

ABSTRACT
Macroautophagy/autophagy contributes to maladaptive kidney repair by inducing pro-fibrotic factors such as FGF2 (fibroblast growth factor 2), but the underlying mechanism remains elusive. Here, we show that EGR1 (early growth response 1) was induced in injured proximal tubules after ischemic acute kidney injury (AKI) and this induction was suppressed by autophagy deficiency in inducible, renal tubule-specific atg7 (autophagy related 7) knockout (iRT-atg7 KO) mice. In cultured proximal tubular cells, TGFB1 (transforming growth factor beta 1) induced EGR1 and this induction was also autophagy dependent. Egr1 knockdown in tubular cells reduced FGF2 expression during TGFB1 treatment, leading to less FGF2 secretion and decreased paracrine effects on fibroblasts. ChIP assay detected an increased binding of EGR1 to the Fgf2 gene promoter in TGFB1-treated tubular cells. Both Fgf2 and Egr1 transcription was inhibited by FGF2 neutralizing antibody, suggesting a positive feedback for EGR1-mediated FGF2 autoregulation. This feedback was confirmed using fgf2-deficient tubular cells and fgf2-deficient mice. Upstream of EGR1, autophagy deficiency in mice suppressed MAPK/ERK (mitogen-activated protein kinase) activation in post-ischemic renal tubules. This inhibition correlated with SQSTM1/p62 (sequestosome 1) aggregation and its sequestration of MAPK/ERK. SQSTM1/p62 interacted with MAPK/ERK and blocked its activation during TGFB1 treatment in autophagy-deficient tubular cells. Inhibition of MAPK/ERK suppressed EGR1 and FGF2 expression in maladaptive tubules, leading to the amelioration of renal fibrosis and improvement of renal function. These results suggest that autophagy activates MAPK/ERK in renal tubular cells, which induces EGR1 to transactivate FGF2. FGF2 is then secreted into the interstitium to stimulate fibroblasts for fibrogenesis.
Abbreviation: 3-MA: 3-methyladenine; ACTA2/α-SMA: actin alpha 2, smooth muscle, aorta; ACTB/β-actin: actin, beta; AKI: acute kidney injury; aa: amino acid; ATG/Atg: autophagy related; BUN: blood urea nitrogen; ChIP: chromatin immunoprecipitation; CKD: chronic kidney disease; CM: conditioned medium; COL1A1: collagen, type I, alpha 1; COL4A1: collagen, type IV, alpha 1; CQ: chloroquine; DBA: dolichos biflorus agglutinin; EGR1: early growth response 1; ELK1: ELK1, member of ETS oncogene family; FGF2: fibroblast growth factor 2; FN1: fibronectin 1; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; HAVCR1/KIM-1: hepatitis A virus cellular receptor 1; IP: immunoprecipitation; LIR: LC3-interacting region; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MAP2K/MEK: mitogen-activated protein kinase kinase; MAPK: mitogen-activated protein kinase; NFKB: nuclear factor kappa B; PB1: Phox and Bem1; PFT: pifithrin α; PPIB/cyclophilin B: peptidylprolyl isomerase B; RT-qPCR: real time-quantitative PCR; SQSTM1/p62: sequestosome 1; TGFB1/TGF-β1: transforming growth factor beta 1; VIM: vimentin
KEYWORDS: Interstitial fibrosis, ischemic acute kidney injury, kidney repair, SQSTM1/p62, tubulo-interstitial communication
Introduction
Acute kidney injury (AKI) is a major kidney disease associated with significant mortality and possible transition or progression to chronic kidney disease (CKD) and end-stage kidney disease [1–3]. Despite an increasing number of molecules and pathways identified as involved in AKI, the mechanism of AKI to CKD transition is much less understood owing to the complexity of involved cell types and lack of optimal experimental models [4]. Studies over the past decade have elaborated the differences between adaptive and maladaptive repair of renal tubules after kidney injury and particularly the concept of maladaptive tubular repair in AKI progression. While adaptive repair may fully restore tubular structure and function, maladaptive repair often leads to CKD characterized by evolving tubular pathologies, chronic inflammation, renal fibrosis, and gradual decline of renal function [5–10]. In maladaptive repair, renal tubules are not only a primary target of damage but also a driving force and determinant of disease progression. By undergoing a secretory phenotype transformation after injury, maladaptive renal tubules can produce and secrete various pro-inflammatory and pro-fibrotic factors to initiate a cascade of downstream events resulting in the transition from AKI to CKD [5–10].
The phenotypic change of renal tubular cells in maladaptive kidney repair is a multifactorial process, involving dedifferentiation, cell cycle arrest, cellular senescence, aberrant reactivation of various developmental pathways, mitochondrial dysfunction, and metabolic change [11–19]. Using both experimental models and human renal biopsies from post-AKI patients, we have recently identified a role for macroautophagy/autophagy in this process [20]. After ischemic AKI in mice, autophagy persisted for weeks in renal tubules and facilitated the transformation of renal tubular cells to a secretory phenotype. Remarkably, autophagy specifically induced the production and secretion of FGF2 (fibroblast growth factor 2) in post-AKI mice and in TGFB1 (transforming growth factor beta 1)-treated renal tubular cells. FGF2 released by tubular cells stimulated renal fibroblasts, acting as a key paracrine factor for tubulo-interstitial communication to promote renal fibrosis during maladaptive repair. In renal biopsies from post-AKI patients, we also detected the increase of tubular autophagy, FGF2 expression and renal interstitial fibrosis [20]. Despite these findings, it was unclear how autophagy promotes tubular production of FGF2.
Unlike most eukaryotic genes, the promoter region of Fgf2 gene does not have the conventional CAAT and TATA box motifs but contains GC-rich overlapping binding sites for two transcription factors, EGR1 (early growth response 1) and specificity/stimulating protein 1. While specificity/stimulating protein 1 is responsible for constitutive Fgf2 expression, EGR1 transactivates Fgf2 in response to cellular stress [21,22]. At the upstream, MAPK (mitogen-activated protein kinase) proteins may phosphorylate the ternary complex factor family member ELK1 (ELK1, member of ETS oncogene family) together with the recruitment of the serum response factor to the serum response element on Egr1 gene promoter to induce Egr1 transcription [23–25]. As a transcription factor, EGR1 contains a highly conserved DNA-binding domain with three zinc fingers that preferentially recognize GC-rich elements present in its target gene promoters [23]. By controlling the transcription of a wide array of genes, EGR1 is implicated in diverse physiological processes. Moreover, abnormal expression of EGR1 have also been linked to cardiovascular disease, inflammatory disease, and fibrosis in different organs [23–25]. In the kidney, EGR1 was firstly reported by Ouellette and colleagues as an immediate early response gene in ischemic AKI in mice [26]. More recent work has suggested the involvement of EGR1 in renal inflammation and fibrosis. For example, Egr1 deficiency attenuated renal inflammation, fibrosis, and function decline in an adenine-induced mouse model of tubulointerstitial nephritis [27]. However, the role of EGR1 in maladaptive repair after AKI remains unclear.
In this study, we found that persistent autophagy contributed to EGR1 induction in renal tubules after ischemic AKI in mice and in TGFB1-treated mouse proximal tubular cells. By directly binding to Fgf2 gene promoter, EGR1 transactivated FGF2 in tubular cells for subsequent paracrine activation of renal fibroblasts to promote fibrosis. Upstream of EGR1, autophagy activated MAPK1/ERK2 (mitogen-activated protein kinase 1)-MAPK3/ERK1 in renal tubular cells by degrading SQSTM1/p62 (sequestosome 1) aggregates and releasing MAPK1/ERK2-MAPK3/ERK1 from SQSTM1/p62 sequestration. Inhibition of MAPK1/ERK2-MAPK3/ERK1 suppressed EGR1 activation and FGF2 induction in maladaptive tubular cells, leading to the alleviation of interstitial fibrosis and improvement of tubular function.
Results
Autophagy deficiency inhibits EGR1 in renal tubular cells during maladaptive kidney repair in post-ischemic AKI mice
To characterize EGR1 in post-ischemic kidneys, we first performed co-immunostaining of EGR1 with HAVCR1/KIM-1 (hepatitis A virus cellular receptor 1), a biomarker for injured proximal tubules [28]. EGR1 was not detected in sham control mice, whereas remarkably induced in the nuclei of renal tubular cells in both renal cortex and outer medulla during post-ischemic kidney repair (Figure 1A). Notably, the majority (roughly 80%) of these EGR1-positive renal tubules were also HAVCR1/KIM-1-postive, suggesting the induction of EGR1 in chronically injured proximal tubules (Figure 1A). Along with EGR1, FGF2 was significantly induced in renal tubules in post-ischemic kidneys whereas only minimal signal was seen in sham control mice (Figure S1). The renal tubules with increased FGF2 expression were also positive for HAVCR1/KIM-1 (Figure S1: FGF2 and HAVCR1/KIM-1 co-staining) and mostly surrounded by thickened interstitial COL4A1 (collagen, type IV, alpha 1) (Figure S1: FGF2 and COL4A1 co-staining). These results are consistent with our recent work showing increased production of FGF2 by atrophic tubules in fibrotic arears of post-ischemic kidneys [20]. More importantly, the appearance of both EGR1 and FGF2 in HAVCR1/KIM-1-positive renal tubules suggest a possible spatial-temporal association between these two proteins. To test this possibility, we did co-immunofluorescence of EGR1 and FGF2. Of interest, in post-injured renal tubules with the induction of nuclear EGR1, FGF2-positive signals were often adjacent to or sometimes even partially co-localized with EGR1 (Figure S1: FGF2 and EGR1 co-staining). Moreover, the induction of EGR1 was mediated by tubular cell autophagy. At 2 weeks after ischemic AKI, wild type (iRT-Atg7 WT) mice had a 3.5-fold increase of Egr1 mRNA over sham control, which was further elevated to 5.5-fold at 4 weeks. The upregulation of Egr1 mRNA was suppressed by tubular autophagy deficiency in iRT-atg7 KO mice, with 2.1-fold at 2 weeks and 3.3-fold at 4 weeks respectively (Figure 1B). Immunoblot analysis also showed that the induction of EGR1 protein in WT kidneys was significantly inhibited in iRT-atg7 KO mice at both 2 and 4 weeks after ischemic AKI (Figure 1C). EGR1 immunofluorescence further verified that at 2 weeks after ischemic AKI, the number of EGR1-positive tubular cells was reduced from 20 per 400× field in WT mice to 12 in iRT-atg7 KO mice (Figure 1D, E). Collectively, these results suggest that EGR1 is induced in an autophagy-dependent manner in renal tubular cells along with increased production of FGF2 in post-ischemic AKI mice.
Figure 1.
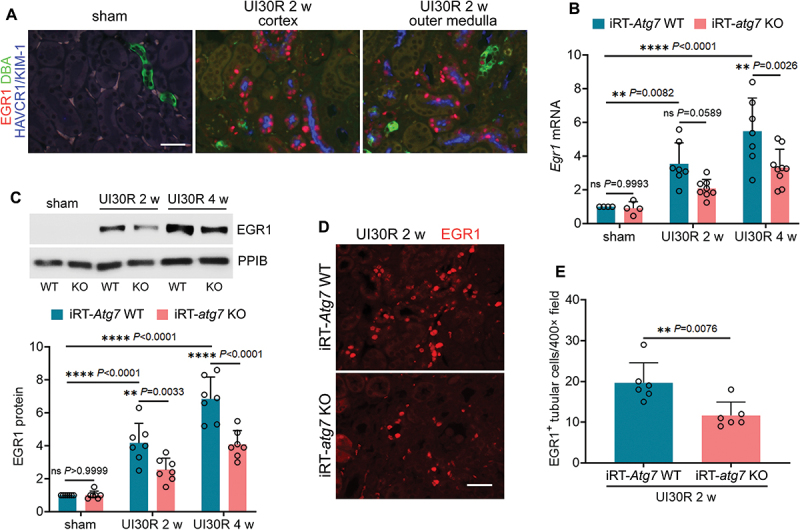
Autophagy deficiency inhibits EGR1 in renal tubular cells during maladaptive kidney repair after ischemic AKI in mice. (A) C57BL/6 mice underwent sham operation (n = 3) or 30-min unilateral renal ischemia followed by reperfusion for 2 weeks (n = 3). Left kidneys were harvested for co-immunofluorescence of EGR1 (red) and HAVCR1/KIM-1 (blue). Fluorescein-DBA (green) was used as a marker for renal distal tubules. Scale bar: 20 µm. (B-E) WT and iRT-atg7 KO mice underwent sham operation or 30-min unilateral renal ischemia followed by reperfusion for up to 4 weeks. Left kidneys were harvested. (B) RT-qPCR of Egr1 mRNA (sham: n = 4; UI30R 2 w: WT n = 7, KO n = 8; UI30R 4 w: WT n = 7, KO n = 9). (C) EGR1 immunoblot and densitometry (n = 7 for each group). PPIB/cyclophilin B was used as an internal loading control. (D) EGR1 immunofluorescence. Scale bar: 20 µm. (E) quantification of EGR1-positive tubular cells (n = 6 for each group). Data in (B), (C) and (E) are presented as mean ± SEM. For statistics, two-way ANOVA with multiple comparisons was used for (B) and (C). 2-tailed, unpaired Student t-test was used for (E).
Autophagy deficiency inhibits EGR1 in cultured mouse renal proximal tubular cells during TGFB1 treatment
Using both pharmacological and genetic approaches, we then examined the role of autophagy in regulating EGR1 in cultured mouse proximal tubular cells. Consistent with the in vivo findings, Egr1 mRNA was induced in BUMPT cells during TGFB1 treatment, and this induction was suppressed by pharmacological inhibition of autophagy with either chloroquine (CQ) or 3 methyladenine (3-MA) (Figure 2A, B). Similar effects were also seen in atg7 KO tubular cells. TGFB1 led to a 5.4-fold increase of Egr1 mRNA and 4.1-fold increase of its protein in WT tubular cells, which was decreased respectively to 3- and 2.4-fold in atg7 KO tubular cells, further supporting a role of autophagy in EGR1 induction during renal fibrosis (Figure 2C-E). Notably, autophagy in renal tubular cells seemed to specifically activate EGR1. Following TGFB1 treatment, three other transcription factors that are well known for regulating fibrotic changes, including SMADs, NFKB (nuclear factor kappa B) and TRP53/p53, were also activated, as indicated by increased phosphorylation of these proteins (Figure 2D). Nevertheless, compared to WT tubular cells, these changes were barely affected by autophagy deficiency in atg7 KO tubular cells (Figure 2D) despite the potential involvement of NFKB and TRP53/p53 in TGFB1-induced tubular production of FGF2 (Figure S2).
Figure 2.
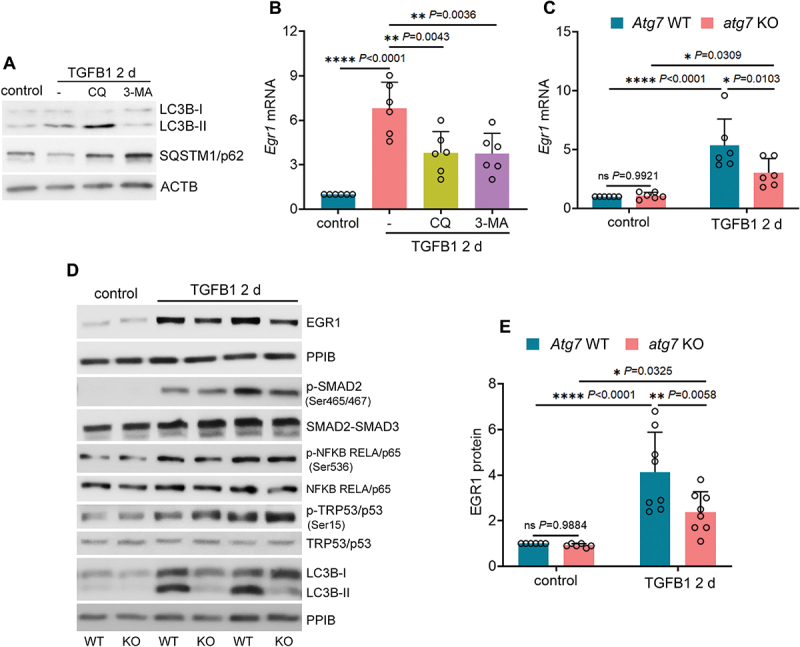
Autophagy deficiency inhibits EGR1 in cultured mouse renal proximal tubular cells during TGFB1 treatment. (A and B) subconfluent BUMPT cells were exposed to 5 ng/ml TGFB1 in serum-free DMEM for 2 days alone or with 5 µM CQ or 1 mM 3-MA. Control cells were kept in serum-free medium without TGFB1. Cells were collected for immunoblot of LC3B and SQSTM1/p62 (A) and RT-qPCR of Egr1 mRNA (B) (n = 6 experiments). (C-E) subconfluent WT and atg7 KO cells were exposed to 5 ng/ml TGFB1 in serum-free DMEM for 2 days. Control cells were kept in serum-free medium without TGFB1. (C) RT-qPCR of Egr1 mRNA (n = 6 experiments). (D) immunoblot of EGR1, p-SMAD2 (Ser465/467), SMAD2-SMAD3, p-NFKB RELA/p65 (Ser536), NFKB RELA/p65, p-TRP53/p53 (Ser15), TRP53/p53 and LC3B. (E) densitometry of EGR1 immunoblot (n = 6 experiments). Data in (B), (C) and (E) are presented as mean ± SEM. For statistics, one-way ANOVA with multiple comparisons was used for (B). Two-way ANOVA with multiple comparisons was used for (C) and (E).
EGR1 transactivates FGF2 in TGFB1-treated mouse proximal tubular cells for subsequent paracrine activation of renal fibroblasts
We next examined the effects of Egr1 knockdown on the production of FGF2 in TGFB1-treated BUMPT cells. Compared to non-target shRNA, Egr1-specific shRNA markedly suppressed Egr1 mRNA expression in both control and TGFB1-treated cells (Figure 3A: Egr1 mRNA). Notably, the induction of FGF2 during TGFB1 treatment was attenuated by Egr1 knockdown, with Fgf2 mRNA reduced from 5.1-fold over control in non-target shRNA-transfected cells to 2.3-fold in Egr1 shRNA-transfected cells (Figure 3A: Fgf2 mRNA). Immunoblot analysis further confirmed the inhibitory effects of Egr1 knockdown on FGF2 protein expression in TGFB1-treated cells (Figure 3B). These results suggest a potential role of EGR1 in transcriptional induction of FGF2. Analysis with the JASPAR database (http://jaspar.genereg.net) predicted 44 putative EGR1-binding sites in the promoter region of mouse Fgf2 gene. Among them, three sites that had the highest scores were either overlapping or next to each other (Table 1). We then examined EGR1 binding to the sequence harboring these three sites by chromatin immunoprecipitation (ChIP) assay. Compared to control cells, TGFB1 induced a 3-fold increase in Fgf2 DNA fragments immunoprecipitated by EGR1 antibody, indicating that EGR1 may directly bind to Fgf2 gene promoter for transcriptional activation (Figure 3C). Associated with inhibited FGF2 production, Egr1 knockdown also suppressed tubular secretion of FGF2 (Figure 3D). To further determine tubular paracrine activity, we collected conditioned medium (CM) from both non-target shRNA- and Egr1 shRNA-transfected BUMPT cells after TGFB1 treatment to incubate subconfluent, serum-starved renal fibroblasts NRK-49F. Compared with non-target control-CM, non-target TGFB1-CM significantly promoted fibroblast proliferation, as indicated by increased cell number and cellular protein (Figure 3E: non-target shRNA). Non-target TGFB1-CM also induced a phenotype transition of NRK-49F from fibroblasts to myofibroblasts with increased expression of VIM (vimentin), FN1 (fibronectin 1) and COL1A1 (collagen, type I, alpha 1) (Figure 3F, G: non-target shRNA). Notably, all these fibrotic changes induced by non-target TGFB1-CM were alleviated in NRK-49F fibroblasts treated with Egr1 shRNA TGFB1-CM (Figure 3E-G: Egr1 shRNA). Together, these results suggest that EGR1 transactivates FGF2 in TGFB1-treated mouse proximal tubular cells for subsequent paracrine activation of renal fibroblasts.
Figure 3.
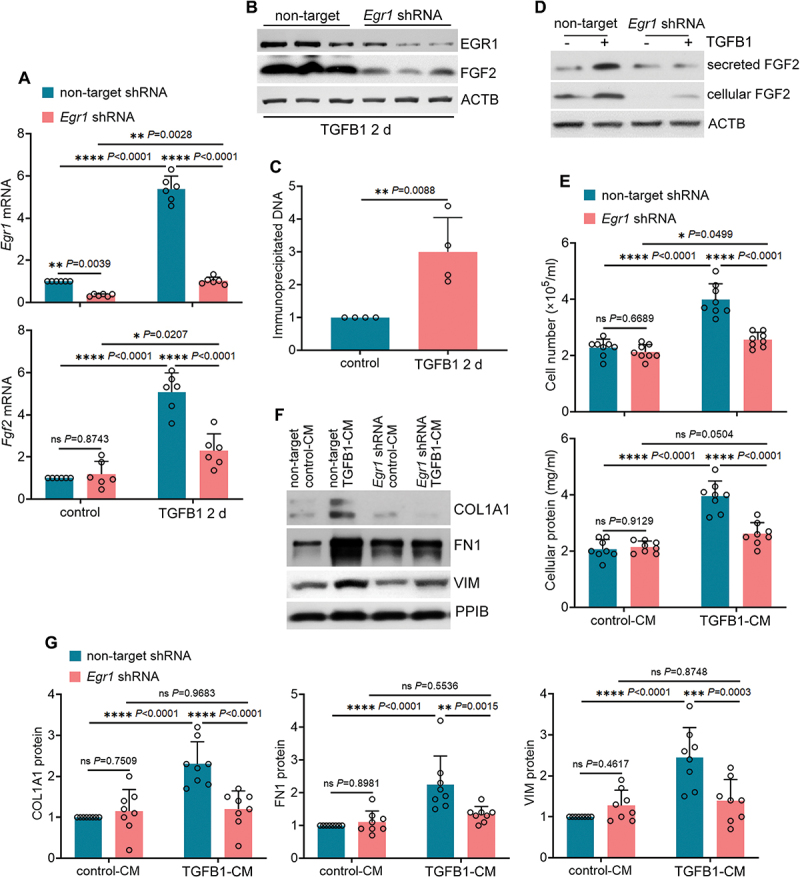
EGR1 transactivates FGF2 in TGFB1-treated mouse proximal tubular cells for subsequent paracrine activation of renal fibroblasts. (A and B) subconfluent, non-target shRNA-transfected or Egr1 shRNA-transfected BUMPT cells were exposed to 5 ng/ml TGFB1 in serum-free DMEM for 2 days. Control cells were kept in serum-free medium without TGFB1. Cells were collected for RT-qPCR of Egr1 and Fgf2 mRNA (A) and immunoblot of EGR1 and FGF2 (B) (n = 6 experiments). (C) subconfluent BUMPT cells were exposed to 5 ng/ml TGFB1 in serum-free DMEM for 2 days. Control cells were kept in serum-free medium without TGFB1. Cells were collected for ChIP assay of EGR1 binding to Fgf2 promoter sequence (n = 4 experiments). (D) subconfluent, non-target shRNA-transfected or Egr1 shRNA-transfected BUMPT cells were exposed to 5 ng/ml TGFB1 in serum-free DMEM for 2 days. Control cells were kept in serum-free medium without TGFB1. The old culture media for both TGFB1-treated and control cells were replaced by fresh media free of TGFB1 at the end of day 2, incubated with the cells for an additional day, and then collected as tubular cell-CM for immunoblot of secreted FGF2 (n = 3 experiments). Cell lysates were also collected in the same experiments for immunoblot of cellular FGF2. ACTB/β-actin was used as an internal loading control. (E-G) subconfluent NRK-49F fibroblasts were incubated with CMs from BUMPT cells (non-target shRNA control-CM, non-target shRNA TGFB1-CM, Egr1 shRNA control-CM, and Egr1 shRNA TGFB1-CM) for 2 days. The fibroblasts were then collected for cell number counting and cellular protein measurement (E), immunoblot of COL1A1, FN1 and VIM (F) and densitometry (G) (n = 7 experiments). Data in (A), (C), (E) and (G) are presented as mean ± SEM. For statistics, 2-tailed, unpaired Student t-test was used for (C). Two-way ANOVA with multiple comparisons was used for (A), (E) and (G).
Table 1.
Three EGR1 binding sites at mouse Fgf2 gene promoter predicted by JASPAR database.
| Fgf2 | JASPAR Score | Start | End | Sequence |
|---|---|---|---|---|
| site 1 | 14.657 | −1378 | −1391 | TCCCCTCCCCCACT |
| site 2 | 13.881 | −1364 | −1377 | CTTCCTCCCCCTCT |
| site 3 | 10.368 | −1377 | −1390 | CCCCTCCCCCACTC |
EGR1 and FGF2 form a positive feedback for FGF2 autoregulation in renal proximal tubular cells during TGFB1 treatment
In addition to paracrine effect on neighboring cells, FGF2 may also act as an autocrine factor to induce its own gene expression [21,29]. To test this autoregulation mechanism in our models, we first examined FGF2 neutralizing antibody. As shown in Figure 4A, FGF2 neutralizing antibody reduced mRNA expression of both Fgf2 and Egr1 during TGFB1 treatment of BUMPT cells, suggesting a positive feedback between FGF2 and EGR1 for FGF2 autoregulation (Figure 4A). This feedback mechanism was further confirmed in fgf2 KO primary tubular cells and fgf2 KO mice. TGFB1 induced EGR1 at both mRNA and protein levels in WT primary tubular cells, which was suppressed by fgf2 KO (Figure 4B, C). Moreover, at 2 weeks after ischemic AKI, EGR1 was induced in the nuclei of renal tubular cells of WT mice. This in vivo induction was also attenuated in fgf2 KO mice (Figure 4D, E), further supporting that EGR1 and FGF2 form a positive feedback for EGR1-mediated FGF2 autoregulation in renal proximal tubular cells. The autoregulation may contribute to the persistent FGF2 induction after TGFB1 treatment and reinforce its paracrine effects on renal fibroblasts.
Figure 4.
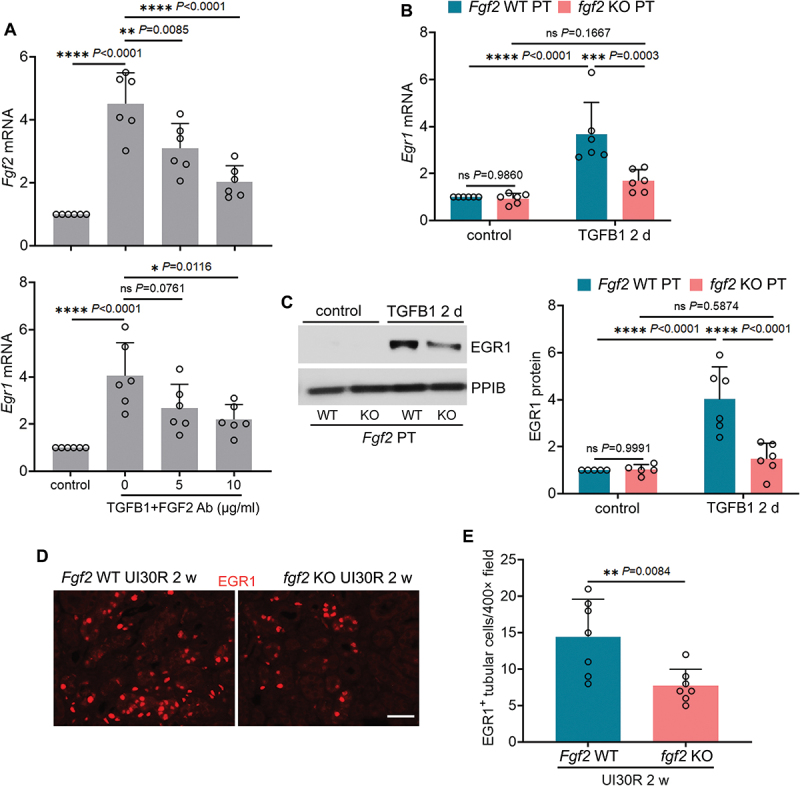
EGR1 and FGF2 form a positive feedback for FGF2 autoregulation in renal proximal tubular cells during TGFB1 treatment. (A) subconfluent BUMPT cells were exposed to 5 ng/ml TGFB1 in serum-free DMEM for 3 days in the presence of FGF2 neutralizing antibody at 5 and 10 µg/ml or mouse IgG (indicated as FGF2 ab of 0 µg/ml) as negative control. Control cells were kept in serum-free medium without TGFB1. Cells were collected for RT-qPCR of Fgf2 and Egr1 mRNA (n = 6 experiments). (B and C) subconfluent isolated primary proximal tubular cells from fgf2 KO mice (fgf2 KO PT) and their WT littermates (Fgf2 WT PT) were exposed to 5 ng/ml TGFB1 in serum-free DMEM for 2 days. Control cells were kept in serum-free medium without TGFB1. Cells were collected for RT-qPCR of Egr1 mRNA (B) and immunoblot of EGR1 (C) (n = 6 experiments). (D) fgf2 KO mice (n = 7) and their WT littermates (n = 7) underwent 30-min unilateral renal ischemia followed by reperfusion for 2 weeks. Left kidneys were collected for EGR1 immunofluorescence. Scale bar: 20 µm. (E) quantification of the numbers of EGR1-positive tubular cells. Data in (A), (B), (C) and (E) are presented as mean ± SEM. For statistics, one-way ANOVA with multiple comparisons was used for (A). Two-way ANOVA with multiple comparisons was used for (B) and (C). 2-tailed, unpaired Student t-test was used for (E).
Activation of MAPKs is accompanied by sustained autophagy during maladaptive renal repair after ischemic AKI
The MAPK signaling pathways play a central role in regulating EGR1 expression [23–25]. To dissect the mechanism that may contribute to autophagy-mediated EGR1 activation and FGF2 production in maladaptive renal tubules, we examined three essential members of the MAPK signaling pathways, including MAPK/ERK, MAPK/p38 and MAPK/JNK. We first monitored the dynamic changes of their activation at different time points of both injury and repair phases following ischemic AKI. Immediately (within 10 min) after renal ischemia, MAPK1/ERK2-MAPK3/ERK1, MAPK/p38 and MAPK/JNK were all activated in kidney tissues, as indicated by the increased phosphorylation of these protein kinases (Figure S3A: UI30R 0 h). While the phosphorylation of both MAPK1/ERK2-MAPK3/ERK1 and MAPK/JNK further enhanced at 6 hours of reperfusion, phosphorylated MAPK/p38 went down rapidly to the basal level (Figure S3A: UI30R 6 h). By 1 day, the phosphorylation of all three kinases was barely detected in ischemic kidneys, suggesting that the activation of MAPK signaling pathways is an early but transient response to acute renal ischemia-reperfusion injury (Figure S3A: UI30R 1 d). Of interest, MAPK1/ERK2-MAPK3/ERK1, MAPK/p38 and MAPK/JNK were all reactivated in kidney tissues at the beginning of post-ischemic renal repair. The phosphorylation of both MAPK1/ERK2-MAPK3/ERK1 and MAPK/JNK reappeared on day 2 (Figure S3A: UI30R 2 d) and visible phosphorylated MAPK/p38 was also detected again on day 3 (Figure S3A: UI30R 3 d). Unlike the transient activation during the acute injury phase, the reactivation of all three MAPK members during the repair phase persisted for weeks (Figure S3A: UI30R 1 w, 2 w and 4 w). In addition to MAPKs, we also measured the timeline of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta)-II accumulation in kidney tissues by immunoblot. Consistent with our previous studies using autophagy reporter mice to monitor the time course and dynamic changes of autophagy and autophagic flux in post-ischemic renal tubules [20,30,31], autophagy was initially inhibited during 30 min of renal ischemia (Figure S3A: UI30R 0 h) and then induced at 6 hours of reperfusion (Figure S3A: UI30R 6 h). Notably, the induction of LC3B-II was not only maintained during the acute injury phase, but also further enhanced throughout the 4-week duration of post-ischemic kidney repair (Figure S3A: UI30R 1 d to 4 w). Together, these results suggest that the activation of MAPK may not relate to autophagy induction during the acute injury phase whereas is likely accompanied and associated with sustained autophagy during kidney repair. Moreover, in line with the induction of EGR1 and FGF2 in maladaptive renal tubules, phosphorylated MAPK1/ERK2-MAPK3/ERK1 was also predominantly induced in HAVCR1/KIM-1-positive renal tubules during post-ischemic kidney repair, with both nuclear and cytoplasmic signals detected in the cells (Figure S3B: p-MAPK1/ERK2-MAPK3/ERK1 and HAVCR1/KIM-1 co-staining). Phosphorylated MAPK/p38 was mainly induced in the nuclei of renal tubular cells during repair, but only a small portion of these p-MAPK/p38-positive renal tubules were co-stained with HAVCR1/KIM-1 (Figure S3B: p-MAPK/p38 and HAVCR1/KIM-1 co-staining). By contrast, the phosphorylation and activation of MAPK/JNK was mostly seen in the cytoplasm of HAVCR1/KIM-1-negative renal tubular cells and interstitial cells in post-ischemic kidneys (Figure S3C).
Autophagy deficiency specifically inhibits MAPK1/ERK2-MAPK3/ERK1 activation in maladaptive renal tubules and this inhibition correlates with SQSTM1/p62 aggregation and sequestration of MAPK1/ERK2-MAPK3/ERK1 in iRT-atg7 KO mice
Using iRT-atg7 KO mice, we then examined the role of autophagy in regulating the activation of MAPK signaling pathways in maladaptive renal tubules. MAPK1/ERK2-MAPK3/ERK1 was phosphorylated and activated in WT kidneys following ischemic AKI, which was suppressed by autophagy deficiency in iRT-atg7 KO mice (Figure 5A: p-MAPK1/ERK2-MAPK3/ERK1). This inhibition appeared to be time dependent. At 2 to 4 weeks after ischemic AKI, WT mice had an 8 to 9-fold increase of phosphorylated MAPK1/ERK2-MAPK3/ERK1 over sham control, which was reduced to 6.5-fold at 2 weeks and further down to 4-fold at 4 weeks in iRT-atg7 KO mice (Figure 5B: p-MAPK1/ERK2-MAPK3/ERK1). Downstream of MAPK1/ERK2-MAPK3/ERK1, the transcription factor ELK1 was also phosphorylated and activated in WT kidneys whereas this phosphorylation was attenuated in iRT-atg7 KO mice (Figure 5A: p-ELK1; Figure S4A). By contrast, the phosphorylation and activation of both MAPK/p38 and MAPK/JNK in post-ischemic kidneys were not affected by autophagy deficiency (Figure S5A, B). More importantly, the inhibition of MAPK1/ERK2-MAPK3/ERK1 phosphorylation in iRT-atg7 KO kidneys was accompanied with a robust, time-dependent accumulation of SQSTM1/p62 (Figure 5A: SQSTM1/p62). While only a slight, statistically insignificant increase of SQSTM1/p62 was seen in WT mice following ischemic AKI, SQSTM1/p62 accumulation in iRT-atg7 KO mice reached to 8.5-fold over sham control at 2 weeks and further elevated to 13-fold by 4 weeks (Figure 5B: SQSTM1/p62). Pearson correlation analysis followed by simple linear regression further confirmed a significant inverse correlation between SQSTM1/p62 accumulation and MAPK1/ERK2-MAPK3/ERK1 activation in post-ischemic kidneys (Figure 5C). Previous studies suggest that during adipogenesis SQSTM1/p62 may sequester MAPK/ERK into cytoplasmic aggregates to prevent the access of MAPK/ERK to its upstream kinase for phosphorylation and activation [32]. In line with this notion, co-immunofluorescence of SQSTM1/p62 and MAPK1/ERK2-MAPK3/ERK1 also revealed a clear colocalization of these two proteins in post-ischemic kidneys of iRT-atg7 KO mice. Autophagy deficiency led to the formation of numerous SQSTM1/p62 aggregates in post-ischemic renal tubules (Figure 5D: iRT-atg7 KO). By quantification, an average of 40 SQSTM1/p62-positive dots per 400× field was detected at 2 weeks after ischemic AKI, and approximately 65% of these SQSTM1/p62-positive dots (26 per 400× field) co-stained with MAPK1/ERK2-MAPK3/ERK1 dots (Figure 5E). Nevertheless, in WT mice with intact autophagy, SQSTM1/p62 aggregates were barely seen in renal tubular cells and a relatively homogenous MAPK1/ERK2-MAPK3/ERK1 staining was detected (Figure 5D: iRT-Atg7 WT). Together, these results suggest that autophagy deficiency specifically inhibits MAPK1/ERK2-MAPK3/ERK1 activation in maladaptive renal tubules and this inhibition correlates with SQSTM1/p62 aggregation and sequestration of MAPK1/ERK2-MAPK3/ERK1 in iRT-atg7 KO mice.
Figure 5.
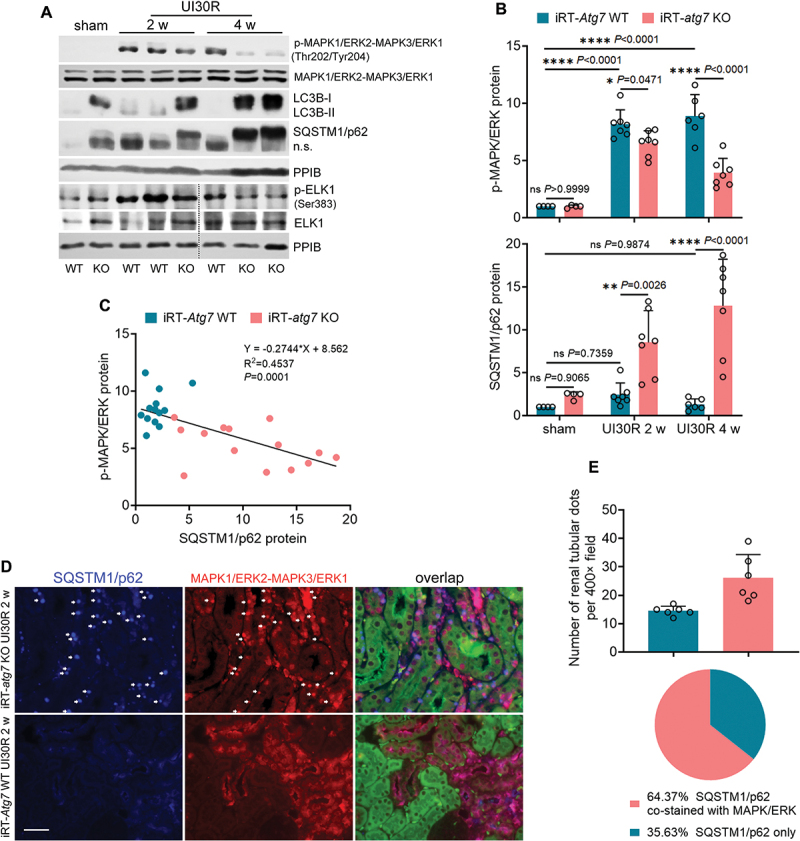
Autophagy deficiency inhibits MAPK1/ERK2-MAPK3/ERK1 activation in maladaptive renal tubules and this inhibition correlates with SQSTM1/p62 aggregation and sequestration of MAPK1/ERK2-MAPK3/ERK1 in iRT-atg7 KO mice. WT and iRT-atg7 KO mice underwent sham operation or 30-min unilateral renal ischemia followed by reperfusion for up to 4 weeks. Left kidneys were harvested. (A) immunoblot of p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204), MAPK1/ERK2-MAPK3/ERK1, LC3B, SQSTM1/p62, p-ELK1 (Ser383) and ELK1. (B) densitometry of p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204) and SQSTM1/p62 immunoblot (sham: n = 4; UI30R 2 w: WT n = 7, KO n = 7; UI30R 4 w: WT n = 6, KO n = 7). (C) correlation analysis of protein fold changes between p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204) and SQSTM1/p62. (D) co-immunofluorescence of MAPK1/ERK2-MAPK3/ERK1 (red) and SQSTM1/p62 (blue). Scale bar: 20 µm. (E) quantification of SQSTM1/p62 aggregates with or without MAPK1/ERK2-MAPK3/ERK1 co-staining in post-ischemic iRT-atg7 KO mice (n = 6). Data in (B) and (E) presented as mean ± SEM. For statistics, two-way ANOVA with multiple comparisons was used for (B). Pearson correlation analysis followed by simple linear regression was used for (C). 2-tailed, unpaired Student t-test and fraction of total analysis were used for (E).
SQSTM1/p62 interacts with MAPK1/ERK2-MAPK3/ERK1 to block its activation by TGFB1 in autophagy-deficient mouse proximal tubular cells
Consistent with the in vivo results, MAPK1/ERK2-MAPK3/ERK1 was also activated by TGFB1 in BUMPT cells, as indicated by an increased phosphorylation (Figure 6A, B). Pharmacological inhibition of autophagy by CQ or 3-MA significantly promoted SQSTM1/p62 accumulation and therefore abolished MAPK1/ERK2-MAPK3/ERK1 activation (Figure 6A, B). Similarly, the phosphorylation of MAPK1/ERK2-MAPK3/ERK1 in TGFB1-treated WT tubular cells was also suppressed in autophagy-defective, SQSTM1/p62-accumulating atg7 KO tubular cells (Figure 6C, D). Consistent with MAPK1/ERK2-MAPK3/ERK1, the activation and phosphorylation of ELK1 in TGFB1-treated tubular cells was inhibited as well (Figure 6A, C: p-ELK1; Figure S4B, C). Once again, under these conditions, the accumulation of SQSTM1/p62 inversely correlated with MAPK1/ERK2-MAPK3/ERK1 phosphorylation and activation in TGFB1-treated renal tubular cells (Figure 6E). Moreover, the activation of both MAPK/p38 and MAPK/JNK in TGFB1-treated renal tubular cells was not affected by autophagy inhibition (Figure S5C-F). These in vitro findings provide further evidence that autophagy may specifically activate MAPK1/ERK2-MAPK3/ERK1 in maladaptive renal tubules and this activation can be blocked by the accumulation of SQSTM1/p62 aggregates in autophagy-deficient tubular cells. Originally described as a scaffold protein to form signaling hubs, SQSTM1/p62 can interact with multiple signaling pathways through its different binding domains [33,34]. To further determine how autophagy activates MAPK1/ERK2-MAPK3/ERK1 and the inhibitory role of SQSTM1/p62, we analyzed protein interaction between SQSTM1/p62 and MAPK1/ERK2-MAPK3/ERK1 by co-immunoprecipitation (IP) assay. No obvious interaction was detected in either control or TGFB1-treated BUMPT cells without autophagy inhibitor CQ. Addition of CQ during TGFB1 treatment remarkably increased the expression of SQSTM1/p62 in MAPK1/ERK2-MAPK3/ERK1 IP products (Figure 6F). Consistently, the interaction of MAPK1/ERK2-MAPK3/ERK1 with SQSTM1/p62 was absent in WT tubular cells. By contrast, a small amount of SQSTM1/p62 was pulled down by MAPK1/ERK2-MAPK3/ERK1 antibody in control atg7 KO tubular cells, which was further enhanced following TGFB1 treatment (Figure 6G). Together, these results suggest that SQSTM1/p62 aggregates may form an inhibitory complex with MAPK1/ERK2-MAPK3/ERK1 to block its activation in autophagy-deficient tubular cells.
Figure 6.
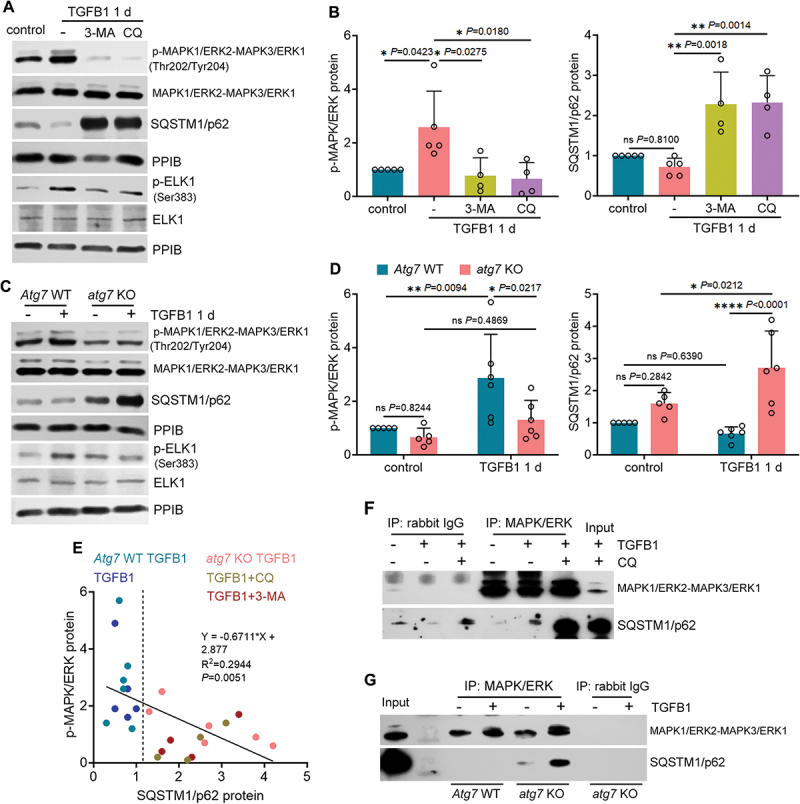
SQSTM1/p62 interacts with MAPK1/ERK2-MAPK3/ERK1 to block its activation by TGFB1 in autophagy-deficient mouse proximal tubular cells. (A and B) subconfluent BUMPT cells were exposed to 5 ng/ml TGFB1 in serum-free DMEM for 1 day alone or with 20 µM CQ or 5 mM 3-MA. Control cells were kept in serum-free medium without TGFB1. Cells were collected for immunoblot of p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204), MAPK1/ERK2-MAPK3/ERK1, SQSTM1/p62, p-ELK1 (Ser383) and ELK1 (n = 5 experiments). (C and D) subconfluent WT and atg7 KO cells were exposed to 5 ng/ml TGFB1 in serum-free DMEM for 1 day. Control cells were kept in serum-free medium without TGFB1. Cells were collected for immunoblot of p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204), MAPK1/ERK2-MAPK3/ERK1, SQSTM1/p62, p-ELK1 (Ser383) and ELK1 (n = 5 experiments). (E) correlation analysis of protein fold changes between p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204) and SQSTM1/p62. (F) co-IP of MAPK1/ERK2-MAPK3/ERK1 and SQSTM1/p62 in BUMPT cells (n = 3 experiments). (G) co-IP of MAPK1/ERK2-MAPK3/ERK1 and SQSTM1/p62 in WT and atg7 KO cells (n = 3 experiments). Data in (B) and (D) are presented as mean ± SEM. For statistics, one-way ANOVA with multiple comparisons was used for (B). Two-way ANOVA with multiple comparisons was used for (D). Pearson correlation analysis followed by simple linear regression was used for (E).
Inhibition of SQSTM1/p62 aggregation further enhances MAPK1/ERK2-MAPK3/ERK1 phosphorylation in TGFB1-treated mouse proximal tubular cells
SQSTM1/p62 is a 440-amino acid (aa) protein consisting of six main domains: Phox and Bem1 domain (PB1, 21–103 aa), zinc finger motif (ZZ, 128–163 aa), TRAF6-binding domain (TB, 225–250 aa), LC3-interacting region (LIR, 321–345 aa), KEAP1-interacting region (KIR, 346–359 aa), and ubiquitin-associated domain (UBA, 386–440 aa) [33,34] (Figure 7A). While the N-terminal PB1 domain-mediated self-oligomerization is needed for SQSTM1/p62 to form cytoplasmic protein aggregates, the LIR domain is responsible for the incorporation of SQSTM1/p62 aggregates into LC3-positive mature autophagosomes for degradation [35,36]. To better understand the role of SQSTM1/p62 aggregation in suppressing MAPK1/ERK2-MAPK3/ERK1 activation, we established BUMPT cells stably expressing wild type GFP-SQSTM1/p62 (WT), its PB1 mutant (K7A D69A), or LIR mutant (D337 338 339A), respectively. Compared with a low level of endogenous SQSTM1/p62, a good expression of exogenous GFP-SQSTM1/p62 or its mutants was detected in all transfected BUMPT cells (Figure 7B). Notably, GFP-SQSTM1/p62 WT formed many cytoplasmic aggregates ranging from small dots with weak GFP signal to large puncta with intense fluorescence in control cells (Figure 7C: WT control). Such aggregation was alleviated following TGFB1 treatment, likely owing to the induction of autophagy (Figure 7C: WT TGFB1). Compared with WT, GFP-SQSTM1/p62 LIR mutant was defective in LC3 binding and consequent autophagic degradation, therefore leading to the accumulation of numerous larger and brighter GFP-SQSTM1/p62 puncta in both control and TGFB1-treated BUMPT cells (Figure 7C: LIR mutant). By contrast, the expression of GFP-SQSTM1/p62 PB1 mutant appeared to be homogenous and diffuse, with no obvious punctate staining detected in either control or TGFB1-treated cells (Figure 7C: PB1 mutant). We then examined the effects of these constructs on MAPK/ERK activation. Compared with control, TGFB1 induced a 2-fold increase of MAPK1/ERK2-MAPK3/ERK1 phosphorylation in BUMPT cells expressing GFP-SQSTM1/p62 WT (Figure 7D, E: WT). Albeit statistically insignificant (P = 0.0552), the aggregates-promoting LTR mutant seemed to suppress MAPK/ERK activation following TGFB1 treatment, reducing the expression of phospho-MAPK1/ERK2-MAPK3/ERK1 to ~ 1.5-fold over control (Figure 7D, E: LIR mutant). Conversely, the PB1 mutant, via eliminating the formation of SQSTM1/p62 cytoplasmic aggregates, further enhanced MAPK1/ERK2-MAPK3/ERK1 phosphorylation to 2.8-fold in TGFB1-treated BUMPT cells (Figure 7D, E: PB1 mutant). These results provide direct evidence that SQSTM1/p62 aggregation and sequestration is essential to its inhibitory effects on MAPK/ERK in renal tubular cells. Persistent induction of autophagy in post-ischemic renal tubules may prevent the accumulation of SQSTM1/p62 aggregates, thereby liberating MAPK1/ERK2-MAPK3/ERK1 from SQSTM1/p62 sequestration for activation during maladaptive kidney repair.
Figure 7.
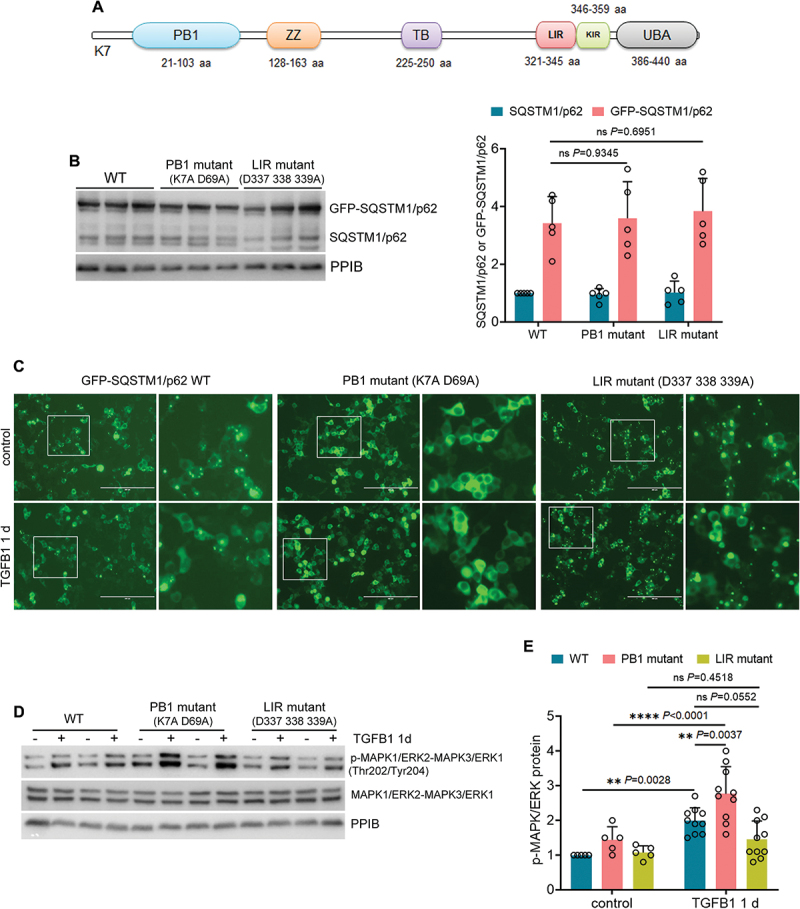
Inhibition of SQSTM1/p62 aggregation further enhances MAPK1/ERK2-MAPK3/ERK1 phosphorylation in TGFB1-treated mouse proximal tubular cells. (A) structural domains in SQSTM1/p62 protein. (B) BUMPT cells stably expressing GFP-SQSTM1/p62 WT, PB1 mutant (K7A D69A), or LIR mutant (D337 338 339A) were collected for immunoblot analysis (n = 5 experiments). (C-E) BUMPT cells stably expressing GFP-SQSTM1/p62 WT, PB1 mutant (K7A D69A), or LIR mutant (D337 338 339A) were incubated for 1 day with or without 5 ng/ml TGFB1 in serum-free DMEM for morphological and immunoblot analyses. (C) Representative fluorescence images showing the formation of GFP-SQSTM1/p62 aggregates. Scale bar: 200 µm. The boxed areas were enlarged and placed on the right side. (D) immunoblots of p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204) and MAPK1/ERK2-MAPK3/ERK1. (E) densitometry of p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204) immunoblot (n = 5 experiments). Data in (B) and (E) are presented as mean ± SEM and two-way ANOVA with multiple comparisons was used for statistics.
Autophagy deficiency does not affect the activation of MAP2K1/MEK1-MAP2K2/MEK2 in renal tubules during post-ischemic kidney repair or in TGFB1-treated mouse proximal tubular cells
Next, we determined whether MAP2K1/MEK1-MAP2K2/MEK2, the upstream kinases of MAPK/ERK are involved in autophagy-mediated MAPK/ERK activation during renal fibrotic stress. TGFB1 activated MAP2K1/MEK1-MAP2K2/MEK2 in WT tubular cells, as indicated by an increased phosphorylation (Figure S6A, B). Unlike MAPK/ERK, the activation of MAP2K1/MEK1-MAP2K2/MEK2 by TGFB1 was not affected in atg7 KO tubular cells (Figure S6A, B), suggesting that tubular autophagy may activate MAPK/ERK independent of its upstream kinase. These in vitro findings were also confirmed in mice. MAP2K1/MEK1-MAP2K2/MEK2 was phosphorylated and activated in WT kidneys following ischemic AKI, which again was not altered by autophagy deficiency in iRT-atg7 KO mice (Figure S6C, D). Moreover, in contrast to the significant colocalization of SQSTM1/p62 aggregates with MAPK1/ERK2-MAPK3/ERK1 in post-ischemic renal tubules of iRT-atg7 KO mice (Figure 5D, E), MAP2K1/MEK1-MAP2K2/MEK2 was barely trapped or sequestered by SQSTM1/p62 aggregation (Figure S6E: iRT-atg7 KO). At 2 weeks after ischemic AKI, only a very small portion (~13%) of SQSTM1/p62-positive aggregates co-stained with MAP2K1/MEK1-MAP2K2/MEK2 whereas the vast majority (~87%) did not (Figure S6F). Together, these results further support that SQSTM1/p62 aggregates may directly inhibit MAPK1/ERK2-MAPK3/ERK1 activation in maladaptive renal tubules without acting on its upstream kinase like MAP2K1/MEK1-MAP2K2/MEK2.
Inhibition of MAPK1/ERK2-MAPK3/ERK1 suppresses EGR1 activation and FGF2 production in TGFB1-treated mouse proximal tubular cells and in post-ischemic renal tubules during maladaptive repair
To further verify the role of MAPK1/ERK2-MAPK3/ERK1 in regulating EGR1 and FGF2 for renal fibrosis, we examined MAPK/ERK inhibitors both in vitro and in vivo. TGFB1 induced activation of MAPK1/ERK2-MAPK3/ERK1 and its downstream target ELK1 in BUMPT cells, which was remarkably attenuated by both MAPK/ERK inhibitors U0126 and PD98959 whereas not affected by U0124, a negative control inhibitor (Figure 8A: p-MAPK1/ERK2-MAPK3/ERK1, p-ELK1; Figure S4D). TGFB1-induced activation of MAPK/p38 and MAPK/JNK was barely altered under these conditions, confirming the specificity of U0126 and PD98959 on MAPK/ERK (Figure 8A: p-MAPK/p38, p-MAPK/JNK). Notably, both U0126 and PD98059 suppressed Egr1 induction in TGFB1-treated BUMPT cells, reducing its mRNA expression from 1.6-fold over control to 0.2- and 0.4-fold, respectively (Figure 8B: Egr1 mRNA). As a result, TGFB1-induced tubular production of Fgf2 at mRNA level was decreased from 3.8-fold over control to 1.9-fold by U0126 and 2.2-fold by PD98059 (Figure 8B: Fgf2 mRNA). Immunoblot analysis further confirmed that the induction of EGR1 and FGF2 at protein level was also significantly inhibited by both U0126 and PD98059 in TGFB1-treated BUMPT cells (Figure 8C, D). By contrast, the negative control inhibitor U0124 was ineffective (Figure 8B-D: U0124). Consistent with the in vitro findings, U0126 specifically suppressed the phosphorylation of MAPK1/ERK2-MAPK3/ERK1 and ELK1 in post-ischemic renal tubules without affecting the activation of MAPK/p38 or MAPK/JNK (Figure 9A, B; Figure S4E). Under these conditions, the induction of both Egr1 and Fgf2 mRNAs was mitigated by U0126 (Figure 9C). At protein level, U0126 reduced the number of EGR1-positive tubular cells in post-ischemic kidneys, which further led to the decrease of tubular expression of FGF2 (Figure 9D-F). Together, these results suggest that MAPK1/ERK2-MAPK3/ERK1 may activate EGR1 and FGF2 in TGFB1-treated mouse proximal tubular cells and in post-ischemic renal tubules during maladaptive repair. It is noteworthy that inhibition of MAPK1/ERK2-MAPK3/ERK1 also suppressed tubular cell autophagy in mice. At 2 weeks after ischemic AKI, the accumulation of LC3B-II in kidney tissues was reduced from 6.2-fold over sham control to 3-fold by U0126 (Figure S7A). Immunohistochemical staining of LC3B further revealed an increased punctate staining of membrane-bound LC3B-II mainly in the cytoplasm of post-ischemic renal tubular cells, which was also attenuated by this MAPK/ERK inhibitor (Figure S7B). These results, together with autophagy activation of MAPK1/ERK2-MAPK3/ERK1, suggest a potential positive feedback regulation between MAPK1/ERK2-MAPK3/ERK1 and autophagy in renal tubular cells that may act in concert to strengthen the maladaptive repair signaling and the production of pro-fibrotic factors such as FGF2.
Figure 8.
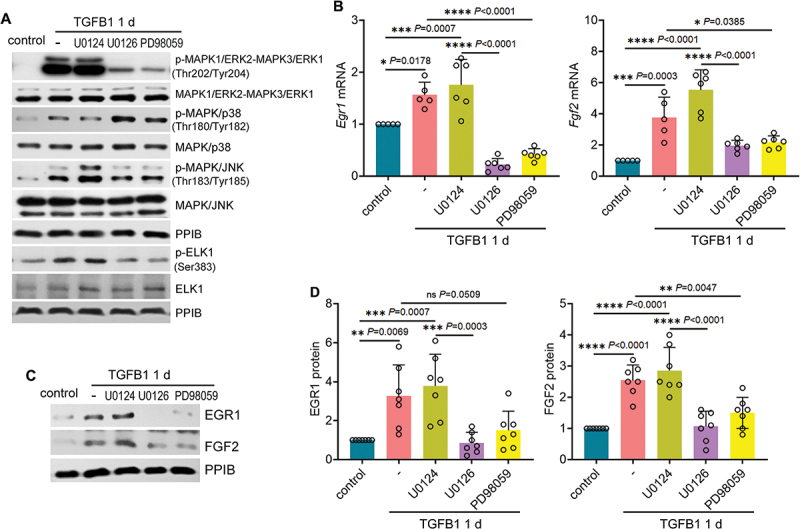
Inhibition of MAPK1/ERK2-MAPK3/ERK1 suppresses EGR1 activation and FGF2 production in TGFB1-treated mouse proximal tubular cells. Subconfluent BUMPT cells were exposed to 5 ng/ml TGFB1 in serum-free DMEM for 1 day alone or with negative control inhibitor (10 µM U0124) or with MAPK1/ERK2-MAPK3/ERK1 inhibitors (10 µM U0126, 50 µM PD98059). Control cells were kept in serum-free medium without TGFB1. (A) immunoblot of p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204), MAPK1/ERK2-MAPK3/ERK1, p-MAPK/p38 (Thr180/Tyr182), MAPK/p38, p-MAPK/JNK (Thr183/Tyr185), MAPK/JNK, p-ELK1 (Ser383) and ELK1 (n = 3 experiments). (B) RT-qPCR of Fgf2 and Egr1 mRNA (n = 5 experiments). (C and D) immunoblot and densitometry of EGR1 and FGF2 (n = 7 experiments). Data in (B) and (D) are presented as mean ± SEM. For statistics, one-way ANOVA with multiple comparisons was used for both (B) and (D).
Figure 9.
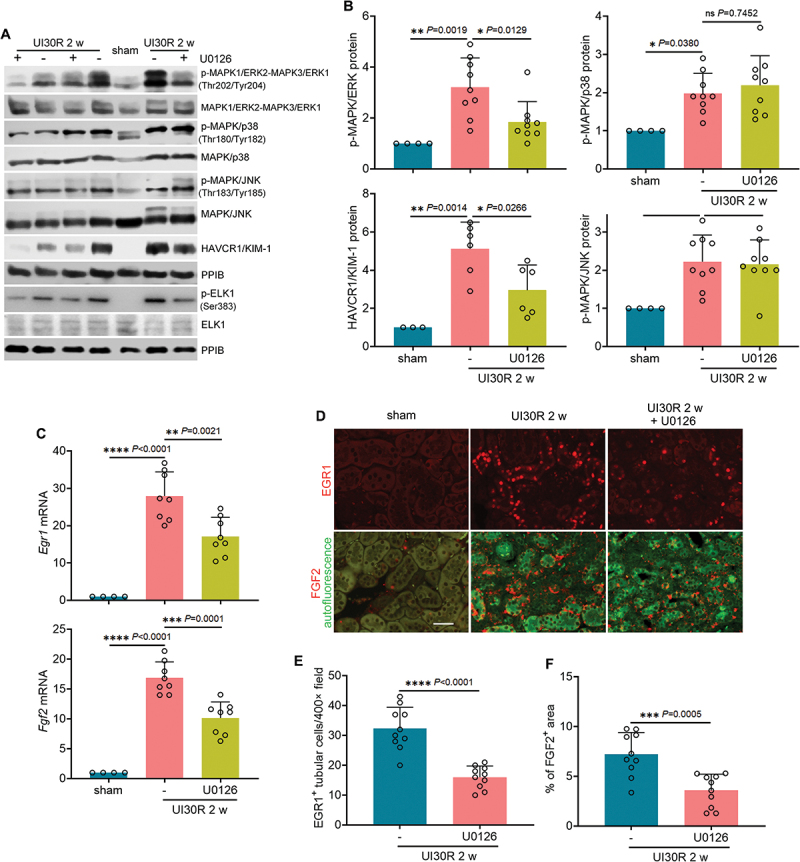
Inhibition of MAPK1/ERK2-MAPK3/ERK1 suppresses EGR1 and FGF2 in renal tubules during maladaptive kidney repair after ischemic AKI in mice. C57BL/6 mice underwent sham operation (n = 4) or 30-min unilateral renal ischemia followed by reperfusion alone (n = 11) or with U0126 (n = 10) (20 mg/kg, i.P., every other day injection, starting from day 2 of reperfusion) for 2 weeks. Left kidneys were harvested. (A) immunoblot of p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204), MAPK1/ERK2-MAPK3/ERK1, p-MAPK/p38 (Thr180/Tyr182), MAPK/p38, p-MAPK/JNK (Thr183/Tyr185), MAPK/JNK, HAVCR1/KIM-1, p-ELK1 (Ser383) and ELK1. (B) densitometry of p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204), p-MAPK/p38 (Thr180/Tyr182), p-MAPK/JNK (Thr183/Tyr185) and HAVCR1/KIM-1 immunoblot. (C) RT-qPCR of Egr1 and Fgf2 mRNA. (D) immunofluorescence of EGR1 and FGF2. Scale bar: 20 µm. (E) quantification of the numbers of EGR1-positive tubular cells. (F) quantification of FGF2-positive staining areas. Data in (B), (C), (E) and (F) are presented as mean ± SEM. For statistics, one-way ANOVA with multiple comparisons was used for (B) and (C). 2-tailed, unpaired Student t-test was used for (E) and (F).
Inhibition of MAPK1/ERK2-MAPK3/ERK1 ameliorates interstitial fibrosis and improves renal function in post-ischemic mice
Along with its inhibition on the axis of MAPK/ERK-EGR1-FGF2 in maladaptive renal tubular cells, U0126 suppressed the development of renal interstitial fibrosis in post-ischemic mice. At 2 weeks after ischemic AKI, massive collagen fibrils either accumulated around small blood vessels in renal cortex or widely spread along atrophic renal tubules in outer medulla. Notably, U0126 alleviated the collagen deposition in renal interstitium, reducing the fibrotic areas from 12.7% to 8% (Figure 10A, B). Consistently, multiple fibrotic markers, including Fn1, Col1a1, Col4a1 and Acta2/α-Sma (actin alpha 2, smooth muscle, aorta), were induced at mRNA level following ischemic AKI, all of which were significantly attenuated by U0126 (Figure 10C). We further examined the changes of these fibrotic markers at protein level by immunostaining. Compared with sham control mice that had minimal positive interstitial signals, increased production of FN1, COL1A1, COL4A1 and ACTA2/α-SMA was detected in renal interstitium throughout cortex and outer medulla in post-ischemic kidneys (Figure 10D). Again, U0126 significantly suppressed tubulointerstitial staining of these fibrotic proteins, further supporting the anti-fibrotic effect of this MAPK/ERK inhibitor (Figure 10D, Figure S8A). Moreover, inhibition of MAPK1/ERK2-MAPK3/ERK1 also partially preserved tubular structure and function to counteract maladaptive repair. The chronic expression of tubular injury marker HAVCR1/KIM-1 in post-ischemic kidneys, measured by both immunoblot (Figure 9A, B: HAVCR1/KIM-1) and immunostaining (Figure S8B, C), was diminished by U0126. To determine renal function of the repaired kidney, right nephrectomy was conducted to remove the contralateral kidney at 2 weeks after unilateral ischemic AKI. One day later, mice without U0126 treatment had 1.99 mg/dl serum creatinine, which was partially but significantly decreased to 1.55 by addition of U0126 (Figure 10E: serum creatinine). Albeit statistically insignificant (P = 0.0599), U0126 also reduced BUN from 121 mg/dl to 105 in the repaired kidney (Figure 10E: BUN). Together, these results suggest that inhibition of MAPK1/ERK2-MAPK3/ERK1 via suppressing EGR1 and FGF2 in maladaptive renal tubules may ameliorate interstitial fibrosis and improve renal function in post-ischemic mice for a better kidney repair.
Figure 10.
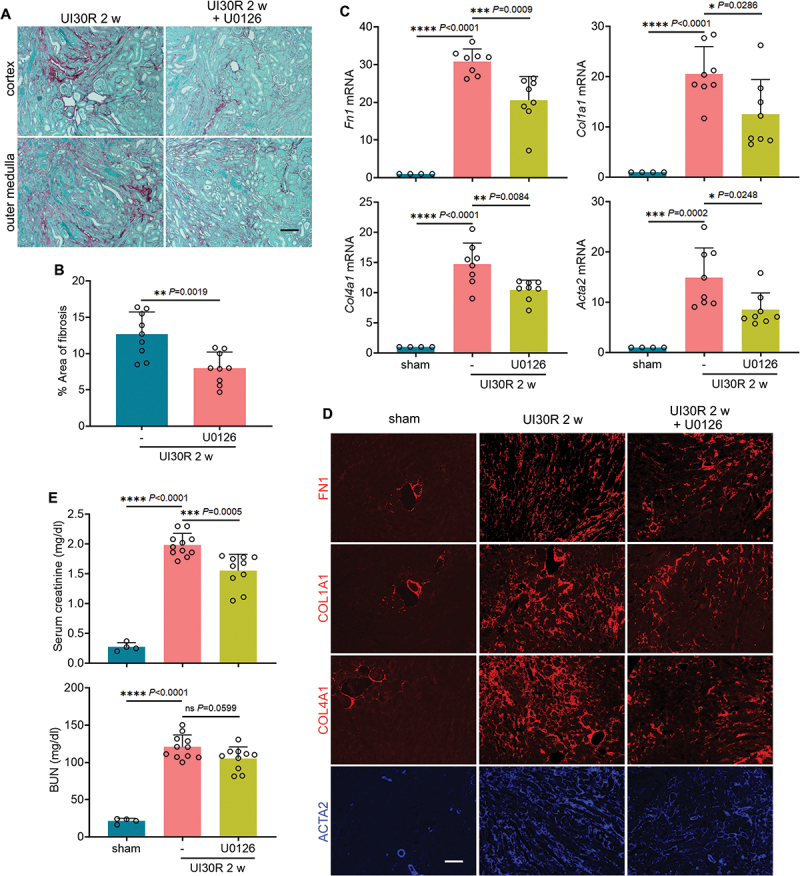
Inhibition of MAPK1/ERK2-MAPK3/ERK1 ameliorates interstitial fibrosis and improves renal function in post-ischemic mice. C57BL/6 mice were subjected to the treatments as described in Figure 9. (A) Sirius red/Fast Green staining. Scale bar: 100 µm. (B) quantification of collagen-stained areas. (C) RT-qPCR of Fn1, Col1a1, Col4a1 and Acta2/α-sma mRNA. (D) immunofluorescence of FN1, COL1A1, COL4A1 and ACTA2/α-SMA. Scale bar: 100 µm. (E) 2 weeks after unilateral ischemia-reperfusion, right kidneys were removed, and blood samples were collected 24 h after right nephrectomy for measurements of BUN and serum creatinine. Data in (B), (C) and (E) are presented as mean ± SEM. For statistics, 2-tailed, unpaired Student t-test was used for (B). One-way ANOVA with multiple comparisons was used for (C) and (E).
Discussion
In contrast to its renoprotective role against acute injury [37,38], tubular cell autophagy contributes to the pathogenesis of maladaptive kidney repair after AKI. Our recent work has demonstrated that autophagy persists at a high level in renal tubular cells after ischemic AKI leading to a secretory phenotype to produce pro-fibrotic cytokines. Among these cytokines, autophagy specifically induces FGF2. Once secreted by renal tubular cells into the interstitium, FGF2 acts as a key paracrine factor for fibroblast activation to promote interstitial fibrosis and maladaptive kidney repair [20]. In the current study, we further investigated the mechanisms underlying autophagy-dependent tubular production of FGF2 during maladaptive kidney repair after AKI (Figure 11). We showed that persistent induction of autophagy in maladaptive renal tubules suppressed the accumulation of SQSTM1/p62 aggregates, thereby preventing SQSTM1/p62 aggregates from interacting with MAPK1/ERK2-MAPK3/ERK1 to form an inhibitory complex. Autophagy-mediated activation of MAPK1/ERK2-MAPK3/ERK1 further induced the transcription factor EGR1. By directly binding to the promoter region of Fgf2 gene, EGR1 then transactivated FGF2 in renal tubular cells for subsequent paracrine activation of renal fibroblasts. EGR1 and FGF2 also formed a positive feedback to mediate FGF2 autoregulation to reinforce its transcriptional induction in tubular cells and paracrine effects on fibroblasts (Figure 11).
Figure 11.
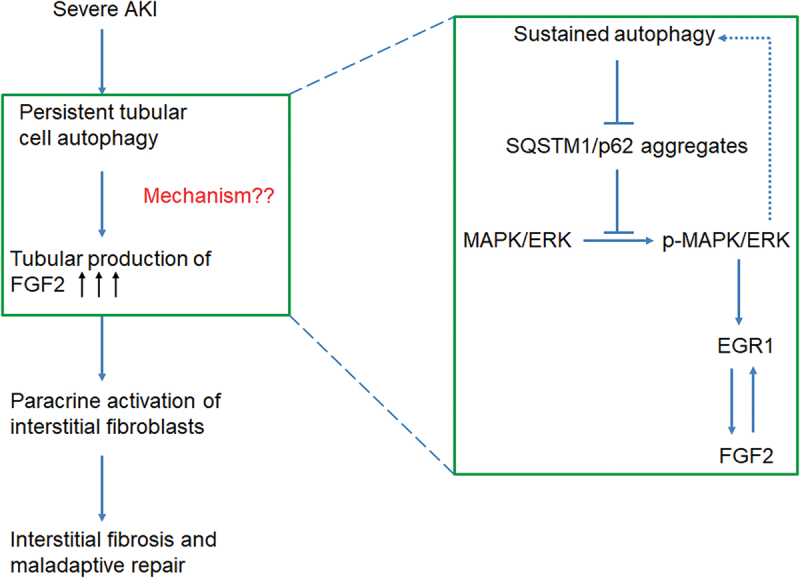
Graphic summary highlighting how tubular cell autophagy promotes interstitial fibrosis for maladaptive kidney repair through the SQSTM1/p62-MAPK/ERK-EGR1-FGF2 signaling cascade.
In this study, we documented a two-phase activation of the MAPK signaling pathways in renal tubules after renal ischemia, with distinct patterns in the injury phase and recovery phase. During the acute injury, a rapid but transient MAPK activation was induced, which did not well overlap with the timeline of autophagy induction. Nevertheless, all three MAPK members (MAPK1/ERK2-MAPK3/ERK1, MAPK/p38 and MAPK/JNK) were reactivated at the beginning of kidney recovery and maintained at high levels together with persistent autophagy throughout 4-week of reperfusion in the repaired kidneys. Moreover, the activation of MAPK1/ERK2-MAPK3/ERK1 but not MAPK/p38 or MAPK/JNK was consistently suppressed by autophagy inhibition in post-ischemic mice and in TGFB1-treated mouse proximal tubular cells, suggesting a specific regulation of MAPK1/ERK2-MAPK3/ERK1 by autophagy during kidney repair. Notably, an increasing number of studies have suggested the involvement of MAPK/ERK signaling pathway in the regulation of autophagy although the mechanism is extremely complex and sometimes the outcome seems contradictory. For example, while MAPK15/ERK8 may stimulate autophagy by interacting with LC3 [39], MAPK1/ERK2-MAPK3/ERK1 phosphorylates RAPTOR/RPTOR (regulatory-associated protein of MTOR) to promote the activation of MTORC1 (mechanistic target of rapamycin complex 1) potentially for autophagy inhibition [40]. Interestingly, in our experimental settings MAPK1/ERK2-MAPK3/ERK1 seemed to contribute to persistent autophagy as inhibition of MAPK1/ERK2-MAPK3/ERK1 by U0126 suppressed tubular cell autophagy in post-ischemic mice. Collectively, these results highlight a bidirectional crosstalk between MAPK/ERK and autophagy that may complicate the inter-regulation of these two pathways and the outcomes.
Mechanistically, we showed that autophagy-dependent MAPK/ERK activation during maladaptive kidney repair was negatively regulated by SQSTM1/p62 aggregation. The accumulation of SQSTM1/p62 inversely correlated with MAPK1/ERK2-MAPK3/ERK1 phosphorylation and activation in post-ischemic kidneys and in TGFB1-treated renal tubular cells. In iRT-atg7 KO mice autophagy deficiency led to the sequestration of MAPK1/ERK2-MAPK3/ERK1 within SQSTM1/p62 aggregates, as indicated by significant colocalization of these two proteins in post-ischemic renal tubules. Co-IP assay further confirmed that autophagy inhibition promoted a direct interaction between SQSTM1/p62 and MAPK1/ERK2-MAPK3/ERK1 in TGFB1-treated proximal tubular cells. Moreover, in BUMPT cells expressing GFP-SQSTM1/p62 constructs, the aggregation-promoting LIR mutant suppressed whereas the aggregation-inhibiting PB1 mutant enhanced MAPK1/ERK2-MAPK3/ERK1 activation following TGFB1 treatment, further supporting the importance of SQSTM1/p62 aggregation and sequestration to MAPK/ERK suppression in autophagy-deficient renal tubular cells. These results are consistent with previous studies showing that SQSTM1/p62 antagonizes basal MAPK/ERK activity and autophagy-mediated degradation of SQSTM1/p62 leads to the hyperactivation of MAPK/ERK in adipocytes [32,41], in hypoxic cancer cells [42], and in podocytes [43]. SQSTM1/p62 contains six domains necessary for its interaction with the autophagic machinery and with signaling pathways. Among them, the N-terminal PB1 domain is believed to be responsible for binding to MAPK/ERK [33]. In addition to SQSTM1/p62, Martinez-Lopez et al. also propose a model in which the cytoplasmic face of autophagic structures, such as LC3-II-positive autophagosomes and ATG5-ATG12-positive phagophores, may serve as scaffolds to exclusively dock the MAPK/ERK cascade components via the interactions between MAPK/ERK and ATG proteins [44]. Distinct kinase-docking domains on MAPK1/ERK2 are suggested to be the binding sites of ATG proteins. The formation of these autophagy-mediated cellular signaling platforms would allow efficient spatial coordination of MAPK/ERK signaling cascade to facilitate MAPK/ERK phosphorylation and activation in response to growth factor [44]. In this model, autophagy inhibition by deleting Atg7 or blocking LC3 lipidation or ATG5-ATG12 conjugation decreases MAPK/ERK phosphorylation without affecting MAP2K/MEK phosphorylation [44]. These results are consistent with our current findings in TGFB1-treated proximal tubular cells and in post-ischemic mouse renal tubules, underscoring not only a specific but also a direct regulation of MAPK/ERK by autophagy in different experimental settings.
Along with sustained MAPK1/ERK2-MAPK3/ERK1 activation, EGR1, a downstream target of MAPK/ERK signaling pathway, was also persistently induced in the repaired kidneys for weeks. Both EGR1 and phosphorylated MAPK1/ERK2-MAPK3/ERK1 were induced predominantly in the nuclei of renal tubules that were HAVCR1/KIM-1-positive, which also happened to be the same tubular segment responsible for FGF2 production, suggesting a spatial-temporal association of MAPK/ERK, EGR1 and FGF2 in chronically injured proximal tubules in favor of maladaptive kidney repair. Of interest, early studies using rodent ischemic AKI models have characterized a rapid but transient EGR1 induction primarily in the nuclei of the thick ascending limbs and principal cells of the collecting ducts [26,45]. Recently, Chen et al. further reveal that a quick and transient upregulation of EGR1 in renal tubules alleviates kidney injury and promotes normal repair by stimulating the proliferation of SOX9+ tubular cells after AKI [46]. Given these findings, we speculate that the activation of EGR1 may also be a two-phase event with significant differences in its duration, localization and functional role between acute injury and kidney repair. Moreover, inhibition of MAPK/ERK suppressed the mRNA and protein expressions of EGR1 both in post-ischemic kidneys and in TGFB1-treated proximal tubular cells, suggesting the involvement of a transcriptional regulation of EGR1 by MAPK/ERK. Indeed, downstream of MAPK/ERK, the transcription factor ELK1 was phosphorylated and activated, and this activation was autophagy-dependent and suppressed by the MAPK/ERK inhibitor U0126 both in vivo and in vitro. These results suggest a mechanism of EGR1 regulation by MAPK/ERK, i.e., MAPK/ERK phosphorylates and activates ELK1, which then induces the transcription of EGR1. Recent studies report that MAPK3/ERK1 may phosphorylate EGR1 at serine 26 in human vascular endothelial cells, which is critical for EGR1 to regulate angiogenesis [47–49]. Whether MAPK3/ERK1 regulates EGR1 by direct phosphorylation in renal fibrosis awaits investigation. In addition to these MAPK/ERK-mediated mechanisms, EGR1 has been reported to undergo epigenetic regulation independent of MAPK/ERK. In rat renal fibroblasts (NRK-49F) microRNA-181 directly targets EGR1 to inhibit fibrotic changes during angiotensin II stimulation [50]. Moreover, MBD2 (methyl-CpG-binding domain protein 2) induces hypomethylation of Egr1 via interacting with the promoter region of the CpG islands of the gene and upregulates Egr1 expression to promote extracellular matrix production in murine embryonic NIH 3T3 fibroblasts [51]. It would be interesting to investigate the involvement of these mechanisms in regulating EGR1 in maladaptive renal tubular cells.
Transcriptional induction of FGF2 by EGR1 has been implicated in different types of cells such as cardiomyocytes [52], vascular endothelial cells [53], vascular smooth muscle cells [54], and astrocytes [22]. In maladaptive renal tubular cells, the persistent induction of EGR1 was dependent on autophagy. Knockdown of Egr1 reduced FGF2 expression at both mRNA and protein levels in TGFB1-treated tubular cells, indicating an important role of EGR1 in promoting autophagy-mediated FGF2 production. ChIP assay further revealed an increased occupancy of Fgf2 gene promoter by EGR1 following TGFB1 treatment, thus confirming a transcriptional regulation. In addition to EGR1, we also examined SMAD2, NFKB and TRP53/p53. Notably, the activation of these three transcription factors in TGFB1-treated renal tubular cells was not affected by autophagy inhibition, suggesting a specific regulation of EGR1 by autophagy for FGF2 transactivation. Using single-nucleus RNA sequencing, recent studies have revealed a dynamic cell cycle response of proximal tubular cells to AKI and identified a failed-repair proximal tubular cell state [55,56]. Lineage tracing of cycling cells and single-nucleus multiomics further highlight a distinct transcriptional program of this failed-repair proximal tubular cell state, which is characterized by upregulation of immediate-early response genes, cis-regulatory elements and transcriptional regulators, and corresponding changes in target gene expression [57]. These results not only offer novel insights into the regulation of post-injury kidney repair but also lay the foundation for further profiling and comparison of the transcriptomes of maladaptive renal tubular cells in autophagy-competent and autophagy-defective kidneys.
Despite the extensive studies in the past decade, the role of tubular cell autophagy in renal interstitial fibrosis remains controversial, with both pro- and anti-fibrotic been documented [38]. In rodent models of unilateral ureteral obstruction (UUO), persistent activation of autophagy in renal tubules may promote interstitial fibrosis via inducing tubular degeneration or stimulating lipid accumulation and lipotoxicity [58,59]; whereas autophagy-mediated degradation of TGFB1 or suppression of G2/M arrest may be anti-fibrotic [60,61]. However, given that UUO is characterized by a rapid and irreversible progression of renal fibrosis, these contradictory findings may not accurately represent the pathological role of autophagy in maladaptive repair after AKI and animal models with defined phases of injury and repair are preferred. Recent studies by us and others have taken the advantage of renal ischemia-reperfusion models and provided novel perspectives in this regard. Baisantry et al. demonstrated that, following ischemic AKI, autophagy promotes the severely damaged tubular cells to survive likely via inducing cellular senescence, which lead to prolonged renal inflammation and increased renal fibrosis [62]. Canaud et al. suggested that, in G2/M-arrested proximal tubular cells, autophagy might participate in the formation of TASCCs (TOR-autophagy spatial coupling compartments) to facilitate the production of pro-fibrotic factors for maladaptive repair [13]. Using the iRT-atg7 KO mouse model with tubular cell autophagy selectively inhibited during kidney repair, we recently provided compelling evidence that persistent tubular cell autophagy promotes interstitial fibrosis after severe ischemic AKI [20]. Under this condition, autophagy may orchestrate other tubular cell stress responses, such as dedifferentiation, senescence, and G2/M arrest, to boost a secretory phenotype transformation to produce pro-fibrotic FGF2 [20]. In the current study, we have further revealed how autophagy in renal tubular cells elevates interstitial fibrosis during maladaptive kidney repair through the SQSTM1/p62-MAPK/ERK-EGR1-FGF2 signaling cascade. These findings advance our understanding of the pathogenesis of AKI to CKD transition and the critical role for tubular cell autophagy in this process. Given the evidence that in kidney biopsies from post-AKI patients the induction of autophagy and FGF2 in renal tubules is positively correlated with the development of interstitial fibrosis [20], targeting autophagy and/or its downstream signaling during kidney repair might represent a potential therapeutic strategy to prevent AKI progression.
Materials and methods
Study approval
All the animal experiments were conducted in compliance with a protocol approved by the Institutional Animal Care and Use Committee of Charlie Norwood VA Medical Center (Animal Component of Research Protocol 23-03-139).
Animals
C57BL/6 mice were originally purchased from the Jackson Laboratory and bred in house for the study. Atg7-floxed (Atg7flox/flox) mice were obtained from Dr. Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science, Japan) [63]. The double-transgenic mouse line (Pax8-rtTA±, LC1±) was provided by Dr. Robert Koester (University of Heidelberg, Germany) [64]. The generation of a doxycycline-inducible, renal tubule-specific atg7 KO mouse model (iRT-atg7 KO) was described in our recent study [20]. Doxycycline hyclate (Sigma-Aldrich, D9891) was dissolved in drinking water containing 5% sucrose and given to mice at 0.2 mg/ml for 1 week to induce Atg7 deletion from renal tubules. To block tubular autophagy during kidney repair without affecting initial injury, doxycycline treatment was started 1 day before renal ischemia surgery and ended 6 days after reperfusion [20]. Fgf2 KO mouse line (Fgf2tm1Doe/J, Stock #003256) was purchased from the Jackson Laboratory. Mice were housed in a pathogen-free animal facility at Charlie Norwood VA Medical Center under a 12/12-h of light/dark pattern with free access to food and water. To avoid confounding effects of age, body weight and strain background, littermate male mice of 10 to 12 weeks old and with 24 to 28 grams of body weight were used for experiments.
Mouse model of unilateral ischemic AKI
Before surgery, mice were given a mix of ketamine (110 mg/kg, i.p.) and xylazine (11 mg/kg, i.p.) for anesthesia and sustained-release buprenorphine (1 mg/kg, s.c.) for analgesia. A homeothermic blanket control unit with rectal probe (Harvard Apparatus, 507220F) was used during surgery to monitor and maintain body temperature at ~ 36.5°C. To induce unilateral renal ischemia, the left renal pedicle was exposed by flank incision and clamped for 30 min. The clamp was then released for reperfusion for up to 4 weeks with the right kidney kept intact. Kidney color changes were observed to indicate sufficient ischemia and reperfusion. Sham control mice underwent the same operation without renal pedicle clamping. To test the effect of MAPK/ERK inhibitor U0126 on post-ischemic AKI repair, starting from day 2 of reperfusion, U0126 (20 mg/kg; Selleckchem, S1102) or vehicle (10% dimethyl sulfoxide, 40% polyethylene glycol 300, 5% Tween 80 [Fisher Scientific, T164–500] in saline) was given to C57BL/6 mice via i.p. every other day for 2 weeks. Upon the completion of U0126 treatment, right kidneys were removed by nephrectomy surgery. Left kidneys and blood samples were collected 24 h after right nephrectomy for further analysis.
Cells
The Boston University mouse proximal tubular (BUMPT) cell line was originally from Dr. Wilfred Lieberthal (Boston University School of Medicine) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; ThermoFisher Scientific 12100046) with 10% fetal bovine serum (FBS) [65]. The immortalized atg7 KO mouse proximal tubular cell line and its floxed control/WT cell line were generated by our lab recently [58]. These 2 stable cell lines were grown in DMEM containing 10% FBS, insulin (10 µg/ml; Sigma-Aldrich, I9278), TRF (transferrin; 5 µg/ml; Sigma-Aldrich, T8158), EGF (epidermal growth factor; 10 ng/ml; Sigma-Aldrich, E9644) and dexamethasone (4 µg/ml; Sigma-Aldrich, D1756). Isolation of primary proximal tubules from 5-week-old WT and fgf2 KO mice was performed according to an established protocol described in our previous work [58]. The primary cells were grown in DMEM/F12 medium (ThermoFisher Scientific 12500062) supplemented with 10% FBS, insulin (5 µg/ml), TRF (5 µg/ml), hydrocortisone (50 nM; Sigma-Aldrich, H0888), and vitamin C (50 µM; Sigma-Aldrich, A4544). The rat kidney fibroblast cell line NRK-49F (CRL-1570) was obtained from the American Type Culture Collection and cultured in DMEM with 10% FBS.
To generate Egr1 knockdown mouse proximal tubular cells, MISSION® shRNA targeting mouse Egr1 (Sigma-Aldrich, TRCN0000231215, sequence: CCGGGCGATGGTGGAGACGAGTTATCTCGAGATAACTCGTCTCCACCATCGCTTTTTG) was packaged into lentivirus and transduced to BUMPT cells following the manufacturer’s instructions. The cells were then selected with 2.5 µg/ml puromycin to have stable Egr1 knockdown cells. Non-target MISSION® shRNA (Sigma-Aldrich, SHC016) was used as a negative control. The efficiency of gene silencing was confirmed by real time-quantitative PCR (RT-qPCR) assay and immunoblot analysis of EGR1.
To generate BUMPT cells stably expressing GFP-SQSTM1/p62, 3 retroviral plasmids were used respectively, including pMXs-puro GFP-SQSTM1/p62 (Addgene 38277), pMXs-puro GFP-SQSTM1/p62 K7A D69A (Addgene 38281) and pMXs-puro GFP-SQSTM1/p62 D337 338 339A (Addgene 38280) (all deposited by Dr. Noboru Mizushima) [36]. 293-pheonix cells were transfected with the plasmids using Lipofectamine 2000 (ThermoFisher Scientific 11668). The culture media containing high titer viruses were collected at 48 h to further infect BUMPT cells that were seeded the night before. The cells were monitored by microscopy the next day for GFP signal to determine the transfection efficiency and then subjected to puromycin selection for about a week to obtain stable cells.
TGFB1 treatment of renal proximal tubular cells
Cells were seeded in 35-mm dishes at a density of 0.4 × 106 cells/dish for BUMPT or 0.5 × 106 cells/dish for WT and atg7 KO mouse proximal tubular cells. The first passage of primary proximal tubular cells (WT and fgf2 KO PT) was grown for 9 to 10 days after isolation and then plated in 35-mm dishes at a density of 0.2 × 106 cells/dish. Cells reached ~ 60% confluence by the next day and were exposed to 5 ng/ml human recombinant TGFB1 (Sigma-Aldrich, T7039) in serum-free DMEM for up to 3 days. In the experiment testing the effect of autophagy inhibitors, CQ (5 µM for 2-day treatment and 20 µM for 1-day treatment; Sigma-Aldrich, C6628) or 3-MA (1 mM for 2-day treatment and 5 mM for 1-day treatment; Sigma-Aldrich, M9281) was added during TGFB1 treatment of BUMPT cells. In the experiments testing the effect of FGF2 neutralizing antibody (Millipore, 05–117), the antibody or mouse IgG was added to BUMPT cells on day 2 of TGFB1 treatment to counteract a robust secretion of FGF2 on day 3 as suggested in our recent work [20]. FGF2 neutralizing antibody was tested at the concentrations of 5 and 10 µg/ml. In the experiments testing the effect of MAPK/ERK inhibitors (Millipore 453710-1SET), U0126 (10 µM), PD98059 (50 µM) or a negative control inhibitor U0124 (10 µM) was added to BUMPT cells during TGFB1 treatment. Moreover, PFT (25 µM, Sigma-Aldrich 506132) or JSH-23 (0.5 or 1 µM, Selleckchem, S7351) was added to BUMPT cells during TGFB1 treatment for the inhibition of TRP53/p53 or NFKB. Cells were monitored morphologically and collected at the indicated time points after treatment for further analysis.
Tubular cell-CM treatment of renal fibroblasts
Collection and purification of tubular cell-CM for treating renal fibroblasts was described in our recent work [20]. Briefly, stable Egr1 knockdown BUMPT cells and negative control shRNA-transfected BUMPT cells were seeded in 60-mm dishes at a density of 1 × 106 cells/dish. Cells reached ~ 60% confluence by the next day and were treated with 5 ng/ml TGFB1 in serum-free medium for 2 days, while control cells were kept in serum-free medium without TGFB1 for 2 days. The culture media for both TGFB1-treated and control cells were then replaced with fresh media free of TGFB1 and incubated for another day to collect tubular cell-CM. To purify and concentrate, 4 ml tubular cell-CM from each 60-mm dish was collected and centrifuged at 1,000 × g for 5 min at room temperature to remove cell debris. The supernatant was then transferred to a Milli-Q® water pre-rinsed centrifugal filter unit (Millipore, UFC8003, Amicon® Ultra-4 centrifugal filter with a 3 kDa molecular weight cutoff). After centrifugation at 7,500 × g on a fixed-angle rotor for 1 h at 4°C, approximately 100 µl concentrated CM was recovered. The secretion of FGF2 in concentrated CM was verified by immunoblot analysis. To treat renal fibroblasts with tubular cell-CM, NRK-49F fibroblasts were seeded in 12-well plate at a density of 0.15 × 106 cells/well to reach ~ 80% confluence by the next day. Following overnight starvation in serum-free medium, subconfluent NRK-49F fibroblasts were incubated with tubular cell-CM (100 µl diluted in 1 ml serum-free DMEM) for 2 days. Cells were monitored morphologically, trypsinized for counting using Bio-Rad TC20 automated cell counter, and then lysed for cellular protein measurement and immunoblot analysis.
RNA isolation and RT-qPCR
Total RNA was extracted from cells or snap-frozen kidney tissues with GeneJET RNA purification kit (ThermoFisher Scientific, K0732) following the manufacturer’s protocol. cDNA was synthesized from 1 µg total RNA using iScript™ cDNA synthesis kit (Bio-Rad 1708891) and RT-qPCR was done with iTaq™ Universal SYBR® Green supermix (Bio-Rad 1725121) following the manufacturer’s instructions. GAPDH (Glyceraldehyde-3-phosphate dehydrogenase) was used as an endogenous control for normalization. Comparative CT (ΔΔCT) was set up on StepOne™ Software for quantification as described in our previous studies [66,67]. The primer sequences used for RT-qPCR were listed in Table S1.
ChIP assay
ChIP assay of EGR1 binding to Fgf2 promoter region was performed according to a method described in our recent studies [66,67]. Briefly, BUMPT cells were fixed in 0.75% formaldehyde, neutralized with glycine, and collected in cold PBS (in g/L: 8.0 NaCl, 0.2 KCl, 1.44 Na2HPO4, 0.24 KH2PO4, pH 7.4). After centrifugation at 1,000 × g for 5 min at 4°C, the cell pellets were lysed and sonicated in a buffer containing 5 mM HEPES, pH 7.9, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100 (Sigma-Aldrich, T9284-500 ML), 0.1% Na-deoxycholate (Sigma-Aldrich, D6750-25 G), 0.1% SDS (Fisher Scientific, S529–500) to shear the DNA. After centrifugation at 8,000 × g for 30 s at 4°C, the supernatants were collected as the chromatin samples. For each chromatin sample, a 50 µl aliquot was incubated with 1 µl RNase A (1 mg/ml; ThermoFisher Scientific, AM2271) and 2 µl proteinase K (20 mg/ml; ThermoFisher Scientific 25530049) overnight at 65°C, and the input DNA was extracted using a QIAquick PCR purification kit (QIAGEN 28104). Based on input DNA concentration, the chromatin samples were diluted with the sonication buffer to 1 µg/ml DNA and incubated with 1 mg/ml BSA (Sigma-Aldrich, A9647-100 G) and 40 µl protein A agarose beads (Santa Cruz Biotechnology, sc-2001) for 2 h at 4°C. After beads removal, the pre-cleaned chromatin samples were incubated overnight with 4 µg anti-EGR1 antibody (Cell Signaling Technology, 4153) to pull down chromatin. The EGR1-chromatin complex was further precipitated with protein A agarose beads and washed sequentially with the sonication buffer, buffer A (50 mM HEPES, pH 7.9, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS), buffer B (20 mM Tris-HCl, pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40 [United States Biological, N3500], 0.5% Na-deoxycholate) and TE (Tris-EDTA) buffer. After eluted with elution buffer (50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% SDS, 50 mM NaHCO3), the purified DNA samples were quantified by RT-qPCR using the following Fgf2 primers: forward (5’-TGGCCTACAGAGTGAGTTCTA-3’) and reverse (5’-TCACCTTCCTCCTCTCTCTTC-3’). The input DNA was used for normalization.
Co-IP of MAPK1/ERK2-MAPK3/ERK1 and SQSTM1/p62
Following treatment, cells were lysed with RIPA buffer (150 mM NaCl, 25 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktail (Sigma-Aldrich, P8340) on ice for 10 min. After centrifugation at 3,000 × g for 10 min at 4°C, the supernatants were collected, and protein concentration was determined by Pierce™ BCA protein assay kit (ThermoFisher Scientific 23225). For each sample, 300 µg cell lysate (diluted with RIPA buffer to 500 µL) was used for IP with PierceTM Direct Magnetic IP/Co-IP kit (ThermoFisher Scientific 88828) following the manufacturer’s protocol. Briefly, 5 µg rabbit anti-MAPK1/ERK2-MAPK3/ERK1 (137F5; Cell Signaling Technology 68303; BSA and azide free) or rabbit IgG was incubated with 25 µL pre-washed magnetic beads on a rotating platform for 1 h at room temperature. After wash, the antibody-coupled beads were then incubated with the diluted cell lysates on a rotating platform for 2 h at room temperature or overnight at 4°C. Precipitated proteins on the beads were washed and eluted for immunoblot analysis of MAPK1/ERK2-MAPK3/ERK1 and SQSTM1/p62.
Immunoblot analysis
Cultured cell or kidney tissue lysates were extracted in SDS buffer containing 62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol supplemented with protease inhibitor cocktail and Benzonase® Nuclease (Millipore 70746). After protein concentration measurement, equal amounts of protein (10 µg for cell lysate, 50 µg for tissue lysate) were loaded in each lane, separated by SDS-PAGE under reducing conditions, and transferred onto PVDF membrane for standard procedure of immunoblot analysis. For concentrated tubular cell-CM, equal volume of 10 µl was loaded in each lane for electrophoresis. Primary antibodies for immunoblot: rabbit anti-EGR1 (15F7; Cell Signaling Technology, 4153; 1:1000), rabbit anti-FGF2 (Abcam, ab106245, discontinued; 1:500), mouse anti-FGF2 (Millipore, 05–118; 1:2000), rabbit anti-LC3B (Novus Biologicals, NB100–2220; 1:1000), mouse anti-SQSTM1/p62 (Abcam, ab56416; 1:2000), rabbit anti-MAPK1/ERK2-MAPK3/ERK1 (137F5; Cell Signaling Technology, 4695; 1:1000), rabbit anti-phospho-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204; Cell Signaling Technology, 9101; 1:1000), rabbit anti-ELK1 (Cell Signaling Technology, 9182; 1:1000), rabbit anti-phospho-ELK1 (Ser383; Cell Signaling Technology, 9181; 1:1000), rabbit anti-MAP2K1/MEK1-MAP2K2/MEK2 (D1A5; Cell Signaling Technology, 8727; 1:1000), rabbit anti-phospho-MAP2K1/MEK1-MAP2K2/MEK2 (Ser217/221, 41G9; Cell Signaling Technology, 9154; 1:1000), rabbit anti-MAPK/p38 (Cell Signaling Technology, 9212; 1:1000), rabbit anti-phospho-MAPK/p38 (Thr180/Tyr182; Cell Signaling Technology, 9211; 1:1000), rabbit anti-MAPK/JNK (Cell Signaling Technology, 9252; 1:1000), rabbit anti-phospho-MAPK/JNK (Thr183/Tyr185, 81E11; Cell Signaling Technology, 4668; 1:1000), goat anti-mouse HAVCR1/KIM-1 (R&D Systems, AF1817; 1:4000), rabbit anti-COL1A1 (Novus Biologicals, NBP1–30054; 1:500), rabbit anti-FN1 (Abcam, ab2413; 1:4000), rabbit anti-VIM (R28; Cell Signaling Technology, 3932; 1:1000), rabbit anti-SMAD2-SMAD3 (Cell Signaling Technology, 3102; 1:1000), rabbit anti-phospho-SMAD2 (Ser465/467, 138D4; Cell Signaling Technology, 3108; 1:1000), rabbit anti-NFKB RELA/p65 (Cell Signaling Technology, 3034, discontinued; 1:1000), rabbit anti-phospho-NFKB RELA/p65 (Ser536, 93H1; Cell Signaling Technology, 3033; 1:1000), rabbit anti-TRP53/p53 (Cell Signaling Technology, 9282; 1:1000), rabbit anti-phospho-TRP53/p53 (Ser15; Cell Signaling Technology, 9284; 1:1000), mouse anti-ACTB/β-actin (Sigma-Aldrich, A5316; 1:10,000), rabbit anti-PPIB/cyclophilin B (Abcam, ab16054; 1:10,000). Secondary antibodies for immunoblot analysis were from ThermoFisher Scientific (A16023, A16011, A15999). Antigens on the blots were revealed with Clarity™ Western ECL substrate kit (Bio-Rad, 1705061).
Renal function and histology
To measure renal function, blood samples were obtained for coagulation and centrifugation at room temperature to collect serum. BUN was determined using Stanbio urea nitrogen test kit (Stanbio Laboratory, 0580). The reaction was conducted at 100°C for 12 min and the absorbance at 520 nm was recorded by the end of reaction. Serum creatinine was measured using creatinine biochemistry reagent (Stanbio Laboratory, 0430). Samples were added to a pre-warmed (37°C) reaction mixture and the absorbance at 510 nm was monitored kinetically at 20 s and 80 s of reaction. The levels of BUN and creatinine (mg/dl) were then calculated according to standard curves. For histology, kidney tissues were fixed with 4% paraformaldehyde, embedded in paraffin, and sectioned at 4 µm. Renal fibrosis was examined by Sirius Red/Fast Green staining (Chondrex, 9046) following the manufacturer’s procedures. Collagen fibrils were stained magenta. 10 to 20 positive collagen-stained fields (100×) were randomly selected from each section and analyzed quantitatively by ImageJ.
Immunofluorescence, co-immunofluorescence and LC3B immunohistochemistry
Immunofluorescence or co-immunofluorescence was performed on 4 µm paraformaldehyde-fixed, paraffin-embedded kidney tissue sections. After rehydration, tissue sections were incubated with either 10 mM sodium citrate, 0.05% Tween 20 (Fisher Scientific, BP337–500), pH 6.0 or 10 mM Tris, 1 mM EDTA, 0.05% Tween 20, pH 9.0 at 95–100°C for antigen retrieval. Non-specific binding was blocked with a buffer containing 2% BSA, 0.2% milk, 2% normal donkey serum (Jackson ImmunoResearch, 017-000-121) and 0.8% Triton X-100. The slides were then exposed to the following primary antibodies for single or double staining at 4°C overnight: rabbit anti-EGR1 (15F7; Cell Signaling Technology, 4153; 1:500), mouse anti-FGF2 (LifeSpan BioSciences, LS-C23590; 1:1000), goat anti-mouse HAVCR1/KIM-1 (R&D Systems, AF1817; 1:400), mouse anti-SQSTM1/p62 (Abcam, ab56416; 1:400), rabbit anti-MAPK1/ERK2-MAPK3/ERK1 (137F5; Cell Signaling Technology, 4695; 1:200), rabbit anti- MAP2K1/MEK1-MAP2K2/MEK2 (D1A5; Cell Signaling Technology, 8727; 1:200), rabbit anti-COL1A1 (Novus Biologicals, NBP1–30054; 1:300), rabbit anti-COL4A1 (Abcam, ab6586; 1:300), rabbit anti-FN1 (Abcam, ab2413; 1:500), rabbit anti-ACTA2/α-SMA (Abcam, ab5694; 1:400), rabbit anti-phospho-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204; Cell Signaling Technology, 9101; 1:200), rabbit anti-phospho-MAPK/p38 (Thr180/Tyr182, D3F9; Cell Signaling Technology, 4511; 1:200), rabbit anti-phospho-MAPK/JNK (Thr183/Tyr185, 81E11; Cell Signaling Technology, 4668; 1:200). After washing, the slides were incubated with corresponding Alexa Fluor™-conjugated secondary antibodies (ThermoFisher Scientific, A10037, A10042, A11057, A21202, A21206, A21081) at room temperature for 1 h. In some experiments, the slides were also stained with fluorescein-dolichos biflorus agglutinin (DBA; Vector Laboratories, FL-1031; 1:100) to indicate distal tubules and collecting ducts. After mounted with ProLong™ Diamond antifade reagent, the slides were examined by fluorescence or confocal microscopy. All quantitative analysis was conducted by ImageJ. For EGR1 staining, 10 to 20 fields (400×) were randomly selected from each section and the number of EGR1-postive renal tubular cells per field was analyzed. For FGF2 staining, 10 to 20 fields (400×) were randomly selected from each section and the percentage of FGF2-postive staining area per field was analyzed. For co-staining of SQSTM1/p62 with MAPK1/ERK2-MAPK3/ERK1 or MAP2K1/MEK1-MAP2K2/MEK2, 10 to 20 fields (400×) were randomly selected from each section and the number of renal tubular SQSTM1/p62 dots with or without MAPK1/ERK2-MAPK3/ERK1 or MAP2K1/MEK1-MAP2K2/MEK2 co-staining per field was counted, respectively. Moreover, 10 to 20 random fields (100×) were selected from each section for quantitative analysis of COL1A1-, COL4A1-, FN1-, ACTA2- and HAVCR1/KIM-1-positive staining areas.
Immunohistochemical staining of LC3B was described in our recent work [20]. Tissue sections (4 µm) were deparaffinized and incubated with 1 mM EDTA, 0.05% Tween 20, pH 8.0 at 95–100°C for antigen retrieval. After a serial blocking in 3% H2O2, streptavidin-biotin (Vector Laboratories, SP-2002) and 2.5% normal horse serum (Vector Laboratories, S-2012), the slides were incubated with rabbit anti-LC3B (Novus Biologicals, NB100–2220; 1:1000) at 4°C overnight and then ImmPRESS™ HRP polymer horse-anti-rabbit secondary antibody (Vector Laboratories, MP-7401) at room temperature for 30 min. A biotin-conjugated Tyramide SuperBoost™ kit (ThermoFisher Scientific, B40931) was used for signal amplification according to the manufacturer’s protocol. The slides were further incubated with VECTASTAIN®
Elite® ABC-HRP kit (Vector Laboratories, PK-6100) at room temperature for 1 h. Signals were revealed with Vector® DAB kit (Vector Laboratories, SK-4100).
Statistics
Qualitative data including immunoblots, co-immunoprecipitation and cell/tissue images are representatives of at least 3 experiments. Quantitative Data were expressed as means ± SEM. Statistical analysis was conducted using the GraphPad Prism 9. Statistical differences between two groups were determined by 2-tailed, unpaired Student t-test. Statistical differences in multiple groups were determined by one-way or two-way ANOVA with multiple comparisons. Pearson correlation analysis followed by simple linear regression was used to determine the association between protein fold changes of p-MAPK1/ERK2-MAPK3/ERK1 (Thr202/Tyr204) and SQSTM1/p62. P values were indicated in the figures. P < 0.05 was considered significantly different.
Supplementary Material
Acknowledgements
We thank Dr. Masaaki Komatsu and Dr. Robert Koester for providing Atg7flox/flox mice and double transgenic Pax8-rtTA±, LC1± mice respectively that were used in this study to generate the inducible, renal tubule-specific Atg7 knockout mouse model.
Funding Statement
The work was supported by the National Institutes of Health of USA [5R01DK058831]; Department of Veterans Affairs of USA [1TK6BX005236]; Department of Veterans Affairs of USA [I01 BX000319]; National Institutes of Health of USA [5R01DK087843].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2281156
References
- [1].Ronco C, Bellomo R, Kellum JA.. Acute kidney injury. Lancet. 2019;394(10212):1949–1964. doi: 10.1016/S0140-6736(19)32563-2 [DOI] [PubMed] [Google Scholar]
- [2].See EJ, Jayasinghe K, Glassford N, et al. Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019;95(1):160–172. doi: 10.1016/j.kint.2018.08.036 [DOI] [PubMed] [Google Scholar]
- [3].Zuk A, Bonventre JV. Recent advances in acute kidney injury and its consequences and impact on chronic kidney disease. Curr Opin Nephrol Hypertens. 2019;28(4):397–405. doi: 10.1097/MNH.0000000000000504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yu SM, Bonventre JV. Acute kidney injury and maladaptive tubular repair leading to renal fibrosis. Curr Opin Nephrol Hypertens. 2020;29(3):310–318. doi: 10.1097/MNH.0000000000000605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11(5):264–276. doi: 10.1038/nrneph.2015.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Venkatachalam MA, Weinberg JM, Kriz W, et al. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol. 2015;26(8):1765–1776. doi: 10.1681/ASN.2015010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Basile DP, Bonventre JV, Mehta R, et al. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol. 2016;27(3):687–697. doi: 10.1681/ASN.2015030309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kumar S. Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int. 2018;93(1):27–40. doi: 10.1016/j.kint.2017.07.030 [DOI] [PubMed] [Google Scholar]
- [9].Liu BC, Tang TT, Lv LL, et al. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 2018;93(3):568–579. doi: 10.1016/j.kint.2017.09.033 [DOI] [PubMed] [Google Scholar]
- [10].Sato Y, Takahashi M, Yanagita M. Pathophysiology of AKI to CKD progression. Semin Nephrol. 2020;40(2):206–215. doi: 10.1016/j.semnephrol.2020.01.011 [DOI] [PubMed] [Google Scholar]
- [11].Taguchi K, Elias BC, Sugahara S, et al. Cyclin G1 induces maladaptive proximal tubule cell dedifferentiation and renal fibrosis through CDK5 activation. J Clin Invest. 2022;132(23):132. doi: 10.1172/JCI158096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Geng H, Lan R, Liu Y, et al. Proximal tubule LPA1 and LPA2 receptors use divergent signaling pathways to additively increase profibrotic cytokine secretion. Am J Physiol Renal Physiol. 2021;320(3):F359–F374. doi: 10.1152/ajprenal.00494.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Canaud G, Brooks CR, Kishi S, et al. Cyclin G1 and TASCC regulate kidney epithelial cell G2-M arrest and fibrotic maladaptive repair. Sci Transl Med. 2019;11(476):11. doi: 10.1126/scitranslmed.aav4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mylonas KJ, O’Sullivan ED, Humphries D, et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci Transl Med. 2021;13(594). doi: 10.1126/scitranslmed.abb0203 [DOI] [PubMed] [Google Scholar]
- [15].Li S, Livingston MJ, Ma Z, et al. Tubular cell senescence promotes maladaptive kidney repair and chronic kidney disease after cisplatin nephrotoxicity. JCI Insight. 2023;8(8). doi: 10.1172/jci.insight.166643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu X, Miao J, Wang C, et al. Tubule-derived exosomes play a central role in fibroblast activation and kidney fibrosis. Kidney Int. 2020;97(6):1181–1195. doi: 10.1016/j.kint.2019.11.026 [DOI] [PubMed] [Google Scholar]
- [17].Yamamoto S, Yamamoto M, Nakamura J, et al. Spatiotemporal ATP Dynamics during AKI predict renal prognosis. J Am Soc Nephrol. 2020;31(12):2855–2869. doi: 10.1681/ASN.2020050580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rinaldi A, Lazareth H, Poindessous V, et al. Impaired fatty acid metabolism perpetuates lipotoxicity along the transition to chronic kidney injury. JCI Insight. 2022;7(18). doi: 10.1172/jci.insight.161783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Balzer MS, Doke T, Yang YW, et al. Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat Commun. 2022;13(1):4018. doi: 10.1038/s41467-022-31772-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Livingston MJ, Shu S, Fan Y, et al. Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy. 2023;19(1):256–277. doi: 10.1080/15548627.2022.2072054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chen CH, Poucher SM, Lu J, et al. Fibroblast growth factor 2: from laboratory evidence to clinical application. Curr Vasc Pharmacol. 2004;2(1):33–43. doi: 10.2174/1570161043476500 [DOI] [PubMed] [Google Scholar]
- [22].Kajitani N, Hisaoka-Nakashima K, Okada-Tsuchioka M, et al. Fibroblast growth factor 2 mRNA expression evoked by amitriptyline involves extracellular signal-regulated kinase-dependent early growth response 1 production in rat primary cultured astrocytes. J Neurochem. 2015;135(1):27–37. doi: 10.1111/jnc.13247 [DOI] [PubMed] [Google Scholar]
- [23].Havis E, Duprez D. EGR1 transcription factor is a multifaceted regulator of matrix production in tendons and other connective tissues. Int J Mol Sci. 2020;21(5):21. doi: 10.3390/ijms21051664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Khachigian LM. Early growth response-1, an Integrative Sensor in cardiovascular and inflammatory disease. J Am Heart Assoc. 2021;10(22):e023539. doi: 10.1161/JAHA.121.023539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bhattacharyya S, Wu M, Fang F, et al. Early growth response transcription factors: key mediators of fibrosis and novel targets for anti-fibrotic therapy. Matrix Biol. 2011;30(4):235–242. doi: 10.1016/j.matbio.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ouellette AJ, Malt RA, Sukhatme VP, et al. Expression of two “immediate early” genes, egr-1 and c-fos, in response to renal ischemia and during compensatory renal hypertrophy in mice. J Clin Invest. 1990;85(3):766–771. doi: 10.1172/JCI114502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ho LC, Sung JM, Shen YT, et al. Egr-1 deficiency protects from renal inflammation and fibrosis. J Mol Med (Berl). 2016;94(8):933–942. doi: 10.1007/s00109-016-1403-6 [DOI] [PubMed] [Google Scholar]
- [28].Humphreys BD, Xu F, Sabbisetti V, et al. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest. 2013;123(9):4023–4035. doi: 10.1172/JCI45361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jimenez SK, Sheikh F, Jin Y, et al. Transcriptional regulation of FGF-2 gene expression in cardiac myocytes. Cardiovasc Res. 2004;62(3):548–557. doi: 10.1016/j.cardiores.2004.01.032 [DOI] [PubMed] [Google Scholar]
- [30].Jiang M, Liu K, Luo J, et al. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol. 2010;176(3):1181–1192. doi: 10.2353/ajpath.2010.090594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Livingston MJ, Wang J, Zhou J, et al. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy. 2019;15(12):2142–2162. doi: 10.1080/15548627.2019.1615822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rodriguez A, Duran A, Selloum M, et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3(3):211–222. doi: 10.1016/j.cmet.2006.01.011 [DOI] [PubMed] [Google Scholar]
- [33].Lippai M, Low P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed Res Int. 2014;2014:832704. doi: 10.1155/2014/832704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Komatsu M. p62 bodies: phase separation, NRF2 activation, and selective autophagic degradation. IUBMB Life. 2022;74(12):1200–1208. doi: 10.1002/iub.2689 [DOI] [PubMed] [Google Scholar]
- [35].Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Bio. 2005;171(4):603–614. doi: 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Itakura E, Mizushima N. p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Bio. 2011;192(1):17–27. doi: 10.1083/jcb.201009067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int. 2016;89(4):779–791. doi: 10.1016/j.kint.2015.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tang C, Livingston MJ, Liu Z, et al. Autophagy in kidney homeostasis and disease. Nat Rev Nephrol. 2020;16(9):489–508. doi: 10.1038/s41581-020-0309-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Colecchia D, Strambi A, Sanzone S, et al. MAPK15/ERK8 stimulates autophagy by interacting with LC3 and GABARAP proteins. Autophagy. 2012;8(12):1724–1740. doi: 10.4161/auto.21857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Carriere A, Romeo Y, Acosta-Jaquez HA, et al. ERK1/2 phosphorylate raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J Biol Chem. 2011;286(1):567–577. doi: 10.1074/jbc.M110.159046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lee SJ, Pfluger PT, Kim JY, et al. A functional role for the p62–ERK1 axis in the control of energy homeostasis and adipogenesis. EMBO Rep. 2010;11(3):226–232. doi: 10.1038/embor.2010.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pursiheimo JP, Rantanen K, Heikkinen PT, et al. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene. 2009;28(3):334–344. doi: 10.1038/onc.2008.392 [DOI] [PubMed] [Google Scholar]
- [43].Li C, Siragy HM. Autophagy upregulates (pro)renin receptor expression via reduction of P62/SQSTM1 and activation of ERK1/2 signaling pathway in podocytes. Am J Physiol Regul Integr Comp Physiol. 2017;313(1):R58–R64. doi: 10.1152/ajpregu.00088.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Martinez-Lopez N, Athonvarangkul D, Mishall P, et al. Autophagy proteins regulate ERK phosphorylation. Nat Commun. 2013;4(1):2799. doi: 10.1038/ncomms3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bonventre JV, Sukhatme VP, Bamberger M, et al. Localization of the protein product of the immediate early growth response gene, egr-1, in the kidney after ischemia and reperfusion. Cell Regul. 1991;2(3):251–260. doi: 10.1091/mbc.2.3.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen JW, Huang MJ, Chen XN, et al. Transient upregulation of EGR1 signaling enhances kidney repair by activating SOX9 + renal tubular cells. Theranostics. 2022;12(12):5434–5450. doi: 10.7150/thno.73426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Santiago FS, Sanchez-Guerrero E, Zhang G, et al. Extracellular signal-regulated kinase-1 phosphorylates early growth response-1 at serine 26. Biochem Biophys Res Commun. 2019;510(3):345–351. doi: 10.1016/j.bbrc.2019.01.019 [DOI] [PubMed] [Google Scholar]
- [48].Santiago FS, Li Y, Khachigian LM. Serine 26 in early growth response-1 is critical for endothelial proliferation, migration, and network formation. J Am Heart Assoc. 2021;10(18):e020521. doi: 10.1161/JAHA.120.020521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Deshpande N, Wilkins MR, Khachigian LM. RNA sequencing identifies genes reliant upon Ser26 in the zinc finger transcription factor, early growth response-1. Vasc Pharmacol. 2022;143:106952. doi: 10.1016/j.vph.2022.106952 [DOI] [PubMed] [Google Scholar]
- [50].Zhang X, Yang Z, Heng Y, et al. MicroRNA‑181 exerts an inhibitory role during renal fibrosis by targeting early growth response factor‑1 and attenuating the expression of profibrotic markers. Mol Med Rep. 2019;19:3305–3313. doi: 10.3892/mmr.2019.9964 [DOI] [PubMed] [Google Scholar]
- [51].Ai K, Li X, Zhang P, et al. Genetic or siRNA inhibition of MBD2 attenuates the UUO- and I/R-induced renal fibrosis via downregulation of EGR1. Mol Ther Nucleic Acids. 2022;28:77–86. doi: 10.1016/j.omtn.2022.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jin Y, Sheikh F, Detillieux KA, et al. Role for early growth response-1 protein in alpha(1)-adrenergic stimulation of fibroblast growth factor-2 promoter activity in cardiac myocytes. Mol Pharmacol. 2000;57(5):984–990. [PubMed] [Google Scholar]
- [53].Fahmy RG, Dass CR, Sun LQ, et al. Transcription factor egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nat Med. 2003;9(8):1026–1032. doi: 10.1038/nm905 [DOI] [PubMed] [Google Scholar]
- [54].Fahmy RG, Khachigian LM. Suppression of growth factor expression and human vascular smooth muscle cell growth by small interfering RNA targeting EGR-1. J Cell Biochem. 2007;100(6):1526–1535. doi: 10.1002/jcb.21145 [DOI] [PubMed] [Google Scholar]
- [55].Kirita Y, Wu H, Uchimura K, et al. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci U S A. 2020;117(27):15874–15883. doi: 10.1073/pnas.2005477117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gerhardt LMS, Liu J, Koppitch K, et al. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proc Natl Acad Sci U S A. 2021;118(27):118. doi: 10.1073/pnas.2026684118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gerhardt LMS, Koppitch K, van Gestel J, et al. Lineage tracing and single-nucleus multiomics reveal novel features of adaptive and maladaptive repair after acute kidney injury. J Am Soc Nephrol. 2023;34(4):554–571. doi: 10.1681/ASN.0000000000000057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Livingston MJ, Ding HF, Huang S, et al. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy. 2016;12(6):976–998. doi: 10.1080/15548627.2016.1166317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yan Q, Song Y, Zhang L, et al. Autophagy activation contributes to lipid accumulation in tubular epithelial cells during kidney fibrosis. Cell Death Discov. 2018;4(1):2. doi: 10.1038/s41420-018-0065-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ding Y, Kim S, Lee SY, et al. Autophagy Regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol. 2014;25(12):2835–2846. doi: 10.1681/ASN.2013101068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li H, Peng X, Wang Y, et al. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy. 2016;12(9):1472–1486. doi: 10.1080/15548627.2016.1190071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Baisantry A, Bhayana S, Rong S, et al. Autophagy induces prosenescent changes in proximal tubular S3 segments. J Am Soc Nephrol. 2016;27(6):1609–1616. doi: 10.1681/ASN.2014111059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Bio. 2005;169(3):425–434. doi: 10.1083/jcb.200412022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Traykova-Brauch M, Schonig K, Greiner O, et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med. 2008;14(9):979–984. doi: 10.1038/nm.1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sinha D, Wang Z, Price VR, et al. Chemical anoxia of tubular cells induces activation of c-src and its translocation to the zonula adherens. Am J Physiol Renal Physiol. 2003;284(3):F488–497. doi: 10.1152/ajprenal.00172.2002 [DOI] [PubMed] [Google Scholar]
- [66].Ma Z, Li L, Livingston MJ, et al. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J Clin Invest. 2020;130(9):5011–5026. doi: 10.1172/JCI135536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wei Q, Sun H, Song S, et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J Clin Invest. 2018;128(12):5448–5464. doi: 10.1172/JCI121859 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.