

ABSTRACT
Growing evidence suggests that macroautophagy/autophagy-lysosomal pathway deficits contribute to the accumulation of amyloid-β (Aβ) in Alzheimer disease (AD). Aerobic exercise (AE) has long been investigated as an approach to delay and treat AD, although the exact role and mechanism are not well known. Here, we revealed that AE could reverse autophagy-lysosomal deficits via activation of ADRB2/β2-adrenergic receptor, leading to significant attenuation of amyloid-β pathology in APP-PSEN1/PS1 mice. Molecular mechanism research found that AE could reverse autophagy deficits by upregulating the AMP-activated protein kinase (AMPK)-MTOR (mechanistic target of rapamycin kinase) signaling pathway. Moreover, AE could reverse V-ATPase function by upregulating VMA21 levels. Inhibition of ADRB2 by propranolol (antagonist, 30 μM) blocked AE-attenuated Aβ pathology and cognitive deficits by inhibiting autophagy-lysosomal flux. AE may mitigate AD via many pathways, while ADRB2-VMA21-V-ATPase could improve cognition by enhancing the clearance of Aβ through the autophagy-lysosomal pathway, which also revealed a novel theoretical basis for AE attenuating pathological progression and cognitive deficits in AD.
KEYWORDS: ADRB2, aerobic exercise, Alzheimer disease, amyloid-β, autophagy, V-ATPase
Introduction
Alzheimer disease (AD), the most common form of dementia, is a progressive neurodegenerative disorder. Due to the aging of the world population, more than 55 million people worldwide suffer from dementia, and the number will increase to approximately 78 million by 2050 [1]. Strong evidence shows that the accumulation of amyloid-β (Aβ) is a critical event in the initiation of pathogenesis and neurodegeneration of AD [2]. Autophagy‒lysosome deficits in Aβ contribute to its accumulation and aggregation, which triggers pathological hallmarks, including hyperphosphorylated MAPT/tau, synaptic impairment and degeneration, mitochondrial dysfunction and neuronal loss [2,3]. Mutations in the APP (amyloid beta precursor protein), PSEN1 (presenilin 1), and PSEN2 genes are responsible for familiar forms of AD. However, more than 95% of AD cases are sporadic without a clear cause. Importantly, increasing evidence indicates that impairment of neurocellular Aβ phagocytosis and clearance is a critical prelude to late-onset AD [3]. Therefore, a large number of studies have focused on discovering effective drugs that can promote Aβ clearance, including Aβ antibodies and Aβ secretase inhibitors. However, most of them fail to attenuate cognitive deficits and cause side effects, including neuroinflammation and other neuropathologies [4,5].
Conversely, great efforts have been made to explore effective lifestyles that could attenuate memory impairments, including cognitive training programs, aerobic exercise (AE), diet, etc [6]. These studies have shed new light on AD prevention and treatment [7–9]. AE has long been investigated as a prevention and adjuvant treatment measure for physical and mental disorders, such as cardiovascular disease, obesity, depression, anxiety and dementia. Several studies have suggested beneficial effects of AE intervention on cognitive impairments in patients with AD [10]. Recent meta-analytical evidence has demonstrated that AE or physical activity might be a potential strategy to attenuate cognitive decline in individuals with potential AD as well as affected patients [11,12]. AE or exercise interventions might reduce the levels of AD-associated pathologic biomarkers such as hyperphosphorylated MAPT/tau and amyloid load [13,14]. However, the protective mechanisms of AE in patients remain unclear [15,16].
Autophagy is a principal pathway for the autolysosome degradation of bulk cytoplasmic contents, abnormal protein aggregates, and obsolete or damaged organelles. It is generally activated by physiological and pathological processes and cell stress from abnormal proteins, aggregates, and damaged organelles [17,18]. It also plays a critical role in the degradation of Aβ, and upregulation of autophagy has been shown to reduce Aβ levels in AD [19]. In fact, autophagy dysfunction plays a role in neurodegenerative diseases including AD. Moreover, autophagy dysfunction also likely contributed by reduced lysosomal vacuolar-type H+-translocating ATPase (V-ATPase) and BECN1 (beclin 1) in AD [20,21].
In AD brains, autophagy processes are reduced in neurons, as evidenced by massive accumulation of autophagic vacuoles, including autophagosomes and autolysosomes, within dystrophic neuritis [22]. However, the molecular mechanism is not known. The V-ATPase assembly factor VMA21 is an essential assembly chaperone of V-ATPase, which plays an important role in autophagic vacuolar myopathy and follicular lymphoma [23,24]. VMA21 deficiency raises lysosomal pH, which induces lysosomal degradative ability deficits and inhibits autophagy. V-ATPase activity is the key condition for autolysosome acidification. Importantly, decreased V-ATPase activity results in Aβ accumulation within enlarged deacidified autolysosomes, forming senile plaques in the brains of AD mouse models [20]. However, the mechanism of decreased V-ATPase activity, especially in AD, is rarely reported.
In this study, we revealed that AE could reverse autophagy-lysosomal deficits, leading to significant attenuation of Aβ pathology and cognitive impairments via ADRB2-mediated V-ATPase assembly factor VMA21 signaling. AE could reverse V-ATPase function by upregulating VMA21 levels, accompanied by inhibition of MTOR (mechanistic target of rapamycin kinase) signaling. Inhibition of ADRB2 by propranolol (prop) (antagonist, 10 μM) blocked AE-attenuated Aβ pathology and cognitive deficits by inhibiting autophagy-lysosomal flux. Our findings suggested that AE could attenuate Aβ pathology by upregulating the ADRB2-VMA21-V-ATPase pathway, which also revealed a novel theoretical basis for AE attenuating pathological progression and cognitive deficits in AD.
Results
AE ameliorated learning and memory deficits in AD mouse models
After AE, the mice were subjected to behavioral tests for learning and memory. First, we measured the spatial learning and memory of mice through a Morris water maze (Figure 1A). APP (amyloid beta precursor protein)-PSEN1/PS1 mice showed learning and memory impairments, as reflected by a longer escape latency in the training trials (3–5 days) than WT mice, whereas AE effectively reduced the escape latency of AD mice (Figure 1B,C). In addition, the AE-induced improvement in cognitive deficits was also observed in the probe trial by removing the platform on day 6 (Figure 1D–F). Compared with the APP-PSEN1 mice, the AE mice took a shorter path Figure 1D) and had a shorter latency (Figure 1E) to probe the platform region, with multiple platform crossings (Figure 1F). The swimming behavior (speed) was similar among these groups (Figure 1G,H). In addition, we further refined the analysis by contextual fear conditioning for associated memory, which involves the hippocampus and amygdala. We observed that AE effectively increased the freezing response of APP-PSEN1 mice in retrieval tests at 24 h (recent memory) (Figure 1J,K). The increased freezing response lasted for 6 days (remote memory) (Figure 1L,M). These data indicated that AE may ameliorate learning and memory deficits in APP-PSEN1 mice.
Figure 1.
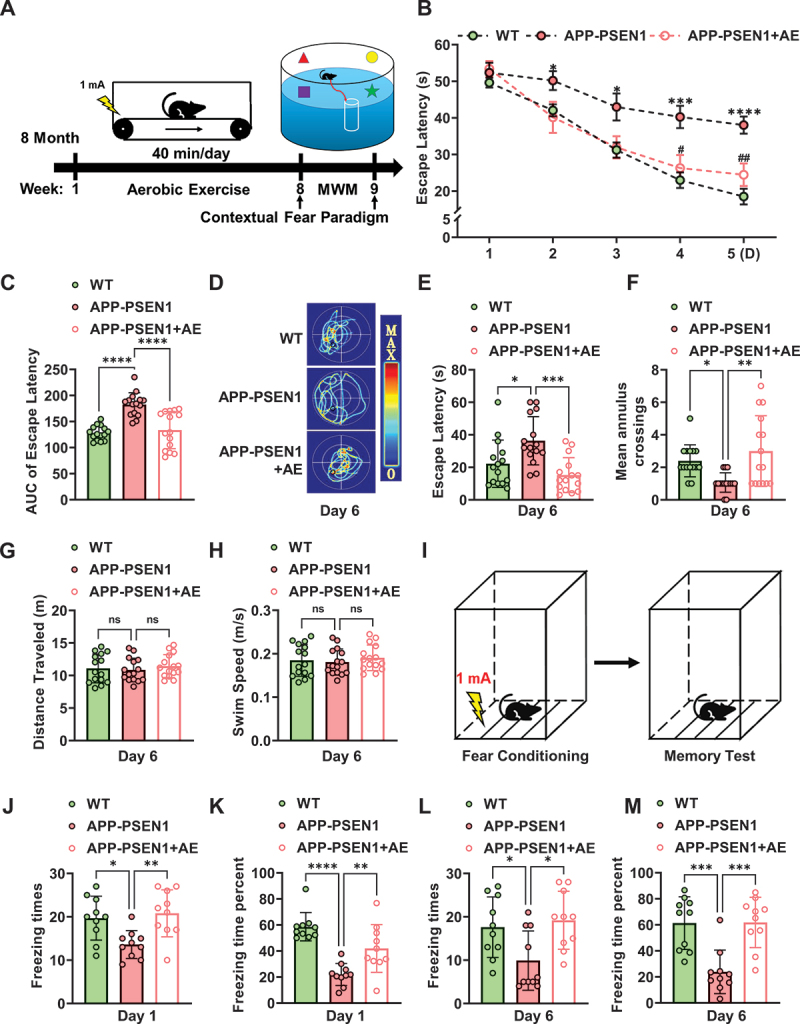
Aerobic exercise ameliorates cognitive dysfunction in APP-PSEN1 mice. (A) schematic of the experimental procedure for the AE and behavioral tests. (B) escape latency to the hidden platform between days 1 and 5. (C) area under the curve (AUC) of fig. 1B. (D) swimming pathway traveled to locate the platform on day 6. (E) the escape latency, (F) number of crossings of the original position of the platform, (G) traveled distance and (H) swimming speed on day 6. (I) schematic of contextual fear freezing test methods. (J-M) effect of AE on the foot shock-induced contextual fear freezing test. n = 15 mice per group in MWM test and n = 10 mice per group in contextual fear freezing test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. The data are presented as the means ± SEMs. Two-way repeated-measures ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (B), and one-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (C-M).
AE alleviated Aβ pathology-independent Aβ production
We further investigated whether AE exerts beneficial effects on the alleviation of Aβ pathology, one of the hallmarks of AD. To measure Aβ pathology in APP-PSEN1 mice, brain sections were immunostained with 4G8 (anti-β-amyloid 17–24), and the number of Aβ plaques was quantified. The data showed that AE effectively decreased Aβ accumulation in the hippocampus and cortex of APP-PSEN1 mice (Figure 2A,B). In addition, Aβ plaques were also examined by thioflavin-S staining. Representative images of brain sections showed a significant decrease in Thioflavin-S-reactive deposition after AE treatment in APP-PSEN1 mice (Figure 2A,C). To further analyze the effects of AE on Aβ in the hippocampus of APP-PSEN1 mice. The protein levels of soluble Aβ were also measured by western blotting. We found that AE significantly decreased total Aβ levels in the hippocampus of APP-PSEN1 mice (Figure 2D,E). We further used ELISA to detect insoluble Aβ and found that AE significantly reduced the amount of insoluble Aβ in APP-PSEN1 in the hippocampus and cortex (Figure 2F).
Figure 2.
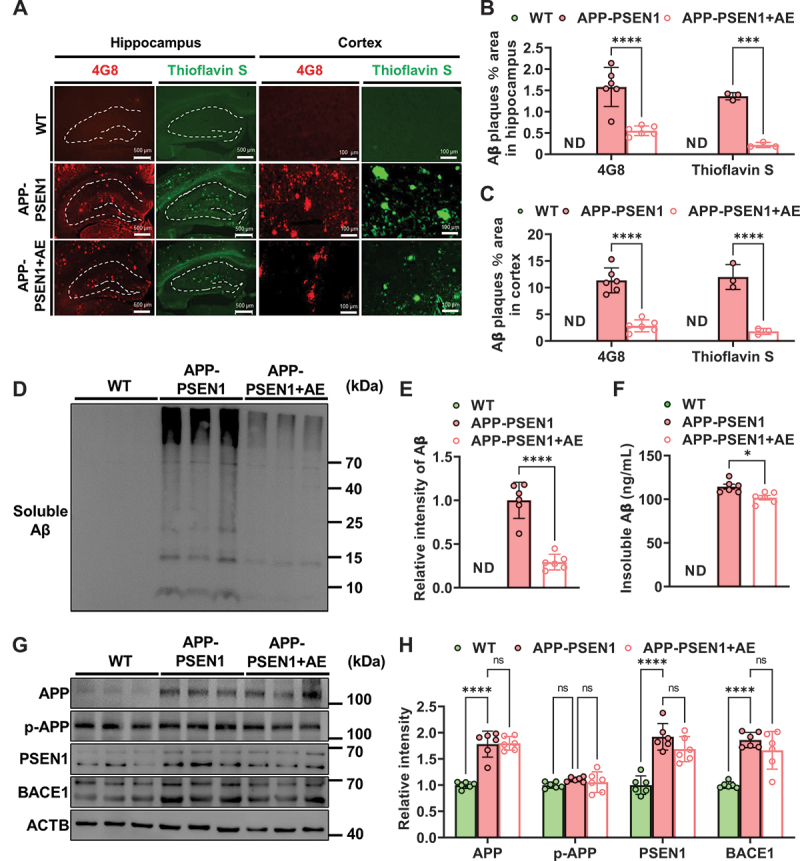
Aerobic exercise ameliorates amyloid-β pathology in APP-PSEN1 mice. (A) Representative images of Aβ plaques by 4G8 staining and thioflavin S staining. (B and C) quantification of the percentage of area occupied by Aβ plaques in the hippocampus and cortex. (D) the amount of soluble Aβ fractions extracted from the hippocampus was examined by western blot analysis. (E) quantification of soluble Aβ intensity using ImageJ. (F) insoluble Aβ detected by ELISA kit. n = 6 mice per group. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. The data are presented as the means ± SEMs. Student’s t tests were used to analyze the data in (B-F), and two-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (H).
Furthermore, we investigated the underlying mechanisms of reduced Aβ pathology by AE. We analyzed the protein levels of APP, p-APP, PSEN1 and BACE1, which are the key factors involved in Aβ production. There was no significant change in the levels of these proteins in the brains of AE mice compared with those of APP-PSEN1 mice (Figure 2G,H). These data suggested that AE alleviated Aβ pathology-independent Aβ production.
AE reversed autophagy-lysosome deficits in APP-PSEN1 mice
Emerging findings suggest that autolysosome acidification decline is closely related to autophagy deficits in AD, which results in lysosome dysfunction and Aβ clearance deficits, eventually leading to extracellular Aβ deposition and intracellular MAPT/tau accumulation [20]. Thus, the autophagy-lysosomal pathway function in APP-PSEN1 mice was detected in the present study. We first examined the effect of AE on the number of LC3-marked autophagic puncta by immunohistochemical staining and found a significant increase in autophagic puncta per cell in the hippocampus and cortex of APP-PSEN1 mice with AE (Figure 3A–C). We further examined the levels of several marker proteins for autophagy by western blotting. In APP-PSEN1 mice, the levels of ATG5, ATG7, and MAP1LC3B/LC3B-II:I in hippocampal extracts all dropped, whereas SQSTM1/p62 increased considerably (Figure 3D,E). However, AE restored the changes in these proteins (Figure 3D,E). The results of electron microscopy were consistent with a decrease in autophagic vesicles in APP-PSEN1 mice and an increase in AE (Figure 3F,G). These data suggested that AE could alleviate Aβ-induced impaired autophagy.
Figure 3.
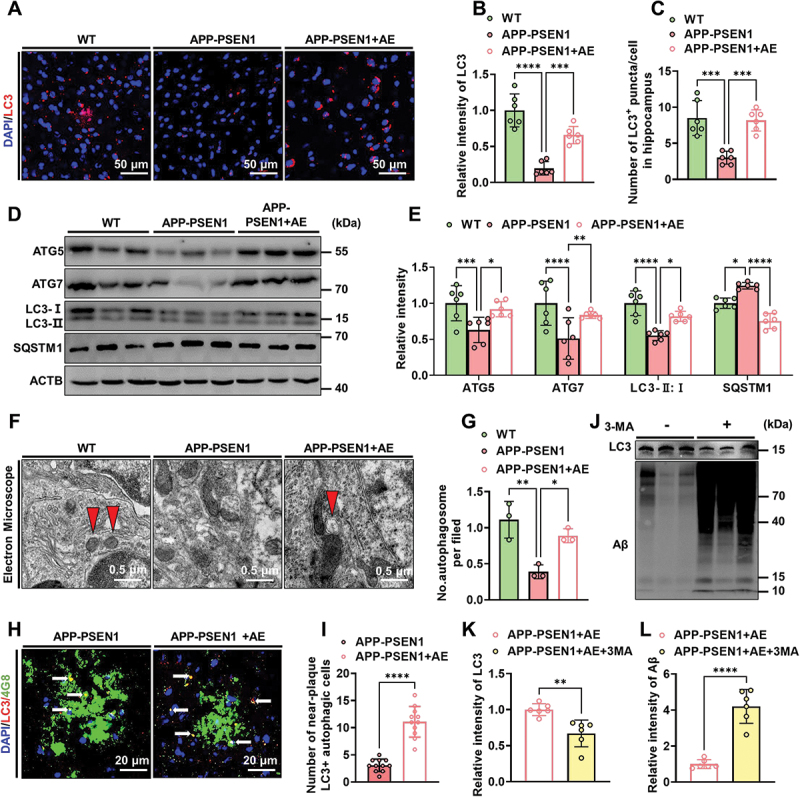
AE enhances autophagy levels in APP-PSEN1 mice. (A) Representative images of the intensity of LC3 in the hippocampus of the mice. (B and C) quantification of LC3 fluorescence intensity and average number of LC3+ puncta per cell in the brain. (D and E) the levels of ATG5, ATG7, LC3-II:I and SQSTM1 in the hippocampus were detected using western blotting and quantitatively analyzed. (F) electron microscopy analysis of autophagosomes. Arrows indicate the autophagosomes (scale bar: 0.5 μm). (G) quantification of autophagosomes. (H) Representative confocal images of LC3 and 4G8 immunofluorescence colabeling. (I) quantification of the number of LC3-positive cells near Aβ plaques. (J-L) Representative images and quantification of the intensity of LC3 and Aβ in the hippocampus of AE mice after 3-MA treatment. n = 6 mice per group. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. The data are presented as the means ± SEMs. One-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (B and C). Two-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (E, G). Student’s t tests were used to analyze the data in (I-L).
To further demonstrate that AE could improve Aβ clearance in AD. We also assessed autophagic flux by analyzing the levels of colocalization of 4G8 and LC3. Consistently, the colocalization of LC3 and 4G8 was significantly increased in AE-treated APP-PSEN1 mice (Figure 3H,I). Furthermore, inhibition of autophagy by 3-methyladenine (3-MA) effectively disturbed the AE-mediated alleviation of Aβ pathology in APP-PSEN1 mice (Figure 3J–L). These findings together indicated that AE ameliorated Aβ pathology by reversing autophagy-lysosome deficits in APP-PSEN1 mice.
AE attenuated autophagy-lysosomal flux deficits by upregulating VMA21 in APP-PSEN1 mice
We further investigated the possible molecular mechanism underlying AE attenuating autophagy-lysosomal flux deficits in APP-PSEN1 mice, and the hippocampus was lysed for mRNA sequence analysis. A volcano plot showing the results of AE versus APP-PSEN1 mice revealed increased transcription in the latter of 252 genes, including Gm43580, which encodes a VMA21-like protein (Figure 4A). AE treatment increased the transcription of Gm43580 (Figure 4A,B). Among them, VMA21 is an essential assembly chaperone of V-ATPase, which plays an important role in autolysosome acidification and autophagy. Therefore, we first measured the mRNA and protein levels of Vma21/VMA21 in the hippocampus of mice. Consistent with the mRNA sequence analysis, the mRNA and protein levels of Vma21/VMA21 were significantly decreased in the hippocampus of APP-PSEN1 mice compared with that of WT mice. However, AE rescued the decreased mRNA and protein levels of Vma21/VMA21 (Figure 4C–E). To further verify the role of decreasing VMA21 in autophagy deficits in AD, we used ELISA kits to assay their V-ATPase activity. V-ATPase activity in the autolysosomes and lysosomes of 8-month-old APP-PSEN1 mice was significantly decreased compared to that of WT mice. AE effectively reversed the decreased V-ATPase activity in APP-PSEN1 mice compared with that in APP-PSEN1 mice (Figure 4F). The decrease in Vma21 mRNA suggested the involvement of transcription. To gain insight into the regulatory mechanism underlying the decrease in Vma21, we screened for the potential binding sites of the Vma21 promoter in the transcription factor binding database [25]. We looked for 2000 bp upstream of the transcription start site of this gene in the NCBI database to obtain a potential promoter sequence for this gene. This promoter sequence was then used in the JASPAR database for the prediction of transcription factors and we found 4 potential transcription factors: EN1, GATA1, RUNX1 and CREB1. We observed that the mRNA and protein levels of Gata1/GATA1 were significantly decreased in the hippocampus of the APP-PSEN1 group compared to the WT group, whereas these changes were mainly restored by AE treatment (Figure 4G). However, the mRNA levels of En1, Runx1 and Creb1 were not significantly changed (Figure 4H–J). Together, these data suggested that the decreased VMA21 might induce autophagy disorder in AD by inducing V-ATPase impairment. AE could increase Vma21 gene transcription via mechanisms involving GATA1 activation.
Figure 4.
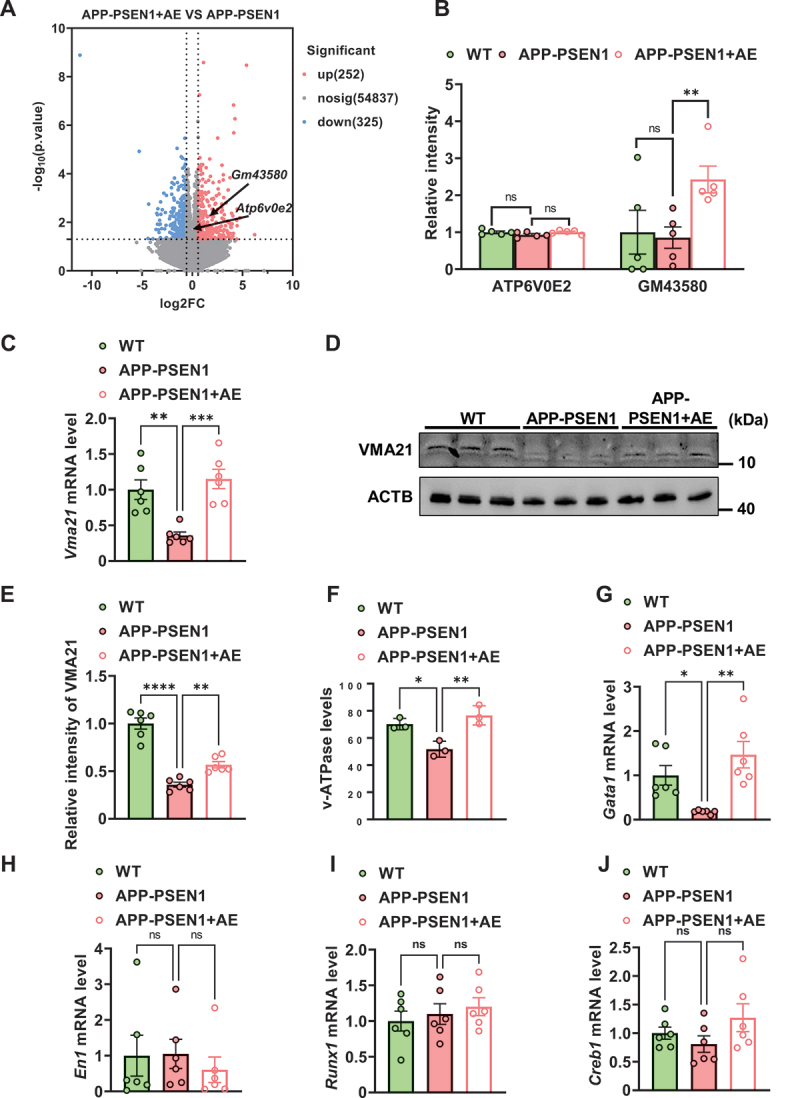
AE attenuates autophagy-lysosomal flux deficits by upregulating VMA21 in APP-PSEN1 mice. (A) volcano plots of AE mice versus APP-PSEN1 mice for autophagy-related proteins: the X-axis represents the ratio between two compared groups, and the Y-axis is the -log10-transformed p value. The red color represents increased expression, and the blue color represents reduced expression. The predicted gene Gm43580 encodes a VMA21-like protein. (B) profile plot of the protein expression of ATP6V0E2 and GM43580. (C-E) the mRNA (C) and protein (D) levels of ADRB2. (F) the level of V-ATPase in each group. (G-J) the mRNA levels of the transcription factors Gata1, En1, Runx1 and Creb1. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. The data are presented as the means ± SEMs. Two-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (B). One-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (C-J).
AE activated ADRB2 in APP-PSEN1 mice
ADRB2 plays a critical role in the regulation of energy metabolism and memory formation and is believed to be involved in AD. Considering the regulation of energy metabolism and noradrenergic systems by AE, we evaluated the activation and expression of ADRB2 in the brains of AD patients and APP-PSEN1 mice. The GSE110298 dataset investigated the relationship between physical activity levels and gene expression patterns in the cognitively-intact aged human hippocampus. This dataset used hippocampus from clinically well-defined cases where late-life physical activity levels (exercise and non-exercise) had been objectively measured on an annual basis. In this dataset, compared with the 11 control subjects (low physical activity) and 12 subjects treated by AE (high physical activity), adrenergic signaling presented upregulation in AE-treated AD patients (Figure 5A). Moreover, in the RNA-seq data from mice, 187 DE proteins overlapped in both APP-PSEN1 vs. WT and AE vs. APP-PSEN1 (Figure 5B), and ADRB2 was associated with 38 of these genes (Figure 5C). The result was confirmed by RT‒PCR and western blotting in APP-PSEN1 mice, as the mRNA and protein levels of Adrb2/ADRB2 were significantly decreased in the hippocampus, while AE reversed these changes (Figure 5D–F). Using the same approach as above, we tried to find transcription factors for ADRB2, and we identified three potential transcription factors for Adrb2: SIX3, SATA6 and IRF1. We found that AE significantly ameliorated the downregulation of Irf1 mRNA level in APP-PSEN1 mice (Figure 5G–I). To verify the role of IRF1 in regulating the expression of Adrb2, we transfected BV2 cells with siIrf1 RNA oligo and subsequently examined the protein content of ADRB2. We found that the ADRB2 level was reduced after silencing Irf1. These data suggested that AE activated ADRB2 through Irf1.
Figure 5.
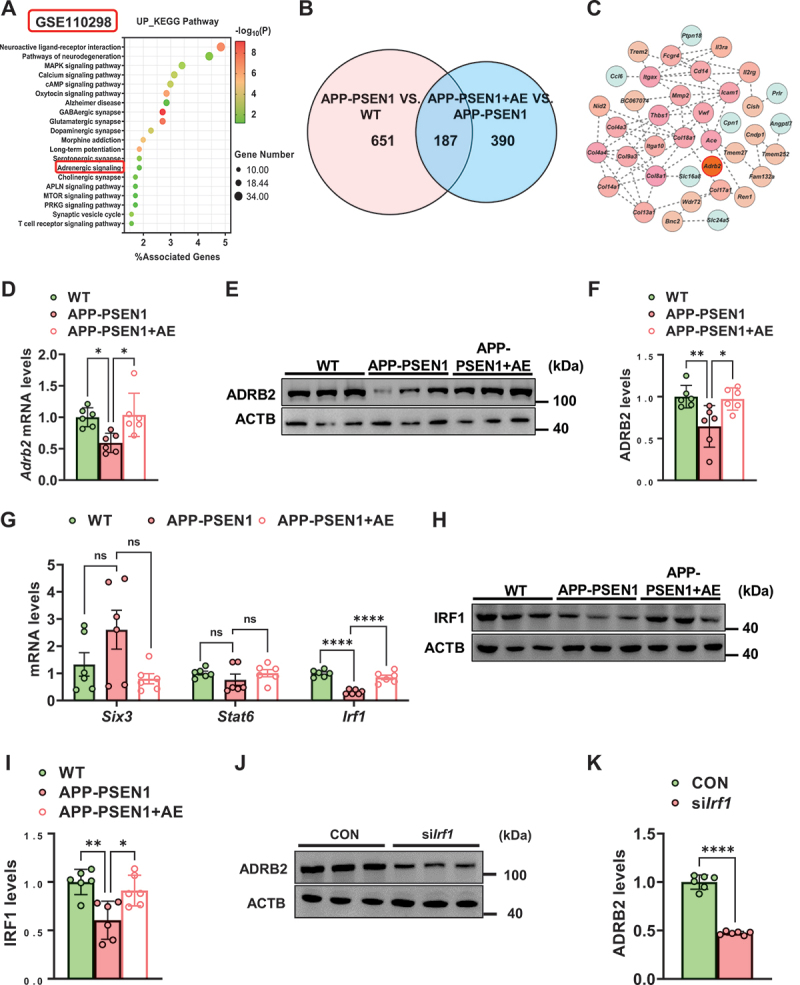
AE activates ADRB2 in APP-PSEN1 mice. (A) top 20 pathways of differentially expressed genes from high physical activity AD patients and low physical activity AD patients. Negative log10 p values are shown. (B and C) 187 overlapping protein changes in both comparison groups and ADRB2 is associated with 38 of these genes (APP-PSEN1 vs. WT and AE vs. APP-PSEN1). (D-F) the mRNA (D) and protein (E) levels of ADRB2. (G) the mRNA levels of the transcription factors Six3, Stat6, and Irf1. (H and I) the protein levels of IRF1. n = 6 mice per group. (J-K) ADRB2 protein levels after silencing Irf1 in BV2 cells. n = 6. *P < 0.05; **P < 0.01; ***P < 0.001 and ****P < 0.0001. The data are presented as the means ± SEMs. One-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (D-J). Student’s t tests were used to analyze the data in (K).
AE ameliorated autophagy impairment by activating MTOR and VMA21 signaling via ADRB2
To investigate whether AE ameliorates autophagy impairment in APP-PSEN1 mice by activating ADRB2, we examined autophagy in each group after propranolol pretreatment. We found that the protein levels of the autophagy-related marker proteins ATG5, ATG7, and LC3-II:I were significantly decreased after propranolol pretreatment relative to those in mice treated with AE alone (Figure 6A,B). In contrast, the protein levels of SQSTM1 were significantly increased (Figure 6A,B). Immunofluorescence results showed that LC3+ cells near Aβ plaques were substantially reduced after propranolol pretreatment (Figure 6C,D). The number of autophagic vesicles was similarly reduced in propranolol-pretreated mice under electron microscopy (Figure 6E,F). To further confirm the direct correlation between activation of ADRB2 and autophagy, we treated N2a cells with the ADRB2-specific agonist terbutaline and introduced the mCherry-GFP-LC3 reporter into N2a cells. mCherry-GFP-LC3 in autolysosomes displayed red fluorescence, as the GFP signal was sensitive to acidic conditions, whereas the mCherry signal was more stable in the lysosome lumen. We observed an increased number of red puncta in the terbutaline-treated N2a cells (Figure 6G,H), indicating increased autophagic flux. To further investigate the roles of ADRB2 in AE-upregulated autophagy function in the brains of AD mice. We also examined classical autophagy upstream signaling pathway-related proteins, including AMP-activated protein kinase (AMPK), MTOR and the corresponding phosphorylated proteins, and we found that the expression of p-PRKAA/AMPK was increased in APP-PSEN1 mice by AE, while that of p-MTOR was decreased (Figure 6I,L). To further investigate the effect of ADRB2 on VMA21, we treated mice and BV2 cells with the ADRB2 inhibitors propranolol and siAdrb2, respectively (Figure 6K,M). We found that propranolol treatment significantly inhibited the effect of AE on the increased expression of VMA21 in APP-PSEN1 mice. Also, in Aβ-treated BV2 cells, we found that the ADRB2 agonist terbutaline ameliorated the reduced Vma21 expression caused by Aβ1–42, whereas this effect was suppressed after siAdrb2 (Figure 6L,N). Together, these results suggested that AE ameliorates autophagy impairment in APP-PSEN1 mice by activating ADRB2 mediates MTOR and VMA21 autophagy signaling.
Figure 6.
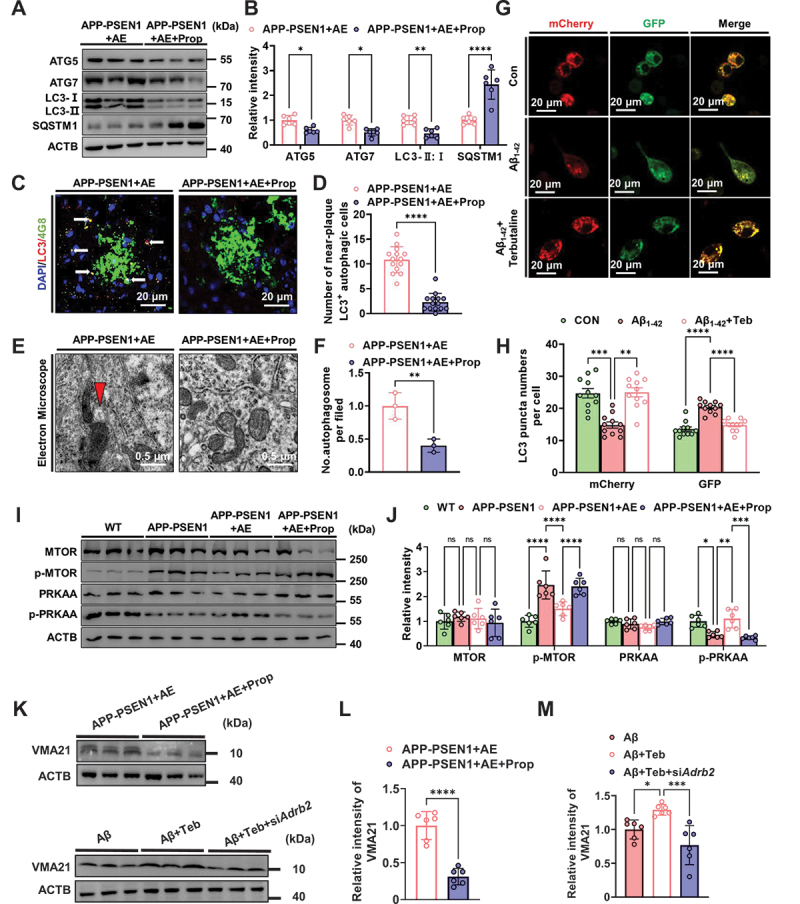
AE ameliorates autophagy impairment in APP-PSEN1 mice by activating ADRB2 mediates MTOR autophagy signaling. (A and B) the levels of ATG5, ATG7, LC3-II:I and SQSTM1/p62 in the hippocampus were detected using western blotting and quantitatively analyzed. (C) Representative confocal images of LC3 and 4G8 immunofluorescence colabeling. (D) quantification of the number of LC3-positive cells near Aβ plaques. (E) electron microscopy analysis of autophagosomes. Arrows indicate the autophagosomes (scale bar: 0.5 μm). (F) quantification of autophagosomes. (G) Representative pictures of autophagic flux and (H) quantification of LC3 puncta in (G). (I-J) the levels of MTOR, p-MTOR, PRKAA and p-PRKAA/AMPK in the hippocampus were detected using western blotting and quantitatively analyzed. (K and L) VMA21 protein levels between the AE and propranolol-inhibited groups. n = 6 mice per group. (K and M) Vma21 protein levels after silencing Adrb2 in BV2 cells. n = 6. *P < 0.05; **P < 0.01; ***P < 0.001 and ****P < 0.0001. The data are presented as the means ± SEMs. Student’s t tests were used to analyze the data in (A-F and L), one-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (H and M) and two-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (J).
The ADRB2 inhibitor propranolol reversed the ameliorative cognitive dysfunction and pathological impairment in APP-PSEN1 mice after AE
To clarify that the ameliorating effect of AE on cognitive impairment as well as pathological damage in APP-PSEN1 mice is exerted through ADRB2, we treated mice with the ADRB2 inhibitor propranolol 1 h before AE. We found that propranolol slightly interfered with the study of AE-treated mice during the learning phase on days 1–5, as evidenced by an increase in the AUC of escape latency (Figure 7A,B). The disruptive effect of propranolol was more pronounced during the 6 days of the testing phase. Propranolol-treated AE mice had a longer path, longer escape latency to find the platform on day 6, and significantly fewer crossings (Figure 7C–E). However, propranolol did not clearly alter the effect of AE on emotional memory in mice and only reduced the freezing times for 24 h (Figure 7F–I). Similarly, we examined the effect of propranolol on pathological changes. The total protein levels of soluble Aβ were similarly increased after propranolol treatment (Figure 7J,K). Consistent with this, the results of 4G8 immunofluorescence staining and thioflavin staining showed that propranolol-pretreated APP-PSEN1 mice had a significant increase in the number of Aβ plaques after AE (Figure 7L,M).
Figure 7.
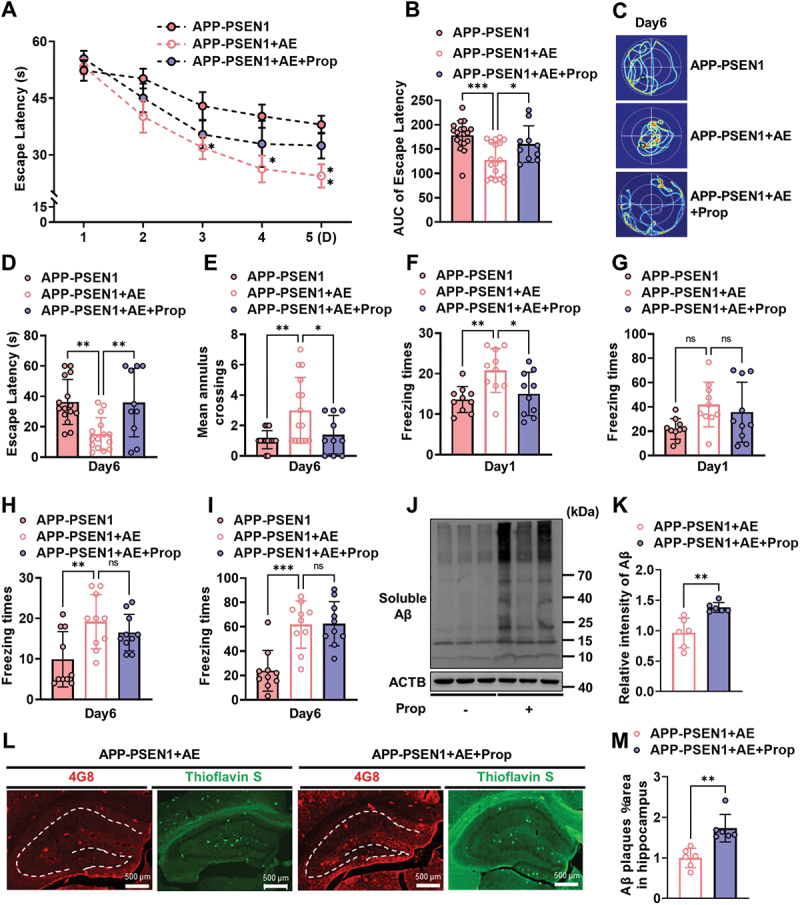
The ADRB2 inhibitor propranolol reverses the ameliorative cognitive dysfunction and pathological impairment in APP-PSEN1 mice after AE. (A and B) escape latency to the hidden platform between days 1 and 5 and the AUC. (C) swimming pathway traveled to locate the platform on day 6. (D) escape latency and (E) number of crossings of the original position of the platform on day 6. (F-I) effect of foot shock-induced contextual fear freezing test. (J and K) the amount of soluble Aβ fractions extracted from the hippocampus was examined by western blot analyses and quantification of soluble Aβ intensity using ImageJ. (L and M) Representative images of Aβ plaques by 4G8 staining and thioflavin staining and quantification of the percentage of area occupied by Aβ plaques in the hippocampus and cortex. n = 6 mice per group. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. The data are presented as the means ± SEMs. Two-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (A), one-way ANOVA followed by Bonferroni’s post hoc test was used to analyze the data in (B-I), and Student’s t tests were used to analyze the data in (K and M).
Discussion
It is recognized that extracellular accumulation of Aβ plays an important role in AD pathological processes and cognition deficits. Accordingly, there are two main aspects of research: one is how to reduce the generation of Aβ, and the other is how to improve the clearance of Aβ. Autophagy has been reported to be involved in the clearance of Aβ, and autophagy deficits are not only an important pathophysiological feature of AD but also play a key role in Aβ accumulation due to clearance deficiency in the brain [26]. In AD, the upstream signaling pathways of the autophagy pathway (e.g., MTOR signaling pathway) are inhibited, thereby suppressing the normal physiological processes of the autophagy pathway [27,28]. Several articles have shown that the number of autophagosomes is significantly reduced in AD brains with structural damage and that activating autophagy is a potential way to improve AD [29,30]. In transgenic mice that express human APP, a model for AD, genetic reduction of Becn1 expression can increase intraneuronal Aβ accumulation, extracellular Aβ deposition, and neurodegeneration and cause microglial changes and profound neuronal ultrastructural abnormalities [21], in addition to studies showing that mitophagy is also reduced in AD [31]. Recent research finds that autolysosome acidification declines is the critical mechanism of autophagy deficits in neurons of AD, which induces autophagic buildup of Aβ as well as senile plaques. Importantly, further study finds that a decline in V-ATPase activity leads to pH upregulation, which results in marked impairment of autolysosomes in neurons [20]. However, the molecular mechanism underlying autophagic dysfunction, especially autophagic lysosomal dysfunction, in AD is currently unclear.
Lysosomes require an acidic lumen between pH 4.5 and 5.0 for the degradative enzyme to effectively digest large molecules in these compartments. The optimal pH value is mainly maintained by proton (H+) influx generated by V-ATPase. Structurally, V-ATPase is a rotating nanomotor composed of multiple subunits, each with multiple isomers [32]. The different demands of intracellular vesicles and the extracellular environment for acidification drive the function and regulation of V-ATPase. The subunits are arranged in two structural domains: the peripheral V1 domain responsible for ATP hydrolysis and the transmembrane domain V0 which plays a role in proton transport. In mammals, the V1 domain has at least eight different subunits (ATP6V1A, ATP6V1B, ATP6V1C, ATP6V1D, ATP6V1E, ATP6V1F, ATP6V1G, and ATP6V1H along with various isoforms), while the V0 domain is composed of up to six different subunits (ATP6V0A, ATP6V0B, ATP6V0C, ATP6V0D, ATP6V0E and certain isoforms) [33,34]. Indeed, the expression and isoform localization of subunits are crucial for the function of V-ATPase. The activity of V-ATPase is controlled by reversible dissociation of the active whole enzyme into its components V1 and V0 domains, which are inactive, occurring in cells and yeast in response to different environments [35].
In addition, VMA21 plays an essential assembly chaperone role in the formation of V-ATPase. The VMA21 gene encodes a small transmembrane chaperone protein composed of 101 amino acids. Topologically, the N and C terminals are cytoplasmic, and the two transmembrane fragments are connected through the lumen (ER, Golgi apparatus, lysosome) domains. At present, it is believed that VMA21 first binds to the V0 subunit on the endoplasmic reticulum, then transports V0 to the Golgi apparatus and assembles V-ATPase with the V1 subunit. VMA21 is then wrapped by vesicles and returned to the endoplasmic reticulum for a new round of assembly and adjustment [36]. Here, we observed autophagy deficits accompanied by a decrease in V-ATPase activity in the brains of APP-PSEN1 mice. Further study confirmed that VMA21 was also significantly reduced. These results suggested that VMA21 May be the key to causing autophagy disorder by inhibiting the formation of V-ATPase.
AE is any physical activity that involves an increased amount of oxygen throughout the body. Studies have demonstrated that AE can help to improve heart and lung function, increase circulation, and decrease high blood pressure (hypertension), inflammation and cholesterol. Of note, there is growing evidence to support the beneficial effects of AE on neurodegenerative diseases such as AD. Meta-analyses have indicated that regular exercise (particularly AE) might be a more effective strategy to ameliorate neurodegeneration and cognitive function, or at least delay neuropathology changes and cognitive decline, in AD patients as well as old individuals. Studies have reported that AE plays a regulatory role in neurogenesis, inflammation, and oxidative stress, all of which play varying roles in the mitigation of AD [37]. Recent studies have shown that greater physical activity is associated with slower Aβ pathology and Aβ-related cognitive decline [38,39]. However, the effects and mechanism of AE on Aβ accumulation and neurodegeneration in AD progression are unclear. In particular, the effects of AE on Aβ clearance by autophagy are rarely reported. We found that AE can reduce the accumulation of pathological Aβ mainly by upregulating autophagy, while the effects of AE on the production process of Aβ were not significant. We believe that this effect may be determined by the expression characteristics of APP and PSEN1 in APP-PSEN1 mice, which are completely different from the mechanism of Aβ generation in sporadic AD patients.
To further clarify the mechanism by which aerobic exercise reverses autophagy disorder in AD, we combined sequencing analysis of AD patients (GSE110298) and identified the potential regulatory receptor ADRB2 of AE. ADRB2 is a prototypical member of the GPCR family of proteins and plays several important roles in physiological systems, including cardiovascular and pulmonary systems. Moreover, ADRB2 is implicated in synaptic plasticity, hemodynamics, and neuroinflammation. ADRB2 in the cortex and hippocampus is critical for learning and memory [40]. Notably, activation of ADRB2 May prevent the impairment of hippocampal LTP by Aβ via the cAMP-PRKA/PKA signaling pathway [41]. Recently, a retrospective inception cohort study finds that the risk of developing AD is elevated among patients exposed to nonselective ADRB antagonists. In contrast, selective ADRB2 agonist exposure is associated with a lower risk of developing AD [42]. Moreover, it has been found that activation of ADRB2 could prevent Aβ-induced microglial inflammation in the brain [43].
Based on our research, we speculate that ADRB2 can be a regulatory receptor for AE, and the downstream pathway may be involved in regulating the autophagy-lysosome pathway. Therefore, we tested the classic autophagy regulatory MTOR pathway and found that AE can effectively inhibit the MTOR pathway. Correspondingly, the use of the ADRB2-specific inhibitor propranolol could significantly reduce AE-induced inhibition of the MTOR pathway and improve autophagy disorders. Taken together, in addition to regulating neurogenesis, inflammation and oxidative stress [37], AE could also mitigate AD by ameliorating autolysosome defects and enhancing Aβ clearance through the ADRB2-VMA21-V-ATPase pathway.
Notably, Nixon’s pioneering work reveals that reduced V-ATPase activity in the AD mouse model leads to decreased lysosomal acidification, resulting in the formation of senile plaques [20]. Meanwhile, it is discerned that phosphorylated APP-βCTF and PSEN1 mutations equally contribute to the reduction of V-ATPase activity, further exacerbating disturbances in the degradation of lysosomal proteins [44,45]. Additionally, β2-adrenergic agonists rescue lysosome acidification and function in PSEN1 deficiency by reversing defective endoplasmic reticulum (ER) to lysosome delivery of CLCN7/CIC-7 (chloride voltage-gated channel 7) [46]. Therefore, our finding that AE via ADRB2-VMA21-V-ATPase enhanced the autophagy-lysosome pathway to clear Aβ may have significant implications for AD treatment.
Here, we want to emphasize that our study focuses on how AE can improve the autophagy-lysosome pathway to clear Aβ, aiming to improve cognition. Aβ is a key pathological feature in both familial and sporadic AD (SAD). Nixon et al. find that lysosomal proteolysis requires PSEN1 and is disrupted by Alzheimer-related PSEN1 mutations, caused by failed PSEN1-dependent targeting of the V-ATPase ATP6V0A1 subunit to lysosomes [45]. It is noteworthy that compromised V-ATPase activity isn’t restricted to familial AD (FAD) alone. Burrinha et al. demonstrates that the reduced expression of the V-ATPase will impact endolysosomal acidification and result in synapse dysfunction in aged wild-type mice [47]. Recent research highlights APOE4 as the most potent genetic risk factor for SAD. Remarkably, brain cells derived from APOE iPSCs display lysosomal defects leading to reduced Aβ clearance [48]. Therefore, we postulated that AE may play a pivotal role in enhancing autophagy-lysosome pathway clearance of Aβ through the ADRB2-VMA21-V-ATPase, which might contribute not only to FAD but also to SAD. However, broader and definitive evidence might necessitate further experimentation in the future.
In summary
Increasing studies suggest that autophagy deficits not only weaken Aβ clearing but also participate in the buildup of senile plaques in AD, while the exact molecular mechanisms are not clear. In the current study, we found that the level of the V-ATPase assembly factor VMA21 was significantly reduced in the brains of AD mice, and it was correlated with a decrease in V-ATPase activity, which regulates lysosomal pH. These data suggested that the decreased VMA21 protein inhibited V-ATPase activity, leading to impaired lysosomal activity and inhibition of autophagy clearance function. VMA21 could be a new autophagy regulatory molecular target in AD (Figure 8). Moreover, AE could activate ADRB2, inhibit the MTOR pathway, regulate VMA21 expression, improve autolysosome disorders, and reduce Aβ accumulation and cognitive impairment (Figure 8). Our findings revealed a novel theoretical basis for AE attenuating pathology progression and cognitive deficits in AD.
Figure 8.
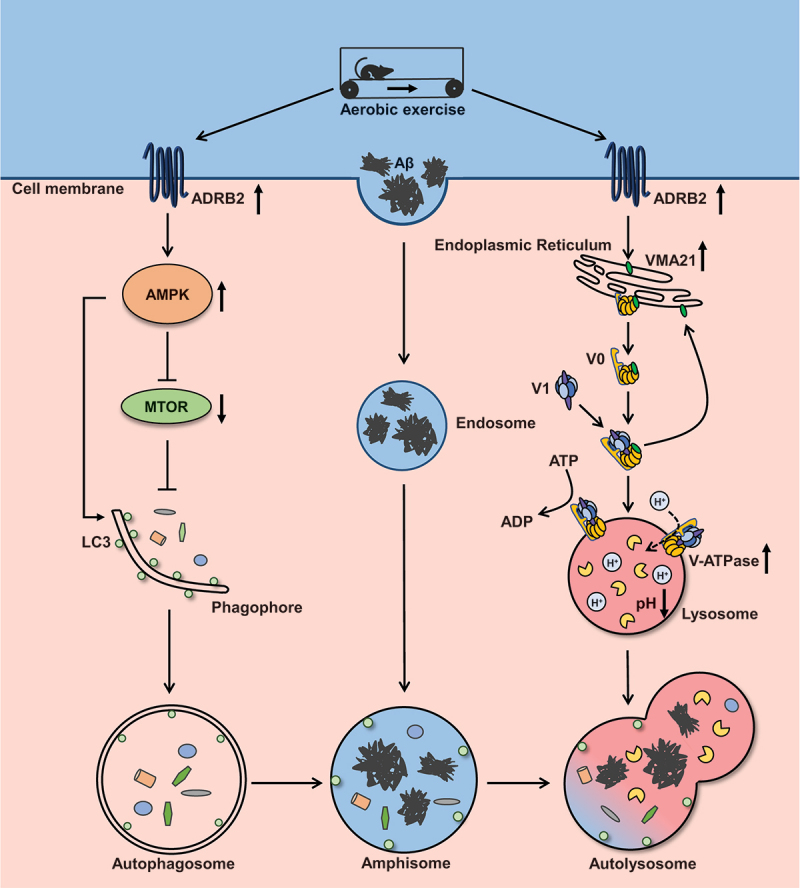
Proposed working model for aerobic exercise attenuates autophagy-lysosomal flux deficits by ADRB2 mediated V-ATPase assembly factor VMA21 signaling and MTOR signaling in Alzheimer disease mice.
Materials and methods
Mice, aerobic exercise, and drug administration
The Animal Ethics Committee of Jiangnan University approved all the animal experimental procedures (JN. No. 20210515c0241130[123]). We used transgenic B6C3-Tg (APPswe, PSEN1dE9) mice (bought from Nanjing University’s Model Animal Central) as AD mouse models and age-matched wild-type (WT) B6C3 mice as controls. All of the mice were 8-months old, half male and half female, and were kept in Jiangnan University’s Experimental Animal Center’s specific-pathogen-free (SPF) environment.
Starting at 8 months of age, mice were run on a six-channel motorized treadmill (TECHMAN, Chengdu, China) for 5 days/week, 40 min/day for 12 weeks[49]. Mice in the aerobic exercise (AE) group were trained on the treadmill at a maximum speed of 15 m/min (6 m/min for 5 min, 9 m/min for 5 min, 12 m/min for 20 min, 15 m/min for 5 min, and 12 m/min for 5 min) with an incline of 0°. 3-Methyladenine (MedChemExpress, HY-19312; autophagy inhibitor) and propranolol (MedChemExpress, HY-B0573B; ADRB2 inhibitor) were administered at a dose of 30 mg/kg by intraperitoneal injection 1 h before the AE [50]. The sedentary mice were transported to the workout facility and subjected to identical circumstances as the AE mice. To guarantee that all mice were able to finish the exercise regimen, animals were evaluated qualitatively during the test.
Behavioral testing
Behavioral testing was carried out as previously described [51] and began at 1 to 5 p.m. on the day the AE experiment were concluded. For the Morris water maze (MWM) test, milk powder was placed in the water in advance until the transparent platform was no longer visible, the water temperature was maintained between 22 and 23°C, and the liquid level was 2 cm higher than the transparent platform. The pool was divided into four quadrants, and the walls of each quadrant were marked with cards of various colors and shapes to help the mice orient themselves. During the first five days of training, mice were trained to find transparent platforms in a water maze, with four trials per day beginning in a different quadrant and proceeding in a random order. Mice were placed in the water maze facing the pool wall from the middle of the quadrant in each trial and had 60 s to find the platform. If the mice did not find the platform within 60 s, they were guided to it to learn for 30 s. The time it took mice to find the platform (escape latency) was recorded in the training section. On the sixth day, mice repeated the test for 60 s with the transparent platform removed, beginning in the contralateral quadrant where the platform was originally located. The entire experiment was captured by a fixed camera above the pool, and connected MWM analysis software (WMT-100S, TECHMAN, China) was used to calculate the escape latency, platform crossing times, target quadrant residence time, and swimming pathway.
The contextual freezing test was conducted between 7 and 9 p.m. on the first and last days of the MWM test. Mice froze intermittently after a previous foot shock when placed in the same environment, and this behavior was associated with contextual memory. The experimental chamber (230 mm × 230 mm × 300 mm) had a stimulated electric grid at the bottom and was housed in a soundproof chamber (600 mm × 600 mm × 600 mm). Before testing, each mouse was placed in the experimental chamber for 3 min to become acquainted with its surroundings. The mice were then given three consecutive electric shocks (0.5 mA, 2 s) with intertrial intervals of 58 s. To eliminate odor, 75% ethanol was used to clean the observation chamber between each pair of mice. During testing, mice were placed in the experimental chamber for 3 min with no electric shocks. The entire experiment was filmed by a fixed camera above the observation room, and the freezing time was calculated using connected analysis software (FCT-100, Tekman Software Information Co., Ltd., Chengdu, China).
Tissue extraction
At the end of the behavioral experiment, mice were placed in the laboratory for 2 h in advance to allow them to acclimatize and then anesthetized with 20% urethane (10 ml/kg, ip). While waiting for the urethane to take effect, the surgical instruments, cardiac perfusion device, and ice pack were prepared. After observing the disappearance of the right reflex, the mice were fixed on a foam board, and then the thoracic cavity was quickly exposed with surgical scissors. The perfusion needle was inserted 1–2 mm from the left side of the heart apex into the left ventricle and carefully placed at the aortic arch. The vessels were then rapidly perfused with 100 mL 0.9% sodium chloride (heated at 37°C in advance) within 5 min and flushed. After perfusion, the mice were decapitated, the brain tissue was placed on ice, and the left hippocampus and cortex were rapidly isolated and preserved in lyophilization boxes in a freezer at −80°C. Another hemisphere was preserved intact in 4% paraformaldehyde solution with picric acid.
Reverse transcription and real-time quantitative PCR
The ultraclean table was prepared by wiping it down with 30% hydrogen peroxide and soaking the instruments in 75% ethanol ahead of time. Hippocampal and cortex tissue was homogenized in TRIzol reagent (Vazyme Biotech, R401–01) using a cryogenic homogenizer (Jingxin, Shanghai, China). After 5 min at room temperature, the samples were centrifuged at 12,000 × g for 5 min at 4°C, and the sediment was discarded. Then, 0.2 mL chloroform was added to the solution, and the mixture was inverted back and forth to make it milky white, shaken and mixed well, and set aside for 2–3 min at room temperature. The upper aqueous phase was collected after centrifugation at 12,000 × g for 15 min at 4°C. The solution was mixed with 0.5 ml isopropanol/ml TRIzol, left at room temperature for 5–10 min, and then centrifuged at 12,000 × g for 10 min at 4°C, and the supernatant was removed. Then, 75% ethanol was added to 1 ml of 75% ethanol/ml TRIzol, the tube was centrifuged with gentle shaking, the precipitate was suspended, the solution was centrifuged at 7,500 × g for 5 min at 4°C and the supernatant was discarded as much as possible. Subsequently, the RNA sample was dissolved in 50 µl RNase-free water. The solution was mixed with 0.5 mL isopropanol and allowed to stand at room temperature for 5–10 min before being centrifuged at 12,000 × g for 10 min at 4°C, and the supernatant was removed. Then, 1 mL 75% ethanol was added, the tube was gently shaken, the precipitate was suspended, and the solution was centrifuged at 7,500 × g for 5 min at 4°C, discarding as much supernatant as possible. After that, the RNA sample was dissolved in 50 µl of RNase-free water. RNA was reverse transcribed in a PCR instrument (Mastercycler, Eppendorf, Germany) using a reverse transcription kit (Vazyme Biotech, R312–01), and the real-time polymerase chain reaction was completed on a real-time PCR cycler (lightcycler480II, Roche LifeScience, Switzerland) following reagent vendor instructions (YEASEN, Shanghai, China). Table 1 shows the sequences of the PCR primers used in this study.
Table 1.
PCR primers employed in the present study.
| Gene | Forward primer (5’ → 3’) | Reverse primer (5’ → 3’) |
|---|---|---|
| Vma21 | ACCCTGAAGACACTGCTGTT | GCCACATAGACAAACAGGGC |
| Gata1 | CTCCCCAGTCTTTCAGGTGT | GCTCTTCCCTTCCTGGTCTT |
| En1 | GACTCACAGCAACCCCTAGT | CTGGGACTCATTCAGGCTGA |
| Runx1 | AACAATGGTACGGATGGGGT | AGGACGCCATAACAACTCCA |
| Creb1 | GCCACCTACCATAGAGCCAT | AGTGGAGTGGTTCAAGGAGG |
| Adrb2 | GAGCGACTACAAACCGTCAC | ATTCTTGGTCAGCAGGCTCT |
Transcriptomic
Transcriptome detection was performed on hippocampal tissues of each group, with 5 replicates in each group. RNA extraction, RNA purification, reverse transcription, library construction and sequencing were performed at Shanghai Majorbio Bio-pharm Biotechnology Co., Ltd. (Shanghai, China) according to the manufacturer’s instructions (Illumina, San Diego, CA). The transcriptome library was prepared following TruSeqTM RNA sample preparation Kit from Illumina (20040534) using 1 μg of total RNA. Briefly, messenger RNA was isolated according to polyA selection method by oligo(dT) beads and then fragmented by fragmentation buffer first. Secondly, double-stranded cDNA was synthesized using a SuperScript double-stranded cDNA synthesis kit (Invitrogen, K1621). Then the synthesized cDNA was subjected to end-repair, phosphorylation and “A” base addition according to Illumina’s library construction protocol. Libraries were size selected for cDNA target fragments of 300 bp on 2% Low Range Ultra Agarose followed by PCR amplified using Phusion DNA polymerase (NEB, M0530) for 15 PCR cycles. After quantified by TBS380, paired-end RNA-seq sequencing library was sequenced with the Illumina NovaSeq 6000 sequencer (2 × 150 bp read length) (www.majorbio.com). The data were analyzed online (www.majorbio.com).
Immunofluorescence and thioflavin S staining
After storing the right brain hemisphere in 4% paraformaldehyde at 4°C for more than 2 weeks, gradient dehydration using 10%, 20%, and 30% sucrose (Sinopharm 10,021,418) solutions was performed for 1 week, with fresh sucrose solution replaced daily. Following dehydration, brains were embedded in optimal cutting temperature compound (OCT) (Sakura, 4583) and cut into 15 μm brain slices using a frozen sectioning machine (CM3050S; Leica Biosystems, USA). PBS (Biosharp, BL601A) solution (including 0.5% Triton X-100 [Sinopharm, LA1604602]) was used to permeabilize brain sections for 30 min at room temperature before blocking in 5% bovine serum albumin (BSA; Beyotime Biotechnology, ST023) for 1 h. Then, the sections were incubated with primary antibodies (Table 2) overnight at 4°C and then incubated for 1 h at 37°C with donkey-anti-rabbit Alexa Fluor 488 (Jackson ImmunoResearch, 711-545-152) or donkey-anti-mouse Alexa Fluor 594 (Jackson ImmunoResearch, 715-585-150). For thioflavin staining, the tissue sections were washed for 5 min 3 times with PBS, and then thioflavin S (Sigma-Aldrich, T1892) was configured with 50% alcohol to a concentration of 0.3% and incubated at room temperature for 8 min. Subsequently, the sections were washed for 5 min 3 times with 50% alcohol and once with PBS for 5 min, shaded and sealed in a dark place. All slides were photographed under a confocal microscope (Axio Imager Z2, Carl Zeiss, Germany).
Table 2.
Antibodies used in the western blotting analysis and their properties.
| Antibody | Specificity | Type | Dilution for WB | Source | Catalog number |
|---|---|---|---|---|---|
| ACTB/β-Actin | ACTB/β-Actin | Poly- | 1:5000 | SAB | 21338 |
| 4G8 | β-Amyloid, 17–24 | Mono- | 1:200 for IF | Biolegend | 800708 |
| Aβ | β-Amyloid | Mono- | 1:1000 for WB | CST | 8243 |
| APP | APP | Mono- | 1:1000 for WB | Biolegend | 800901 |
| p-APP | p-APP | Mono- | 1:1000 for WB | CST | 6986 |
| PSEN1 | PSEN1 | Poly- | 1:1000 for WB | SAB | 24526 |
| BACE1 | BACE1 | Poly- | 1:1000 for WB | SAB | 54901 |
| IRF1 | IRF1 | Mono- | 1:1000 for WB | CST | 8478 |
| LC3 | Total LC3B | Poly- | 1:1000 for WB 1:200 for IF |
CST | 2775 |
| ATG5 | Total ATG5 | Mono- | 1:1000 for WB | CST | 12994 |
| ATG7 | Total ATG7 | Mono- | 1:1000 for WB | CST | 8558 |
| SQSTM1/p62 | Total SQSTM1/p62 protein | Mono- | 1:1000 for WB | CST | 23214 |
| VMA21 | VMA21 vacuolar-type H+-ATPase homolog | Mono- | 1:500 for WB | Proteintech | 21921 |
| ADRB2 | adrenergic receptor, beta 2 | Mono- | 1:1000 for WB | Abcam | 182136 |
| MTOR | Total MTOR | Mono- | 1:1000 for WB | CST | 2983 |
| p-MTOR | Phospho-MTOR (Ser2448) | Mono- | 1:1000 for WB | CST | 5536 |
| PRKAA/AMPK | PRKAA1/AMPK alpha 1 | Mono- | 1:1000 for WB | SAB | 48827 |
| p-PRKAA/AMPK | Phospho-PRKAA/AMPKα (Thr172) | Mono- | 1:1000 for WB | CST | 2535 |
Mono-, monoclonal; poly-, polyclonal.
Transmission electron microscopy
Hippocampal tissues were fixed with 2% glutaraldehyde and commissioned to the Shiyanjia laboratory (www.shiyanjia.com) for sectioning. In brief, the specimen was first fixed with 2.5% glutaraldehyde (Sinopharm 30,092,436) in phosphate buffer (0.1 M, pH 7.0) for more than 4 h; washed three times in the phosphate buffer for 15 min at each step; then postfixed with 1% OsO4 in phosphate buffer for 1–2 h and washed three times in the phosphate buffer for 15 min at each step. Then the specimen was first dehydrated by a graded series of ethanol (30, 50, 70, 80, 90, 95 and 100%) for about 15 to 20 min at each step, then transferred to absolute acetone for 20 min. Subsequently, the specimen was placed in 1:1 mixture of absolute acetone and the final Spurr resin mixture (SPI-CHEM 02,680-AB) for 1 h at room temperature, then transferred to 1:3 mixture of absolute acetone and the final resin mixture for 3 h and to final Spurr resin mixture for overnight. For the embedding and ultrathin sectioning, the specimen was placed in a microcentrifuge tube containing Spurr resin and heated at 70°C for more than 9 h. The specimen was sectioned in LEICA EM UC7 ultratome and sections were stained by uranyl acetate and alkaline lead citrate for 5 to 10 min, respectively, and observed in Hitachi Model H-7650 TEM.
Western blotting
Tissues were homogenized in radioimmunoprecipitation assay lysis buffer (Beyotime Biotechnology, P0013B) using a cryogenic homogenizer (Jingxin, Shanghai, China). Tissue homogenates were mixed with 2X SDS‒PAGE sample loading buffer (Beyotime Biotechnology, P0015B) and then boiled at 95°C for 10 min. The proteins were separated for approximately 1 h using 4–20% Precast Protein Plus Gel (Yeasen Biotechnology, 36270ES10) and transferred onto nitrocellulose membranes. The membranes were blocked for 1 h room temperature with 5% BSA before being incubated with primary antibody at 4°C (at least 8 h). The membranes were then incubated for 1 h at room temperature with a secondary antibody (anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase, 1:5000). ImageJ was used to perform quantitative analysis on the blots. All the primary antibodies used are listed in Table 2.
Cell culture and treatment
Mouse neuroblastoma N2a cells (Procell Life, CL-0168) and BV2 microglial cells (Procell Life, CL-0493A) were cultured in DMEM (Cytiva, SH30023.01) containing 10% fetal bovine serum (FBS; Tianhang Biotechnology 11,011–8611) at 37°C in a humidified incubator with 5% CO2. N2a cells were cultured in confocal petri dishes (Biosharp, BS-15-GJM) and BV2 cells were plated in 12-well plates. Aβ1–42 (Synthesized by ChinaPeptides, Shanghai, China) and terbutaline (MedChemExpress, HY-B0802A) were added to the culture medium at 10 μM. The mouse-specific siRNA targeting Irf1 was synthesized by OBiO Technology (Shanghai, China) and the sequence of the siRNA targeting murine Irf1 was as follows: CCAAGACATGGAAGGCAAA. The mouse-specific siRNA targeting Adrb2 was designed and synthesized by Ribobio Technology (Guangzhou, China).
Tandem mCherry-GFP fluorescence microscopy
A tandem monomeric Cherry-GFP-tagged LC3 (mRFP-GFP-LC3) plasmid was donated by Prof. Jianzhi Wang and Prof. Gongping Liu from Huazhong University of Science and Technology. N2a cells were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum. The cell transfection concentration was 0.1 µg/µl. To evaluate tandem fluorescent LC3 puncta, 48 h after mCherry-GFP-LC3 transfection, cells were captured using a confocal microscope (Axio Imager Z2, Carl Zeiss, Germany).
Statistical analyses
All numerical data are expressed as the means ± SEMs, Two-way repeated-measures ANOVA followed by Bonferroni’s post hoc test, two-way ANOVA followed by Bonferroni’s post hoc test, one-way ANOVA followed by Bonferroni’s post hoc test and student’s t tests were used. The statistical analysis was performed using SPSS 15.0 statistical software (SPSS Inc., Chicago, IL, USA).
Acknowledgements
The authors would like to thank to Prof. Jianzhi Wang and Prof. Gongping Liu from Huazhong University of Science and Technology for the mCherry-GFP-LC3 plasmids he provided.
Funding Statement
This work was supported by grants from the Natural Science Foundation of Jiangsu Province [BK20211238, BK20231050], China Postdoctoral Science Foundation [2020M681491], Jiangsu Province Postdoctoral Science Foundation [2020Z037] and Six Talent Peaks Project in Jiangsu Province [SZCY-003] and Fundamental Research Funds for the Central Universities [JUSRP123070].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].World Alzheimer Report 2021. Alzheimer’s Disease International, 2021. [Google Scholar]
- [2].Hardy J, Selkoe DJ.. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- [3].Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science. 2010;330(6012):1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311–321. [DOI] [PubMed] [Google Scholar]
- [5].Liu YH, Giunta B, Zhou H-D, et al. Immunotherapy for Alzheimer disease—the challenge of adverse effects. Nat Rev Neurol. 2012;8(8):465–469. [DOI] [PubMed] [Google Scholar]
- [6].Ball K, Berch DB, Helmers KF, et al. Effects of cognitive training interventions with older adults: a randomized controlled trial. JAMA. 2002;288(18):2271–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sobol NA, Hoffmann K, Frederiksen KS, et al. Effect of aerobic exercise on physical performance in patients with Alzheimer’s disease. Alzheimers Dement. 2016;12(12):1207–1215. [DOI] [PubMed] [Google Scholar]
- [8].Jiang X, Chai G-S, Wang Z-H, et al. Spatial training preserves associative memory capacity with augmentation of dendrite ramification and spine generation in Tg2576 mice. Sci Rep. 2015;5(1):9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chauhan PS, Yadav D, Arukha AP. Dietary Nutrients and Prevention of Alzheimer’s Disease. CNS Neurol Disord Drug Targets. 2022;21(3):217–227. doi: 10.2174/1871527320666210405141123 [DOI] [PubMed] [Google Scholar]
- [10].Pitkälä KH, Pöysti, MM, Laakkonen, ML, et al. Effects of the Finnish Alzheimer disease exercise trial (FINALEX): a randomized controlled trial. JAMA Intern Med. 2013;173(10):894–901. [DOI] [PubMed] [Google Scholar]
- [11].Du Z, Li Y, Li J, et al. Physical activity can improve cognition in patients with Alzheimer’s disease: a systematic review and meta-analysis of randomized controlled trials. Clin Interv Aging. 2018;13:1593–1603. doi: 10.2147/CIA.S169565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Panza GA, Taylor BA, MacDonald HV, et al. Can exercise improve cognitive symptoms of Alzheimer’s disease? J Am Geriatr Soc. 2018;66(3):487–495. [DOI] [PubMed] [Google Scholar]
- [13].Tarumi T, Rossetti H, Thomas BP, et al. Exercise training in amnestic mild cognitive impairment: a one-year randomized controlled trial. J Alzheimers Dis. 2019;71(2):421–433. [DOI] [PubMed] [Google Scholar]
- [14].Liang KY, Mintun MA, Fagan AM, et al. Exercise and Alzheimer’s disease biomarkers in cognitively normal older adults. Ann Neurol. 2010;68(3):311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Erickson KI, Voss MW, Prakash RS, et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A. 2011;108(7):3017–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brown BM, Peiffer JJ, Martins RN. Multiple effects of physical activity on molecular and cognitive signs of brain aging: can exercise slow neurodegeneration and delay Alzheimer’s disease? Mol Psychiatry. 2013;18(8):864–874. doi: 10.1038/mp.2012.162 [DOI] [PubMed] [Google Scholar]
- [17].Boland B, Yu WH, Corti O, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2018;17(9):660–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Menzies FM, Fleming A, Caricasole A, et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron. 2017;93(5):1015–1034. [DOI] [PubMed] [Google Scholar]
- [20].Lee JH, Yang D-S, Goulbourne CN, et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat Neurosci. 2022;25(6):688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest. 2008;118(6):2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64(2):113–122. [DOI] [PubMed] [Google Scholar]
- [23].Ramachandran N, Munteanu I, Wang P, et al. VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol. 2013;125(3):439–457. [DOI] [PubMed] [Google Scholar]
- [24].Wang F, Yang Y, Boudagh G, et al. Follicular lymphoma-associated mutations in the V-ATPase chaperone VMA21 activate autophagy creating a targetable dependency. Autophagy. 2022;18(8):1982–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sandelin A, Alkema W, Engström P, et al. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32(Database issue):D91–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nixon RA, Yang DS. Autophagy failure in Alzheimer’s disease–locating the primary defect. Neurobiol Dis. 2011;43(1):38–45. doi: 10.1016/j.nbd.2011.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cai Z, Zhao B, Li K, et al. Mammalian target of rapamycin: a valid therapeutic target through the autophagy pathway for Alzheimer’s disease? J Neurosci Res. 2012;90(6):1105–1118. [DOI] [PubMed] [Google Scholar]
- [28].Fang EF, Hou Y, Palikaras K, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22(3):401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28(27):6926–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997. doi: 10.1038/nm.3232 [DOI] [PubMed] [Google Scholar]
- [31].Cummins N, Tweedie A, Zuryn S, et al. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019;38(3):38(3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nishi T, Forgac M. The vacuolar (H+)-ATPases–nature’s most versatile proton pumps. Nat Rev Mol Cell Biol. 2002;3(2):94–103. doi: 10.1038/nrm729 [DOI] [PubMed] [Google Scholar]
- [33].Wilkens S, Zhang Z, Zheng Y. A structural model of the vacuolar ATPase from transmission electron microscopy. Micron. 2005;36(2):109–126. doi: 10.1016/j.micron.2004.10.002 [DOI] [PubMed] [Google Scholar]
- [34].Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10(9):623–635. doi: 10.1038/nrm2745 [DOI] [PubMed] [Google Scholar]
- [35].McGuire C, Stransky L, Cotter K, et al. Regulation of V-ATPase activity. Front Biosci (Landmark Ed). 2017;22(4):609–622. [DOI] [PubMed] [Google Scholar]
- [36].Malkus P, Graham LA, Stevens TH, et al. Role of Vma21p in assembly and transport of the yeast vacuolar ATPase. Mol Biol Cell. 2004;15(11):5075–5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Valenzuela PL, Castillo-García A, Morales JS, et al. Exercise benefits on Alzheimer’s disease: state-of-the-science. Ageing Res Rev. 2020;62:101108. doi: 10.1016/j.arr.2020.101108 [DOI] [PubMed] [Google Scholar]
- [38].Rabin JS, Klein H, Kirn, DR, et al. Associations of physical activity and β-amyloid with longitudinal cognition and neurodegeneration in clinically normal older adults. JAMA Neurol. 2019;76(10):1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-β plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18(8):875–881. [DOI] [PubMed] [Google Scholar]
- [40].Gibbs ME, Summers RJ. Contrasting roles for beta1, beta2 and beta3-adrenoceptors in memory formation in the chick. Neuroscience. 2005;131(1):31–42. doi: 10.1016/j.neuroscience.2004.10.036 [DOI] [PubMed] [Google Scholar]
- [41].Li S, Jin M, Zhang D, et al. Environmental novelty activates β2-adrenergic signaling to prevent the impairment of hippocampal LTP by Aβ oligomers. Neuron. 2013;77(5):929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hutten DR, Bos JHJ, de Vos S, et al. Targeting the beta-2-adrenergic receptor and the risk of developing Alzheimer’s disease: A retrospective inception cohort study. J Alzheimers Dis. 2022;87(3):1089–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xu H, Rajsombath MM, Weikop P, et al. Enriched environment enhances β-adrenergic signaling to prevent microglia inflammation by amyloid-β. EMBO Mol Med. 2023;15(1):e17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Im E, Jiang Y, Stavrides PH, et al. Lysosomal dysfunction in down syndrome and Alzheimer mouse models is caused by v-ATPase inhibition by Tyr 682 -phosphorylated APP βCTF. Sci Adv. 2023;9(30):eadg1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lee JH, Yu, WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lee JH, Wolfe DM, Darji S, et al. β2-adrenergic agonists rescue lysosome acidification and function in PSEN1 deficiency by reversing defective ER-to-lysosome delivery of ClC-7. J Mol Biol. 2020;432(8):2633–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Burrinha T, Cunha C, Hall, MJ, et al. Deacidification of endolysosomes by neuronal aging drives synapse loss. Traffic. 2023;24(8):334–354. [DOI] [PubMed] [Google Scholar]
- [48].Cao J, Huang M, Guo L, et al. MicroRNA-195 rescues ApoE4-induced cognitive deficits and lysosomal defects in Alzheimer’s disease pathogenesis. Mol Psychiatry. 2021;26(9):4687–4701. [DOI] [PMC free article] [PubMed] [Google Scholar]