

ABSTRACT
Tripartite motif (TRIM) proteins are a large family of E3 ubiquitin ligases implicated in antiviral defense systems, tumorigenesis, and protein quality control. TRIM proteins contribute to protein quality control by regulating the ubiquitin-proteasome system, endoplasmic reticulum-associated degradation, and macroautophagy/autophagy. However, the detailed mechanisms through which various TRIM proteins regulate downstream events have not yet been fully elucidated. Herein, we identified a novel function of TRIM22 in the regulation of autophagy. TRIM22 promotes autophagosome-lysosome fusion by mediating the association of GABARAP family proteins with PLEKHM1, thereby inducing the autophagic clearance of protein aggregates, independent of its E3 ubiquitin ligase activity. Furthermore, a TRIM22 variant associated with early-onset familial Alzheimer disease interferes with autophagosome-lysosome fusion and autophagic clearance. These findings suggest TRIM22 as a critical autophagic regulator that orchestrates autophagosome-lysosome fusion by scaffolding autophagy-related proteins, thus representing a potential therapeutic target in neurodegenerative diseases.
Abbreviations: AD: Alzheimer disease; ADAOO: AD age of onset; AICD: APP intracellular domain; APP: amyloid beta precursor protein; BSA: bovine serum albumin; cDNAs: complementary DNAs; CQ: chloroquine; CTF: carboxyl-terminal fragment; EBSS: Earle’s balanced salt solution; GABARAP: GABA type A receptor-associated protein; GST: glutathione S-transferase; HA: hemagglutinin; HOPS: homotypic fusion and protein sorting; IFN: interferon; IL1A/IL-1α: interleukin 1 alpha; KO: knockout; MTORC1: mechanistic target of rapamycin kinase complex 1; NFKBIA/IκBα: NFKB inhibitor alpha; NFE2L2/NRF2: NFE2 like bZIP transcription factor; PBS: phosphate-buffered saline; PI3K: class I phosphoinositide 3-kinase; PLA: proximity ligation assay; PLEKHM1: pleckstrin homology and RUN domain containing M1; PSEN1: presenilin 1; SEM: standard errors of the means; SNAREs: soluble N-ethylmaleimide-sensitive factor attachment protein receptors; SNCA: synuclein alpha; SNP: single nucleotide polymorphism; TBS: tris-buffered saline; TNF/TNF-α: tumor necrosis factor; TRIM: tripartite motif; ULK1: unc-51 like autophagy activating kinase 1; WT: wild-type
KEYWORDS: Alzheimer disease, autophagosome-lysosome fusion, autophagy, PLEKHM1, TRIM22
Introduction
Autophagy is a lysosome-mediated degradation process that clears misfolded proteins and damaged subcellular organelles [1]. Autophagy is constitutively active at low levels to maintain cellular homeostasis. In response to cellular stress, autophagy is upregulated via MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) signaling, thereby resolving cellular stress [2]. Under nutrient starvation, ULK1 (unc-51 like autophagy activating kinase 1), normally inhibited by MTORC1-dependent phosphorylation under nutrient-rich conditions, is activated by AMP-activated protein kinase-dependent phosphorylation [3]. ULK1 is a part of an eponymous complex, also known as the autophagy pre-initiation complex, along with RB1CC1/FIP200, ATG101, and ATG13 [4,5]. Through direct phosphorylation or indirect ubiquitination, the ULK1 complex promotes the formation of a class III phosphatidylinositol 3-kinase (PtdIns3K) complex, also known as the PIK3C3/VPS34 or autophagy initiation complex, which is composed of PIK3C3, BECN1, ATG14, NRBF2 and PIK3R4/VPS15 [5,6]. The class III PtdIns3K complex promotes autophagosome formation by phosphorylating phosphatidylinositol in phospholipid-enriched membranes to phosphatidylinositol-3-phosphate [7]. The recruitment of autophagy-initiating factors to complex assembly sites is regulated by several proteins, including RAB2 and CALCOCO2/NDP52 for the ULK1 complex as well as DACT1/Dpr1, and RACK1 for the PIK3C3 complex [8–12]. The fusion of a mature autophagosome with a lysosome is mediated by RAB GTPases, soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs), and tethering proteins [13]. PLEKHM1 (pleckstrin homology and RUN domain containing M1) plays a key role in autophagosome-lysosome fusion by interacting with RAB7 in lysosomes as well as with the homotypic fusion and protein sorting (HOPS) complex, STX17 (syntaxin 17) of SNAREs, LC3, and GABARAP (GABA type A receptor-associated protein) in autophagosomes [14,15].
Over the last two decades, studies have revealed that autophagic defects contribute to the pathogenesis of various human diseases, including cancer, autoimmune diseases, and neurodegenerative diseases. In the brain, complete loss of core autophagy proteins, such as ATG5 or ATG7, leads to neurodegeneration characterized by progressive motor and behavioral defects and the accumulation of polyubiquitinated inclusion bodies [16,17]. The accumulation of aggregate-prone proteins, such as amyloid β, MAPT/tau, and mutant HTT (huntingtin), is a hallmark of neurodegenerative diseases. Aggregate clearance depends mainly on the autophagy-lysosome degradation pathway [18]. The activation of autophagy removes aggregate-prone proteins, ameliorates cognitive deficits [19–21], and reduces neuronal vulnerability to stress [22]. Furthermore, the depletion of autophagic receptors disturbs the clearance of pathogenic proteins, accelerating the onset and progression of neurodegenerative diseases in mouse models [23–25]. Promoting the autophagy-mediated intracellular clearance of aggregate-prone proteins thus represents a promising therapeutic strategy for neurodegenerative diseases.
Tripartite motif (TRIM) proteins are a large family of E3 ubiquitin ligases that possess an N-terminal RING domain, followed by one or two B-box-type zinc finger domains and a coiled-coil domain [26,27]. The E3 ubiquitin ligase activity of TRIM proteins is responsible for the regulation of various signaling cascades, including antiviral defense mechanisms, and inflammation. The coiled-coil domain and SPRY domains of TRIM proteins mediate their interaction with various partners. Recent studies have shown that TRIM proteins can regulate autophagic activity by interacting with autophagy-related proteins, such as in the case of TRIM5/TRIM5α interacting with Atg8-family proteins, ULK1, and BECN1; TRIM16 with ULK1 and BECN1; TRIM32 with ULK1 and AMBRA1; TRIM37 with MTORC1; and TRIM50 with BECN1 [28–32]. These interactions likely enable TRIM proteins to act as autophagic adaptors, that is, platforms for core autophagy regulators, collectively termed TRIMosomes [27,33].
TRIM22 belongs to the same subfamily as TRIM5, which functions as a core subunit of TRIMosomes, suggesting that TRIM22 could also act as an autophagic adaptor. Previous studies have shown that TRIM22 also interacts with autophagy-related proteins, and siRNA screening results revealed that TRIM22 may be a positive autophagy regulator [28,34,35]. However, the exact role of TRIM22 in autophagy remains unclear. TRIM22 is an antiviral effector whose expression is strongly induced by interferon (IFN) [36,37]. In addition, the association of TRIM22 with Alzheimer disease (AD) has been reported in a pedigree bearing a PSEN1 (presenilin 1) mutation, which causes early onset familial AD [38]. However, the etiological mechanisms underlying this genetic association remain unclear.
Herein, we aimed to determine the role of TRIM22 in autophagy based on its interactions with core autophagy regulators. To this end, we examined the association of TRIM22 with autophagic degradation and revealed the molecular mechanism underlying TRIM22-mediated autophagic regulation. We also set out to determine the clinical significance of the AD-associated TRIM22 variant based on its role in the clearance of aggregate-prone proteins.
Results
TRIM22 deficiency interferes with intracellular clearance
We generated TRIM22-knockout (KO) HeLa cell lines to elucidate the regulatory roles of TRIM22 in autophagy (Figure 1A and S1A). IFNG/IFN-γ-induced TRIM22 upregulation was abolished in TRIM22-KO cells, indicating a total loss of endogenous TRIM22 protein (Figure 1B and S1B). Compared with wild-type (WT) cells, TRIM22-KO cells contained more LC3A/B-II-positive autophagic compartments (Figure 1C,D), and the level of autophagic substrate SQSTM1/p62 was elevated in TRIM22-KO cells. These effects could be rescued via re-expression of TRIM22 (Figure 1C). In TRIM22-KO cells, the accumulation of LC3A/B-II mediated by lysosome inhibition was less efficient compared to that in WT cells (Figure 1E). The level of polyubiquitinated proteins, which are also autophagic substrates, was elevated in TRIM22-KO cells but decreased in TRIM22-overexpressing cells (Figure 1F and S1C). Similarly, knocking down TRIM22 expression using siRNAs in hTERT-RPE1 and HeLa cells also elevated levels of SQSTM1 and polyubiquitinated proteins (Figure 1G and S1D,E). Puromycin treatment can induce the formation of aggresome-like induced structures, which are degraded through autophagy [39]. Clearance of ubiquitin-positive aggresome-like induced structures was also impeded in TRIM22-KO cells (Figure 1H). Loss of TRIM22 resulted in similar defects in the condition of induced autophagy via MTOR inhibition (Figure 1I). However, it is intriguing to note that changes in TRIM22 protein levels had no effect on the activities of intracellular clearance induced through starvation with Earle’s balanced salt solution (EBSS), suggesting that TRIM22 may play a role in basal and MTOR inhibition-induced autophagy (Figure 1I,J).
Figure 1.
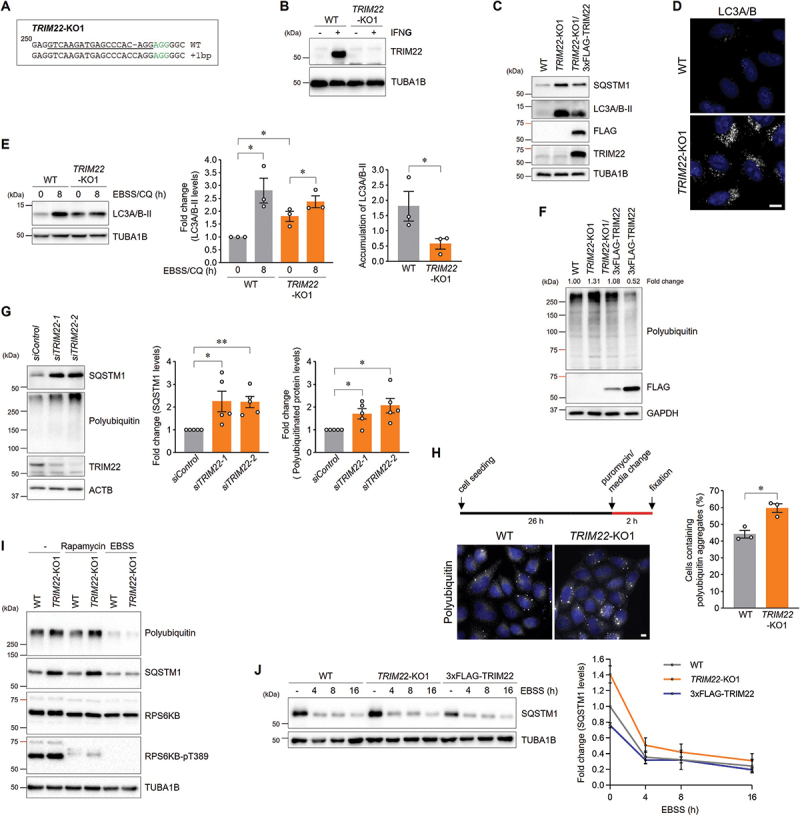
TRIM22 deficiency interferes with intracellular clearance. (A,B) The generation of a TRIM22-knockout cell line was validated via genomic DNA sequencing (A) and immunoblotting (B). (A) Sequence alignment of the allele from wild-type HeLa cells and TRIM22-KO1 cells identified through sequencing. Nucleotide numbers are expressed relative to the initiation codon. Sequences corresponding to the 19-nt sgRNA target are underlined and the 3-nt PAM is highlighted in green. (B) Wild-type HeLa cells or TRIM22-KO1 cells were grown in the absence or presence of 50 ng/mL of IFNG for 24 h and then analyzed via immunoblotting with the indicated antibodies. (C) Wild-type HeLa cells, TRIM22-KO1 cells, or TRIM22-KO1 cells stably expressing 3×FLAG-TRIM22 cells were subjected to immunoblotting with the indicated antibodies. (D) Wild-type HeLa cells or TRIM22-KO1 cells were immunostained for LC3A/B. (E) Wild-type HeLa cells or TRIM22-KO1 cells were incubated with EBSS containing 20 μM of CQ for 8 h and then analyzed via immunoblotting with the indicated antibodies. Band intensities for LC3A/B-II were measured and normalized to the mean intensity of the untreated wild-type cell group. The accumulation of LC3A/B-II was calculated by subtracting the intensity of the untreated group from that of the EBSS/CQ-treated group. Graphs show means ± SEM of three independent experiments. (F) Wild-type HeLa cells, TRIM22-KO1 cells, TRIM22-KO1 cells stably expressing 3×FLAG-TRIM22 cells, or 3×FLAG-TRIM22-expressing cells were subjected to immunoblotting with the indicated antibodies. Band intensities of polyubiquitinated proteins were measured and normalized to the intensity of wild-type cells. (G) hTERT-RPE1 cells were transfected with either control or TRIM22 siRNAs, and subsequently subjected to immunoblotting with the indicated antibodies. Band intensities of SQSTM1 and polyubiquitinated proteins were measured and normalized to the mean intensity of siControl-transfected cells. Graphs show means ± SEM of five independent experiments. (H) Wild-type HeLa cells or TRIM22-KO1 cells were treated with 5 μg/mL of puromycin for 2 h and immunostained with polyubiquitin-specific antibodies. Cells containing more than five polyubiquitin aggregates were quantified from more than 200 cells per group. Graphs show means ± SEM of three independent experiments. (I) Wild-type HeLa cells or TRIM22-KO1 cells were incubated with either 100 nM of rapamycin or with EBSS for 8 h. Cell lysates were subjected to immunoblotting with indicated antibodies. (J) Wild-type HeLa cells, TRIM22-KO1 cells, or 3×FLAG-TRIM22-expressing cells were incubated with EBSS for the indicated times, and cell lysates were subjected to immunoblotting with the indicated antibodies. Band intensities of SQSTM1 were measured and normalized to the mean intensity of untreated wild-type cell group. Graphs show means ± SEM of three independent experiments. *p < 0.05; **p < 0.01. Scale bars: 10 μm.
Meanwhile, recent research has suggested that TRIM22 plays a role in suppressing autophagy in M5-treated HaCat cells by activating the PI3K-AKT-MTOR pathway [40]. In this regard, we examined whether alterations in TRIM22 expression, either through knockout or overexpression, would affect the PI3K-AKT-MTOR pathway. Remarkably, key markers of this signaling pathways, including levels of phospho-MTOR, phospho-RPS6KB/S6K, and phospho-AKT levels, remained largely unaffected by changes in TRIM22 expression (Figure S2A). This finding was consistently observed under conditions to induce autophagy, such as treatment of rapamycin or EBSS (Figure S2B,C). Hence, we speculate that the observed differences could potentially be ascribed to the specialized conditions of M5, which contains cytokines such as TNF/TNF-α, IL1A/IL-1α, IL17A, and IL22, mimicking the excessive proliferation of keratinocytes in psoriasis. Taken together, these results suggest that TRIM22 functions in autophagic degradation.
TRIM22 positively regulates autophagosome-lysosome fusion by mediating the association of GABARAPs with PLEKHM1
The acidic environment of lysosomes is critical for the activity of lysosomal hydrolases, which degrade autophagic substrates within autolysosomes. The intensity of lysosomal pH indicators did not significantly change in TRIM22-KO cells (Figure 2A). The activity of cathepsin D, which is a major lysosomal hydrolase, was also not affected by TRIM22 depletion (Figure 2B). Next, we monitored cells stably expressing mRFP-EGFP-LC3B, a tandem fluorescent-tagged LC3 system that enables visual evaluation of autophagosome-lysosome fusion. Compared with WT cells, TRIM22-KO cells exhibited more yellow LC3B puncta and less red LC3B puncta, representing autophagosomes and autolysosomes, respectively (Figure 2C and S3A). The decrease in the autolysosome:autophagosome ratio was also observed in the cells with depleted TRIM22 via siRNA transfection (Figure S3B). In contrast, the ratio was increased by TRIM22 overexpression (Figure 2D). Greater mRFP signals, representing an increase in the autolysosome:autophagosome ratio in TRIM22-overexpressing cells, were confirmed through fluorescence-activated cell sorting analysis (Figure 2E). Although the above results suggest a positive role of TRIM22 in autophagosome-lysosome fusion, these data should be carefully interpreted for the following reasons. First, TRIM22 could facilitate autophagosome formation, which precedes fusion. Second, autophagosome-lysosome fusion is a dynamic event that exhibits variation between cells with regard to timing. Therefore, we synchronized the fusion event via treatment with bafilomycin A1, which inhibits autophagosome-lysosome fusion prior to release, and then analyzed autolysosome formation in a time-dependent manner (Figure 2F). The ratio of autolysosomes (red-only puncta) to total LC3B-positive puncta was less than 5% before release in all experimental groups, whereas the fusion rate was faster in TRIM22-overexpressing cells and slower in TRIM22-KO cells when compared to WT cells. The impaired autophagosome-lysosome fusion in TRIM22-KO cells was also observed, albeit slightly weaker, under the MTOR inhibition condition (Figure 2G). Above results indicate that TRIM22 is a positive regulator of autophagosome-lysosome fusion.
Figure 2.
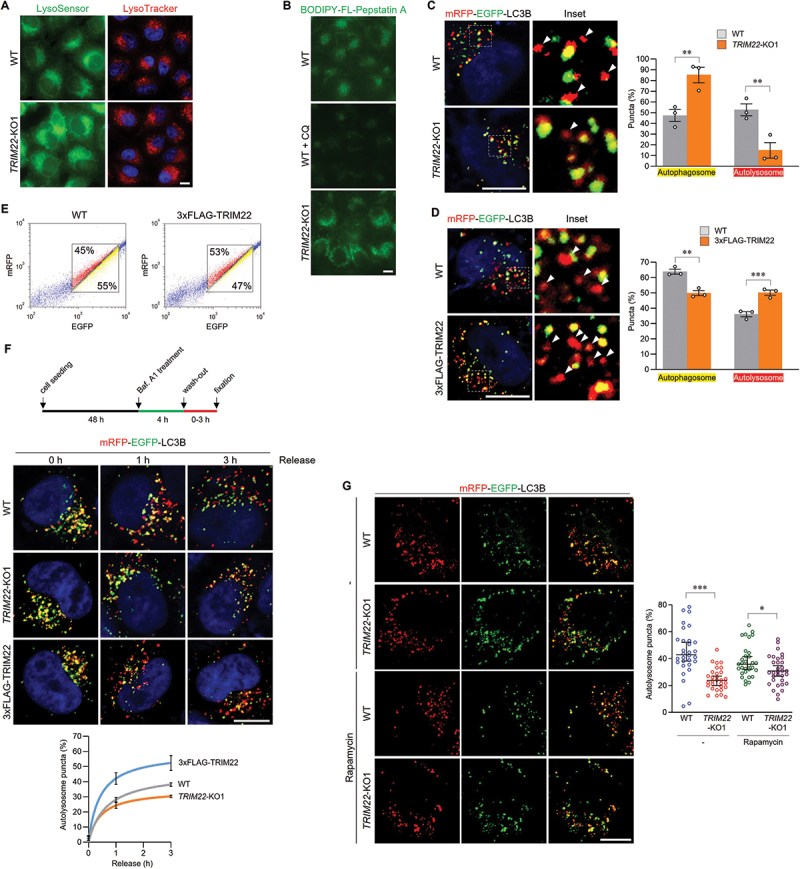
TRIM22 positively regulates autophagosome-lysosome fusion. (A) Wild-type HeLa cells or TRIM22-KO1 cells were treated with LysoSensor green DND-189 or LysoTracker red DND-99 for 30 min before fixation. (B) Wild-type HeLa cells or TRIM22-KO1 cells were treated with BODIPY-FL-Pepstatin A for 30 min. As a control, wild-type HeLa cells were cultured with CQ for 4 h before BODIPY-FL-Pepstatin A treatment. (C) mRFP-EGFP-LC3B was stably expressed in wild-type HeLa cells or TRIM22-KO1 cells, and fluorescent LC3B puncta were then analyzed in more than 30 cells per group. Puncta with mRFP+/EGFP+ indicate autophagosomes, and puncta with mRFP+/EGFP− (arrowheads) indicate autolysosomes. Graphs show means ± SEM of three independent experiments. (D,E) mRFP-EGFP-LC3B was stably expressed in wild-type HeLa cells or cells expressing 3×FLAG-TRIM22. (D) Fluorescent LC3B puncta were analyzed in more than 30 cells per group. Arrowheads indicate autolysosomes. Graphs show means ± SEM of three independent experiments. (E) Cells were analyzed using flow cytometry. The intensities of mRFP and EGFP signals from more than 10,000 cells were analyzed, and the percentage of cells in corresponding gates is shown. (F) mRFP-EGFP-LC3B was stably expressed in wild-type HeLa cells, TRIM22-KO1 cells, or 3×FLAG-TRIM22-expressing cells. The cells were treated with bafilomycin A1 (40 nM), followed by washing out and fixation as indicated, and the ratio of autolysosomes was then analyzed. Graphs show means ± SEM of more than 20 cells per group. (G) mRFP-EGFP-LC3B was stably expressed in wild-type HeLa cells or TRIM22-KO1 cells. The cells were treated with 100 nM of rapamycin for 8 h, and then the cells with fluorescent LC3B puncta were analyzed. Graphs show median with 95% confidence interval of more than 30 cells per group. *p < 0.05; **p < 0.01; *** p < 0.001. Scale bars: 10 μm.
TRIM proteins have been reported to interact with the LC3 and GABARAP subfamilies, which are required for autophagosome formation and autophagosome-lysosome fusion, respectively [28,41,42]. We found that TRIM22 specifically bound to in vitro-purified GABARAP family proteins, but not LC3 family proteins (Figure 3A). To determine the interaction motif of TRIM22 with GABARAPs, we generated truncation mutants of TRIM22 (Figure 3B). GABARAPs specifically interacted with carboxyl terminus of TRIM22, which contains a SPRY domain (Figure 3C), but failed to interact with the TRIM22-10A mutant in which potential LC3-interacting region (LIR) motifs have been mutated (Figure 3D). The interaction is mediated via the LIR docking site (Figure 3E), which is generally associated with autophagic receptors [43]. In addition, overexpressed TRIM22 colocalized with GABARAPs, but not with LC3s, in puncta structures (Figure 3F). Furthermore, lysosomal localization of GABARAP and GABARAPL1 was inhibited in TRIM22-KO cells, indicating that TRIM22 mediates the recruitment of GABARAPs to lysosomes (Figure 3G). These results suggest that TRIM22 may regulate autophagosome-lysosome fusion by recruiting GABARAP subfamily proteins.
Figure 3.
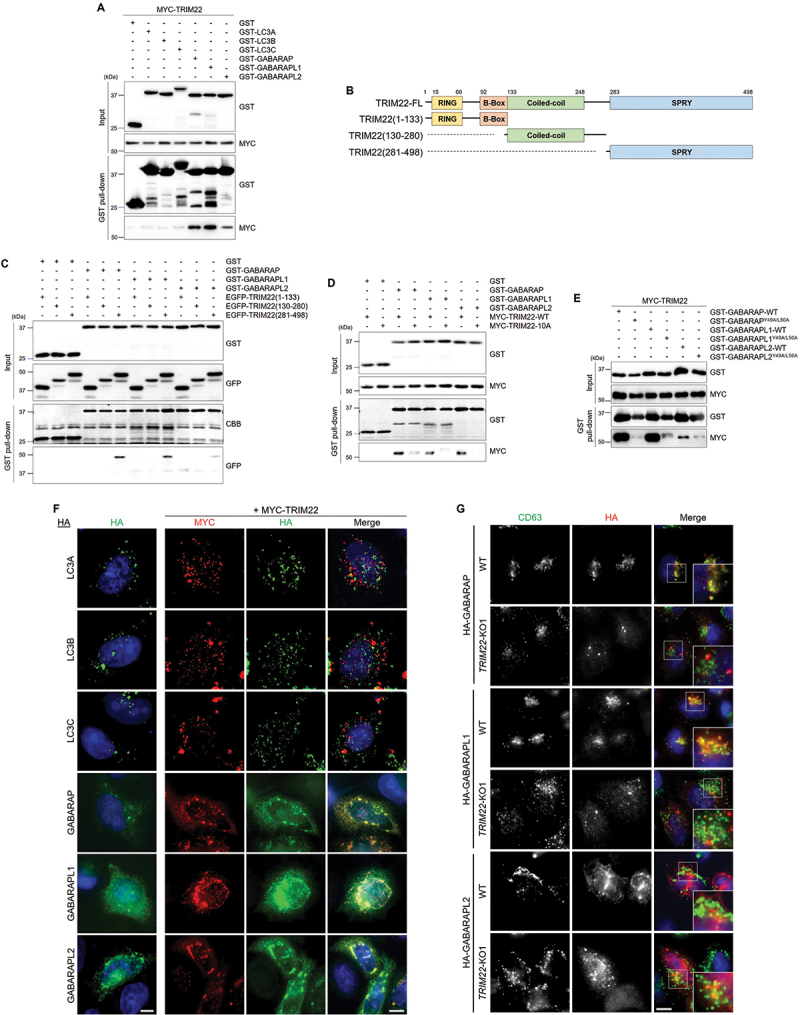
TRIM22 interacts with GABARAPs and mediates their lysosomal localization. (A) HEK293T cells were transfected with MYC-TRIM22, and lysates were incubated with the indicated GST-tagged proteins and precipitated with glutathione sepharose 4B beads. Precipitates were then immunoblotted as shown. (B) Schematic domain structure of TRIM22 and its deletion mutants. (C–E) HEK293T cells were transfected as indicated, and lysates were incubated with the indicated GST-tagged proteins and precipitated with glutathione sepharose 4B beads. Precipitates were then immunoblotted as shown. (F) HeLa cells were transfected with HA-tagged LC3A, LC3B, LC3C, GABARAP, GABARAPL1, or GABARAPL2 along with MYC-TRIM22. The cells were immunostained for HA (green) and MYC (red). (G) Wild-type HeLa cells or TRIM22-KO1 cells were transfected as indicated, and the cells were immunostained for CD63 (green) and HA (red). Scale bars: 10 μm.
PLEKHM1 is a critical protein that mediates autophagosome-lysosome fusion by interacting with the HOPS complex and GABARAP proteins [15]. We identified an interaction between TRIM22 and PLEKHM1 under both overexpression and endogenous conditions (Figure 4A,B). These results led us to hypothesize that TRIM22 may regulate the association between GABARAPs and PLEKHM1. We found that colocalization of PLEKHM1 with GABARAP or GABARAPL1 was significantly reduced by TRIM22 depletion (Figure 4C and S3C,D). Conversely, TRIM22 overexpression enhanced colocalization (Figure 4C). We further investigated the cellular association between PLEKHM1 and GABARAPs using a proximity ligation assay (PLA). The number of PLA dots, indicating the interaction between the two proteins, was significantly reduced in TRIM22-KO cells but increased in TRIM22-overexpressing cells, as compared to in WT cells (Figure 4D and S3E). Under the MTOR inhibition condition, a reduction in the colocalization of PLEKHM1 with GABARAP in TRIM22-KO cells was also observed (Figure 4E). These results show that TRIM22 positively regulates autophagosome-lysosome fusion by mediating the association of GABARAPs with PLEKHM1.
Figure 4.
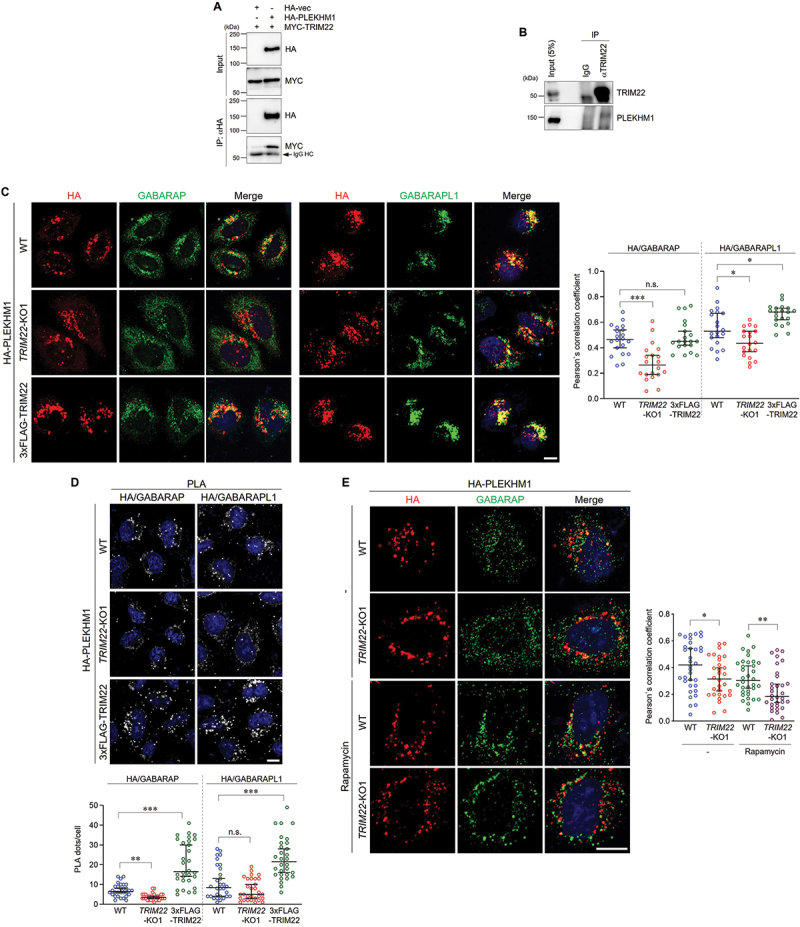
TRIM22 positively regulates autophagosome-lysosome fusion by mediating the association of GABARAPs with PLEKHM1. (A) HEK293T cells were transfected as indicated, and lysates were immunoprecipitated with antibodies against HA. Precipitates were then immunoblotted as shown. An arrow denotes IgG heavy chain. (B) HeLa cells were treated with 50 ng/mL of IFNG for 24 h, followed by starvation with EBSS for 2 h. Lysates were immunoprecipitated with rabbit IgG or antibodies against TRIM22. Precipitates were then immunoblotted as shown. (C,D) Wild-type HeLa cells, TRIM22-KO1 cells, or 3×FLAG-TRIM22-expressing cells were transfected with HA-PLEKHM1. (C) The cells were immunostained for HA (red) and GABARAP (left, green) or GABARAPL1 (right, green). Colocalization of HA-PLEKHM1 with GABARAP or GABARAPL1 was analyzed by calculation of Pearson’s correlation coefficients. Graphs show median with 95% confidence interval of more than 20 cells per group. (D) The cells were subjected to PLA with anti-HA antibodies along with antibodies against GABARAP (left) or GABARAPL1 (right). The number of PLA dots was quantified. Graphs show median with 95% confidence interval of more than 30 cells per group. (E) Wild-type HeLa cells or TRIM22-KO1 cells were transfected with HA-PLEKHM1 and immunostained for HA (red) and GABARAP (green). Colocalization of HA-PLEKHM1 with GABARAP was analyzed by calculation of Pearson’s correlation coefficients. Graphs show median with 95% confidence interval of more than 30 cells per group. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant. Scale bars: 10 μm.
Next, we sought to determine how the interaction of TRIM22 with PLEKHM1 and GABARAPs regulates autophagosome-lysosome fusion. We found that the B-box of TRIM22 is critical for its binding with PLEKHM1 (Figure 5A,B). The B-box-deleted form of TRIM22 (TRIM22-ΔB) mostly lost the binding affinity with PLEKHM1, while it interacted with GABARAPs likely as the full-length form (Figure 5B,C). We also found that the colocalization of GABARAP with PLEKHM1, which is critical for autophagosome-lysosome fusion, was significantly compromised by TRIM22-ΔB expression (Figure 5D). Eventually, TRIM22-ΔB failed to rescue the impaired autophagosome-lysosome fusion in TRIM22-KO cells, indicating that TRIM22 facilitates autophagosome-lysosome fusion through its interaction with PLEKHM1 (Figure 5E). A failure in autophagosome-lysosome fusion under TRIM22-ΔB expression impeded clearance of autophagic substrates which was enhanced by TRIM22-WT expression (Figure 5F). Altogether, these results indicate that TRIM22 promotes autophagosome-lysosome fusion by mediating the association of PLEKHM1 with GABARAPs.
Figure 5.
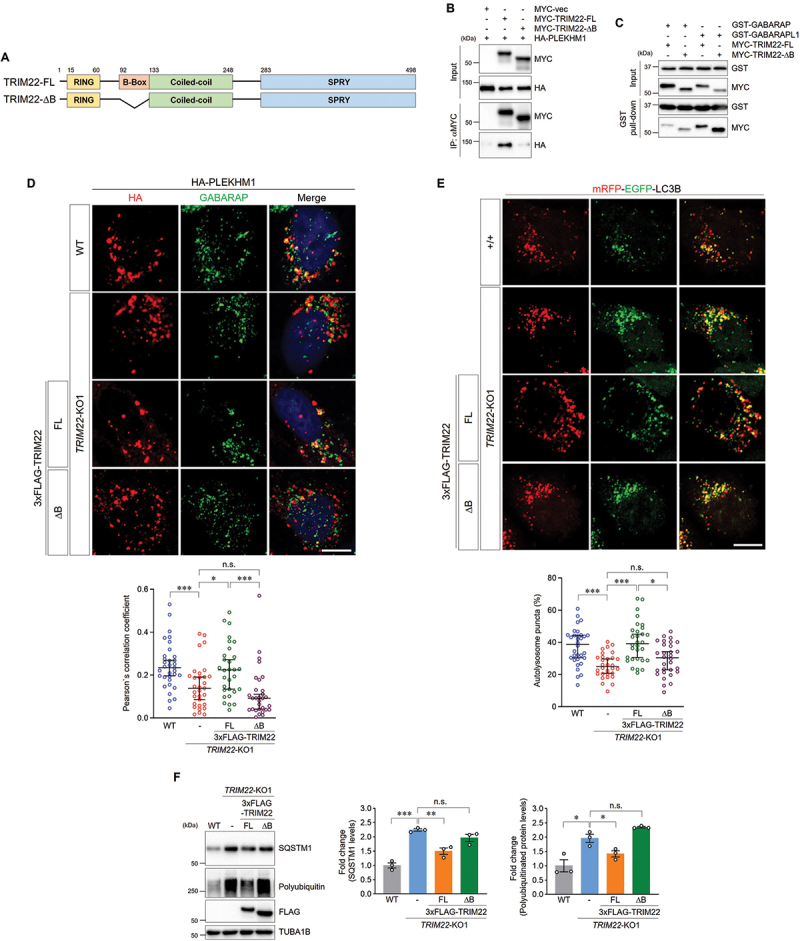
TRIM22 facilitates autophagosome-lysosome fusion by interacting with PLEKHM1. (A) Schematic domain structure of TRIM22 and the B-box deletion mutant. (B) HEK293T cells were transfected as indicated, and lysates were immunoprecipitated with antibodies against MYC. Precipitates were then immunoblotted as shown. (C) HEK293T cells were transfected with MYC-TRIM22-FL or ΔB, and lysates were incubated with the indicated GST-tagged proteins and precipitated with glutathione sepharose 4B beads. Precipitates were then immunoblotted as shown. (D) Wild-type HeLa cells, TRIM22-KO1 cells, and TRIM22-KO1 cells expressing 3×FLAG-TRIM22-FL or ΔB were transfected with HA-PLEKHM1. The cells were immunostained as indicated. Colocalization of HA-PLEKHM1 with GABARAP was analyzed by calculation of Pearson’s correlation coefficient. Graphs show median with 95% confidence interval of more than 30 cells per group. (E) mRFP-EGFP-LC3B was stably expressed in wild-type HeLa cells, TRIM22-KO1 cells, and TRIM22-KO1 cells expressing 3×FLAG-TRIM22-FL or ΔB. The cells with fluorescent LC3B puncta were analyzed. mRFP+/EGFP− puncta were counted as autolysosomes. Graphs show median with 95% confidence interval of more than 30 cells per group. (F) Wild-type HeLa cells, TRIM22-KO1 cells, and TRIM22-KO1 cells expressing 3×FLAG-TRIM22-FL or ΔB were subjected to immunoblotting with the indicated antibodies. Band intensities of SQSTM1 or polyubiquitinated proteins were measured and normalized to the mean intensity of wild-type cells. Graphs show means ± SEM of three independent experiments. FL, full-length. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant. Scale bars: 10 μm.
The function of TRIM22 as an E3 ubiquitin ligase is distinct from its role in the regulation of autophagy
Next, we investigated the role of TRIM22 as an E3 ubiquitin ligase in its involvement with the regulation of autophagy. First, we examined whether the autophagic defects resulting from TRIM22 depletion could be caused by quantitative alterations in its substrates. Notably, the levels of autophagic substrates remained unaffected upon the overexpression of NFKBIA/IκBα or NFE2L2/NRF2 (Figure S4A,B), both of which have been previously identified as targets of degradation through the TRIM22-mediated ubiquitin-proteasome system [44,45]. Moreover, the depletion of these substrates exhibited no impact on either the levels of autophagic substrates or the efficiency of autophagosome-lysosome fusion controlled by TRIM22 (Figure S4C-E). These results suggest that the regulatory function of TRIM22 in autophagy does not appear to be directly linked to the level of its substrates.
Second, we directly examined whether the E3 ubiquitin ligase activity of TRIM22 is required for its function in the regulation of autophagy. Similar to the WT form of TRIM22, the catalytically inactive TRIM22C15,18A (CCAA) mutant also induced a reduction in autophagic substrates upon overexpression (Figure S5A) [44,45], and this mutant almost completely restored the elevated levels of autophagic substrates in TRIM22-KO cells (Figure S5B). Moreover, there was no notable difference in binding affinity between WT and CCAA mutant of TRIM22 to PLEKHM1 or GABARAPs (Figure S5C,D). Additionally, the CCAA mutant effectively rescued the impaired autophagosome-lysosome fusion and inefficient colocalization of PLEKHM1 with GABARAP in TRIM22-KO cells (Figure S5E-H). Taken together, above results indicate that the function of TRIM22 as an E3 ubiquitin ligase is distinct from its role in the regulation of autophagy.
AD-related TRIM22 variant interferes with intracellular clearance by inhibiting autophagosome-lysosome fusion
Given that the R321K single nucleotide polymorphism (SNP) in TRIM22 was previously identified as an AD age of onset (ADAOO) modifier based on a pedigree carrying the E280A mutation in PSEN1, which caused early onset familial AD [38], we hypothesized that TRIM22 could be associated with AD risk through its role in intracellular clearance. Including a 695-amino acid full-length protein, the levels of all other APP (amyloid beta precursor protein) species, such as carboxyl-terminal fragment (CTF) and APP intracellular domain (AICD), were elevated in TRIM22-KO cells, an effect which was abolished by re-expression of TRIM22 (Figure 6A). Next, we examined whether the R321K SNP in TRIM22 affected autophagic degradation. While TRIM22-WT expression rescued the abnormal accumulation of polyubiquitinated proteins in TRIM22-KO cells, TRIM22R321K expression enhanced the defects in autophagic degradation (Figure 6B). Failure to recover from autophagic degradation defects caused by the R321K variant was confirmed through the analysis of SNCAA30P or SNCAA53T aggregates (Figure 6C,D), which are normally degraded by autophagy [18].
Figure 6.
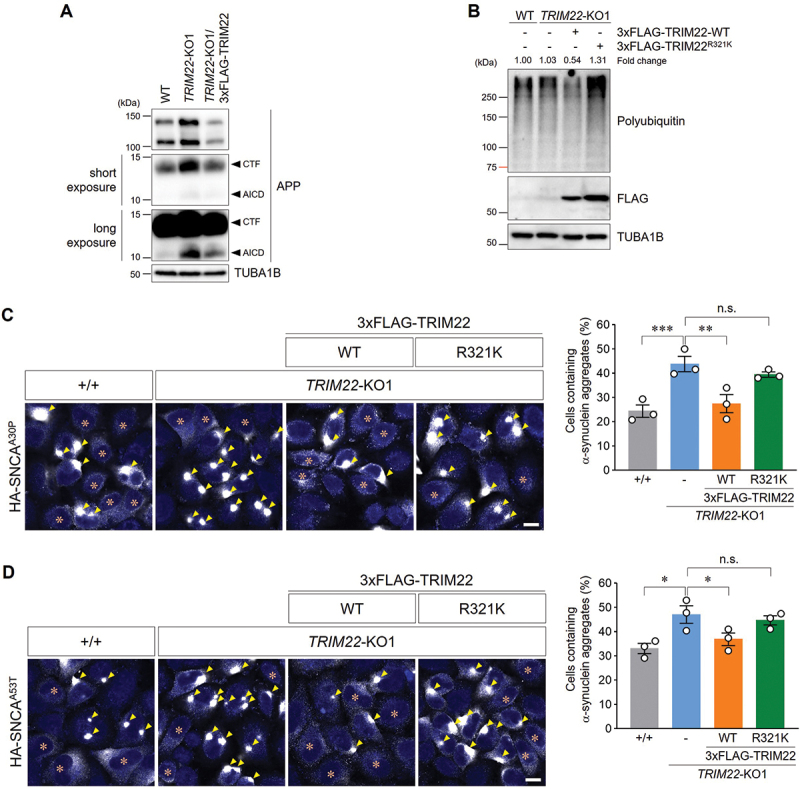
An AD-related TRIM22 variant interferes with intracellular clearance. (A) Wild-type HeLa cells, TRIM22-KO1 cells, or TRIM22-KO1 cells stably expressing 3×FLAG-TRIM22 were subjected to immunoblotting with the indicated antibodies. Arrowheads denote the CTF of APP or the AICD. (B) Wild-type HeLa cells, TRIM22-KO1 cells, or TRIM22-KO1 cells stably expressing 3×FLAG-TRIM22-WT or R321K were subjected to immunoblotting with the indicated antibodies. Band intensities for polyubiquitinated proteins were measured and normalized to the intensity of wild-type cells. (C,D) Wild-type HeLa cells, TRIM22-KO1 cells, and TRIM22-KO1 cells expressing 3×FLAG-TRIM22-WT or R321K were transfected with HA-SNCAA30P (C) or HA-SNCAA53T (D). Cells were immunostained for HA, and cells containing HA-SNCA aggregates were then analyzed as described (more than 500 cells per group). Arrowheads indicate aggregated α-synuclein, and asterisks indicate the cells with no aggregates. Graphs show means ± SEM of three independent experiments. Representative images are shown. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant. Scale bars: 10 μm.
Autophagosome-lysosome fusion, which is a prerequisite for autophagic degradation, was regulated by TRIM22 (Figures 2 and 4). Therefore, we investigated the effects of the TRIM22R321K variant on autophagosome-lysosome fusion. Failure to revert the reduced autophagosome-lysosome fusion phenotype was confirmed through analyzing the ratio of autolysosomes (Figure 7A) and the fluorescent intensities of mRFP-EGFP-LC3B in TRIM22R321K variant-expressing cells (Figure 7B). These results suggest that the TRIM22R321K variant inhibits autophagosome-lysosome fusion.
Figure 7.
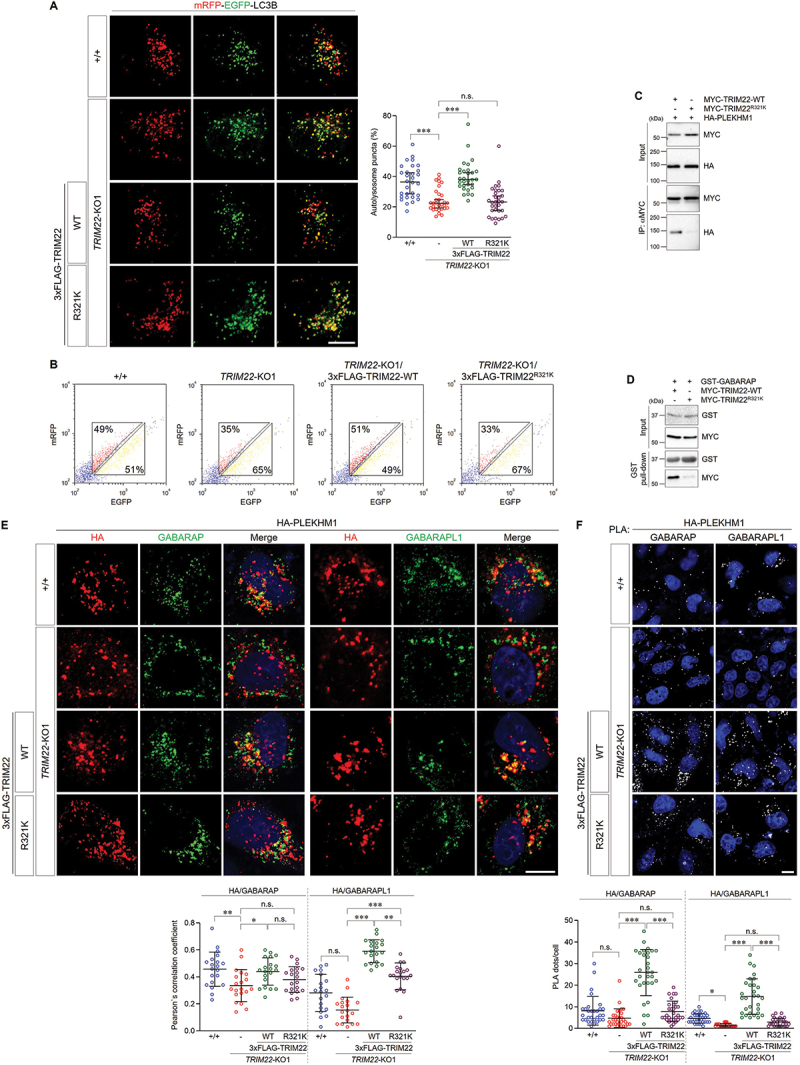
The R321K variant inhibits the role of TRIM22 in autophagosome-lysosome fusion. (A,B) mRFP-EGFP-LC3B was stably expressed in wild-type HeLa cells, TRIM22-KO1 cells, and TRIM22-KO1 cells expressing 3×FLAG-TRIM22-WT or R321K. (A) The cells with fluorescent LC3B puncta were analyzed. Puncta with mRFP+/EGFP+ indicate autophagosomes, and puncta with mRFP+/EGFP− indicate autolysosomes. Graphs show median with 95% confidence interval of more than 30 cells per group. (B) Cells were analyzed using flow cytometry. The intensities of mRFP and EGFP signals from more than 2,000 cells were analyzed, and the percentages of cells in corresponding gates are indicated. (C) HEK293T cells were transfected as indicated, and lysates were immunoprecipitated with antibodies against MYC. Precipitates were then immunoblotted as shown. (D) HEK293T cells were transfected as indicated, and lysates were incubated with GST-GABARAP proteins and precipitated with glutathione sepharose 4B beads. Precipitates were then immunoblotted as shown. (E,F) Wild-type HeLa cells, TRIM22-KO1 cells, and TRIM22-KO1 cells expressing 3×FLAG-TRIM22-WT or R321K were transfected with HA-PLEKHM1. (E) The cells were immunostained as indicated. Colocalization of HA-PLEKHM1 with GABARAP (left) or GABARAPL1 (right) was analyzed via calculation of Pearson’s correlation coefficient. Graphs show median with 95% confidence interval of more than 20 cells per group. (F) Cells were subjected to PLA with anti-HA antibodies along with antibodies against GABARAP (left) or GABARAPL1 (right). The number of PLA dots was quantified. Graphs show median with 95% confidence interval of more than 30 cells per group. *p < 0.05; **p < 0.01; *** p < 0.001; n.s., not significant. Scale bars: 10 μm.
Given that TRIM22 regulated autophagosome-lysosome fusion by mediating the association of GABARAPs with PLEKHM1, we then examined whether the TRIM22R321K variant affects interactions with the aforementioned proteins. Interestingly, we found that the interaction between TRIM22 and PLEKHM1 or GABARAP was inhibited by the TRIM22R321K variant (Figure 7C,D). In addition, colocalization of PLEKHM1 with GABARAP or GABARAPL1 was significantly reduced in TRIM22R321K-expressing cells compared to that in TRIM22-WT-expressing cells (Figure 7E). The association of PLEKHM1 with GABARAP or GABARAPL1 in cells was also examined via PLA, with results showing that the TRIM22R321K variant interfered with protein interactions (Figure 7F). These findings suggest that TRIM22R321K-mediated inhibition of the PLEKHM1-GABARAP association may suppress autophagosome-lysosome fusion, thereby interfering with autophagic intracellular clearance.
Discussion
Recent studies have suggested that TRIM proteins are pivotal autophagic adaptors that orchestrate a series of autophagic processes by scaffolding autophagy-related proteins. In this study, we found that TRIM22 acts as a core regulator of autophagy by promoting autophagy and intracellular clearance. We demonstrated that TRIM22 facilitates autophagosome-lysosome fusion by mediating the association between PLEKHM1 and GABARAP. Importantly, we found that the R321K AD-associated SNP in TRIM22 interferes with autophagosome-lysosome fusion, thereby inhibiting the intracellular clearance of neurotoxic protein aggregates. These findings suggest a protective role for TRIM22 in the pathogenesis of neurodegenerative diseases, including PSEN1E280A-associated AD (Figure 8).
Figure 8.
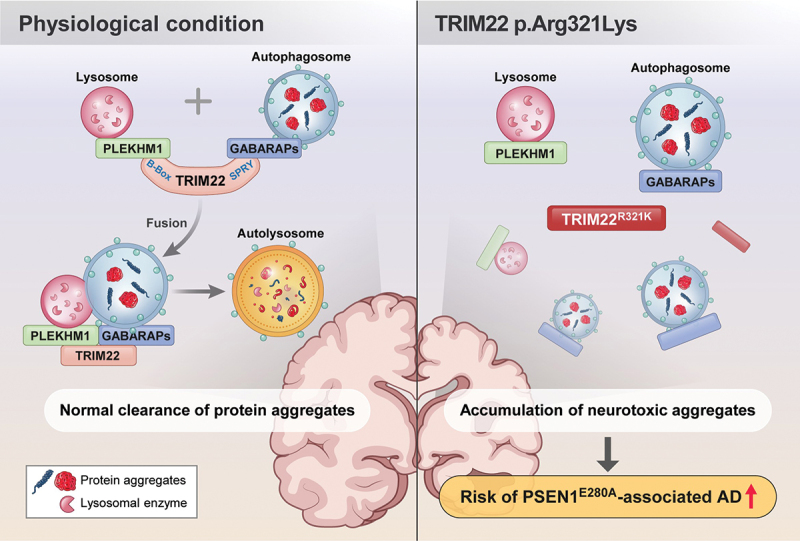
Roles of TRIM22 in intracellular clearance. Under physiological conditions, TRIM22 facilitates the association of GABARAPs and PLEKHM1, thereby inducing autophagosome-lysosome fusion. The autolysosomes effectively remove protein aggregates. However, the R321K SNP in TRIM22 fails to mediate the association between GABARAPs and PLEKHM1. As a result, clearance of neurotoxic aggregates is inhibited, which finally potentially contributes to increasing the risk of neurodegenerative diseases, including PSEN1E280A-associated AD.
Accumulating evidence suggests that several TRIM proteins serve as central platforms for recruiting cargos and autophagy core regulators for efficient autophagic degradation. In humans, most TRIM proteins interact with ATG8s, which consist of LC3s and GABARAPs [27]. LC3s and GABARAPs have distinct functions in autophagy. LC3s function in autophagosome formation, whereas GABARAPs function in autophagosome-lysosome fusion [41,42]. The interaction of TRIM proteins with LC3s facilitates the recruitment of TRIMosomes to the phagophore, as well as its binding to autophagic receptors, such as SQSTM1 [27]. However, the significance of these interactions between TRIM proteins and GABARAPs remains unclear. In this study, we uncovered, for the first time, how the interaction between TRIM proteins and GABARAPs regulates autophagy. We demonstrated that TRIM22 directly binds to GABARAPs and PLEKHM1, which are essential for interaction with the HOPS complex that is required for autophagosome-lysosome fusion, thereby positively regulating autophagosome-lysosome fusion. Furthermore, we presented compelling evidence demonstrating that the function of TRIM22 as an E3 ubiquitin ligase is distinct from its role in the regulation of autophagy.
Previously, TRIM22 was suggested to regulate the clearance of Mycobacterium tuberculosis in macrophages [34,35]. Because TRIM22 depletion altered the levels of autophagy-related proteins, such as LC3-II, SQSTM1, NFKB/NF-κB (nuclear factor kappa B), and BECN1, TRIM22 may augment autophagy via the NFKB-BECN1 pathway in macrophages. These results are in part consistent with our finding that TRIM22 mediates autophagic degradation. However, there was a discrepancy in the response to TRIM22 depletion. In TRIM22-KO HeLa cells, LC3A/B-II levels or the number of LC3A/B-II-positive autophagic compartments was significantly increased (Figure 1), possibly due to failure of autophagosome-lysosome fusion. In contrast, LC3A/B-II levels or the number of LC3A/B-II-positive autophagic compartments was decreased in TRIM22-depleted macrophages [34,35]. Based on these results, we predict that the role of TRIM22 in autophagy regulation, particularly in facilitating autophagosome-lysosome fusion, may vary among tissues and cell types.
The E280A mutation in the PSEN1 gene, which encodes a γ-secretase component required for APP processing, is the most frequently occurring mutation in patients with familial AD. A recent genome-wide association study of the “Paisa” pedigree, from which the PSEN1E280A mutation had been segregated, revealed nine genomic variants that affect ADAOO [38]. Among them, rs7412 (APOE*E2) delayed ADAOO to the greatest extent, while rs12364019 (TRIM22), which results in the substitution of arginine321 with lysine, accelerated ADAOO the most. Although this study suggested that TRIM22 may affect the etiology of AD, the association of the TRIM22R321K variant with AD is still unclear. In the present work, we showed that the R321K variant interfered with the role of TRIM22 in autophagosome-lysosome fusion. The PSEN1E280A mutation also interferes with lysosomal acidification and autophagic degradation [46]. Our results suggest that the TRIM22R321K variant may exacerbate inefficient autophagy in individuals carrying the PSEN1E280A mutation, which could in turn accelerate ADAOO. Thus, further studies are needed to elucidate the complex relationships of TRIM22 and PSEN1 in autophagy and AD pathogenesis.
Collectively, our findings showed that TRIM22 is an autophagy regulator that facilitates autophagosome-lysosome fusion by scaffolding GABARAPs and PLEKHM1. We also showed that a genetic variation in TRIM22, which increases the risk of familial AD, was closely related to inefficient autophagic clearance of neurotoxic substrates, suggesting that TRIM22 could be a potential target for autophagy modifiers. We hope that our insights into the pathogenesis of specific types of familial AD will further expand the current understanding of the pathways driving neurodegenerative diseases caused by aggregate accumulation.
Materials and methods
DNA constructs
Human GABARAP (MHS6278–211689836), GABARAPL1 (MHS6278–202800914), GABARAPL2 (MHS6278–202806924), LC3A (MHS6278–202757001), LC3B (MHS6278–202808448), LC3C (MHS6278–213243290), and TRIM22 (MHS6278–202759096) complementary DNAs (cDNAs) were obtained from Horizon Discovery. Human PLEKHM1 (73593; deposited by Paul Odgren) and mRFP-EGFP-LC3B (21074; deposited by Tamotsu Yoshimori) cDNAs were obtained from Addgene. The cDNAs were subcloned into pEGFP-C2, pGW1-FLAG, -hemagglutinin (HA), or -MYC vector [47] for transient expression in mammalian cells. The LIR null mutant of TRIM22 (TRIM22-10A) was generated through sequential mutagenesis of ten putative LIR motifs (Y175, I178, Y298, V301, Y394, V397, F435, V438, W485, L488) to alanine. For lentivirus production, the cDNAs were subcloned into pCDH-CMV-MCS-EF1-Puro (System Biosciences, CD510B–1) or pCDH-CMV-MCS-EF1-Puro-3×FLAG [47] lentiviral vectors. For glutathione S-transferase (GST) fusion protein purification, cDNAs of the ATG8 gene family were subcloned into pGEX-6P-1 (Merck, 27-4597-01). All point mutations were introduced via polymerase chain reaction (PCR) and DpnI-based in vitro mutagenesis, then confirmed via DNA sequencing.
Antibodies
The following antibodies were used in this study: anti-FLAG (M2, mouse; Sigma-Aldrich, F1804), anti-FLAG (polyclonal, rabbit; ProteinTech Group, 20543–1-AP), anti-GFP (7.1 and 13.1, mouse; Roche, 11814460001), anti-GST (3G12B10, mouse; ProteinTech Group, 66001–2-Ig), anti-GST (B-14, mouse; Santa Cruz Biotechnology, sc-138), anti-HA (F-7, mouse; Santa Cruz Biotechnology, sc-7392), anti-HA (polyclonal, rabbit; Genetex, GTX115044), anti-MYC (9E10, mouse; Santa Cruz Biotechnology, sc-40), anti-MYC (polyclonal, goat; Bethyl Laboratories, A190-104A), anti-ACTB/β-actin (2D4H5, mouse; ProteinTech Group, 66009–1-Ig), anti-APP (Y188, rabbit; Abcam, ab32136), anti-CD63 (H5C6, mouse; BD Biosciences, 556019), anti-GABARAP (E1J4E, rabbit; Cell Signaling Technology, 13733S), anti-GABARAPL1 (D5R9Y, rabbit; Cell Signaling Technology, 26632S), anti-GAPDH (GT239, mouse; Genetex, GTX627408), anti-LC3A/B (polyclonal, rabbit; Sigma-Aldrich, L8918), anti-polyubiquitin (FK2, mouse; Cayman Chemical, 14220), anti-PLEKHM1 (E9S3Q, rabbit; Cell Signaling Technology, 77092S), anti-RPS6KB (polyclonal, rabbit; Cell Signaling Technology, 9202S), anti-RPS6KB-pT389 (polyclonal, rabbit; Cell Signaling Technology, 9205S), anti-SQSTM1 (3, mouse; BD Biosciences, 610832), anti-TRIM22 (polyclonal, rabbit; ProteinTech Group, 13744–1-AP), anti-TUBA1B/α-tubulin (1E4C11, mouse; ProteinTech Group, 66031–1-Ig), and anti-TUBA1B (polyclonal, rabbit; Abcam, ab18251).
Chemicals and reagents
The following chemicals and reagents were used in this study: bafilomycin A1 (Sigma-Aldrich, B1793), chloroquine (Santa Cruz Biotechnology, sc-217882), EBSS (Welgene, LB002–03), IFNG (Sino Biological, 11725-HNAS), LysoTracker Red DND-99 (Invitrogen, L7528), LysoSensor Green DND-189 (Invitrogen, L7535), pepstatin A, BODIPY FL conjugate (Invitrogen, P12271), puromycin (Gold Biotechnology, P-600-100), and rapamycin (Sigma-Aldrich, R8781).
Immunoblotting
Proteins in Laemmli sample buffer were resolved via sodium dodecyl sulfate-polyacrylamide gel electrophoresis/SDS-PAGE and then electrophoretically transferred to nitrocellulose membranes. Membranes were blocked with 3% non-fat milk in Tris-buffered saline (20 mM Tris-HCl, 137.5 mM NaCl, pH 7.6) with 0.05% Tween-20 (LPS Solution, TW2001; TBST) for 30 min and incubated with primary antibodies in TBST with 3% bovine serum albumin (BSA; Bovogen, BSAS-NZ) at 4°C overnight. Blots were washed three times with TBST, incubated with horseradish peroxidase-conjugated secondary antibodies (GenScript Biotech, A00098, A00160, and A00178) for 1 h, and washed three times with TBST. Clarity Western ECL Substrate (Bio-Rad Laboratories, 1705061) was used to detect immunoreactive proteins. Images were obtained and processed using a ChemiDoc XRS+ System (Bio-Rad Laboratories, Hercules, CA, USA) with Image Lab software (Bio-Rad Laboratories). All the bands were in the linear range of detection.
Immunoprecipitation
Cells were washed with phosphate-buffered saline (PBS; Biosesang, PR2007-100-74) and then lysed in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100 [LPS Solution, TRX01], 1.5 mM EDTA, pH 8.0) with Pierce Protease and Phosphatase Inhibitor Mini Tablets (Thermo Fisher Scientific, A32959) on ice for 30 min. Lysates were clarified via centrifugation (12,000 × g) for 10 min at 4°C and incubated with 0.5 μg of antibodies at 4°C for 16 h, followed by incubation with Protein A/G PLUS-agarose (Santa Cruz Biotechnology, sc-2003) for an additional 2 h. Beads were washed three times with lysis buffer, and bound proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis for further analysis.
Cell culture
HEK293T and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (GenDEPOT, CM001–050) supplemented with 10% fetal bovine serum (GenDEPOT, F0900–050) at 37°C in a 5% CO2 humidified incubator. hTERT-RPE1 cells were cultured in Dulbecco’s modified Eagle’s Medium/F-12 1:1 mixture (Welgene, LM002–04) supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 humidified incubator. The cell lines used in the experiments were regularly tested for mycoplasma contamination.
Plasmid transfection and RNA interference
Cells were transfected with Avalanche-Everyday (EZ Biosystems, EZT-EVDY-1) for plasmid DNAs and Lipofectamine RNAiMax (Invitrogen, 13778075) for siRNAs according to the manufacturer’s instructions. The following siRNAs were used: siControl (Bioneer, SN-1003); siTRIM22-1 (Bioneer), 5′-CAGAACCUCUGAGCCUAGA-3′; siTRIM22-2 (Bioneer), 5′-GGUCACCAAACAUUCCGCA-3′.
Generation of stable cell lines
Stable cell lines were generated as previously described [48]. In detail, HeLa cells stably expressing TRIM22-WT, TRIM22 mutants, or mRFP-EGFP-LC3B were generated via lentiviral infection. pCMV-dR8.2 dvpr (Addgene, 8455; deposited by Bob Weinberg) and pCMV-VSV-G (Addgene, 8454; deposited by Bob Weinberg) were cotransfected into HEK293T cells along with their respective pCDH-CMV-MCS-EF1-Puro-based constructs. On the next day, lentivirus was harvested from the culture medium via centrifugation and frozen at −80°C. HeLa cells were infected with the indicated viruses, and two days later, infected cells were incubated with 2 μg/mL of puromycin for selection over an additional day.
Immunofluorescence staining and confocal microscopy
HeLa cells cultured on coverslips were fixed with cold methanol for 10 min or 4% paraformaldehyde for 15 min. The fixed cells were blocked with 3% BSA in PBST (PBS with 0.1% Triton X-100) or PBSS (PBS with 0.05% saponin [Sigma-Aldrich, 47036]) for 15 min. Cells were then incubated with primary antibodies diluted in blocking solution for 2 h, washed three times with PBST or PBSS, incubated with Alexa Fluor-conjugated secondary antibodies (Molecular Probes, A21206, A31570, A32754, and A32766) diluted in blocking solution for 1 h, and washed three times with PBST or PBSS. For nuclear staining, Hoechst 33342 (ImmunoChemistry Technologies, 639) was applied for 2 min. Coverslips were mounted on glass slides using Fluoromount-G (SouthernBiotech, 0100–01). Cells were visualized using an LSM 800 confocal microscope (Carl Zeiss, Oberkochen, Germany) with a 63 × 1.4 numerical aperture Plan-Apochromat lens or an IX83 fluorescent microscope (Olympus Corporation, Tokyo, Japan) with a 60 × 1.42 apochromatic lens. Images were processed using ZEN software (Carl Zeiss).
Generation of knockout cell lines
For TRIM22 gene editing, targets were selected from the list recommended by the online CRISPR Design Tool, E-CRISP, as previously described [49]. To generate the TRIM22-KO1 cell line, a small guide RNA targeting 5′-GTCAAGATGAGCCCACAGGAG-3′ of TRIM22 was cloned into pSpCas9(BB)-2A-Puro (pX459) (Addgene, 48139; deposited by Feng Zhang). The constructs were transfected into HeLa cells with Avalanche-Everyday (EZ Biosystems, EZT-EVDY-1), and the cells were incubated with 2 μg/mL of puromycin for 24 h. After further incubation for 24 h, the cells were transferred to a 96-well plate for single-cell isolation. Genomic DNAs from cells cultured in 6-well plates were isolated using a HiGene Genomic DNA Prep Kit (Biofact Biofactory, GD264–060), and PCR was performed to amplify the target site using specific primers (forward: 5′-TGCAGGAGTTTGTGACCAAG-3′, reverse: 5′-CATTCCTTGACCACCTCGTT-3′). The PCR products were subcloned into an A-tailed pBluescript-SK (+) vector (Agilent Technologies, 212205) for plasmid sequencing. At least 10 sequencing results from different bacterial colonies were evaluated to verify the CRISPR-CAS9-mediated knockout of TRIM22.
Flow cytometry
mRFP-EGFP-LC3B-expressing HeLa cells grown on the 12-well cell culture plate were trypsinized. The cells were washed and resuspended in 400 μL of PBS. Over 10,000 cells were loaded to FACS AriaTM III (BD Biosciences, Franklin Lakes, NJ, USA), and the data were analyzed using FACSDivaTM software (BD Biosciences).
Protein purification and in vitro affinity-isolation assays
Protein purification and in vitro affinity-isolation assays were performed as previously described [50]. In detail, pGEX-6P-1 or pGEX-6P-1-ATG8 plasmids were transformed into Escherichia coli strain BL21 (DE3) (Enzynomics, CP110). E. coli cells were grown and induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (Gold Bio, I2381C5) at 30°C for 2 h. Bacterial cells were collected, and proteins were purified with glutathione resin (GenScript Biotech, L00206) according to the manufacturer’s instructions. Purified GST fusion proteins were mixed with HEK293T cell lysates expressing indicated TRIM22 constructs in binding buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100, 1.5 mM EDTA, pH 8.0) with Pierce Protease and Phosphatase Inhibitor Mini Tablets at 4°C for 16 h, followed by incubation with glutathione resin for an additional 2 h. Beads were washed three times with binding buffer, and bound proteins were analyzed via immunoblotting and Coomassie Brilliant Blue staining.
Quantification of colocalization and statistical analysis
Colocalization analysis was performed on confocal sections showing the maximum signals of HA-PLEKHM1, GABARAP, or GABARAPL1. ZEN software (Carl Zeiss) was used to calculate Pearson’s correlation coefficient for more than 20 cells per group. GraphPad Prism (GraphPad Software) or Microsoft Excel (Microsoft Corporation) was used to analyze statistical data and draw graphs. All statistical data were tested for normality using the Shapiro-Wilk test. The significance of differences between two groups was determined using Student’s t-tests or Mann-Whitney U tests. Significance levels for comparisons among groups were determined using the Kruskal-Wallis test with Dunn’s post-hoc test. Results with P values lower than 0.05 were considered significant.
PLA
PLA was performed using a PLA probe (Duolink In Situ Red Starter Kit Mouse/Rabbit; Sigma-Aldrich, DUO92101). In detail, cells cultured on coverslips were fixed with cold methanol and permeabilized with 0.1% PBST. The cells were blocked with a blocking solution for 30 min at 37°C before incubation with the primary antibody overnight at 4°C. Cells were then washed twice for 5 min each time in wash buffer A, incubated with the PLA probe solution for 1 h at 37°C, washed again in wash buffer A, and incubated in ligation solution for 30 min at 37°C. The cells were then washed in wash buffer A, incubated in amplification solution for 100 min at 37°C, and washed twice for 10 min each time in wash buffer B. Finally, the coverslips were mounted on a slide glass for imaging.
Supplementary Material
Funding Statement
This study was supported by grants from the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT to J.C. (NRF-2019R1A5A2026045, NRF-2020R1A2C1010399) and to S.L. (NRF-2020R1A2C1101827, NRF-2021R1A5A2031612).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2287925
References
- [1].Gatica D, Lahiri V, Klionsky DJ.. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018. Mar;20(3):233–242. doi: 10.1038/s41556-018-0037-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rabanal-Ruiz Y, Otten EG, Korolchuk VI., et al. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017 Dec 12;61(6):565–584. doi: 10.1042/EBC20170027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011. Feb;13(2):132–141. doi: 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Turco E, Fracchiolla D, Martens S. Recruitment and activation of the ULK1/Atg1 kinase complex in selective autophagy. J Mol Biol. 2020 Jan 3;432(1):123–134. doi: 10.1016/j.jmb.2019.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Antonioli M, Di Rienzo M, Piacentini M, et al. Emerging mechanisms in initiating and terminating autophagy. Trends In Biochemical Sciences. 2017. Jan;42(1):28–41. doi: 10.1016/j.tibs.2016.09.008 [DOI] [PubMed] [Google Scholar]
- [6].Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013. Jul;15(7):741–750. doi: 10.1038/ncb2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Molino D, Zemirli N, Codogno P, et al. The journey of the autophagosome through mammalian Cell organelles and membranes. Journal Of Molecular Biology. 2017 Feb 17;429(4):497–514. doi: 10.1016/j.jmb.2016.12.013 [DOI] [PubMed] [Google Scholar]
- [8].Ding X, Jiang X, Tian R, et al. RAB2 regulates the formation of autophagosome and autolysosome in mammalian cells. Autophagy. 2019. Oct;15(10):1774–1786. doi: 10.1080/15548627.2019.1596478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shi X, Chang C, Yokom AL, et al. The autophagy adaptor NDP52 and the FIP200 coiled-coil allosterically activate ULK1 complex membrane recruitment. Elife. 2020 Aug 10;9. doi: 10.7554/eLife.59099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ma B, Cao W, Li W, et al. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res. 2014. Aug;24(8):912–924. doi: 10.1038/cr.2014.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lu J, He L, Behrends C, et al. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat Commun. 2014 May 22;5:3920. doi: 10.1038/ncomms4920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhao Y, Wang Q, Qiu G, et al. RACK1 promotes autophagy by enhancing the Atg14L-Beclin 1-Vps34-Vps15 complex formation upon phosphorylation by AMPK. Cell Rep. 2015 Nov 17;13(7):1407–1417. doi: 10.1016/j.celrep.2015.10.011 [DOI] [PubMed] [Google Scholar]
- [13].Nakamura S, Yoshimori T. New insights into autophagosome-lysosome fusion. J Cell Sci. 2017 Apr 1;130(7):1209–1216. doi: 10.1242/jcs.196352 [DOI] [PubMed] [Google Scholar]
- [14].Jiang P, Nishimura T, Sakamaki Y, et al. The HOPS complex mediates autophagosome–lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014. Apr;25(8):1327–1337. doi: 10.1091/mbc.e13-08-0447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].McEwan DG, Popovic D, Gubas A, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015 Jan 8;57(1):39–54. doi: 10.1016/j.molcel.2014.11.006 [DOI] [PubMed] [Google Scholar]
- [16].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006 Jun 15;441(7095):885–889. doi: 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- [17].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006 Jun 15;441(7095):880–884. doi: 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- [18].Park H, Kang JH, Lee S. Autophagy in neurodegenerative diseases: a Hunter for aggregates. Int J Mol Sci. 2020 May 10;21(9):3369. doi: 10.3390/ijms21093369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002. May;11(9):1107–1117. doi: 10.1093/hmg/11.9.1107 [DOI] [PubMed] [Google Scholar]
- [20].Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse Model of Alzheimer’s disease. PLoS One. 2010 Apr 1;5(4):e9979. doi: 10.1371/journal.pone.0009979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Berger Z, Ravikumar B, Menzies FM, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006 Feb 1;15(3):433–442. doi: 10.1093/hmg/ddi458 [DOI] [PubMed] [Google Scholar]
- [22].Silva MC, Nandi GA, Tentarelli S, et al. Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat Commun. 2020 Jun 26;11(1):3258. doi: 10.1038/s41467-020-16984-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sarraf SA, Shah HV, Kanfer G, et al. Loss of TAX1BP1-directed autophagy results in protein aggregate accumulation in the brain. Mol Cell. 2020 Dec 3;80(5):779–795 e10. doi: 10.1016/j.molcel.2020.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fox LM, Kim K, Johnson CW, et al. Huntington’s disease pathogenesis is modified in vivo by Alfy/Wdfy3 and selective macroautophagy. Neuron. 2020 Mar 4;105(5):813–821 e6. doi: 10.1016/j.neuron.2019.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Choi I, Zhang Y, Seegobin SP, et al. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020 Mar 13;11(1):1386. doi: 10.1038/s41467-020-15119-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Di Rienzo M, Romagnoli A, Antonioli M, et al. TRIM proteins in autophagy: selective sensors in cell damage and innate immune responses. Cell Death Differ. 2020. Mar;27(3):887–902. doi: 10.1038/s41418-020-0495-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hatakeyama S. TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochem Sci. 2017. Apr;42(4):297–311. doi: 10.1016/j.tibs.2017.01.002 [DOI] [PubMed] [Google Scholar]
- [28].Mandell MA, Jain A, Arko-Mensah J, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014 Aug 25;30(4):394–409. doi: 10.1016/j.devcel.2014.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chauhan S, Kumar S, Jain A, et al. Trims and Galectins Globally Cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016 Oct 10;39(1):13–27. doi: 10.1016/j.devcel.2016.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Di Rienzo M, Antonioli M, Fusco C, et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci Adv. 2019. May;5(5):eaau8857. doi: 10.1126/sciadv.aau8857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang W, Xia Z, Farre JC, et al. TRIM37 deficiency induces autophagy through deregulating the MTORC1-TFEB axis. Autophagy. 2018;14(9):1574–1585. doi: 10.1080/15548627.2018.1463120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fusco C, Mandriani B, Di Rienzo M, et al. TRIM50 regulates Beclin 1 proautophagic activity. Biochim Biophys Acta, Mol Cell Res. 2018. Jun;1865(6):908–919. doi: 10.1016/j.bbamcr.2018.03.011 [DOI] [PubMed] [Google Scholar]
- [33].Kimura T, Mandell M, Deretic V. Precision autophagy directed by receptor regulators - emerging examples within the TRIM family. J Cell Sci. 2016 Mar 1;129(5):881–891. doi: 10.1242/jcs.163758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kimura T, Jain A, Choi SW, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Bio. 2015 Sep 14;210(6):973–989. doi: 10.1083/jcb.201503023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lou J, Wang Y, Zheng X, et al. TRIM22 regulates macrophage autophagy and enhances mycobacterium tuberculosis clearance by targeting the nuclear factor-multiplicity kappaB/beclin 1 pathway. J Cell Biochem. 2018. Nov;119(11):8971–8980. doi: 10.1002/jcb.27153 [DOI] [PubMed] [Google Scholar]
- [36].Barr SD, Smiley JR, Bushman FD, et al. The interferon response inhibits HIV particle production by induction of TRIM22. PLOS Pathog. 2008 Feb 29;4(2):e1000007. doi: 10.1371/journal.ppat.1000007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lim KH, Park ES, Kim DH, et al. Suppression of interferon-mediated anti-HBV response by single CpG methylation in the 5’-UTR of TRIM22. Gut. 2018. Jan;67(1):166–178. doi: 10.1136/gutjnl-2016-312742 [DOI] [PubMed] [Google Scholar]
- [38].Velez JI, Lopera F, Sepulveda-Falla D, et al. APOE*E2 allele delays age of onset in PSEN1 E280A Alzheimer’s disease. Mol Psychiatry. 2016. Jul;21(7):916–924. doi: 10.1038/mp.2015.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Szeto J, Kaniuk NA, Canadien V, et al. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy. 2006. Jul-Sep;2(3):189–199. doi: 10.4161/auto.2731 [DOI] [PubMed] [Google Scholar]
- [40].Ren Y, Dong H, Jin R, et al. TRIM22 actives PI3K/Akt/mTOR pathway to promote psoriasis through enhancing cell proliferation and inflammation and inhibiting autophagy. Cutan Ocul Toxicol. 2022. Dec;41(4):304–309. doi: 10.1080/15569527.2022.2127750 [DOI] [PubMed] [Google Scholar]
- [41].Nguyen TN, Padman BS, Usher J, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Bio. 2016 Dec 19;215(6):857–874. doi: 10.1083/jcb.201607039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Weidberg H, Shvets E, Shpilka T, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29(11):1792–1802. doi: 10.1038/emboj.2010.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. 2020 Jan 3;432(1):80–103. doi: 10.1016/j.jmb.2019.07.016 [DOI] [PubMed] [Google Scholar]
- [44].Liu W, Zhao Y, Wang G, et al. TRIM22 inhibits osteosarcoma progression through destabilizing NRF2 and thus activation of ROS/AMPK/mTOR/autophagy signaling. Redox Biol. 2022. Jul;53:102344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ji J, Ding K, Luo T, et al. TRIM22 activates NF-kappaB signaling in glioblastoma by accelerating the degradation of IkappaBalpha. Cell Death Differ. 2021. Jan;28(1):367–381. doi: 10.1038/s41418-020-00606-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010 Jun 25;141(7):1146–1158. doi: 10.1016/j.cell.2010.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lee S, Park H, Zhu P, et al. Hereditary spastic paraplegia SPG8 mutations impair CAV1-dependent, integrin-mediated cell adhesion. Sci Signal. 2020. [January/7/2020];13(613):eaau7500. doi: 10.1126/scisignal.aau7500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chang J, Lee S, Blackstone C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J Clin Invest. 2014. Dec;124(12):5249–5262. doi: 10.1172/JCI77598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kang J, Kim JW, Heo H, et al. Identification of BAG2 and Cathepsin D as plasma biomarkers for Parkinson’s disease. Clin Transl Sci. 2021. Mar;14(2):606–616. doi: 10.1111/cts.12920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lee S, Chang J, Blackstone C. FAM21 directs SNX27-retromer cargoes to the plasma membrane by preventing transport to the golgi apparatus. Nat Commun. 2016 Mar 9;7:10939. doi: 10.1038/ncomms10939 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.