

Abstract
Rubinstein-Taybi syndrome (RTS) is an archetypical genetic syndrome that is characterised by intellectual disability, well-defined facial features, distal limb anomalies and atypical growth, among numerous other signs and symptoms. It is caused by variants in either of two genes (CREBBP, EP300) which encode for the proteins CBP and p300, which both have a function in transcription regulation and histone acetylation. As a group of international experts and national support groups dedicated to the syndrome, we realised that marked heterogeneity currently exists in clinical and molecular diagnostic approaches and care practices in various parts of the world. Here, we outline a series of recommendations that document the consensus of a group of international experts on clinical diagnostic criteria for types of RTS (RTS1: CREBBP; RTS2: EP300), molecular investigations, long-term management of various particular physical and behavioural issues and care planning. The recommendations as presented here will need to be evaluated for improvements to allow for continued optimisation of diagnostics and care.
Keywords: Genetic Diseases, Inborn; Mental Disorders; Genetics, Medical; Phenotype
Introduction
Rubinstein-Taybi syndrome (RTS) (MIM (Mendelian Inheritance in Man) #180849; #613684; #610543) is a multisystem disorder with physical, cognitive and behavioural characteristics, which can be caused by variants in two genes that regulate transcription via chromatin remodelling. The condition is named after the US paediatrician Jack Rubinstein and Iranian radiologist Hooshang Taybi who described seven affected infants in 1963.1 There are >800 publications on RTS and related topics. Within the framework of the European Reference Network Ithaca a group of international experts recognised the importance of equal practices regarding diagnostic procedures and care for individuals with RTS. To address this issue, an international consensus group was established, which performed a literature review, evaluated data critically, formulated conclusions and held a face-to-face meeting in the presence of patient group representatives. This has led to the present series of guidelines for diagnostics and care for individuals with RTS. For Methods see online supplemental materials.
jmg-2023-109438supp001.pdf (332KB, pdf)
Clinical diagnostic criteria
Definition
The goal of defining an entity is that affected individuals and their caregivers who face similar signs, symptoms and health problems, can meet one another, share knowledge, emotions and experiences about the disorder, support one another, and, this way, facilitate care and research. So the essence of a definition is to allow grouping together individuals with the same diagnosis.
Currently, variants in the genes CREBBP and EP300 are known to cause RTS.2 3 One may argue that the diagnosis of RTS should be based on these molecular findings and clinical diagnostic criteria are no longer needed. Several issues argue against this: there are individuals with a phenotype classically fitting RTS, but without detectable cytogenetic or molecular anomaly; there are individuals with a genuine variant in CREBBP or EP300 but with a phenotype different from the RTS phenotype,4 which can have major consequences in counselling patients and families; there are individuals with either a CREBBP or an EP300 variant of uncertain pathogenicity, and whose phenotype resembles RTS only to a limited extent, leaving it uncertain whether or not the variant is causative for the phenotype; there are many countries worldwide in which the availability of molecular studies is limited, and in which caregivers have to rely on a clinical diagnosis for counselling. For these reasons, we concluded that a clinical definition of the RTS phenotype is still needed and will remain needed.
There is no widely accepted set of clinical diagnostic criteria for RTS. We used the largest published set of data on individuals with RTS and either a CREBBP (n=308) or an EP300 variant (n=52),5 to determine the sensitivity of signs and symptoms (table 1).
Table 1.
Main clinical findings in percentages of individuals with molecularly confirmed Rubinstein-Taybi syndrome
| HPO ID* | CREBBP (n=308) | EP300 (n=52) | |
| Growth | |||
| Intrauterine growth retardation | 0001511 | 49 | 42 |
| Postnatal growth retardation | 0004322 | 75 | 66 |
| Obesity | 0001513 | 29 | 39 |
| Microcephaly | 0000252 | 54 | 87 |
| Craniofacial features | |||
| Highly arched eyebrows | 0002253 | 85 | 65 |
| Long eyelashes | 0000527 | 89 | 90 |
| Epicanthal folds | 0000286 | 44 | 15 |
| Strabismus | 0000486 | 71 | 39 |
| Myopia | 0000545 | 56 | 24 |
| Downslanted palpebral fissures | 0000494 | 79 | 56 |
| Convex nasal ridge | 0000444 | 81 | 44 |
| Columella below alae nasi | 0009765 | 88 | 92 |
| Typical smile† | 0000273 | 94 | 47 |
| Highly arched palate | 0002705 | 77 | 67 |
| Talon cusps‡ | 0011087 | 73 | 4 |
| Micrognathia | 0000347 | 61 | 42 |
| Low-set ears | 0000369 | 44 | 27 |
| Trunk and limbs | |||
| Broad thumbs | 0011304 | 96 | 69 |
| Angulated thumbs | § | 49 | 2 |
| Broad finger tips | 0011300 | 87 | 22 |
| Broad halluces | 0010055 | 95 | 81 |
| Hypertrichosis | 0000998 | 76 | 51 |
| Keloids | 0010562 | 23 | 10 |
| Scoliosis | 0002650 | 18 | 25 |
| Cardiovascular anomalies | 0002564 | 35 | 26 |
| Constipation | 0002019 | 76 | 54 |
| Urinary tract anomalies | 0000079 | 28 | 24 |
| Neuromuscular | |||
| Seizures | 0001250 | 25 | 10 |
| Cognition and behaviour | |||
| Intellectual disability (any degree) | 0001249 | 99 | 94 |
| Autism/autism spectrum disorder | 0000729 | 49 | 25 |
*HPO ID, Human Phenotype Ontology Identifier.
†Smile characterized by crescent-moon shaped palpebral fissures, deepening of labionasal folds, upturned corners of the mouth, usually mouth almost closed, tight upper vermillion and pouting lower vermillion.
‡Permanent dentition.
§No HPO identifier available; we used as definition: angulation of the distal phalanx of a thumb towards the anterior axis (radial side) of the limb.
We used the scored features as available, to avoid a bias. Signs present in at least 75% of either of the two groups were accepted as being sufficiently characteristic of the condition. In addition, we added three features with a lower frequency but which are highly specific for RTS: (1) radially deviated thumbs; (2) keloid formation; and (3) maternal pre-eclampsia. We considered adding talon cusps to these criteria but refrained from doing so as this sign is not yet present in the age group during which typically a diagnostic question arises. When developing the scoring system, it was observed that the presence or absence of the sign ‘long eyelashes’ did not contribute to sensitivity, and given the low intra-observer reliability of this feature it was excluded from the scoring criteria. Furthermore, the features known to be highly specific for RTS (radially deviated thumbs typical smile; columella below alae nasi, maternal pre-eclampsia keloids) were given a higher weighted value in the scoring system to reflect their diagnostic importance. Features were then subdivided into Cardinal Features, which we considered to be essential for RTS, and Suggestive Features, which are present less frequently but should raise suspicion for RTS (table 2; figure 1). Subsequent discussion of these criteria allowed consensus for the clinical diagnostic criteria, based on the presence of both Cardinal and Suggestive Features (online supplemental R1). If an individual scores 12 or higher, including meeting a score for the Cardinal Features, the diagnosis of RTS can be clinically confirmed irrespective of the results of molecular testing. A score of 8–11 including a positive score for the Cardinal Features indicates a likely diagnosis of RTS which requires further confirmation by molecular testing. A score of 5–7, with or without a Cardinal Feature, indicates that the diagnosis of RTS is still possible and molecular studies are indicated. A score of 0–4 indicates that the diagnosis is unlikely, and other explanations of the phenotype should be explored.
Table 2.
Clinical diagnostic criteria for Rubinstein-Taybi Syndrome
| Cardinal | Supportive |
| 1.Face (at least three of six). | a. Maternal pre-eclampsia. |
| a. Highly arched eyebrows. | b. Keloids. |
| b. Downslanted palpebral fissures. | c. Hypertrichosis. |
| c. Convex nasal ridge. | 1 point if c is positive, or |
| d. Columella below alae nasi. | 3 points if a and/or b (with or without c) are positive. |
| e. Highly arched palate. | |
| f. Typical smile. | |
| 3 points or | |
| 4 points if d and/or f are positive. | |
| 2.Skeletal. | |
| a. Angulated thumbs and/or halluces. | |
| b. Broad thumbs. | |
| c. Broad halluces. | |
| 3 points if b and/or c is positive, or | |
| 4 points if a (with or without b/c) is positive. | |
| 3. Growth. | |
| a. Microcephaly. | |
| b. Postnatal growth retardation. | |
| 2 points if a and/or b are positive. | |
| 4. Development. | |
| Delayed development/intellectual disability | |
| 2 points. |
Cardinal Score is positive only if two of the four groups score positiscores ve and also at least either skeletal or craniofacial scores positive
Definitive clinical diagnosis of Rubinstein-Taybi syndrome: Score ≥12 and positive cardinal score.
Likely clinical diagnosis of Rubinstein-Taybi syndrome: Score 8–11 and positive cardinal score. This score warrants molecular analyses of CREBBP and EP300.
Possible clinical diagnosis of Rubinstein-Taybi syndrome: Score 5–7 and negative cardinal score. This score warrants molecular analyses of CREBBP and EP300.
Unlikely clinical diagnosis of Rubinstein-Taybi syndrome: Score 0–4 and negative cardinal score. Further studies for other aetiologies indicated.
Figure 1.
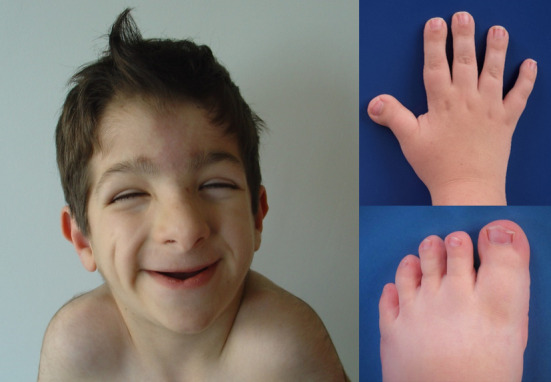
Cardinal features of the clinical diagnostic criteria of face and limbs for Rubinstein-Taybi syndrome (RTS).
jmg-2023-109438supp002.pdf (158.2KB, pdf)
We realise that the presence of unusual signs and symptoms is not incorporated in the score as a negative feature. Still, these should always also be taken into account. Especially the presence of an unusual sign or symptom in someone with a score indicating a likely or definitive diagnosis of RTS should lead to considering the presence of a co-existing second (possibly Mendelian) disorder. In addition, in scoring signs, especially low-hanging columella, the ethnic background should be taken into account as in some ethnicities a low-hanging columella is a common variant. If uncertainty remains it is often useful to evaluate both parents and other relatives as well (online supplemental R2). Lastly, in the first months of life delayed development and disturbed postnatal growth may not yet present and a definitive score may be only possible at an age when this can be reliably ascertained.
Subsequently, we evaluated whether the set of diagnostic features allowed establishing the diagnosis reliably in a group of 100 individuals with molecularly confirmed RTS, that had not been part of the group of patients on which the criteria were built (online supplemental table S1). All individuals scored 5 or higher, indicating none would have been missed as having RTS based on clinical criteria (complete sensitivity). Only seven patients scored in the group Possibly RTS, others scored in the group Likely RTS (n=38) or Definitively RTS (n=55). Furthermore, we evaluated whether 45 individuals with a specific group of pathological CREBBP or EP300 variants, who have been considered to have a separate entity (Menke-Hennekam syndrome (MKHK); MIM #618332 / #618333),4 would be correctly distinguished from RTS (online supplemental table S1). Results showed that none scored as definitive or likely RTS, 9 as possibly RTS and 36 as unlikely RTS, so the entities could correctly be discerned. To determine the specificity, we reasoned that three entities that may resemble RTS and are not uncommon, that is, Floating Harbour syndrome (FHS; MIM #136140) (n=45), Wiedemann-Steiner syndrome (WDSTS; MIM #605130) (n=46) and Cornelia de Lange syndrome (CDLS; MIM #122470) (n=100), should be reliably discerned from RTS based on the set of weighted clinical features (online supplemental table S2). Results showed that none of the individuals with FHS and CDLS fulfilled the criteria for a definitive diagnosis of RTS, but one of the patients with WDSTS had such a score. In addition, one of the patients with WDSTS had a score within the Likely RTS group but was found by the present authors to have a classical RTS facial gestalt. This has to be expected as RTS is a chromatinopathy, and variants in other genes acting in the same pathway are likely to have consequences for the phenotype as well and rarely may even alter the phenotype significantly. Further studies to explain this unusual phenotype are planned. Furthermore, 8 of the 46 WDSTS individuals, and 1 of the 100 CDLS individuals fulfilled the criteria for Likely RTS, indicating that specificity was very high, but not complete. Due to the overlap in function of the genes involved in the four entities, this is to be expected.6 The results are in agreement with our joint clinical experience that, infrequently, discrimination between RTS and WDSTS based on clinical criteria can be extremely difficult. This happens less frequently in patients with CDLS and in FHS, but the phenotypic overlap is still marked. Obviously, this has consequences for the molecular analyses in someone with such scores (see Molecular diagnostic criteria). We realise that prospective studies will be needed to determine more reliable specificity and sensitivity. In addition, such studies should include individuals of non-European descent, to evaluate whether the scoring system will be equally valid as in individuals of European descent.
Severity score
A major issue for families, especially at the time of diagnosis, is an indication of the severity of RTS. No severity score for RTS has been published to date. In our opinion a comparison and weighing of the severity and influences that various signs and symptoms have on the quality of life of an affected individual can only be made by the affected individuals and their families, and not just by physicians. We suggest that a group of family members should be facilitated to indicate which set of physical, cognitive and behavioural issues influence the life of individuals with RTS most. Ideally, such criteria should be stratified according to the nature of the molecular genetic cause (online supplemental R3).
Molecular diagnostic criteria
RTS has been subdivided into type 1 (RTS1; OMIM #180849) and type 2 (RTS2; OMIM #613684) associated with heterozygous pathogenic variants or re-arrangements in the genes CREBBP and EP300, respectively, typically leading to haploinsufficiency. Both genes encode paralogous transcriptional coactivators with lysine acetyltransferase activity.7 8 The proteins CBP and p300 play a crucial role in transcription initiation by acting as a bridge, linking transcription factors to the transcription machinery, and through acetylation of histones9 10 (figure 2).
Figure 2.
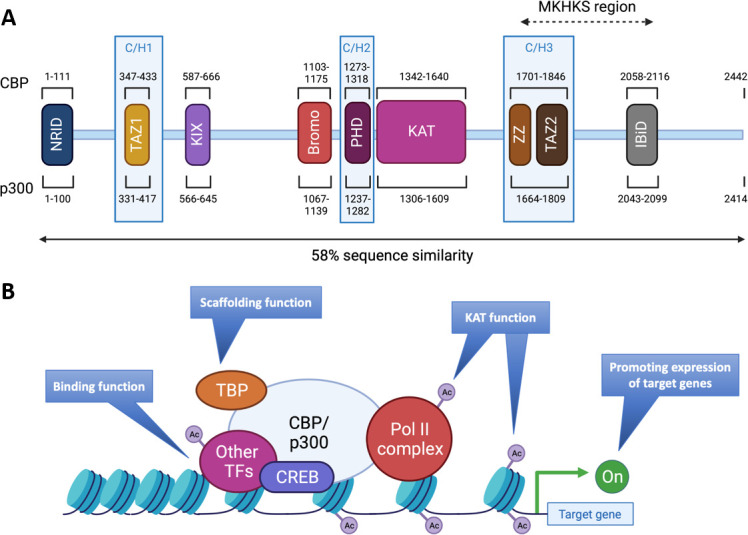
Structures and functions of CBP/p300. (A) The proteins CBP and p300 are composed of 2442 amino acids (AA) and 2414 AA, respectively, with 58% of sequence similarity within their domains. The various domains are represented by their position in the AA sequence: N-terminal nuclear receptor interaction domain (NRID or RID), cysteine-histidine rich region 1 (C/H1) containing the transcriptional adapter zinc finger 1 (TAZ1), kinase-inducible domain (KID) interacting domain (KIX), Bromodomain, C/H2 containing a plant homeodomain (PHD), lysine acetyltransferase domain (KAT), C/H3 containing the zinc finger (ZZ) and TAZ2 domains and interferon-binding transactivation domain (IBiD). The Menke-Hennekam syndrome (MKHKS) region corresponds to the location of the missense variants leading to the MKHKS. (B) CBP and p300 act as transcriptional co-activators of target genes by different mechanisms: (1) Binding function by facilitating the physical and functional interactions of TF; (2) scaffolding function allowing the recruitment of TF and in particular CREB; (3) KAT function by catalysing the transfer of acetyl groups on lysine residues of both histone tails and non-histone proteins such as the RNApolII complex and TF. Ac, acetyl group; TBP, TATA binding protein; TF, transcription factors. Adapted from a study by Van Gils et al.15
Mutation spectrum
Variants in CREBBP and EP300 have been identified in 55–75%,2 3 11 12 and 8–11%,3 5 13 14 of individuals with RTS, respectively, of whom 2–3% have deletions of the complete gene. In 5–20% no molecular anomaly can be detected (online supplemental R4). To date, over 500 CREBBP and over 100 EP300 pathogenic variants are known, distributed along all 31 exons (figure 3). Several recurrent CREBBP variants have been reported, ~50% of missense variants are localised in the KAT domain,15 and recurrent rearrangements occur between introns 1 and 2 of CREBBP due to the high frequency of repeated or palindromic sequences in this region.16 17
Figure 3.
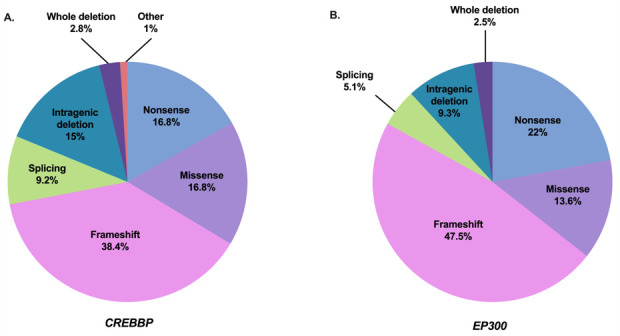
Mutation spectrum of CREBBP and EP300 in individuals with Rubinstein-Taybi syndrome (RTS) (referenced in HGMDPro variant database and/or LOVD). (A) Repartition of 500 pathogenic variants in CREBBP referenced as causing RTS1 including 84 non-sense variants, 192 frameshift variants, 46 splicing variants, 84 missense variants, 75 intragenic deletions, 14 deletions including CREBBP completely, 2 intragenic duplications and 3 complex rearrangements. (B) Repartition of 118 pathogenic variants in EP300 referenced as causing RTS2 including 26 non-sense variants, 56 frameshift variants, 6 splicing variants, 16 missense variants, 11 intragenic deletions and 3 deletions encompassing EP300 completely. Adapted from a study by Van Gils et al.15
Genotype-phenotype correlation
Individuals with RTS1 and RTS2 both may show the classical phenotype but this may also vary. Individuals with RTS2 demonstrate in general less marked typical facial characteristics, no radial deviation of the thumbs, have infrequently keloids and a higher average cognitive level.5 13 14 However, maternal pre-eclampsia, intrauterine growth retardation and microcephaly are more common in RTS2 compared with RTS1.5
The type and site of variants in CREBBP and EP300 do not associate with a specific phenotype with respect to external morphology, malformations, cognition or behaviour,5 11 13 18 19 (online supplemental R5). The exception is formed by missense variants between the end of exon 30 and the beginning of exon 31 of both CREBBP and EP300, which both lead to a phenotype that differs from RTS (table 1) and has been designated as MKHK (OMIM #618332, #618333).4 20 These missense variants hypothesised to affect specifically the binding properties of the ZNF2 (zinc finger, ZZ type) and ZNF3 (zinc finger, TAZ type) domains to different CBP partners by affecting their own folding.21 22
RTS shows broad phenotypic overlap with other Mendelian disorders affecting the structure of chromatin genome-wide called ‘chromatinopathies’, such as FHS (OMIM #136140), CDLS (OMIM #122470, #300590, #610759, #614701, #300882, #608749), WDSTS (OMIM #605130), Kabuki syndrome (OMIM #147920, #300867), genitopatellar syndrome (OMIM #606170), Biesecker-Young-Simpson syndrome (OMIM #603736) and Gabriele-de Vries syndrome (OMIM #617557).
Diagnostic approach
There are two main entry points for molecular genetic testing in RTS: clinical suspicion of RTS or no clinical suspicion (figure 4). If clinical presentation suggests RTS, the first-line tests are either targeted analysis of CREBBP and EP300 by Sanger sequencing and Multiplex Ligation-dependent Probe Amplification (MLPA) or by high throughput analysis (array Comparative Genomic Hybridisation (aCGH); whole-exome sequencing (WES) if accessible). If RTS is not suspected in an individual with intellectual disability and/or malformations, the first tier is high throughput analyses (aCGH; WES or whole-genome sequencing). Evaluation of variant should be performed using the ACMG (American College of Medical Genetics and Genomics) classification.23 Additional RNA studies are needed in case of unknown splicing variants. Suspicion of somatic mosaicism should be confirmed in more than a single tissue (buccal swab; bladder epithelium cells; skin biopsy). The phenotype should be re-evaluated after the identification of a (possibly) pathogenic variant to confirm that the molecular finding fits the clinical phenotype. If targeted analyses yield negative results and high throughput analyses are not available, the diagnosis remains dependent on the clinical phenotype and a definitive diagnosis may not be possible.
Figure 4.
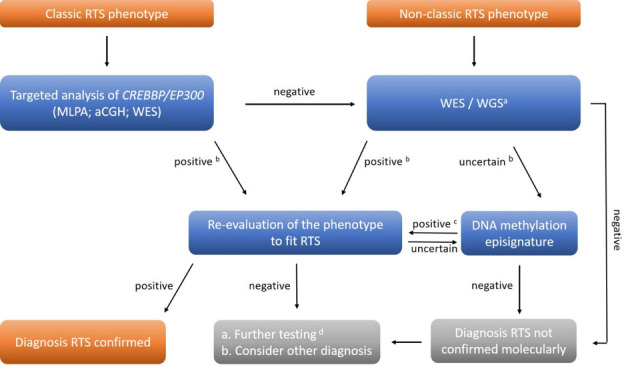
Molecular diagnostic pathways for Rubinstein-Taybi syndrome. In individuals with clinically classic RTS phenotype, the first-line molecular diagnostic approach is targeted analysis of CREBBP and EP300 by Sanger sequencing and MLPA or by high throughput analysis (aCGH; WES). In individuals in whom RTS is not suspected, aCGH and WES or WGS are performed. a Including analysis of CREBBP / EP300 and genes causing related entities; b evaluation of results using ACMG classification23; c episignature specific for RTS24 ; d RNA studies; searches for mosaicism. aCGH, array Comparative Genomic Hybridisation; ACMG, American College of Medical Genetics and Genomics; MLPA, Multiplex Ligation-dependent Probe Amplification; RTS, Rubinstein-Taybi syndrome; WES, whole-exome sequencing; WGS, whole-genome sequencing
If the clinical diagnosis cannot be confirmed molecularly, molecular analyses yield a variant of unknown significance, or the phenotype does not fit the molecular finding, analysis of a genome-wide methylation pattern (epigenetic signature) can be performed as individuals with RTS have a specific pattern.24
If all studies are negative, one should consider other diagnoses. Still, currently, not all molecular mechanisms leading to RTS are known, and if the clinical diagnostic criteria for RTS are met (see Clinical diagnostic criteria), the diagnosis of RTS remains the standard in guiding management and follow-up of the patient.
Recurrence risk
RTS is inherited as an autosomal dominant trait and occurs de novo in over 99% of patients. However, familial occurrence does occur, either if a parent is relatively mildly affected or due to somatic or germ-line mosaicism.25 26 To date, eight instances of somatic or germ-line mosaicism and seven instances of parent-to-child transmission have been described in over 2000 reported affected individuals, indicating the empirical recurrence risk is 0.5–1%.27 The recurrence risk for offspring of an affected individual is 50%, although it may be lower due to a spontaneous miscarriage (online supplemental R6).
Prenatal diagnosis
Without a positive family history, the prenatal diagnosis of RTS is only infrequently made as there are few reliable antenatal signs. Truly detailed three-dimensional ultrasonography may allow suggestive facial characteristics, but the morphology of the extremities, and specifically the radially deviated thumbs, are the main diagnostic handles.28 29 Additional findings that may be helpful are intrauterine growth retardation, polyhydramnios, underdevelopment of the cerebellum and gallbladder anomalies.26
The main reason to perform prenatal diagnostics for RTS is the birth of a previous child with RTS in the family. If a causative variant in CREBBP or EP300 has been detected, reliable molecular prenatal diagnostics can be performed in samples obtained by chorionic villus sampling or amniocentesis, or in embryonic cells obtained by in vitro fertilisation (online supplemental R7).
Prenatal testing in families without a previous child with RTS and a known pathogenic variant, by non-invasive cell-free fetal DNA screening, is not advocated, as an interpretation of the pathogenicity of variants detected this way may be extremely difficult. This limits the validity and informative value of the prenatal testing and may cause ethical issues for the families in deciding whether or not a pregnancy should be continued. Any prenatal testing needs to be discussed carefully with the couple before the procedure and should take into account the differences in perspective of couples and national legislation.
Neonatal care
Recognition
86% of children present within the first month of life and 70% of these on the first day of life; prolonged hospital admission after birth was reported in 61%.30 Early recognition of RTS may help identify complications and assist families to cope.31 The typical facial features of RTS evolve with time.32 The characteristic appearance in the neonatal period differs somewhat as it is mainly characterised by a prominent forehead with haemangiomas (‘stork-bite naevus’) in the glabella region, (apparent) hypertelorism, epicanthi and at that age up-slanting palpebral fissures. The nasal bridge tends to be straight, the tip short and upturned, and the nasal septum is not or only slightly extending beyond the alae.32 A small mouth, highly arched palate and small mandible are also present. Additional features can be unusually thick, black hair, a large anterior fontanelle and long eyelashes. Newborns with a variant in EP300 tend to have a less obvious phenotype.5 The distal limb anomalies are the most characteristic of RTS in the neonatal period and are similar to those at an older age (see Clinical diagnostic criteria). Cryptorchidism is common.
Feeding
Neonatal feeding difficulties are common (71–80%), due to swallowing incoordination, poor nipple grasp, hypotonia and gastro-oesophageal reflux.33 Nutritional supplementation including gastric tube feeding is required in 40% of cases, as are occasionally percutaneous tubes, but most feeding challenges will have resolved within the first year of life.30 Should feeding difficulties persist, additional professionals should be consulted (see Gastroenterology). Still, half of the mothers report a sufficient suck and were pleased with their breastfeeding experience.33 Adequate breastfeeding instructions, proper positioning and ongoing encouragement are indicated (online supplemental R8).
Birth parameters
At birth most infants fall within the normal range for weight, length and head circumference,34 although a higher incidence of microcephaly and growth restriction has been reported in infants with EP300 variants, possibly related to the frequently occurring pre-eclampsia.5 There is no increased risk of preterm birth.35 The use of RTS-specific growth charts is encouraged to monitor growth adequately (online supplemental R9).
Systemic manifestations
The various systemic manifestations of RTS are described elsewhere in the guidelines. The work-up of every newborn with suspected or confirmed RTS should include ophthalmological exams (glaucoma; coloboma); cardiac assessment (malformations); and renal ultrasound (malformations) (online supplemental R10). Obviously, further care such as the baseline newborn hearing screening and vaccinations should be performed as per the general population.
Endocrinology
Hypoglycaemia
Transient hypoglycaemia occurs with a low frequency in newborns with RTS and responds well to usual management schemes (online supplemental R11). Hypoglycaemic hyperinsulinism (HH) is very rare, may occur after birth or in the first years of life, is sometimes associated with concurrent illness, and can be transient or permanent.36 37 It has mainly been described in children with EP300 variants.5 Early diagnosis and treatment of HH is crucial to avoid permanent brain damage.38 Treatment is as in the general population: frequent enteral feeding, continuous glucose infusion and diazoxide). Usually, specialist consultation is needed.39
Growth
Postnatal growth retardation is a hallmark of RTS.34 Usually within months after birth, the length, weight and head circumference drop from normal values to ~ −2SDS. Neither boys nor girls show a pubertal growth spurt, which contributes to a subsequent average adult height of −3SDS for both men and women.34 The use of growth charts specific for RTS, based on molecularly confirmed patients, facilitates adequate monitoring of growth (online supplemental R9). Growth hormone (GH) deficiency is infrequent but has been reported in few individuals, in whom GH therapy resulted in an increase in height SDS.40 Every child in whom growth differs markedly from the growth pattern of the dedicated growth charts needs to be evaluated for GH deficiency (online supplemental R12). If present, treatment is as in the general population. Prepubertal boys and girls may develop an unusual body shape due to increased fat tissue around the abdomen and hips, which disappears in puberty in boys, but often persists throughout life in girls.41
Puberty
The timing of puberty and the development of secondary sex characteristics usually falls within normal limits. The mean age of onset of puberty was 12.2 years,35 with a mean age of menarche at 13.6 years.41 There is no indication that fertility has decreased, although formal studies are lacking. About 25% of adult males and females with RTS are sexually active.42 Sexual education should be proposed according to the level of emotional and cognitive functioning,43 and contraceptive options are recommended as in the general population taking the level of developmental functioning into account (online supplemental R13).
Gastroenterology
Malformations of the gastrointestinal tract such as a duodenal web and malrotation occur at a low frequency in newborns with RTS, although the frequency of the malrotation may be higher than in the general population.44 45 Symptomatology is similar to in newborns without RTS and should be managed as in the general population.41 45
Feeding problems are very frequently present at birth and may remain present for a prolonged period of time.41 45 46 Oral feeding is preferred if it is safe and feasible, while tube feeding may be needed and a gastrostomy for long-term use. The involvement of dieticians is often helpful (online supplemental R14). Although feeding problems are in part explained by recurrent respiratory infections and hypotonia, also gastro-oesophageal reflux (GOR) may play a role.46 Limited GOR occurs in all healthy infants and children; if causing excessive symptoms it is referred to as GOR disease (GORD).47 The symptomatology of GORD may vary widely, from feeding problems, dental enamel erosions and recurrent pneumonias to restlessness and poor sleep. The pathogenesis remains uncertain.46 GOR(D) should be differentiated from excessive regurgitation after feeds in otherwise asymptomatic infants, which is usually indicated as infant rumination syndrome.48 Extremely rarely, oeosinophilic oesophagitis may develop.49 Given the lack of evidence for the management of GORD specifically in RTS, management of GORD should be as in the general population,47: thickening of food and reassurance of parents as a first step. If symptoms persist, an initial trial with PPI (Proton Pump Inhibitors) treatment can be considered. If problems continue, further evaluation should be considered. If a PPI trial improves symptomatology, this does not conclusively prove acid-related GORD. Long-term use of PPI may cause side-effects,50 thus in successful PPI trials individuals should undergo weaning trials regularly (eg, after 6 months and yearly thereafter) to evaluate the utility of continuing PPI treatment, while mitigating rebound effects by dose tapering. If symptoms persist or recur, additional testing, such as pH-impedance testing and/or endoscopy can be considered (online supplemental R15). Fundoplication and other surgical interventions are not recommended in an early phase of management, as these have a relatively high failure rate, commonly cause complications and can induce dysphagia and subsequent feeding problems; it should be reserved for patients with proven GORD unresponsive to optimal nutritional and medical therapy.51 Fortunately, complications of long-term GORD such as Barrett’s oesophagus are rare in RTS,52 and oesophageal cancer has not been reported.
Constipation is extremely prevalent in RTS across all age groups throughout the lifespan.41 45 The cause remains unknown, Hirschsprung disease or other identifiable aetiologies do not occur more frequently than in the general population. Additional investigations are only indicated if symptomatology suggests an underlying disease. Long-term treatment with increased dietary fibres and fluid intake, and oral osmotic laxatives remain the cornerstone of treatment53 (online supplemental R16). In severe cases, stimulant laxatives may be added, and further management schemes are as in the general population.
Cardiology and pulmonology
Cardiovascular system
Congenital heart defects (CHDs) occur in 30% of cases, without a genotype–phenotype correlation.18 54–56 The reported differences in incidence according to ethnicity can be explained by ascertainment bias and differences in methodology.57 The typical CHDs are patent ductus arteriosus, persistent foramen ovale and atrial and ventricular septal defect.5 13 19 55 58–60 Individuals with a CHD do not have a higher rate of other malformations or are associated with impaired cognitive function.
The cardiovascular system should be evaluated at diagnosis, including cardiac sonography (online supplemental R17). Treatment is as in the general population, including endocarditis prophylaxis as indicated. Surgery is needed in 15–22% of patients.42 61 CHDs do not cause unexpected complications in adults.42
Cardiovascular problems typical for the general adult population occur in adults with RTS at a lower frequency. Hypertension is reported in 10% of adults,42 and surveillance and treatment are as in the general population (online supplemental R18).
Pulmonary system
Mild respiratory distress in the first hours of life is common in RTS neonates. Treatment is only needed if other risk factors such as prematurity are present. Upper respiratory infections are common (see Immunology). Infections of the lower respiratory system are uncommon,42 and are explained by feeding problems, microaspirations and gastro-oesophageal reflux. Exceptionally, immunodeficiency may play a role; the reported higher frequency of lower respiratory infections was caused by a study bias.62 In case of recurrent pneumonia with wheezing, hoarseness or stridor, the patient should first be evaluated for microaspirations and gastro-oesophageal reflux49 (online supplemental R19). If negative, a search for immunodeficiency is indicated. Bronchiectasis has been described only in individuals with severe immunological malfunctioning.63
Interstitial lung disease that becomes evident either in childhood,64 or adulthood,65 is uncommon but potentially severe. The diagnosis is made through the radiological characteristics of CT and can be confirmed by biopsy.64 Management is as in the general population and is problematic.
Pulmonary functioning can also be compromised secondary to restrictive pulmonary diseases related to scoliosis,66 and pulmonary hypertension caused by chronic sleep apnoea (obstructive sleep apnoea (OSA))67 (see Otolaryngology and Anaesthesiology).
Ophthalmology
Ocular abnormalities and/or reduced vision are reported in 20–80% of individuals with RTS.55 57 61 68–72 An overview of ocular anomalies is presented in online supplemental table S3 (online supplemental materials). Every child with RTS needs to be referred for ophthalmological evaluation once the diagnosis is suspected (online supplemental R20).
Eye abnormalities were reported to be more common in individuals from Asia and Latin America than those from Africa and the Middle East, but this may be biased.57 Both individuals with CREBBP and EP300 variants present ocular anomalies, but due to small numbers of data on individuals with EP300 variants differences in occurrence remain uncertain.
Anatomical anomalies
Congenital nasolacrimal duct obstruction by a persistent membranous obstruction at the entrance of the duct into the nose causes a watery eye from birth. It is mostly unilateral, with the incidence between 11% and 47%.55 57 59 71–74 Treatment follows international guidelines (Nasolacrimal Duct Obstruction in Children - American Academy of Ophthalmology aao.org) but the surgeon should be aware of the thicker bones and brittle lacrimal sacs in children with RTS.75
The reported frequency of congenital glaucoma varies from 4% to 11%.55 57 61 72 75 The glaucoma can be unilateral or bilateral and be associated with anterior segment anomalies such as iris coloboma or lens luxation. Symptoms include tearing, blepharospasm and photophobia, and enlargement of the eye, manifesting as megalocornea and rapidly increasing myopia. Treatment should be as soon as possible after birth as it can lead to a marked loss of vision (www.eugs.org. Congenital Glaucoma - Europe - American Academy of Ophthalmology aao.org).
Cataracts has been reported in 6–25% of individuals with RTS,19 57 61 72 75 and is usually congenital.72 Reliable incidence figures are lacking. Early diagnosis and treatment in the first 2 months of life are mandatory to avoid visual deprivation, treatment is as in the general population (Pediatric Cataracts: Overview - American Academy of Ophthalmology aao.org). Frequent follow-up is needed for appropriate refractive correction and monitoring of secondary complications. Cataracts may also develop later in life71 (online supplemental R21).
Coloboma is reported in 10% of individuals.19 57 59 71–73 The coloboma can affect the iris, choroid, retina and/or optic nerve. Symptoms depend on location and size and may include visual field loss, reduced vision and photophobia. There is no curative therapy, but sometimes glare can be reduced by wearing sunglasses.
Retinal abnormalities occur frequently,72 but are often subtle, so may go unnoticed, without severe loss of vision, except for macular degeneration secondary to high myopia (online supplemental R21). Evidence may be present in the abnormal distribution of pigment in the macula and a subnormal electroretinogram. In some patients, the abnormal aspect of the macula is caused by foveal hypoplasia (Van Genderen, unpublished).
Functional anomalies
Visual impairment (best corrected binocular visual acuity <6/18) occurs in 20% of individuals,72 and typically is caused by anatomical abnormalities. Bilateral severe anomalies may lead to infantile nystagmus because of decreased sensory input from birth. Refractive errors and strabismus are very common, both occurring in 50–75% of individuals, and may change rapidly with age indicating the need for frequent controls, especially under 5 years of age13 55 57 61 71 72 (online supplemental R21). In young children, high refractive errors need correction to prevent amblyopia. Children may however refuse to wear glasses if improvement of vision is not immediately evident. Gradual introduction in situations in which the child benefits most from glasses may allow the child to get accustomed to wearing spectacles (online supplemental R22).
Treatment of strabismus to prevent amblyopia is as in the general population, provided the affected eye has no congenital anomaly that inhibits amelioration of vision.
Photophobia is common due to cataracts, glaucoma or trichiasis.72 Treatment is by treating the cause. Photophobia secondary to coloboma or retinal dysfunction can be ameliorated by shielding the eyes from direct (sun) light or wearing sunglasses.
Otolaryngology and anaesthesiology
Hearing
The typical facial characteristics in individuals with RTS include a small chin and small oral cavity which can result in airway difficulties and, together with gastro-oesophageal reflux, can result in complications such as recurrent middle ear infections.76 Conductive, sensorineural and mixed hearing loss may result.77–79 Regular auditory evaluation is therefore recommended (online supplemental R23).
Sleep
Abnormal facial anatomy and increased collapsibility of the laryngeal walls predispose individuals with RTS to higher rates of sleep-disordered breathing and OSA.80 81 Sleep disorders are frequent in children, and occur in 62% of adults.42 61 OSA is typically characterised by snoring and excessive daytime sleepiness and affects 25% of adults with RTS.42 61 If present in children the facial anatomy is often markedly abnormal and accompanied by obesity, hypotonia and adenotonsillar hypertrophy.81 As with the general population, management should take into account the various causal factors as well as potential difficulties in treating both children and adults with RTS67 (online supplemental R24). Assessment of the sleep patterns using a validated questionnaire, such as the Sleep Disturbance Scale for Children,82 may offer information on both sleep patterns and response to therapy (online supplemental R25). Prior to a major surgical intervention, polysomnography should be considered.83 Management of sleep disorders is aimed at implementing healthy sleep practices, particularly position during sleep, behavioural strategies and the use of and education on pharmacological interventions. Melatonin should be used appropriately in individuals with specific types of insomnias and sleep rhythm disturbances.
Anaesthesiology
Approximately 48% of adults with RTS require surgery at least once, with half of those requiring two or more surgeries during their lifetime.42 Children with RTS are no exception as they receive a higher fraction of anaesthetics relative to their age-matched cohorts.35 As a result of the multisystemic manifestations of RTS, anaesthesiologists should be prepared to provide a tailored anaesthetic for this population (online supplemental R26).
Premedication and behavioural therapy support may prove beneficial in the preoperative setting. A single case series described complications such as cardiac arrhythmias associated with intraoperative administration of atropine and succinylcholine, but other studies have shown the safe and efficacious use in RTS,84 85 and this is also our joint personal experience. The altered facial anatomy may make mask ventilation, laryngoscopy and intubation challenging, and coupled with positioning limitations that may be present due to scoliosis, kyphosis, hypermobility and obesity, may warrant the use of video-laryngoscopy or fiberoptic intubation.35 86 Rarely, transnasal placement of a nasopharyngeal airway or nasogastric tube is inhibited due to narrow or atretic choanae.
Intraoperative management of ventilation and postextubation care can be complicated by the presence of laryngotrachomalacia and augmented airway reactivity. In the immediate postoperative period, opioid use, while not contraindicated, should be used judiciously to prevent exacerbation of obstructive symptoms and hasten potential apnoeas. The perioperative use of analgesic and anxiolytic adjuncts such as non-steroidal anti-inflammatory drugs, acetaminophen and dexmedetomidine are encouraged, if not contraindicated secondary to other comorbidities or surgical considerations. Initiation of transient, non-invasive positive airway pressure may be helpful. Secondary to the elevated risk of complications with anaesthesia and airway manipulation, particular efforts should be made to bundle non-emergent procedures into a single anaesthetic to mitigate potential morbidity (online supplemental R27).
Dermatology
The main skin problem in RTS is the propensity to develop keloid. Keloids are non-malignant fibrous growths resulting from an abnormal response to skin injuries or inflammation that extend beyond the borders of the original wound. The pathogenesis of keloids is thought to involve multiple patient-specific factors (genetics, age, hormones, ethnicity), and environmental factors (trauma, surgery, inflammation) which collectively stimulate wound healing and persistent inflammation.87 Spontaneous keloids occur only in genetic syndromes,88 raising the question whether they are truly spontaneous, or whether unrecognised triggering environmental factors occur.
RTS is the syndrome considered to have the highest risk of keloid development.89 The frequency of Dutch and UK RTS individuals developing keloids was 24%.89 While keloids are most frequently occurring in association with CREBBP variants, around 10% of individuals with EP300-related disease develop such changes.5 13 56 Compared with the general population keloids develop earlier in life in individuals with RTS,57 89 and increase with age: up to 60% was reported in a cohort of adults.42 Up to 100 keloids have been recorded in the same individual.90 In RTS keloids are most frequently seen on the shoulders and chest.89 Development of keloids is not associated with other traits of the phenotype within RTS.89
Apart from aesthetic issues, keloids cause pain, itching and reduced mobility of the involved region, thus seriously affecting the quality of patients’ lives89 (online supplemental R28). Prevention is difficult and keloids may be unavoidable as minimal trauma such as rubbing of clothes may be sufficient to induce keloid formation. There are no standardised treatment protocols for keloids in individuals with RTS. Therapy options include repeated intra-lesion steroid injections, laser therapy, compression, local radiation, cryotherapy and surgery, either individually or in combination, but no treatment is fully satisfactory, and the recurrence rate remains high.91 There is no detectable association between keloids and cancer risk, suggesting different aetiologies or pathogeneses.92
Another skin problem in RTS occurring in 17% of a series of molecularly proven Dutch cases,93 are multiple pilomatricomas: benign skin tumours derived from hair matrix, often harbouring activating mutations of beta-catenin.94 These skin-coloured, red or white lesions typically occur on the head and neck in children and adolescents, but do occur elsewhere and may arise at older ages as well. Pilomatricomas typically calcify, causing them to feel like hard lumps. They may coexist with keloids.19 95 Similar to keloids there are often multiple pilomatricomas, and puberty may act as a triggering factor. Complete surgical excision has been recommended,96 but others suggested surgical removal only in case of discomfort89 (online supplemental R29).
Ingrowing nails occur regularly in both fingers and toes, especially in the partially duplicated thumbs and halluces,35 and may cause pain and skin infections. Adequate instructions regarding nail care and avoiding narrow shoes may prevent ingrowing nails (online supplemental R30). Treatment is as in the general population. Further skin findings in RTS are congenital generalised hypertrichosis, both in individuals with CREBBP and EP300 variants,97 apparently more frequent in individuals from Latin America and the Middle East and less frequently in those from Africa.57 Usually, it becomes less marked with age. Other changes are angiomas, melanocytic naevi, white papulae on the trunk and limbs, supernumerary nipples and sometimes lentigines and café-au-lait spots.
Urogenital system
Urinary tract
Urinary tract anomalies occur in 23% of individuals with RTS,5 13 19 35 55 57 and include horseshoe kidney, renal duplication, renal agenesis, renal dysplasia, hydronephrosis, nephrolithiasis and vesicoureteral reflux. Symptomatology and treatment follow the general population management. Individuals with CREBBP and EP300 variants are equally affected and there is no known genotype–phenotype correlation.
The high prevalence of renal anomalies warrants at least one renal ultrasound and blood pressure measurement when the diagnosis of RTS has been made (online supplemental R31). If renal anomalies or elevated blood pressure are detected, consultation with a specialist ((paediatric) nephrologist and urologist) is recommended (online supplemental R32). Hypertension in children with RTS is rare but can occur, and is then caused by renal artery stenosis (Hennekam, unpublished observations).
Genitalia
The most common genital anomaly is unilateral or bilateral cryptorchidism, which occurs in 59% of men.13 17 19 35 55 57 All men should be checked by careful physical examination after diagnosis (online supplemental R33). Treatment is as in the general population following international guidelines.98 Other external anomalies occurring in less than 10% of individuals are hypospadias in both men and women, and fusion of labia minora,19 35 which can be treated as in the general population. Shawl scrotum formation is common in RTS and needs no treatment.
Uterine malformations have been reported rarely.99 Women may have hypermenorrhagia or metrorrhagia. A questionnaire survey among 76 women (online supplemental materials Menses Survey) yielded that 10 of them did not yet or did no longer menstruate, 21 of the remaining 66 (32%) used medication (typically contraceptives) because of menses problems, 19 of the 45 (42%) without this medication has metrorrhagia and 10 of 45 (22%) has menorrhagia. Contraceptives were invariably successfully treating the menses problems (online supplemental R34).
Musculoskeletal system
Musculoskeletal anomalies in RTS vary widely. They are somewhat more frequent in individuals with CREBBP variants than in those with EP300 variants.5 Using the data from several large series of patients,5 11 13 17–19 100–103 major limb anomalies (CREBBP variants vs EP300 variants) are broad thumbs (343/360; 95% vs 51/81; 63%), radially-deviated thumbs (183/343; 53% vs 5/71; 7%) and broad halluces (278/290; 96% vs 55/81; 68%). The broadness of the thumbs hardly ever causes problems, but the broadness of the halluces may cause problems in walking or wearing shoes, especially if the halluces are medially deviated. In a minority of patients, surgical correction is needed. Several methods for surgical correction have been reported.104–107 However, often the deviated thumbs have good function and recurrence of the deviation after surgery is common. In our experience, a decision regarding surgery is best postponed until the function of the hands in the patient can be accurately evaluated, which typically can be done around 3–4 years of age. If surgery is indicated, it should be performed by a surgeon familiar with the procedure in RTS (online supplemental R35).
Other findings include limitation of mobility between the proximal and distal phalanx of the thumbs, broadness of distal phalanges of fingers, limited syndactylies, and rarely camptodactyly, but these do not require treatment.
Hypermobility in the hip, elbow, fingers and thumbs, knee and patella is common.35 80 108 109 In combination with other not well-known factors (muscular, bony, neurological), this may cause stiffness and the typical waddling gait in some adolescents and adults. A detailed evaluation of motor skills is indicated110 (online supplemental R36). Further studies describing gait problems in RTS are lacking.
Regular evaluation of the gait is indicated since patella dislocation and Perthes-like hip problems may need therapy (online supplemental R36). In particular, patella problems can cause major mobility challenges and, if untreated, can cause problems like genua valga and knee contractures. These issues may ultimately necessitate wheelchair use. Recurrent patella dislocation may require physical therapy, orthotics or surgical correction,111 112 although procedures are not always successful.
An emerging gait disturbance in older children and adolescents may be caused by an aseptic hip joint inflammation resembling Perthes disease, which occurs in 3% of patients, is often marked, and may take 2 or 3 years to resolve spontaneously.80 It may be difficult to distinguish this from slipped capital femoral epiphyses.113 Management is symptomatic.
Other uncommon limb problems such as congenital hip dislocation, tight heel cords and increased risk for fractures, should be treated as in the general population.
Scoliosis is reported in 34/184 (18%) of individuals with CREBBP variants and 15/78 (19%) of those with EP300 variants,5 and develops in late childhood and puberty. Treatment is as in the general population (online supplemental R37). Significant thoracic kyphosis and lumbar lordosis can occur and typically do not need treatment.41 45 Radiologically the spine may show changes resembling an early ankylosing spondylitis (M Bechterew) but progression into a true ankylosing spondylitis has not been reported.35 Other infrequent spine anomalies include instability of C1-C2, underdevelopment of the dens and cervical vertebral fusions, which should be managed as in the general population.114 Occult spina bifida is detected regularly but does not cause clinical manifestations and may be left untreated.
Children and adults have an increased fracture risk, and 8% of adults have osteoporosis indicating a potentially disturbed ossification in RTS,42 (Simpson et al unpublished observations) (online supplemental R38). Clues for this abnormal ossification in radiographies of the upper spine have been reported.35
Intraoral characteristics
The main non-dental oral characteristic of RTS is the narrow, highly arched palate, which may rarely show clefting of either the complete palate (sometimes submucous), the soft palate or only the uvula, which may or may not be accompanied by a cleft lip.5 A careful evaluation of the palate is indicated in every newborn or child with RTS (online supplemental R39). The treatment of clefting is as in the general population. Other, less frequent characteristics are a relatively large tongue, the bifid tip of the tongue, a short frenulum, and wide alveolar ridges.35
Dental characteristics are almost universally present and may exist as abnormalities in tooth number (15–30%; hyperdontia, hypodontia, mesiodens), structure (23–29%; enamel hypoplasia, discolouration), eruption (5%; neonatal teeth, persistence of primary teeth, delayed eruption), position (62–64%; malocclusion, malalignment, crowded teeth, crossbite) and abnormal tooth shape including talon cusps, a diagnostic hallmark for RTS.61 115 116 Talon cusps are accessory cusps on the lingual side of incisors. CREBBP and EP300 are strongly expressed in both incisors and molars117 and influence the formation of the secondary and (to a lesser extent) primary enamel knots, allowing, if mutated, for talon cusp formation in 27% of primary incisors and 70–92% of permanent (upper) incisors.115 116 Sealing the fissures around the talon cusps may prevent caries. Treatment is only needed if it interferes with mouth closure and occlusion or leads to marked caries (online supplemental R40).
Dental anomalies may also be secondary, that is, difficulties in maintaining adequate oral health leading to caries and periodontal disease, and also to enamel demineralisation due to gastro-oesophageal reflux.115 116 Children and adults with RTS often demonstrate also anxieties when facing dental assessments and treatments, stressing the need for early intervention.118 Informing parents and other caregivers of the importance of early adequate oral hygiene, and subsequent advice, is paramount. Regular dental evaluation and treatment, preferably by a dentist with experience in caring for individuals with special needs, can prevent further problems, and treatment may be aided with sedation or general anaesthesia,119 (online supplemental R41). Orthodontic assessments and treatments are as in the general population. However, some procedures may not be well tolerated and should be considered in close collaboration with the individual and family.
Immunology
Infections
Recurrent infections of organs or organ systems do not typically occur in RTS, except for respiratory infections (70% of children, <20% of adults), including otitis media.35 42 61 Explanations include microaspiration and gastro-oesophageal reflux, but dysfunction of the immune response may also contribute. B cell defects have been reported.62 If a child with RTS has recurrent unexplained infections, a baseline immune workup including complete blood count with differential, immunoglobulin (Ig) levels (IgG, IgA and IgM), vaccine titres and lymphocyte subsets with B-cell phenotyping should be performed (online supplemental R42). In lower airway infections microaspiration or gastro-oesophageal reflux should be considered (online supplemental R19). If the immune workup yields abnormal results, consultation with an immunologist is indicated (online supplemental R42). Although a reduction of T cell or specific T-cell subtypes has been found in some cases, combined immune defects such as viral or opportunistic infections, have not been reported and specific antiviral or antifungal prophylaxis is not indicated.62 Vaccination can be performed as in the general population, causing the typical level of protection (online supplemental R43).
Oncology
CREBBP and EP300 are involved in several basic cellular activities, such as DNA repair, growth, differentiation, apoptosis and tumour suppression. Early surveys suggested an increased frequency of malignancies in case reports on individuals with RTS.120 However, a more recent population-based study found no evidence of an increased risk for malignancies in individuals below 40 years of age.93 Data for older individuals are too limited to allow conclusions. Benign tumours, however, were more common: meningiomas and pilomatricomas were present in 8% and 17% of molecularly-proven patients, respectively.93 Surveillance for malignancies below 40 years of age is not recommended; the value of additional surveillance at an older age remains uncertain, and these individuals should follow surveillance schemes according to national standards (online supplemental R44).
Neurology
Central nervous system anomalies
The most common intracranial malformations (74%) in individuals with RTS are corpus callosum-related malformations. Periventricular posterior white matter abnormalities (63%), cerebellar vermis malformations (58%) and small or absent olfactory bulb (32%) are also regularly observed.28 54 121–124 Infrequent findings are Arnold-Chiari malformation, underdeveloped pituitary gland and Dandy-Walker malformation.28 35 40 41 54 55 125 126 None of these findings has direct consequences for regular medical care and routine cerebral brain MRI is not recommended and indications for brain MRI studies should follow the standard of care for the general population (online supplemental R45), with the exception of microcephaly without other neurological manifestations. Spinal cord malformations such as tethered cord, syringomyelia, lipomas and spina bifida have also been observed.13 35 121 124 127 Spinal MRI is indicated if neurological signs or symptoms are present. Studies for genotype—brain phenotype association have suggested an association of microcephaly and low-positioning of the conus with an altered KAT function,121 and no other association.
Epilepsy
Non-specific electroencephalogram (EEG) abnormalities are observed in around 58–76% of individuals with RTS2 but clinical epileptic manifestations are infrequent, ranging from 9% to 33%.5 13 57 121 128–130 In individuals with RTS type 2, epilepsy is reported in 0–10%.5 13 Specific EEG findings also in individuals without a history of seizures have been suggested,121 122 but have no consequences for medical care. Routine EEGs are therefore not recommended, and EEGs should remain limited to individuals with RTS with epileptic seizures. Treatment and surveillance should follow national standards of care (online supplemental R46).
Neurodevelopment
The early symptoms of delayed development are the delay in achieving basic motor skills (table 3).35 131 First words are typically spoken at 2 years of age, sentences of two words or three words at 4 years of age or later on, with a wide variability across individuals. IQ ranges from 25 to 79, non-verbal performance IQ generally being higher than verbal IQ.41 121 132 133 Individuals with a CREBBP variant typically have a moderate-to-severe intellectual disability (ID), while individuals with EP300 variants have mainly a mild ID and only rarely severe ID.5 There is no correlation between the type and site of variants and cognitive abilities.5 11
Table 3.
Developmental milestones of children with Rubinstein-Taybi syndrome compared with typically developing children
| Milestone | Rubinstein-Taybi syndrome | General population (Dowman 2012) | ||
| Mean age (months) |
Range | Mean age (months) |
Range | |
| Laughing | 2.5 | 2–6 | 2 | 2–6 |
| Rolling over | 10 | 4–18 | 6 | 5–9 |
| Sitting | 16 | 9–24 | 7 | 6–12 |
| Crawling | 19 | 12–36 | 9 | 8–12 |
| Standing | 29 | 11–80 | 9 | 8–18 |
| Walking | 35 | 18–54 | 14 | 12–18 |
| First words | 24 | 6–84 | 12 | 8–18 |
Intellectual disability involves related impairments of cognitive function, learning attainment, expressive language, symbolic play and adaptive behaviour. The role of reduced neuronal histone acetylation in the aetiology of ID has been pointed out by mouse models of RTS showing deficits in long-term memory, but not in short-term memory on a variety of learning and memory tasks.134 135 Weaker memory impairments were found in Ep300 mutant mice136 in keeping with the milder ID of EP300- compared with CREBBP-mutated individuals. Consolidation of learnt information into long-term memories through stimuli-driven transcription is mainly imputed to CBP given its interaction with CREB, a key transcription factor involved in memory formation which diminished levels impair spatial memory,137 as observed in RTS children. Mice with Cbp mutation(s) disrupting CBP-CREB interaction, besides memory deficits, exhibit impaired motor skill learning,138 similar to the difficulties in planning and executing motor acts experienced by CREBBP-mutated patients.
Early assessment of cognitive abilities will benefit each child to access care earlier and for optimal stimulation of development (online supplemental R47). Non-verbal children may benefit from non-symbolic communication, such as non-speech vocalisation and gestures, which help them in their social interactions, and augmentative communication should be prioritised from early on, also in the preverbal stage (online supplemental R48). Early physiotherapy may enhance rehabilitation as well, focusing on the most weakened skills, which have been identified as those requiring a high level of visuomotor coordination.110 Early implementation and maintenance of communication strategies to catalyse preverbal and verbal language development and socialisation skills. Follow-up should include also repeated neuropsychological testing to ensure continuous optimal stimulation, especially at sensitive life phases (school entry, puberty, traumatic events, adulthood and ageing)42 (online supplemental R49).
Behaviour
Recommendations for clinical practice
Interventions for behaviours, cognition and emotion specifically for individuals with RTS are lacking. Applying strategies and intervention approaches designed for individuals with intellectual disability in general, as well as interventions for individuals with a diagnosis of autism, may be helpful (online supplemental table S4 summarises key recommendations).
Self-injurious and aggressive behaviour
The prevalences of self-injurious and aggressive behaviour vary markedly in children and adults with RTS (between 7–48% and 10–16%, respectively).18 139 These figures are similar to the prevalences in individuals with intellectual disability and autism in general.140 Aggressive behaviours may increase in older individuals.132 141 Our joint experience indicates that the self-injurious behaviour and aggression do not show specific characteristics. However, formal studies assessing individuals over time and describing specific topographies of behaviour using standardised measures, are lacking.
Emotions
Emotional outbursts, often severe and weekly, were noted in 7/31 children.139 However, a questionnaire study measuring ‘temper tantrums or hot temper’ found no differences between children with RTS and typically developing children.142 Emotional outbursts were reported in 5/13 adults with RTS,139 seemingly indicating an increase with age, as reported by others.132
On the Child Behaviour Checklist, 64.5% of individuals above 13 years of age and 27.5% of younger individuals were reported to be very anxious.141 The anxiety is not correlated with genotypes.59 For some anxiety subtypes, scores did not differ from children diagnosed with an anxiety disorder.143 Screening for anxieties using a questionnaire validated for individuals with intellectual disability will benefit many individuals with RTS (online supplemental R50). Subsequent interventions should follow best practice guidance for individuals with intellectual disability.
Repetitive behaviours
Repetitive behaviours in individuals with RTS include body, hand and object stereotypy, adherence to routines, repetitive phrases and repetitive questioning.66 142 144 Repetitive behaviour, in particular, repetitive questioning, has been associated with inhibitory control and working memory difficulties,145 146 which has led to the hypothesis that individuals may have difficulties suppressing questioning behaviour, and retaining information in their working memory.145 146 Co-occurrence of adherence to routines and temper outbursts in older individuals has led to the suggestion that executive function difficulties may contribute to these characteristics.142 146
Autism spectrum characteristics
Prevalence rates of autism range from 37–44% on standardised screening assessments.139 142 The estimates for individuals with a CREBBP variant have been higher (49%) compared with those with an EP300 variant (25%).5 Studies using direct assessments of children with a CREBBP variant and a severe intellectual disability, demonstrate areas of cognitive and socio-emotional differences similar to those in children with a diagnosis of autism matched for degree of disability.133 Therefore, families can make use of strategies designed for autism populations, specifically with respect to strategies for language delays, imitation and symbolic activities42 (online supplemental R51).
Caregivers need to be aware that most screening questionnaires use both repetitive behaviour and social behaviour in their scoring, and individuals with RTS may reach the cut-off for autism only because of their repetitive behaviour.
Social characteristics
Social behaviour is typically characterised by motivation to interact with others, and enhanced social skills,142 and ‘over-friendliness’ have been reported in >70% of individuals,132 139 while other studies using observational measures have suggested social motivation is aligned with typical development.143
Parents have reported that their children are vulnerable to social exploitation, particularly as they age and gain independence.147 While social motivation is likely to be heightened or preserved, social understanding (eg, the ability to think about what another may be thinking) is a relative weakness.147 Individuals with RTS may benefit from learning appropriate skills to manage complex social situations, understand others’ intentions, and reduce impulsivity (online supplemental R52).
Self-regulation, impulsivity and overactivity
Distractibility, impulsivity and overactivity have been noted from early descriptions of RTS.1 35 41 A short attention span was found in 76–90%,35 41 irrespective of the cognitive level.142 Studies yielded varying results regarding hyperactivity, and sometimes underactivity was noticed.1 35 61 147
Increased pain threshold
Our joint experience indicates that many parents report their child has not shown evidence of pain or discomfort following a fall or an accident, even for gallstones, fractures, burns or other significant injuries and illnesses. Consequently, it is important not to underestimate subtle changes in behaviour. Medical professionals should be receptive to parent reports, and investigate proactively, even if the presence of a major health problem seems unlikely.
Adult care
Over 90% of individuals with RTS reportedly survive to adulthood,71 and progress in diagnostics, knowledge and management abilities allows improved care for older individuals.61 Adults with RTS enjoy both social and occupational activities and show a varied experience of everyday life. A recently reported cohort of adults underscored the importance of continued management and follow-up.42 Half of all individuals required multispecialist follow-up and surgery during adulthood, usually more than once. Fortunately, significant morbidity in adulthood is not frequent. The adult natural history of RTS is defined by behavioural/psychiatric problems (83%), gastrointestinal problems (73%), skin and adnexa problems (65%), sleep problems (62%) and further concerns of high pain threshold, decreased mobility, hypersensitivity to noise and crowded places and vision difficulties or loss (approximately 50%).
The behavioural pattern remains broad but includes frequently rigid, repetitive and inflexible behaviours and emotional dysregulation (anxiety, aggression, frustration and/or a mood disorder) with reported age-dependent progression.141 144 Sleep problems show a consistent pattern of sleep apnoea, difficulty staying asleep and an increased need for sleep.42
Clinical concerns include gastrointestinal problems with the highest frequency of constipation and in much lower frequency, other problems including oeosinophilic oesophagitis. Retinal dysplasia increases with age,72 but does not cause severe loss of vision. Skin problems are variable but typically progressive, such as keloid formation, ingrowing fingernails and/or toenails (with infections) and poor wound healing.42 Hypertension, overweight, diabetes mellitus and cardiovascular problems do occur in adults but in a lower frequency compared with the general population.42 Treatment is as in the general population (online supplemental R53).
Data on fertility are limited but likely fertility is not impacted. Adults with RTS may be sexually active (25%).42 The risk to offspring is 50% with each pregnancy and familial recurrence has been reported. Thus, developmentally appropriate sexual education throughout the lifespan and especially at transition to adulthood is indicated43 61 (online supplemental R54). Contraceptive options should be discussed with the individual and family.
Reliable data on other adult problems such as dementia are not available.
Clinical trials
CBP and p300 have multiple actions and functions, and clinical trials are aimed at decreasing or correcting abnormal functioning. Prenatally, variants in CREBBP/EP300 can cause malformations unamenable to postnatal change (online supplemental R55). Variants can also cause dysplasias, and these may still be influenced postnatally. CBP/p300 are the ‘master co-activators’ of transcription in humans,148 due to their involvement in many important pathways related to development and differentiation, and postnatal functions such as calcium signalling, nutrient metabolism, hypoxia and stress response.149–151 The latter may be influenced postnatally, thus obvious candidate dysfunctions are memory problems, behaviour, keloids and gastrointestinal problems (online supplemental R56).
Cognition
CREBBP/EP300 mutations cause epigenetic modifications that impact brain development and postnatal brain function of cbp+/cbp− mice.150 Histone deacetylases inhibitors (HDACi) lead to an increase in the acetylation in mice. The HDACi suberoylanilide hydroxamic acid and trichostatin A have been shown to influence neurological functioning and long-term memory in mice.135
Inhibitors of phosphodiesterase 4 (PDE4) prevent the hydrolysis of cAMP-enhancing PKA-dependent signalling upstream of CREBBP. The PDE4 inhibitor rolipram abolishes the long-term memory defects of cbp+/cbp− mice.152 Rolipram is currently tested in Fragile X syndrome and Alzheimer’s disease (ClinicalTrials.gov Identifier: NCT03817684) that may be associated with reduced histone acetylation.153 If successful it is a candidate to be used in individuals with RTS as well.
The HDAC inhibitor sodium valproate can pass the blood-brain barrier. A monocentric, double-blind, randomised, phase 2 trial, primary endpoint long-term memory, investigated the efficiency of sodium valproate after 1 year of treatment (30 mg/kg/d) in 41 children with RTS (ClinicalTrials.gov NCT01619644). Results using subtests of a neuropsychological test battery specifically designed for memory evaluation did not demonstrate a significant difference between the verum and placebo groups. As a side effect, a slight amelioration of some motor functions was found, and a trial with sodium valproate using motor skills as a primary outcome should be considered.
Keloids
Keloids develop most likely following an inciting stimulus (environmental factor) in genetically predisposed individuals. The unremitting accumulation of thick fibres of collagen I and III in the extracellular matrix of connective tissue places keloids among fibrotic disorders. Keloids are unique to humans, there are no adequate animal models, and a high interlesional and intralesional heterogeneity impairs the comparison of in vitro models.87
The principal cell type responsible for keloids is the myofibroblast derived from resident skin fibroblasts through transdifferentiation or pluripotent stem cells,154 but also keratinocytes play a distinct role based on their stemness signature.155 Fibroblasts from keloids overexpress transforming growth factor (TGF)-β1/2 and their receptors that interact with intracellular SMADs (Signaling Mothers Against Decapentaplegic), stimulate transcription of genes intervening in wound healing and cause persistent inflammation through continuous cell division, growth of extracellular matrix beyond the wound boundary, and abnormal vascularisation. Inhibition of the TGF-β1/2 signalling pathway is, therefore, the main target of keloids therapeutics. Indeed, the TGF-β receptor inhibitor LY2109761 has been shown to suppress the secretion of keloid matrix components and to slow down the proliferation of derived fibroblasts.156
Within keloids, several pathways are dysregulated epigenetic modifications including DNA methylation, histone modification and non-coding RNAs.157 158 Reverting these epigenetic anomalies to those of normal skin may also lead to successful treatment. Mutated CBP/p300 causes abnormal histone acetylation which may cause the epigenetic signature of keloids in individuals with RTS to be different from that of keloids from individuals with other disorders. Much of the work on histone modifications on keloids has been focused on the use of the HDAC inhibitor trichostatin A.159 An increase in keloids of HDAC2 (and not of other HDACs),160 suggests the topical application of an HDAC2 inhibitor to be a potential treatment.157 CUDC-907 is an inhibitor of HDAC and also of the PI3K/AKT/mTOR pathway, and has been proposed as a candidate systemic drug.161
Another approach is using upregulation of the mitochondrial oxidative stress response and protein processing in the endoplasmic reticulum (ER).162 Treatment with an inhibitor of ER stress tauroursodeoxycholic acid (TUDCA) reduced scar formation in the rabbit ear.162 The potential use in men is favoured by the clinical approval of TUDCA in cholestasis, and its effective inhibition of ER stress in fibropulmonary disease in mice.163 Single-cell RNA sequencing of keloid tissue has shown significant expansion of fibroblast and vascular endothelial cell subpopulations, responsible for the aberrant keloid fibrogenesis and angiogenesis. In fibroblasts TWIST1 and SMAD3 are top-upregulated genes and TWIST1 inhibition has been proposed as a therapeutic target (Liu 2021). Tumour-related pathways are activated in fibroblast and endothelial cell subpopulations, accounting for the excessive proliferation and resistance to apoptosis of keloids,164 and indicating the transferability and efficiency of medical therapies applied in tumours for the clinical treatment of keloids.
Acknowledgments
We are indebted and thank the ERN-ITHACA and all the Rubinstein-Taybi syndrome support groups and families.
Footnotes
Correction notice: The article has been corrected since it was published online. An author's name and affiliation have been amended.
Contributors: DL, AB-Z, CB, EBC, SDH, SG-M, HK, LL, VLG, LAM, DM, FS, CAS, LT, JAVdZ, MMVG, JV-G, JW and RH researched data for the article. DL, AB-Z, CB, EBC, SDH, SG-M, HK, LL, VLG, LAM, DM, FS, CAS, LT, JAVdZ, MMVG, JV-G, JW, J-LA, OB, PB, AHMB, AMC-G, ED-G, FAD, PF, CR-F, EH, SAH, CM, JM, SMN, CO, EP, AR, IR, RRP, JR, AS, FS-S, BNS, DFS, MFS, KS, ET, NT, IVP, DCMVDK, MPVW, KV, SW and RH contributed to discussion of the content. DL, AB-Z, CB, EBC, SDH, SG-M, HK, LL, VLG, LAM, DM, FS, CAS, LT, JAVdZ, MMVG, JV-G, JW and RH wrote the article. DL and RH reviewed and/or edited the article. AB-Z, CB, EBC, SDH, SG-M, HK, LL, VLG, LAM, DM, FS, CAS, LT, JAVdZ, MMVG, JVG, JW are the second authors.
Funding: This publication has been supported by the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability (ERN-ITHACA). ERN-ITHACA is partly co-funded by the Health Program of the European Union.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Author note: The recommendations are included as online supplemental file 2. They have been part of the peer-reviewed manuscript files accepted for publication, and constitute an integral part of the article.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Ethics statements
Patient consent for publication
Consent obtained from parent(s)/guardian(s).
Ethics approval
The authors affirm that human research participants provided informed consent, for publication of the images in Figure 1.
References
- 1. Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child 1963;105:588–608. 10.1001/archpedi.1963.02080040590010 [DOI] [PubMed] [Google Scholar]
- 2. Petrij F, Giles RH, Dauwerse HG, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 1995;376:348–51. 10.1038/376348a0 [DOI] [PubMed] [Google Scholar]
- 3. Roelfsema JH, White SJ, Ariyürek Y, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and Ep300 genes cause disease. Am J Hum Genet 2005;76:572–80. 10.1086/429130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Menke LA, Gardeitchik T, Hammond P, et al. Further delineation of an entity caused by CREBBP and Ep300 mutations but not resembling Rubinstein-Taybi syndrome. Am J Med Genet A 2018;176:862–76. 10.1002/ajmg.a.38626 [DOI] [PubMed] [Google Scholar]
- 5. Fergelot P, Van Belzen M, Van Gils J, et al. Phenotype and genotype in 52 patients with Rubinstein-Taybi syndrome caused by Ep300 mutations. Am J Med Genet A 2016;170:3069–82. 10.1002/ajmg.a.37940 Available: https://onlinelibrary.wiley.com/toc/15524833/170/12 [DOI] [PubMed] [Google Scholar]
- 6. Larizza L, Finelli P. Developmental disorders with intellectual disability driven by chromatin dysregulation: clinical overlaps and molecular mechanisms. Clin Genet 2019;95:231–40. 10.1111/cge.13365 [DOI] [PubMed] [Google Scholar]
- 7. Chrivia JC, Kwok RP, Lamb N, et al. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 1993;365:855–9. 10.1038/365855a0 [DOI] [PubMed] [Google Scholar]
- 8. Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell 2007;131:633–6. 10.1016/j.cell.2007.10.039 [DOI] [PubMed] [Google Scholar]
- 9. Bedford DC, Kasper LH, Fukuyama T, et al. Target gene context influences the transcriptional requirement for the Kat3 family of CBP and P300 Histone Acetyltransferases. Epigenetics 2010;5:9–15. 10.4161/epi.5.1.10449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang L, Tang Y, Cole PA, et al. Structure and chemistry of the P300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Curr Opin Struct Biol 2008;18:741–7. 10.1016/j.sbi.2008.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pérez-Grijalba V, García-Oguiza A, López M, et al. New insights into genetic variant spectrum and genotype-phenotype correlations of Rubinstein-Taybi syndrome in 39 CREBBP-positive patients. Mol Genet Genomic Med 2019;7:e972. 10.1002/mgg3.972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cross E, Duncan-Flavell PJ, Howarth RJ, et al. Screening of a large Rubinstein-Taybi cohort identified many novel variants and emphasizes the importance of the CREBBP histone acetyltransferase domain. Am J Med Genet A 2020;182:2508–20. 10.1002/ajmg.a.61813 [DOI] [PubMed] [Google Scholar]
- 13. Cohen JL, Schrier Vergano SA, Mazzola S, et al. Ep300-related Rubinstein-Taybi syndrome: highlighted rare phenotypic findings and a genotype-phenotype meta-analysis of 74 patients. Am J Med Genet A 2020;182:2926–38. 10.1002/ajmg.a.61883 [DOI] [PubMed] [Google Scholar]
- 14. Negri G, Magini P, Milani D, et al. From whole gene deletion to point mutations of Ep300-positive Rubinstein–Taybi patients: new insights into the mutational spectrum and peculiar clinical hallmarks. Hum Mutat 2016;37:175–83. 10.1002/humu.22922 [DOI] [PubMed] [Google Scholar]
- 15. Van Gils J, Magdinier F, Fergelot P, et al. Rubinstein-Taybi syndrome: a model of epigenetic disorder. Genes (Basel) 2021;12:968. 10.3390/genes12070968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coupry I, Monnet L, Attia AAEM, et al. Analysis of CBP (CREBBP) gene deletions in Rubinstein-Taybi syndrome patients using real-time quantitative PCR. Hum Mutat 2004;23:278–84. 10.1002/humu.20001 [DOI] [PubMed] [Google Scholar]
- 17. Rusconi D, Negri G, Colapietro P, et al. Characterization of 14 novel deletions underlying Rubinstein–Taybi syndrome: an update of the CREBBP deletion repertoire. Hum Genet 2015;134:613–26. 10.1007/s00439-015-1542-9 [DOI] [PubMed] [Google Scholar]
- 18. Schorry EK, Keddache M, Lanphear N, et al. Genotype–phenotype correlations in Rubinstein–Taybi syndrome. Am J Med Genet A 2008;146A:2512–9. 10.1002/ajmg.a.32424 Available: https://onlinelibrary.wiley.com/toc/15524833/146A/19 [DOI] [PubMed] [Google Scholar]
- 19. Spena S, Milani D, Rusconi D, et al. Insights into genotype–phenotype correlations from CREBBP point Mutation screening in a cohort of 46 Rubinstein–Taybi syndrome patients. Clin Genet 2015;88:431–40. 10.1111/cge.12537 [DOI] [PubMed] [Google Scholar]
- 20. Banka S, Sayer R, Breen C, et al. Genotype-phenotype specificity in Menke-Hennekam syndrome caused by missense variants in exon 30 or 31 of CREBBP. Am J Med Genet A 2019;179:1058–62. 10.1002/ajmg.a.61131 [DOI] [PubMed] [Google Scholar]
- 21. De Guzman RN, Liu HY, Martinez-Yamout M, et al. Solution structure of the Taz2 (Ch3) domain of the transcriptional adaptor protein Cbp1. J Mol Biol 2000;303:243–53. 10.1006/jmbi.2000.4141 [DOI] [PubMed] [Google Scholar]
- 22. Ponting CP, Blake DJ, Davies KE, et al. ZZ and TAZ: new putative zinc fingers in dystrophin and other proteins. Trends Biochem Sci 1996;21:11–3. [PubMed] [Google Scholar]
- 23. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the Association for molecular pathology. Genet Med 2015;17:405–24. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aref-Eshghi E, Kerkhof J, Pedro VP, et al. Evaluation of DNA methylation episignatures for diagnosis and phenotype correlations in 42 mendelian neurodevelopmental disorders. Am J Hum Genet 2020;106:356–70. 10.1016/j.ajhg.2020.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gervasini C, Castronovo P, Bentivegna A, et al. High frequency of mosaic CREBBP deletions in Rubinstein–Taybi syndrome patients and mapping of somatic and germ-line Breakpoints. Genomics 2007;90:567–73. 10.1016/j.ygeno.2007.07.012 [DOI] [PubMed] [Google Scholar]
- 26. Chiang P-W, Lee N-C, Chien N, et al. Somatic and germ-line mosaicism in Rubinstein–Taybi syndrome. Am J Med Genet A 2009;149A:1463–7. 10.1002/ajmg.a.32948 [DOI] [PubMed] [Google Scholar]
- 27. Bartsch O, Kress W, Kempf O, et al. Inheritance and variable expression in Rubinstein–Taybi syndrome. Am J Med Genet A 2010;152A:2254–61. 10.1002/ajmg.a.33598 Available: https://onlinelibrary.wiley.com/toc/15524833/152A/9 [DOI] [PubMed] [Google Scholar]
- 28. Van-Gils J, Naudion S, Toutain J, et al. Fetal phenotype of Rubinstein-Taybi syndrome caused by CREBBP mutations. Clin Genet 2019;95:420–6. 10.1111/cge.13493 [DOI] [PubMed] [Google Scholar]
- 29. Cardalliac C, Vincent M, Joubert M, et al. Rubinstein-Taybi syndrome in a fetus: contribution of 2- and 3-dimensional ultrasonography. J Ultrasound Med 2018;37:531–4. 10.1002/jum.14342 [DOI] [PubMed] [Google Scholar]
- 30. Levetan C, Van Gils J, Saba A, et al. Rubinstein-Taybi syndrome: presentation in the first month of life. J Pediatr 2022;249:106–10. 10.1016/j.jpeds.2022.06.038 [DOI] [PubMed] [Google Scholar]
- 31. Lewis C, Skirton H, Jones R. Living without a diagnosis: the parental experience. Genet Test Mol Biomarkers 2010;14:807–15. 10.1089/gtmb.2010.0061 [DOI] [PubMed] [Google Scholar]
- 32. Allanson JE, Hennekam RC. Rubinstein-Taybi syndrome: objective evaluation of craniofacial structure. Am J Med Genet 1997;71:414–9. [DOI] [PubMed] [Google Scholar]
- 33. Moe JK, Holland MD, Johnson RK. Breastfeeding practices of infants with Rubinstein-Taybi syndrome. J Hum Lact 1998;14:311–5. 10.1177/089033449801400415 [DOI] [PubMed] [Google Scholar]
- 34. Beets L, Rodríguez-Fonseca C, Hennekam RC. Growth charts for individuals with Rubinstein–Taybi syndrome. Am J Med Genet A 2014;164A:2300–9. 10.1002/ajmg.a.36654 [DOI] [PubMed] [Google Scholar]
- 35. Hennekam RC, Van Den Boogaard MJ, Sibbles BJ, et al. Rubinstein-Taybi syndrome in the Netherlands. Am J Med Genet Suppl 1990;6:17–29. 10.1002/ajmg.1320370604 Available: http://doi.wiley.com/10.1002/ajmg.v37%3A6%2B [DOI] [PubMed] [Google Scholar]
- 36. Wild KT, Nomakuchi TT, Sheppard SE, et al. Hyperinsulinism in an individual with an Ep300 variant of Rubinstein-Taybi syndrome. Am J Med Genet A 2021;185:1251–5. 10.1002/ajmg.a.62085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Welters A, El-Khairi R, Dastamani A, et al. Persistent hyperinsulinaemic hypoglycaemia in children with Rubinstein–Taybi syndrome. Eur J Endocrinol 2019;181:121–8. 10.1530/EJE-19-0119 [DOI] [PubMed] [Google Scholar]
- 38. Muukkonen L, Männistö J, Jääskeläinen J, et al. The effect of hypoglycaemia on neurocognitive outcome in children and adolescents with transient or persistent congenital hyperinsulinism. Dev Med Child Neurol 2019;61:451–7. 10.1111/dmcn.14039 [DOI] [PubMed] [Google Scholar]
- 39. Thornton PS, Stanley CA, De Leon DD, et al. Recommendations from the pediatric endocrine society for evaluation and management of persistent hypoglycemia in neonates, infants, and children. J Pediatr 2015;167:238–45. 10.1016/j.jpeds.2015.03.057 [DOI] [PubMed] [Google Scholar]
- 40. Marzuillo P, Grandone A, Coppola R, et al. Novel cAMP binding protein-BP (CREBBP) mutation in a girl with Rubinstein-Taybi syndrome, GH deficiency, Arnold Chiari malformation and pituitary hypoplasia. BMC Med Genet 2013;14:28. 10.1186/1471-2350-14-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stevens CA, Carey JC, Blackburn BL. Rubinstein-Taybi syndrome: a natural history study. Am J Med Genet Suppl 1990;6:30–7. 10.1002/ajmg.1320370605 Available: http://doi.wiley.com/10.1002/ajmg.v37%3A6%2B [DOI] [PubMed] [Google Scholar]
- 42. Douzgou S, Dell’Oro J, Fonseca CR, et al. The natural history of adults with Rubinstein-Taybi syndrome: a families-reported experience. Eur J Hum Genet 2022;30:841–7. 10.1038/s41431-022-01097-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Quint EH. Menstrual and reproductive issues in adolescents with physical and developmental disabilities. Obstet Gynecol 2014;124:367–75. [DOI] [PubMed] [Google Scholar]
- 44. Stevens CA. Intestinal malrotation in Rubinstein–Taybi syndrome. Am J Med Genet A 2015;167A:2399–401. 10.1002/ajmg.a.37167 Available: https://onlinelibrary.wiley.com/toc/15524833/167/10 [DOI] [PubMed] [Google Scholar]
- 45. Rubinstein JH. Broad thumb-hallux (Rubinstein-Taybi) syndrome 1957-1988. Am J Med Genet 1990;37:3–16. 10.1002/ajmg.1320370603 Available: http://doi.wiley.com/10.1002/ajmg.v37%3A6%2B [DOI] [PubMed] [Google Scholar]
- 46. Grunow JE. Gastroesophageal reflux in Rubinstein--Taybi syndrome. J Pediatr Gastroenterol Nutr 1982;1:273–4. 10.1097/00005176-198201020-00019 [DOI] [PubMed] [Google Scholar]
- 47. Rosen R, Vandenplas Y, Singendonk M, et al. Pediatric gastroesophageal reflux clinical practice guidelines: joint recommendations of the North American society for pediatric gastroenterology, hepatology, and nutrition and the European society for pediatric gastroenterology, hepatology, and nutrition. J Pediatr Gastroenterol Nutr 2018;66:516–54. 10.1097/MPG.0000000000001889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nikaki K, Rybak A, Nakagawa K, et al. Rumination syndrome in children presenting with refractory gastroesophageal reflux symptoms. J Pediatr Gastroenterol Nutr 2020;70:330–5. 10.1097/MPG.0000000000002569 [DOI] [PubMed] [Google Scholar]
- 49. Noble A, Drouin E, Faure C. Eosinophilic esophagitis and gastritis in Rubinstein-Taybi syndrome. J Pediatr Gastroenterol Nutr 2007;44:498–500. 10.1097/MPG.0b013e31802c41cd [DOI] [PubMed] [Google Scholar]
- 50. Vaezi MF, Yang Y-X, Howden CW. Complications of proton pump inhibitor therapy. Gastroenterology 2017;153:35–48. 10.1053/j.gastro.2017.04.047 [DOI] [PubMed] [Google Scholar]
- 51. Hassall E. Decisions in diagnosing and managing chronic gastroesophageal reflux disease in children. J Pediatr 2005;146(3 Suppl):S3–12. 10.1016/j.jpeds.2004.11.034 [DOI] [PubMed] [Google Scholar]
- 52. Kumar P, Thota PN. Barrett’s esophagus in Rubinstein-Taybi syndrome. Cureus 2020;12:e11709. 10.7759/cureus.11709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tabbers MM, DiLorenzo C, Berger MY, et al. Evaluation and treatment of functional constipation in infants and children: evidence-based recommendations from ESPGHAN and NASPGHAN. J Pediatr Gastroenterol Nutr 2014;58:258–74. 10.1097/MPG.0000000000000266 [DOI] [PubMed] [Google Scholar]
- 54. Lee JS, Byun CK, Kim H, et al. Clinical and mutational spectrum in Korean patients with Rubinstein–Taybi syndrome: the spectrum of brain MRI abnormalities. Brain Dev 2015;37:402–8. 10.1016/j.braindev.2014.07.007 [DOI] [PubMed] [Google Scholar]
- 55. Yu S, Wu B, Qian Y, et al. Clinical exome sequencing identifies novel CREBBP variants in 18 Chinese Rubinstein-Taybi syndrome kids with high frequency of polydactyly. Mol Genet Genomic Med 2019;7:e1009. 10.1002/mgg3.1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Choi N, Kim HY, Lim BC, et al. Genetic and clinical heterogeneity in Korean patients with Rubinstein-Taybi syndrome. Mol Genet Genomic Med 2021;9:e1791. 10.1002/mgg3.1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tekendo-Ngongang C, Owosela B, Fleischer N, et al. Rubinstein-Taybi syndrome in diverse populations. Am J Med Genet A 2020;182:2939–50. 10.1002/ajmg.a.61888 [DOI] [PubMed] [Google Scholar]
- 58. Hanauer D, Argilla M, Wallerstein R. Rubinstein-Taybi syndrome and hypoplastic left heart. Am J Med Genet 2002;112:109–11. 10.1002/ajmg.10617 [DOI] [PubMed] [Google Scholar]
- 59. Negri G, Milani D, Colapietro P, et al. Clinical and molecular characterization of Rubinstein-Taybi syndrome patients carrying distinct novel mutations of the Ep300 gene. Clin Genet 2015;87:148–54. 10.1111/cge.12348 [DOI] [PubMed] [Google Scholar]
- 60. Bentivegna A, Milani D, Gervasini C, et al. Rubinstein-Taybi syndrome: spectrum of crebbp mutations in Italian patients. BMC Med Genet 2006;7:77. 10.1186/1471-2350-7-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stevens CA, Pouncey J, Knowles D. Adults with Rubinstein–Taybi syndrome. Am J Med Genet A 2011;155A:1680–4. 10.1002/ajmg.a.34058 Available: https://onlinelibrary.wiley.com/toc/15524833/155/7 [DOI] [PubMed] [Google Scholar]
- 62. Saettini F, Herriot R, Prada E, et al. Prevalence of immunological defects in a cohort of 97 Rubinstein-Taybi syndrome patients. J Clin Immunol 2020;40:851–60. 10.1007/s10875-020-00808-4 [DOI] [PubMed] [Google Scholar]
- 63. Lougaris V, Facchini E, Baronio M, et al. Progressive severe B cell deficiency in pediatric Rubinstein-Taybi syndrome. Clin Immunol 2016;173:181–3. 10.1016/j.clim.2016.10.019 [DOI] [PubMed] [Google Scholar]
- 64. Bradford L, Ross MK, Minso J, et al. Interstitial lung disease in children with Rubinstein-Taybi syndrome. Pediatr Pulmonol 2022;57:264–72. 10.1002/ppul.25709 [DOI] [PubMed] [Google Scholar]
- 65. Kosaki R, Kikuchi S, Koinuma G, et al. Two patients with Rubinstein-Taybi syndrome and severe pulmonary interstitial involvement. Am J Med Genet A 2010;152A:1844–6. 10.1002/ajmg.a.33382 Available: https://onlinelibrary.wiley.com/toc/15524833/152A/7 [DOI] [PubMed] [Google Scholar]
- 66. Bounakis N, Karampalis C, Sharp H, et al. Surgical treatment of scoliosis in Rubinstein-Taybi syndrome type 2: a case report. J Med Case Rep 2015;9:10. 10.1186/1752-1947-9-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Choi HS, Yu JJ, Kim Y-H, et al. Pulmonary hypertension due to obstructive sleep apnea in a child with Rubinstein-Taybi syndrome. Korean J Pediatr 2012;55:212–4. 10.3345/kjp.2012.55.6.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Candan S, Ornek C, Candan F. Ocular anomalies in Rubinstein–Taybi syndrome: a further case report and review of the literature. Clin Dysmorphol 2014;23:138–42. 10.1097/MCD.0000000000000047 [DOI] [PubMed] [Google Scholar]
- 69. DaCosta J, Brookes J. Infantile glaucoma in Rubinstein–Taybi syndrome. Eye (Lond) 2012;26:1270–1. 10.1038/eye.2012.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Huang F-R, Zhang A-M, Xu J, et al. A clinical characteristics and genetic analysis of a case of Rubinstein-Taybi syndrome with glaucoma. Eur Rev Med Pharmacol Sci 2021;25:1447–54. 10.26355/eurrev_202102_24852 [DOI] [PubMed] [Google Scholar]
- 71. Milani D, Manzoni FMP, Pezzani L, et al. Rubinstein-Taybi syndrome: clinical features, genetic basis, diagnosis, and management. Ital J Pediatr 2015;41:4. 10.1186/s13052-015-0110-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. van Genderen MM, Kinds GF, Riemslag FC, et al. Ocular features in Rubinstein-Taybi syndrome: investigation of 24 patients and review of the literature. Br J Ophthalmol 2000;84:1177–84. 10.1136/bjo.84.10.1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Enomoto Y, Yokoi T, Tsurusaki Y, et al. Divergent variant patterns among 19 patients with Rubinstein-Taybi syndrome uncovered by comprehensive genetic analysis including whole genome sequencing. Clin Genet 2022;101:335–45. 10.1111/cge.14103 [DOI] [PubMed] [Google Scholar]
- 74. Naik JM, Naik MN, Ali MJ. Lacrimal drainage anomalies in Rubinstein-Taybi syndrome: case report and review of literature. Orbit 2019;38:335–7. 10.1080/01676830.2018.1515961 [DOI] [PubMed] [Google Scholar]
- 75. Yu PT, Luk H-M, Lo IFM. Rubinstein-Taybi syndrome in Chinese population with four novel mutations. Am J Med Genet A 2021;185:267–73. 10.1002/ajmg.a.61922 [DOI] [PubMed] [Google Scholar]
- 76. Stocks RMS, Egerman R, Thompson JW, et al. Airway management of the severely retrognathic child: use of the laryngeal mask airway. Ear Nose Throat J 2002;81:223–6. [PubMed] [Google Scholar]
- 77. Peñaranda A, Cerón M. Rubinstein-Taybi syndrome and mixed bilateral hypoacousia case report. Otol Neurotol 2007;28:501–3. 10.1097/MAO.0b013e31803115d4 [DOI] [PubMed] [Google Scholar]
- 78. Naimi DR, Munoz J, Rubinstein J, et al. Rubinstein–Taybi syndrome: an immune deficiency as a cause for recurrent infections. Allergy Asthma Proc 2006;27:281–4. 10.2500/aap.2006.27.2864 [DOI] [PubMed] [Google Scholar]
- 79. Torres LC, Sugayama SMM, Arslanian C, et al. Evaluation of the immune humoral response of Brazilian patients with Rubinstein-Taybi syndrome. Braz J Med Biol Res 2010;43:1215–24. 10.1590/s0100-879x2010007500119 [DOI] [PubMed] [Google Scholar]
- 80. Hennekam RCM. Rubinstein–Taybi syndrome. Eur J Hum Genet 2006;14:981–5. 10.1038/sj.ejhg.5201594 [DOI] [PubMed] [Google Scholar]
- 81. Zucconi M, Ferini-Strambi L, Erminio C, et al. Obstructive sleep apnea in the Rubinstein-Taybi syndrome. Respiration 1993;60:127–32. 10.1159/000196186 [DOI] [PubMed] [Google Scholar]
- 82. Bruni O, Ottaviano S, Guidetti V, et al. The sleep disturbance scale for children (SDSC). construction and validation of an instrument to evaluate sleep disturbances in childhood and adolescence. J Sleep Res 1996;5:251–61. 10.1111/j.1365-2869.1996.00251.x [DOI] [PubMed] [Google Scholar]
- 83. Roland PS, Rosenfeld RM, Brooks LJ, et al. Clinical practice guideline: polysomnography for sleep-disordered breathing prior to tonsillectomy in children. Otolaryngol Head Neck Surg 2011;145(1 Suppl):S1–15. 10.1177/0194599811409837 [DOI] [PubMed] [Google Scholar]
- 84. Stirt JA. Succinylcholine in Rubinstein-Taybi syndrome. Anesthesiology 1982;57:429. 10.1097/00000542-198211000-00025 [DOI] [PubMed] [Google Scholar]
- 85. Darlong V, Pandey R, Garg R, et al. Perioperative management of a patient of Rubinstein-Taybi syndrome with ovarian cyst for laparotomy. J Anaesthesiol Clin Pharmacol 2014;30:422–4.:422. 10.4103/0970-9185.137285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gathuya Z, Bosenberg A. Anaesthesia and Rubinstein-Taybi syndrome. South Afr J Anaesth Analg 2005;11:135–7. 10.1080/22201173.2005.10872414 [DOI] [Google Scholar]
- 87. Limandjaja GC, Niessen FB, Scheper RJ, et al. The Keloid disorder: heterogeneity, Histopathology, mechanisms and models. Front Cell Dev Biol 2020;8:360. 10.3389/fcell.2020.00360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jfri A, Rajeh N, Karkashan E. A case of multiple spontaneous keloid scars. Case Rep Dermatol 2015;7:156–60. 10.1159/000437249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. van de Kar AL, Houge G, Shaw AC, et al. Keloids in Rubinstein-Taybi syndrome: a clinical study. Br J Dermatol 2014;171:615–21. 10.1111/bjd.13124 Available: https://onlinelibrary.wiley.com/toc/13652133/171/3 [DOI] [PubMed] [Google Scholar]
- 90. Bienias W, Pastuszka M, Gutfreund K. Letter to the editor. Arch Med Sci 2015;11:232–4. 10.5114/aoms.2015.49210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Betarbet U, Blalock TW. Keloids: a review of etiology, prevention, and treatment. J Clin Aesthet Dermatol 2020;13:33–43. [PMC free article] [PubMed] [Google Scholar]
- 92. Siraganian PA, Rubinstein JH, Miller RW. Keloids and neoplasms in the Rubinstein-Taybi syndrome. Med Pediatr Oncol 1989;17:485–91. 10.1002/mpo.2950170526 [DOI] [PubMed] [Google Scholar]
- 93. Boot MV, van Belzen MJ, Overbeek LI, et al. Benign and malignant tumors in Rubinstein-Taybi syndrome. Am J Med Genet A 2018;176:597–608. 10.1002/ajmg.a.38603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Rokunohe D, Nakano H, Akasaka E, et al. Rubinstein-Taybi syndrome with multiple pilomatricomas: the first case diagnosed by CREBBP mutation analysis. J Dermatol Sci 2016;83:240–2. 10.1016/j.jdermsci.2016.06.005 [DOI] [PubMed] [Google Scholar]
- 95. Yagi Y, Kuwatsuka Y, Asai M, et al. Coexistence of keloids and pilomatricoma in a patient with Rubinstein-Taybi syndrome. Dermatol Online J 2018;24:13030/qt4rq2k5fr. [PubMed] [Google Scholar]
- 96. Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol 2018;40:631–41. 10.1097/DAD.0000000000001118 [DOI] [PubMed] [Google Scholar]
- 97. Pavone P, Praticò AD, Falsaperla R, et al. Congenital generalized hypertrichosis: the skin as a clue to complex malformation syndromes. Ital J Pediatr 2015;41:55. 10.1186/s13052-015-0161-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Uroweb - European Association of Urology . EAU guidelines on paediatric urology - INTRODUCTION - Uroweb. Available: https://uroweb.org/guidelines/paediatric-urology [Accessed 11 Oct 2023].
- 99. de Castro Coelho F, Câmara S, Alves I, et al. Septate uterus in a girl with Rubinstein–Taybi syndrome. Case Rep Pediatr 2018;2018:7878156. 10.1155/2018/7878156 Available: 10.1155/2018/7878156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hamilton MJ, Newbury-Ecob R, Holder-Espinasse M, et al. Rubinstein-Taybi syndrome type 2: report of nine new cases that extend the phenotypic and genotypic spectrum. Clin Dysmorphol 2016;25:135–45. 10.1097/MCD.0000000000000143 [DOI] [PubMed] [Google Scholar]
- 101. López M, García-Oguiza A, Armstrong J, et al. Rubinstein-Taybi 2 associated to novel Ep300 mutations: deepening the clinical and genetic spectrum. BMC Med Genet 2018;19:36. 10.1186/s12881-018-0548-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sharma N, Mali AM, Bapat SA. Spectrum of crebbp mutations in Indian patients with Rubinstein-Taybi syndrome. J Biosci 2010;35:187–202. 10.1007/s12038-010-0023-5 [DOI] [PubMed] [Google Scholar]
- 103. Wincent J, Luthman A, van Belzen M, et al. Crebbp and Ep300 mutational spectrum and clinical presentations in a cohort of Swedish patients with Rubinstein-Taybi syndrome. Mol Genet Genomic Med 2016;4:39–45. 10.1002/mgg3.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wood VE, Rubinstein JH. Surgical treatment of the thumb in the Rubinstein-Taybi syndrome. J Hand Surg Br 1987;12:166–72. 10.1016/0266-7681_87_90005-2 [DOI] [PubMed] [Google Scholar]
- 105. Cerqueiro-Mosquera J, Fleming AN. The bilobed flap: a new application in the reconstruction of congenital thumb deviation. J Hand Surg Br 2000;25:262–5. 10.1054/jhsb.2000.0377 [DOI] [PubMed] [Google Scholar]
- 106. Jain A, Rehman S, Smith G. Long-term results following osteotomy of the thumb delta phalanx in Rubinstein-Taybi syndrome. J Hand Surg Eur Vol 2010;35:296–301. [DOI] [PubMed] [Google Scholar]
- 107. Le Mapihan M, Badina A, Pannier S, et al. Non-vascularized toe phalanx transfer for correction of severe clinodactyly of the thumb in Rubinstein-Taybi syndrome. J Hand Surg Eur Vol 2020;45:715–21. 10.1177/1753193420909784 [DOI] [PubMed] [Google Scholar]
- 108. Mehlman CT, Rubinstein JH, Roy DR. Instability of the patellofemoral joint in Rubinstein-Taybi syndrome. J Pediatr Orthop 1998;18:508–11. [PubMed] [Google Scholar]
- 109. Lázaro JS, Herráez SS, Gállego LD, et al. Spontaneous patella dislocation in Rubinstein Taybi syndrome. Knee 2007;14:68–70. 10.1016/j.knee.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 110. Cazalets JR, Bestaven E, Doat E, et al. Evaluation of motor skills in children with Rubinstein-Taybi syndrome. J Autism Dev Disord 2017;47:3321–32. 10.1007/s10803-017-3259-1 [DOI] [PubMed] [Google Scholar]
- 111. Stevens CA. Patellar dislocation in Rubenstein-Taybi syndrome. Am J Med Genet 1997;72:188–90. [PubMed] [Google Scholar]
- 112. Mirzatolooei F. Patellofemoral ligament reconstruction in a patient with Rubinstein-Taybi syndrome. Acta Med Iran 2014;52:228–30. [PubMed] [Google Scholar]
- 113. Shah H, Singh G, Vijayan S, et al. Second report of slipped capital femoral epiphysis in Rubinstein–Taybi syndrome. Clin Dysmorphol 2011;20:55–7. 10.1097/MCD.0b013e32833f000b [DOI] [PubMed] [Google Scholar]
- 114. Yamamoto T, Kurosawa K, Masuno M, et al. Congenital anomaly of cervical vertebrae is a major complication of Rubinstein–Taybi syndrome. Am J Med Genet A 2005;135:130–3. 10.1002/ajmg.a.30708 [DOI] [PubMed] [Google Scholar]
- 115. Hennekam RCM, Van Doorne JM. Oral aspects of Rubinstein-Taybi syndrome. Am J Med Genet 1990;37:42–7. Available http://doi.wiley.com/10.1002/ajmg.v37%3A6%2B [DOI] [PubMed] [Google Scholar]
- 116. Bloch-Zupan A, Stachtou J, Emmanouil D, et al. Oro-dental features as useful diagnostic tool in Rubinstein–Taybi syndrome. Am J Med Genet A 2007;143A:570–3. 10.1002/ajmg.a.31622 [DOI] [PubMed] [Google Scholar]
- 117. Laugel-Haushalter V, Paschaki M, Thibault-Carpentier C, et al. Molars and incisors: show your Microarray IDS. BMC Res Notes 2013;6:113. 10.1186/1756-0500-6-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Clinical Affairs Committee-behavior management subcommittee, American Academy of pediatric dentistry . Guideline on behavior guidance for the pediatric dental patient. Pediatr Dent 2015;37:57–70. [PubMed] [Google Scholar]
- 119. Ashley P, Anand P, Andersson K. Best clinical practice guidance for conscious sedation of children undergoing dental treatment: an EAPD policy document. Eur Arch Paediatr Dent 2021;22:989–1002. 10.1007/s40368-021-00660-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Miller RW, Rubinstein JH. Tumors in Rubinstein-Taybi syndrome. Am J Med Genet 1995;56:112–5. Available https://onlinelibrary.wiley.com/toc/10968628/56/1 [DOI] [PubMed] [Google Scholar]
- 121. Ajmone PF, Avignone S, Gervasini C, et al. Rubinstein-Taybi syndrome: new neuroradiological and neuropsychiatric insights from a multidisciplinary approach. Am J Med Genet B Neuropsychiatr Genet 2018;177:406–15. 10.1002/ajmg.b.32628 [DOI] [PubMed] [Google Scholar]
- 122. Giacobbe A, Ajmone PF, Milani D, et al. Electroclinical phenotype in Rubinstein-Taybi syndrome. Brain Dev 2016;38:563–70. 10.1016/j.braindev.2015.12.003 [DOI] [PubMed] [Google Scholar]
- 123. Mishra S, Agarwalla SK, Potpalle DR, et al. Rubinstein-Taybi syndrome with agenesis of corpus callosum. J Pediatr Neurosci 2015;10:175–7. 10.4103/1817-1745.159207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hadzsiev K, Gyorsok Z, Till A, et al. Rubinstein-Taybi syndrome 2 with cerebellar abnormality and neural tube defect. Clin Dysmorphol 2019;28:137–41. 10.1097/MCD.0000000000000262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Guion-Almeida ML, Richieri-Costa A. Callosal agenesis, Iris coloboma, and megacolon in a Brazilian boy with Rubinstein-Taybi syndrome. Am J Med Genet 1992;43:929–31. 10.1002/ajmg.1320430604 Available: https://onlinelibrary.wiley.com/toc/10968628/43/6 [DOI] [PubMed] [Google Scholar]
- 126. Tsai AC-H, Dossett CJ, Walton CS, et al. Exon deletions of the Ep300 and CREBBP genes in two children with Rubinstein–Taybi syndrome detected by aCGH. Eur J Hum Genet 2011;19:43–9. 10.1038/ejhg.2010.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tanaka T, Ling BC, Rubinstein JH, et al. Rubinstein–Taybi syndrome in children with tethered spinal cord. J Neurosurg 2006;105(4 Suppl):261–4. 10.3171/ped.2006.105.4.261 [DOI] [PubMed] [Google Scholar]
- 128. Cantani A, Gagliesi D. Rubinstein-Taybi syndrome. Review of 732 cases and analysis of the typical traits. Eur Rev Med Pharmacol Sci 1998;2:81–7. [PubMed] [Google Scholar]
- 129. Hou J-W. Rubinstein-Taybi syndrome: clinical and molecular cytogenetic studies. Acta Paediatr Taiwan 2005;46:143–8. [PubMed] [Google Scholar]
- 130. Kumar S, Suthar R, Panigrahi I, et al. Rubinstein-Taybi syndrome: clinical profile of 11 patients and review of literature. Indian J Hum Genet 2012;18:161–6. 10.4103/0971-6866.100751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dosman CF, Andrews D, Goulden KJ. Evidence-based milestone ages as a framework for developmental surveillance. Paediatr Child Health 2012;17:561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Hennekam RC, Baselier AC, Beyaert E, et al. Psychological and speech studies in Rubinstein-Taybi syndrome. Am J Ment Retard 1992;96:645–60. [PubMed] [Google Scholar]
- 133. Taupiac E, Lacombe D, Thiébaut E, et al. Psychomotor, cognitive, and socio-emotional developmental profiles of children with Rubinstein-Taybi syndrome and a severe intellectual disability. J Intellect Dev Disabil 2021;46:80–9. 10.3109/13668250.2020.1776455 [DOI] [PubMed] [Google Scholar]
- 134. Alarcón JM, Malleret G, Touzani K, et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 2004;42:947–59. 10.1016/j.neuron.2004.05.021 [DOI] [PubMed] [Google Scholar]
- 135. Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 2004;42:961–72. 10.1016/j.neuron.2004.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Viosca J, Lopez-Atalaya JP, Olivares R, et al. Syndromic features and mild cognitive impairment in mice with genetic reduction on P300 activity: differential contribution of P300 and CBP to Rubinstein-Taybi syndrome etiology. Neurobiol Dis 2010;37:186–94. 10.1016/j.nbd.2009.10.001 [DOI] [PubMed] [Google Scholar]
- 137. Benito E, Barco A. CREB's control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci 2010;33:230–40. 10.1016/j.tins.2010.02.001 [DOI] [PubMed] [Google Scholar]
- 138. Oliveira AMM, Abel T, Brindle PK, et al. Differential role for CBP and P300 CREB-binding domain in motor skill learning. Behav Neurosci 2006;120:724–9. 10.1037/0735-7044.120.3.724 [DOI] [PubMed] [Google Scholar]
- 139. Boer H, Langton J, Clarke D. Development and behaviour in genetic syndromes: Rubinstein‐Taybi syndrome. Res Intellect Disabil 1999;12:302–7. 10.1111/j.1468-3148.1999.tb00086.x Available: https://onlinelibrary.wiley.com/toc/14683148/12/4 [DOI] [Google Scholar]
- 140. Arron K, Oliver C, Moss J, et al. The prevalence and phenomenology of self-injurious and aggressive behaviour in genetic syndromes. J Intellect Disabil Res 2011;55:109–20. 10.1111/j.1365-2788.2010.01337.x [DOI] [PubMed] [Google Scholar]
- 141. Yagihashi T, Kosaki K, Okamoto N, et al. Age-dependent change in behavioral feature in Rubinstein-Taybi syndrome. Congenit Anom (Kyoto) 2012;52:82–6. 10.1111/j.1741-4520.2012.00356.x [DOI] [PubMed] [Google Scholar]
- 142. Galéra C, Taupiac E, Fraisse S, et al. Socio-behavioral characteristics of children with Rubinstein-Taybi syndrome. J Autism Dev Disord 2009;39:1252–60. 10.1007/s10803-009-0733-4 [DOI] [PubMed] [Google Scholar]
- 143. Crawford H, Waite J, Oliver C. Diverse profiles of anxiety related disorders in fragile X, Cornelia de Lange and Rubinstein-Taybi syndromes. J Autism Dev Disord 2017;47:3728–40. 10.1007/s10803-016-3015-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Waite J, Moss J, Beck SR, et al. Repetitive behavior in Rubinstein–Taybi syndrome: parallels with autism spectrum Phenomenology. J Autism Dev Disord 2015;45:1238–53. 10.1007/s10803-014-2283-7 [DOI] [PubMed] [Google Scholar]
- 145. Perry V, Ellis K, Moss J, et al. Executive function, repetitive behaviour and restricted interests in neurodevelopmental disorders. Res Dev Disabil 2022;122:104166. 10.1016/j.ridd.2021.104166 [DOI] [PubMed] [Google Scholar]
- 146. Waite J, Beck SR, Powis L, et al. The executive function account of repetitive behavior: evidence from Rubinstein-Taybi syndrome. Am J Intellect Dev Disabil 2023;128:49–65. 10.1352/1944-7558-128.1.49 [DOI] [PubMed] [Google Scholar]
- 147. Ellis K, Moss J, Stefanidou C, et al. The development of early social cognitive skills in neurogenetic syndromes associated with autism: Cornelia de Lange, fragile X and Rubinstein-Taybi syndromes. Orphanet J Rare Dis 2021;16:488. 10.1186/s13023-021-02117-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Breen ME, Mapp AK. Modulating the masters: chemical tools to dissect CBP and P300 function. Curr Opin Chem Biol 2018;45:195–203. 10.1016/j.cbpa.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Dancy BM, Cole PA. Protein lysine acetylation by P300/CBP. Chem Rev 2015;115:2419–52. 10.1021/cr500452k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Valor LM, Pulopulos MM, Jimenez-Minchan M, et al. Ablation of CBP in forebrain principal neurons causes modest memory and transcriptional defects and a dramatic reduction of histone acetylation but does not affect cell viability. J Neurosci 2011;31:1652–63. 10.1523/JNEUROSCI.4737-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wang F, Marshall CB, Ikura M. Transcriptional/epigenetic regulator CBP/P300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol Life Sci 2013;70:3989–4008. 10.1007/s00018-012-1254-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Bourtchouladze R, Lidge R, Catapano R, et al. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci U S A 2003;100:10518–22. 10.1073/pnas.1834280100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Schueller E, Paiva I, Blanc F, et al. Dysregulation of histone acetylation pathways in hippocampus and frontal cortex of Alzheimer’s disease patients. Eur Neuropsychopharmacol 2020;33:101–16. 10.1016/j.euroneuro.2020.01.015 [DOI] [PubMed] [Google Scholar]
- 154. Macarak EJ, Wermuth PJ, Rosenbloom J, et al. Keloid disorder: fibroblast differentiation and gene expression profile in fibrotic skin diseases. Exp Dermatol 2021;30:132–45. 10.1111/exd.14243 Available: https://onlinelibrary.wiley.com/toc/16000625/30/1 [DOI] [PubMed] [Google Scholar]
- 155. Yan L, Cao R, Liu Y, et al. Mir-21-5p links epithelial-mesenchymal transition phenotype with stem-like cell signatures via AKT signaling in keloid keratinocytes. Sci Rep 2016;6:28281. 10.1038/srep28281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Wang X, Gu C, Shang F, et al. Inhibitory effect of the Ly2109761 on the development of human keloid fibroblasts. Anal Cell Pathol (Amst) 2021;2021:e8883427. 10.1155/2021/8883427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Stevenson AW, Deng Z, Allahham A, et al. The epigenetics of keloids. Exp Dermatol 2021;30:1099–114. 10.1111/exd.14414 [DOI] [PubMed] [Google Scholar]
- 158. Lv W, Ren Y, Hou K, et al. Epigenetic modification mechanisms involved in keloid: current status and prospect. Clin Epigenetics 2020;12:183. 10.1186/s13148-020-00981-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Diao J-S, Xia W-S, Yi C-G, et al. Histone deacetylase inhibitor reduces hypertrophic scarring in a rabbit ear model. Plast Reconstr Surg 2013;132:61e–9e. 10.1097/PRS.0b013e318290f698 [DOI] [PubMed] [Google Scholar]
- 160. Fitzgerald O’Connor EJ, Badshah II, Addae LY, et al. Histone deacetylase 2 is upregulated in normal and keloid scars. J Invest Dermatol 2012;132:1293–6. 10.1038/jid.2011.432 [DOI] [PubMed] [Google Scholar]
- 161. Tu T, Huang J, Lin M, et al. CUDC-907 reverses pathological phenotype of keloid fibroblasts in vitro and in vivo via dual inhibition of Pi3K/AKT/mTOR signaling and Hdac2. Int J Mol Med 2019;44:1789–800. 10.3892/ijmm.2019.4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kim S, Lee SE, Yi S, et al. Tauroursodeoxycholic acid decreases keloid formation by reducing endoplasmic reticulum stress as implicated in the pathogenesis of keloid. Int J Mol Sci 2021;22:10765. 10.3390/ijms221910765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Tanaka Y, Ishitsuka Y, Hayasaka M, et al. The exacerbating roles of CCAAT/enhancer-binding protein homologous protein (CHOP) in the development of bleomycin-induced pulmonary fibrosis and the preventive effects of tauroursodeoxycholic acid (TUDCA) against pulmonary fibrosis in mice. Pharmacol Res 2015;99:52–62. 10.1016/j.phrs.2015.05.004 [DOI] [PubMed] [Google Scholar]
- 164. Liu X, Chen W, Zeng Q, et al. Single-cell RNA-sequencing reveals lineage-specific regulatory changes of fibroblasts and vascular endothelial cells in keloids. J Invest Dermatol 2022;142:124–35. 10.1016/j.jid.2021.06.010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmg-2023-109438supp001.pdf (332KB, pdf)
jmg-2023-109438supp002.pdf (158.2KB, pdf)