

Abstract
Cushing’s syndrome is an endocrine disorder caused by excess production of the stress hormone cortisol. Precision medicine strategies have identified single allele mutations within the PRKACA gene that drive adrenal Cushing’s syndrome. These mutations promote perturbations in the catalytic core of protein kinase A (PKAc) that impair autoinhibition by regulatory subunits and compartmentalization via recruitment into AKAP signaling islands. PKAcL205R is found in ~45% of patients, whereas PKAcE31V, PKAcW196R, and L198insW and C199insV insertion mutants are less prevalent. Mass spectrometry, cellular, and biochemical data indicate that Cushing’s PKAc variants fall into two categories: those that interact with the heat-stable protein kinase inhibitor PKI, and those that do not. In vitro activity measurements show that wild-type PKAc and W196R activities are strongly inhibited by PKI (IC50 < 1 nM). In contrast, PKAcL205R activity is not blocked by the inhibitor. Immunofluorescent analyses show that the PKI-binding variants wild-type PKAc, E31V, and W196R are excluded from the nucleus and protected against proteolytic processing. Thermal stability measurements reveal that upon co-incubation with PKI and metal-bound nucleotide, the W196R variant tolerates melting temperatures 10°C higher than PKAcL205. Structural modeling maps PKI-interfering mutations to a ~20 Å diameter area at the active site of the catalytic domain that interfaces with the pseudosubstrate of PKI. Thus, Cushing’s kinases are individually controlled, compartmentalized, and processed through their differential association with PKI.
Introduction
Endocrine diseases have a substantial impact on public health [1]. They can cause long-term disability, significantly alter quality-of-life, and are a leading cause of death worldwide [2]. Cushing’s syndrome occurs when the body experiences too much of the stress hormone cortisol. There are two types of Cushing syndrome: exogenous disease that is frequently caused by over prescription of corticosteroids, and endogenous endocrine disorders [3,4]. Adrenal or non-ACTH Cushing’s syndrome is most often caused by adrenal or pituitary tumors and is considerably more common in women [5,6]. Approximately 15 per million people are diagnosed with adrenal Cushing’s syndrome annually [7]. Clinical presentation can be variable, and diagnosis can be difficult. Symptoms include mid-section weight gain, moon face, and a buffalo hump [8,9]. Excess cortisol also increases glucose in the bloodstream and elevates blood pressure [10]. Hence, diabetes and hypertension are common comorbidities that result from Cushing’s syndrome [2].
Adrenal Cushing’s syndrome is often a consequence of defective cAMP signaling in the zona fasciculata of the adrenal cortex [4,11]. Exome sequencing of adrenal tissue from patients conducted by five independent groups identified mutations in protein kinase A catalytic subunits (PKAc) that drive adrenal Cushing’s syndrome [12–18]. Most PKAc mutations cluster where the kinase domain interfaces with its regulatory (R) subunits [19]. This protein–protein interaction is not only necessary for autoinhibition of kinase activity, but also directs subcellular targeting of PKA holoenzymes through association with A kinase anchoring proteins (AKAPs; [20–22]). Cushing’s mutations may also influence PKAc association with other effectors including the heat-stable inhibitor PKI and the A-kinase interacting protein AKIP [23–26].
Protein kinase A is a broad-spectrum kinase that phosphorylates hundreds of cellular substrates [27–29]. Accordingly, lesions in PRKAC and PRKAR genes are linked to a growing number of diseases [30–33]. Adrenal Cushing’s syndrome is an autosomal dominant disease that emanates from lesions in a single allele of the PRKACA gene [2]. PKAc-L205R is the most prevalent mutant found in patients, accounting for ~45% of cases [19]. However, other disease causing PKAc mutations have been reported in the PKAc alpha isoform including E31V, W196R and the L198insW and C199insV insertion mutants (Figure 1A & [19]). A single Cushing’s mutation, S54L has been identified in the beta isoform of PKAc [18,34]. These amino acid changes promote subtle structural perturbations in the catalytic core of the kinase that impair autoinhibition by the type I or type II regulatory subunits of PKA and compartmentalization via recruitment into AKAP signaling islands [4,35–37]. Accordingly, shifts in the phosphorylation of compartment specific substrates and changes in substrate selectivity of PKAc variants have been proposed as molecular hallmarks of adrenal Cushing’s syndrome [18,19,38,39]. Yet, despite their apparent similarities, Cushing’s mutants can have distinct impacts on physiology. We previously demonstrated that PKAcL205R and PKAcW196R are associated with different downstream pathways. PKAcL205R up-regulates YAP/TAZ whereas PKAcW196R activates ERK [40]. In this report, we show that Cushing’s PKAc variants can be classified based on their differential association with the endogenous heat-stable inhibitor PKI. This ATP dependent pseudosubstrate inhibitor protein blocks kinase activity with nanomolar potency and is instrumental in the nuclear export of PKAc under conditions of chronic exposure to cAMP [41–43]. PKI is expressed in almost all tissues and has been shown to be localized in the nucleus. We show that interaction with PKI influences subcellular redistribution and protein stability of certain PKAc Cushing’s variants. Molecular modeling of PKI-resistant Cushing’s variants reveals a cluster of residues in the catalytic core of the protein kinase that map to the interface with the pseudosubstrate region of PKI.
Figure 1. Cushing’s PKAc mutants have distinct biochemical profiles.
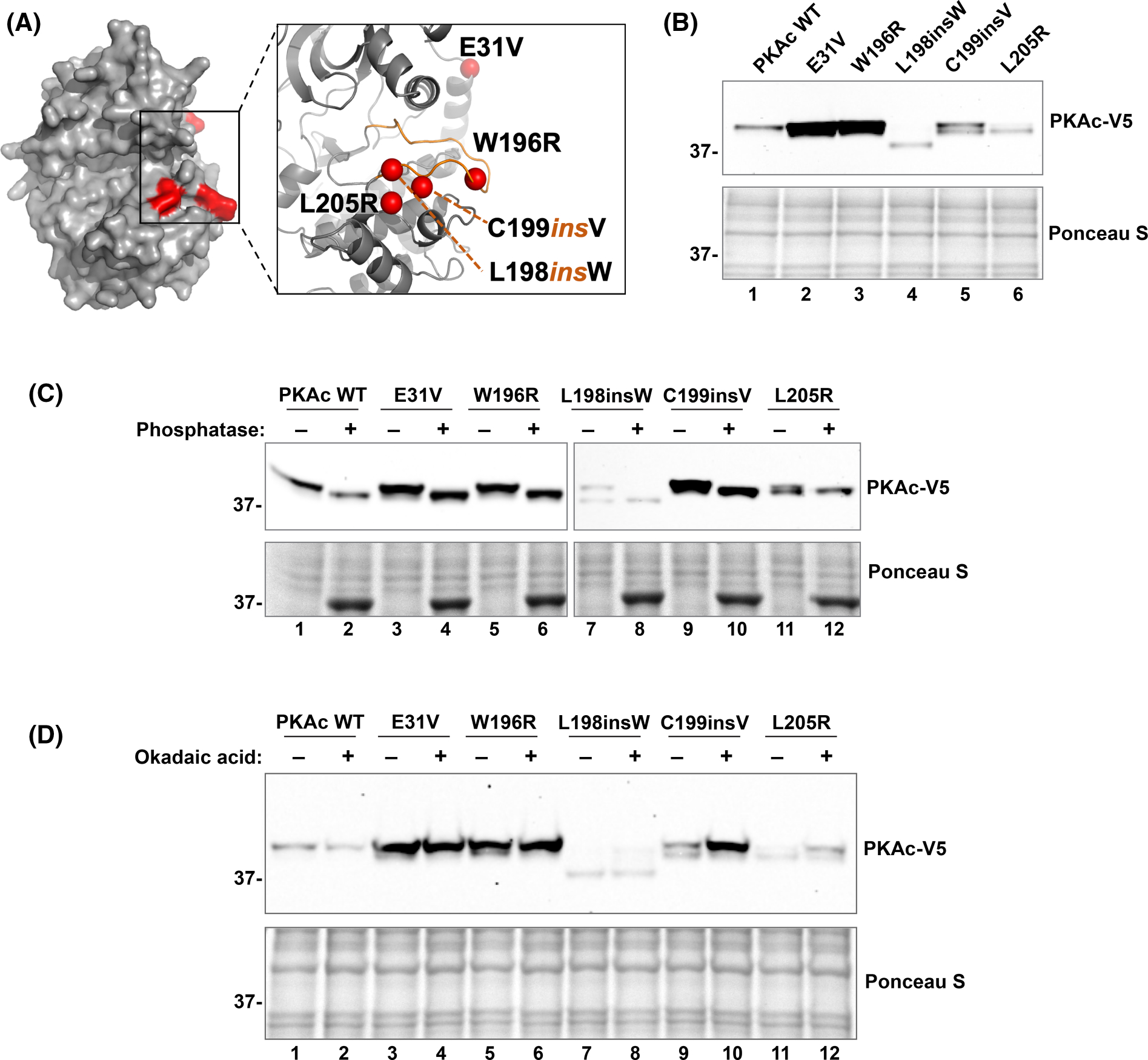
(A) Structural model (Pymol) representation showing the location of Cushing’s mutations (red) on PKAc. Zoomed inset demonstrates that most mutations are located on the substrate binding catalytic groove. (B) Immunoblot of lysates from U2OSPKAc−/− cells transfected with equal amounts (0.6 μg) of each PKAc variant; n = 3. (C) λ-phosphatase assay. Immunoblot of lysates from U2OSPKAc−/− cells transfected with PKAc variants. Untreated (−) and treated (+) conditions are shown side-by-side. Top right panel for lanes 7–12 is a longer exposure to visualize weaker signals for these mutants. n = 3. (D) Okadaic acid treatment assay: U2OSPKAc−/− cells transfected with PKAc variants were treated with okadaic acid (100 nM) for 6 h, then lysed and analyzed using SDS–PAGE; n = 3.
Results
Most adrenal Cushing’s syndrome mutations map to the catalytic core of the kinase (Figure 1A). To compare the impact of different Cushing’s PKAc mutations, we transfected V5-tagged kinase variants into CRISPR gene-edited U2OS cells lacking a functional PRKACA allele (CαKO cells). Immunoblot analysis demonstrated varied intensities and electrophoretic mobility among disease causing PKAc mutants (Figure 1B). PKAc, like most serine/threonine kinases, is phosphorylated on multiple sites in the fully mature form [44,45]. In our previous studies, we found 11 phosphosites almost completely devoid of phosphorylation in PKAcL205R as compared with the WT kinase [40]. Based on this data, we reasoned that differences in phosphorylation status of Cushing’s PKAc variants could be responsible for the observed electrophoretic mobility shifts. To test this notion, cell lysates were incubated with or without purified lambda phosphatase and evaluated for changes in electrophoretic mobility by immunoblot. While untreated samples displayed varied intensities between upper and lower doublets, phosphatase treatment collapsed all PKAc forms to the faster migrating band (Figure 1C). Complementary experiments utilized the phosphatase inhibitor okadaic acid. Consistently, immunoblot analysis showed a diminished or absent lower band in all conditions when okadaic acid was present (Figure 1D). In both experiments the L198insW mutant exhibited an even faster electrophoretic mobility that could not be explained solely by phosphorylation. Together, these results indicate that Cushing’s PKAc mutants possess differential phosphorylation statuses that are contingent on the nature of the specific point mutation.
The range of immunoblot intensities of Cushing’s PKAc mutants suggests that these proteins are subject to differential degradation mechanisms in cells (Figure 1B). We performed studies using the proteasomal inhibitor MG132 and the lysosomal antagonist bafilomycin A1 (BafA1) to determine the mechanism of degradation (Figure 2A). MG132 is a peptide aldehyde that inhibits the proteolytic activity of the 26S proteasome. BafA1 is an antibiotic that targets vacuolar-type H+-ATPase (V-ATPase), thus preventing lysosome acidification necessary for the function of lysosomal proteases. Both drug treatments were performed in the presence of the translation inhibitor cycloheximide (CHX) to allow for visualization of PKAc steady-state kinetics. Immunoblot analyses of U2OS cells transfected with each Cushing’s mutant demonstrate that MG132 treatment enhanced stability of all PKAc variants, as compared with untreated controls (Figure 2B). Conversely, BafA1 had little or no effect on the stability of all variants (Figure 2C). These results suggest that PKAc degradation, including that of the Cushing’s kinases, proceeds through the proteasome.
Figure 2. Proteasomal inhibition stabilizes PKAc variants.
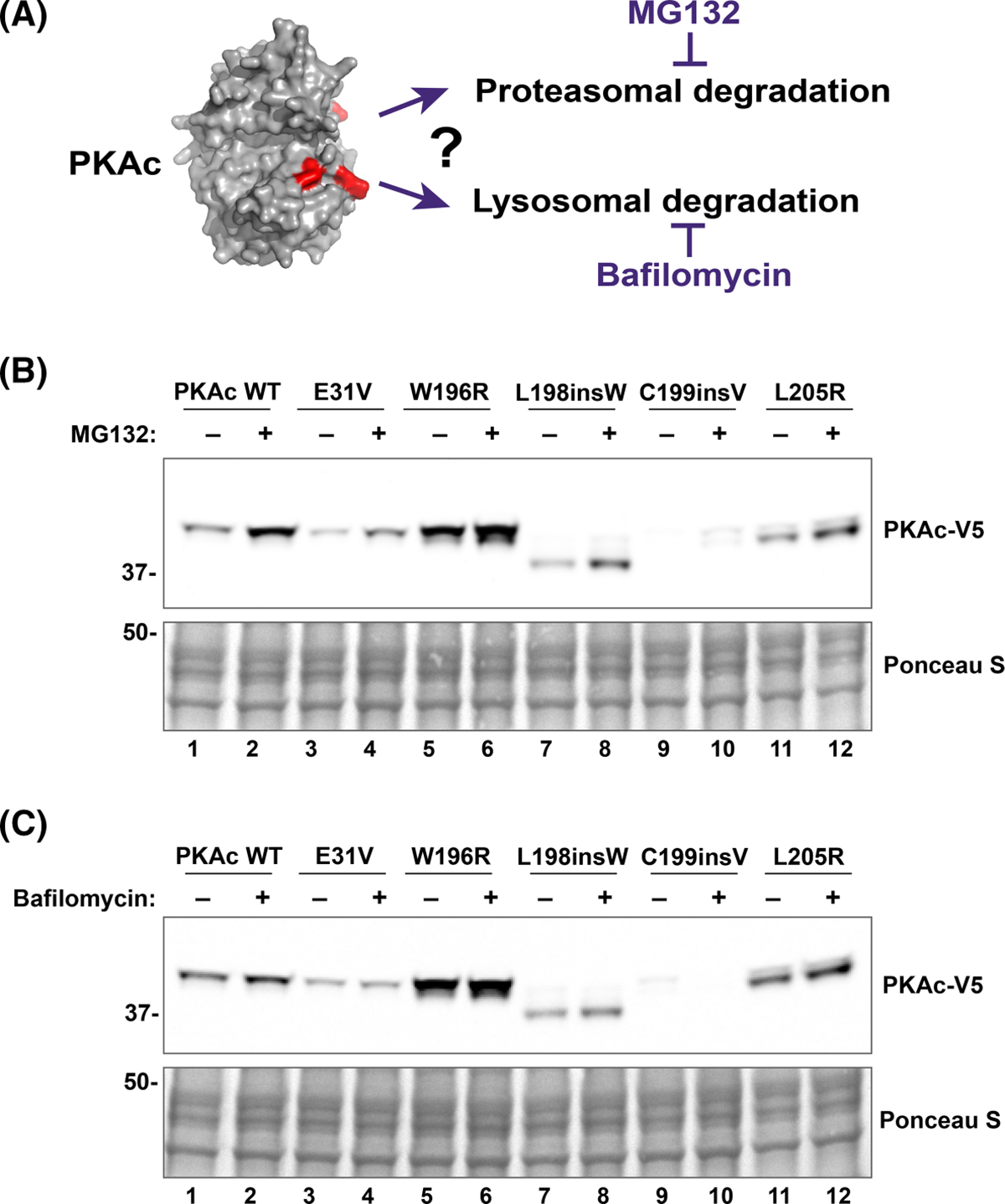
(A) Schematic highlighting mechanistic approach used to establish the processing pathway that are potentially implicated in PKAc variants degradation. (B and C) Representative immunoblots showing the effect of proteasome and lysosome inhibition on PKAc stability. U2OSPKAc−/− cells transfected with PKAc variants were treated with either MG132 (1 μM, (B) or BafA1 (100 nM, (C). After 1 h, CHX (50 μg) was added for an additional 7 h. Cells were then lysed, and lysates analyzed using SDS–PAGE. n = 3 replicates for each inhibitor experiment.
Native PKAc is largely excluded from the nuclear compartment [46–48]. Yet mislocalization of the kinase and omission from AKAP signaling islands in the adrenal cortex of Cushing’s syndrome patients is a pathogenic trait of certain PKAc mutants [12,13,15,40,49,50]. To examine how Cushing’s mutations impact this intracellular localization, immunofluorescent staining was conducted in CαKO cells (Figure 3A,C,E,G,I,K). Line scan analysis was used to visualize PKAc signal profiles (Figure 3B,D,F,H,J,L) and determine relative (nuclear to cytoplasmic) PKAc intensity per pixel for each variant (Figure 3M). In all conditions, signal for transfected RIIα-GFP displayed a strong enrichment in the cytosolic compartment (Figure 3A,C,E,G,I,K, right panels). As expected, wild-type (WT) PKAc was excluded from the nucleus (Figure 3A,B,M). Among the Cushing’s syndrome mutants, both PKAc E31V and W196R were predominantly detected in the cytosol, though to a lesser degree than wild-type kinase (Figure 3C–F,M). Interestingly, the remaining mutants, L198insW, C199insV, and L205R, exhibited an even distribution between cytosol and nucleus. Thus, the five PKAc variants tested clustered into two groups, those exhibiting nuclear exclusion (WT, E31V, W196R), and those that do not display preference for cytosol or nucleus (L198insW, C199insV, L205R).
Figure 3. PKAc variants are differentially localized inside cells.
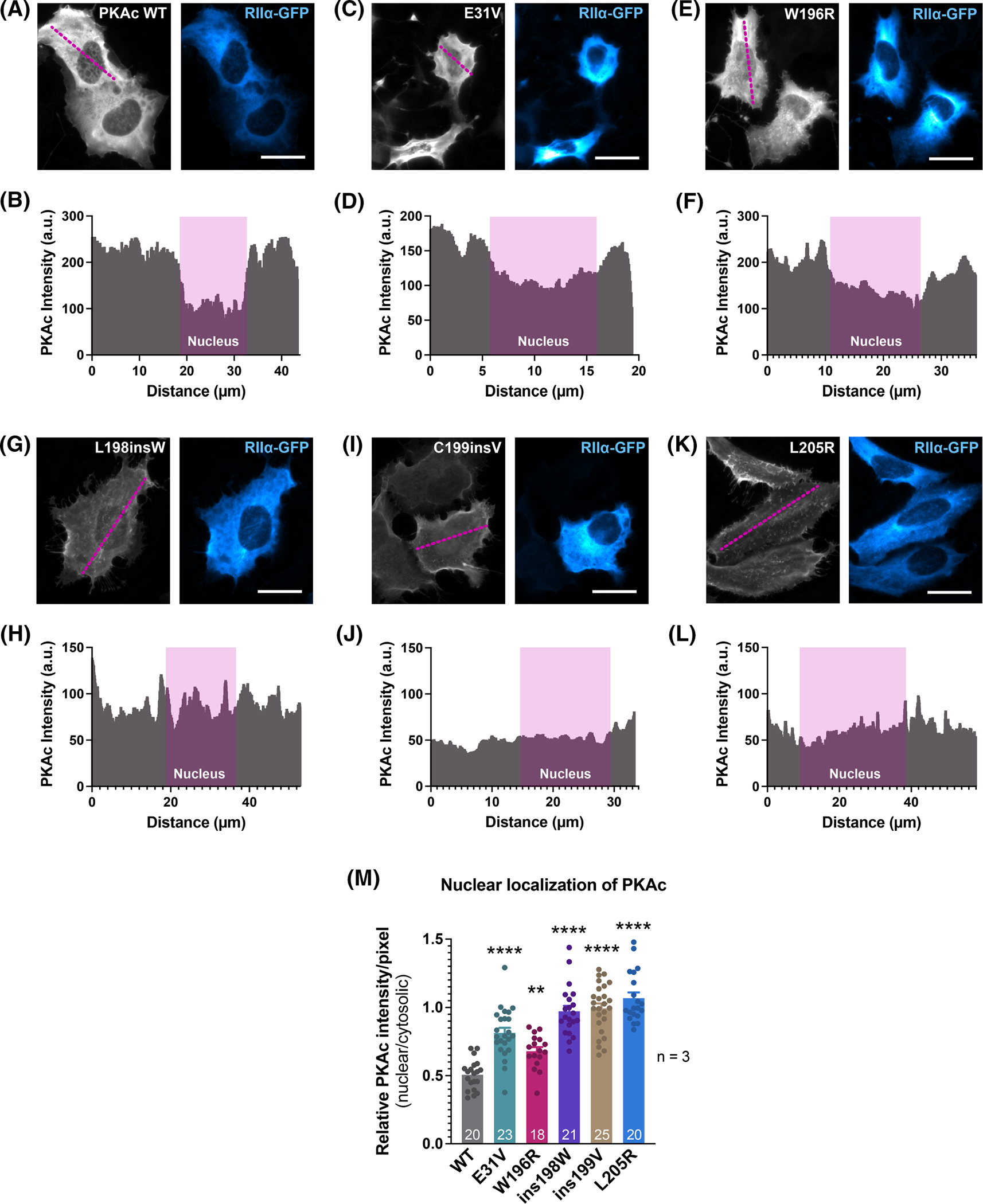
(A–L) Representative immunofluorescence images of U2OSPKAc−/− cells co-transfected with PKAc-V5 variants and RIIα-GFP. Cells were fixed and immunostained for V5 (grey-scale, PKAc) and GFP (blue, RIIα). Line-scan analyses (panels shown below the images) were performed to determine relative nuclear: cytoplasmic PKAc-V5 signal. (M) Quantification of relative nuclear: cytoplasmic PKAc. ImageJ software was used to determine the integrated density of PKAc-V5 fluorescence in the nucleus versus the cytoplasm. DAPI signal (not shown) was used to define the nucleus. The number of analyzed cells is shown at the base of each bar.
A possible mechanism to explain these distinct classes of Cushing’s PKAc mutants involves interaction with the endogenous protein kinase A inhibitor, PKI. This heat-stable protein resides in the nucleus and was one of the first identified proteins with a nuclear export signal [43]. Its sequence also contains a pseudosubstrate motif, which occupies the PKAc substrate-binding site with low nanomolar affinity [23–26]. This PKAc-PKI complex can then interact with nuclear export machinery to be shuttled back to the cytosol [28,51]. We hypothesized that differential interactions with PKI could determine nuclear localization variability in PKAc mutants. Evidence for this hypothesis was found in our proximity proteomic analyses of the L205R and W196R mutations [40]. In these experiments, a promiscuous biotin ligase tag on PKAc variants labeled proximal proteins in live adrenal cells [39]. Mass spectrometry analysis yielded datasets containing proteins intimately associated with each kinase (Figure 4A). As expected, PKI peptides were identified in both WT and W196R datasets (Figure 4B). In contrast, no PKI peptides were found in the L205R screen. Interestingly, while the wild-type PKAc dataset contained a total of 2 PKI peptides, the W196R condition included 16 peptides (Figure 4C). These findings suggest that PKAcW196R has more opportunities than the wild-type kinase to interact with PKI. This is likely due to the mutant kinase’s poor association with AKAP complexes and a resulting increase in its diffusion into the nucleus.
Figure 4. PKI interaction classifies PKAc Cushing’s mutants.
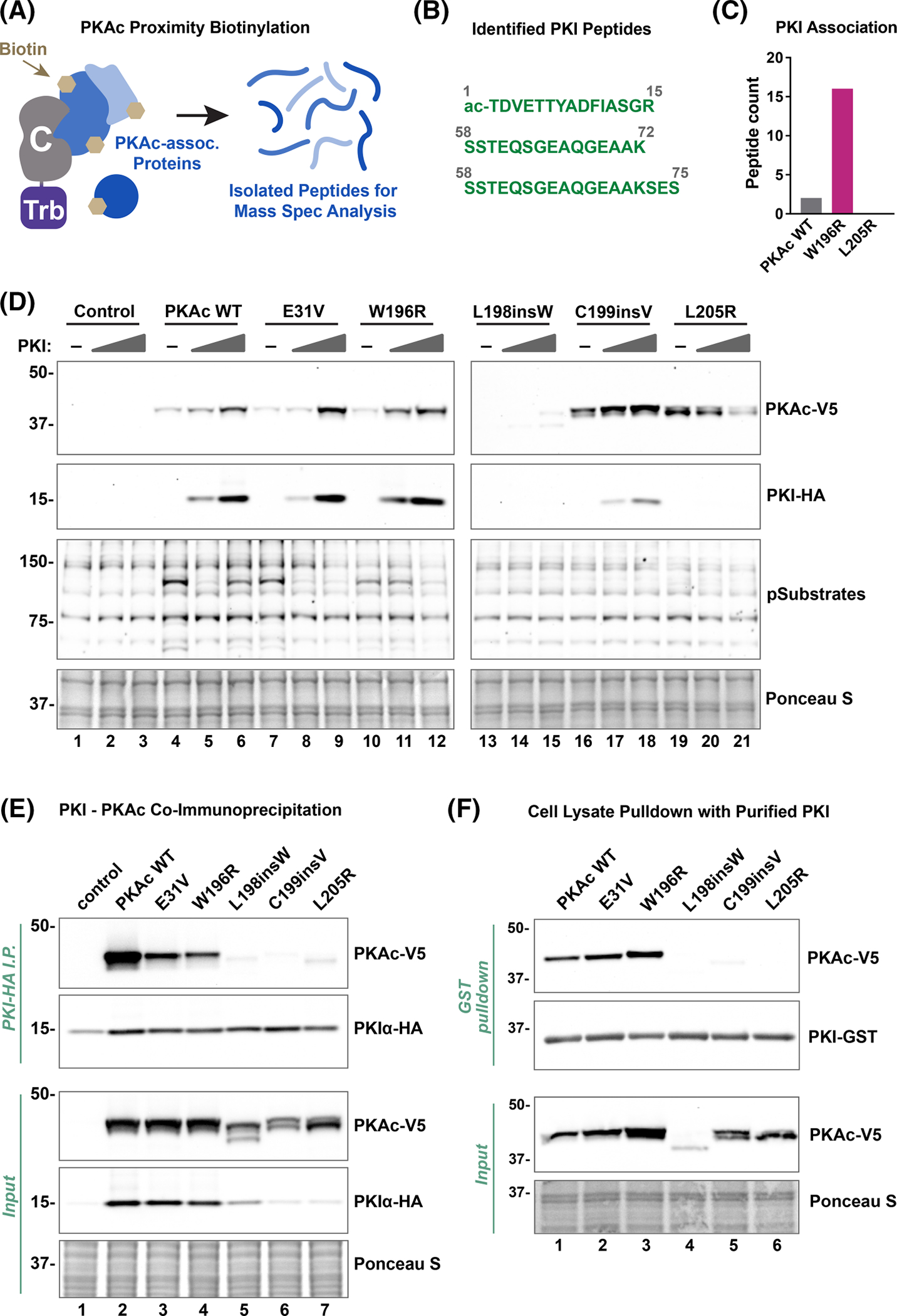
(A) Schematic of the proximity-biotinylation proteomics approach used to identify interacting proteins. Mini-turbo (Trb) enzyme, fused to PKAc, labels associating or proximal (5–10 nm) proteins with biotin. Biotinylated proteins are affinity enriched with streptavidin beads and identified using mass spectrometry. (B) PKI peptides found to be associated with PKAcW196R from adrenal H295R cells. (C) PKAcW196R was associated with more PKI peptides [16] as compared with PKAcWT [2]. (D) Immunoblot showing effect of PKI on PKAc stability and activity (PKAc phospho-substrates). Top two panels for lanes 13–21 are longer exposures to visualize weaker signals for these mutants. Lysates were from U2OSPKAc−/− cells co-transfected with PKI-HA and PKAc-V5 variants. Figures are representative of at least three experimental replicates. (E) PKI-HA immunoprecipitation from HEK293T cell lysates co-transfected with PKI-HA and PKAc-V5 variants. (F) In vitro immunoprecipitation experiments using purified PKI-GST on lysates from U2OSPKAc−/− cells transfected with PKAc-V5 variants (indicated above each lane).
Next, cell-based assays were performed in CαKO cells to test whether the PKI-binding profiles of Cushing’s mutants correlate with their nuclear localization. Co-expression of PKAc variants along with increasing amounts of PKI demonstrated a clear co-stabilization among wild-type PKAc, E31V, and W196R conditions, as visualized by immunoblotting (Figure 4D, top 2 panels). Probing with an antibody that recognizes phosphorylated consensus PKA substrate motifs (RRXpS/pT) demonstrated variable ability of PKI to inhibit phosphorylation of PKA substrates, with clear effects in only the wild-type PKAc, E31V, and W196R conditions (Figure 4D, third panel). Co-immunoprecipitation experiments in HEK293T cells corroborated the PKI interaction pattern, demonstrating a division among the kinase variants. While PKI was immunoprecipitated from all samples (Figure 4E, second panel) only wild-type PKAc, E31V, and W196R co-immunoprecipitated with the endogenous inhibitor (Figure 4E, lanes 2–4). The L198insW, C199insV, and L205R Cushing’s variants all failed to associate with PKI (Figure 4E, lanes 5–7). Interestingly, we observed a concomitant loss of PKI signal for cells expressing binding deficient L198insW, C199insV, and L205R PKAc variants (Figure 4E, input middle panel, lanes 5–7). To ensure that differential stabilization of PKI in cells was not impacting co-immunoprecipitation experiments, a hybrid pulldown was conducted. Purified GST-PKI was loaded onto glutathione (GSH) resin and incubated with cell lysates containing different PKAc variants (Figure 4F). Only WT PKAc, E31V, and W196R were detected in the binding fraction, thereby confirming our co-immunoprecipitation results (Figure 4F, top panel, lanes 1–3). Together, these data indicate that Cushing’s PKAc mutants display varied binding to PKI that inversely correlates with nuclear localization.
Further interrogation of PKI-binding was conducted using purified proteins in vitro. The W196R and L205R variants served as representatives of cytoplasmic- and nuclear-localized PKAc mutants, respectively. These two kinases, along with wild-type PKAc and a PKI-GST fusion, were purified to homogeneity from E. coli and then incubated together. As expected, the GST controls failed to bind PKAc (Figure 5A, lanes 5–8). Experiments performed with PKI-GST demonstrated strong pulldown of both wild-type PKAc and W196R variants, and no detectable L205R (Figure 5A, lanes 1–3). Since PKI’s ability to inhibit PKAc activity relies upon its physical association with the kinase, Cushing’s variants that do not associate with PKI should be refractory to inhibition of catalytic activity. We conducted kinase assays using the synthetic PKA substrate Kemptide and measured percentage activity at increasing concentrations of PKI relative to the buffer-only control for each kinase variant. While both WT PKAc and W196R kinases were potently inhibited by PKI (IC50 < 1 nM), L205R was very poorly inhibited (IC50 ~ 100 nM; Figure 5B). Thermal profiling of representative Cushing’s mutants demonstrated a similar pattern. Wild-type PKAc and the W196R variant can withstand much higher melting temperatures upon co-incubation with Mg-ATP and PKI, with Tm values increasing by 10°C (Figure 5C,D,F). In contrast, PKAcL205R was not stabilized by PKI under the same conditions (Figure 5E,F). Thus, in vitro experiments confirm that cytoplasmic PKAcW196R displays a much stronger association with PKI than does the more nuclear L205R mutant, recapitulating our cell-based assays.
Figure 5. PKI displays selectivity toward PKAc variants in vitro.
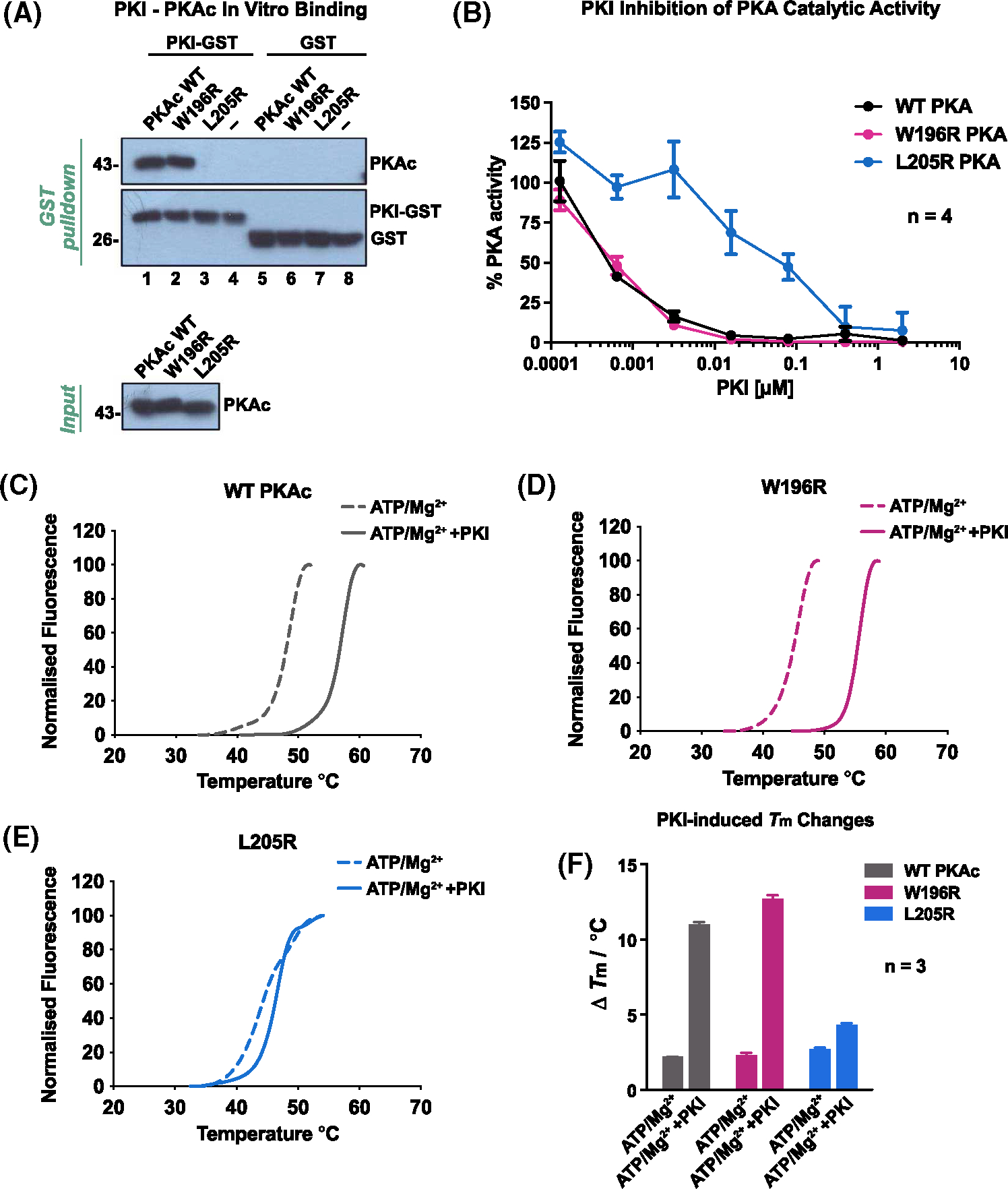
(A) In vitro pulldown using PKI-GST on PKAc. Both proteins were purified from E. coli. (B) PKAc kinase activity measurements in the presence of the inhibitory peptide, PKI. % activity was determined by measuring the amount of phosphorylated kemptide substrate. (C–F) Thermal stability measurements. Representative thermal denaturation profiles of PKAc variants were generated for (C) wild-type PKAc (grey). (D) PKAcW196R(magenta). (E) PKAcW196R (blue) in the presence of ATP/Mg2+ ±PKI. (F) Average ΔTm/°C values of three independent experiments are shown for each experimental condition.
Discussion
Protein Kinase A is the inaugural member of the AGC family of protein kinases and targets substrates containing an Arg–Arg–Xaa–Ser/Thr–Xaa consensus sequence [29,52]. PKA mediated phosphorylation events control vital physiological processes including heartbeat, renal function, and hormone action [22]. Accordingly, genomic alterations in the PKA signaling pathway underlie defects in cardiac contractility, certain neuroendocrine cancers, polycystic kidney disease, and various endocrine disorders [26,48]. In this report, we compare the molecular consequences of mutations in the catalytic subunit of protein kinase A (PKAc) that drive the stress hormone disorder adrenal Cushing’s syndrome [49,53]. Most Cushing’s PKA mutations cluster where the kinase domain interfaces with its regulatory subunits ([19]; Figure 1A). This protein–protein interaction is not only necessary for autoinhibition of kinase activity, but also directs compartmentalization of the kinase holoenzyme through association with A kinase anchoring proteins [20–22,37]. In other cellular contexts, PKA activity is modulated by its heat-stable inhibitor protein PKI [23,54]. We have used this latter protein–protein interaction to define two distinct classes of Cushing’s PKAc variants: those that bind PKI and those that are refractory to the heat-stable inhibitor.
The heat-stable inhibitor is an intrinsically disordered protein of 75 amino acids that functions as a pseudosubstrate for protein kinase A [51,55,56]. A substrate mimicking motif (Arg–Arg–Asn–Ala–Ile) in the aminoterminal region targets the active site of PKA to block the phosphotransferase reaction whereas a canonical nuclear export sequence facilitates the removal of excess PKAc from the nucleus [23,46]. Peptides encompassing pseudosubstrate region (PKI residues 5–24) have become the gold standard reagents for exclusive inhibition of PKA, both in vitro and inside living cells [23–25]. While the PKAcL205R variant is known to be refractory to PKI binding, we show that among the 5 Cushing’s mutants tested only PKAcE31V and PKAcW196R bind PKI [53,57]. Since PKI is important for removal of free PKAc from the nuclear compartment, these findings argue that nuclear export of PKAcL205R, PKAcL198insW, and PKAcC199insV is abrogated, [46,58]. Hence, interaction with the heat-stable inhibitor protein may alter the subcellular redistribution of some, but not all, PKAc Cushing’s variants.
Our data suggest that PKI not only affects the activity and subcellular location of Cushing’s kinases but influences protein stability as a function of complex formation. While PKAcL205R, PKAcL198insW, and PKAcC199insV are barely detectable in cell extracts, association with PKI enhances detection of wild-type PKAc and the E31V and W196R mutants. Our studies also highlight a previously unrecognized feature of PKI: interaction with the kinase reciprocally stabilizes the heat-stable inhibitor. This is perhaps not too surprising as free PKI is an intrinsically disordered protein and exquisitely sensitive to a spectrum of proteinases [23,51,56,59]. Nonetheless, this raises a conundrum. Although PKI increases the stability of PKAcE31V and PKAcW196R, its primary role is to block the catalytic activity of these enzymes. Two plausible explanations are offered that circumvent this apparent paradox. First, while PKI is a nanomolar inhibitor of native PKAc, its inhibitory potency may be reduced toward Cushing’s variants [24,60]. Such changes in the equilibrium between bound and free kinase could release enough PKAcE31V or PKAcW196R to phosphorylate proteins within their immediate vicinity. Indirect support for this scenario is provided by our previous phosphoproteomic evidence, which found that PKAcW196R targets a distinct subset of substrates as compared with the wild-type kinase [39,40]. Second, PKI is present in cells at vanishingly low levels [55]. Indeed, it has been reported that the heat-stable inhibitor only has the capacity to block 20% of the total PKA activity generated during a sustained cAMP response [26,42,61]. Thus, there may be too much free PKAcE31V and PKAcW196R protein in adrenal cortex cells to evade total inhibition by PKI.
Stability is also determined by phosphorylation status of a protein [45,62]. Multisite phosphorylation is a prerequisite to full maturation of PKAc, with up to 11 sites occupied in the native kinase [45,63]. We have previously shown that PKAcL205R and PKAcW196R exhibit decreased phosphate incorporation at most sites [39,40]. The same seems true for PKAcE31V, PKAcL198insW, and PKAcC199insV variants based on their altered electrophoretic mobilities on SDS gels and faster migration after incubation with protein phosphatases. Thus, incomplete phosphorylation renders PKAc more sensitive to proteolytic processing, with proteasomal pathways dominating the intracellular clearance for all versions of the kinase (Figure 2). However, it is possible, even likely, that other protein processing pathways contribute to the targeted destruction of Cushing’s PKAc variants. Interestingly, the three PKAc variants that exhibit reduced protein stability and phosphorylationdependent maturity are the most nuclear of the mutants. These differences in physiochemical profiles and localization are likely crucial elements in the pathology of this endocrine disorder and likely explain why both categories of Cushing’s kinases cause disease. Although the stable and fully active PKAcW196R is dynamically exported from the nucleus, its brief time in that subcellular compartment is quite likely to be more productive than the catalytically impaired PKAcL205R. In contrast, the sustained nuclear residence of PKAcL205R affords more time to phosphorylate transcription factors such as CREB or ATF-1 to prime cortisol-production machinery. Such differences in catalytic efficiency and compartmentalization may be key to understanding how distinct downstream consequences emanate from individual Cushing’s mutations. For example, we have shown that PKAcL205R elevates YAP/TAZ levels while PKAcW196R activates the ERK pathway [40].
Detailed biochemical characterization of PKAcL205R and PKAcW196R proteins in Figures 4 and 5 offers a comprehensive comparative analysis of PKI inhibited and uninhibited Cushing’s variants. Elegant structural and molecular modeling studies by Veglia and colleagues argue that the added bulk and charge of arginine at position 205 sterically hiders binding of this Cushing’s variant to the pseudosubstrate site on PKI [53,64]. Others have suggested that introducing arginine at this location alters the selectivity of PKAcL205R to induce a substrate rewiring of the Cushing’s kinase [19,38]. In keeping with this latter theory, we have shown that PKAcL205R is a catalytically inefficient enzyme that exhibits ~20-fold lower phosphotransferase activity towards the Kemptide substrate than wild-type PKAc or PKAcW196R [40]. Hence, PKAcL205R may be both physiochemically and biologically distinct from other Cushing’s kinases.
In silico AlphaFold modeling studies presented in Figure 6A postulate that insertion of Tryptophan 198 or Valine 199 in the context of the PKAcL198insW and PKAcC199insV perturb binding to the pseudosubstrate region of PKI [65]. Conversely, Glu 31 is located on the opposite face of the kinase core. This implies that PKAcE31V retains its capacity for PKI binding and autoinhibition by R subunits (Figure 6A). Consequently, this Cushing’s variant must exert its pathological action through a distinct mechanism. The same may also be true for PKAcS54L, the only reported Cushing’s mutation described in the PRKACB gene [18,34]. Interestingly, this phosphorylated residue is conserved in all isoforms of PKAc [18]. As shown in Figure 6A our molecular modeling predicts that the extra aliphatic content associated with a leucine side chain at this position appears to have minimal impact on the PKI-C subunit interface. However, loss of a negatively charged phosphate could further disrupt substrate interactions. Taken together, these structural predictions offer a compelling molecular explanation as to why PKAcL205R is mislocalized and free to ‘roam around’ the cell, leading to indiscriminate phosphorylation of substrates. In keeping with this notion, we have previously reported that PKAcL205R preferentially phosphorylates nuclear and mitochondrial substrates as compared with wild-type PKAc [39,40]. Another adverse consequence can be enhanced transcription of cAMP responsive genes encoding the steroidogenic acute regulatory protein (StAR), a mitochondrial PKA substrate that regulates the rate-limiting step in the production of cortisol [40,48,66,67]. Thus, nuclear accumulation of PKAcL205R, PKAcL198insW, or PKAcC199insV may potentiate pathological cAMP responses at the wrong place and at the wrong time in adrenal cells.
Figure 6. PKI binding determines PKAc nuclear localization and stability.
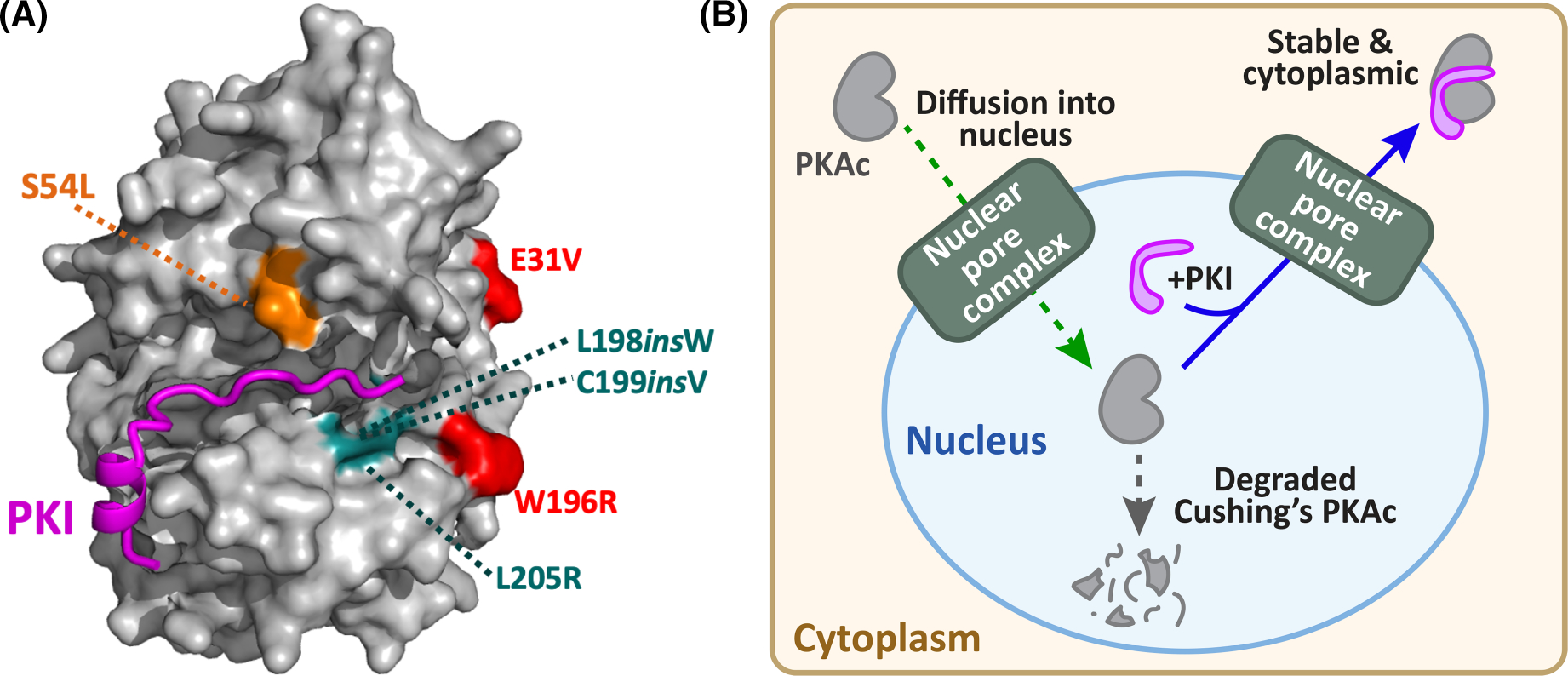
(A) Structural model (AlphaFold) showing location of PKI refractory Cushing’s PKAc mutations (cyan) in positions key for interaction with PKI (magenta). Cushing’s mutations (red) that that do not impact PKI association are distal to the binding groove for the heat-stable inhibitor. The location of S54L (orange) PKAc (β-isoform) mutation above the kinase-PKI binding interface. (B) Schematic depicting the cellular conclusions of this study. Free PKAc subunits (grey) diffuse into the nucleus whereupon they bind PKI (magenta) and are exported back to the cytoplasm. PKAc variants unable to bind PKI remain nuclear and are susceptible to protein degradation.
Conclusion
Whole exome sequencing of adrenal tissue from patients has identified mutations and insertions in PKAc that are drivers of Adrenal Cushing syndrome [12–18]. Somatic mutations in a single allele of the PRKACA or PRKACB locus are sufficient to cause overproduction of the stress hormone cortisol. The impact of single mutations on PKAc action are typified by mutations that encode PKAcL205R and PKAcW196R. Superficially, both kinase variants appear virtually identical as they encode substitutions for arginine only nine residues apart. Yet each mutant kinase propagates a distinct pathogenic response. PKAcL205R attenuates the Hippo pathway, whereas MAP kinase signaling is elevated in adrenal-specific heterozygous PKAcW196R knock-in mice, resulting in adrenal hyperplasia [40]. These findings emphasize how distinct pathological signaling events can be mobilized through subtle but distinct changes in the physiochemical properties of PKAc. We extend this concept by showing that PKAc Cushing’s variants can be classified based upon their ability to interact with the heat-stable protein kinase inhibitor PKI. Retaining an ability to bind PKI favors cytoplasmic retention and the stability of PKAcE31V and PKAcW196R protein whereas loss of this protein–protein interaction drives nuclear accumulation and reduced stability of PKAcL205R PKAcL198insW, or PKAcC199insV variants (Figure 6B). As genomic sequencing advances, it will be interesting to evaluate how newly discovered Cushing’s PKAc variants are classified in terms of their interaction with PKI and whether they engage common or distinct signaling effectors to propagate cortisol hypersecretion.
Methods
Structural models
PKAc-PKI structures were modeled using AlphaFold2_Advanced (ColabNotebook). Sequences used for modeling were derived from Uniprot (PRKACA-P17612, PKIA-P61925) and modified to include relevant Cushing’s PKAc mutations. Modeling was performed using default settings and the highest scoring model visualized using Pymol (2.5.4).
Microbe strains
All purified proteins were produced in BL21 (DE3) pLysS E. coli cells (Novagen) grown in Luria-Bertani (LB) broth supplemented with the appropriate antibiotic. Expression induced with 0.5 mM IPTG for 18 h at 18°C and 220 RPM. Amplification of non-viral mammalian expression plasmids was performed in GC10 competent cells (Genesee) grown in LB broth at 37°C. Amplification of viral vectors was performed in either Stbl3 (Invitrogen) or Stable (NEB) competent cells grown in LB broth with ampicillin at 30°C.
Cell lines
All mammalian cells were grown under 37°C, 5% CO2 incubation conditions. Female NCI-H295R cells (CVCL_0458) were maintained in ATCC DMEM: F12 medium containing 2.5% Corning NuSerum I and 1% Corning ITS+ supplement. Female U2OS CRISPR PRKACA−/− cells [37] and HEK293T cells were maintained in DMEM medium with 10% fetal bovine serum.
Antibodies
The following antibodies were used in our studies: GFP Rockland 600–101-215 (IF, IP, WB); V5-tag Thermo Fisher R96025 (IF, IP, WB); HA-Tag CST 3724 (IP); HA-Peroxidase Roche 12013819001 (WB); phospho-PKA Substrate CST 9624 (WB).
Plasmid generation
The PDEST-PKAc and pDEST-PKIA plasmids were generated by inserting the PRKACA coding sequence and the PKIA coding sequence into the pDest backbone by Gateway cloning. Cushing’s PKAc plasmid variants were generated by PCR mutagenesis cloning using primers listed on Table 1. PCR reactions were set up using 36.2 μl ddH2O, 10 μl 5× HF Phusion buffer (NEB), 1 μl of 10 μM mixed dNTPs, 1.5 μl combined primers at 10 μM each, 1 μl template DNA at 5 ng/μl, and 0.3 μl Hot-Start Phusion polymerase (NEB) in a Bio-Rad thermocycler. Thermocycling protocols varied depending on primer conditions and length of target region. For mutagenesis protocols, the PCR product was digested with DpnI restriction enzyme to get rid of the template DNA and then purified using a PCR clean-up kit (GeneJet, K0702). To enhance re-ligation, the ends of the PCR product were phosphorylated with T4 polynucleotide kinase enzyme (NEB, M0201S) for 1 h at 37°C. Ligation was then performed with T4 DNA ligase (M0202S) for 30 min at RT. Transformation into competent TOP10 cells was performed on ice for 30 min before heat shock for 45 s at 42°C followed by 2 min incubation on ice, and 1 h recovery in antibiotic-free LB broth before plating on the appropriate selection plates. Colonies were expanded in LB broth supplemented with the appropriate antibiotic. Plasmids were purified by minipreps (Omega Bio-TEK, D6945–02).
Table 1.
Classification of Cushing’s syndrome PKAc mutants based upon their ability to bind PKI
| Primer Name | Sequence |
|---|---|
|
| |
| E31V_mut_FWD | GTAAGTCCCGCTCAGAACACAG |
| E31V_mut_REV | CCATTTTTTAAGAAAATCTTCTTTGGCTTTGG |
| W196R_mut_FWD | AGGACCTTGTGCGGCAC |
| W196R_mut_REV | AGTGCGGCCCTTCACG |
| 198TrpIns_mut_FWD | TGGTGCGGCACCCCTGAGTA |
| 198TrpIns_mut_REV | CAAGGTCCAAGTGCGGCC |
| 199ValIns_mut_FWD | GTAGGCACCCCTGAGTACCTG |
| 199ValIns_mut_REV | GCACAAGGTCCAAGTGCG |
| L205R mut FWD | CGTGCCCCTGAGATTATCCTGAGCAAAG |
| L205 mut REV | GTACTCAGGGGTGCCGCACAAG |
Immunoprecipitations
Cell lysates were made using lysis buffer containing 1% (v/v) Triton X-100, 130 mM NaCl, 20 mM NaF, 2 mM EDTA, and 50 mM Tris pH 7.4 (at 4°C) along with 1 μM AEBSF, 10 μM leupeptin, and 1 μM benzamidine. Lysates were incubated (5 min) on ice, pipetted up and down 10x with a P200 tip, and clarified by centrifugation at 15,000×g for 8 min at 4°C. Protein concentration was measured by BCA (Thermo Scientific) and adjusted to 1 mg/ml using lysis buffer. Samples (300–500 ml) were precleared by rotating with 20 μl protein A agarose for 30 min at 4°C. Supernatants were then incubated overnight with 1–2 μg of the appropriate antibody for 18 h (4°C). Following day, proteins were precipitated by incubation with 30 μl of protein A agarose for 1 h with gentle agitation. Beads were washed (3x) with lysis buffer and bound protein was eluted with PAGE sample buffer (3% β-mercaptoethanol, final) at 80°C for 10 min.
Immunoblotting
Cell lysates were prepared using either Triton −100 lysis buffer (see Immunoprecipitations) or RIPA lysis buffer (1% (v/v) NP-40 Tergitol, 0.5% (w/v) deoxycholate, 0.1% (w/v) SDS, 130 mM NaCl, 20 mM NaF, 2 mM EDTA, and 20 mM Tris pH 7.5 (at 4°C), and supplemented with 1 mM AEBSF, 10 μM leupeptin, and 1 mM benzamidine. For experiments to detect S/T phosphoproteins, 10 mM beta glycerophosphate was added. Samples were incubated for 5 min on ice and centrifuged at 15,000×g for 8 min at 4°C. Protein concentration was measured by BCA (Thermo Scientific). SDS–PAGE gels were loaded with 10–30 mg protein after heating for 10 min at 80°C with PAGE sample buffer containing 3% β-mercaptoethanol. Proteins were transferred to nitrocellulose or PVDF, with the latter being used to ensure capture and detection of the small PKI protein. Membranes were then incubated with ponceau S to measure total protein loading, blocked in 5% (w/v) milk TBST for 30 min at RT, and probed with antibodies in 5% (w/v) BSA TBST for 18 h at 4°C. Membranes were washed 3 times in TBST and then incubated with secondary antibodies conjugated to HRP diluted in 5% (w/v) milk TBST for 1 h at RT. After washing again 3x in TBST, signals were visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher) on an Invitrogen iBright Imaging System.
Drug treatments
Cells were transfected with a mixture of 2 μl TransIT®-LT1 reagent (Mirus Bio 107676–120) and 100 μl optimum (Gibco 11058021) per 1 μg of plasmid DNA. After 48 h, cells were treated with either 1 μM MG-132 (Millipore 474788) or 100 nM BafA1 (Sigma–Aldrich B1793). After 1 h, 50 μg/ml CHX (Sigma–Aldrich C1988) was added for all conditions and incubated for an additional 7 h (8 h, total treatment time) before harvesting. For okadaic acid treatments, cells were treated with 100 nM okadaic acid (CST 59345) and incubated for 6 h before harvesting.
λ-phosphatase assay
Lysates were prepared with RIPA buffer (described above) excluding NaF. Phosphatase treatments were set up with 0.5 μl lambda protein phosphatase (NEB P0753S), MnCl2 and protein metallo-phosphatase (PMP) supplied in the kit. The mixture was incubated at 30°C for 2 h and the lysates then processed for immunoblotting as described above.
In vitro pull-down assays
Purified N-terminal GST-tagged PKI fusion proteins were conjugated to GSH Sepharose beads by incubating for 2 h at RT in the presence of 1 mM DTT, washed three times in lysis buffer (50 mM Tris pH 7.4, 0.1 M NaCl, 2 mM EDTA, 20 mM NaF, 0.5% (v/v) Triton X-100, benzamidine, AEBSF, leupeptin) and resuspended at a final protein concentration of ~0.2 μM. This was added to 0.25 mg of lysates from U2OS CRISPR PRKACA−/− cells transfected with PKAc-V5 Cushing’s variants. The mixtures were incubated for 18 h at 4°C with constant agitation. The supernatant was then removed, and beads washed (3×) with lysis buffer. Complexes were eluted from the beads by incubation with reduced GSH (20 mM) and NaOH (60 mM) in lysis buffer for 10 min at RT. PAGE sample buffer supplemented with BME was added and complexes denatured at 95°C for 10 min. Proteins were analyzed by SDS–PAGE on a 12% polyacrylamide gel. For pull-down assays using purified recombinant PKAc proteins, 5 μg GST-PKI (or control GST) was conjugated to GSH-beads, equilibrated in 50 mM Tris pH 7.4, 0.1 M NaCl, 1 mM DTT, 1 mM ATP, 10 mM MgCl2 and 5% (v/v) glycerol, and incubated with 2 μg PKAc for 1 h at 30°C with gentle agitation.
Mass spectrometry data
Mass spectrometry data were derived from studies as detailed in references [39,40]. The PKI association graph used total PKIα peptide counts amalgamated from four biological replicates.
Recombinant protein purification
PKA catalytic (C) subunits and PKI (with N-terminal 6× His [pET30; kanamycin] and GST tags [pOPINJ; ampicillin] respectively) were produced in BL21 (DE3) pLysS E. coli cells (Novagen), grown in LB broth (C) supplemented with appropriate antibiotics at 37°C (220 rpm) until OD600nm ~ 0.6 and expression induced with 0.5 mM IPTG for 18 h at 18°C and purified by affinity chromatography using Ni-NTA agarose resin (GenScript) and size exclusion chromatography using a HiLoad 16/600 Superdex 200 column (GE Healthcare) equilibrated in 50 mM Tris–HCl, pH 7.4, 100 mM NaCl, 1 mM DTT and 10% (v/v) glycerol. Recombinant PKAc and PKI expressed in bacteria as described [68]. Purification procedures are described further in [44,45].
Protein kinase assays
PKA kinase assays were performed using a real-time mobility shift-based microfluidic system, as described previously [45], in the presence of 2 μM of the fluorescent-tagged ‘Kemptide’ substrate (LRRASLG) and 1 mM ATP (as standard). Pressure and voltage settings were −1.8 (PSI), −2250 V (upstream voltage), and −500 V (downstream voltage) respectively. All assays were performed in 50 mM HEPES (pH 7.4), 0.015% (v/v) Brij-35, 1 mM DTT and 5 mM MgCl2, and peptide phosphorylation was detected in real time as the ratio of phosphopeptide:peptide. Changes in PKA activity in the presence of PKI was quantified as the rate of phosphate incorporation into the substrate peptide (pmol phosphate · min−1 · μM enzyme−1), and then normalized with respect to control assays. To prevent ATP depletion and consequential loss of assay linearity, phosphate incorporation into the peptide was generally limited to <20%. PKA WT and W196R mutants were assayed at a final concentration of 0.3 nM, and PKA L205R was assayed at 6 μM to account for the lower rate of activity.
Differential scanning fluorimetry
Thermal-shift assays were performed using a StepOnePlus Real-Time PCR machine (Life Technologies) using Sypro-Orange dye (Invitrogen) and thermal ramping (0.3°C in step intervals between 25 and 94°C). PKA proteins were diluted to a final concentration of 5 μM in 50 mM Tris–HCl, pH 7.4 and 100 mM NaCl in the presence or absence of 1 mM ATP (and 10 mM MgCl2), or 10 μM PKI with ATP-MgCl2, and were assayed as described previously [45]. Normalized data were processed using the Boltzmann equation to generate sigmoidal denaturation curves, and average Tm/ΔTm values calculated using GraphPad Prism software, as previously described [69].
Immunofluorescent staining
Cells were plated on acid-washed coverslips and transfected with plasmids using Mirus TransIT-LT 48 h before harvest. Cells were fixed in 4% (w/v) PFA for 15 min at RT and washed 3× in PBS. Coverslips were moved to a humidity chamber and blocked for 1 h at RT in 3% (w/v) BSA, 0.3% (v/v) Triton X-100. Primary antibodies were diluted in blocking solution and applied to coverslips overnight at 4°C. Coverslips were washed 3× with PBS, incubated with fluorescent secondary antibodies (1 : 1000) and DAPI (~1 : 10 000), and washed 3× in PBS again before mounting. Images were taken on a Keyence BZ-X710 microscope and processed/analyzed using Image J analysis software (FIJI). Line scan analysis was performed on a z-section identified as the clearest DAPI signal, indicating focus on the vertical center of the cell. Lines covered the complete length of a cell, crossing the nucleus. Fluorescent intensity was measured for PKAc and DAPI signals on composite images. DAPI values were used to identify nuclear boundaries. Average pixel intensity values for PKAc signal were calculated for nuclear and non-nuclear compartments and the ratio of nuclear to non-nuclear signal was determined in each cell.
Quantification and statistical analyses
Data quantification and statistical analyses were performed with GraphPad Prism 9 for Mac as indicated in each figure legend. All data are presented with individual values displayed when possible along with mean ± SEM unless otherwise noted in the figure legend. Individual figure legends contain specific information on statistical parameters. Experiments involving more than three conditions used one-way ANOVA with subsequent t-tests corrected for multiple comparisons. Specific statistical approaches were determined based on the distributions and parameters for each dataset.
Acknowledgements
The authors would like to thank T. Lakey, E. Butcher, and K. Forbush for technical assistance.
Funding
This work was supported by National Institutes of Health (NIH) grants F32DK121415 (M.H.O.), R01DK119186 and R01DK119192 (J.D.S) and funding from BBSRC BB/T018127/1 and BB/S018514/1 (P.A.E.).
Abbreviations
- CHX
cycloheximide
- GSH
glutathione
- LB
Luria-Bertani
- PKAc
protein kinase A catalytic subunits
- PKI
protein kinase inhibitor
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
CRediT Author Contribution
John D. Scott: Supervision, Funding acquisition, Visualization, Writing — original draft, Writing — review and editing. Mitchell H. Omar: Conceptualization, Supervision, Funding acquisition, Investigation, Visualization, Writing — original draft, Writing — review and editing. Maryanne Kihiu: Investigation, Visualization, Writing — original draft, Writing — review and editing. Dominic Byrne: Resources, Investigation, Writing — review and editing. Kyung-Soon Lee: Investigation, Writing — review and editing. Tyler M. Lakey: Investigation. Erik Butcher: Investigation. Patrick A. Eyers: Supervision, Funding acquisition, Writing — review and editing.
Data Availability
Mass spectrometry data used for this paper is available at massive.ucsd.edu under the identifier MSV000088654.
References
- 1.Golden SH, Brown A, Cauley JA, Chin MH, Gary-Webb TL, Kim C et al. (2012) Health disparities in endocrine disorders: biological, clinical, and nonclinical factors–an Endocrine Society scientific statement. J. Clin. Endocrinol. Metab. 97, E1579–E1639 10.1210/jc.2012-2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golden SH, Robinson KA, Saldanha I, Anton B and Ladenson PW (2009) Clinical review: prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J. Clin. Endocrinol. Metab. 94, 1853–1878 10.1210/jc.2008-2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tiwari A, Goel M, Pal P and Gohiya P (2013) Topical-steroid-induced iatrogenic Cushing syndrome in the pediatric age group: a rare case report. Indian J. Endocrinol. Metab. 17, S257–S258 10.4103/2230-8210.119593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hernandez-Ramirez LC and Stratakis CA (2018) Genetics of Cushing’s syndrome. Endocrinol. Metab. Clin. North Am. 47, 275–297 10.1016/j.ecl.2018.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fleseriu M and Castinetti F (2016) Updates on the role of adrenal steroidogenesis inhibitors in Cushing’s syndrome: a focus on novel therapies. Pituitary 19, 643–653 10.1007/s11102-016-0742-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cuevas-Ramos D, Lim DST and Fleseriu M (2016) Update on medical treatment for Cushing’s disease. Clin. Diabetes Endocrinol. 2, 16 10.1186/s40842-016-0033-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broder MS, Neary MP, Chang E, Cherepanov D and Ludlam WH (2015) Incidence of Cushing’s syndrome and Cushing’s disease in commercially-insured patients <65 years old in the United States. Pituitary 18, 283–289 10.1007/s11102-014-0569-6 [DOI] [PubMed] [Google Scholar]
- 8.Lacroix A, Feelders RA, Stratakis CA and Nieman LK (2015) Cushing’s syndrome. Lancet (London, England) 386, 913–927 10.1016/S0140-6736(14)61375-1 [DOI] [PubMed] [Google Scholar]
- 9.Sunwoo SH, Lee JS, Bae S, Shin YJ, Kim CS, Joo SY et al. (2019) Chronic and acute stress monitoring by electrophysiological signals from adrenal gland. Proc. Natl Acad. Sci. U.S.A. 116, 1146–1151 10.1073/pnas.1806392115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cicala MV and Mantero F (2010) Hypertension in Cushing’s syndrome: from pathogenesis to treatment. Neuroendocrinology 92 Suppl 1, 44–49 10.1159/000314315 [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg D, Groussin L, Jullian E, Perlemoine K, Bertagna X and Bertherat J (2002) Role of the PKA-regulated transcription factor CREB in development and tumorigenesis of endocrine tissues. Ann. N. Y. Acad. Sci. 968, 65–74 10.1111/j.1749-6632.2002.tb04327.x [DOI] [PubMed] [Google Scholar]
- 12.Beuschlein F, Fassnacht M, Assie G, Calebiro D, Stratakis CA, Osswald A et al. (2014) Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N. Engl. J. Med. 370, 1019–1028 10.1056/NEJMoa1310359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao Y, He M, Gao Z, Peng Y, Li Y, Li L et al. (2014) Activating hotspot L205R mutation in PRKACA and adrenal Cushing’s syndrome. Science 344, 913–917 10.1126/science.1249480 [DOI] [PubMed] [Google Scholar]
- 14.Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G et al. (2014) Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J. Clin. Endocrinol. Metab. 99, E2093–E2100 10.1210/jc.2014-2152 [DOI] [PubMed] [Google Scholar]
- 15.Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C et al. (2014) Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat. Genet. 46, 613–617 10.1038/ng.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y et al. (2014) Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 344, 917–920 10.1126/science.1252328 [DOI] [PubMed] [Google Scholar]
- 17.Ronchi CL, Di Dalmazi G, Faillot S, Sbiera S, Assié G, Weigand I et al. (2016) Genetic landscape of sporadic unilateral adrenocortical adenomas without PRKACA p.Leu206Arg mutation. J. Clin. Endocrinol. Metab. 101, 3526–3538 10.1210/jc.2016-1586 [DOI] [PubMed] [Google Scholar]
- 18.Espiard S, Knape MJ, Bathon K, Assié G, Rizk-Rabin M, Faillot S et al. (2018) Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI insight 3, e98296 10.1172/jci.insight.98296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bathon K, Weigand I, Vanselow JT, Ronchi CL, Sbiera S, Schlosser A et al. (2019) Alterations in protein kinase a substrate specificity as a potential cause of cushing syndrome. Endocrinology 160, 447–459 10.1210/en.2018-00775 [DOI] [PubMed] [Google Scholar]
- 20.Tasken K and Aandahl EM (2004) Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev. 84, 137–167 10.1152/physrev.00021.2003 [DOI] [PubMed] [Google Scholar]
- 21.Wong W and Scott JD (2004) AKAP signalling complexes: focal points in space and time. Nat. Rev. Mol. Cell Biol. 5, 959–970 10.1038/nrm1527 [DOI] [PubMed] [Google Scholar]
- 22.Scott JD and Pawson T (2009) Cell signaling in space and time: where proteins come together and when they’re apart. Science 326, 1220–1224 10.1126/science.1175668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott JD, Fischer EH and Krebs EG (1985) The inhibitory region of the heat-stable protein inhibitor of the cAMP-dependent protein kinase. Proc. Natl Acad. Sci. U.S.A. 84, 703–708 10.1073/pnas.82.13.4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott JD, Glaccum MB, Fischer EH and Krebs EG (1986) Primary-structure requirements for inhibition by the heat-stable inhibitor of the cAMP-dependent protein kinase. Proc. Natl Acad. Sci. U.S.A. 83, 1613–1616 10.1073/pnas.83.6.1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng HC, Kemp BE, Pearson RB, Smith AJ, Misconi L, Van Patten SM et al. (1986) A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. J. Biol. Chem. 261, 989–992 10.1016/S0021-9258(17)36041-6 [DOI] [PubMed] [Google Scholar]
- 26.Collins KB and Scott JD (2023) Phosphorylation, compartmentalization, and cardiac function. IUBMB Life 75, 353–369 10.1002/iub.2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shabb JB (2001) Physiological substrates of cAMP-dependent protein kinase. Chem. Rev. 101, 2381–2411 10.1021/cr000236l [DOI] [PubMed] [Google Scholar]
- 28.Turnham RE and Scott JD (2016) Protein kinase A catalytic subunit isoform PRKACA; history, function and physiology. Gene 577, 101–108 10.1016/j.gene.2015.11.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor SS, Ilouz R, Zhang P and Kornev AP (2012) Assembly of allosteric macromolecular switches: lessons from PKA. Nat. Rev. Mol. Cell Biol. 13, 646–658 10.1038/nrm3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stratakis CA (2018) Cyclic AMP-dependent protein kinase catalytic subunit A (PRKACA): the expected, the unexpected, and what might be next. J. Pathol. 244, 257–259 10.1002/path.5014 [DOI] [PubMed] [Google Scholar]
- 31.Ma S, Liu W, Zhang A, Pan L, Tang W, Jiang B et al. (2019) Identification of a PRKAR1A mutation (c.491_492delTG) in familial cardiac myxoma: a case report. Medicine (Baltimore) 98, e14866 10.1097/md.0000000000014866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palencia-Campos A, Aoto PC, Machal EMF, Rivera-Barahona A, Soto-Bielicka P, Bertinetti D et al. (2020) Germline and mosaic variants in PRKACA and PRKACB cause a multiple congenital malformation syndrome. Am. J. Hum. Genet. 107, 977–988 10.1016/j.ajhg.2020.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turnham RE, Smith FD, Kenerson HL, Omar MH, Golkowski M, Garcia I et al. (2019) An acquired scaffolding function of the DNAJ-PKAc fusion contributes to oncogenic signaling in fibrolamellar carcinoma. eLife 8, e44187 10.7554/eLife.44187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Espiard S and Bertherat J (2015) The genetics of adrenocortical tumors. Endocrinol. Metab. Clin. North Am. 44, 311–334 10.1016/j.ecl.2015.02.004 [DOI] [PubMed] [Google Scholar]
- 35.Piggott LA, Bauman AL, Scott JD and Dessauer CW (2008) The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc. Natl Acad. Sci. U.S.A. 105, 13835–13840 10.1073/pnas.0712100105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith FD, Reichow SL, Esseltine JL, Shi D, Langeberg LK, Scott JD et al. (2013) Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. Elife 2, e01319 10.7554/eLife.01319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith FD, Esseltine JL, Nygren PJ, Veesler D, Byrne DP, Vonderach M et al. (2017) Local protein kinase A action proceeds through intact holoenzymes. Science 356, 1288–1293 10.1126/science.aaj1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lubner JM, Dodge-Kafka KL, Carlson CR, Church GM, Chou MF and Schwartz D (2017) Cushing’s syndrome mutant PKAL205R exhibits altered substrate specificity. FEBS Lett. 591, 459–467 10.1002/1873-3468.12562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Omar MH, Lauer SM, Lau HT, Golkowski M, Ong SE and Scott JD (2023) Proximity biotinylation to define the local environment of the protein kinase A catalytic subunit in adrenal cells. STAR Protoc. 4, 101992 10.1016/j.xpro.2022.101992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Omar MH, Byrne DP, Jones KN, Lakey TM, Collins KB, Lee KS et al. (2022) Mislocalization of protein kinase A drives pathology in Cushing’s syndrome. Cell Rep. 40, 111073 10.1016/j.celrep.2022.111073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitehouse S, Feramisco JR, Casnellie JE, Krebs EG and Walsh DA (1983) Studies on the kinetic mechanism of the catalytic subunit of the cAMP-dependent protein kinase. J. Biol. Chem. 258, 3693–3701 10.1016/S0021-9258(18)32720-0 [DOI] [PubMed] [Google Scholar]
- 42.Whitehouse S and Walsh DA (1982) Purification of a physiological form of the inhibitor protein of the cAMP-dependent protein kinase. J. Biol. Chem. 257, 6028–6032 10.1016/S0021-9258(20)65100-6 [DOI] [PubMed] [Google Scholar]
- 43.Wen W, Meinkoth JL, Tsien RY and Taylor SS (1995) Identification of a signal for rapid export of proteins from the nucleus. Cell 82, 463–473 10.1016/0092-8674(95)90435-2 [DOI] [PubMed] [Google Scholar]
- 44.Keshwani MM, Klammt C, von Daake S, Ma Y, Kornev AP, Choe S et al. (2012) Cotranslational cis-phosphorylation of the COOH-terminal tail is a key priming step in the maturation of cAMP-dependent protein kinase. Proc. Natl Acad. Sci. U.S.A. 109, E1221–E1229 10.1073/pnas.1202741109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Byrne DP, Vonderach M, Ferries S, Brownridge PJ, Eyers CE and Eyers PA (2016) cAMP-dependent protein kinase (PKA) complexes probed by complementary differential scanning fluorimetry and ion mobility-mass spectrometry. Biochem. J. 473, 3159–3175 10.1042/bcj20160648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wen W, Harootunian AT, Adams SR, Feramisco J, Tsien RY, Meinkoth JL et al. (1994) Heat-stable inhibitors of cAMP-dependent protein kinase carry a nuclear export signal. J. Biol. Chem. 269, 32214–32220 10.1016/S0021-9258(18)31623-5 [DOI] [PubMed] [Google Scholar]
- 47.Meinkoth JL, Ji Y, Taylor SS and Feramisco JR (1990) Dynamics of the distribution of cyclic AMP-dependent protein kinase in living cells. Proc. Natl Acad. Sci. U.S.A. 87, 9595–9599 10.1073/pnas.87.24.9595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Omar MH and Scott JD (2020) AKAP signaling islands: venues for precision pharmacology. Trends Pharmacol. Sci. 41, 933–946 10.1016/j.tips.2020.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calebiro D, Di Dalmazi G, Bathon K, Ronchi CL and Beuschlein F (2015) cAMP signaling in cortisol-producing adrenal adenoma. Eur. J. Endocrinol. 173, M99–106 10.1530/EJE-15-0353 [DOI] [PubMed] [Google Scholar]
- 50.Calebiro D, Hannawacker A, Lyga S, Bathon K, Zabel U, Ronchi C et al. (2014) PKA catalytic subunit mutations in adrenocortical Cushing’s adenoma impair association with the regulatory subunit. Nat. Commun. 5, 5680 10.1038/ncomms6680 [DOI] [PubMed] [Google Scholar]
- 51.Taylor SS, Herberg FW, Veglia G and Wu J (2023) Edmond Fischer’s kinase legacy: history of the protein kinase inhibitor and protein kinase A. IUBMB Life 75, 311–323 10.1002/iub.2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manning G, Whyte DB, Martinez R, Hunter T and Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 10.1126/science.1075762 [DOI] [PubMed] [Google Scholar]
- 53.Walker C, Wang Y, Olivieri C, Karamafrooz A, Casby J, Bathon K et al. (2019) Cushing’s syndrome driver mutation disrupts protein kinase A allosteric network, altering both regulation and substrate specificity. Sci. Adv. 5, eaaw9298 10.1126/sciadv.aaw9298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scott JD (1993) Cyclic nucleotide-dependent protein kinases review. Pharmacol. Ther. 139, 137–156 10.1016/0163-7258(91)90075-w [DOI] [PubMed] [Google Scholar]
- 55.Walsh DA, Ashby CD, Gonzalez C, Calkins D, Fischer EH and Krebs EG (1971) Purification and characterization of a protein inhibitory of adenosine 3′,5′-monophosphate-dependent protein kinases. J. Biol. Chem. 246, 1977–1985 10.1016/S0021-9258(19)77177-4 [DOI] [PubMed] [Google Scholar]
- 56.Scott JD, Fischer EH, Takio K, DeMaille JB and Krebs EG (1985) Amino acid sequence of the heat-stable inhibitor of the cAMP-dependent protein kinase from rabbit skeletal muscle. Proc. Natl Acad. Sci. U.S.A. 82, 5732–5736 10.1073/pnas.82.17.5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luzi NM, Lyons CE, Peterson DL and Ellis KC (2018) Kinetics and inhibition studies of the L205R mutant of cAMP-dependent protein kinase involved in Cushing’s syndrome. FEBS Open Bio 8, 606–613 10.1002/2211-5463.12396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fantozzi DA, Taylor SS, Howard PW, Maurer RA, Feramisco JR and Meinkoth JL (1992) Effect of the thermostable protein kinase inhibitory on intracellular localization of the catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem. 267, 16824–16828 10.1016/S0021-9258(18)41857-1 [DOI] [PubMed] [Google Scholar]
- 59.Malencik D, Scott JD, Fischer EH, Krebs EG and Anderson SA (1986) Association of calmodulin with peptide analogues of the inhibitory region of the heat-stable protein inhibitor of adenosine cyclic 3′,5′-phosphate dependent protein kinase. Biochemistry 25, 3502–3508 10.1021/bi00360a004 [DOI] [PubMed] [Google Scholar]
- 60.Scott JD, Faux MC (1998) Use of synthetic peptides in the dissection of protein-targeting interactions. In Protein Targeting Protocols. Methods in Molecular Biology (Clegg RA ed), vol 88. Humana Press, Totowa, NJ, USA. [DOI] [PubMed] [Google Scholar]
- 61.Krebs EG Blumenthal DK, Edelman AM, and Hales CN (1985) The functions of the cAMP-dependent protein kinase. In Mechanisms of Receptor Regulation (Crooke ST and Poste G, eds), pp. 324–367, Plenum, New York [Google Scholar]
- 62.Nishi H, Shaytan A and Panchenko AR (2014) Physicochemical mechanisms of protein regulation by phosphorylation. Front. Genet. 5, 270 10.3389/fgene.2014.00270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Byrne DP, Omar MH, Kennedy EJ, Eyers PA and Scott JD (2022) Biochemical analysis of AKAP-anchored PKA signaling complexes methods. Mol. Biol. 2483, 297–317 10.1007/978-1-0716-2245-2_19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olivieri C, Wang Y, Li GC, V SM, Kim J, Stultz BR et al. (2020) Multi-state recognition pathway of the intrinsically disordered protein kinase inhibitor by protein kinase A. Elife 9, e55607 10.7554/eLife.55607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mirdita M, Schutze K, Moriwaki Y, Heo L, Ovchinnikov S and Steinegger M (2022) Colabfold: making protein folding accessible to all. Nat. Methods 19, 679–682 10.1038/s41592-022-01488-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Selvaraj V, Stocco DM and Clark BJ (2018) Current knowledge on the acute regulation of steroidogenesis. Biol. Reprod. 99, 13–26 10.1093/biolre/ioy102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stocco DM (2001) StAR protein and the regulation of steroid hormone biosynthesis. Annu. Rev. Physiol. 63, 193–213 10.1146/annurev.physiol.63.1.193 [DOI] [PubMed] [Google Scholar]
- 68.Eyers PA, Liu J, Hayashi NR, Lewellyn AL, Gautier J and Maller JL (2005) Regulation of the G(2)/M transition in Xenopus oocytes by the cAMP-dependent protein kinase. J. Biol. Chem. 280, 24339–24346 10.1074/jbc.M412442200 [DOI] [PubMed] [Google Scholar]
- 69.Byrne DP, Shrestha S, Galler M, Cao M, Daly LA, Campbell AE et al. (2020) Aurora A regulation by reversible cysteine oxidation reveals evolutionarily conserved redox control of Ser/Thr protein kinase activity. Sci. Signal. 13, eaax2713 10.1126/scisignal.aax2713 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Mass spectrometry data used for this paper is available at massive.ucsd.edu under the identifier MSV000088654.