

Abstract
Objectives
To assess the safety and efficacy of upadacitinib versus adalimumab from SELECT-COMPARE over 5 years.
Methods
Patients with rheumatoid arthritis and inadequate response to methotrexate were randomised to receive upadacitinib 15 mg once daily, placebo or adalimumab 40 mg every other week, all with concomitant methotrexate. By week 26, patients with insufficient response to randomised treatment were rescued; patients remaining on placebo switched to upadacitinib. Patients completing the 48-week double-blind period could enter a long-term extension. Safety and efficacy were assessed through week 264, with radiographic progression analysed through week 192. Safety was assessed by treatment-emergent adverse events (TEAEs). Efficacy was analysed by randomised group (non-responder imputation (NRI)) or treatment sequence (as observed).
Results
Rates of TEAEs were generally similar with upadacitinib versus adalimumab, although numerically higher rates of herpes zoster, lymphopenia, creatine phosphokinase elevation, hepatic disorder and non-melanoma skin cancer were reported with upadacitinib. Numerically greater proportions of patients randomised to upadacitinib versus adalimumab achieved clinical responses (NRI); Clinical Disease Activity Index remission (≤2.8) and Disease Activity Score based on C reactive protein <2.6 were achieved by 24.6% vs 18.7% (nominal p=0.042) and 31.8% vs 23.2% (nominal p=0.006), respectively. Radiographic progression was numerically lower with continuous upadacitinib versus adalimumab at week 192.
Conclusion
The safety profile of upadacitinib through 5 years was consistent with the known safety profile of upadacitinib, with no new safety risks. Clinical responses were numerically higher with upadacitinib versus adalimumab at 5 years. Upadacitinib demonstrates a favourable benefit–risk profile for long-term rheumatoid arthritis treatment.
Trial registration number
Keywords: Antirheumatic Agents; Arthritis, Rheumatoid; Biological Therapy; Inflammation
WHAT IS ALREADY KNOWN ON THIS TOPIC.
Upadacitinib has demonstrated efficacy with an acceptable safety profile in the phase 3 SELECT clinical trial programme of patients with rheumatoid arthritis (RA), including in the SELECT-COMPARE long-term extension (LTE) through 3 years.
WHAT THIS STUDY ADDS
The SELECT-COMPARE LTE is the first open-label study of upadacitinib in RA to assess the safety and efficacy of upadacitinib compared with an active comparator (adalimumab) for >1 year. The safety profile of upadacitinib through 5 years was consistent with the 3-year safety profile, with no new safety signals observed and no evidence of an increase in the risk of adverse events with longer exposure to upadacitinib. Upadacitinib also continued to show greater efficacy compared with adalimumab through 5 years.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
These results support a favourable benefit–risk profile for upadacitinib in the long-term treatment of RA, up to at least 5 years.
Introduction
Rheumatoid arthritis (RA) is a chronic, systemic, inflammatory disease that primarily affects the joints, with the development of significant disability and pain, and reduced quality of life, in most patients.1 A considerable number of patients with RA have an inadequate response or intolerance to the initial recommended treatment of conventional synthetic disease-modifying antirheumatic drugs (csDMARDs).2 3 For these patients, advanced therapies, including biological DMARDs (bDMARDs) and targeted synthetic DMARDs (tsDMARDs), are usually initiated as per the treat-to-target strategy.
Upadacitinib, an oral, reversible Janus kinase (JAK) inhibitor (tsDMARD),4 has demonstrated efficacy in the phase 3 SELECT clinical trial programme of patients with RA, with an acceptable and generally well-characterised safety profile.5–9
SELECT-COMPARE is an ongoing double-blind, phase 3 study evaluating upadacitinib 15 mg once daily versus adalimumab 40 mg every other week (EOW), both in combination with methotrexate (MTX), in patients with an inadequate response to MTX alone; the first 26 weeks of the study were placebo controlled. The study met both primary endpoints of achievement of ≥20% improvement in American College of Rheumatology response criteria (ACR20; for US/Food and Drug Administration regulatory purposes) and 28-joint Disease Activity Score based on C reactive protein (DAS28 (CRP)) <2.6 (for EU/European Medicines Agency regulatory purposes) in the upadacitinib group compared with the placebo group at week 12.6 In addition, the study met the ranked key secondary endpoint of radiographic progression (change from baseline in modified Total Sharp Score (mTSS)) inhibition in the upadacitinib group compared with the placebo group at week 26. ACR20 and DAS28 (CRP) <2.6 were achieved by a significantly greater proportion of patients, and radiographic progression was inhibited in a significantly greater proportion of patients, in the upadacitinib group versus the placebo group at weeks 12 and 26, respectively. Upadacitinib was also superior to adalimumab based on achievement of ≥50% improvement in ACR response criteria (ACR50), change from baseline in patient’s assessment of pain and change from baseline in the Health Assessment Questionnaire-Disability Index (HAQ-DI) at week 12 and achievement of DAS28 (CRP) ≤3.2 at week 12.6
Through 26 weeks, the safety profile of upadacitinib was similar to that of adalimumab, except for higher rates of herpes zoster and creatine phosphokinase (CPK) elevation in the upadacitinib group. All patients either remained on, or were rescued or switched to, upadacitinib or adalimumab before or at week 26. Through 3 years in the ongoing open-label long-term extension (LTE) of SELECT-COMPARE, upadacitinib consistently showed better clinical and functional responses across all efficacy endpoints compared with adalimumab, and the safety profile of upadacitinib was consistent with previous reports in RA and the known safety profile of upadacitinib in other indications.6 10–13
Here, we report 5-year data on the safety and efficacy of upadacitinib 15 mg once daily versus adalimumab 40 mg EOW, both in combination with MTX, from the SELECT-COMPARE LTE study.
Methods
Study design
The study design and eligibility of SELECT-COMPARE, including the LTE, has been described previously.6 12 13 Briefly, SELECT-COMPARE is a phase 3 randomised study comprising a 26-week double-blind, placebo-controlled period that was part of a 48-week double-blind active comparator-controlled period and an ongoing open-label LTE for a total of up to 10 years (online supplemental figure 1). Patients were eligible if they were ≥18 years of age with a diagnosis of RA (meeting the 2010 ACR/European Alliance of Associations for Rheumatology classification criteria)14 and had been on MTX for ≥3 months and at a stable dose of 15–25 mg/week for ≥4 weeks prior to the first dose of study drug (or ≥10 mg/week if intolerant to ≥12.5 mg/week). Patients had active disease, defined as ≥6 swollen joints (of 66 examined), ≥6 tender joints (of 68 examined), high-sensitivity CRP (hsCRP)≥5 mg/L and ≥1 of the following at screening: ≥3 erosions on X-rays of hands and feet, or ≥1 erosion and seropositivity for rheumatoid factor or anti-cyclic citrullinated peptide antibody, and an inadequate response or intolerance to MTX. Exclusion criteria included inadequate response to a prior bDMARD or prior exposure to a JAK inhibitor or adalimumab. Patients were randomised in a 2:2:1 ratio to upadacitinib 15 mg once daily, placebo or adalimumab 40 mg EOW, in combination with MTX. Patients could be rescued from placebo to upadacitinib, upadacitinib to adalimumab or adalimumab to upadacitinib within the first 26 weeks of the study by predefined criteria. Patients who did not achieve ≥20% improvement in tender joint count (TJC) and swollen joint count (SJC) were rescued at weeks 14, 18 or 22 while patients who did not achieve low disease activity (LDA) as defined by Clinical Disease Activity Index (CDAI) criteria (CDAI≤10) were rescued at week 26. All patients receiving placebo who were not rescued by week 26 were switched to upadacitinib from that time point.
rmdopen-2023-004007supp001.pdf (914.5KB, pdf)
From week 26, initiation of, or change in, glucocorticoids, non-steroidal anti-inflammatory drugs and acetaminophen/paracetamol were allowed at the investigator’s discretion. From week 48, modification or initiation of csDMARDs was allowed at the investigator’s discretion. csDMARDs were restricted to up to two of the following: MTX, sulfasalazine, hydroxychloroquine, chloroquine and leflunomide, except for the combination of MTX and leflunomide, which was not permitted. Patients who completed the 48-week double-blind period could enter the LTE study to continue receiving upadacitinib 15 mg once daily or adalimumab 40 mg EOW (open-label after the last patient completed their week 48 visit).
Safety
Safety was assessed up to week 264, through the cut-off date of 5 October 2022. Treatment-emergent adverse events (TEAEs), serious TEAEs, TEAEs leading to discontinuation, TEAEs of special interest and proportions of patients experiencing grade 3 or grade 4 laboratory abnormalities were summarised up to 5 years in all patients who received ≥1 dose of upadacitinib or adalimumab. TEAEs of special interest included serious infection, opportunistic infection (excluding herpes zoster and tuberculosis), herpes zoster, active tuberculosis, malignancy (excluding non-melanoma skin cancer (NMSC)), NMSC, lymphoma, adjudicated major adverse cardiovascular events (MACEs), adjudicated venous thromboembolic events (VTEs), adjudicated gastrointestinal perforation, renal dysfunction, anaemia, lymphopenia, neutropenia, CPK elevation and hepatic disorder. In addition, malignancies (excluding NMSC), MACE and VTE were summarised in the subset of patients receiving continuous upadacitinib or continuous adalimumab from randomisation. TEAEs are presented as exposure-adjusted event rates (EAERs; events per 100 patient-years (E/100 PY)).
Safety assessments were performed as have been described previously.6 12 13 TEAEs were coded according to the Medical Dictionary for Regulatory Activities, V.25.0. TEAEs and laboratory changes (other than CPK and creatinine levels) were graded using the Rheumatology Common Toxicity Criteria, V.2.0.15 Changes in CPK and creatinine levels were graded using the Common Toxicity Criteria of the National Cancer Institute (NCI CTC).16 MACE and VTE were adjudicated in a blinded manner by an independent Cardiovascular Adjudication Committee. Assessment of TEAE severity was made at the investigator’s discretion (mild (grade 1), moderate (grade 2) or severe (grade 3 or 4)). TEAEs were categorised as transient based on the availability of an end date for that TEAE.
Efficacy endpoints
Efficacy endpoints assessed through week 264 included the proportions of patients achieving CDAI LDA (CDAI≤10) or remission (CDAI≤2.8), DAS28 (CRP) ≤3.2 or <2.6 and ≥20/50/70% improvement in ACR criteria (ACR20/50/70 responses). Changes from baseline in ACR components (TJC based on 68 joints (TJC68), SJC based on 66 joints (SJC66), patient’s global assessment of disease activity, physician’s global assessment of disease activity, patient’s assessment of pain (0–100 mm scale), (HAQ-DI; on a 0–3 scale) and hsCRP (mg/L)), as well as change from baseline in severity of morning stiffness (0–10 Numerical Rating Scale; 0=no morning stiffness, 10=worst possible morning stiffness) and duration of morning stiffness (minutes), were also assessed.
Radiographic progression was assessed up to 192 weeks (latest available collected time point) based on X-rays of hands and feet at weeks 0, 26, 96 and 192. X-rays were assessed by two independent readers who were blinded to treatment and visit, with an additional adjudicator if there was a discrepancy of ≥8 between the two readers’ change in mTSS scores. Radiographic endpoints included the proportion of patients who showed no radiographic progression (patients with a change from baseline in mTSS≤0), change from baseline in mTSS,17 18 change from baseline in joint erosion scores and change from baseline in joint space narrowing.
Statistical analysis
Safety data were assessed in all patients who received at least one dose of upadacitinib or adalimumab and summarised under two drug exposure groups (any upadacitinib and any adalimumab); assignment of TEAEs was based on the drug being received at the time of the event. Any upadacitinib group included patients who received continuous upadacitinib from randomisation through 5 years as well as upadacitinib exposure from patients who were rescued/switched from placebo or adalimumab to upadacitinib; any adalimumab group included patients who received continuous adalimumab from randomisation through 5 years as well as adalimumab exposure from patients who were rescued from upadacitinib to adalimumab. Event rates for adjudicated MACE, adjudicated VTE and malignancy (excluding NMSC), were also assessed in continuous upadacitinib and continuous adalimumab subgroups.
Efficacy analyses were conducted in all randomised patients who received ≥1 dose of study drug. Efficacy data up to week 264 were analysed by treatment sequence (ie, placebo to upadacitinib; continuous upadacitinib; continuous adalimumab; adalimumab to upadacitinib; and upadacitinib to adalimumab), as well as by randomised treatment group (intention-to-treat analysis). For binary endpoints and continuous endpoints analysed by treatment sequence, as well as radiographic endpoints, as observed (AO) analyses were conducted without imputation for missing data; AO analyses were based on the number of patients continuing in the LTE. For binary endpoints analysed by randomised treatment group, non-responder imputation (NRI) was used for visits after rescue, premature discontinuation of the study drug and missing data. Treatments were compared using the Cochran-Mantel-Haenszel test, adjusting for the stratification factor of prior bDMARD use (yes, no). Nominal p values are presented.
Results
Patient disposition and demographics
Overall, 1629 patients were randomised and received ≥1 dose of study drug (upadacitinib: 651, adalimumab: 327 and placebo: 651) (online supplemental figure 2). Of the 651 patients randomised to upadacitinib, 252 (38.7%) were rescued to adalimumab by week 26 while a greater proportion of patients (159/327; 48.6%) randomised to adalimumab were rescued to upadacitinib. Over half of patients (342/651; 52.5%) randomised to upadacitinib completed week 48 on randomised therapy and entered the LTE study compared with 126/327 (38.5%) of those randomised to adalimumab. A greater proportion of patients randomised to upadacitinib also completed 5 years on continuous therapy (261/651; 40.1%) compared with those randomised to adalimumab (96/327; 29.4%) (online supplemental figure 2). Between weeks 48 and 264, the proportions of patients receiving continuous upadacitinib and continuous adalimumab who discontinued due to AEs were 9.6% and 7.9%, respectively, and due to lack of efficacy were 0.9% and 0.8%, respectively.
Patient demographics and baseline disease characteristics have been reported previously6; they were generally well balanced across treatment arms (online supplemental table 1). Of the proportion of patients receiving glucocorticoids at baseline, 22.2% and 25.3% had discontinued glucocorticoid use at week 156 in the continuous upadacitinib and continuous adalimumab groups, respectively, and this increased to 28.8% and 35.9%, respectively, at week 264.
Safety
Through 5 years, 1417 patients received any upadacitinib and 579 received any adalimumab. Overall exposure to any upadacitinib and any adalimumab through 5 years was 4496.6 PY and 1472.4 PY, respectively. The overall EAERs of any TEAE in patients receiving any upadacitinib were similar to any adalimumab (187.2 E/100 PY and 202.3 E/100 PY, respectively). Similar rates were also seen in patients receiving any upadacitinib compared with any adalimumab for serious TEAEs (11.5 E/100 PY and 13.3 E/100 PY, respectively). TEAEs leading to discontinuation of the study drug, any COVID-19-related AEs and deaths are summarised in table 1 (further details of COVID-19 adverse events are described in online supplemental table 2). The most common TEAEs (>5 E/100 PY) reported in both groups were urinary tract infection, upper respiratory tract infection and nasopharyngitis (online supplemental table 3).
Table 1.
TEAEs through 264 weeks
| EAER | Any UPA 15 mg QD (n=1417; PY=4496.6) E (E/100 PY) |
Any ADA 40 mg EOW (n=579; PY=1472.4) E (E/100 PY) |
| Any TEAE | 8419 (187.2) | 2978 (202.3) |
| Serious TEAE | 517 (11.5) | 196 (13.3) |
| TEAE leading to discontinuation of study drug | 195 (4.3) | 81 (5.5) |
| Any COVID-19-related adverse event | 239 (5.3) | 62 (4.2) |
| Any death* | 41 (0.9) | 14 (1.0) |
| TEAEs of special interest | ||
| Serious infection | 167 (3.7) | 49 (3.3) |
| Opportunistic infection† | 13 (0.3) | 2 (0.1) |
| Herpes zoster | 127 (2.8) | 17 (1.2) |
| Active tuberculosis | 3 (<0.1) | 3 (0.2) |
| Malignancy (excluding NMSC) | 26 (0.6) | 12 (0.8) |
| NMSC | 24 (0.5) | 2 (0.1) |
| Lymphoma | 0 | 3 (0.2) |
| Adjudicated MACE‡ | 12 (0.3) | 4 (0.3) |
| Adjudicated VTE§ | 12 (0.3) | 6 (0.4) |
| Adjudicated gastrointestinal perforation | 1 (<0.1) | 0 |
| Renal dysfunction | 12 (0.3) | 6 (0.4) |
| Anaemia | 113 (2.5) | 49 (3.3) |
| Lymphopenia | 115 (2.6) | 13 (0.9) |
| Neutropenia | 95 (2.1) | 34 (2.3) |
| CPK elevation | 179 (4.0) | 26 (1.8) |
| Hepatic disorder | 476 (10.6) | 101 (6.9) |
Safety was assessed up to week 264, through the cut-off date of 5 October 2022. TEAEs included any adverse event with an onset date on or after the first dose of study drug and up to 30 days after the last dose of placebo or UPA and 70 days for ADA, if patients discontinued prematurely. Data through 264 weeks include all patients receiving UPA or ADA, including rescue groups, with assignment based on drug exposure at the time of the event.
*Includes treatment emergent (occurring ≤30 days after the last dose of UPA or ≤70 days after the last dose of ADA) and non-treatment-emergent (occurring >30 days after the last dose of UPA or >70 days after the last dose of ADA) deaths.
†Excluding herpes zoster and tuberculosis.
‡MACE defined as cardiovascular death (includes acute myocardial infarction, sudden cardiac death, heart failure, cardiovascular procedure-related death, death due to cardiovascular haemorrhage, fatal stroke, pulmonary embolism and other cardiovascular causes), non-fatal myocardial infarction and non-fatal stroke.
§VTE included deep vein thrombosis and pulmonary embolism (fatal and non-fatal).
ADA, adalimumab; CPK, creatine phosphokinase; E, Event; EAER, exposure-adjusted event rate; EOW, every other week; MACE, major adverse cardiovascular event; NMSC, non-melanoma skin cancer; PY, patient-years; QD, once daily; TEAE, treatment-emergent adverse event; UPA, upadacitinib; VTE, venous thromboembolic event.
Overall rates of TEAEs of special interest were generally similar between groups, except for numerically higher rates of herpes zoster, lymphopenia, CPK elevation, hepatic disorder (mostly transaminase elevations) and NMSC in patients receiving any upadacitinib compared with any adalimumab (table 1). In any upadacitinib and adalimumab groups, similar rates of serious infections (3.7 E/100 PY and 3.3 E/100 PY, respectively) and opportunistic infections (excluding herpes zoster and tuberculosis) (0.3 E/100 PY and 0.1/100 PY, respectively) were observed. Rates of anaemia were also similar in any upadacitinib and any adalimumab groups. Herpes zoster infection rates were 2.8 E/100 PY and 1.2 E/100 PY in any upadacitinib and any adalimumab groups, respectively. Most herpes zoster infections were non-serious, involved one dermatome and were non-disseminated; rates of serious herpes zoster events were 0.3 E/100 PY and 0 in any upadacitinib and any adalimumab groups, respectively. In any upadacitinib group, 17.6% of patients had unilateral presentation involving multiple dermatomes. One event had central nervous system involvement in any upadacitinib group (Ramsay Hunt syndrome); 10 events had ophthalmic involvement (2 in any adalimumab group and 8 in any upadacitinib group). Rates of lymphopenia were 2.6 E/100 PY and 0.9 E/100 PY in any upadacitinib and any adalimumab groups, respectively. The majority of lymphopenia events were mild or moderate, with severe events accounting for 37.4% (grade 3 or 4 (as assessed by the investigator)) and 15.4% (grade 3) in any upadacitinib and any adalimumab groups, respectively. Of 45 severe (grade 3 or 4) events of lymphopenia, 3 events (pharyngitis, varicella and post-procedural infection; all in any upadacitinib group) were associated with infection, and 1 additional event had a questionable association because COVID-19 infection was reported 1 month prior to lymphopenia in the patient involved. Higher proportions of patients with grade 3/4 reductions (as assessed by Outcome Measures in Rheumatology (OMERACT) criteria) in lymphocytes (grade 3: 0.5 to <1.0×109/L; grade 4: <0.5×109/L) were reported in any upadacitinib group compared with any adalimumab group (table 2). Higher proportions of patients with grade 3 reductions in neutrophils (8.5% and 5.9%) and haemoglobin (1.8% and 0.9%) were reported in any upadacitinib group compared with any adalimumab group, respectively. No pattern or trend in types of infection occurring within 30 days of a grade 3 or 4 event of neutropenia was observed.
Table 2.
Grade 3 or 4 laboratory abnormalities through 264 weeks
| Parameter | Any UPA 15 mg QD (n=1417) % of patients |
Any ADA 40 mg EOW (n=579) % of patients |
|
| Haemoglobin (g/L) | Grade 3 (decrease 21–29* or Hb ≥70 to <80) | 8.5 | 5.9 |
| Grade 4 (decrease ≥30* or Hb <70) | 3.7 | 4.0 | |
| Lymphocytes (×109/L) | Grade 3 (0.5 to <1.0) | 33.3 | 11.3 |
| Grade 4 (<0.5) | 4.7 | 1.0 | |
| Neutrophils (×109/L) | Grade 3 (0.5 to <1.0) | 1.8 | 0.9 |
| Grade 4 (<0.5) | 0.4 | 0.3 | |
| ALT (U/L) | Grade 3 (3.0–8.0×ULN) | 6.9 | 2.8 |
| Grade 4 (>8.0×ULN) | 0.9 | 0.7 | |
| AST (U/L) | Grade 3 (3.0–8.0×ULN) | 4.0 | 1.9 |
| Grade 4 (>8.0×ULN) | 0.8 | 0.9 | |
| CPK (U/L) | Grade 3 (>5.0–10.0×ULN) | 2.5 | 0.9 |
| Grade 4 (>10.0×ULN) | 1.1 | 0.5 | |
| Creatinine (µmol/L) | Grade 3 (>3.0–6.0×ULN) | 0.1 | 0.3 |
| Grade 4 (>6.0×ULN) | 0.1 | 0 |
Safety was assessed up to week 264, through the cut-off date of 5 October 2022. Data are for patients with worsening in grade severity for laboratory parameters. Grading is based on Outcome Measures in Rheumatology criteria, except for CPK and creatinine, for which National Cancer Institute Common Terminology Criteria were used.
*Decrease from baseline. Baseline is defined as the last observation on or before the date of the first dose of study drug in the corresponding treatment group.
ADA, adalimumab; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CPK, creatine phosphokinase; EOW, every other week; Hb, Haemoglobin; QD, once daily; ULN, upper limit of normal; UPA, upadacitinib.
Rates of CPK elevations were 4.0 E/100 PY and 1.8 E/100 PY in any upadacitinib and any adalimumab groups, respectively (table 1). The majority of CPK elevations were mild or moderate, with severe events (grade 3 (as assessed by the investigator)) accounting for 5.0% and 3.8% in any upadacitinib and any adalimumab groups, respectively. The majority of CPK elevations were also transient (79.9% and 96.2% in any upadacitinib and any adalimumab groups, respectively) and did not lead to discontinuation of study drug. Higher proportions of patients with grade 3 and grade 4 elevations (as assessed by NCI CTC criteria) in CPK (>5.0 to 10.0×upper limit of normal (ULN) and >10.0×ULN) were reported in any upadacitinib group compared with any adalimumab group. One case of rhabdomyolysis, related to alcohol withdrawal and seizure, was reported in any upadacitinib group and was deemed by the investigator to have no reasonable possibility of being related to the study drug. Rates of hepatic disorder were 10.6 E/100 PY and 6.9 E/100 PY in any upadacitinib and any adalimumab groups, respectively (table 1). The majority of hepatic disorder events involved alanine aminotransferase (ALT) or aspartate aminotransferase (AST) elevations, with no cases of Hy’s law identified and were mild or moderate, with severe events accounting for 9.5% (grade 3 or 4 (as assessed by the investigator)) and 8.9% (grade 3) of hepatic disorders and 8.9% (grade 3 or 4) and 12.3% (grade 3) of ALT/AST elevations in any upadacitinib and any adalimumab groups, respectively. The majority of hepatic disorder events were also transient (78.4% and 76.2% in any upadacitinib and any adalimumab groups, respectively). Higher proportions of patients with grade 3 elevations (as assessed by OMERACT criteria) in ALT/AST (3.0–8.0×ULN) were reported in any upadacitinib group compared with any adalimumab group; similar proportions of patients with grade 4 elevations (>8×ULN) in ALT/AST were reported in both groups. 14 patients discontinued study drug due to ALT/AST elevations (11 in any upadacitinib group and 3 in any adalimumab group). One event of adjudicated gastrointestinal perforation occurred in a patient receiving any upadacitinib.
Rates of malignancies (excluding NMSC) were similar in any upadacitinib and any adalimumab groups (0.6 E/100 PY and 0.8 E/100 PY, respectively) (table 1). Rates of NMSC were numerically higher in any upadacitinib group compared with any adalimumab group (0.5 E/100 PY and 0.1 E/100 PY, respectively), while rates of lymphoma were numerically higher in any adalimumab group compared with any upadacitinib group (0.2 E/100 PY and 0 E/100 PY, respectively). Rates of melanoma were similar in any upadacitinib and any adalimumab groups (<0.1 E/100 PY for both groups). A full list of malignancies can be found in online supplemental table 4. Rates of adjudicated MACE and adjudicated VTE were similar in any upadacitinib and any adalimumab groups (0.3 E/100 PY and 0.3 E/100 PY for MACE and 0.3 E/100 PY and 0.4 E/100 PY for VTE, respectively; table 1 and online supplemental table 4). All patients with MACE or VTE had ≥1 risk factor including hypertension, diabetes mellitus, smoking and obesity (online supplemental tables 5 and 6). In the subgroups of patients receiving continuous upadacitinib (n=398; 1573.0 PY) or continuous adalimumab (n=168; 582.4 PY), the rates of malignancies (excluding NMSC) were 0.8 E/100 PY vs 0.9 E/100 PY, rates of adjudicated MACE were 0.1 E/100 PY vs 0.3 E/100 PY and rates of adjudicated VTE were 0.2 E/100 PY vs 0.5 E/100 PY, respectively (table 3).
Table 3.
EAERs of malignancy (excluding NMSC), adjudicated MACE and VTE through 264 weeks
| EAER | Continuous UPA 15 mg QD (n=398; PY=1573.0) E (E/100 PY)(95% CI) |
Continuous ADA 40 mg EOW (n=168; PY=582.4) E (E/100 PY) (95% CI) |
| Malignancy (excluding NMSC) | 12 (0.8) (0.4 to 1.3) | 5 (0.9) (0.3 to 2.0) |
| Adjudicated MACE* | 2 (0.1) (0.0 to 0.5) | 2 (0.3) (0.0 to 1.2) |
| Adjudicated VTE† | 3 (0.2) (0.0 to 0.6) | 3 (0.5) (0.1 to 1.5) |
Safety was assessed up to week 264, through the cut-off date of 5 October 2022.
*MACE defined as cardiovascular death (includes acute MI, sudden cardiac death, heart failure, cardiovascular procedure-related death, death due to cardiovascular haemorrhage, fatal stroke, pulmonary embolism and other cardiovascular causes), non-fatal MI and non-fatal stroke.
†VTE included fatal and non-fatal deep vein thrombosis and pulmonary embolism.
ADA, adalimumab; E, events; EAER, exposure-adjusted event rate; EOW, every other week; MACE, major adverse cardiovascular event; MI, myocardial infarction; NMSC, non-melanoma skin cancer; PY, patient-years; QD, once daily; UPA, upadacitinib; VTE, venous thromboembolic event.
55 deaths (41 of which were treatment emergent) occurred in patients receiving any upadacitinib (41 deaths) and any adalimumab (14 deaths). The rate of deaths in any upadacitinib and any adalimumab groups were similar (0.9 E/100 PY and 1.0 E/100 PY, respectively; table 1 and online supplemental table 7). The most common causes of death reported were COVID-19 or pneumonia associated with COVID-19 (12 events); rates of any COVID-19 infection, serious COVID-19 infection and fatal COVID-19 infection were numerically higher in any upadacitinib group compared with any adalimumab group (online supplemental table 2).
Efficacy
Over 5 years, the proportions of patients achieving LDA and remission as defined by CDAI criteria (CDAI≤10 and CDAI≤2.8), and the proportions of patients achieving DAS28 (CRP) ≤3.2 and DAS28 (CRP) <2.6, were consistently higher in patients randomised to upadacitinib (n=651) compared with adalimumab (n=327) as analysed by NRI (figure 1). At week 264, CDAI≤10 was achieved by 36.4% vs 26.9% (NRI; nominal p=0.004) of patients randomised to upadacitinib versus adalimumab, respectively; CDAI≤2.8 was achieved by 24.6% vs 18.7% (NRI; nominal p=0.042) of patients randomised to upadacitinib versus adalimumab, respectively. At week 264, DAS28 (CRP) ≤3.2 was achieved by 34.7% vs 24.8% (NRI; nominal p=0.002) of patients randomised to upadacitinib versus adalimumab, respectively; DAS28 (CRP) <2.6 was achieved by 31.8% vs 23.2% (NRI; nominal p=0.006) of patients randomised to upadacitinib versus adalimumab, respectively.
Figure 1.

Proportions of patients achieving CDAI LDA/remission and DAS28 (CRP) ≤3.2/<2.6 through 264 weeks (NRI). #p<0.05, ##p<0.01, ###p<0.001 for UPA 15 mg once daily vs ADA 40 mg every other week. All p values are nominal. Treatment groups are by initial randomisation. NRI was used for patients who were rescued or prematurely discontinued study drug, as well as for missing data. Data points plotted here are shown in online supplemental table 8. ADA, adalimumab; CDAI, Clinical Disease Activity Index; DAS28 (CRP), 28-joint Disease Activity Score based on C reactive protein; LDA, low disease activity; NRI, non-responder imputation; UPA, upadacitinib.
Summarising by treatment sequence (AO), at week 264, similar proportions of patients receiving continuous upadacitinib versus continuous adalimumab achieved CDAI≤10 and CDAI≤2.8 (figure 2). CDAI≤10 and CDAI≤2.8 were achieved by numerically higher proportions of patients who switched from adalimumab to upadacitinib versus upadacitinib to adalimumab. At week 264, similar proportions of patients receiving continuous upadacitinib versus continuous adalimumab also achieved DAS28 (CRP) ≤3.2 and DAS28 (CRP) <2.6 (figure 2). DAS28 (CRP) ≤3.2 and DAS28 (CRP) <2.6 were achieved by numerically higher proportions of patients who switched from adalimumab to upadacitinib versus upadacitinib to adalimumab.
Figure 2.

Proportions of patients achieving CDAI LDA/remission and DAS28 (CRP) ≤3.2/<2.6 through 264 weeks (AO). Groups are by treatment sequence AO, without imputation for missing data. All patients in the PBO group who were not previously rescued were switched to UPA at week 26. Data points plotted here are shown in online supplemental table 9. ADA, adalimumab; AO, as observed; CDAI, Clinical Disease Activity Index; DAS28 (CRP), 28-joint Disease Activity Score based on C reactive protein; LDA, low disease activity; PBO, placebo; UPA, upadacitinib.
Over 5 years, the proportions of patients achieving ACR20, ACR50 and ACR70 were consistently higher in patients randomised to upadacitinib (n=651) compared with adalimumab (n=327) as analysed by NRI (figure 3). At week 264, ACR20 was achieved by 38.4% vs 28.4% (NRI; nominal p=0.002) of patients randomised to upadacitinib versus adalimumab, respectively; ACR50 was achieved by 35.3% vs 25.7% (NRI; nominal p=0.003) of patients randomised to upadacitinib versus adalimumab, respectively; and ACR70 was achieved by 28.6% vs 22.3% (NRI; nominal p=0.042) of patients randomised to upadacitinib versus adalimumab, respectively.
Figure 3.

Proportions of patients achieving ACR20, ACR50 and ACR70 through 264 weeks (NRI). #p<0.05, ##p<0.01, ###p<0.001 for UPA 15 mg once daily vs ADA 40 mg every other week. All p values are nominal. Treatment groups are by initial randomisation. NRI was used for patients who were rescued or prematurely discontinued study drug, as well as for missing data. Data points plotted here are shown in online supplemental table 8. ACR20/50/70, ≥20/50/70% improvement in American College of Rheumatology response criteria; ADA, adalimumab; NRI, non-responder imputation; UPA, upadacitinib.
Summarising by treatment sequence (AO), ACR20 rates were generally similar in patients receiving continuous upadacitinib versus continuous adalimumab throughout 5 years, including at week 264 (97.0% vs 96.0%) (figure 4). ACR50 and ACR70 rates were generally numerically higher in patients receiving continuous upadacitinib versus continuous adalimumab until week 216, but at week 264, similar proportions of patients receiving continuous upadacitinib versus continuous adalimumab achieved ACR50 and ACR70. At week 264, ACR20 was achieved by similar proportions of patients who switched from adalimumab to upadacitinib compared with those who switched from upadacitinib to adalimumab, respectively, while ACR50 and ACR70 were achieved by numerically higher proportions of patients who switched from adalimumab to upadacitinib compared with those who switched from upadacitinib to adalimumab (figure 4).
Figure 4.

Proportions of patients achieving ACR20, ACR50 and ACR70 through 264 weeks (AO). Groups are by treatment sequence AO, without imputation for missing data. All patients in the PBO group who were not previously rescued were switched to UPA at week 26. Data points plotted here are shown in online supplemental table 9. ACR20/50/70, ≥20/50/70% improvement in American College of Rheumatology response criteria; ADA, adalimumab; AO, as observed; PBO, placebo; UPA, upadacitinib.
For ACR components over 264 weeks analysed by treatment sequence (AO), patients receiving continuous upadacitinib generally achieved numerically greater improvements compared with those receiving continuous adalimumab, except for TJC68 in which responses were generally similar (online supplemental figure 3).
Numerically greater improvements in severity and duration of morning stiffness were also achieved in patients receiving continuous upadacitinib compared with those receiving continuous adalimumab (online supplemental figure 4A,B).
At week 192, as analysed by treatment sequence (AO), similar proportions of patients demonstrated no radiographic progression (ΔmTSS≤0 from baseline; 80.9% vs 78.0% for continuous upadacitinib vs continuous adalimumab and 77.5% vs 74.0% for adalimumab to upadacitinib vs upadacitinib to adalimumab) (figure 5A). Additionally, at week 192, patients receiving continuous upadacitinib showed numerically lower radiographic progression compared with the other treatment sequence groups) (ΔmTSS; 0.53 vs 1.18 for continuous upadacitinib vs continuous adalimumab, respectively, and 0.88 vs 1.72 for adalimumab to upadacitinib vs upadacitinib to adalimumab, respectively) (figure 5B and online supplemental figure 5A). In addition, changes from baseline in joint erosion and joint space narrowing were numerically lower in patients receiving continuous upadacitinib versus continuous adalimumab and in patients who switched from adalimumab to upadacitinib versus upadacitinib to adalimumab at week 192 (online supplemental figure 5B,C).
Figure 5.
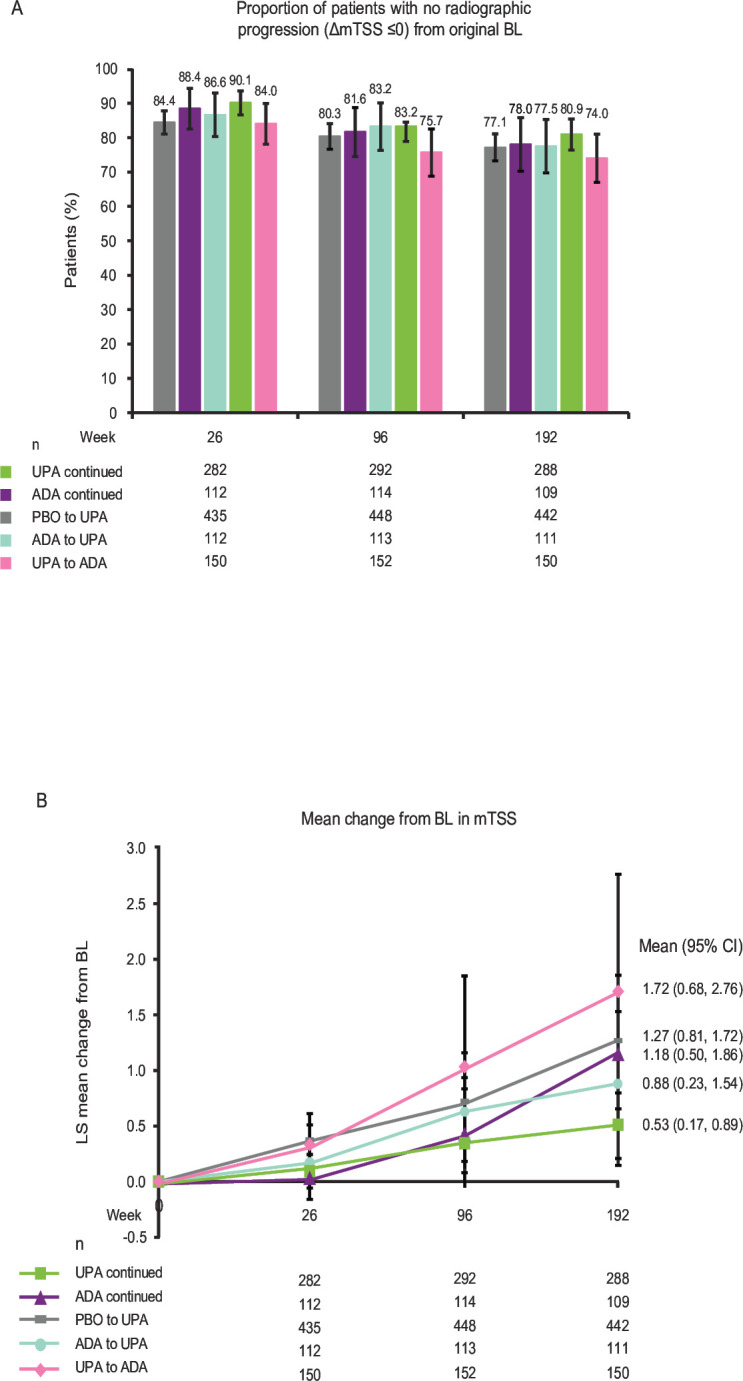
Radiographic outcomes at 26, 96 and 192 weeks (AO). Groups are by treatment sequence AO, without imputation for missing data. All patients in the PBO group who were not previously rescued were switched to UPA at week 26. Data points plotted here are shown inonline supplemental tables 9,10. Δ, change from baseline; ADA, adalimumab; AO, as observed; BL, baseline; LS, least squares; PBO, placebo; mTSS, modified Total Sharp Score; UPA, upadacitinib.
Discussion
These 5-year data from the ongoing, open-label LTE of the SELECT-COMPARE study provide a longer-term update on the safety and efficacy of upadacitinib since the published 3-year data.13 Open-label LTE studies such as this provide systematic information on long-term safety profiles, which are of key clinical relevance in chronic conditions such as RA.13 19 SELECT-COMPARE is unique in that it assesses the safety and efficacy of upadacitinib compared with the active comparator adalimumab for up to 10 years; other head-to-head studies of upadacitinib in RA only compared upadacitinib to active comparators for up to 1 year.20–22
The safety profile of upadacitinib 15 mg once daily through 5 years was consistent with the 3-year safety profile and the integrated safety analysis from the SELECT phase 3 programme,11 13 with no new safety signals observed. Rates of any TEAE, serious TEAEs and TEAEs leading to discontinuation were similar with upadacitinib and adalimumab. Rates of any COVID-19 infection, serious COVID-19 infection and fatal COVID-19 infection were numerically higher in patients receiving upadacitinib compared with adalimumab. These results are consistent with the findings of a long-term safety study of upadacitinib using pooled data across indications.10 Overall, rates of TEAEs of special interest were generally similar between patients receiving upadacitinib and adalimumab, except for numerically higher rates of herpes zoster, lymphopenia, CPK elevation, hepatic disorders (mostly transaminase elevations) and NMSC with upadacitinib. The proportions of patients with grade 3/4 laboratory abnormalities were generally higher with upadacitinib compared with adalimumab and were similar to the results reported at 3 years. Higher rates of herpes zoster and CPK elevation with upadacitinib are expected based on the known safety profile of JAK inhibitors.11 23–28 However, most cases of herpes zoster were non-serious, involved one dermatome and did not lead to discontinuation of study drug; there were three events of ophthalmic herpes zoster (two in any upadacitinib group and one in any adalimumab group). Herpes zoster vaccination is recommended for all patients prior to initiating upadacitinib treatment.29–32 The majority of CPK elevations were transient, and the rates at 5 years versus those at 3 years, were numerically lower in the upadacitinib group and similar in the adalimumab group (4.0 E/100 PY vs 4.7 E/100 PY and 1.8 E/100 PY vs 1.7 E/100 PY), respectively. Rates of NMSC were numerically higher in patients receiving upadacitinib compared with adalimumab, as expected based on the known risk of NMSC in patients with inflammatory diseases receiving JAK inhibitors.33–35 Although SELECT-COMPARE was not designed as a head-to-head safety study, the rates of TEAEs of special interest, including MACE, VTE and malignancies (excluding NMSC), which have emerged for the JAK inhibitor class of drugs, were prespecified based on findings in previous studies and other JAK inhibitors.36 The observed rates of TEAEs of special interest, including MACE, VTE and malignancies (excluding NMSC), were consistent with the SELECT-COMPARE 3-year results,13 demonstrating no evidence of an increase in risk with longer exposure to upadacitinib. This is also consistent with results from a long-term safety analysis of upadacitinib across indications.10 No differences were observed in rates of MACE, VTE or malignancies (excluding NMSC) between upadacitinib and adalimumab through 5 years when analysed by treatment at time of event and in subgroups receiving continuous upadacitinib or adalimumab treatment from randomisation. While there appears to be some numerical imbalance between upadacitinib and adalimumab in malignancies per organ system (online supplemental table 4), particularly for basal cell carcinoma, the rates are generally comparable between the treatment groups. The event rate for malignancies (excluding NMSC) was consistent with RA populations37–39 and, as expected, most malignancies occurred in patients ≥50 years of age. Although rates of MACE/VTE and rates of death were similar between the two groups, the rate of deaths due to cardiac disorders was higher in the upadacitinib group. However, all cases of MACE or VTE (including deaths due to cardiac disorders) occurred in patients with ≥1 known risk factor. A post hoc analysis of the SELECT RA clinical programme, which evaluated the safety of upadacitinib in patients with increased cardiovascular risk, found that while herpes zoster and NMSC were observed at higher rates in patients receiving upadacitinib compared with adalimumab or MTX, rates of MACE, VTE and malignancy (excluding NMSC) were similar across treatments, although they were numerically higher in patients at increased cardiovascular risk.40 Further long-term studies are required to fully determine whether there is an association between JAK inhibitors and NMSC,10 and VTE given that conditions for which JAK inhibitors are indicated carry inherent risks for VTE.
At 264 weeks, upadacitinib continued to show greater efficacy than adalimumab when analysed by NRI while analysis by treatment sequence (AO) showed similar responses between patients receiving continuous upadacitinib and continuous adalimumab. The AO analysis included all patients who continued therapy, likely because they experienced clinical benefit without significant toxicity; therefore, the similar AO results between continuous upadacitinib and continuous adalimumab are as expected and suggest that if patients experience clinical benefit and tolerate the study drug, their benefit is likely to continue. On the other hand, the NRI analysis comprised all randomised patients, including patients who did not experience a significant response or who discontinued study drug. The NRI results in this population suggest that patients are more likely to benefit from, and tolerate, upadacitinib compared with adalimumab. Due to rescue handling, in the NRI analysis, patients who did not achieve CDAI LDA and were rescued at week 26 were considered non-responders for all binary endpoints at all visits after rescue, therefore; NRI response rates are very conservative after week 26 for endpoints less stringent than CDAI LDA.
Patients receiving upadacitinib showed greater achievement of LDA and remission as defined by CDAI criteria, DAS28 (CRP) ≤3.2/<2.6 and ACR20/50/70, as well as greater improvements in the patient-reported outcomes of HAQ-DI and morning stiffness severity and duration at week 264. Inhibition of radiographic progression (change from baseline in mTSS ≤0) remained high and consistently similar between patients receiving continuous upadacitinib and continuous adalimumab. The mean change from baseline in mTSS was numerically lower with continuous upadacitinib than continuous adalimumab from week 96 to week 192. The rate of radiographic progression was not clinically significant in patients receiving continuous upadacitinib or continuous adalimumab and is, therefore, likely to translate to very little functional decline in these patients.
In line with improved efficacy, the use of corticosteroids continued to decline in the continuous upadacitinib and continuous adalimumab groups.
Limitations of this analysis include that SELECT-COMPARE was not designed or powered to detect statistical differences in safety events; nor was it designed with parallel groups in the LTE because of rescue switching.13 Therefore, these data should be interpreted with respect to this. The nature of LTE studies means that AO data may be biased by patients who remain in the study being better able to tolerate the drug and show response; however, all binary endpoints were also analysed by NRI, which provides insight into an intention-to-treat comparison between those randomised to upadacitinib versus adalimumab. Also, the analysis of treatment response did not take into consideration how modifications to background RA medications may have contributed to the ability of these patients to achieve treatment goals. Specifically, no analysis was conducted to assess whether a higher proportion of patients who switched to adalimumab also discontinued MTX, which could have affected efficacy in the upadacitinib to adalimumab arm.
In summary, the safety profile of upadacitinib 15 mg once daily through 5 years in the SELECT-COMPARE study was consistent with previous reports in RA and the known safety profile of upadacitinib in other indications, with no new safety risks observed.6 10–13 Upadacitinib 15 mg once daily continued to show numerically better clinical responses in terms of CDAI LDA and remission, DAS28 (CRP) ≤3.2/<2.6 and ACR20/50/70 compared with adalimumab 40 mg EOW at 5 years, and radiographic progression remained low with both treatments at week 192. These results continue to support a favourable benefit–risk profile for upadacitinib 15 mg once daily in the long-term treatment of RA.
Acknowledgments
AbbVie and the authors thank the participants, study sites and investigators who are participating in this clinical trial. Medical writing support was provided by Laura Chalmers, PhD, of 2 the Nth (Cheshire, UK).
Footnotes
Contributors: RF, YL and CGP contributed to the study conception and design. RF, JS, PD, LB, CGP, YT and EM participated in data acquisition. SKP and NK were involved in the interpretation of the data. XB and YL conducted the statistical analyses. All authors analysed and interpreted the data and contributed to the critical revision of the manuscript. All named authors met the International Committee of Medical Journal Editors criteria for authorship for this article, take responsibility for the integrity of the work as a whole and have given their approval for this version to be published. RF acts as guarantor and accepts full responsibilty for the work, had access to the data, and controlled the decision to publish.
Funding: This work is supported by AbbVie, who is funding this trial and participated in the trial design, research, analysis, data collection, interpretation of data, and the review and approval of the publication. All authors had access to relevant data and participated in the drafting, review and approval of this publication. No honoraria or payments were made for authorship.
Competing interests: RF has received consulting fees and/or grant/research support from AbbVie, Amgen, BI, Biosplice, BMS, Flexion, Galapagos, Galvani, Gilead, GSK, Horizon, Janssen, Lilly, Novartis, Pfizer, Sanofi-Aventis, Selecta, UCB, Viela, Vorso and Vyne and has participated on a data safety monitoring board or advisory board for Kiniksa. JS has received speaking fees, consulting fees and grant/research support from AbbVie, Accord, BMS, Janssen, MSD, Pfizer, Roche, Sandoz and UCB. SKP, XB, NK and YL are employees of AbbVie and may hold stock or options. PD has received speaker fees from AbbVie, Galapagos, Lilly, Nordimed and Thermofischer. LB has received speaking fees, consulting fees and grant/research support from AbbVie, Amgen, BMS, Celgene, Lilly, Fresenius Kabi, Gilead, Janssen, Novartis, Organon, Pfizer, Sanofi-Aventis, Teva and UCB. CGP is an employee and shareholder of Spire Sciences and has served as a consultant for Aclaris, AstraZeneca, Daiichi-Sankyo, Five Prime, Genentech, Gilead, GSK, Istesso, Labcorp, Lilly, Pacira, Paradigm, SetPoint, Sorrento, SynOx and UCB. YT has received speaker fees and/or honoraria from AbbVie, Asahikasei, AstraZeneca, BI, BMS, Chugai, Eisai, Gilead, GSK, Lilly, Pfizer, Taiho and Taisho and research grants from Asahikasei, Chugai, Eisai, Mitsubishi-Tanabe and Taisho. EM has received speaking fees, consulting fees and grant/research support from AbbVie, Amgen, AstraZeneca, BMS, Hi-Bio, Janssen, Lilly, Novartis, Pfizer, Roche, Sandoz and Sanofi, and has received payment for expert testimony from AbbVie.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available on reasonable request. AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymised, individual-level and trial-level data (analysis datasets), as well as other information (eg, protocols and clinical study reports), provided the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan, and execution of a Data Sharing Agreement. Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
The study is being conducted according to the International Council for Harmonization guidelines, local regulations and guidelines governing clinical study conduct, and the Declaration of Helsinki. All patients provided written informed consent, and the study protocol and consent forms were approved by an institutional review board or independent ethics committee at each study site. The coordinating investigator (Charles Birbara of the University of Massachusetts, Worcester, Massachusetts, USA) received approval from the Advarra Institutional Review Board.
References
- 1. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet 2016;388:2023–38. 10.1016/S0140-6736(16)30173-8 [DOI] [PubMed] [Google Scholar]
- 2. Fraenkel L, Bathon JM, England BR, et al. 2021 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken) 2021;73:924–39. 10.1002/art.41752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smolen JS, Landewé RBM, Bergstra SA, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis 2023;82:3–18. 10.1136/ard-2022-223356 [DOI] [PubMed] [Google Scholar]
- 4. Parmentier JM, Voss J, Graff C, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol 2018;2:23. 10.1186/s41927-018-0031-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burmester GR, Kremer JM, Van den Bosch F, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2018;391:2503–12. 10.1016/S0140-6736(18)31115-2 [DOI] [PubMed] [Google Scholar]
- 6. Fleischmann R, Pangan AL, Song I-H, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double-blind, randomized controlled trial. Arthritis Rheumatol 2019;71:1788–800. 10.1002/art.41032 [DOI] [PubMed] [Google Scholar]
- 7. Genovese MC, Fleischmann R, Combe B, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet 2018;391:2513–24. 10.1016/S0140-6736(18)31116-4 [DOI] [PubMed] [Google Scholar]
- 8. Rubbert-Roth A, Enejosa J, Pangan AL, et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N Engl J Med 2020;383:1511–21. 10.1056/NEJMoa2008250 [DOI] [PubMed] [Google Scholar]
- 9. van Vollenhoven R, Takeuchi T, Pangan AL, et al. Efficacy and safety of upadacitinib monotherapy in methotrexate-naive patients with moderately-to-severely active rheumatoid arthritis (SELECT-EARLY): a multicenter, multi-country, randomized, double-blind, active comparator-controlled trial. Arthritis Rheumatol 2020;72:1607–20. 10.1002/art.41384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burmester GR, Cohen SB, Winthrop KL, et al. Safety profile of upadacitinib over 15 000 patient-years across rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis and atopic dermatitis. RMD Open 2023;9:e002735. 10.1136/rmdopen-2022-002735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cohen SB, van Vollenhoven RF, Winthrop KL, et al. Safety profile of upadacitinib in rheumatoid arthritis: integrated analysis from the SELECT phase III clinical programme. Ann Rheum Dis 2021;80:304–11. 10.1136/annrheumdis-2020-218510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fleischmann RM, Genovese MC, Enejosa JV, et al. Safety and effectiveness of upadacitinib or adalimumab plus methotrexate in patients with rheumatoid arthritis over 48 weeks with switch to alternate therapy in patients with insufficient response. Ann Rheum Dis 2019;78:1454–62. 10.1136/annrheumdis-2019-215764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fleischmann R, Mysler E, Bessette L, et al. Long-term safety and efficacy of upadacitinib or adalimumab in patients with rheumatoid arthritis: results through 3 years from the SELECT-COMPARE study. RMD Open 2022;8:e002012. 10.1136/rmdopen-2021-002012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. 10.1002/art.27584 [DOI] [PubMed] [Google Scholar]
- 15. Woodworth T, Furst DE, Alten R, et al. Standardizing assessment and reporting of adverse effects in rheumatology clinical trials II: the Rheumatology Common Toxicity Criteria V.2.0. J Rheumatol 2007;34:1401–14. [PubMed] [Google Scholar]
- 16. NCI . Common terminology criteria for adverse events (CTCAE) version 4.0 NIH publication, 2009. Available: https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/Archive/CTCAE_4.0_2009-05-29_QuickReference_8.5x11.pdf
- 17. van der Heijde D. How to read radiographs according to the Sharp/van der Heijde method. J Rheumatol 2000;27:261–3. [PubMed] [Google Scholar]
- 18. van der Heijde DM, van Leeuwen MA, van Riel PL, et al. Biannual radiographic assessments of hands and feet in a three-year prospective followup of patients with early rheumatoid arthritis. Arthritis Rheum 1992;35:26–34. 10.1002/art.1780350105 [DOI] [PubMed] [Google Scholar]
- 19. Wollenhaupt J, Lee E-B, Curtis JR, et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: final results of a global, open-label, long-term extension study. Arthritis Res Ther 2019;21:89. 10.1186/s13075-019-1866-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Combe B, Kivitz A, Tanaka Y, et al. Filgotinib versus placebo or adalimumab in patients with rheumatoid arthritis and inadequate response to methotrexate: a phase III randomised clinical trial. Ann Rheum Dis 2021;80:848–58. 10.1136/annrheumdis-2020-219214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fleischmann R, Mysler E, Hall S, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL strategy): a phase 3B/4, double-blind, head-to-head, randomised controlled trial. Lancet 2017;390:457–68. 10.1016/S0140-6736(17)31618-5 [DOI] [PubMed] [Google Scholar]
- 22. Taylor PC, Keystone EC, van der Heijde D, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med 2017;376:652–62. 10.1056/NEJMoa1608345 [DOI] [PubMed] [Google Scholar]
- 23. Bieber T, Thyssen JP, Reich K, et al. Pooled safety analysis of baricitinib in adult patients with atopic dermatitis from 8 randomized clinical trials. J Eur Acad Dermatol Venereol 2021;35:476–85. 10.1111/jdv.16948 [DOI] [PubMed] [Google Scholar]
- 24. Cohen SB, Tanaka Y, Mariette X, et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheum Dis 2017;76:1253–62. 10.1136/annrheumdis-2016-210457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee EB, Yamanaka H, Liu Y, et al. Efficacy and safety of tofacitinib for the treatment of rheumatoid arthritis in patients from the Asia-Pacific region: post-hoc analyses of pooled clinical study data. Int J Rheum Dis 2019;22:1094–106. 10.1111/1756-185X.13516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smolen JS, Genovese MC, Takeuchi T, et al. Safety profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J Rheumatol 2019;46:7–18. 10.3899/jrheum.171361 [DOI] [PubMed] [Google Scholar]
- 27. Sunzini F, McInnes I, Siebert S. JAK inhibitors and infections risk: focus on herpes Zoster. Ther Adv Musculoskelet Dis 2020;12:1759720X20936059. 10.1177/1759720X20936059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamaoka K, Tanaka Y, Kameda H, et al. The safety profile of upadacitinib in patients with rheumatoid arthritis in Japan. Drug Saf 2021;44:711–22. 10.1007/s40264-021-01067-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. EMA . RINVOQ Summary of Product Characteristics, Available: https://www.ema.europa.eu/en/documents/product-information/rinvoq-epar-product-information_en.pdf
- 30. US FDA . RINVOQ Prescribing Information, Available: https://www.rxabbvie.com/pdf/rinvoq_pi.pdf
- 31. CDC . Shingrix recommendations, Available: https://www.cdc.gov/vaccines/vpd/shingles/hcp/Shingrix/recommendations.html
- 32. Bass AR, Chakravarty E, Akl EA, et al. 2022 American College of Rheumatology guideline for vaccinations in patients with rheumatic and musculoskeletal diseases. Arthritis Care Res (Hoboken) 2023;75:449–64. 10.1002/acr.25045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jalles C, Lepelley M, Mouret S, et al. Skin cancers under Janus kinase inhibitors: a World Health Organization drug safety database analysis. Therapie 2022;77:649–56. 10.1016/j.therap.2022.04.005 [DOI] [PubMed] [Google Scholar]
- 34. Liu R, Wan Q, Zhao R, et al. Risk of non-melanoma skin cancer with biological therapy in common inflammatory diseases: a systemic review and meta-analysis. Cancer Cell Int 2021;21:614. 10.1186/s12935-021-02325-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang J-L, Yin W-J, Zhou L-Y, et al. Risk of non-melanoma skin cancer for rheumatoid arthritis patients receiving TNF antagonist: a systematic review and meta-analysis. Clin Rheumatol 2020;39:769–78. 10.1007/s10067-019-04865-y [DOI] [PubMed] [Google Scholar]
- 36. Ytterberg SR, Bhatt DL, Mikuls TR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med 2022;386:316–26. 10.1056/NEJMoa2109927 [DOI] [PubMed] [Google Scholar]
- 37. Askling J, Berglind N, Franzen S, et al. How comparable are rates of malignancies in patients with rheumatoid arthritis across the world? A comparison of cancer rates, and means to optimise their comparability, in five RA registries. Ann Rheum Dis 2016;75:1789–96. 10.1136/annrheumdis-2015-208105 [DOI] [PubMed] [Google Scholar]
- 38. Gross RL, Schwartzman-Morris JS, Krathen M, et al. A comparison of the malignancy incidence among patients with psoriatic arthritis and patients with rheumatoid arthritis in a large US cohort. Arthritis Rheumatol 2014;66:1472–81. 10.1002/art.38385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wolfe F, Michaud K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: analyses from a large US observational study. Arthritis Rheum 2007;56:2886–95. 10.1002/art.22864 [DOI] [PubMed] [Google Scholar]
- 40. Fleischmann R, Curtis JR, Charles-Schoeman C, et al. Safety profile of upadacitinib in patients at risk of cardiovascular disease: integrated post hoc analysis of the SELECT phase III rheumatoid arthritis clinical programme. Ann Rheum Dis 2023;82:1130–41. 10.1136/ard-2023-223916 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
rmdopen-2023-004007supp001.pdf (914.5KB, pdf)
Data Availability Statement
Data are available on reasonable request. AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymised, individual-level and trial-level data (analysis datasets), as well as other information (eg, protocols and clinical study reports), provided the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan, and execution of a Data Sharing Agreement. Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.