

ABSTRACT
Macroautophagy/autophagy is a tightly regulated cellular process integral to homeostasis and innate immunity. As such, dysregulation of autophagy is associated with cancer, neurodegenerative disorders, and infectious diseases. While numerous factors that promote autophagy have been characterized, the key mechanisms that prevent excessive autophagy are less well understood. Here, we identify CSNK2/CK2 (casein kinase 2) as a negative regulator of autophagy. Pharmacological inhibition of CSNK2 activity or siRNA-mediated depletion of CSNK2 increased basal autophagic flux in cell lines and primary human lung cells. Vice versa, ectopic expression of CSNK2 reduced autophagic flux. Mechanistically, CSNK2 interacted with the FLN (filamin)-NHL domain-containing tripartite motif (TRIM) family members TRIM2, TRIM3 and TRIM71. Our data show that recruitment of CSNK2 to the C-terminal NHL domain of TRIM3 lead to its robust phosphorylation at serine 661 by CSNK2. A phosphorylation-defective mutant of TRIM3 was unable to reduce autophagosome numbers indicating that phosphorylation by CSNK2 is required for TRIM-mediated autophagy inhibition. All three TRIMs facilitated inactivation of the ULK1-BECN1 autophagy initiation complex by facilitating ULK1 serine 757 phosphorylation. Inhibition of CSNK2 promoted autophagy upon influenza A virus (IAV) and measles virus (MeV) infection. In line with this, targeting of CSNK2 or depletion of TRIM2, TRIM3 or TRIM71 enhanced autophagy-dependent restriction of IAV, MeV and human immunodeficiency virus 1 (HIV-1). Thus, our results identify the CSNK2-TRIM2, -TRIM3, -TRIM71 axis as a key regulatory pathway that limits autophagy. Targeting this axis may allow for therapeutic induction of autophagy against viral infections and in diseases associated with dysregulated autophagy.
Abbreviation: ATG5: autophagy related 5; BafA1: bafilomycin A1; BECN1: beclin 1; CCD: coiled-coil domain; CSNK2/CK2: casein kinase 2; CSNK2A1: casein kinase 2 alpha 1; CSNK2A2: casein kinase 2 alpha 2; CSNK2B: casein kinase 2 beta; FLN: filamin; HeLa GL: HeLa cells stably expressing eGFP-LC3B; HIV-1: human immunodeficiency virus 1; IAV: influenza A virus; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3; MeV: measles virus; MTOR: mechanistic target of rapamycin kinase; RING: really interesting new gene; SQSTM1/p62: sequestosome 1; TRIM: tripartite motif; ULK1: unc-51 like autophagy activating kinase 1
KEYWORDS: Autophagy, casein kinase, tripartite motif proteins, virus, ULK1, phosphorylation
Introduction
Macroautophagy (hereafter called autophagy) is a crucial cellular homeostatic pathway that governs the turnover of obsolete proteins or organelles. During autophagy, cytoplasmic cargo is engulfed by double-membrane vesicles called autophagosomes and then degraded through fusion of the mature autophagosome with lysosomes. Cargo can either be subjected to unselective “bulk” degradation (e.g., upon starvation) or it can be precisely earmarked making it recognizable by specific autophagy receptors such as SQSTM1/p62 (sequestosome 1), which is degraded along the cargo [1]. Impaired autophagy leads to the accumulation of defective organelles and proteins, and eventually contributes to limiting nutrient availability [2]. Conversely, excessive autophagy promotes cell death via auto-digestion, called autosis [3]. Thus, as a central homeostatic pathway, autophagy has to be tightly regulated [4].
Autophagy is regulated by cellular kinases including ULK1 (unc-51 like autophagy activating kinase 1) and 5' AMP-activated protein kinase (AMPK) that are required for its initiation. While many autophagy promoting kinases have been characterized, little is currently known about kinases that restrict autophagy. A notable exception is the MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1), which reduces ULK1 activation by phosphorylation and thus suppresses autophagy initiation. Negative regulators of autophagy are highly clinically relevant therapeutic targets, enabling an increase of autophagic flux. For example, targeting MTOR using rapamycin or its derivatives is a clinically approved treatment against cancer [5]. The human genome encodes for more than 500 protein kinases, and many of those were found to contribute to cellular homeostasis, although their functions in autophagy remain little understood. Among those is CSNK2/CK2 (casein kinase 2) [6], a highly conserved, constitutively active Ser/Thr protein kinase that is usually present in a tetrameric complex made up by two catalytically active subunits, either CSNK2A1/CSNK2α and/or CSNK2A2/CSNK2α', and two CSNK2B/CSNK2β regulatory subunits. While the CSNK2A1 and CSNK2A2 subunits alone are sufficient to confer kinase functionality, CSNK2B increases the stability and activity of the complex and modulates substrate specificity [7]. The CSNK2 complex is found both in the nucleus and the cytoplasm and is described to also associate with the plasma membrane, mitochondria, and the cytoskeleton [7]. CSNK2 activity influences many cellular pathways via hundreds of proposed substrates [8]. However, the exact role(s) and molecular mechanism(s) of CSNK2 in autophagy regulation have just begun to be elucidated.
Besides kinases, tripartite motif (TRIM) proteins are well-established to regulate autophagy [9,10]. TRIMs are highly versatile E3 ligases comprised of a conserved N-terminal Really Interesting New Gene – BBox - coiled-coil domain (RING-BBox-CCD) motif and a variable C-terminal region, containing domains with a wide range of functions [9,11]. The RING domain confers E3 ligase activity and mediates post translational modification (PTM) of targets, usually conjugation of ubiquitin or ubiquitin-like proteins. These PTMs can influence the stability and/or activity of the target, and thus the associated pathways [12]. The function mediated by the BBox is largely unknown and the CCD is thought to be involved in oligomerization of the TRIMs, which is often required for full activity [13]. The variable domains at the C-terminus usually define substrate specificity through protein-protein interactions but may also have other functions such as binding of nucleic acids or confer enzymatic activity [9]. For example, TRIM5/TRIM5α induces autophagy through an interaction with mammalian Atg8-family proteins such as MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3), whereas TRIM23 interacts with TBK1 (TANK binding kinase 1), promoting SQSTM1-induced autophagy [10,14]. While several members of the TRIM family have been identified to promote basal or virus-induced autophagy, little is currently known about specific TRIM proteins that reduce autophagic flux.
Emerging evidence established autophagy as an integral part of innate immune defenses by targeting viruses and viral components for lysosomal degradation and by exposing viral pathogen-associated molecular patterns (PAMPs) to pattern recognition receptors (PRRs) like TLR (toll like receptor) proteins [2]. Furthermore, autophagic degradation can provide peptide fragments for presentation on major histocompatibility complex/MHC molecules in antigen-presenting cells. Viral infection induces autophagy; however, pathogenic viruses like influenza A virus (IAV), measles virus (MeV), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or human immunodeficiency virus 1 (HIV-1) have evolved strategies to avoid the restriction by autophagy [15–19]. Moreover, exogenous induction of autophagy, e.g., by rapamycin, has been shown to restrict replication of diverse viruses [16,19].
Here, we establish that CSNK2 negatively regulates both basal and virus-induced autophagy. Our mechanistic studies revealed that CSNK2 interacts with the FLN-NHL containing TRIM proteins TRIM2, TRIM3 and TRIM71, which reduces autophagic flux. To prevent autophagy induction, CSNK2 activates TRIM3 by phosphorylation of Ser661 in the NHL repeats. Finally, we show that pharmacological inhibition of CSNK2 enhances virus-induced autophagy and consequently restricts replication of IAV, MeV or HIV-1. Our data identifies the CSNK2-TRIM2, -TRIM3, -TRIM71 axis as a key negative regulatory pathway of basal and virus-induced autophagy that may be targeted for therapeutic modulation of autophagy induction.
Results
Identification of CSNK2 as negative regulator of autophagy
To identify kinases that negatively regulate autophagy, we determined the impact of 81 kinase inhibitors on autophagy (Figures 1A, S1A, Table S1). To probe for autophagy, we applied a previously established flow cytometry-based assay using HeLa cells stably expressing eGFP-LC3B (HeLa GL) to measure autophagy induction [20]. Mild permeabilization of the cells retains eGFP-tagged, autophagosome-bound LC3B-II in the cells so that the eGFP MFI can be used as a proxy for autophagosome abundance per cell. To selectively probe for kinases that reduce autophagic flux, we treated the cells with saturating concentrations of bafilomycin A1, which leads to an accumulation of autophagosomes. Thus, treatment with inhibitors targeting kinases that negatively regulate autophagy would increase autophagosome levels beyond the bafilomycin A1 base line (dotted line, Figure 1A). Wortmannin, that inhibits autophagy initiation and rapamycin that increases autophagic flux served as negative and positive control, respectively. This analysis revealed that two kinase inhibitors markedly increased autophagic flux: the PRKC (protein kinase C) inhibitor PKC412 and the CSNK2 inhibitor CX-4945. Notably, both inhibitors did not alter cell viability at the concentrations used (10 µM) and increased autophagosome abundance also in the absence of bafilomycin A1 (Figures 1B, S1A). Whereas PKC412 is a broad-spectrum inhibitor of PRKC and PRKC-like kinases, CX-4945 is well known to specifically prevent CSNK2 activity [21]. We confirmed this using an endogenous CSNK2 activity assay (Figure S1B). In line with the assessment of autophagosome levels by flow cytometry, quantification of eGFP-LC3B-positive puncta in HeLa GL cells showed that treatment with CX-4945 increased the number of autophagosomes per cell in a dose-dependent manner (Figure 1C,D). To assess the half maximum effective concentration 50 (EC50) of CSNK2 inhibition for autophagy induction, we performed dose-response experiments in the presence and absence of bafilomycin A1. CX-4945, which inhibits CSNK2 activity at an half maximum inhibitory concentration 50 (IC50) of 1 nM in vitro and ~3 µM in cell-based assays [21], induced autophagy at an EC50 of 3 µM (Figures 1E, S1C,D). To corroborate this, we used another specific CSNK2 inhibitor, GO289, which was reported to inhibit CSNK2 in vitro with an IC50 of 7 nM [22]. Similar to CX-4945, the EC50 of GO289 for autophagy induction was ~1.5 µM (Figures 1E, S1E,F).
Figure 1.
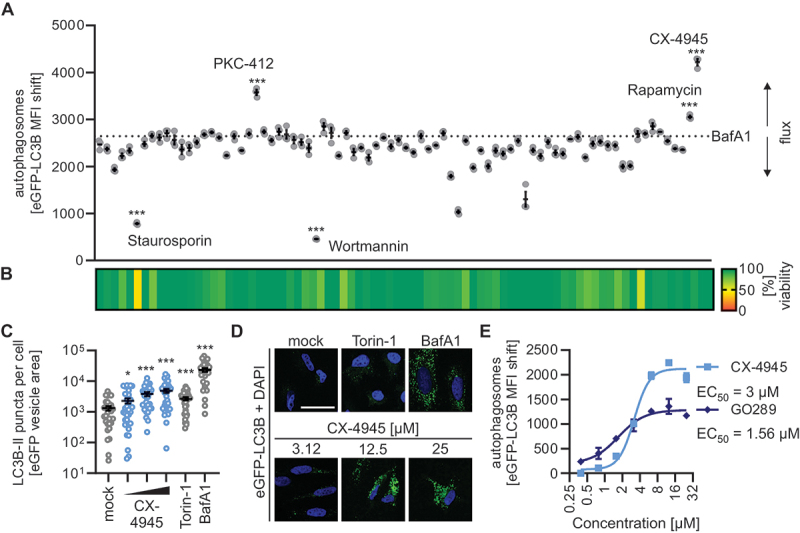
CSNK2 inhibition induces autophagy. (A) Autophagosome levels in HeLa GL cells treated with kinase inhibitors (10 µM) and bafilomycin A1 (BafA1, co-treatment, 250 nM) as assessed 4 h post stimulation by flow cytometry (eGFP-LC3B MFI). Selected agents are annotated, full list in Table S1. Lines represent the mean ± SEM, n = 3. (B) Cell metabolic activity in samples from (A) was assessed via MTT assay 4 h post-treatment depicted as a heatmap of means as indicated, n = 3. (C) eGFP-LC3B puncta quantification in HeLa GL cells treated with increasing concentrations of CX-4945 (3.12–25 µM) for 24 h as indicated. Treatment with torin-1 (5 µM) and bafilomycin A1 (BafA1, 250 nM) for 24 h served as control. Lines represent the mean ± SEM, n = 29–33 (individual cells). (D) Representative fluorescence confocal laser scanning fluorescence microscopy images from (C). eGFP (green), nuclei (DAPI, blue). Scale bar: 25 µm. (E) Autophagosome levels (eGFP-LC3B MFI) in HeLa GL cells treated with increasing concentrations of CX-4945 or GO289 (0.25–32 µM), co-treated with bafilomycin A1 (BafA1, 250 nM) for 4 h. Dots represent the mean ± SEM, n = 3. EC50 of CX-4945 and GO289 are indicated next to the respective legend. Student’s t-test with Welch correction. *, p < 0.05; ***, p < 0.001.
Taken together, these results show that pharmacological inhibition of CSNK2 by CX-4945 and GO289 induces autophagic flux at a low micromolar concentration.
CSNK2 restricts autophagic flux in cell culture and primary human cells
To corroborate our finding that autophagic flux is enhanced by CSNK2 inhibition, we depleted the endogenous CSNK2 complex in HeLa cells using a pan-CSNK2 siRNA pool. Knockdown efficiency was ~ 90% for the individual subunits (Figure S2A). Upon depletion of the CSNK2 complex autophagosome levels were significantly increased and co-treatment with bafilomycin A1 confirmed that de novo autophagic flux was induced (Figure 2A). Similarly, targeting of MTOR (positive control) also resulted in increased levels of autophagosomes (Figure 2A). Inversely, overexpression of the individual subunits of CSNK2 (CSNK2A1, CSNK2A2 and CSNK2B), or the whole CSNK2 complex (where the three subunits were expressed to similar levels), led to a robust reduction of autophagic flux in a dose-dependent manner (Figures 2B, S2B). To establish the relevance of CSNK2 in the regulation of autophagy in primary cells, we monitored endogenous LC3B processing and SQSTM1 protein abundance-two major hallmarks of autophagy – in primary human lung fibroblasts (NHLF). Treatment of NHLF cells with CX-4945 resulted in a dose-dependent increase of lipidated, processed LC3B-II levels and the ratio of LC3B-II to LC3B/LC3B-I, similar to the positive controls torin-1 and bafilomycin A1 (Figure 2C). Concurrently, CX-4945 treatment reduced SQSTM1 protein levels (compared to the mock control) in a dose-dependent manner (Figure 2D), indicating an increase of autophagic flux by CSNK2 inhibition. To corroborate that CSNK2 regulates classical autophagy, we compared the impact of CSNK2 inhibition in ATG5 wild type (WT) and knockout (KO) A549 cells. Whereas CX-4954 treatment effectively reduced SQSTM1 levels in WT cells, it did not lead to a marked decrease of SQSTM1 levels in ATG5 KO cells (Figure S2C).
Figure 2.
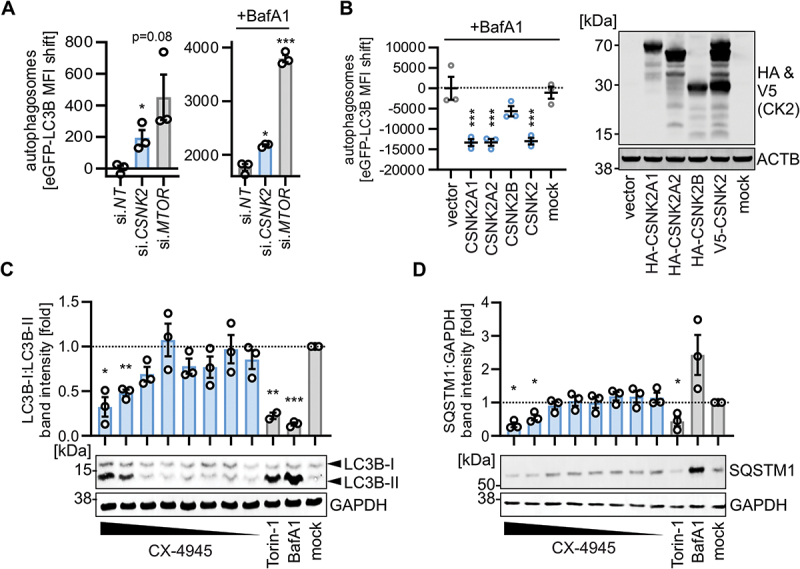
CSNK2 activity reduces autophagy. (A) Autophagosome levels (eGFP-LC3B MFI) in HEK293T GL cells depleted by siRNA of CSNK2 or MTOR quantified 48 h post transfection with/without co-treatment of bafilomycin A1 (BafA1, 250 nM, 4 h) using flow cytometry. Bars represent mean ± SEM, n = 3. (B) Autophagosome levels (eGFP-LC3B MFI) in HEK293T GL cells transiently expressing indicated CSNK2 subunits or the whole complex as assessed by flow cytometry. (right panel) Representative immunoblot showing expression of the HA or V5 tagged CSNK2 subunits. Mock, no transfection. (C) Quantification of LC3-II:LC3-I levels relative to the untreated control (mock) in whole cell lysates of normal human lung fibroblasts (NHLF) treated with CX-4945 (50–0.39 µM) for 24 h as indicated. Bars represent mean ± SEM, n = 3, (independent experiments). (bottom panel) Representative immunoblots. (D) Quantification of SQSTM1:GAPDH in whole cell lysates of NHLF treated with CX-4945 (50–0.39 µM) for 24 h as indicated, relative to the control. (bottom panel) Representative immunoblots. Torin-1 (5 µM) and bafilomycin A1 (BafA1, 250 nM) served as positive controls. Bars represent mean ± SEM, n = 3 (independent experiments). Student’s t-test with Welch correction. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Taken together, these data demonstrate that CSNK2 reduces autophagic flux in cell lines and primary human cells.
The FLN-NHL containing TRIM proteins TRIM2, TRIM3 and TRIM71 reduce autophagic flux
There is accumulating evidence that TRIM proteins are key regulators of autophagy [10]. Our previous findings suggested that among all 61 TRIMs tested, TRIM2 and TRIM3 may have the strongest inhibitory effect on autophagy [14]. Both are part of the same subfamily of TRIMs that is characterized by a FLN-NHL domain at their C terminus. Of note, the third member of this subfamily, TRIM71, also inhibited autophagic flux upon overexpression, albeit to a lesser extent than TRIM2 and TRIM3 [14]. Thus, these FLN-NHL containing TRIM proteins emerged from our screen as a TRIM subfamily of which all members inhibit autophagy. Of note, their closest relatives, TRIM32, TRIM45 and TRIM56, lack either the NHL repeats or the FLN domain, or both [23] (Figure 3A). Flow cytometry analysis in HEK293T cells stably expressing eGFP-LC3B (HEK293T GL) and transiently transfected with the individual TRIM proteins showed that only FLN-NHL containing TRIMs, but not closely related TRIMs, robustly reduced autophagosome abundance (Figure 3B). Consistent with this, all three FLN-NHL containing TRIM proteins significantly reduced the number of eGFP-LC3B-positive puncta (indicative of autophagosomes) area per cell (Figure 3C,D). Treatment with chloroquine (CQ) served as a positive control. In line with this, expression of TRIM2, TRIM3 or TRIM71 in HEK293T cells led to a significant accumulation of SQSTM1 protein, indicative of reduced autophagic flux (Figure 3E,F). Vice versa, depletion of endogenous TRIM2, TRIM3 or TRIM71 by siRNA increased eGFP-LC3B-positive puncta area per cell in HeLa GL cells (Figure 3G,H). Knockdown efficiencies of TRIM2 and TRIM3 were above 75% (Figure S3A). Low levels of TRIM71 mRNA prevented efficient detection in HeLa cells. Of note, unlike many other TRIM proteins, TRIM2 and TRIM3 mRNAs are not induced, but slightly reduced upon interferon treatment in HEK293T cells (Figure S3B). To understand the impact of TRIM2, TRIM3 and TRIM71 on autophagy, we analyzed their effect on core components of autophagy, as well as on autophagy initiation complex activity. Endogenous protein levels of BECN1/Beclin-1 and other key components of the autophagic machinery (i.e., ULK1, ATG5, ATG16L1, PRKAA/AMPKa, and ZFYVE1/DFCP1) were unaffected by TRIM2, TRIM3 or TRIM71 depletion or overexpression (Figures 3I, S3C-E). Autophagy initiation is guided by the ULK1, BECN1 and PIK3C3/VPS34 complex [24,25]. Complex activity is regulated by several PTMs including activating Ser15 phosphorylation on BECN1 and inhibitory Ser757 phosphorylation on ULK1 [25,26]. Depletion of TRIM2, TRIM3 or TRIM71 led to a robust reduction of ULK1 Ser757 phosphorylation, while there was no appreciable effect on BECN1 Ser15 phosphorylation (Figures 3I,J, S3F). Conversely, overexpression of TRIM3 increased ULK1 S757 phosphorylation but had no effect on BECN1 Ser15 phosphorylation (Figure 3K,L).
Figure 3.
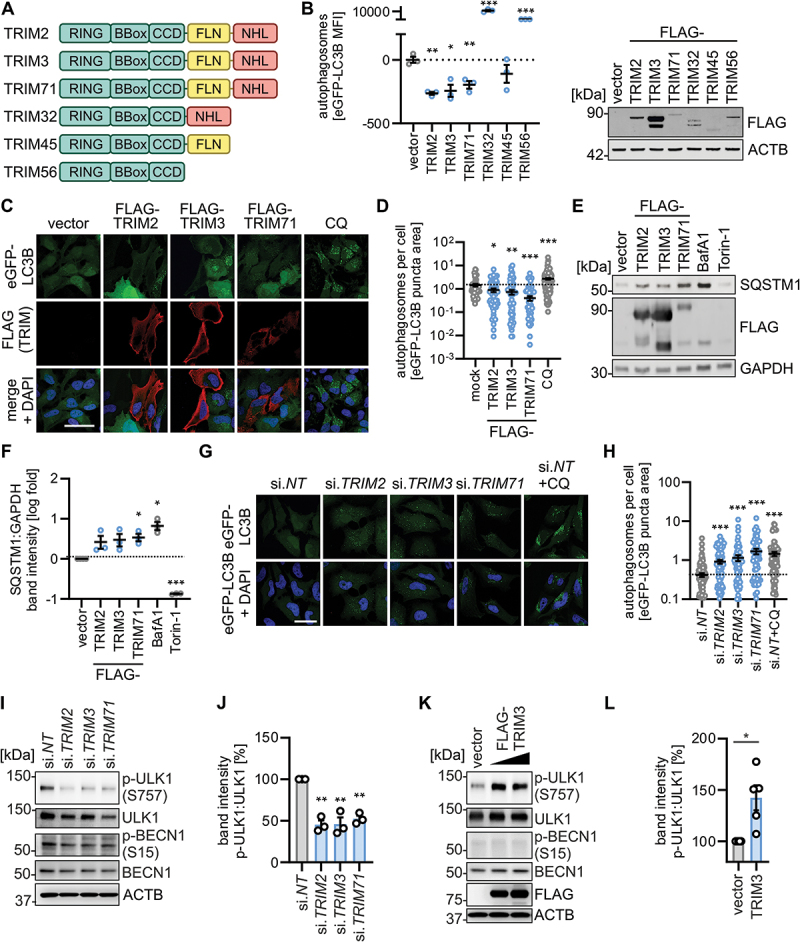
TRIM2, TRIM3 and TRIM71 inhibit autophagy initiation. (A) Schematic depiction of the domain organization of the FLN-NHL TRIMs (TRIM2, TRIM3, TRIM71) and closely related TRIM proteins. TRIM, tripartite motif. (B) Quantification of autophagosome levels in HEK293T GL cells transiently expressing the indicated FLAG-tagged TRIMs at 48 h post transfection by flow cytometry. Lines represent mean ± SEM, n = 3. (right panel) Representative immunoblot of whole cell lysates. (C) Representative confocal laser scanning fluorescence microscopy images of HeLa GL cells transiently expressing indicated TRIMs (48 h) or treated with chloroquine (CQ, 10 µM, overnight). TRIMs (FLAG, red), eGFP-LC3B (green), nuclei (DAPI, blue). Scale bar: 10 µm. (D) Quantification of eGFP-puncta area per cell in the images from (C). Lines represent mean ± SEM, n = 61–139 (individual cells). (E) Representative immunoblots of whole cell lysates of HEK293T cells transiently expressing indicated TRIMs (48 h post transfection). Blots were stained with anti-SQSTM1 and anti-GAPDH. Torin-1 (5 µM) and bafilomycin A1 (BafA1, 250 nM) treatment were used as positive controls. (F) Quantification of the immunoblotting data of (E). Lines represent mean ± SEM, n = 3. (G) Representative confocal laser scanning fluorescence microscopy images of HeLa GL cells (48 h post transfection) depleted of the indicated TRIMs by siRNA or treated with chloroquine (CQ, 10 µM, overnight). eGFP-LC3B (green), nuclei (DAPI, blue). Scale bar: 10 µm. (H) Quantification of the eGFP-puncta area per cell in the images from (F), Lines represent mean ± SEM, n = 61–117 (individual cells). (I) Representative immunoblots of whole cell lysates of HDF cells depleted of the indicated TRIMs by siRNA at 40 h post transfection. Blots were stained with anti-p-ULK1 (S757), anti-ULK1, anti-p-BECN1 (Ser15), anti-BECN1 and anti-ACTB. (J) quantification of the band intensities of p-ULK1 (Ser757) to total ULK1 of the immunoblotting data of (I). Bars represent mean ± SEM, n = 3. (K) Representative immunoblots of whole cell lysates of HEK293T cells that transiently expressed empty vector or TRIM3 at 40 h post transfection. Blots were stained with anti-p-ULK1 (Ser757), anti-ULK1, anti-p-BECN1 (Ser15), anti-BECN1, anti-FLAG and anti-ACTB. (L) Quantification of the band intensities of p-ULK1 (Ser757):ULK1 levels of the immunoblotting data of (K) at highest TRIM3 transfection levels. Bars represent mean ± SEM, n = 5. Student’s t-test with Welch correction. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
These results show that the three FLN-NHL domain containing TRIM2, TRIM3 and TRIM71 proteins, but not other closely related TRIMs, negatively regulate autophagy likely due to sustained inhibitory phosphorylation of ULK1 at S757.
FLN-NHL containing TRIM proteins interact with the CSNK2 complex
To investigate the molecular mechanism underlying autophagy inhibition by CSNK2 and FLN-NHL domain containing TRIMs, we analyzed if these proteins interact. Co-immunoprecipitation of ectopically expressed CSNK2A1, CSNK2A2 and CSNK2B in HEK293T cells indicated an interaction of TRIM2, TRIM3 and TRIM71 with the CSNK2 complex (Figure 4A). In line with this, TRIM2 and TRIM3 lacking the RING domain (TRIM2ΔRING and TRIM3ΔRING) readily interacted with the whole endogenous CSNK2 complex (comprising CSNK2A1, CSNK2A2 and CSNK2B), as assessed by mass spectrometry upon large-scale purification from HEK293T cells (Figures 4B,C, S4A,B, Table S2). TRIM2ΔRING and TRIM3ΔRING were used to avoid putative degradation of the interaction targets by the E3 ubiquitin ligase function of the RING domain. CSNK2A1 and CSNK2A2 were among the top interacting proteins (rank 2 and 5), 88- and 48-fold enriched in the TRIM2ΔRING pulldown and 33- and 6-fold enriched by the TRIM3ΔRING purification, respectively. Confocal microscopy studies examining co-localization indicated that all components of the CSNK2 complex colocalized with the three FLN-NHL containing TRIM proteins in the cytoplasm of HeLa cells (Figure S4C). Proximity ligation assays (PLA) of FLAG-tagged TRIM2, TRIM3 or TRIM71 and the three subunits of CSNK2 transiently expressed in HeLa cells indicated that the TRIMs and CSNK2 are in close proximity in the cytoplasm (Figure 4D). Next, we examined the interaction between endogenous CSNK2 and TRIM3. PLA and co-immunoprecipitation studies in NHLF and HEK293T cells, respectively, showed binding and colocalization of endogenous TRIM3 with endogenous CSNK2 (Figures 4E, S4D) confirming the results of the overexpression experiments.
Figure 4.
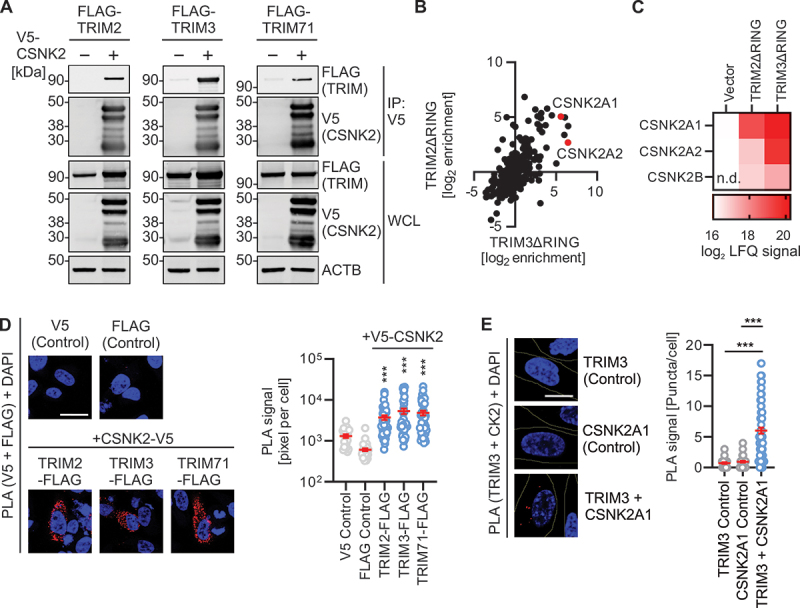
CSNK2 interacts with FLN-NHL TRIMs. (A) Representative Immunoblots of an anti-V5 immunoprecipitation of whole cell lysates of HEK293T cells transiently expressing FLAG-tagged TRIM2, TRIM3 or TRIM71, and V5-tagged CSNK2 subunits as indicated. Blots were stained with anti-FLAG, anti-HA and anti-ACTB. WCL, whole cell lysate. (B) Scatter plot depicting enrichment of proteins co-purifying FLAG-tagged RING deleted mutants of TRIM2 and TRIM3 of a large-scale affinity isolation in HEK293T cells. Enrichments calculated relative to the vector control as label free quantification signal. CSNK2 subunits are highlighted in red. (C) Co-enrichment of the CSNK2 subunits with RING deleted versions of TRIM2 and TRIM3 extracted from the data in (B). Log2 of the label free quantification signal is shown as a heatmap as indicated. n.D., not detected. (D) Representative confocal microscopy images showing the colocalization of overexpressed CSNK2-V5 with each FLAG-tagged TRIM2, TRIM3 and TRIM71 in HeLa cells compared to the single stained mock controls by PLA (red). Nuclei, DAPI (blue). Scale bar: 25 µm. (right panel) quantification of the PLA signal as pixel per cell. Lines represent mean ± SEM, n = 22–47 (individual cells). (E) Representative confocal microscopy images showing the colocalization of endogenous CSNK2A1 and TRIM3 in NHLF cells compared to the single stained mock controls by PLA (red). Nuclei, DAPI (blue). Scale bar: 10 µm. (right panel) quantification of the PLA signal as puncta per cell. Lines represent mean ± SEM, n = 41–94 (individual cells). Student’s t-test with Welch correction. ***, p < 0.001.
These experiments show that the CSNK2 complex interacts with the FLN-NHL containing TRIM proteins 2, 3, and 71 in the cytoplasm.
CSNK2 binding to the NHL domain of TRIM3 is required for autophagy regulation
TRIM2, 3, and 71 have high sequence homology, with ~ 70% of the amino acid identical in TRIM2 and TRIM3 and ~ 35% between TRIM3 and TRIM71 [27]. Expression profiles of the Human Protein Atlas (proteinatlas.org) indicate that TRIM3 is ubiquitously expressed, whereas TRIM2 is predominantly found in the brain, and TRIM71 in male tissues (Figures 5A, S5A) [28]. Thus, we focused on TRIM3 to understand the mechanistic details of the functional connection between the CSNK2 complex and FLN-NHL containing TRIM proteins. As expected, inhibition of CSNK2 by CX-4945 alleviated the negative impact of ectopic TRIM3 expression on autophagosome abundance (Figure 5B). To map the domains of TRIM3 that are required for autophagy inhibition and interaction with CSNK2, we constructed deletion variants (Figure 5C). TRIM3ΔRING is truncated from the N terminus (aa 64–744), whereas TRIM3ΔNHL is truncated from the C-terminus (aa 1–473), thus lacking the full E3 ligase RING domain or the six NHL repeats, respectively. Co-immunoprecipitation assays revealed that the NHL repeats of TRIM3 are required for interaction with CSNK2A2, whereas the RING domain is dispensable for the interaction (Figure 5D). This is in line with studies from many different groups showing that the C-terminal domains of TRIM proteins (e.g., the NHL repeats) usually serve as protein-protein interaction platforms [11]. To assess the function of the domain deletion TRIM3 mutants, we performed autophagosome analysis using flow cytometry and immunofluorescence. Analysis of autophagosome levels by flow cytometry showed that only full-length TRIM3 reduced autophagosome levels significantly; in contrast, TRIM3ΔRING had no impact on autophagy while TRIM3ΔNHL increased autophagosome levels (Figure 5E). Expression of TRIM32 served as a positive control [20,29]. In accord with the flow cytometry analyses, only expression of WT TRIM3 decreased the number of eGFP-LC3B-positive puncta per cell, while TRIM3ΔNHL drastically increased eGFP-LC3B-positive puncta and TRIM3ΔRING having no significant effect (Figure 5F,G). In line with this, accumulation of SQSTM1 was only induced by WT TRIM3 (Figure S5B).
Figure 5.
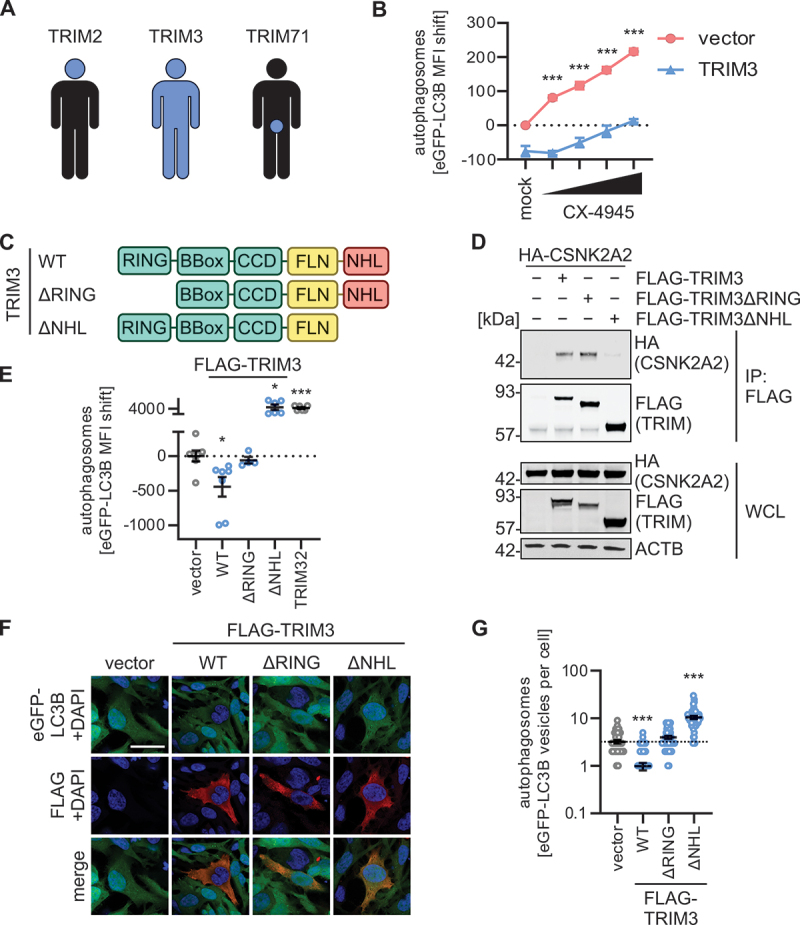
TRIM3 mediated autophagy modulation is dependent on its NHL domain. (A) Schematic depiction of the expression patterns of TRIM2, TRIM3 and TRIM71 across the human body. See Figure S5A. (B) Quantification of autophagosome levels (eGFP-LC3B MFI) in HEK293T GL cells transiently expressing TRIM3 or a vector control. 48 h post transfection, cells were treated with increasing concentration of CX-4945 (4 h) and autophagosome levels (eGFP-LC3B MFI) were quantified using flow cytometry. Treatment with bafilomycin A1 (BafA1, 250 nM) was used as a positive control. Lines represent mean ± SEM, n = 3. (C) Schematic depiction of the domain organization of TRIM3 and the ΔRING and ΔNHL truncation mutants. (D) Anti-FLAG immunoprecipitation (IP) in whole cell lysates of HEK293T cells transiently expressing HA-tagged CSNK2A2 and indicated FLAG-tagged TRIM3 constructs. 48 h post transfection whole cell lysates (WCLs) and precipitates were analyzed by immunoblotting. Blots were stained with anti-HA, anti-FLAG and anti-ACTB. (E) Quantification of autophagosome levels (eGFP-LC3B MFI, 48 h post transfection) in HEK293T GL cells transiently expressing indicated TRIM3 truncation mutants. Lines represent mean ± SEM, n = 3. (F) Representative confocal laser scanning fluorescence microscopy images of HeLa GL cells transiently expressing indicated FLAG-tagged TRIM3 constructs (48 h post transfection). TRIMs (FLAG, red), eGFP-LC3B (green), nuclei (DAPI, blue). Scale bar: 10 µm. (G) Quantification of the eGFP-puncta area per cell in the images from (F). Lines represent mean ± SEM, n = 42–57 (individual cells). Student’s t-test with Welch correction. *, p < 0.05; ***, p < 0.001.
These results indicate that the CSNK2-NHL interaction regulates the function of TRIM3, whereas the RING domain serves as the effector domain. Of note, the NHL domain may have an auto-inhibitory effect.
CSNK2 activates TRIM3 via phosphorylation of the C-terminal domain
As both CSNK2 and TRIMs have enzymatic activities – phosphorylation and ubiquitination, respectively – we sought to understand the hierarchy between CSNK2 and the TRIMs (Figure S6A). Ubiquitination of a target can have several functions, most commonly their degradation (via K48-linked polyubiquitination) or activation (mediated by various polyubiquitin-chain linkages other than K48-polyubiquitin). Western blot analysis showed that ectopic expression of TRIM3 did not induce ubiquitination of endogenous CSNK2A1 or CSNK2A2 (Figure 6A), nor affected their endogenous expression levels in HEK293T cells (Figure S6B). In vitro kinase activity assays revealed that the presence of the TRIMs did not significantly alter CSNK2 activity (Figures 6B, S6C). In contrast, treatment with CX-4945 (control) reduced CSNK2 activity. This suggested that the TRIMs do not alter the enzymatic activity or stability of the CSNK2 complex. Thus, we asked whether CSNK2 phosphorylates TRIM3. Strikingly, analysis of TRIM3 phosphorylation using PhosTag gels showed that almost 100% of cellular TRIM3 in HEK293T cells is phosphorylated (Figure 6C). This was confirmed by western blot analysis of immunoprecipitated TRIM3 using phospho-Ser and phospho-Tyr antibodies (Figure 6D). To clarify whether CSNK2 mediates the phosphorylation of TRIM3, we depleted CSNK2A1, CSNK2A2 and CSNK2B from HEK293T cells using specific siRNAs. Phosphorylation levels of immunoprecipitated TRIM3 were markedly reduced upon knockdown of CSNK2 subunits (Figure 6E). Kinase consensus site prediction analysis revealed three putative CSNK2 phosphorylation motifs in TRIM3: T40 in the RING domain of TRIM3 (also found in TRIM2), S83 and S84 located just before the BBox of TRIM3 (also found in TRIM2), and S661 in the fifth of six NHL repeats of TRIM3 (also found in TRIM2 and TRIM71) (Figure 6F). Mutation of residue S661, but not T40 or S83 S84, to alanine rendered TRIM3 unable to reduce autophagosome levels in immunofluorescence analysis (Figure 6G,H). In line with this, expression of WT TRIM3 and the T40A and S83A S84A mutants were able to reduce autophagosome levels as assessed by flow cytometry, whereas TRIM3 S661A failed to do so (Figure 6I). This suggests that S661 is crucial for TRIM3’s autophagy regulatory activity. In line with this, mutation of S661 drastically reduced the phosphorylation of TRIM3 (Figure 6J).
Figure 6.
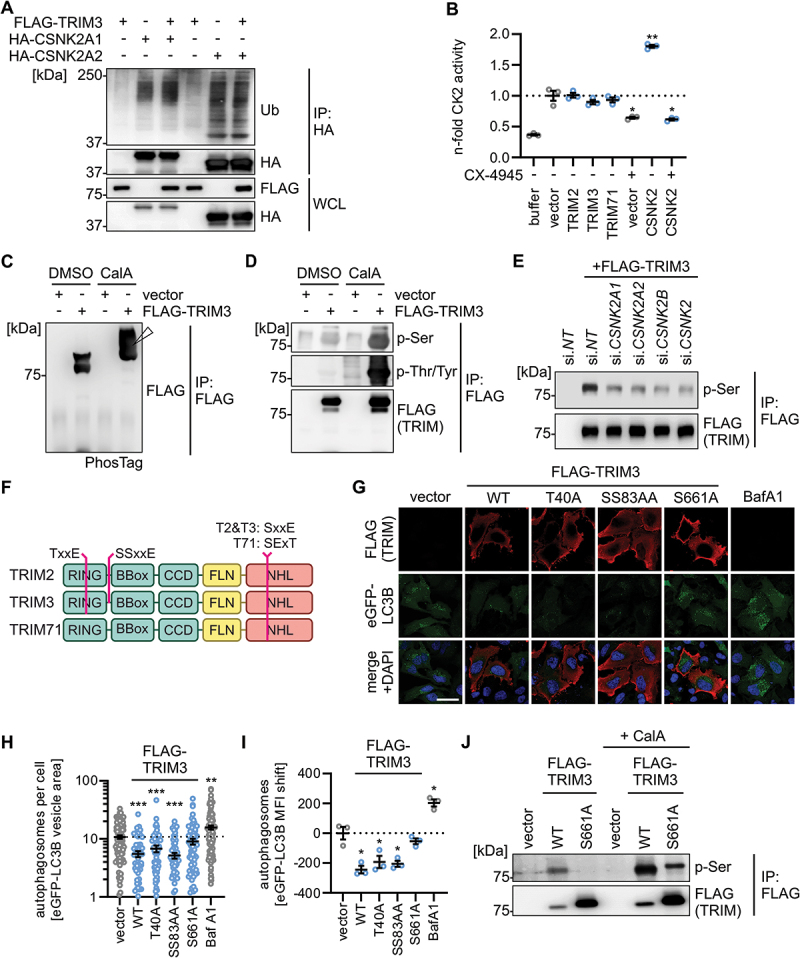
CSNK2 phosphorylates TRIM3 at S661 to inhibit autophagy. (A) Ubiquitination status of indicated HA-tagged CSNK2 subunits purified by anti-HA immunoprecipitation from whole cell lysates (WCLs) of transfected HEK293T cells at 40 h post transfection. IPs and WCLs were analyzed by immunoblotting. Blots were stained with anti-ub (ubiquitin), anti-FLAG and anti-HA. (B) Quantification of CSNK2 activity in whole cell lysates of HEK293T cells transiently expressing indicated proteins and/or treated with CX-4945 (10 µM) by ELISA. Data are relative to the vector control. Lines represent mean ± SEM, n = 3. (C and D) Analysis of the phosphorylation status of TRIM3 in HEK293T cells transiently expressing TRIM3 or a vector control by (C) PhosTag acrylamide gel analysis (the white arrow indicates the shifted phosphorylated TRIM3) and (D) enrichment of the FLAG-tagged TRIM3 and immunoblot analysis using phospho-specific antibodies. Blots were stained with anti-FLAG, anti-p-Ser and anti-p-Thr/Tyr. CalA, Calyculin A. (E) Analysis of the phosphorylation status of TRIM3 purified from HEK293T cells expressing TRIM3 or a vector control and depleted of the indicated proteins using siRNAs targeting individual components of the CSNK2 complex (as indicated) or the whole complex (si.CSNK2) as assessed by immunoblotting. Blots were stained with anti-FLAG and anti-p-Ser. (F) Schematic depiction of the domain organization of the indicated TRIMs and the position and sequence context of the predicted putative CSNK2 phosphorylation sites. (G) Exemplary representative laser scanning confocal microscopy images of HEK293T GL cells transiently expressing the indicated TRIM3 mutants or a vector control 48 h post transfection. TRIMs (FLAG, red), eGFP-LC3B (green), nuclei (DAPI, blue). Scale bar: 10 µm. (H) eGFP-LC3B-puncta area per cell from the pictures in (G) was quantified. Lines represent mean ± SEM, n = 59–112 (individual cells) (I) Quantification of autophagosome levels (eGFP-LC3B MFI, 48 h post transfection) in HEK293T GL cells transiently expressing indicated TRIM3 mutants or a vector control. Bafilomycin A1 (BafA1, 250 nM, overnight) was used a positive control. Lines represent mean ± SEM, n = 3. (J) Analysis of the phosphorylation status of TRIM3 WT and TRIM3 S661A purified by anti-FLAG IP from whole cell lysates of HEK293T cells transiently expressing indicated TRIM3 constructs or a vector control and analyzed by immunoblots. Blots were staiend with anti-FLAG and anti-p-Ser. Student’s t-test with Welch correction. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Taken together, these data show that CSNK2 phosphorylates S661 in the NHL domain of TRIM3, and that this phosphorylation event is required for TRIM3 to reduce autophagic flux.
Targeting the CSNK2-TRIM2, -TRIM3, -TRIM71 axis promotes anti-viral autophagy
Autophagy is induced during viral infection as part of an innate immune defense program. To determine whether CSNK2 suppresses anti-viral autophagy, we infected HeLa GL cells with IAV and MeV [30,31] that induce low levels of autophagy 5 h and 24 h post infection (Figure 7A,B). Treatment with CX-4945 drastically increased the number of autophagosomes in IAV or MeV infected HeLa GL cells by 6- or 3-fold, respectively (Figure 7A,B). To understand whether this additional induction of autophagy is anti-viral, we treated A549 cells with CX-4945 1 h prior to infection and monitored the replication of IAV-GFP or MeV-GFP, using high-throughput live-cell microscopy (Figures 7C,D). Quantification of GFP-positive (i.e., infected) cells showed that CX-4945 treatment reduced the replication of IAV-GFP and MeV-GFP in a dose-dependent manner up to 5-fold (Figures 7C,D, S7A,B). As CSNK2 is a kinase known for its many targets [8], we determined the contribution of autophagy to the anti-viral effect, by comparing WT and ATG5 KO A549 cells (Figure S7C). Of note, treatment with CX-4945 affected cell viability of both cell lines (WT and ATG5 KO A549) at highest concentrations in a similar manner (Figure S7D). While CX-4945 treatment inhibited IAV-GFP and MeV-GFP replication in both WT and ATG5 KO cells in a dose-dependent manner, the magnitude of the inhibitory effect was significantly reduced in ATG5 KO cells (Figure 7C,D). Treatment with CX-4945 had reduced anti-viral effects in ATG5 KO A549 cells (Figure 7E,F), suggesting that autophagy contributes to the anti-viral impact of CSNK2 inhibition. Analysis of the infectious titers of IAV-GFP and MeV-GFP confirmed the anti-viral impact and partial autophagy dependency of CX-4945 treatment (Figures 7G,H, S7E,F). In WT A549 cells, CX-4945 reduced the infectious titers of IAV or MeV by 8.5-fold and ~ 40-fold, respectively, at the highest concentration used (25 µM). To further corroborate our finding that targeting the CSNK2-TRIM2, -TRIM3, -TRIM71 axis induces anti-viral autophagy, we tested the impact of CSNK2 inhibition on HIV-1. CX-4945 treatment reduced infectious virus yields of HIV-1 NL4-3 in a dose-dependent manner in WT but not ATG5 KO HEK293T cells (Figure 7I). CX-4945 also inhibited the primary HIV-1 strain CH077 (Figure S7G). In line with this, siRNA-mediated depletion of TRIM2, TRIM3 or TRIM71 reduced HIV-1 replication in TZM-bl cells by 60–90% (Figure 7J).
Figure 7.
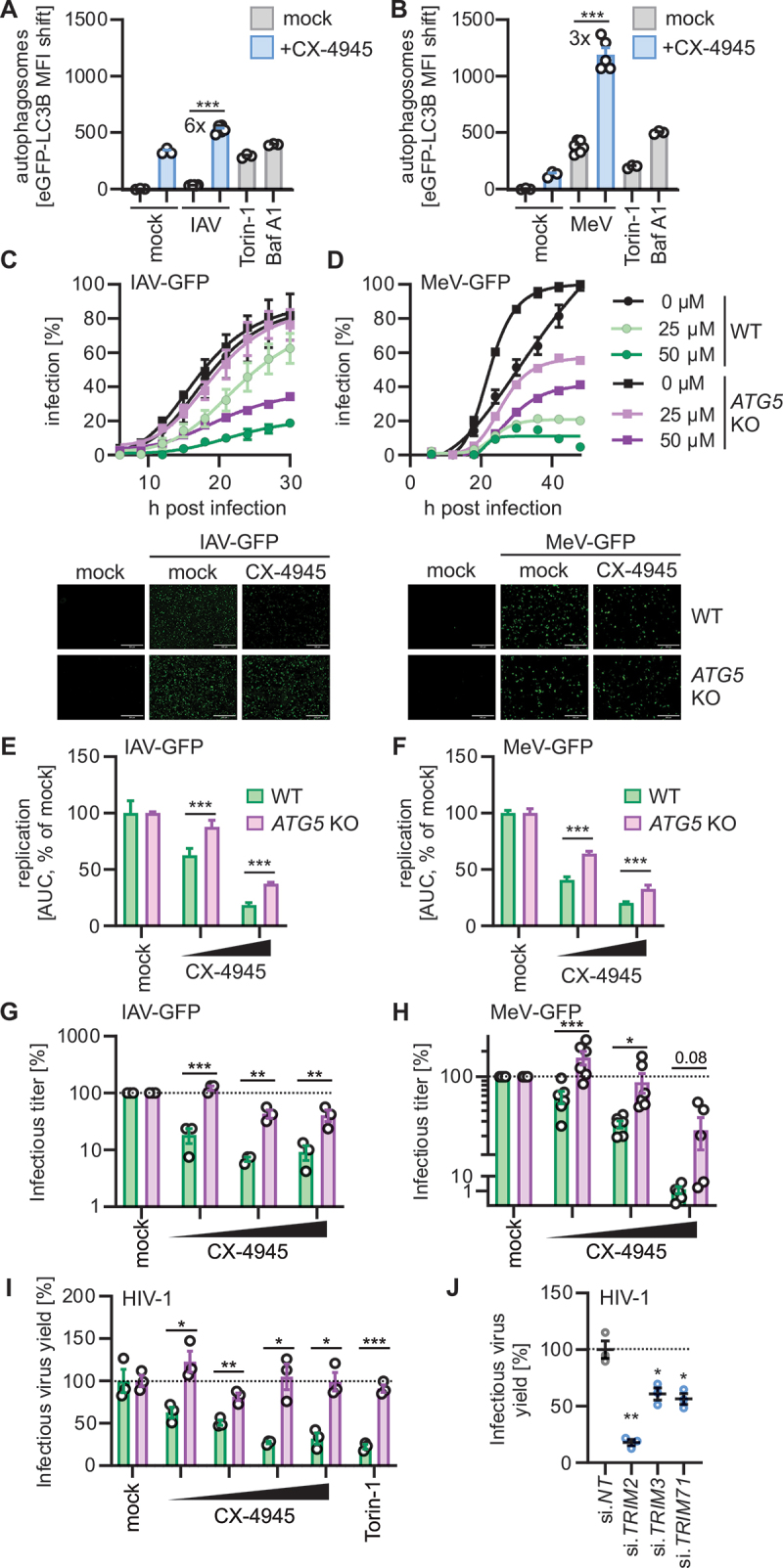
Pharmacological targeting of the CSNK2-TRIM2, -TRIM3, -TRIM71 axis restricts virus growth. (A and B) Quantification of autophagosome levels (eGFP-LC3B MFI) in HeLa GL cells, with and without CX-4945 (1 µM, 1 h) treatment were infected with (A) influenza a virus (IAV, MOI 5, 5 h) or (B) measles virus (MeV, MOI 1, 24 h) by flow cytometry. Torin-1 (1 µM, 4 h) and bafilomycin A1 (BafA1, 250 nM, 4 h) treatment was used as controls. Lines represent mean ± SEM, n = 3–6. (C and D) A549 WT and A549 ATG5 KO cells were treated with CX-4945 (25 µM, 50 µM, 1 h) or left untreated and infected with (C) IAV-GFP or (D) MeV-GFP. Infection kinetics were monitored via fluorescence microscopy for 30 h (IAV-GFP) or 40 h (MeV-GFP) by quantifying GFP+ cell count every 3 h and normalized to maximum infection (100%). Representative fluorescence microscopy images are shown of time-point 24 h. Infected cells (GFP, green). Scale bars: 100 µm. Dots represent the mean ± SEM, n = 3. (E and F) Relative comparison of (E) IAV-GFP or (F) MeV-GFP replication in A549 WT and A549 ATG5 KO cells using area under the curve analysis of the data in (C) and (D). Bars represent mean ± SEM, n = 3. (G and H) A549 WT and A549 ATG5 KO cells were treated with CX-4945 (6.25 µM −25 µM, 2 h) or left untreated, and infected with (G) IAV-GFP or (H) MeV-GFP. Supernatants were collected 24 h (IAV-GFP) or 48 h (MeV-GFP) post infection. Infectious virus titers from A549 cells treated with IAV-GFP infected supernatants (G) or from Vero cells treated with MeV-GFP infected supernatants (H). TCID50 (IAV-GFP) and FFU/mL (MeV-GFP) were determined at 72 h post infection. Relative infectious virus titers are normalized to the mock control (DMSO). Bars represent mean ± SEM, n = 3 (IAV) or 6 (MeV). (I) Infectious virus yields of HIV-1 NL4–3 proviral DNA transfected HEK293T WT or HEK293T ATG5 KO cells treated with increasing concentrations of CX-4945 (1.56–12.5 µM) and quantified by TZM-bl reporter assay 24 h post transfection. Relative infectious virus yield is normalized to the untreated control (mock). Treatment with torin-1 (1 µM) was used as a control. Bars represent mean ± SEM, n = 3. (J) Quantification of infectious virus yield of HIV-1 CH077 in TZM-bl cells depleted of TRIM2, TRIM3 and TRIM71 by siRNA and infected with CH077 HIV-1 one day post transfection. Three days post infection, the infectious virus yield in the supernatant was determined by TZM-bl reporter assay. Relative infectious virus yield is normalized to the non-targeting (NT) control (100%). Lines represent mean ± SEM, n = 3 (technical replicates). Student’s t-test with Welch correction or two-way ANOVA (G, H). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
In summary, these data indicate that pharmacological inhibition of CSNK2, or depletion of TRIM2, TRIM3 and TRIM71, exert anti-viral activity against IAV, MeV and HIV-1. Our results also show that the anti-viral effect of CX-4945 is partially dependent on autophagy.
Discussion
Key factors of the core machinery of autophagy and many cellular factors that promote autophagy especially during viral infection have been characterized in the recent decades. In comparison, little is currently known about cellular pathways that keep autophagy in check. Here, we identified that the CSNK2-TRIM2, -TRIM3, -TRIM71 axis is a major signaling cascade in limiting autophagic flux. Our mechanistic analyses revealed that CSNK2 binds and phosphorylates TRIM2, TRIM3 and TRIM71 in their common NHL domain, which activates these TRIM proteins to inhibit autophagy induction. Thus, our data identifies CSNK2 as a pivotal kinase for maintaining innate immune homeostasis.
While our studies clearly established a negative impact of CSNK2 on autophagy in multiple cell lines and also primary human cells, the role of CSNK2 in autophagy was previously debated [32–34] and the mechanistic details have remained unknown. In agreement with our data, it had been proposed that downregulation of CSNK2 levels induces cell death by excessive autophagy [32]. Along these lines, pharmacological inhibition of CSNK2 was shown to promote autophagy in cancer cells [33]. However, it has also been reported that CSNK2 upregulation by calorie restriction induces autophagy [35] and that CSNK2 promotes autophagic degradation of KEAP1 via AMPK in human cancer cells [36]. These data may, however, be due to different means of autophagy induction (calorie restriction vs viral infection) or influenced by CSNK2’s role in cellular senescence.
In our kinase inhibitor screen (Figure 1A), only three kinase inhibitors increased autophagic flux: rapamycin targeting MTOR, PKC-412 targeting PKC and CX-4945 targeting CSNK2. Besides CSNK2, there are only few other kinases known to negatively impact autophagy. Most prominently, the MTOR complex suppresses autophagy by phosphorylating the pro-autophagy kinase ULK1 [37]. Selected isoforms of the cellular kinase PKC, such as PKCα, were also reported to reduce autophagy induction via modulation of the PI3K-AKT-MTOR-MAPK/ERK axis [38]. However, it needs to be considered that our panel of kinase inhibitors, while containing 81 different compounds targeting ~ 50 different kinases did not comprehensively target all cellular kinases (of note, >500 kinases exist in humans) [39]. Thus, future work is clearly required to identify additional kinases that dampen autophagic flux.
Several TRIM proteins, such as TRIM5α and TRIM23, were previously identified to promote autophagy induction [9,10,14,40]. Here, we identified TRIM2, 3, and 71 as the first TRIMs described to negatively regulate autophagy. While these three TRIM proteins exhibit like most TRIMs the common N-terminal RING-BBox-Coiled Coil domain structure, they uniquely possess a composite FLN and NHL repeat domain organization at the C-terminus. Our data indicates that the combination of an FLN and NHL domain is required for autophagy inhibition. Interestingly, expression of TRIM3 constructs with deletion of the NHL domain even caused a drastic increase in autophagosomes. This suggests that the NHL domain not only serves as a binding and phosphorylation platform of CSNK2 but may also be required to repress autophagosome-inducing capabilities of other domains. However, future studies are required to understand this in molecular detail. In a study relying on overexpression experiments, TRIM3 was previously suggested to target a core component of the autophagic machinery, BECN1, for degradation thus preventing autophagy induction [41]. However, our data show that endogenous BECN1 levels are unaffected by TRIM2, TRIM3 and TRIM71 depletion or overexpression. Instead, the activity of the autophagy initiation complex containing BECN1 and ULK1 was influenced by the three FLN-NHL TRIMs, as indicated by altered phosphorylation of ULK1 at the inhibitory phospho-site Ser757. It is tempting to speculate that ULK1 Ser757 phosphorylation may be increased due to enhanced activity of the MTOR complex, that confers this phosphorylation [26]. However, the exact mechanism(s) of how these TRIMs inhibit autophagy initiation, as well as the precise target(s) of their ubiquitin ligase activity, remain to be identified in future studies.
Why would three proteins have redundant functions? Protein expression data (Figure 5A, S5A) indicates that TRIM2 is expressed primarily in neuronal tissue, while TRIM3 is ubiquitously present, and TRIM71 is preferentially found in male tissues and upregulated during development [28]. Thus, these three TRIMs may have tissue-specific and/or temporal roles in autophagy regulation. Of note, it was reported that TRIM2 and TRIM3 may interact via their FLN and coiled-coil domains, suggesting also the presence of TRIM2-TRIM3 hetero-oligomers [27]. In addition, recent evidence has shown that the ubiquitin E3 ligase domains of TRIM2 and 3 have different self-association and activity profiles [27]. Taken together, this would argue against redundancy of the three FLN-NHL TRIMs, but future work is clearly needed to dissect the potentially distinct or tissue-specific functions of TRIM2, TRIM3 and TRIM71.
Our data show that the anti-viral effect of autophagy induction by CX-4945 on viral replication was within 10–100-fold. However, we likely underestimate the anti-viral impact of autophagy due to viral evasion mechanisms [15]. Many viruses even evolved to exploit the autophagic machinery, thus requiring specific components for efficient replication. For example, MeV was reported to depend on sustained autophagy for virion production [42]. In line with this, our results show that MeV replicates slower in ATG5 KO cells as compared to WT cells. Furthermore, IAV infectious virus release and virion stability are thought to be promoted by the autophagic machinery. However, it was also shown that IAV components are degraded in a lysosome-autophagosome-dependent manner [18,31,43]. HIV-1 may depend on autophagy to assemble membranes, but its proteins are also targets of autophagic degradation [44,45]. Altogether, this suggests that while specific components of the autophagy machinery may be hijacked by the virus, exogenous induction of autophagy, e.g., by CSNK2-targeting compounds, confers an anti-viral effect, overriding viral evasion and exploitation mechanisms.
Our results indicate that autophagy is only partially responsible for the anti-viral effect of CSNK2 inhibition. It also needs to be considered that as the CSNK2-TRIM2, -TRIM3, -TRIM71 axis regulates autophagy as part of innate immunity, it is a plausible target of viral pathogens. Thus, the impact of CSNK2 inhibition on viral replication may have been underestimated in this study. Future studies are required to delineate other pro-viral functions of CSNK2 and define whether the CSNK2-TRIM2, -TRIM3, -TRIM71 axis is targeted by viruses to avoid innate immune restriction.
Our data shows that targeting of CSNK2 by pharmacological inhibitors has an anti-viral role in IAV, MeV and HIV-1 infection. CSNK2 inhibition was previously proposed as an anti-viral treatment against coronaviruses including SARS-CoV-2 [46–48]. Importantly, therapeutic induction of autophagy is not only relevant in infectious diseases but also for cancer treatment. Targeting of MTOR by, e.g., rapamycin or its homolog everolimus to induce autophagy is currently used in the clinics to inhibit cancer growth. However, CSNK2 inhibition to modulate cellular autophagy needs to be carefully considered as CSNK2 infamously regulates over a 1,000 cellular proteins [8], and thus adverse effects of the treatment may need to be considered. Of note, MTOR has a similarly large substrate spectrum as CSNK2 [37]. Thus, inhibition of CSNK2 is in clinical trials against various cancers [49]. Future work to explore pharmacological inhibition of CSNK2 for the treatment of viral infections and other human diseases seems highly warranted.
Materials and methods
Cell culture, viruses and reagents
HEK293T GL and HeLa GL were constructed as previously described [20]. NHLF (CC-2512) were obtained from Lonza, HEK293T (CRL3216), A549 (CCL-185) and Vero E6 (CRL1586) cells were purchased from American type culture collection (ATCC). Human dermal fibroblasts, hTERT immortalized (HDF) were provided by Patrick Hearing (Stony Brook University, New York, USA) [50,51]. Cells were kept in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, 41965039) supplemented with 10% fetal bovine serum (FBS; Gibco, A5256701), 13 µM gentamicin (Gibco, 15710) and 2 mM L-glutamine (PAN-Biotech, P04–80050), hereafter referred to as DMEMxxx. Influenza A virus (strain PR8/34 H1N1, VR-95) was obtained from ATCC, measles virus (MeV, Schwarz strain) and MeV-GFP were a gift from K.-K. Conzelmann (Max von Pettenkofer-Institute, Ludwig-Maximilians-University Munich, Germany) [52]. IAV-GFP (SC35M) was kindly provided by M Schwemmle (University of Freiburg, Germany) [53]. Reagents used: Bafilomycin A1 (Santa Cruz Biotechnology, sc-201550), chloroquine (Santa Cruz Biotechnology sc-205629), InSolution Rapamycin (Sigma-Aldrich, 553211), (E/Z)-GO289 (Selleckchem, S9850), CX-4945 (Selleckchem, S2248), Kinase Inhibitor Library (Screen-Well; Enzo Life Sciences BML-2832), EZSolution torin-1 (BioVision, 2353)
Transient transfection of cell lines
Cells were seeded to reach a confluency of 60–80% for transfection. Plasmid DNA was suspended in Opti-MEM (Gibco 31985070), and in parallel, PEI (Sigma-Aldrich; 408727) (2 µg PEI per 1 µg plasmid DNA) or TransIT-LT1 (Mirus, MIR 2300) (3 µl transfection reagent per 1 µg plasmid DNA) was suspended in Opti-MEM. Both suspensions were mixed 1:1. After incubation at RT for 20 min (PEI) or 30 min (TransIT-LT1), the transfection mix was added to each well. The medium can be replaced with fresh medium after 6–16 h.
Transient reverse transfection of cells
Plasmid DNA (140 ng) was suspended in 8.5 µl Opti-MEM (Gibco, 31985070) per well in a 96-well F-bottom plate. PEI (Sigma-Aldrich, 408727) (2.5 µg PEI per 1 µg plasmid DNA) was diluted in 8.5 µl Opti-MEM per well. Both solutions were combined 1:1 and mixed by vortexing. The transfection mix was incubated at RT for 20 min, and 45,000 detached HEK293T GL cells in 83 µl DMEMxxx were added to each well and gently mixed. The next day, the medium was replaced with 100 µl fresh DMEMxxx.
siRNA-mediated knock down
Cells were transfected with siRNA using Lipofectamine RNAiMAX (Invitrogen, 13778075). All siRNAs used were purchased from Horizon Discovery and ordered as SMARTPools: MTOR (M-003008-03), CSNK2A1 (M-003475-03), CSNK2A2 (M-004752-00), CSNK2B (M-007679-00), TRIM2 (M-006955-00), TRIM3 (M-006931-00), TRIM71 (M-023459-01), Non-targeting siRNA Control Pool#1 (D-001206-13) was used as negative control. The cells were seeded to reach a confluency of 60–80% for transfection. siRNA (e.g., 5 pmol per 24-well) was diluted Opti-MEM (Gibco, 31985070), and in parallel, RNAiMAX reagent (e.g., 1.5 µl per 24-well) was diluted in Opti-MEM. The solutions were mixed 1:1 and incubated at RT for 5 min. The transfection mix was added to each well. The cells were further processed for analysis 48–72 h post-transfection.
Generation of ATG5 KO cells
HEK293T cells were seeded in 6-well plates and cultured overnight. The cells were transfected with a lentiviral backbone expressing Cas9 and a sgRNA and the three helper plasmids pMDLg, pRSV-Rev, pMD2.G using PEI-mediated transfection. 46 h after the transfection, the medium was replaced with 2 ml DMEMxxx [20]. 48 h after the transfection, the lentiviral particles were harvested. The target cells (A549 and HEK293T) were seeded in 6-well plates and transduced by adding 300 µl of the lentiviral particle stock. 72 h after transduction, the cells were washed twice with PBS (Gibco, 14190144) and cultured. After two passages, the cells were sorted into individual cells by FACS, grown into clonal cell lines and screened for ATG5 knock out in western blot. Cryo stocks of verified knock outs were prepared.
Whole cell lysates and immunoblotting
Whole cell lysates and immunoblotting were performed as previously described [20]. In brief, cells were lysed in TM-lysis buffer (150 mM NaCl (Merck, 106404), 5 mM HEPES (Sigma-Aldrich, H3375), 1% (v:v) Triton X-100 (Sigma-Aldrich, T8787), 5 mM EDTA (Sigma-Aldrich, E9884), and 0.2% (v:v) Protease Inhibitor Cocktail (Sigma-Aldrich, P2714)) and the debris pelleted (4°C 20,000 g for 20 min). The cleared lysates were transferred to fresh reaction tubes and the protein concentrations were measured using the Rapid Gold BCA Protein Assay Kit (Pierce) according to the manufacturer’s instructions and adjusted. 6× loading dye (187.5 mM Tris-HCL [AppliChem GmbH, A2264, pre-adjusted to pH 6.8], 75% [v:v] glycerol [Sigma-Aldrich, G5516], 6% [w:v] SDS [Sigma-Aldrich 822,050] and 0.3% [w:v] Orange G [Sigma-Aldrich, O3756]) and dithiothreitol (DTT; Thermo Scientific, R0862) were added to a final concentration of 1× and 0.1 M, respectively, and the samples were stored at −20°C. Prior to SDS-PAGE, the samples were heated to 95°C for 10 min. 5–25 µg total protein was loaded onto a NuPAGE 4 to 12% Bis-Tris gel (Invitrogen, NP0321BOX) running in MES-SDS running buffer (Alfa Aesar, J62138.K2) at constant voltage (80 V) for 2 h. Following SDS-PAGE, the separated proteins were transferred onto a PVDF-membrane (Millipore 05,317-10EA) by semi-dry blotting at a constant voltage (30 V) for 30 min. After blotting, the membrane was stained using the Revert 700 total protein stain kit (LI-COR, 926–11016) as per instructions and whole protein signal was measured using a LI-COR Odyssey and the Image Studio software. After blocking in 25 ml Blocker Casein in PBS (Thermo Scientific, 37528) for 1 h at RT, the membrane was incubated with the primary antibody diluted in PBS-T (PBS supplemented with 0.2% [v:v] Tween 20 [Sigma-Aldrich, P9416]) supplemented by 0.1% casein (Thermo Scientific, 37528) for 1.5 h at RT or overnight at 4°C. Subsequently, the PVDF-membrane was washed three times with PBS-T and then incubated in IRDye secondary antibody (see below) diluted 1:20,000 in PBS-T with 0.1% casein for 0.5 h, followed by three washing steps with PBS-T. The fluorescent antibodies were then detected using a LI-COR odyssey and the Image Studio software. Quantification and image processing were performed using the Image Studio lite software.
Primary Antibodies: Polyclonal rabbit anti-LC3 antibody (Sigma-Aldrich, L8918; 1:1000), monoclonal mouse anti-SQSTM1/p62 antibody (abcam, ab56416; 1:1000), anti-GAPDH (BioLegend, 607902; 1:1000), monoclonal mouse anti-ACTB/β-actin antibody (Sigma-Aldrich, A5441; 1:5000), anti-HA: HA-Tag (C29F4) rabbit monoclonal antibody (mAb; Cell Signaling Technology, 3722S; 1:1000), anti-V5: Monoclonal rabbit anti-V5-tag antibody (Cell Signaling Technology, 13202S; 1:1000), monoclonal mouse anti-FLAG-tag antibody (Sigma-Aldrich, F1804; 1:5000), anti-CSNK2A1/CSNK2α (casein kinase 2 alpha) antibody (GeneTex, GTX107897), anti-CSNK2A2/CSNK2α’ (abcam, ab10474), CSNK2A/CSNK2 alpha antibody (Cell Signaling Technology, 2656S; 1:1000), ubiquitin antibody (P4D1; Santa Cruz Biotechnology, sc-8017; 1:1000), pSer/phosphoserine antibody (16B4; Santa Cruz Biotechnology, sc -81,514; 1:1000), phospho-threonine/tyrosine antibody (Cell Signaling Technology, 9381S; 1:1000), BECN1/Beclin-1 Antibody (Cell Signaling Technology, 3738S; 1:1000), p-BECN1/Beclin-1 (Ser15, D4B7R; (Cell Signaling Technology, 84966; 1:1000), ULK1 (D8H5; Cell Signaling Technology, 8054S; 1:1000), p-ULK1 (Ser757) antibody (Cell Signaling Technology, 6888S, 1:1000), TRIM3 antibody (Santa Cruz Biotechnology, sc-136363; 1:1000), TRIM3 antibody (Proteintech, 28392–1-AP; 1:1000) BECN1/Beclin-1 Antibody (Santa Cruz Biotechnology, sc-48381; 1:1000). ATG16L1 antibody rabbit mAb (D6D5; Cell Signaling Technology, 8089S; 1:1000), ZFYVE1/DFCP1 rabbit mAb (E9R6P; Cell Signaling Technology, 85156S; 1:1000), PRKAA/AMPKα Antibody (Cell Signaling Technology, 2532S; 1:1000), ATG5 rabbit mAb (D5F5U; Cell Signaling Technology, 12994S; 1:1000).
Secondary Antibodies: IRDye 800CW Donkey anti-Rabbit IgG Secondary Antibody (LI-COR, 926–32213), IRDye 680RD Donkey anti-Mouse IgG Secondary Antibody (LI-COR, 926–68072), IRDye 680RD Donkey anti-Rabbit IgG Secondary Antibody (LI-COR, 926–68073), IRDye 800CW Donkey anti-Mouse IgG Secondary Antibody (LI-COR, 926–32212), IRDye 680RD Goat anti-Rabbit IgG Secondary Antibody (LI-COR, 926–68071), IRDye 800CW Goat anti-Mouse IgG Secondary Antibody (LI-COR, 926–32210), IRDye 800CW Goat anti-Rat IgG Secondary Antibody (LI-COR, 926–32219).
Confocal laser scanning fluorescence microscopy
Cells were seeded on glass coverslips in 24-well plates. Following the respective experimental procedure, the cells were washed twice with ice-cold PBS (Gibco, 14190144) and fixed in 4% paraformaldehyde (PFA; Santa Cruz Biotechnology, sc-281692) in PBS for 20 min at RT. Next, the cells were washed three times with PBS, and blocked and permeabilized by incubating them in PBS supplemented with 0.5% Triton X-100 (Sigma-Aldrich, T8787), 5% FBS (Gibco 10,270,106) for 30 min at RT. The cells were then washed twice using PBS followed by incubation with primary antibody (see below) diluted in 150 µl PBS at 4°C for 2 h. The cells were washed three times with PBS containing 0.1% Tween 20 (Sigma-Aldrich, P9416) and incubated with secondary antibody (see below) and 4’,6-diamidino-2-phenylindole (DAPI, 1 mg/l; Invitrogen, D1306) diluted in PBS at 4°C for 2 h. Finally, the cells were washed three times with PBS containing 0.1% Tween 20 and once with deionized water. The glass coverslips were then mounted on microscopy slides using self-hardening Mowiol mounting medium (10% [w:v] Mowiol 4–88 [Carl Roth, 0713], 25% [w:v] Glycerol [Sigma-Aldrich, G5516], 50% [v:v] Tris-HCl [at 0.2 mM, pre-adjusted to pH 8.5, AppliChem GmbH, A2264] and 2.5% [w:v] DABCO [Carl Roth, 0718]). After drying at 4°C overnight, the samples were analyzed using a Zeiss LSM 710 confocal microscope. The Images were processed using the ImageJ software.
Primary antibodies: Monoclonal mouse anti-FLAG-tag antibody, M2 (1:400, Sigma-Aldrich, F1804), Monoclonal rabbit anti-V5-tag antibody, D3H8Q (1:200, Cell Signaling Technology, 13202S).
Secondary antibodies: Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 (Invitrogen, A-11004), Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 (Invitrogen, A-11011), Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Invitrogen, A-11001), Goat anti-mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 (Invitrogen, A-21235).
Quantification of autophagy in confocal laser scanning fluorescence microscopy images
The autophagosome area per cell was analyzed in images that were acquired as described above using ImageJ and a semi-automated workflow. First, the GFP channel was separated from the other channels. Next, the background was subtracted using a rolling ball radius of 3, and the image was smoothed. An automatic threshold was applied to the image using the “Moments” threshold algorithm and assuming a dark background. Finally, the area of a cell was selected by hand, and the particles inside the area were analyzed using the “analyze particles” function.
Rapid quantification of autophagosomes by flow cytometry
Quantification was performed as previously descrtibed [20]. The cells were harvested by removing the medium and detaching them using trypsin (PAN Biotech, P10–023100) (37°C for 5 min). The detached cells were suspended in FBS supplemented medium and transferred to 96-well V-bottom plates. The cells were washed with PBS (Gibco, 14190144) containing 1% FBS (Gibco, 10270106), suspended in PBS containing 0.05% saponin (Sigma-Aldrich, 47036) and incubated for 10 min at 4°C. Subsequently, the cells were washed twice with PBS containing 1% FBS. Next, the cells were fixed by in 4% PFA in PBS (Santa Cruz Biotechnology, sc-281692) for 30 min at RT. The supernatant was discarded and the cells were stored in PBS supplemented with 1% FBS at 4°C until analysis. Following fixation, the cells could be stained by incubating them in PBS supplemented with the primary antibody (see confocal laser scanning fluorescence microscopy) at RT for 30 min. The cells were then washed and incubated in PBS supplemented with the secondary at RT for 30 min. The cells were stored in PBS supplemented with 1% FBS at 4°C until analysis.
Co-immunoprecipitation
HEK293T cells were transiently transfected with the indicated expression plasmids using PEI-mediated transfection. 48 h post-transfection, the cells were harvested in ice-cold PBS (Gibco, 14190144) and lysed in ice-cold TM-lysis buffer (see whole cell lysates and immunoblotting). The lysates were vortexed and incubated on ice for 10 min. Next, the lysates were cleared by centrifugation (4°C 20,000 g for 20 min) and the protein concentration of the lysates was determined using the Pierce Rapid Gold BCA Protein Assay Kit (Thermo Scientific, A53225) as per instructions. The protein concentrations were adjusted, and input samples were taken for later western blot analysis. To prepare the magnetic beads (anti-FLAG M2 Magnetic Beads, Merck, M8823; V5-Trap Magnetic Agarose, ChromoTek, v5tma; Pierce Anti-HA Magnetic Beads, Thermo Scientific, 88837), they were washed twice with ice-cold TM-lysis buffer using a magnetic rack and added to the cleared lysates. The lysates were incubated on the beads at 4°C for 4 h on a rotor. Next, the supernatant was removed from the beads using a magnetic rack, and the beads were washed 5 times with ice-cold TM-lysis buffer. Then, 100 µl of 1× SDS loading buffer (see whole cell lysates and immunoblotting) was added to the beads, and the samples were heated to 95°C for 10 min. The samples were then stored at −20°C. When applicable, mass spectrometric analysis was performed at the Core Unit Mass Spectrometry and Proteomics at Ulm University. The proteins contained in the samples were identified, and their abundance was quantified using label-free quantification. For Co-IP of endogenous proteins Dynabeads Protein G (Invitrogen, 10004D) were incubated with designated antibodies and isotype controls and incubated for 2 h at 4°C. The beads were washed twice with ice-cold TM-lysis buffer using a magnetic rack. HEK293T cells were harvested as described above for transfected cells and applied as described above. Antibodies used: CSNK2A (Cell Signaling Technology, 2656S), TRIM3 (Santa Cruz Biotechnology, sc-136363), Normal Rabbit IgG Isotype Control (EMD Millipore, 12–370), Normal Mouse IgG Isotype Control (EMD Millipore, 12–371).
CSNK2 activity assay
CSNK2 activity was quantified using the CSNK2 (casein kinase 2) Kinase Assay/Inhibitor Screening Kit (CycLex, CY-1170) as per instructions. To this end, cleared cell lysates were adjusted for total protein content using the Pierce Rapid Gold BCA Protein Assay Kit (Thermo Scientific, A53225) as per instructions. The lysates were then processed using the CSNK2 Kinase Assay Kit and the absorption at 450 nm was measured using a Molecular Devices V max kinetic microplate reader.
In silico prediction of putative CSNK2 phosphorylation sites
Using the Eukaryotic Linear Motif resource [54] for Functional Sites in Proteins, putative CSNK2 phosphorylation sites in the amino acid sequences of TRIM2, TRIM3 and TRIM71 were identified. Three promising sites were chosen based on their score and the conservation across the three TRIMs.
MTT assay
Cells were treated with the respective compounds for the indicated time frame. Supernatants were replaced with MTT-reagent (methylthiazolyldiphenyl-tetrazolium bromide, 0.5 mg/ml; Sigma-Aldrich, M2128) in PBS (Gibco, 14190144) and incubated for 3 h. The supernatant was discarded, the formazan crystals were dissolved in DMSO-Ethanol solution (1:1, Thermo Scientific Chemicals, A13280.36) and the absorption recorded at 490–650 nm using a Molecular Devices V max kinetic microplate reader.
Cell viability assay
Cells were treated with the respective compounds for the indicated time frame. The cells were harvested by removing the medium and detaching them using trypsin (PAN Biotech, P10–023100) (37°C for 5 min). The detached cells were suspended in FBS supplemented medium and transferred to 96-well V-bottom plates. The cells were washed with PBS (Gibco, 14190144), resuspended in PBS supplemented with Viability Dye (eBioscience Fixable Viability Dye eFluor 780, Invitrogen, 65–0865, 1:1000) and incubated for 30 min at 4°C. The cells were washed with PBS twice and fixed with 2% PFA in PBS for 20 min at room temperature. The cells were kept in PBS supplemented with 1% FBS until analysis by flow cytometry.
qRT-PCR
Total RNA was extracted from cells using the Quick-RNA Microprep Kit (Zymo Research, R1051) according to the manufacturer’s instructions. Reverse transcription and qRT – PCR were performed in one step using 2 µl (~400 ng) of the purified RNA samples as templates (SuperScript III Platinum one step qRT-PCR-Kit; Invitrogen, 11732088) on a StepOnePlus Real-Time PCR System according to the manufacturer’s instructions. TaqMan probes for each individual gene were acquired as premixed TaqMan Gene Expression Assays (Thermo Fisher Scientific, see below) and added to the reaction. Expression level for each target gene was calculated by normalizing against GAPDH using the ΔΔCT method and represented relative to the values for mock-transfected cells, which were set to 1.
TaqMan Gene Expression Assays: CSNK2A/CSNK2α (Thermo Fisher Scientific, Hs00751002_s1), CSNK2A2/CSNK2α’ (Thermo Fisher Scientific, Hs00176505_m1), CSNK2B/CSNK2β (Thermo Fisher Scientific, Hs00365835_m1), MTOR (Thermo Fisher Scientific, Hs00234508_m1), TRIM2 (Thermo Fisher Scientific, Hs07287153_m1), TRIM3 (Thermo Fisher Scientific, Hs01548703_m1), IFIT2 (Thermo Fisher Scientific, Hs01584837_s1), CXCL10 (Thermo Fisher Scientific, Hs00171042_m1), GAPDH (Thermo Fisher Scientific, Hs02786624_g1).
Time resolved monitoring of virus infection
A549 and A549 ATG5 KO cells were seeded in 96-well F-bottom plates in DMEMxxx and cultured overnight. The cells were treated with CX-4945 at the indicated concentrations 2 h before infection with MeV-GFP or IAV-GFP (MOI 0.1). Virus infection was monitored by counting of the GFP+ cells in 3 h steps for 48 h using the Cytation 3 microplate reader.
Quantification of virus-induced autophagy
HeLa GL cells were seeded in 96-well F-bottom plates in DMEMxxx and cultured overnight. The cells were treated with CX-4945 (1 µM) for 1 h before infection with IAV (MOI 5, harvest 5 h post infection) and MeV (MOI 1, harvest 24 h post infection). The cells were harvested and analyzed as described in the section Rapid quantification of autophagosomes by flow cytometry.
Quantification of viral titers
A549 and A549 ATG5 KO cells in 24-well plates were pre-treated for 2 h with different concentrations of CX-4945 and controls as indicated and infected with MeV or IAV at a MOI of 0.1. Cells were washed twice with PBS 2.5 h or 5 h after infection with IAV and MeV respectively and treated with Medium supplemented with CX-4945 and controls. 24 h post infection Supernatant of IAV infected cells was frozen to −80°C and cells were harvested in TM-lysis buffer and the lysates prepared for western Blot analysis. 48 h post infection MeV infected cells were lysed in the supernatant by freezing at −80°C. The supernatants were cleared prior to further analysis by centrifugation (10 min at 4°C 14,000 rpm).
IAV infected cell supernatants were subjected to TCID50 analysis. A549 cells were seeded in 96-well plates and cultured overnight. The cells were washed twice with PBS, infected with a 10-fold dilution series of the IAV-infected supernatants diluted in DMEM and 1 h post infection DMEM supplemented with 2% FBS was added. 72 h post infection CPE was monitored for all samples and the TCID50 calculated.
MeV infected supernatants were subjected to foci forming units assay. Vero cells were seeded in DMEMxxx 3 h before infection with a 10-fold dilution series of the MeV-infected supernatants. 72 h post infection GFP-positive foci were counted and the foci forming units per mL calculated.
Analyzing protein phosphorylation status by phos-tag SDS-PAGE
Whole cell lysate were generated from transfected HEK293T cells as described in the section whole cell lysates and immunoblotting. SDS-PAGE, blotting and analysis was performed as described in the section whole cell lysates and immunoblotting using Phos-tag Acrylamide gels (Fujifilm Wako Chemicals, AAL-107). In Phos-tag gels the retention time of phosphorylated proteins is increased, separating them from non-phosphorylated proteins during gel electrophoresis.
Staining total protein in gels by silver staining
Whole cell lysate was generated from transfected HEK293T cells and SDS-PAGE was performed as described in the section whole cell lysates and immunoblotting. The gel was stained for total protein using the SilverQuest Silver Staining Kit (Invitrogen, LC6070) as per instructions. Images were taken using a BIO-RAD Gel Doc XR+ and the Image Lab software. Image processing was performed using the ImageJ software.
Proximity ligation assay (PLA)
PLA for endogenous and overexpressed proteins was performed as previously described [51,55]. In brief, confluent HeLa cells were transiently transfected with plasmids coding for TRIM2, TRIM3 and TRIM71 harboring a FLAG tag and CSNK2A1, CSNK2A2 and CSNK2B harboring a V5 tag. After 48 h the cells were fixed with 3.7% formaldehyde, permeabilized in 0.5% Triton and blocked with 5% BSA. Primary antibody staining was performed for 1.5 h at 37°C. Primary antibodies used: Monoclonal mouse anti-FLAG-tag, M2 (Sigma-Aldrich, F1804; 1:400), Monoclonal rabbit anti-V5-tag (D3H8Q; Cell Signaling Technology, 13202S; 1:600), TRIM3 (Santa Cruz Biotechnology, sc-136363; 1:1000), CSNK2A (Cell Signaling Technology, 2656S; 1:1000). PLA staining was performed according to the manufacturers protocol for the Duolink PLA Fluorescence Protocol. PLA Probes and Reagents used: Duolink In Situ PLA Probe Anti-Rabbit PLUS (Sigma-Aldrich, DUO92002), Duolink In Situ PLA Probe Anti-Mouse MINUS (Sigma-Aldrich, DUO92004), Duolink In Situ Detection Reagents FarRed (Sigma-Aldrich, DUO92013).
HIV-1 production
HEK293T and HEK293T ATG5 KO cells were seeded in 48-well F-bottom plate in DMEMxxx and cultured overnight. Cells were treated 2 h pre-transfection with CX-4945 (50–1.56 µM) and torin-1 (1 µM, BioVision, 2353–5). Cells were transfected, using TransIT-LT1 (Mirus, MIR 2306) with the plasmid encoding HIV-1 NL4–3 (pNL4–3, ARP-114) proviral constructs (obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: Human Immunodeficiency Virus 1 (HIV-1), Strain NL4–3 Infectious Molecular Clone (pNL4–3), ARP-2852, contributed by Dr. M. Martin) and the transmitted founder virus CHO77 (kindly provided by the Beatrice Hahn and John Kappes Laboratories [56]). 6 h post transfection the medium was changed. The virus was harvested 24 or 48 h post transfection and transferred onto TZM-bl cells.
TZM-bl reporter assay
To quantify relative infectious virus yield of HIV-1 containing supernatants 10,000 TZM-bl cells per well were seeded in 96-well F-bottom plates in 100 µl DMEMxxx. The next day, the cells were infected with HIV-1 containing supernatants and cultured for 72 h. Then, the supernatants were discarded and 40 µl Gal-Screen β-galactosidase Reporter Gene Assay substrate (Invitrogen, T1028) diluted 1:4 in PBS (Gibco, 14190144) was added to the cells. After incubation at RT for 30 min, 35 µl substrate was transferred to white 96well F-bottom plates and the β-galactosidase signal was measured using an Orion microplate reader.
Cloning and origin of plasmids
A plasmid containing TRIM2 was a kind Gift from Adolfo García-Sastre. The ORF of TRIM2 was subcloned into the pIRES_FLAG vector using NEBuilder HiFi DNA Assembly (NEB, E2621X) according to the manufacturer’s instructions. The insert was amplified by PCR using the Phusion High-Fidelity PCR Kit (NEB, E0553L) according to the manufacturer’s instructions with the primers T2f and T2r purchased from Biomers. The pIRES_FLAG vector was linearized with NotI and MluI restriction enzymes. A cDNA clone of TRIM3 was purchased from SourceBioscience. The ORF of TRIM3 was subcloned into the pIRES_FLAG vector using NEBuilder HiFi DNA Assembly (NEB, E2621X) according to the manufacturer’s instructions. The insert was amplified by PCR using the Phusion High-Fidelity PCR Kit (NEB, E0553L) according to the manufacturer’s instructions with the primers T3f and T3r purchased from Biomers. The pIRES_FLAG vector was linearized with NotI and MluI restriction enzymes. pMXS-hs-3×HA-LIN-41 was a gift from Shinya Yamanaka (Addgene, 52717184). The ORF of TRIM71 was subcloned into the pIRES_FLAG vector using NEBuilder HiFi DNA Assembly (NEB, E2621X) according to the manufacturer’s instructions. The insert was amplified by PCR using the Phusion High-Fidelity PCR Kit (NEB, E0553L) according to the manufacturer’s instructions with the primers T71f and T71r purchased from Biomers. The pIRES_FLAG vector was linearized with NotI and MluI restriction enzymes. pEGFP-N1_hTRIM32 was a gift from Martin Dorf (Addgene, 6954147). The ORF of TRIM32 was subcloned into the pIRES_FLAG vector using NEBuilder HiFi DNA Assembly (NEB, E2621X) according to the manufacturer’s instructions. The insert was amplified by PCR using the Phusion High-Fidelity PCR Kit (NEB, E0553L) according to the manufacturer’s instructions with the primers T32f and T32r purchased from Biomers. The pIRES_FLAG vector was linearized with NotI and MluI restriction enzymes. A plasmid containing the transcript variant 2 of TRIM45 was a kind Gift from Walther Mothes. The ORF of TRIM45 was subcloned into the pIRES_FLAG vector using NEBuilder HiFi DNA Assembly (NEB, E2621X) according to the manufacturer’s instructions. The insert was amplified by PCR using the Phusion High-Fidelity PCR Kit (NEB, E0553L) according to the manufacturer’s instructions with the primers T45f and T45r purchased from Biomers. The pIRES_FLAG vector was linearized with NotI and MluI restriction enzymes. A plasmid containing TRIM56 was a kind Gift from Walther Mothes. The ORF of TRIM56 was subcloned into the pIRES_FLAG vector using NEBuilder HiFi DNA Assembly (NEB, E2621X) according to the manufacturer’s instructions. The insert was amplified by PCR using the Phusion High-Fidelity PCR Kit (NEB, E0553L) according to the manufacturer’s instructions with the primers T56f and T56r purchased from Biomers. The pIRES_FLAG vector was linearized with NotI and MluI restriction enzymes. pIRES_FLAG-TRIM2ΔRING was generated using pIRES_FLAG_TRIM2 as a template. The truncated insert was amplified by PCR using the Phusion High-Fidelity PCR Kit (NEB, E0553L) according to the manufacturer’s instructions with the primers T2dRf and T2dRr purchased from Biomers. The insert was digested with XhoI and MluI restriction enzymes. The pIRES_FLAG vector was linearized with XhoI and MluI restriction enzymes. The insert and linearized vector were ligated using the DNA Ligation Kit Ver.2.1 (Takara, 6022) according to the manufacturer’s instructions. pIRES_TRIM3ΔRING-FLAG was generated using pIRES_TRIM3_FLAG as a template and NEBuilder HiFi DNA Assembly (NEB, E2621X) according to the manufacturer’s instructions. The truncated insert was amplified by PCR using the Phusion High-Fidelity PCR Kit (NEB, E0553L) according to the manufacturer’s instructions with the primers T3dRf and T3dRr purchased from Biomers. The pIRES_FLAG vector was linearized with NotI and MluI restriction enzymes. pIRES_TRIM3ΔBBox-FLAG was generated using pIRES_TRIM3_FLAG as a template and Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) according to the manufacturer’s instructions. The truncated vector was amplified by PCR using the primers T3dBf and T3dBr purchased from Biomers. pIRES_TRIM3ΔCCD-FLAG was generated using pIRES_TRIM3_FLAG as a template and Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) according to the manufacturer’s instructions. The truncated vector was amplified by PCR using the primers T3dCf and T3dCr purchased from Biomers. pIRES_TRIM3ΔFLN-FLAG was generated using pIRES_TRIM3_FLAG as a template and Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) according to the manufacturer’s instructions. The truncated vector was amplified by PCR using the primers T3dFf and T3dFr purchased from Biomers. pIRES_TRIM3ΔNHL-FLAG was generated using pIRES_TRIM3_FLAG as a template and Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) according to the manufacturer’s instructions. The truncated vector was amplified by PCR using the primers T3dNf and T3dNr purchased from Biomers. pIRES_TRIM3ΔFilNHL-FLAG was generated using pIRES_TRIM3_FLAG as a template and Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) according to the manufacturer’s instructions. The truncated vector was amplified by PCR using the primers T3dFNf and T3dFNr purchased from Biomers. pIRES_TRIM3ΔTripartite-FLAG was generated using pIRES_TRIM3_FLAG as a template and Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) according to the manufacturer’s instructions. The truncated vector was amplified by PCR using the primers T3dTf and T3dTr purchased from Biomers. pIRES_TRIM3ΔC25A-FLAG was generated using pIRES_TRIM3_FLAG as a template and Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) according to the manufacturer’s instructions. The truncated vector was amplified by PCR using the primers T3dCf and T3dCr purchased from Biomers. pZW16 (CSNK2A2) was a gift from David Litchfield (Addgene, 27087177). The ORF of CSNK2A2 was subcloned into the pIRES_HA vector using NEBuilder HiFi DNA Assembly (NEB, E2621X) according to the manufacturer’s instructions. The insert was amplified by PCR using the Phusion High-Fidelity PCR Kit (NEB, E0553L) according to the manufacturer’s instructions with the primers CSNK2a’f and CSNK2a’r purchased from Biomers. The pIRES_HA vector was linearized with NotI and MluI restriction enzymes. pRS2 (CSNK2A2, CSNK2B) was a gift from David Litchfield (Addgene, 27093177). The ORF of CSNK2B was subcloned into the pIRES_V5 vector using NEBuilder HiFi DNA Assembly (NEB, E2621X) according to the manufacturer’s instructions. The insert was amplified by PCR using the Phusion High-Fidelity PCR Kit (NEB, E0553L) according to the manufacturer’s instructions with the primers CSNK2bf and CSNK2br purchased from Biomers. The pIRES_V5 vector was linearized with NotI and MluI restriction enzymes. pTwist_3×FLAG_optTRIM3 was purchased from Twist Biosciences. The T40A mutation was introduced using the Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) according to the manufacturer’s instructions. The PCR was performed with the primers T3-40f and T3-40 r purchased from Biomers. pTwist_3×FLAG_optTRIM3 was purchased from Twist Biosciences. The S83A S84A mutation was introduced using the Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) according to the manufacturer’s instructions. The PCR was performed with the primers T3-83f and T3-83 r purchased from Biomers. pTwist_3×FLAG_optTRIM3 was purchased from Twist Biosciences. The S661A mutation was introduced using the Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) according to the manufacturer’s instructions. The PCR was performed with the primers T3-661f and T3-661 r purchased from Biomers. All primers are listed in Table 1.
Table 1.
Oligonucleotides for cloning.
| Name | Sequence |
|---|---|
| CSNK2a’f | 5’-AGC TAG CGC CCT CGA GGC CAC CAT GCC CGG CCC GGC-3’ |
| CSNK2a’r | 5’-GGC GGA ATT GGG CTA GAG CGG CCG CTT AAG CGT AAT CTG GAA CGT CAT ATG GAT AGG ATC CTG CAT AGT CCG GGA CGT CAT AGG GAT AGC CCG CAT AGT CAG GAA CAT CGT ATG GGT ATC GTG CTG CCG TGA GAC CA-3’ |
| CSNK2bf | 5’-GGC AGC TAG CGC CCT CGA GGC CAC CAT GGG TAA GCC TAT CCC TAA CCC TCT CCT CGG TCT CGA TTC TAC GGG CGG CGG CGG GAG CGG AGG AGG TGG AAG CAG CAG CTC AGA GGA GGT GT-3’ |
| CSNK2br | 5’-GGG CGG AAT TGG GCT AGA GCG GCC GCT CAG CGA ATC GTC TTG ACT-3’ |
| T2dRf | 5’-GAG CTC GAG GCC ACC ATG TGT CTG CAC ACT TTC TGC GAG AGG-3’ |
| T2dRr | 5’-CTC ACG CGT TTA CTG TAA GTA TCG ATA G-3’ |
| T2f | 5’-GCT CTT AAG GCA GCT AGC GCC CTC GAG GCC ACC ATG GAC TAC AAG GAC GAC GAT GAC AAG ACG CGT GCA TCT AGA CAC AGG AGT GGC CGT TAT G-3’ |
| T2r | 5’-GGG GCG GAA TTG GGC TAG AGC GGC CGC TCA CTG TAA GTA TCG ATA GAC TTT GAA ACA G-3’ |
| T32f | 5’-GCT CTT AAG GCA GCT AGC GCC CTC GAG GCC ACC ATG GCT GCA GCA GC-3’ |
| T32r | 5’-CTT GTA GTC TCT AGA TGC ACG CGT TGG GGT GGA ATA TCT TC-3’ |
| T3-40f | 5’-GTG CCT GCA CGC CTT CTG CGA-3’ |
| T3-40 r | 5’-GGC AGC ACC TTA GGA CAC TG-3’ |
| T3-661f | 5’-CAA GTT CGG CGC CCA TGG CGA AG-3’ |
| T3-661 r | 5’-AAC AGG AAC TCG CCA TCA-3’ |
| T3-83f | 5’-TTT CTT CAT CGC CGC CCT GAT GGA AGC CAT GCA GC-3’ |
| T3-83 r | 5’-TTG TTT TGC AGG GCG CTT-3’ |
| T3dBf | 5’-AGG GAT GTG GTG GAG CAG-3’ |
| T3dBr | 5’-AGC CAC TAC ACT GAG GGG-3’ |
| T3dCf | 5’-ACC ATT TGT GGG GCC AAA C-3’ |
| T3dCf | 5’-ATG CAG CAT CGC CCT GGA TCG G-3’ |
| T3dCr | 5’-CCT CAG CAG CAC TGT GCC-3’ |
| T3dCr | 5’-ACC AGG AAC TGC TTG TCC-3’ |
| T3dFf | 5’-CTG CGT CCG GGG GAC CTG-3’ |
| T3dFr | 5’-GAG CAG TGC GCC CAG ATT G-3’ |
| T3dFNf | 5’-ACG CGT GCA TCT AGA GAC-3’ |
| T3dFNr | 5’-GAC GAG CTC ATC CTC AAT TGG G-3’ |
| T3dNf | 5’-ACG CGT GCA TCT AGA GAC-3’ |
| T3dNr | 5’-GAC GAG CTC ATC CTC AAT TGG G-3’ |
| T3dRf | 5’-GCT CTT AAG GCA GCT AGC GCC CTC GAG GCC ACC ATG GAG AGA TGT CTC C-3’ |
| T3dRr | 5’-CCT TGT AGT CTC TAG ATG CAC GCG TCT GGA GGT AGC GAT AGG C-3’ |
| T3dTf | 5’-ACC ACG AGC GCC ACT GCA-3’ |
| T3dTr | 5’-TGC CAT GGT GGC CTC GAG-3’ |
| T3f | 5’-GCT CTT AAG GCA GCT AGC GCC CTC GAG GCC ACC ATG GCA AAG AGG GAG G-3’ |
| T3r | 5’-CCT TGT AGT CTC TAG ATG CAC GCG TCT GGA GGT AGC GAT AGG C-3’ |
| T45f | 5’-AGC TCT TAA GGC AGC TAG CGC CCT CGA GGC CAC CAT GTC AGA AAA C-3’ |
| T45r | 5’-GTA GTC TCT AGA TGC ACG CGT GAG AGC CAC AGT CCT AAG-3’ |
| T56f | 5’-TCT TAA GGC AGC TAG CGC CCT CGA GGC CAC CAT GGT TTC CCA CGG GT-3’ |
| T56r | 5’-TCC TTG TAG TCT CTA GAT GCA CGC GTA CTG TCC GGA GAA CGG A-3’ |
| T71f | 5’-GCT CTT AAG GCA GCT AGC GCC CTC GAG GCC ACC ATG GCT TCG TTC CCC G-3’ |
| T71r | 5’-CCT TGT AGT CTC TAG ATG CAC GCG TGA AGA CGA GGA TTC-3’ |
Statistical analyses
Statistical analyses were performed using GraphPad Prism 9. P-values were determined using a two-tailed Student’s t-test with Welch’s correction or One-way ANOVA for multiple comparisons (Mann-Whitney test). Unless otherwise stated, data are shown as the mean of at least three biological replicates ± SEM. Significant differences are indicated as: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Not significant differences are not indicated. Specific statistical parameters are specified in the figure legends.
Supplementary Material
Acknowledgements
We thank Daniela Krnavek, Martha Mayer, Kerstin Regensburger, Regina Burger, Jana-Romana Fischer, Birgit Ott, Meta Volcic and Fabian Zech for assistance and discussions.
Funding Statement
This study was supported by DFG grants to F.K. and K.M.J.S. (CRC 1279 and SPP1923), the BMBF to K.M.J.S. (IMMUNOMOD-01KI2014), and the U.S. National Institutes of Health (NIH) grants R01 AI148534 and R37 AI087846 (to M.U.G). L.K., H.H., V.H. and D.F. are part of the International Graduate School for Molecular Medicine, Ulm (IGradU).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2281128
Data availability statement
The mass spectrometry data generated during this study was deposited at Massive ProteomeExchange with the accession number MSV000091956.
References
- [1].Gatica D, Lahiri V, Klionsky DJ.. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20(3):233–242. doi: 10.1038/s41556-018-0037-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi: 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22(3):367–376. doi: 10.1038/cdd.2014.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].He C, Klionsky DJ. Regulation Mechanisms and Signaling Pathways of Autophagy. Ann Rev Genet. 2009;43(1):67–93. doi: 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xie J, Wang X, Proud CG. mTOR inhibitors in cancer therapy. F1000Res. 2016;(5):F1000. Faculty Rev-2078 10.12688/f1000research.9207.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Koch S, Capaldo CT, Hilgarth RS, et al. Protein kinase CK2 is a critical regulator of epithelial homeostasis in chronic intestinal inflammation. Mucosal Immunol. 2013;6(1):136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Faust M, Montenarh M. Subcellular localization of protein kinase CK2. Cell Tissue Res. 2000;301(3):329–340. doi: 10.1007/s004410000256 [DOI] [PubMed] [Google Scholar]
- [8].Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17(3):349–368. doi: 10.1096/fj.02-0473rev [DOI] [PubMed] [Google Scholar]
- [9].Koepke L, Gack MU, Sparrer KM. The antiviral activities of TRIM proteins. Curr Opin Microbiol. 2021;59:50–57. doi: 10.1016/j.mib.2020.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sparrer KMJ, Gack MU. TRIM proteins: new players in virus-induced autophagy. PLOS Pathog. 2018;14:e1006787. doi: 10.1371/journal.ppat.1006787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].van Gent M, Sparrer KMJ, Gack MU. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu Rev Virol. 2018;5:385–405. doi: 10.1146/annurev-virology-092917-043323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hatakeyama S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem Sci. 2017;42(4):297–311. doi: 10.1016/j.tibs.2017.01.002 [DOI] [PubMed] [Google Scholar]
- [13].Sanchez JG, Chiang JJ, Sparrer KMJ, et al. Mechanism of TRIM25 catalytic activation in the antiviral RIG-I pathway. Cell Rep. 2016;16:1315–1325. doi: 10.1016/j.celrep.2016.06.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sparrer KMJ, Gableske S, Zurenski MA, et al. TRIM23 mediates virus-induced autophagy via activation of TBK1. Nat Microbiol. 2017;2:1543–1557. doi: 10.1038/s41564-017-0017-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Choi Y, Bowman JW, Jung JU. Autophagy during viral infection — a double-edged sword. Nat Rev Microbiol. 2018;16(6):341–354. doi: 10.1038/s41579-018-0003-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hayn M, Hirschenberger M, Koepke L, et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021;35:109126. doi: 10.1016/j.celrep.2021.109126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Koepke L, Hirschenberger M, Hayn M, et al. Manipulation of autophagy by SARS-CoV-2 proteins. Autophagy. 2021;17:2659–2661. doi: 10.1080/15548627.2021.1953847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Beale R, Wise H, Stuart A, et al. A LC3-interacting motif in the influenza a virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe. 2014;15(2):239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Castro-Gonzalez S, Shi Y, Colomer-Lluch M, et al. HIV-1 Nef counteracts autophagy restriction by enhancing the association between BECN1 and its inhibitor BCL2 in a PRKN-dependent manner. Autophagy. 2021;17(2):553–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Koepke L, Winter B, Grenzner A, et al. An improved method for high-throughput quantification of autophagy in mammalian cells. Sci Rep. 2020;10(1):12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pierre F, Chua PC, O’Brien SE, et al. Discovery and SAR of 5-(3-Chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J Med Chem. 2011;54(2):635–654. [DOI] [PubMed] [Google Scholar]
- [22].Oshima T, Niwa Y, Kuwata K, et al. Cell-based screen identifies a new potent and highly selective CK2 inhibitor for modulation of circadian rhythms and cancer cell growth. Sci Adv. 2019;5:eaau9060. doi: 10.1126/sciadv.aau9060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sardiello M, Cairo S, Fontanella B, et al. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol Biol. 2008;8(1):225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Ann Rev Genet. 2009;43(1):67–93. doi: 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–750. doi: 10.1038/ncb2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Esposito D, Dudley-Fraser J, Garza-Garcia A, et al. Divergent self-association properties of paralogous proteins TRIM2 and TRIM3 regulate their E3 ligase activity. Nat Commun. 2022;13(1):7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- [29].Rienzo MD, Piacentini M, Fimia GM. A TRIM32-AMBRA1-ULK1 complex initiates the autophagy response in atrophic muscle cells. Autophagy. 2019;15:1674–1676. doi: 10.1080/15548627.2019.1635385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang R-H, Zhang H-L, Li P-Y, et al. Autophagy is involved in the replication of H9N2 influenza virus via the regulation of oxidative stress in alveolar epithelial cells. Virol J. 2021;18:22. doi: 10.1186/s12985-020-01484-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang R, Zhu Y, Zhao J, et al. Autophagy promotes replication of influenza a virus in vitro. J Virol. 2019;93:e01984–18. doi: 10.1128/JVI.01984-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].BB Olsen, TH Svenstrup, B Guerra. Downregulation of protein kinase CK2 induces autophagic cell death through modulation of the mTOR and MAPK signaling pathways in human glioblastoma cells. Int J Oncol. 2012;41:1967–1976. doi: 10.3892/ijo.2012.1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Su Y-W, Cheng J-J, Huang W-Y, et al. Inhibition of CK2 by CX4945 to induce autophagy-mediated cell death through dephosphorylating acetyl-CoA carboxylase in squamous cell carcinoma of head and neck cancer. JCO. 2017;35(15_suppl):e17503–e17503. [Google Scholar]
- [34].Di Lorenzo G, Iavarone F, Maddaluno M, et al. Phosphorylation of FAM134C by CK2 controls starvation-induced ER-phagy. Sci Adv. 2022;8:eabo1215. doi: 10.1126/sciadv.abo1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Park J-W, Jeong J, Bae Y-S. Protein kinase CK2 is upregulated by calorie restriction and induces autophagy. Mol Cells. 2022;45(3):112–121. doi: 10.14348/molcells.2021.0183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jang DE, Song J, Park J-W, et al. Protein kinase CK2 activates Nrf2 via autophagic degradation of Keap1 and activation of AMPK in human cancer cells. BMB Rep. 2020;53:272–277. doi: 10.5483/BMBRep.2020.53.5.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kim YC, Guan K-L. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32. doi: 10.1172/JCI73939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kaleli HN, Ozer E, Kaya VO, et al. Protein Kinase C Isozymes and Autophagy during Neurodegenerative Disease Progression. Cells. 2020;9(3):553. doi: 10.3390/cells9030553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Johnson JL, Yaron TM, Huntsman EM, et al. An atlas of substrate specificities for the human serine/threonine kinome. Nature. 2023;613(7945):759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mandell MA, Jain A, Arko-Mensah J, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014;30(4):394–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dong W, Luo B, Qiu C, et al. TRIM3 attenuates apoptosis in Parkinson’s disease via activating PI3K/AKT signal pathway. Aging. 2020;13:735–749. doi: 10.18632/aging.202181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Richetta C, Grégoire IP, Verlhac P, et al. Sustained autophagy contributes to measles virus infectivity. PLOS Pathog. 2013;9(9):e1003599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhou A, Zhang W, Dong X, et al. The battle for autophagy between host and influenza a virus. Virulence. 2022;13(1):46–59. doi: 10.1080/21505594.2021.2014680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Killian MS. Dual role of autophagy in HIV-1 replication and pathogenesis. AIDS Res Ther. 2012;9(1):16. doi: 10.1186/1742-6405-9-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Nardacci R, Ciccosanti F, Marsella C, et al. Role of autophagy in HIV infection and pathogenesis. J Intern Med. 2017;281:422–432. doi: 10.1111/joim.12596 [DOI] [PubMed] [Google Scholar]
- [46].Miranda J, Bringas R, Fernandez-de-Cossio J, et al. Targeting CK2 mediated signaling to impair/tackle SARS-CoV-2 infection: a computational biology approach. Mol Med. 2021;27(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Borgo C, D’Amore C, Sarno S, et al. Protein kinase CK2: a potential therapeutic target for diverse human diseases. Sig Transduct Target Ther. 2021;6:1–20. doi: 10.1038/s41392-021-00567-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ramón AC, Pérez GV, Caballero E, et al. Targeting of protein kinase CK2 elicits antiviral activity on bovine coronavirus infection. Viruses. 2022;14:552. doi: 10.3390/v14030552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].D’Amore C, Borgo C, Sarno S, et al. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy – potential clinical relevance. Cell Oncol. 2020;43:1003. doi: 10.1007/s13402-020-00566-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yu J, Boyapati A, Rundell K. Critical role for SV40 small-t antigen in human cell transformation. Virology. 2001;290(2):192–198. doi: 10.1006/viro.2001.1204 [DOI] [PubMed] [Google Scholar]
- [51].Acharya D, Reis R, Volcic M, et al. Actin cytoskeleton remodeling primes RIG-I-like receptor activation. Cell. 2022;185(19):3588–3602.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Braun E, Hotter D, Koepke L, et al. Guanylate-binding proteins 2 and 5 exert broad antiviral activity by inhibiting furin-mediated processing of viral envelope proteins. Cell Rep. 2019;27:2092–2104.e10. doi: 10.1016/j.celrep.2019.04.063 [DOI] [PubMed] [Google Scholar]
- [53].Reuther P, Göpfert K, Dudek AH, et al. Generation of a variety of stable influenza a reporter viruses by genetic engineering of the NS gene segment. Sci Rep. 2015;5(1):11346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kumar M, Michael S, Alvarado-Valverde J, et al. The eukaryotic linear motif resource: 2022 release. Nucleic Acids Res. 2022;50(D1):D497–D508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Volcic M, Sparrer KMJ, Koepke L, et al. Vpu modulates DNA repair to suppress innate sensing and hyper-integration of HIV-1. Nat Microbiol. 2020;5:1247–1261. doi: 10.1038/s41564-020-0753-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ochsenbauer C, Edmonds TG, Ding H, et al. Generation of transmitted/founder HIV-1 infectious Molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol. 2012;86(5):2715–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data generated during this study was deposited at Massive ProteomeExchange with the accession number MSV000091956.