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | CRL-3216 |
| Human: K562-dCas9-KRAB | Gilbert et al.3 | N/A |
| Recombinant DNA | ||
| pCMV-dR8.91 packaging vector | Creative Biogene | Cat. #OVT2971 |
| pMD2.G envelope vector | Addgene | RRID:Addgene_12259 |
| pU6-sgRNA-EF1Alpha-puro-T2A-BFP | Addgene | RRID:Addgene_60955 |
| Software and algorithms | ||
| CRISPRi-v2.1 algorithm | Horlbeck et al.4 | N/A |
| FlowJo | Becton Dickinson & Company | https://www.flowjo.com |
| GraphPad Prism 6 | GraphPad | https://graphpad.com/ |
| Skyline-daily | MacLean et al.10 | Skyline version 21.1.1.223; https://skyline.ms |
| SnapGene | Dotmatics | https://www.snapgene.com/ |
| Spectronaut | Biognosys | Spectronaut version 15.1.210713.50606; https://biognosys.com/software/spectronaut/ |
| Other | ||
| Acclaim PepMap 100 C18 trap column (75 μm × 2 cm, 3 μm particle size | Thermo Fisher Scientific | PN 164535 |
| Acclaim PepMap 100 C18 analytical column (75 μm × 50 cm, 3 μm particle size | Thermo Fisher Scientific | PN 164570 |
| Benchtop centrifuge | N/A | N/A |
| Empore 2215 C18 47-mm | CDS Analytical | Cat. #98-0604-0217-3EA |
| Flat-ended 200-μL pipette tips | N/A | N/A |
| Microtubes 0.65 mL | N/A | N/A |
| Microtubes 1.5 mL | N/A | N/A |
| Microtubes 2 mL | N/A | N/A |
| Oasis HLB 1 cc cartridges, 30 mg sorbent | Waters | Cat. #WAT058882 |
| Orbitrap Eclipse Tribrid mass spectrometer | Thermo Fisher Scientific | N/A |
| Probe sonicator | N/A | N/A |
| Rotating mixer | N/A | N/A |
| SpeedVac vacuum concentrator | N/A | N/A |
| Thermomixer | N/A | N/A |
| UltiMate 3000 HPLC system | Thermo Fisher Scientific | N/A |
| Vacuum manifold | N/A | N/A |
Materials and equipment
FACS buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS (1 ×) | 1 × | 9760 μL |
| FBS | 2% | 200 μL |
| EDTA (0.5 M) | 2 mM | 40 μL |
| Total | N/A | 10 mL |
Note on storage conditions: FACS buffer can be stored at 4°C for a week after preparation.
Puromycin selection RPMI 1640 medium
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 (1 ×) | 1 × | 44.5 mL |
| FBS | 10% | 5 mL |
| Pen-Strep (100 ×) | 1 × | 500 μL |
| Puromycin (10 mg/mL) | 1 μg/mL | 5 μL |
| Total | N/A | 50 mL |
Note on storage conditions: Cell culture media can be stored at 4°C for a week after preparation.
Lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Urea | 8 M | 4.8 mg |
| Water | N/A | 6.748 mL |
| TEAB, pH 8 (1 M) | 200 mM | 2 mL |
| Nicotinamide (30 mM) | 3 mM | 1 mL |
| Sodium chloride (5 M) | 75 mM | 150 μL |
| HALT protease/phosphatase single-use inhibitor cocktail (100x) | 1x | 100 μL |
| Trichostatin A (5 mM) | 1 μM | 2 μL |
| Total | N/A | 10 mL |
Note on storage conditions: The lysis buffer should be prepared immediately before use.
8 M urea, 50 mM TEAB, pH 8 buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Urea | 8 M | 4.8 g |
| Water | N/A | 9.5 mL |
| TEAB, pH 8 (1 M) | 50 mM | 500 μL |
| Total | N/A | 10 mL |
Note on storage conditions: The 8 M urea solution should be prepared immediately before use.
50 mM TEAB, pH 8 solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Water | N/A | 9.5 mL |
| TEAB, pH 8 (1 M) | 50 mM | 500 μL |
| Total | N/A | 10 mL |
Note on storage conditions: TEAB solution can be stored at 4°C for a month after preparation.
250 mM DTT solution
| Reagent | Final concentration | Amount |
|---|---|---|
| DTT | 250 mM | 38.6 mg |
| 50 mM TEAB, pH 8 | N/A | 1 mL |
| Total | N/A | 1 mL |
Note on storage conditions: The DTT solution should be prepared immediately before use.
200 mM IAA solution
| Reagent | Final concentration | Amount |
|---|---|---|
| IAA | 200 mM | 37.0 mg |
| 50 mM TEAB, pH 8 | N/A | 1 mL |
| Total | N/A | 1 mL |
Note on storage conditions: The IAA solution should be prepared immediately before use and kept away from light.
10% formic acid solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Water | N/A | 9 mL |
| Formic acid | 10% | 1 mL |
| Total | N/A | 10 mL |
Note on storage conditions: 10% formic acid solution can be stored at 20°C–25°C for up to a year.
CRITICAL: Concentrated formic acid solutions are corrosive and flammable, and can cause severe burns to the skin, eyes, mouth, respiratory and GI tract during even short exposures. Proper gloves, goggles, and a chemical-resistant smock should be worn, and a chemical fume hood should be used when working with these solutions.
0.2% formic acid solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Water | N/A | 9.98 mL |
| Formic acid | 0.2% | 20 μL |
| Total | N/A | 10 mL |
Note on storage conditions: 0.2% formic acid solution can be stored at 20°C–25°C for up to a year.
50% acetonitrile, 0.2% formic acid solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Acetonitrile | 50% | 5 mL |
| Water | N/A | 4.98 mL |
| Formic acid | 0.2% | 20 μL |
| Total | N/A | 10 mL |
Note on storage conditions: 50% acetonitrile/0.2% formic acid solution can be stored at 20°C–25°C for up to a year.
0.15% trifluoroacetic acid solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Water | N/A | 9.85 mL |
| Trifluoroacetic acid | 0.15% | 15 μL |
| Total | N/A | 10 mL |
Note on storage conditions: 0.15% trifluoroacetic acid solution can be stored at 20°C–25°C for up to a year.
HPLC solvent A
| Reagent | Final concentration | Amount |
|---|---|---|
| LC-MS-grade water | 98% | 490 mL |
| Acetonitrile | 2% | 10 mL |
| Formic acid | 0.1% | 500 μL |
| Total | N/A | 500 mL |
HPLC solvent B
| Reagent | Final concentration | Amount |
|---|---|---|
| Acetonitrile | 98% | 490 mL |
| LC-MS-grade water | 2% | 10 mL |
| Formic acid | 0.1% | 500 μL |
| Total | N/A | 500 mL |
Note on storage conditions: HPLC Solvents A and B can be stored at 20°C–25°C for up to a year.
Step-by-step method details
Cloning the sgRNA into the lentiviral vector
This section aims to generate the gRNA-expressing transfer plasmid for the production of gRNA-expressing lentivirus.
-
1.
Anneal the sense and anti-sense sgRNA DNA oligos (Table 1) by heating on a metal block at 95°C for 5 min, then gradually cool to 20°C–25°C on bench.
Note: The cooling step may take an hour or more.
Note: The annealed oligos can be placed on ice, or stored at −20°C, until used in the ligation step, Step 3.
-
2.Linearize the pU6-sgRNA-EF1Alpha-puro-T2A-BFP backbone using restriction endonucleases BstXI and BlpI.
-
a.BstXI digestion at 37°C for 1 h (Table 2).
-
b.Heat the reaction mixture at 80°C for 10 min to inactivate BstXI.
-
c.Purify the singly digested backbone vector using a DNA prep kit (Zymo Research, Cat. #D3020, https://zymoresearch.eu/products/quick-dna-microprep).
-
d.Measure the DNA concentration using a NanoDrop spectrophotometer.
-
e.BlpI digestion at 37°C for 1 h (Table 3).
-
f.BlpI cannot be inactivated by heating. Recover the digested backbone vector using a DNA prep kit (Zymo Research, Cat. #D3020, https://zymoresearch.eu/products/quick-dna-microprep).
-
g.Measure the DNA concentration using a NanoDrop spectrophotometer.
-
a.
Note: For the optimal results, instead of doing double digestion, perform single digestion sequentially (37°C, 1 h each) using the optimal NEBuffers for BstXI and BlpI, respectively.
Note: The linearized backbone vector can be stored at −20°C for future use.
Note: Alternatively, perform double digestion in NEBuffer r2.1 (or NEBuffer 2 + rAlbumin) at 37°C with additional units of enzyme and/or longer incubation time.
-
3.
Ligate the annealed oligos with the linearized backbone vector. Gently mix the ligation mixture (Table 4) by pipetting up and down, and then microfuge briefly. Incubate at 20°C–25°C for 5 min.
-
4.Transform ligated vector into competent E. coli cells.
-
a.Chill the ligation mixture on ice, and transform 1 μL of the reaction into 5 μL competent 5-alpha E. coli.
-
b.Incubate on ice for 30 min.
-
c.Heat shock cells at 42°C for 45 s, put immediately back on ice for ∼1 min.
-
d.Add 10 μL liquid LB medium, then immediately plate all 6 μL of transformed bacteria onto an ampicillin LB agarose plate.
-
e.Incubate in a 37°C incubator for 16 h.
-
a.
Note: We used 5-alpha E. coli (New England Biolabs, Cat. #C2987) in our studies, but other commonly used cloning competent cell strains, including 10-beta E. coli (New England Biolabs, Cat. #C3019), could also be used.
-
5.Pick 1–4 colonies per sgRNA construct.
-
a.Culture each in 4 mL ampicillin LB liquid medium at 37°C, shaking at 220 rpm for 12–18 h.
-
b.Purify plasmids using an endo-toxin free miniprep kit (Zymo Research, Cat. #D4210, https://zymoresearch.eu/collections/zymopure-plasmid-kits/products/zymopure-ii-plasmid-miniprep-kit).
-
a.
Note: Save the plate at 4°C for a few days until the construct is confirmed by Sanger sequencing.
-
6.
Verify the construct by Sanger sequencing through the service provider of your choice (e.g., Elim Biopharm, CA) using the sgRNA-R-seq primer (CCTTCTCTAGGCACCGGTTC). Align the sequence with the backbone vector and the protospacer targeting your gene of interest using the software of your choice (e.g., SnapGene).
Note: The desired construct should be the backbone vector inserted with the correct protospacer. If none of the plasmids is correct, repeat Step 5 by picking new colonies, or start over from Step 1. Troubleshooting 1.
Pause point: The correct sgRNA-expressing lentiviral vector can be stored at −20°C for future use.
Table 1.
Annealing mixture
| Component | Vol. |
|---|---|
| 100 μM sense oligo | 1 μL |
| 100 μM anti-sense oligo | 1 μL |
| 10 × rCutSmart | 5 μL |
| Nuclease-free Water | to 50 μL |
Table 2.
BstXI digestion mixture
| Component | Vol. |
|---|---|
| Purified DNA | 1 μg (can be scaled up) |
| NEBuffer r3.1 (10 ×) | 5 μL |
| BstXI | 1 μL (10 units) |
| Nuclease-free Water | to 50 μL |
Table 3.
BlpI digestion mixture
| Component | Vol. |
|---|---|
| DNA | 1 μg (can be scaled up) |
| rCutSmart (10 ×) | 5 μL |
| BlpI | 1 μL (10 units) |
| Nuclease-free Water | to 50 μL |
Table 4.
Ligation mixture
| Component | Vol. |
|---|---|
| 1:20 diluted annealed oligos | 1 μL |
| Linearized backbone vector | 50 ng |
| Quick Ligase Reaction Buffer (2 ×) | 5 μL |
| Quick Ligase | 0.5 μL |
| Nuclease-free Water | to 10 μL |
Lentiviral production and infection
This section aims to produce gRNA-expressing lentivirus, which is then used to infect the dCas9-KRAB K562 cells.
-
7.
Prepare the sgRNA-expressing lentiviral transfer plasmid and the packaging (pCMV-dR8.91) and envelope (pMD2.G) plasmids.
-
8.
The day before transfection, plate 5–6 × 106 293T cells in 10 mL complete DMEM (containing 10% FBS) in a 10 cm tissue culture treated plate, so that the cells reach ∼80% confluence right before plasmid transfection.
-
9.
Replace media with fresh media 2 h before transfection. Co-transfect the gRNA-expressing transfer vector with packaging (pCMV-dR8.91) and envelope (pMD2.G) plasmids, using Lipofectamine 3000 (Invitrogen, Cat. #L3000001, https://www.thermofisher.com/order/catalog/product/L3000001) (Table 5).
Note: Since the lentivirus transfer plasmid encodes BFP, blue fluorescence should be seen 48 h after successful transfection.
-
10.Collect lentivirus-containing media.
-
a.72 h after transfection, collect the media containing lentivirus to a 15 mL tube.
-
b.Filter the supernatant through 0.45 μm PVDF syringe filter to remove cell debris.
-
c.Aliquot viral supernatant to 1.5 mL tube (500 μL each).
-
d.Snap-freeze on dry ice.
-
e.Store at −80°C, and avoid freeze/thaw cycles.
-
a.
-
11.
Culture dCas9-KRAB K562 cells in complete RPMI 1640 medium (containing 10% FBS).
-
12.Prepare K562 cells for lentivirus spinoculation.
-
a.For each sgRNA construct, aliquot 5 × 105 cells into a 1.5 mL eppendorf tube for lentiviral spinoculation.
-
b.Centrifuge the cells at 400 g at 20°C–25°C for 5 min.
-
c.Remove supernatant media, and resuspend the cells with 500 μL lentiviral supernatant without making bubbles.
-
a.
-
13.
Spinoculate the cells with lentivirus by centrifugation at 1000 g at 32°C for 2 h.
-
14.Culture lentivirus-spinoculated cells.
-
a.Remove the supernatant media, resuspend the cells with 1 mL pre-warmed fresh complete RPMI 1640 media, and seed the cells into 1 well of a 6-well plate.
-
b.Add 3 mL pre-warmed fresh complete RPMI 1640 media to the well (a total of 4 mL media in the well). Culture for 72 h before FACS or antibiotic selection.
-
a.
Note: Lentiviral infection parameters should be established for each cell line, and polybrene (2–10 μg/mL) may be used to increase infection efficiency. For sgRNA constructs in K562 cells, we spin-infected 5 × 105 cells with 500 μL of fresh viral supernatant which results in ∼20% infection.
Table 5.
Transfection mixture
| Component | Amount | Notes |
|---|---|---|
| Opti-MEM | 125 μL | Vortex 2–3 s. |
| lipo3000 | 5 μL | |
| Opti-MEM | 125 μL | Mix well, then add diluted DNA + P3000 reagent to diluted lipo3000. Incubate at 20°C–25°C for 15 min. Then add the 250 μL mixture to cells. |
| P3000 reagent | 5 μL | |
| pCMV-dR8.91 | 1 μg | |
| pMD2.G | 0.5 μg | |
| gRNA transfer vector | 1 μg |
CRISPRi cell selection
This section aims to purify CRISPRi K562 cell lines by FACS and/or antibiotic selection.
-
15.Prepare K562 cells for FACS and/or antibiotic selection.
-
a.72 h after lentiviral inoculation, collect ∼5 × 105 cells from each well.
-
b.Wash the cells by resuspension and pelleting with 500 μL ice cold FACS buffer twice.
-
c.Resuspend the cells in a third aliquot of 500 μL ice cold FACS buffer.
-
a.
-
16.
Analyze the cells on the flow cytometer (e.g., BD FACS Aria2) for BFP expression as quickly as possible to keep the cells healthy. Collect BFP (+) cells. Subculture the cells. Troubleshooting 2.
Optional: Since the lentiviral vector also contains a puromycin-resistance gene, non-infected cells can be further removed by culturing in complete RPMI 1640 media containing 1 μg/mL puromycin.
Note: To achieve a stable cell population, the antibiotic selection should last at least as long as it takes the non-infected cells to completely die. Typically, 2 days of puromycin selection at 1 μg/mL will enrich the population to ∼90% infected. We treated the cells with 1 μg/mL puromycin for 5 days. After that, if the antibiotic is removed from the culture, check the cells regularly to confirm the gene knockdown efficiency or BFP expression. Once the knockdown efficiency is confirmed, the purified CRISPRi cell lines can be used for subsequent lysine malonylation examination.
-
17.
Evaluate the gene knockdown efficiency of the CRISPRi cell lines by comparing the target gene expression levels in the gene-targeting sgRNA-expressing cell line with the scramble sgRNA-expressing negative control cell line through RNA extraction, reverse transcription, and qPCR.
Note: Gene knockdown efficiency should also be confirmed by western blotting if possible. Troubleshooting 3.
Proteomics sample preparation
This section includes three procedures: cell lysis, in solution protein digestion with Trypsin, and proteolytic peptide C18 hydrophilic–lipophilic balance (HLB) desalting.
Note: All the following steps are performed using ∼4 × 106 human CRISPRi K562 cells.
-
18.Cell lysis.
-
a.Add 300 μL of lysis buffer into each sample obtained in Step 16.Note: The volume of lysis buffer may need to be adjusted depending on the number of cells used to ensure that the pellet is fully immersed and the cells are fully homogenized and lysed. If needed, we recommend to add 100 μL of lysis buffer at the time to avoid over-diluting the protein material.
-
b.Sonicate 2 times for 15 s on 30% amplitude using a probe sonicator.Note: Let the sample cool on ice for 30 s in between each round of sonication.
-
c.Centrifuge at 15,700 g at 4°C for 15 min.
-
d.Aspirate and transfer the supernatant (solubilized protein lysate) to a clean 1.5-mL microcentrifuge tube, avoiding any debris at the bottom of the tube.
-
a.
-
19.Protein reduction and alkylation.
-
a.Determine the protein concentration using Thermo Fisher Scientific Pierce BCA Protein Assay (Cat. #23225).Note: Any other protein concentration determination assays can be used that is compatible with the lysis buffer components.
-
b.Transfer the same amount of proteins for each protein lysate to a clean 1.5-mL microcentrifuge tube.Note: All the following steps were performed with 800 μg proteins. The remaining protein lysates can be long-term stored at −80°C.CRITICAL: Transfer 800 μg proteins minimum to ensure successful affinity enrichment of malonylated peptides. Troubleshooting 4.
-
c.Add 8 M urea, 50 mM TEAB, pH 8 buffer to bring every sample to the same volume.
-
d.Reduce the proteins by adding 250 mM DTT solution to make a final concentration of 20 mM DTT. Incubate for 30 min at 37°C and 1,400 rpm in a thermomixer.
-
e.Incubate for 10 min at 20°C–25°C.
-
f.Alkylate the proteins by adding 200 mM IAA solution to make a final concentration of 40 mM IAA. Incubate for 30 min at 20°C–25°C in the dark.
-
a.
-
20.In-solution protein digestion.
-
a.Dilute the alkylated protein lysate by adding 50 mM TEAB solution to a final concentration of 0.8 M urea.CRITICAL: Check the pH using a pH paper strip to ensure that the pH of the solution is 8. Both urea dilution down to 0.8 M and pH at 8 are required for optimal activity of Trypsin.
-
b.Add the solution of sequencing-grade trypsin to a 1:50 (wt:wt) enzyme:protein ratio.
-
c.Incubate 12–18 h at 37°C and 1,400 rpm in a thermomixer.
-
d.Acidify with 10% FA solution to a final concentration of 1% FA to quench the trypsin digestion.CRITICAL: Check the pH using a pH paper strip to ensure that the pH of the solution is 2–3.
-
a.
-
21.Peptide desalting with a C18 hydrophilic–lipophilic balance (HLB) cartridge.
-
a.Centrifuge at 1,800 g for 15 min at 20°C–25°C to pellet any insoluble material.
-
b.Desalt the proteolytic peptides with Oasis HLB 1 cc cartridges, 30 mg sorbent (Waters, Cat. #WAT058882).
-
i.Wet the cartridge two times with 800 μL of 50% ACN, 0.2% FA.
-
ii.Equilibrate the cartridge three times with 0.2% FA.
-
iii.Load the peptide sample on the cartridge.
-
iv.Wash the cartridge three times with 0.2% FA.
-
v.Elute the peptide sample one time with 800 μL of 50% ACN, 0.2% FA and one time with 400 μL of 50% ACN, 0.2% FA into a clean 1.5-mL microcentrifuge tube.Note: Any other C18 desalting devices can be used that is compatible with the solution pH and binding capacity requirement.
-
i.
-
c.Transfer 100 μg of eluted peptide solution to a clean 0.65-mL microcentrifuge tube for determining the protein level changes and normalizing the malonyl level changes. The remaining 700-μg eluted peptide solution is used for the affinity enrichment of malonylated peptides (Figure 1).Note: The peptide concentration can be verified at this step using appropriate assays, before transferring 100 μg of eluted peptide solution.Pause point: The protocol can be paused here by long-term storing the samples at −80°C.
-
d.Vacuum dry the two eluted peptide solutions in a Speedvac.
-
e.Resuspend the 100-μg dried peptides in 100 μL of 0.2% FA for a final concentration of 1 μg/μL and thoroughly mix the solution. Centrifuge at 12,000 g at 20°C–25°C for 2 min.Note: The 100-μg samples can be used directly for LC-MS/MS analysis or short-term stored at −20°C before LC-MS/MS analysis. Before transferring each sample to an autosampler vial, indexed retention time peptides (iRT; Biognosys)5 are spiked into the sample according to manufacturer’s instructions.
-
a.
Figure 1.

Strategy for the enrichment of malonylated peptides from a solution of proteolytic peptides
After protein reduction, alkylation, trypsin digestion, and proteolytic peptide HLB desalting, 100-μg of the peptide solution is aliquoted for protein-level analysis to identify protein changes and to normalize malonylated peptide abundance. The remaining material is used to enrich the malonylated peptides by immunoprecipitation using antibody-coated beads. The resulting enrichment samples are used to determine malonyl-level changes.
Malonyl-lysine immunoaffinity enrichment
This section includes two procedures: affinity-based enrichment of malonylated peptides and preparation of C18 stage tips for enriched malonylated peptide desalting. The affinity-based enrichment procedure is based on the instructions from the PTMScan Malonyl-Lysine [Ma-K] Kit (Cell Signaling Technology, Cat. #93872). For more details and troubleshooting, please refer to the Cell Signaling Technology website:
https://www.cellsignal.com/products/proteomic-analysis-products/malonyl-lysine-mal-k-kit/93872.
Note: Keep IAP buffer, 1x PBS buffer, and water on ice until use.
-
22.Affinity-based enrichment of malonylated peptides: pull down.
-
a.Resuspend the 700-μg dried peptides from Step 21.d. with 1.4 mL of cold IAP buffer and thoroughly mix the solution.CRITICAL: Check the pH using a pH paper strip to ensure that the pH of the solution is ∼7.
-
b.Centrifuge at 10,000 g at 4°C for 10 min. A small insoluble pellet may appear.
-
c.Wash the antibody beads by adding 1 mL of cold 1x PBS to a tube of antibody-bead slurry and thoroughly mix by pipetting. Transfer the solution to a clean 1.5-mL microcentrifuge tube. Spin down the antibody beads with a mini centrifuge at 20°C–25°C for 30 s and wait for 1 min to ensure efficient bead settling to the bottom of the tube.
-
d.Carefully remove the buffer from the antibody beads.Note: Avoid removing the last few microliters of PBS buffer to avoid aspirating the antibody beads.
-
e.Wash the antibody beads three more times by repeating three times Steps 22.c. and 22.d for a total of 4 × 1 mL washes.
-
f.Prepare a pre-cut 200-μL pipette tip by cutting off 7 mm of the lower end of a 200-μL pipette tip to obtain a wide-bore, 1-mm orifice tip.Note: Wide-bore, 1-mm orifice 200-μL pipette tips are commercially available as well.
-
g.Resuspend the washed beads in 440 μL of cold 1x PBS by pipetting up and down several times with a pre-cut 200-μL pipette tip to mix. Transfer 100 μL each into 4 clean 1.5-mL microcentrifuge tubes ensuring that the antibody bead solution is well mixed.Note: The leftover antibody bead solution (40 μL) ensures the same volume of beads per sample.CRITICAL: Depending on the starting amount of material, the volume of beads per sample must be adjusted.
-
h.Spin down the antibody beads with a mini centrifuge at 20°C–25°C for 30 s and wait for 1 min to ensure efficient bead settling to the bottom of the tube.
-
i.Carefully remove all buffer from the antibody beads with a gel loading 200-μL pipette tip with a flat 0.2-mm thick end.Note: The gel loading 200-μL pipette tip with a flat 0.2-mm thick end helps preventing aspirating antibody beads.CRITICAL: Ensure that each tube has a similar antibody bead pellet at the bottom.
-
j.Aspirate and transfer the supernatant of the peptide sample from Step 22.b. directly onto the washed antibody beads.
-
k.Incubate 12–18 h at 4°C in a rotating mixer.
-
a.
-
23.Affinity-based enrichment of malonylated peptides: elution.
-
a.Remove the sample from the rotating mixer and allow the antibody beads to settle for 30 min at 4°C.
-
b.Centrifuge at 2,000 g at 4°C for 30 s.
-
c.Aspirate and transfer the supernatant solution (non-malonylated peptide solution) to a clean 1.5-mL microcentrifuge tube. Save the flow-through solution at −80°C.Note: Avoid removing the last few microliters of supernatant solution to avoid aspirating the antibody beads.
-
d.Wash the antibody beads by adding 1 mL of cold IAP buffer. Carefully mix by inverting the tube five times and allow the antibody beads to settle for 5 min at 4°C.
-
e.Centrifuge at 2,000 g at 4°C for 30 s.
-
f.Carefully remove the buffer from the antibody beads.Note: Avoid removing the last few microliters of IAP buffer to avoid aspirating the antibody beads.
-
g.Wash the antibody beads two more times by repeating Steps 23.d. to 23.f for a total of 3 × 1 mL IAP buffer washes.
-
h.Wash the antibody beads by adding 1 mL of cold water. Carefully mix by inverting the tube five times and allow the antibody beads to settle for 5 min at 4°C.
-
i.Centrifuge at 2,000 g at 4°C for 30 s.
-
j.Carefully remove the water from the antibody beads.Note: Avoid removing the last few microliters of water to avoid aspirating the antibody beads.
-
k.Wash the antibody beads two more times by repeating Steps 23.h. to 23.j for a total of 3 × 1 mL water washes.
-
l.Centrifuge at 2,000 g at 4°C for 30 s.
-
m.Carefully remove all water from the antibody beads with a gel loading 200-μL pipette tip with a flat 0.2-mm thick end.
-
n.Elute the enriched malonylated peptides by carefully adding 55 μL of 0.15% TFA to the antibody beads. Incubate for 10 min at 20°C–25°C. Gently tap the bottom of the tube on the benchtop to mix intermittently.
-
o.Spin down the antibody beads with a mini centrifuge at 20°C–25°C for 30 s and wait for 1 min to ensure efficient bead settling to the bottom of the tube.
-
p.Transfer the eluted peptide solution to a clean 0.65-mL microcentrifuge tube using a wide-bore 200-μL pipette tip.
-
q.Elute the beads again by carefully adding 45 μL of 0.15% TFA to the antibody bead pellet. Incubate for 10 min at 20°C–25°C. Gently tap the bottom of the tube to mix on the benchtop intermittently.
-
r.Spin down the antibody beads with a mini centrifuge at 20°C–25°C for 30 s and wait for 1 min to ensure efficient bead settling to the bottom of the tube.
-
s.Use a wide-bore 200-μL pipette tip to transfer the second eluate and combine it with the first peptide elution.
-
t.Centrifuge the eluates at 12,000 g at 20°C–25°C for 5 min to pellet any antibody residual beads that carried over.
-
a.
-
24.Malonylated peptide desalting with C18 StageTip.
-
a.Prepare the StageTip by inserting three Empore 2215 C18 47-mm (CDS Analytical, Cat. # 98-0604-0217-3EA) disks into a 200-μL pipette tip using an 18-gauge blunt-ended syringe needle. Snugly pack the C18 disks in the tip using a thin LC tubing. Place the StageTip into a tip holder (for instance a bottom-pierced clean 0.65-mL microcentrifuge tube) and assemble into a clean 2-mL microcentrifuge tube.Note: Any other small-scale C18 desalting devices can be used that is compatible with the solution pH and binding capacity requirement.
-
b.Wet the StageTip with 100 μL of 100% ACN and centrifuge at 3,000 g at 20°C–25°C for 1 min.
-
c.Wash the StageTip with 100 μL of 50% ACN, 0.2% FA and centrifuge at 3,000 g at 20°C–25°C for 1 min.
-
d.Equilibrate the StageTip two times with 100 μL of 0.2% FA and centrifuge at 3,000 g at 20°C–25°C for 1 min.
-
e.Load the peptide sample on the StageTip, centrifuge at 3,000 g at 20°C–25°C for 1 min, and discard the flow-through.
-
f.Wash the StageTip two times with 100 μL of 0.2% FA and centrifuge at 3,000 g at 20°C–25°C for 1 min.
-
g.Elute the peptide sample with 50 μL of 50% ACN, 0.2% FA into a clean 1.5-mL microcentrifuge tube and centrifuge at 3,000 g at 20°C–25°C for 1 min.
-
h.Transfer all eluted peptide solution into a clean 0.65-mL microcentrifuge tube.Pause point: The protocol can be paused here by long-term storing the samples at −80°C.
-
i.Vacuum dry the eluted peptide solution in a Speedvac.
-
j.Resuspend the dried peptides in 20 μL of 0.2% FA and thoroughly mix the solution. Centrifuge at 12,000 g at 20°C–25°C for 2 min.Note: The samples can be used directly for LC-MS/MS analysis or short-term stored at −20°C before LC-MS/MS analysis. Before transferring each sample to an autosampler vial, 1 μL of indexed retention time peptides (iRT; Biognosys)5 is spiked into the sample according to manufacturer’s instructions.
-
a.
LC-MS/MS analysis: Data-independent acquisitions
This section describes the settings for the LC-MS/MS acquisition in data-independent acquisition (DIA) mode6,7,8 of the two samples obtained in the Step 21.e. (protein-level analysis) and 24.b. (malonyl-level analysis). A detailed step-by-step procedure for building the DIA method is also described in the Supplementary Appendix of Bons et al.2
-
25.Inject samples onto a Dionex UltiMate 3000 system coupled to an Orbitrap Eclipse Tribrid mass spectrometer (both from Thermo Fisher Scientific).
-
a.Inject 200 ng of peptides for the protein-level analysis (Figure 2A).
-
b.Inject 4 μL of enriched malonylated peptides for the malonyl-level analysis (Figure 2B).
-
c.Solvent A is composed of 2% acetonitrile, 0.1% formic acid in LC-MS-grade water and solvent B is composed of 98% acetonitrile, 0.1% formic acid in LC-MS-grade water.
-
d.Load peptides on an Acclaim PepMap 100 C18 trap column (75 μm × 2 cm, 3 μm particle size; Thermo Fisher Scientific PN 164535) over 10 min at 5 μL/min with 100% solvent A.
-
a.
Note: Trap columns with different specifications can be employed with eventual slight adjustments of the loading conditions. For instance, peptides can be loaded on a 5-μm particle size trap column over 5 min at 5 μL/min with 100% solvent A.
-
26.Elute peptides with an Acclaim PepMap 100 C18 analytical column (75 μm × 50 cm, 3 μm particle size; Thermo Fisher Scientific PN 164570) heated at 35°C and at a flow rate of 300 nL/min over 215 min using the following gradient of solvent B:
-
a.2% for 10 min,
-
b.From 2% to 20% in 125 min,
-
c.From 20% to 32% in 40 min,
-
d.From 32% to 80% in 1 min,
-
e.80% for 9 min,
-
f.From 80% to 2% in 1 min,
-
g.2% for 29 min.
-
a.
-
27.
Acquire the data in DIA mode using the settings described in Table 6. One cycle consists of one full MS scan collected in the Orbitrap analyzer followed by DIA-MS/MS scans collected in the Orbitrap analyzer based on an isolation scheme of 26 variable windows covering the 350–1,650 m/z range with an overlap of 1 m/z8 (Table 7).
Note: The settings need to be adjusted if a different LC-MS/MS system is used.
Figure 2.

Representative total ion chromatograms of a K562 cell whole protein lysate and malonyl enrichment sample
Total ion chromatogram of a replicate of a K562 cell (A) whole protein lysate sample and (B) malonyl enrichment sample acquired in data-independent acquisition (DIA)-MS on an Orbitrap Eclipse Tribrid mass spectrometer using a 210-min chromatographic gradient.
Table 6.
Settings for data-independent acquisition on an Orbitrap Eclipse Tribrid mass spectrometer
| Ion source properties | |
| Spray Voltage (Positive ion) | 2100 V |
| Sweep Gas | 0 |
| Ion transfer tube Temperature | 300°C |
| MS scan properties | |
| Detector type | Orbitrap |
| Orbitrap Resolution | 120,000 |
| Scan range | 350-1,650 m/z |
| AGC target | Custom |
| Normalized AGC target | 750% (i.e., 3e6 ions) |
| Maximum Injection time mode | Custom |
| Maximum Injection time | 60 ms |
| Microscans | 1 |
| Data type | Profile |
| Polarity | Positive |
| Targeted MSnproperties | |
| MSn level | 2 |
| Isolation Mode | Quadrupole |
| Isolation Window | 1.6 m/z |
| Activation Type | HCD |
| Collision Energy Mode | Fixed |
| HCD Collision Energy | 27% |
| Detector Type | Orbitrap |
| Orbitrap Resolution | 30,000 |
| Scan Range Mode | Define First Mass |
| First Mass | 200 m/z |
| AGC Target | Custom |
| Normalized AGC Target | 6000% (i.e.,, 3e6 ions) |
| Maximum Injection Time Mode | Auto |
| Maximum Injection Time | 60 ms |
| Microscans | 1 |
| Data Type | Profile |
| Polarity | Positive |
| Loop Control | N |
| N (Number of Spectra) | 26 |
| Dynamic Retention Time | Off |
| Time Mode | Start/End Time |
Table 7.
Isolation scheme of the data-independent acquisition method
| Window | Start m/z | Stop m/z | Center m/z | z | Isolation width |
|---|---|---|---|---|---|
| 1 | 350 | 383 | 366.5 | 3 | 33 |
| 2 | 382 | 408 | 395 | 3 | 26 |
| 3 | 407 | 429 | 418 | 3 | 22 |
| 4 | 428 | 448 | 438 | 3 | 20 |
| 5 | 447 | 467 | 457 | 3 | 20 |
| 6 | 466 | 484 | 475 | 3 | 18 |
| 7 | 483 | 503 | 493 | 3 | 20 |
| 8 | 502 | 521 | 511.5 | 3 | 19 |
| 9 | 520 | 539 | 529.5 | 3 | 19 |
| 10 | 538 | 557 | 547.5 | 3 | 19 |
| 11 | 556 | 575 | 565.5 | 3 | 19 |
| 12 | 574 | 594 | 584 | 3 | 20 |
| 13 | 593 | 614 | 603.5 | 3 | 21 |
| 14 | 613 | 634 | 623.5 | 3 | 21 |
| 15 | 633 | 656 | 644.5 | 3 | 23 |
| 16 | 655 | 678 | 666.5 | 3 | 23 |
| 17 | 677 | 701 | 689 | 3 | 24 |
| 18 | 700 | 726 | 713 | 3 | 26 |
| 19 | 725 | 756 | 740.5 | 3 | 31 |
| 20 | 755 | 787 | 771 | 3 | 32 |
| 21 | 786 | 823 | 804.5 | 3 | 37 |
| 22 | 822 | 862 | 842 | 3 | 40 |
| 23 | 861 | 914 | 887.5 | 3 | 53 |
| 24 | 913 | 979 | 946 | 3 | 66 |
| 25 | 978 | 1077 | 1027.5 | 3 | 99 |
| 26 | 1076 | 1650 | 1363 | 3 | 574 |
DIA data processing: Protein-level and malonyl-level analysis
This section describes how to analyze the DIA data acquired on the malonyl enrichment samples to determine malonyl-level changes and on the whole lysate samples to determine protein-level changes and normalize the malonylated peptide abundances. We used directDIA implemented in Spectronaut software (Biognosys)9 to process the raw MS files. directDIA relies on a library-free spectrum-centric strategy with the use of deep-learning approaches and the built-in Pulsar search engine to (i) build a spectral library and (ii) perform a targeted extraction to retrieve reproducible and highly quantitative information from the same DIA files. This fully automated pipeline also supports PTM analysis to provide confident and precise PTM site localization and accurate quantification of the proteome-wide profiled modified peptides. A detailed step-by-step procedure for processing the collected DIA data is also described in the Supplementary Appendix of Bons et al.2 For more details, please refer to the user manual of Spectronaut (latest version: version 18, https://biognosys.com/content/uploads/2023/06/Spectronaut18_UserManual.pdf).
We recommend using the latest version of directDIA/Spectronaut as it includes the latest improvements and innovations. Here, we used version 15.1.210713.50606. Other software tools can be used with adjusted settings, such as the open-source Skyline,10 DIA-NN,11 and MSFragger-DIA12 tools.
-
28.Protein-level analysis with directDIA.
-
a.Use directDIA with default settings unless specified in Table 8 to search the DIA data acquired on the whole lysate samples against the Human UniProt-SwissProt proteome database (https://www.uniprot.org/proteomes).Note: The settings need to be adjusted if different LC-MS/MS system and acquisition settings are used.Note: The directDIA – DIA Analysis includes a Post-Analysis section for statistical analysis. Select the appropriate statistical test for the experimental design.
-
b.The candidates.tsv output file contains the list of all identified and quantified protein groups as well as the results of the statistical analysis. We recommend to filter out the protein groups quantified with one unique peptide.
-
a.
-
29.Malonyl-level analysis with directDIA (Figure 3).
-
a.Use directDIA with default settings unless specified in Table 8 to search the DIA data acquired on the malonyl enrichment samples against the same Human UniProt-SwissProt proteome database (https://www.uniprot.org/proteomes) as used for Step 28.Note: The settings need to be adjusted if different LC-MS/MS system and acquisition settings are used.Note: The directDIA – DIA Analysis includes a Post-Analysis section for statistical analysis. Select the appropriate statistical test to the experimental design.
-
b.The candidates.tsv output file contains the list of all identified and quantified peptides (malonylated and non-modified) as well as the results of the statistical analysis. Normalize the abundance of the malonylated peptides in each experimental condition by dividing by the abundance of the corresponding protein group in the same experimental condition obtained in Step 28.b.
-
a.
-
30.Additional malonyl-level analysis with Skyline (Figure 3).Note: To strengthen the confidence in the malonyl results for this study, we further manually investigated the identified malonylated peptides in the open-source software Skyline.10 The procedure consists in (i) generating a spectral library from the DIA acquisitions of the malonyl enrichment samples in Spectronaut, (ii) import the spectral library in Skyline, (iii) import the DIA files in Skyline. For more details, please refer to the Skyline website (https://skyline.ms/project/home/software/Skyline/begin.view).Note: We recommend using the latest version of Skyline as it includes the latest improvements and innovations. Here, we used version 21.1.1.223.
-
a.Generation of the spectral library in Spectronaut.
-
i.Use Spectronaut with default settings unless specified in Table 9 to search the DIA data acquired on the malonyl enrichment samples against the same Human UniProt-SwissProt proteome database (https://www.uniprot.org/proteomes) as used for Step 28.Note: The settings need to be adjusted if different LC-MS/MS system and acquisition settings are used.
-
ii.Export the spectral library as an .xls file and convert it as a .csv file.
-
i.
-
b.Data inspection in Skyline.
-
i.Use Skyline with the settings detailed in Table 10 and select Settings > Integrate All:Note: The settings need to be adjusted if different LC-MS/MS system and acquisition settings are used.
-
ii.Import the spectral library obtained in step 30.a. by selecting File > Import > Assay Library.
-
iii.Import the DIA raw files of the malonyl enrichment samples by selecting File > Import > Results.
-
iv.Visualize the extracted ion chromatograms obtained for the malonylated peptides. If needed, manually adjust the peak integration boundaries and remove interfering transitions while ensuring to keep at least three transitions per precursor ion. The statistical analysis can be performed in Skyline. Troubleshooting 5.
-
i.
-
a.
Table 8.
Settings for the protein-level and malonyl-level analysis in Spectronaut
| Protein-level analysis | Malonyl-level analysis | ||
|---|---|---|---|
| Pulsar search | |||
| Peptides | Enzymes/Cleavage Rules | Trypsin/P | Trypsin/P |
| Digest Type | Specific | Specific | |
| Missed Cleavagesa | 2 | 4 | |
| Modifications | Max Variable Modificationsa | 5 | 8 |
| Fixed Modifications | Carbamidomethyl (C) | Carbamidomethyl (C) | |
| Variable Modificationsa | Acetyl (Protein N-term) Oxidation (M) |
Acetyl (Protein N-term) Oxidation (M) Malonyl (K) |
|
| Identification | PSM FDR | 0.01 | 0.01 |
| Peptide FDR | 0.01 | 0.01 | |
| Protein Group FDR | 0.01 | 0.01 | |
| Result Filters |
Fragment Ions | ||
| Ion AA Length (min) | N = 3 | N = 3 | |
| m/z | 200 - 3,000 | 200 - 3,000 | |
| Relative Intensity | Min = 5 | Min = 5 | |
| Precursors | |||
| Best N Fragments per Peptide | 3–6 | 3–6 | |
| Modifications | None | None | |
| DIA analysis | |||
| XIC Extraction | MS1 Mass Tolerance Strategy | Dynamic | Dynamic |
| Correction Factor | 1 | 1 | |
| MS2 Mass Tolerance Strategy | Dynamic | Dynamic | |
| Correction Factor | 1 | 1 | |
| Calibration | MZ Extraction Strategy | Maximum Intensity | Maximum Intensity |
| Allow source-specific iRT Calibration | True | True | |
| Precision iRT | True | True | |
| iRT <-> RT Regression Type | Local (Non-Linear) Regression | Local (Non-Linear) Regression | |
| MS1 Mass Tolerance Strategy | System Default | System Default | |
| MS2 Mass Tolerance Strategy | System Default | System Default | |
| Identification | Precursor PEP Cutoffa | 0.2 | 1 |
| Precursor Qvalue Cutoff | 0.01 | 0.01 | |
| Protein Qvalue Cutoff (Experiment) | 0.01 | 0.01 | |
| Protein Qvalue Cutoff (Run)a | 0.05 | 1 | |
| Exclude Duplicate Assays | True | True | |
| Generate Decoys | True | True | |
| Decoy Generation Method | Mutated | Mutated | |
| Preferred Fragment Source | NN Predicted Fragments | NN Predicted Fragments | |
| Decoy Limit Strategy | Dynamic | Dynamic | |
| Library Size Fraction | 0.1 | 0.1 | |
| Quantification | Precursor Strategy | Identified (QValue) | Identified (QValue) |
| Imputation Strategy | Use Background Signal | Use Background Signal | |
| Quantify MS Level | MS2 | MS2 | |
| Quantify Type | Area | Area | |
| Cross-Run Normalizationa | True | None | |
| Normalization Filter Typea | None | N/A | |
| Normalization Strategya | Local Normalization | N/A | |
| Row Selectiona | Qvalue sparse | N/A | |
| Interference Correction | True | True | |
| Only Identified Peptides | True | True | |
| MS1 Min | 2 | 2 | |
| MS2 Min | 3 | 3 | |
| Major (Protein) Groupinga | by Protein Group Id | by Protein Group Id | |
| Major Group Quantitya | Sum peptide quantity | Mean peptide quantity | |
| Major Group Top Na | Min = 1; Max = 7 | Min = 1; Max = 3 | |
| Minor (Peptide) Groupinga | by Stripped Sequence | by Modified Sequence | |
| Minor Group Quantitya | Sum precursor quantity | Mean precursor quantity | |
| Minor Group Top Na | Min = 1; Max = 10 | Min = 1; Max = 3 | |
| Workflow | Profiling Strategya | iRT Profiling | None |
| Carry-over exact Peak Boundariesa | None | N/A | |
| Profiling Row Selectiona | Minimum QValue Row Selection | N/A | |
| Qvalue Thresholda | 0.01 | N/A | |
| Profiling Target Selectiona | Automatic Selection | N/A | |
| PTM Workflow | PTM Localizationa | None | True |
| Probability Cutoffa | N/A | 0.75 | |
| PTM Analysisa | N/A | True | |
| Multiplicitya | N/A | True | |
| Flanking Regiona | N/A | 7 | |
| PTM Consolidationa | N/A | Sum | |
Parameters that differ between the protein-level and malonyl-level analysis.
Figure 3.

Malonyl-lysine analysis in K562 cells by data-independent acquisition-mass spectrometry (DIA-MS)
(A) Retention time calibration obtained for a replicate of the malonyl enrichment DIA data set.
(B) Relative identification count of the modifications set for the DIA search.
(C) Content of the spectral library generated in Spectronaut from the DIA acquisitions of the malonyl enrichment samples.
(D) Pseudo-MS/MS spectrum of the PDPA5KmalSAPAPK peptide (m/z 582.80, z = 2+) (histone H2B2F) contained in the spectral library and visualized in Skyline.
(E) Extracted ion chromatogram of the PDPA5KmalSAPAPK peptide (m/z 582.80, z = 2+) (histone H2B2F) visualized in Skyline.
Table 9.
Settings for the generation of the spectral library in Spectronaut
| Pulsar search | ||
| Peptides | Enzymes/Cleavage Rules | Trypsin/P |
| Digest Type | Specific | |
| Missed Cleavages | 4 | |
| Modifications | Max Variable Modifications | 5 |
| Fixed Modifications | Carbamidomethyl (C) | |
| Variable Modifications | Acetyl (Protein N-term) Oxidation (M) Malonyl (K) |
|
| MS2 | Ion Types | b; y |
| Library generation | ||
| Tolerances | Thermo Orbitrap | |
| Calibration Search | Dynamic | |
| MS1 Correction Factor | 1 | |
| MS2 Correction Factor | 1 | |
| Main Search | Dynamic | |
| MS1 Correction Factor | 1 | |
| MS2 Correction Factor | 1 | |
| Identification | Pulsar | |
| PSM FDR | 0.01 | |
| Peptide FDR | 0.01 | |
| Protein FDR | 0.01 | |
| PTM Localization Filter | None | |
| Spectral Library Filters | Fragment Ions | |
| Ion AA Length (min) | N = 3 | |
| m/z | 200 - 3,000 | |
| Relative Intensity | Min = 5 | |
| Precursors | ||
| Best N Fragments per Peptide | 3–6 | |
| Modifications | None | |
Table 10.
Settings for inspecting the data in Skyline
| Peptide settings | ||
| Digestion | Enzymes | Trypsin [KR|P] |
| Max missed cleavages | 4 | |
| Filters | Min length | 5 |
| Max length | 35 | |
| Auto-select all matching peptides | True | |
| Library | Pick peptides matching | Library |
| Modifications |
Structural modifications | Carbamidomethyl (C, fixed) Acetyl (N-term, variable) Oxidation (M, variable) Malonyl (K, variable) |
| Max variable mods | 3 | |
| Transition settings | ||
| Filter | Precursor charges | 2, 3, 4 |
| Ion charges | 1, 2, 3 | |
| Ion types | p, y, b | |
| Product ions | ||
| From | ion 2 | |
| To | last ion | |
| Auto-select all matching transitions | True | |
| Library | Ion match tolerance | 0.5 m/z |
| If a library spectrum is available, pick its most intense ions | True | |
| Pick | 10 product ions | |
| From filtered ion charges and types | True | |
| Instrument | Min m/z | 200 |
| Max m/z | 1500 | |
| Method match tolerance m/z | 0.055 m/z | |
| Full-scan | MS1 filtering | |
| Isotope peaks included | Count | |
| Precursor mass analyzer | Orbitrap | |
| Peaks | 3 | |
| Resolving power | 120,000 at 200 m/z | |
| MS/MS filtering | ||
| Acquisition method | DIA | |
| Product mass analyzer | Orbitrap | |
| Isolation scheme | cf. Step 27 | |
| Resolving power | 30,000 m/z at 200 m/z | |
| Retention time filtering | ||
| Include all matching scans | True | |
Expected outcomes
Spin-infection of 5 × 105 cells with 500 μL of fresh viral supernatant results in ∼20% infection as shown by flow cytometry 72 h after lentiviral infection (Figure 4A). The gene knockdown efficiency can be examined at RNA level through qPCR and Western blotting (Figures 4B and 4C).
Figure 4.

CRISPRi K562 cell purification by FACS and gene knockdown efficiency examination by qPCR and western blotting
(A) sgRNA-expressing cells were BFP (+), and collected after FACS. Gating strategies are shown here. The percentage of BFP (+) cells were about 14–27% of live cells. FSC, forward scatter; SSC, side scatter; BFP, blue fluorescent protein.
(B) Bar graph shows the target RNA expression level in the knockdown K562 cell line (sgTarget) relative to the scramble sgRNA-expressing negative control (sgNC). The lower the percentage, the higher the knockdown efficiency.
(C) Western blotting results showing the protein expression levels of KATs and SIRT5 in knockdown cell lines.
Our proteomic workflow, that relies on affinity-based enrichment of malonylated peptides, comprehensive and label-free data-independent acquisition (DIA)-MS, library-free DIA data processing with directDIA/Spectronaut followed by optional inspection/verification in Skyline, was applied to ∼4 × 106 human CRISPRi K562 cells, and a successful outcome is exemplified on Figure 3. This procedure yielded >220 malonylated peptides corresponding to >210 unique malonylation sites from >150 protein groups from the malonyl enrichment samples. The efficient retention time calibration achieved by using the same DIA files to both generate the spectral library and to perform ion chromatogram extractions improved the specificity of the assay (Figure 3A). Overall, >12,000 peptides (malonylated or not) were identified, which represents an enrichment in malonylated peptides of ∼2% (Figure 3B). This proportion can be expected as the malonyl group is a labile PTM, that is subject to neutral loss with collision-induced dissociation (CID)-type fragmentation (see limitations section). To increase the confidence of the results, the signal for the malonylated peptides were verified in Skyline using the spectral library generated in Spectronaut from the multiplexed DIA MS/MS spectra (Figures 3C and 3D). As an example, the detected fragment ion y9 serves a direct evidence of the malonyl site on the peptide PDPA5KmalSAPAPK of the human histone H2B type 2-F (H2B2F) (Figure 3E). In addition, the protein-level analysis revealed that over 3,400 protein groups were identified and quantified with at least two unique peptides. The depth of the malonylome coverage yielded by this protocol depends on several factors, including the amount of starting protein material used for the affinity-based PTM enrichment, the amount of peptide material loaded onto the column, and the LC-MS system and settings. It is worth noting that this procedure can be applied to other sample types (e.g., other cell types, tissues2,13) and other organisms with some adjustments, such as the protein database.
Limitations
Although most of the K562 dCas9-KRAB cell lines have target genes efficiently silenced, some of them were not. qPCR results suggested that 4 of 22 KAT genes that we targeted had a knockdown efficiency < 80%. So, several different gRNA sequences may have to be tested to succeed. Also, to rule out the potential off-target effect of gRNAs, we would recommend to generate multiple (at least two) lines of CRISPRi cells expressing different gRNA sequences targeting the same gene of interest for the examination of the gene’s function in lysine malonylation regulation or any other phenotype.
The CRISPRi system is a powerful tool to stably knockdown gene expression. However, it may not work so well for every gene in every cell type. So far, we have successfully generated multiple CRISPRi K562, Jurkat, and 293T cell lines in house, but we experienced difficulties in making CRISPRi HepG2 cell lines using the same gRNAs. Although the mechanism is not entirely clear to us, cell type-specific lack of accessibility of target gene promoter for the gRNA-driven dCas9-fused KRAB silencing machinery might be part of the reason. In this case, shRNA-mediated RNA interference, or CRISPR-mediated gene knockout, may be considered as an alternative approach for achieving gene loss-of-function.
The analysis of lysine malonylation by mass spectrometry presents challenges due to the low stoichiometry of the malonylated peptides and the lability of the malonyl group. Indeed, it can undergo a neutral loss of a CO2 (−44 m/z) group during the ion fragmentation, typically applying collision-induced dissociation-based technique in the mass spectrometer. Consequently, this results in a mass increment of +42 m/z as for acetyl group, instead of +86 m/z. Altogether, these factors may limit the depth of the profiling of lysine malonylation.
Troubleshooting
Problem 1
Sanger sequencing results suggest that the sgRNA sequence was not inserted into the lentiviral backbone vector (related to Step 6).
Potential solution
-
•
Pick more colonies from the plate generated at Step 5, then miniprep for Sanger sequencing.
-
•
In case the backbone is not linearized or properly digested by both of the endonucleases, perform agarose gel electrophoresis and purify linearized plasmids from gel each time after endonuclease digestion.
-
•
In case the DNA oligonucleotides were not properly annealed, double check the DNA sequences and repeat Step 1.
-
•
In case the annealed oligonucleotides were not properly ligated with linearized backbone vector, repeat Step 3.
-
•
Double check the LB agarose plate to make sure that it contains ampicillin, and was newly prepared.
Problem 2
No BFP (+) K562 cells are collected through FACS, or no K562 cells remain viable after antibiotic selection (related to Step 16).
Potential solution
-
•
Ensure that an appropriate channel (e.g., Ex./Em. = 405/450 nm) is chosen for BFP detection using flow cytometry. If antibiotic selection was performed, ensure that the right type of antibiotic, i.e., puromycin, is used for gRNA-expressing K562 cell selection.
-
•
For lentivirus production, ensure that the 293T cells used for lentivirus production are healthy and in a log growth phase. Plate well-trypsinized single 293T cells, and avoid over-confluency.
-
•
Ensure the proper ratios of transfer, envelope, and packaging plasmids are used to transfect 293T cells to achieve optimal titers.
Problem 3
The knockdown efficiency is low (related to Step 17).
Potential solution
-
•
Ensure sgRNA-expressing K562 cells are selected for BFP through FACS, or puromycin selection at the concentration around 1 μg/mL for at least 48 h before examination.
-
•
Ensure that the PCR primer set (qPCR experiment) or antibody (western blotting) is specific to the gene/protein of interest. And ensure the good quality of the reagents for the experiments.
-
•
We recommend to use two or more gRNA sequences predicted to target your gene of interest to ensure the a high (>70%) knockdown efficiency in at least one line of K562 cells. In case that none of the gRNA sequences is efficient enough to mediate the CRISPRi, which is rare, use another gRNA sequence, or several other gRNA sequences, for oligonucleotide synthesis, and start over from Step 1.
Problem 4
The amount of recovered protein material is insufficient after cell lysis (related to Step 19).
Potential solution
-
•
Increase the number of cells.
-
•
Ensure the good quality of the reagents that compose the lysis buffer and the protein concentration determination assay.
Problem 5
The depth of the proteome and lysine malonylome is low, and reproducibility is poor between the replicates (related to Steps 28–30).
Potential solution
Lower performance of the proteomic analysis can be due to insufficient amount of starting protein material, issues during the sample preparation, and/or suboptimal performance of the LC-MS/MS system.
-
•
Ensure that the reagents are freshly prepared, properly manipulated and not contaminated. Reagents include, but are not limited to, protease and de-acylase inhibitors, DTT, IAA, trypsin, and PTMScan Malonyl-Lysine [Ma-K] Kit.
-
•
Ensure that the samples are kept on ice and the reagents of the PTMScan Malonyl-Lysine [Ma-K] Kit are cold to prevent protease and de-acylase activity.
-
•
Adjust the volume of antibody beads per sample.
-
•
Ensure that the same volume of antibody beads is used for all samples.
-
•
Monitor the performance of the LC-MS/MS system using quality control samples.
-
•
Inject a higher amount of material onto the column for LC-MS/MS analysis if low intensity peaks are observed as this may prevent the confident interpretation of the MS/MS spectra.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Eric Verdin (EVerdin@buckinstitute.org).
Technical contact
Technical questions about this protocol should be directed to the technical contact, Ran Zhang (RZhang@buckinstitute.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw data and complete MS datasets have been uploaded to the Center for Computational Mass Spectrometry, to the MassIVE repository at UCSD, and can be downloaded using the following link: ftp://massive.ucsd.edu/v06/MSV000093564/. The accession numbers for the raw data and complete MS datasets reported in this paper are MassIVE ID number: MSV000093564 and ProteomeXchange ID: PXD047514.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank Drs. Marius Walter and Rosalba Perrone for assistance with CRISPRi K562 cell construction and Dr. Herbert Kasler for fluorescence-activated cell sorting. This study was supported by NIH grant R24DK085610 (E.V.), the Glenn Foundation for Medical Research (R.Z.), the National Natural Science Foundation of China (no. 82301607, R.Z.), NCRR shared instrumentation grants 1S10 OD016281 (TripleTOF 6600, Buck Institute) and 1S10 OD028654 (Orbitrap Eclipse Tribrid, B.S.).
Author contributions
R.Z. generated CRISPRi K562 cell lines. J.B., J.P.R., and B.S. performed malonylation mass spectrometry analysis. R.Z. and J.B. wrote the initial manuscript. All authors together conceptualized the study, discussed the results, and commented on the manuscript.
Declaration of interests
E.V. is a scientific co-founder of Napa Therapeutics and serves on the scientific advisory board of Seneque.
Contributor Information
Ran Zhang, Email: rzhang@buckinstitute.org.
Eric Verdin, Email: everdin@buckinstitute.org.
References
- 1.Zhang R., Bons J., Scheidemantle G., Liu X., Bielska O., Carrico C., Rose J., Heckenbach I., Scheibye-Knudsen M., Schilling B., Verdin E. Histone malonylation is regulated by SIRT5 and KAT2A. iScience. 2023;26 doi: 10.1016/j.isci.2023.106193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bons J., Rose J., Zhang R., Burton J.B., Carrico C., Verdin E., Schilling B. In-depth analysis of the Sirtuin 5-regulated mouse brain malonylome and succinylome using library-free data-independent acquisitions. Proteomics. 2023;23 doi: 10.1002/pmic.202100371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H., Guimaraes C., Panning B., Ploegh H.L., Bassik M.C., et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horlbeck M.A., Gilbert L.A., Villalta J.E., Adamson B., Pak R.A., Chen Y., Fields A.P., Park C.Y., Corn J.E., Kampmann M., Weissman J.S. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife. 2016;5 doi: 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Escher C., Reiter L., MacLean B., Ossola R., Herzog F., Chilton J., MacCoss M.J., Rinner O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 2012;12:1111–1121. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillet L.C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., Aebersold R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. ARTN O111.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins B.C., Hunter C.L., Liu Y., Schilling B., Rosenberger G., Bader S.L., Chan D.W., Gibson B.W., Gingras A.C., Held J.M., et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017;8 doi: 10.1038/s41467-017-00249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruderer R., Bernhardt O.M., Gandhi T., Xuan Y., Sondermann J., Schmidt M., Gomez-Varela D., Reiter L. Optimization of Experimental Parameters in Data-Independent Mass Spectrometry Significantly Increases Depth and Reproducibility of Results. Mol. Cell. Proteomics. 2017;16:2296–2309. doi: 10.1074/mcp.RA117.000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruderer R., Bernhardt O.M., Gandhi T., Reiter L. High-precision iRT prediction in the targeted analysis of data-independent acquisition and its impact on identification and quantitation. Proteomics. 2016;16:2246–2256. doi: 10.1002/pmic.201500488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacLean B., Tomazela D.M., Shulman N., Chambers M., Finney G.L., Frewen B., Kern R., Tabb D.L., Liebler D.C., MacCoss M.J. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demichev V., Messner C.B., Vernardis S.I., Lilley K.S., Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 2020;17:41–44. doi: 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu F., Teo G.C., Kong A.T., Fröhlich K., Li G.X., Demichev V., Nesvizhskii A.I. Analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform. Nat. Commun. 2023;14:4154. doi: 10.1038/s41467-023-39869-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishida Y., Rardin M.J., Carrico C., He W., Sahu A.K., Gut P., Najjar R., Fitch M., Hellerstein M., Gibson B.W., Verdin E. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell. 2015;59:321–332. doi: 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data and complete MS datasets have been uploaded to the Center for Computational Mass Spectrometry, to the MassIVE repository at UCSD, and can be downloaded using the following link: ftp://massive.ucsd.edu/v06/MSV000093564/. The accession numbers for the raw data and complete MS datasets reported in this paper are MassIVE ID number: MSV000093564 and ProteomeXchange ID: PXD047514.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.