

Abstract

Gallium trichloride (GaCl3) was used as a solvent for the oxidative dissolution of the lanthanide (Ln) metals cerium (Ce) and holmium (Ho). Reactions were performed at temperatures above 100 °C in sealed vessels to maintain the liquid phase for GaCl3 during the oxidizing reactions. The best results were obtained from reactions using 8 equiv of GaCl3 to metal where the inorganic complexes [Ga][Ln(GaCl4)4] [Ln = Ce (1), Ho (2)] could be isolated. Recrystallization of 1 and 2 employing fluorobenzene (C6H5F) produced [Ga(η6-C6H5F)2][Ln(GaCl4)4] [Ln = Ce (3), Ho (4)] where reversible η6 coordination of C6H5F to [Ga]+ was observed. All complexes were characterized through elemental analysis (F and Cl), IR and UV–vis–near-IR spectroscopies, and both solution and solid-state NMR techniques.
Short abstract
Employing molten GaCl3 as a solvent for the oxidative dissolution of the lanthanide metals cerium (Ce) or holmium (Ho) results in chlorine transfer from the gallium to the lanthanide, generating Ce3+ and Ho3+ salts concurrent with [Ga][GaCl4]. The lanthanide products were found to be [Ga][Ln(GaCl4)4] (Ln = Ce, Ho), salts that have a noncoordinated [Ga]+ and eight-coordinated lanthanide centers. Solid-state NMR has proven to be particularly useful as a characterization technique for this series of complexes.
Oxidative dissolution is a nascent field of metal recycling and purification with a high potential impact.1−3 Outside of aqueous acid solutions, many of the reaction media used in these transformations bear trihalide counterions that perform two-halide oxidation reactions, concomitantly oxidizing and dissolving metals.4,5 Both ionic liquid and molten salt matrixes offer variation with liquid temperatures, redox stability, and polarity, all of which could benefit lanthanide recycling. Employing ionic liquid and molten salt matrixes with 4f metals allows for unique transformations,6−11 separations,12−21 and redox behaviors22−24 that encourage further development to expand inorganic reaction chemistry. Continued investigations into lanthanide recycling may also expose untapped sources of these critical materials.
The oxidative dissolution of lanthanide metals using gallium trichloride (GaCl3) may provide new avenues for recycling while generating novel molecular motifs. With an accessible melting point of 77.9 °C, GaCl3 has properties that exist between ionic liquids and molten salts, making it an unconventional solvent to investigate oxidative chemical transformations. Reports have detailed investigations of GaCl3 as a solvent matrix for chalcogen oxidative dissolution25,26 and as a Lewis acidic solvent for the dissolution of the elements bismuth27−29 and antimony.30 Our prior work demonstrated the potential of GaCl3 as a chlorine-transfer reagent for uranium and plutonium,31 furthering our interests in exploring GaCl3 as a solvent in metal dissolution chemistry. No prior reports detail its use as a solvent for f-block metals. Here, a mild-temperature, molten binary-salt flux of GaCl3 was explored as a nonclassical solvent for the oxidative dissolution of Ln metals (Scheme 1). The reaction provided us access to rare coordination motifs that standard reaction solvents would be unlikely to provide.
Scheme 1. Reaction Scheme Depicting the Oxidative Dissolution of Ce and Ho in GaCl3 and Picture of the Resultant Crystal of [Ga][Ho(GaCl4)4].

The metals cerium (Ce) and holmium (Ho) were selected for this study. Ce was chosen for its prominence in lanthanide chemistry as a plutonium surrogate and its accessible 3+/4+ redox couple.32−34 The accessibility of Ce4+ is a useful probe to study potential overoxidation under these reaction conditions. Ho represents a middle-to-late lanthanide with an inherently high magnetic moment. Additionally, Ho3+ complexes can adopt a pink coloration, which is valuable for monitoring the reaction progress and facilitating the technically challenging purification. This report describes the oxidative dissolution of Ce and Ho and the product isolation and characterization via solid-state and solution NMR spectroscopy, elemental analysis, UV–vis–near-IR (NIR) and IR techniques, and single-crystal X-ray diffraction (SC-XRD).
The synthetic investigation was performed in sealed gas chromatography vials or vacuum-sealed ampules containing Ce or Ho metal with excess GaCl3. The reactions were heated to temperatures above the melting point of GaCl3 (ca. 110 °C) for 3–5 days. Over this time, the metals dissolved and were oxidized by GaCl3. The reaction of Ce remained colorless, while the Ho reaction acquired a pink hue (Figure S1). No evidence of Ce4+ formation was observed during this study. Slow cooling of the reactions led to the formation of large crystals interspersed with a fine white powder. Rinsing the material with pentane removed the excess GaCl3 and resulted in a crude purification where small amounts of Ga2Cl4, found as [Ga][GaCl4], were observed along with the desired products, as determined by 71Ga NMR. The large crystals were physically separated, analyzed by SC-XRD, and assigned as [Ga][Ln(GaCl4)4] [Ln = Ce (1), Ho (2)]. The formation of [Ga][GaCl4] was confirmed through a comparison of the solid-state 71Ga NMR spectra of the crude products to reference samples of [Ga][GaCl4] (Figure S4). The reaction stoichiometry proposed for these observations is found in eq 1.
| 1 |
SC-XRD studies of the crystalline product revealed salt pairs of [Ga]+ and [Ln(GaCl4)4]−. The Ce and Ho structures were isomorphous, crystallizing in the monoclinic space group P2/c. The asymmetric unit cell comprises half of the [Ln(GaCl4)4]− anion and half of the [Ga]+ cation. The Ln in the anionic fragment was found in a distorted square-antiprismatic geometry (Figure 1a). The average Ga–Cl distances of [GaCl4]− were found to be 2.1818(2) and 2.1820(7) Å for the Ce and Ho complexes, respectively, similar to that of other [Ga][GaCl4] main-group salts [Ga–Clavg = 2.1721(4) Å].35−38 The bridging Ga–Cl values are longer, with an average of 2.2285(4) Å compared to that of the terminal chlorides, at 2.1360(6) Å. The Ln–Cl bond also contracts with a decreasing ionic radius from 2.8774(10) Å (Ce) to 2.7728(7) Å (Ho).
Figure 1.

(a) Grown unit cell of 2 presented at 50% probability ellipsoids. 1 is isostructural. (b) Expansion of the chloride environment around [Ga]+.
A coordination sphere of eight chlorine atoms is rare in f-metal chemistry. One example, published by Schwotzer and co-workers, detailed a trimeric U4+ complex, [η6-C6Me6)UCl2(μ-Cl)3UCl2(η6-C6Me6)], where the central uranium atom displays similar chloride saturation.39 A few examples are also reported of f-metals bound directly to tetrachlorogallate [GaCl4]− anions, where the lanthanide metal (La, Ce, or Yb) is bound to three [GaCl4]− anions and an η6-arene ligand.40,41 The [Ga]+ cation is found to be unbound in 1 and 2 but is surrounded by chloride atoms in the extended salt lattice (Figure 1b). The average Ga3–Cl bond distances [3.4382(11) Å (Ce); 3.4332(8) Å (Ho)] are significantly longer than the average Ga–Cl bond lengths of the GaCl4 fragments by approximately 1.2 Å, indicating minimal interaction and the presence of a free [Ga]+.
The generation of noncoordinated [Ga]+ under the reaction conditions is a notable result. An exciting trend in monovalent gallium began in 2010 when a report detailed an accessible synthesis of [(η6-arene)Ga][Al(OC(CF3)3)4] using the noncoordinating tetrakis(perfluoro-tert-butoxide)aluminate anion.42 This and related [Ga]+ materials have most commonly been synthesized by the oxidation of Ga metal with silver salts or through the reductive elimination of gallium hydride materials.42−52 The results obtained from this study can be explained by the presence of [Ga][GaCl4] in the reaction mixture. These products can be considered to be a “true” salt pair [Ga]+[Ln(GaCl4)4]− or, alternatively, an adduct of [Ga][GaCl4] and Ln(GaCl4)3.
The further purification and removal of residual [Ga][GaCl4] was achieved through recrystallization of 1 and 2 from fluorobenzene (C6H5F). Dissolution of 1 and 2 in C6H5F allowed for filtration of the poorly soluble [Ga][GaCl4], where crystallization produced a bis(η6-arene)-coordinated [Ga]+ derivative that was identified as [Ga(η6-C6H5F)2][Ln(GaCl4)4] [Ln = Ce (3), Ho (4)]. Elemental analysis of the material confirmed complete conversion, with the expected F– and Cl– content consistent with the calculated values. Holding complexes 3 and 4 under reduced pressure induced C6H5F loss and the regeneration of 1 and 2, respectively. This reversible coordination of the arene ring was leveraged to purify 1 and 2.
SC-XRD experiments performed on 3 and 4 confirmed their assignment (Figure 2). The crystals of the two homologues were solved in the orthorhombic space group Pbca, with the asymmetric unit cell comprising half of the [Ln(GaCl4)4]− fragment and with an outer-sphere [Ga]+ sandwiched by two half C6H5F molecules. The observed arene–centroid distances to [Ga]+ were fairly long [3, 2.8990(13) and 3.0807(14) Å; 4, 2.905(2) and 3.117(2) Å] and on the order of previously reported arene–centroid distances between 3.145(3) and 2.536(2) Å.42−48 The centroid–[Ga]+–centroid angle was nearly linear at 175.15(4)° for 3 and 174.68(6)° for 4 (Figure 2b). The nearest chloride distances to the free [Ga]+ were contracted significantly to 3.1558(6) and 3.2138(6) Å (3) and 3.1448(12) and 3.2020(11) Å (4). The Ln–Cl distances of the [Ln(GaCl4)4]− anion for 3 and 4 showed a similar decrease of 0.1047(9) Å, with a decreasing ionic radius of the Ln3+ ion from Ce to Ho.
Figure 2.
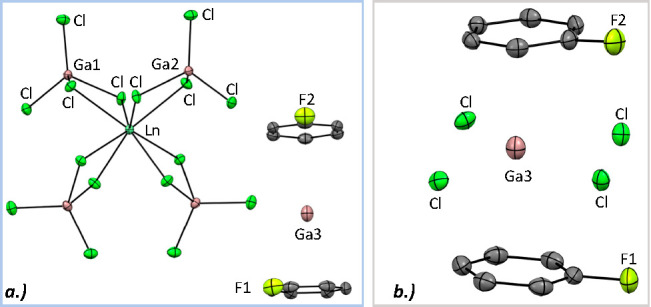
(a) Grown unit cell of 4 presented at 50% probability ellipsoids, with hydrogen and disordered fluorine atoms removed for clarity. 3 is isostructural. (b) Expansion of the arene and chloride environments of [Ga]+.
UV–vis–NIR and IR spectroscopies were performed. The expected transitions in the UV–vis–NIR spectrum for 3 were hidden under the solvent absorbance at ∼300 nm–1, while the spectrum of 4 contained the expected f → d and f → f transitions (Figures S6 and S7). The IR spectra for complexes 1 and 2 show large broad bands at approximately 3440 and 3420 cm–1, which are shifted from a similar feature of [Ga][GaCl4] found at 3316 cm–1. The other bands are broad and large, also reminiscent of those of [Ga][GaCl4]. The C6H5F adducts 3 and 4 reveal that these prominent features are replaced with small sharp bands for each complex found around 2975 cm–1 for the C–H bonds of the arene (Figures S8 and S9).
A series of 71Ga NMR experiments were performed to further characterize 1–4. The coordinated [GaCl4]− fragments were unobserved due to their proximity to the paramagnetic metal center. Being further removed from the paramagnetic center, [Ga]+ appeared as a sharp resonance observed at δ = −594.6 ppm (1) and δ = −603.7 ppm (2) in dichloromethane (CH2Cl2). The 71Ga NMR resonances attributed to [Ga]+ for compounds 3 and 4 were observed at δ = −700.9 ppm (3) and δ = −690.9 ppm (4) in C6H5F; purified 3 and 4 were observed at δ = −629.9 and −644.9 ppm in CH2Cl2. These complexes were unstable in CH2Cl2; the solution turned dark brown in color, and the sharp [Ga]+ resonance diminished over the course of minutes. The use of solid-state NMR techniques provided a more complete characterization.
The 71Ga magic-angle-spinning (MAS) NMR spectra of 1–4 were collected, and the observed resonances were assigned as [Ga]+ (Figure 3). Variation of the spin rate up to 12 kHz was performed to identify the peak arising from the isotropic shift from the spinning side bands. The low magnetic moment of the Ce3+ center did not affect the observed [Ga]+ resonance in 1 and 3. Compared to [Ga][GaCl4], the MAS spectra contained no observed resonances attributable to the Ln-coordinated [GaCl4]− units. The spectrum of purified 1 consisted of a resonance centered at δ = −693.6 ppm, which shifted to δ = −756.2 ppm upon coordination to C6H5F. The 71Ga MAS NMR spectra of 2 and 4 appeared to be much more complex due to the paramagnetic Ho3+. This complexity arose from increased shift anisotropy from hyperfine interactions with the Ho metal center.53−56 The results of the 71Ga NMR experiments are summarized in Table S1. To characterize the [GaCl4]− units, a series of static 71Ga NMR spectra were collected (Figure S5). The signal from these units appeared as a broad signal underlying the sharper peak for the [Ga]+ species. Again, the line widths of the spectra for compounds 2 and 4 were substantially broader than those of compounds 1 and 3 due to the larger hyperfine interactions with Ho3+. Spectral deconvolutions revealed the relative contributions to the line shapes from quadrupolar coupling and the paramagnetic chemical shift (Table S2). We observed a significantly larger shift contribution to the [GaCl4]− line shape compared to [Ga]+ due to their proximity to the lanthanide center. The [GaCl4]− units also had a large contribution from quadrupole coupling, but in 2 and 4, this contribution was minor compared with that from the paramagnetic chemical shift. The quadrupole coupling constant (CQ) for [Ga]+ showed a substantial increase upon removal of C6H5F, which was attributed to a change in the relative motion of [Ga]+ that results in an averaging of the quadrupole coupling.
Figure 3.

Solid-state 71Ga NMR spectra presented for the Ce complexes 1 (a) and 3 (c) and the Ho complexes 2 (b) and 4 (d).
By exploring the reactivity of molten GaCl3 as an oxidizing solvent for Ce and Ho, this work incorporates several impactful facets of chemistry, including oxidative dissolution with nonclassical solvents, lanthanide reactivity and bonding, and low-valent gallium chemistry. The reactions of Ce and Ho in a molten GaCl3 matrix were successful and the products were high yielding, generating coordination motifs previously unobserved for lanthanide metals. The solid-state NMR experiments provided characterization of complexes 1–4 without the solvent effects or undesired reactivity that was observed using solution-phase NMR. We will continue to explore the oxidative dissolution of f-block metals using GaCl3, focusing on uranium and plutonium, and pursue further characterization and reactivity of the resultant products.
Acknowledgments
Los Alamos National Laboratory is operated by Triad National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (2020LANLE372 under Contract 89233218CNA000001). This work was supported by the U.S. Department of Energy, NNSA, Plutonium Modernization Program (NA-191). A.M.T. thanks Sarah K. Tondreau for assistance with manuscript organization and copyediting. T.V.F. and S.H.C. are grateful for additional postdoctoral support provided by the Glenn T. Seaborg Institute. B.K.C. was supported through a LANL LDRD project (20220389ER). We acknowledge and thank Jesse Murillo and Andrew J. Gaunt for allowing us the use of their lab space and instrumentation, which was provided by the U.S Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry Program (2020LANLE372).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.3c02774.
Experimental conditions, crystallographic tables and correlated CCDC deposition numbers, and spectroscopic characterization of all compounds synthesized (PDF)
Author Contributions
This work was conceived by N.H.A. and A.M.T. and executed at Los Alamos National Laboratory.
The authors declare no competing financial interest.
Supplementary Material
References
- Drobot N. F.; Trifonova E. N.; Krenev V. A.; Drobot D. V. Oxidative dissolution of refractory metals by chlorination in aqueous organic media. Russ. J. Coord. Chem. 2005, 31 (4), 243–246. 10.1007/s11173-005-0084-4. [DOI] [Google Scholar]
- Schiavi P. G.; Altimari P.; Sturabotti E.; Giacomo Marrani A.; Simonetti G.; Pagnanelli F. Decomposition of Deep Eutectic Solvent Aids Metals Extraction in Lithium-Ion Batteries Recycling. ChemSusChem 2022, 15 (18), e202200966 10.1002/cssc.202200966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson K.; Binnemans K. Selective extraction of metals using ionic liquids for nickel metal hydride battery recycling. Green Chem. 2014, 16 (10), 4595–4603. 10.1039/C3GC41930D. [DOI] [Google Scholar]
- Li X.; Van den Bossche A.; Vander Hoogerstraete T.; Binnemans K. Ionic liquids with trichloride anions for oxidative dissolution of metals and alloys. Chem. Commun. 2018, 54 (5), 475–478. 10.1039/C7CC08645H. [DOI] [PubMed] [Google Scholar]
- Van den Bossche A.; De Witte E.; Dehaen W.; Binnemans K. Trihalide ionic liquids as non-volatile oxidizing solvents for metals. Green Chem. 2018, 20 (14), 3327–3338. 10.1039/C8GC01061G. [DOI] [Google Scholar]
- Billard I.; Gaillard C. Actinide and lanthanide speciation in imidazolium-based ionic liquids. Radiochim. Acta 2009, 97 (7), 355–359. 10.1524/ract.2009.1617. [DOI] [Google Scholar]
- Binnemans K. Lanthanides and Actinides in Ionic Liquids. Chem. Rev. 2007, 107 (6), 2592–2614. 10.1021/cr050979c. [DOI] [PubMed] [Google Scholar]
- Mudring A.-V.; Tang S. Ionic Liquids for Lanthanide and Actinide Chemistry. Eur. J. Inorg. Chem. 2010, 18, 2569–2581. 10.1002/ejic.201000297. [DOI] [Google Scholar]
- Mudring A.-V.Ionic liquids as versatile media in lanthanide chemistry. In Ionic Liquids IV; Brennecke J. F., Rogers R. D., Seddon K. R., Eds.; ACS Symposium Series 975; American Chemical Society, 2007; pp 172–185. 10.1021/bk-2007-0975.ch012. [DOI] [Google Scholar]
- Pereira C. C. L.; Carretas J. M.; Monteiro B.; Leal J. P. Luminescent Ln-Ionic Liquids beyond Europium. Molecules 2021, 26 (16), 4834. 10.3390/molecules26164834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos E.; Albo J.; Irabien A. Magnetic ionic liquids: synthesis, properties and applications. RSC Adv. 2014, 4 (75), 40008–40018. 10.1039/C4RA05156D. [DOI] [Google Scholar]
- Belova V. V. Tendencies in Application of Ionic Liquids and Binary Extractants in Extraction and Separation of Lanthanides and Actinides. Radiochemistry 2021, 63 (1), 1–10. 10.1134/S106636222101001X. [DOI] [Google Scholar]
- Bhatt A. I.; Kinoshita H.; Koster A. L.; May I.; Sharrad C.; Steele H. M.; Volkovich V. A.; Fox O. D.; Jones C. J.; Lewin B. G.; Hennig C.. Actinide, lanthanide, and fission product speciation and electrochemistry in ionic melts. In Separations for the Nuclear Fuel Cycle in the 21st Century; Lumetta G. J., Nash K. L., Clark S. B., Friese J. I., Eds.; ACS Symposium Series 933; American Chemical Society, 2006; pp 219–231. 10.1021/bk-2006-0933.ch014. [DOI] [Google Scholar]
- Billard I.Ionic liquids: new hopes for efficient lanthanide/actinide extraction and separation? In Handbook on the Physics and Chemistry of the Rare Earths; Bünzil J. G., Pecharsky V. K., Eds.; Elsevier, 2013; Vol. 43, pp 213–273. 10.1016/B978-0-444-59536-2.00003-9. [DOI] [Google Scholar]
- Billard I.; Ouadi A.; Gaillard C. Liquid-liquid extraction of actinides, lanthanides, and fission products by use of ionic liquids: from discovery to understanding. Anal. Bioanal. Chem. 2011, 400 (6), 1555–1566. 10.1007/s00216-010-4478-x. [DOI] [PubMed] [Google Scholar]
- De Jesus K.; Rodriguez R.; Baek D. L.; Fox R. V.; Pashikanti S.; Sharma K. Extraction of lanthanides and actinides present in spent nuclear fuel and in electronic waste. J. Mol. Liq. 2021, 336, 116006. 10.1016/j.molliq.2021.116006. [DOI] [Google Scholar]
- Iqbal M.; Waheed K.; Rahat S. B.; Mehmood T.; Lee M. S. An overview of molecular extractants in room temperature ionic liquids and task specific ionic liquids for the partitioning of actinides/lanthanides. J. Radioanal. Nucl. Chem. 2020, 325 (1), 1–31. 10.1007/s10967-020-07199-1. [DOI] [Google Scholar]
- Kolarik Z. Ionic Liquids: How Far Do they Extend the Potential of Solvent Extraction of f-Elements?. Solvent Extr. Ion Exch. 2013, 31 (1), 24–60. 10.1080/07366299.2012.700589. [DOI] [Google Scholar]
- Kumari A.; Sinha M. K.; Sahu S. K.; Pandey B. D. Solvent Extraction and Separation of Trivalent Lanthanides Using Cyphos IL 104, a Novel Phosphonium Ionic Liquid as Extractant. Solvent Extr. Ion Exch. 2016, 34 (5), 469. 10.1080/07366299.2016.1207459. [DOI] [Google Scholar]
- Venkatesan K. A.; Jagadeeswara Rao C.; Nagarajan K.; Vasudeva Rao P. R. Electrochemical behavior of actinides and fission products in room-temperature ionic liquids. Int. J. Electrochem. 2012, 2012, 841456. 10.1155/2012/841456. [DOI] [Google Scholar]
- Yudaev P. A.; Chistyakov E. M. Ionic Liquids as Components of Systems for Metal Extraction. ChemEngineering 2022, 6 (1), 6. 10.3390/chemengineering6010006. [DOI] [Google Scholar]
- Billard I.; Gaillard C.; Mekki S.; Ouadi A.; Stumpf S.; Hesemann P.; Moutiers G.; Trubert D.; Le Naour C.. Ionic liquids for actinides and lanthanides chemistry. In Recent Advances in Actinide Science; May I., Bryan N. D., Alvares R., Eds.; The Royal Society of Chemistry, 2006; pp 656–658. 10.1039/9781847555366. [DOI] [Google Scholar]
- MacInnes M. M.; Jones Z. R.; Li B.; Anderson N. H.; Batista E. R.; DiMucci I. M.; Eiroa-Lledo C.; Knope K. E.; Livshits M. Y.; Kozimor S. A.; Mocko V.; Pace K. A.; Rocha F. R.; Stein B. W.; Wacker J. N.; Yang P. Using molten salts to probe outer-coordination sphere effects on lanthanide(III)/(II) electron-transfer reactions. Dalton Trans 2021, 50 (43), 15696–15710. 10.1039/D1DT02708E. [DOI] [PubMed] [Google Scholar]
- Liu K.; Tang H.-B.; Pang J.-W.; Liu Y.-L.; Feng Y.-X.; Chai Z.-F.; Shi W.-Q. Electrochemical Properties of Uranium on the Liquid Gallium Electrode in LiCl-KCl Eutectic. J. Electrochem. Soc. 2016, 163 (9), D554–D561. 10.1149/2.1191609jes. [DOI] [Google Scholar]
- Eich A.; NejatyJahromy Y.; Schiemann O.; Beck J. Se4CuX, Te4CuX and Cu2Se7X2 (X = GaCl4) — Coordination compounds of neutral infinite chalcogen chains. Inorg. Chem. Commun. 2015, 58, 20–23. 10.1016/j.inoche.2015.04.025. [DOI] [Google Scholar]
- Eich A.; Hoffbauer W.; Schnakenburg G.; Bredow T.; Daniels J.; Beck J. Double-Cube-Shaped Mixed Chalcogen/Pentele Clusters from GaCl3 Melts. Eur. J. Inorg. Chem. 2014, 2014 (19), 3043–3052. 10.1002/ejic.201402230. [DOI] [Google Scholar]
- Åkerstedt J.; Gorlov M.; Kloo L. Room-Temperature Synthesis of the Bi5[GaCl4]3 Salt From Three Different Classes of Ionic Liquids. J. Cluster Sci. 2013, 24 (1), 157–164. 10.1007/s10876-012-0526-3. [DOI] [Google Scholar]
- Freudenmann D.; Feldmann C. [Bi3GaS5]2[Ga3Cl10]2[GaCl4]2·S8 containing heterocubane-type [Bi3GaS5]2+, star-shaped [Ga3Cl10]−, monomeric [GaCl4]− and crown-like S8. Dalton Trans 2011, 40 (2), 452–456. 10.1039/C0DT00985G. [DOI] [PubMed] [Google Scholar]
- Ulvenlund S.; Bengtsson-Kloo L.; Stahl K. Formation of subvalent bismuth cations in molten gallium trichloride and benzene solutions. J. Chem. Soc., Faraday Trans. 1995, 91 (23), 4223–4234. 10.1039/FT9959104223. [DOI] [Google Scholar]
- Eich A.; Schnakenburg G.; Beck J. Ga2SbCl7O – A Molecular Gallium Antimony Chloride Oxide Synthesized from a GaCl3 Melt. Z. Anorg. Allg. Chem. 2014, 640 (12–13), 2431–2434. 10.1002/zaac.201400260. [DOI] [Google Scholar]
- Carpenter S. H.; Klamm B. E.; Fetrow T. V.; Scott B. L.; Gaunt A. J.; Anderson N. H.; Tondreau A. M. Chlorination of Pu and U Metal Using GaCl3. Inorg. Chem. 2023, 62, 8462–8466. 10.1021/acs.inorgchem.3c00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piro N. A.; Robinson J. R.; Walsh P. J.; Schelter E. J. The electrochemical behavior of cerium(III/IV) complexes: Thermodynamics, kinetics and applications in synthesis. Coord. Chem. Rev. 2014, 260, 21–36. 10.1016/j.ccr.2013.08.034. [DOI] [Google Scholar]
- Vanýsek P.Electrochemical Series. In CRC Handbook of Chemistry and Phyics, 95th ed.; Haynes W. M., Lide D. R., Eds.; CRC Press: Boca Raton, FL, 2014; pp 5-80–5-89. [Google Scholar]
- Trinidad P.; Ponce de Leon C.; Walsh F. C. The use of electrolyte redox potential to monitor the Ce(IV)/Ce(III) couple. J. Environ. Manage. 2008, 88 (4), 1417–1425. 10.1016/j.jenvman.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Bourque J. L.; Nanni R. A.; Biesinger M. C.; Baines K. M. Synthesis and Reactivity of Cationic Gallium(I) [12]Crown-4 Complexes. Inorg. Chem. 2021, 60 (19), 14713–14720. 10.1021/acs.inorgchem.1c01801. [DOI] [PubMed] [Google Scholar]
- Kunze A.; Gleiter R.; Bethke S.; Rominger F. Complexes of π-Prismands with Gallium(I). Organometallics 2006, 25 (20), 4787–4791. 10.1021/om060470n. [DOI] [Google Scholar]
- Schmidbaur H.; Thewalt U.; Zafiropoulos T. Isolation and crystal structure of [(C6H6)2Ga·GaCl4]2·3C6H6, a bis(η6-benzene)gallium(I) complex. Organometallics 1983, 2 (11), 1550–1554. 10.1021/om50005a012. [DOI] [Google Scholar]
- Schmidbaur H.; Thewalt U.; Zafiropoulos T. Synthese und Struktur von Bis(mesitylen)gallium(I)-tetrachlorogallat(III). Chem. Ber. 1984, 117 (12), 3381–3387. 10.1002/cber.19841171207. [DOI] [Google Scholar]
- Campbell G. C.; Cotton F. A.; Haw J. F.; Schwotzer W. Syntheses, structures, and solid-state carbon-13 NMR of two η6-arene uranium(IV) complexes, [U(C6Me6)Cl2(μ-Cl)3UCl2(C6Me6)]AlCl4 and (C6Me6)UCl2(μ-Cl)3UCl2(μ-Cl)3UCl2(C6Me6). Organometallics 1986, 5 (2), 274–279. 10.1021/om00133a018. [DOI] [Google Scholar]
- Filatov A. S.; Rogachev A. Y.; Petrukhina M. A. Lanthanum(III) chloroaluminate and chlorogallate complexes with toluene and hexamethylbenzene: The effect of arene methylation on the structure. J. Mol. Struct. 2008, 890 (1), 116–122. 10.1016/j.molstruc.2008.04.035. [DOI] [Google Scholar]
- Gorlov M.; Hussami L. L.; Fischer A.; Kloo L. Mononuclear η6-Arene Complexes of Lanthanides: One-Step Syntheses, Crystal Structures, and Arene Exchange. Eur. J. Inorg. Chem. 2008, 2008, 5191–5195. 10.1002/ejic.200800807. [DOI] [Google Scholar]
- Slattery J. M.; Higelin A.; Bayer T.; Krossing I. A Simple Route to Univalent Gallium Salts of Weakly Coordinating Anions. Angew. Chem., Int. Ed. 2010, 49 (18), 3228–3231. 10.1002/anie.201000156. [DOI] [PubMed] [Google Scholar]
- Buchin B.; Gemel C.; Cadenbach T.; Schmid R.; Fischer R. A. The [Ga2(C5Me5)]+ Ion: Bipyramidal Double-Cone Structure and Weakly Coordinated Monovalent Ga+. Angew. Chem., Int. Ed. 2006, 45 (7), 1074–1076. 10.1002/anie.200503028. [DOI] [PubMed] [Google Scholar]
- Dagorne S.; Bellemin-Laponnaz S.; Maisse-François A.; Rager M.-N.; Jugé L.; Welter R. Synthesis and Structure of Neutral and Cationic Gallium Complexes Incorporating Bis(oxazolinato) Ligands. Eur. J. Inorg. Chem. 2005, 2005, 4206–4214. 10.1002/ejic.200500478. [DOI] [Google Scholar]
- Higelin A.; Haber C.; Meier S.; Krossing I. Isolated cationic crown ether complexes of gallium(I) and indium(I). Dalton Trans 2012, 41 (39), 12011–12015. 10.1039/c2dt30379e. [DOI] [PubMed] [Google Scholar]
- Higelin A.; Keller S.; Göhringer C.; Jones C.; Krossing I. Unusual Tilted Carbene Coordination in Carbene Complexes of Gallium(I) and Indium(I). Angew. Chem., Int. Ed. 2013, 52 (18), 4941–4944. 10.1002/anie.201209757. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler M. R.; Higelin A.; Kraft A.; Hughes S.; Steffani A.; Plattner D. A.; Slattery J. M.; Krossing I. Univalent Gallium Salts of Weakly Coordinating Anions: Effective Initiators/Catalysts for the Synthesis of Highly Reactive Polyisobutylene. Organometallics 2013, 32 (22), 6725–6735. 10.1021/om4005516. [DOI] [Google Scholar]
- Lichtenthaler M. R.; Maurer S.; Mangan R. J.; Stahl F.; Mönkemeyer F.; Hamann J.; Krossing I. Univalent Gallium Complexes of Simple and ansa-Arene Ligands: Effects on the Polymerization of Isobutylene. Chem. Eur. J. 2015, 21 (1), 157–165. 10.1002/chem.201404833. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler M. R.; Stahl F.; Kratzert D.; Benkmil B.; Wegner H. A.; Krossing I. σ- or π-Coordination? Complexes of Univalent Gallium Salts with Aromatic Nitrogen Bases. Eur. J. Inorg. Chem. 2014, 2014, 4335–4341. 10.1002/ejic.201402360. [DOI] [Google Scholar]
- Macdonald C. L. B.; Gorden J. D.; Voigt A.; Filipponi S.; Cowley A. H. Group 13 decamethylmetallocenium cations. Dalton Trans 2008, 1161–1176. 10.1039/B716220K. [DOI] [PubMed] [Google Scholar]
- Schorpp M.; Tamim R.; Krossing I. Oxidative addition, reduction and reductive coupling: the versatile reactivity of subvalent gallium cations. Dalton Trans 2021, 50 (42), 15103–15110. 10.1039/D1DT02682H. [DOI] [PubMed] [Google Scholar]
- Wehmschulte R. J.; Peverati R.; Powell D. R. Convenient Access to Gallium(I) Cations through Hydrogen Elimination from Cationic Gallium(III) Hydrides. Inorg. Chem. 2019, 58 (18), 12441–12445. 10.1021/acs.inorgchem.9b02136. [DOI] [PubMed] [Google Scholar]
- Grey C. P.; Dobson C. M.; Cheetham A. K.; Jakeman R. J. B. Studies of rare-earth stannates by 119Sn MAS NMR. The use of paramagnetic shift probes in the solid state. J. Am. Chem. Soc. 1989, 111 (2), 505–511. 10.1021/ja00184a017. [DOI] [Google Scholar]
- Brough A. R.; Grey C. P.; Dobson C. M. Paramagnetic ions as structural probes in solid-state NMR: distance measurements in crystalline lanthanide acetates. J. Am. Chem. Soc. 1993, 115 (16), 7318–7327. 10.1021/ja00069a034. [DOI] [Google Scholar]
- Palke A. C.; Stebbins J. F. Paramagnetic interactions in the 31P NMR spectroscopy of rare earth element orthophosphate (REPO4, monazite/xenotime) solid solutions. Am. Mineral. 2011, 96 (8–9), 1343–1353. 10.2138/am.2011.3816. [DOI] [Google Scholar]
- Palke A. C.; Stebbins J. F.; Boatner L. A. 31P Magic Angle Spinning NMR Study of Flux-Grown Rare-Earth Element Orthophosphate (Monazite/Xenotime) Solid Solutions: Evidence of Random Cation Distribution from Paramagnetically Shifted NMR Resonances. Inorg. Chem. 2013, 52 (21), 12605–12615. 10.1021/ic401757z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.