

Abstract
The DPYSL2/CRMP2 gene encodes a microtubule-stabilizing protein crucial for neurogenesis and is associated with numerous psychiatric and neurodegenerative disorders including schizophrenia, bipolar disorder, and Alzheimer’s disease. DPYSL2 generates multiple RNA and protein isoforms, but few studies have differentiated between them. We previously reported an association of a functional variant in the DPYSL2-B isoform with schizophrenia (SCZ) and demonstrated in HEK293 cells that this variant reduced the length of cellular projections and created transcriptomic changes that captured schizophrenia etiology by disrupting mTOR signaling-mediated regulation. In the present study, we follow up on these results by creating, to our knowledge, the first models of endogenous DPYSL2-B knockout in human induced pluripotent stem cells (iPSCs) and neurons. CRISPR/Cas9-faciliated knockout of DPYSL2-B in iPSCs followed by Ngn2-induced differentiation to glutamatergic neurons showed a reduction in DPYSL2-B/CRMP2-B RNA and protein with no observable impact on DPYSL2-A/CRMP2-A. The average length of dendrites in knockout neurons was reduced up to 58% compared to controls. Transcriptome analysis revealed disruptions in pathways highly relevant to psychiatric disease including mTOR signaling, cytoskeletal dynamics, immune function, calcium signaling, and cholesterol biosynthesis. We also observed a significant enrichment of the differentially expressed genes in SCZ-associated loci from genome-wide association studies (GWAS). Our findings expand our previous results to neuronal cells, clarify the functions of the human DPYSL2-B isoform and confirm its involvement in molecular pathologies shared between many psychiatric diseases.
INTRODUCTION
Dihydropyramidase-like 2 (DPYSL2; also known as CRMP2/TOAD-64/Ulip2/DRP-2/unc33/CRMP62/TUC-2) is a ~ 140 kb gene on chromosome 8p21.2 that encodes collapsin response mediator protein 2 (CRMP2). DPYSL2 belongs to a family of homologous genes (DPYSL1–5/CRMP1–5) that are conserved across vertebrates and highly expressed during early brain development in neurons and glia [1, 2]. DPYSL2 was first identified as an intracellular respondent of Semaphorin 3A, a repulsive axon guidance molecule [3]. CRMP2 is a microtubule-associating protein which as a tetramer binds to and stabilizes assembled microtubules, and as a monomer traffics tubulin dimers to their plus end [4]. Consequently, CRMP2 is a mitigator of diverse cytoskeletal-related functions including mitosis, cell migration [5, 6], endocytosis, calcium homeostasis and neurotransmission [2, 7, 8], mitochondrial morphology and motility [9], kinesin and dynein-facilitated molecular transport, cell polarity, and dendritic and axonal elongation [2, 10, 11]. Phosphorylation at various amino acids by the CDK5/GSK3-β and Rho pathways affects CRMP2’s affinity for assembled microtubules and tubulin dimers, which subsequently results in fluid elongation or retraction of axons [2]. CRMP2 activity is also regulated by the mammalian target of rapamycin (mTOR), a kinase which mediates translation efficiency of CRMP2 by phosphorylating translation initiation proteins that bind to a 5’ Terminal OligoPyrimidine (TOP) motif in the 5’ untranslated region (UTR) of CRMP2 mRNA [12–14]. Disruptions in the expression and regulation of CRMP2 have been documented in numerous psychiatric and neurological disorders, including schizophrenia [2, 10, 12, 15–19], bipolar disorder [2, 15, 16, 19], Alzheimer’s disease [20–22], autism spectrum disorder [23], intellectual disability [24], major depressive disorder [2, 16], Down’s syndrome [2, 25], neurofibromatosis [2], multiple sclerosis [2, 26, 27], and amyotrophic lateral sclerosis [2, 26]. Variation in CRMP2 phosphorylation was recently determined to be altered in the blood and brain of patients with bipolar disorder and schizophrenia [19], indicating that CRMP2 may be a novel biomarker that could aid diagnosis.
Despite the large amount of interest and research on CRMP2, few studies have been isoform-specific. The human DPYSL2 gene has 16 exons which are alternatively spliced into 8 transcripts, 3 of which have a consensus coding sequence: DPYSL2-A/CRMP2-A (ENST00000521913/NM_001197293); DPYSL2-B/CRMP2-B (ENST00000311151/NM_001386), and DPYSL2-C/CRMP2-C (ENST00000523027/NM_001244604). These three isoforms share most of their sequence but are differentiated by alternative first exons. DPYSL2-B is the most well-documented and is considered the canonical transcript because it was discovered first and is the most conserved across species [2, 3, 28, 29]. Most studies of DPYSL2/CRMP2 refer to the DPYSL2-B/CRMP2-B gene sequence/mRNA/cDNA/protein; but either do not acknowledge the existence of multiple isoforms or manipulate regions of DPYSL2/CRMP2 that are common to multiple isoforms. Given that isoforms can have distinct functions, it is necessary to distinguish the role of each transcript to determine which are relevant to mental illness.
In previous work we showed that the DPYSL2-B isoform carries a SCZ-associated polymorphic CT dinucleotide repeat (DNR) in the 5’ untranslated region (UTR). This polymorphic region responded to mTOR signaling and showed allelic differences in reporter assays [17]. We then introduced this repeat to HEK293 cells and observed a significant reduction of the corresponding CRMP2-B isoform, shortening of the HEK293 cellular projections, and marked differences in transcriptome analysis. The changing transcriptome also showed a reverse expression signature from that seen with antipsychotic drugs from ConnectivityMap (CMAP) and other schizophrenia gene perturbations supporting the importance DPYSL2-B and a role for mTOR signaling in schizophrenia [12]. We now report further support for our conclusions in a better model system.
In the present study, we use CRISPR/Cas9 in induced pluripotent stem cells (iPSCs) to create the first genetic knockout of the endogenous DPYSL2-B isoform in human cells. We then differentiate these edited iPSCs into excitatory glutamatergic neurons and investigate the impact of the knockout on the transcript, protein, and morphology levels.
MATERIALS AND METHODS
sgRNA design and cloning
Two sgRNAs were designed using CRISPOR (http://crispor.tefor.net/) to delete the first exon (ENSE00002094501) of DPYSL2-B. sgRNA oligos (IDT; California, USA; Supplementary Table 1) were ligated into pDG459 as previously described [30] and transformed into E.Coli (ThermoFisher; Massachusetts, USA; C404003) following the manufacturer’s recommendations. Plasmids were isolated (Qiagen; Maryland, USA; 27104) and screened for correct insertion via Sanger sequencing with specialized primers (IDT, Supplementary Table 1).
iPSC quality testing, culture and maintenance
The iPSC line used for this study (MH0185922 aka RU13, African male control) was obtained from the NIMH/RUCDR repository (https://www.nimhgenetics.org/). Upon receipt, the cell line was authenticated and screened for chromosomal abnormalities with the Illumina Infinium Global Screening Array-24 v2.0 by the Johns Hopkins Genetic Resources Core Facility. Mycoplasma screens were negative. For culture, all plates were coated with 5ug/mL laminin (Biolamina; Sweden; LN521) in DPBS (Quality Biological; Maryland, USA; 114–059-101) and iPSCs were grown feeder-free in StemFlex media with supplement (ThermoFisher A3349401) at 37 °C unless otherwise specified. On days of thawing, passaging, transduction or transfection the media was supplemented with 10 μM Rock Inhibitor (Y-27632 dihydrochloride, Tocris; UK; 1254).
CRISPR/Cas9 transfection, screening, and single-cell cloning of iPSCs
iPSCs were transfected with Lipofectamine Stem (ThermoFisher STEM00003) and pDG459 following the manufacturer’s instructions. Starting 40 hours after transfection, puromycin was added for 24 h to select for transfected cells. The iPSCs were then fed daily until confluent and shedding into the media. To genotype the “bulk” transfected cells, the floating cells in each well were pelleted and combined with 20 μL of Quick Extract buffer (Lucigen; Wisconsin, USA; QE0905T). DNA was extracted according to the manufacturer’s recommendations. 5 μL of DNA was used for PCR with the Accuprime PCR kit (ThermoFisher 12339016) supplemented with 10x PCRx Enhancer (ThermoFisher 11495017) with primers in Supplementary Table 1. PCR products were electrophoresed then imaged with a BioRad (California, USA) GelDoc EZ Imager with Image Lab Software. Bulk wells positive for the deletion were sparse plated in 6-well plates at a density of 500 cells/well and grown for 7–10 days to form distinct colonies. Half of each colony was transferred to a 96-well plate for continued culture. The other half was transferred to a PCR tube with 10 μL of Quick Extract buffer and screened as described above. Colonies positive for the deletion by PCR and gel were confirmed by Sanger sequencing with the PCR primers.
Re-genotyping of positive deletion clones revealed repeated emergence of a band at the expected size for unedited DNA. Sequencing revealed that this DNA contained a 1 bp insertion in the sgRNA3 protospacer at the start of the coding sequence. After confirming that this insertion should cause a frameshift and a premature termination codon, we used the same single-cell cloning and genotyping protocols to isolate 6 clones with the insertion for further analyses.
Off-target analysis and Global diversity array of frameshift and control iPSCs
Candidate off-target sites for the two sgRNAs were obtained from CRISPOR and filtered to those with 3 or fewer mismatches with the on-target protospacer. The resulting sites were run through a Perl script that identified those in exons or overlapping with DNase I hypersensitivity peaks (www.encodeproject.org, file name hg38_wgEncodeRegDnaseClustered.txt). This analysis revealed a single off-target candidate site at chr14:67889176– 67889198. All clones were genotyped for this site with primers listed in Supplementary Table 1.
After Ngn2-rtTA transduction (see below), all clones were again authenticated and screened for chromosomal abnormalities via the Illumnia Infinium Global Diversity Array by the Johns Hopkins Genetic Resources Core Facility. No chromosomal abnormalities were found. Identity by state analysis with plink [31] found the clones were 99.9416–99.9537% identical, within the array error rate.
Transduction and Ngn2-induced differentiation of frameshift and control iPSCs into glutamatergic neurons
iPSCs were transduced with rtTA and Ngn2 lentiviruses from Cellomics (Pennsylvania, USA; CLVP-109–200UL) or the Children’s Hospital of Philadelphia Clinical Vector Core (Pennsylvania, USA; LV260/LV259; Supplementary Table 2) as previously described [32]. The untransfected iPSC line was transduced six times to create six control “clones”. To make neurons for RNA-seq and Western Blot, six frameshift clones and six control clones were differentiated across 7 batches (Supplementary Table 2) as previously described [32].
For dendrite morphology analysis, three frameshift and three control clones were differentiated on rat astrocytes to encourage the cells to spread out. On DIV −7, astrocytes were thawed into T75 flasks and fed once every 2–3 days with glial media [33]. On DIV −1, chamber slides (ThermoFisher 62407–294) were coated with poly-L-ornithine (0.01%, Sigma-Aldrich; Massachusetts, USA; P4957)/laminin. On DIV 0, 9 × 104 rat astrocytes were plated per chamber in glial media. On DIV 2, the glial media was removed and 1.5 × 105 neurons were plated/ chamber on top of the astrocytes in iN-NBM/B27 media. Differentiation proceeded as cited above except the iN-NBM/27 media was replaced with Neuronal Medium [33] and doxycycline was removed on Day 12. Immunocytochemistry was performed on DIV21.
Immunocytochemistry and dendrite morphology analysis of glutamatergic neurons on rat astrocytes
Chambers were rinsed with 1xPBS, then fixed with 4% paraformaldehyde (Alfa Aesar; Massachusetts, USA; J61899) for 30 min. Samples were washed with 1xPBS, then blocked in 1xPBS with 5% goat serum (Cell Signaling Technology; Massachusetts, USA; 5425 S) and 0.3% Triton X-100 (Sigma-Aldrich 93443–100 mL) for 1 hour; then incubated overnight at 4 °C with primary antibodies (Supplementary Table 3) in antibody solution {1xPBS with 1% BSA (Millipore Sigma A4161–1G) and 0.3% Triton X-100}. The next day samples were washed, incubated at 37 °C for 1 hour with secondary antibodies (Supplementary Table 3) in antibody solution, then washed again. Glass cover slips (Fisher 1254 M) were mounted with Prolong Gold antifade reagent with DAPI (Invitrogen P36931). Slides were imaged on a Zeiss Axio Observer with Zen Blue software. The images were blinded and analyzed using the MAP2 and NEUN channels in Image J (v 1.53) with the Neurphology J [34] plugin.
RNA extraction and qRT-PCR
RNA was extracted immediately after harvesting using the Quick-RNA Miniprep Kit (Zymo; California, USA; R1054). cDNA was generated from two RU13 control iPSC clones, four other control iPSC lines, and four RU13 frameshift iPSC clones with the Superscript III kit (ThermoFisher 18080–051) following manufacturer’s recommendations using 1 μg of RNA per sample and random hexamer primers. cDNA was diluted 1:10 in UltraPure water and used for qRT-PCR with iTaq Universal SYBR Green Supermix (BioRad 172–5121) with primers targeting the first exon of DPYSL2-B, GAPDH, and β-actin (Supplementary Table 1). Samples were run on a CFX Connect with CFX Maestro software.
Protein extraction and Western blot of iPSCs and glutamatergic neurons
iPSC lysates were prepared by thawing cell pellets on ice, then adding cold RIPA Buffer with 1 mM PMSF (Cell Signaling Technology 8553 S) at a ratio of 160 μL per 1 million cells. Samples were rotated for one hour then centrifuged at 21,000 × g for 30 min at 4 °C. The cleared lysates were quantified with BCA analysis (ThermoFisher 23225). 20 μg of protein was combined with XT Sample Buffer (Bio-Rad 1610791), XT Reducing Agent (Bio-Rad 1610792), and water to a final volume of 45 uL. Samples were boiled at 95 °C for 5 min, then cooled on ice for 10 min.
Neuron lysates were prepared by adding cold Phosphosafe Extraction Reagent (MilliporeSigma 71296–3) with PhosStop (MilliporeSigma 4906845001) and cOmplete protease inhibitor (Sigma 11697498001) to frozen neuron pellets on ice at a ratio of 100 μL per 1 million cells. Once thawed, pellets were incubated on ice for 15 min with vortexing every 5 min, followed by centrifugation at 21,000 × g for 15 min at 4 °C. The cleared lysates were neutralized by adding 1/3 volume XT Sample Buffer and heating to 98 °C for 10 min, then 20 °C for 1 min. Neutralized lysates were combined with XT Reducing Agent, then heated at 65 °C for 10 min and 20 °C for 1 min.
Samples were loaded into XT Tricine gels (BioRad 3450129) with EZ-Run Pre-Stained Rec Protein Ladder (ThermoFisher BP3603–500) and Blue Prestained Protein Standard (NEB; Massachusetts, USA; P7718S). Gels were electrophoresed in XT Tricine Running Buffer (Bio-Rad 1610790) at 93 V for 30 min, then 80 V for 75 min, then 120 V for 75 min. Protein was transferred onto PVDF membrane (BioRad 162–0262) with the BioRad Trans-Blot Turbo system on the mixed MW program. After transfer, the membrane was stained with Ponceau and cut at 55kD into two pieces, which were then rinsed, blocked for 1 h in 1xTBS (Quality Biological 351–086-101) with 5% milk, and incubated in antibody solution {1xTBS with 1% milk and 0.2% Tween 20 (Sigma P7949)} with primary antibodies against GAPDH and CRMP2 (Supplementary Table 3) overnight at 4 °C. The next day membranes were washed four times with 1xTBST, followed by incubation with secondary antibodies (Supplementary Table 3) for 1 hour at 37 °C. Membranes were washed another 4 times with 1xTBST, then once with 1xTBS before imaging on a Licor Odyssey Clx with Image Studio 3 software. Images were quantified with Image J.
For the neuron blot, 64–72 kD bands and 72–85 kD band were interpreted to represent CRMP2-B and CRMP2-A respectively with or without post-translational modifications. This is because CRMP2 is well-documented to undergo many post-translational modifications [2]; the multiple banding pattern was consistent across multiple replicate blots; the lysis buffer we used preserves post-translational modifications including phosphorylation; and other blots done in our lab of HEKs [12] and neurons (data not shown) that were extracted with RIPA (which does not maintain phosphorylation) detected CRMP2-B at 64kD and CRMP2A at 72kD.
Transcriptome analyses
Six frameshift and six control neuron samples were submitted to Novogene for paired-end RNA-sequencing. The twelve libraries were sequenced in one batch, and all passed Novogene’s post-sequencing quality control. We received on average 25.3 million reads per sample with a maximum of 25.8 and a minimum of 24.3 million. The 150 bp paired-end reads were aligned to human reference genome GRCH38 by Hisat2 version 2.1.0 [35]. Samtools 1.9 [36] produced corresponding BAM files and StringTie 2.0.3 [37] was used to assemble and estimate the abundance of transcripts based on GRCh38 human gene annotations [38]. Bioconductor package tximport computed raw counts by reversing the coverage formula used by StringTie with the input of read length [39]. The output then was imported to another Bioconductor package DESeq2 for differential gene expression analysis [40].
Schizophrenia GWAS Enrichment –
We downloaded all Ensembl transcripts and their coordinates (hg19) from UCSC. These were merged into genes with their start and end position corresponding to the furthest extending transcripts. We downloaded the results of the largest schizophrenia GWAS to date [41] from the Psychiatric Genomics Consortium at https://pgc.unc.edu/for-researchers/download-results/ (file name PGC3_SCZ_wave3_public.v2.tsv.gz) and filtered them to retain all SNPs with a p-value less than 1 × 10−7. This threshold which is slightly higher than genome wide significance (5 × 10−8) was used to maximize power while maintaining a strong enrichment for true signals. Those were then assembled into linkage disequilibrium groups that were at least 500 kb apart. We then identified all Ensembl genes that were within 500 kb from either direction of the linkage disequilibrium groups and considered all these genes as likely to drive the association.
RESULTS
CRISPR/Cas9 editing and characterization of iPSCs
In order to specifically knock out DPYSL2-B, we targeted its unique first exon (Fig. 1a). Using CRISPR/Cas9 we generated six iPSC clones with a 1 bp insertion in exon 1 (p.Iso12AsnC->CT) predicted to cause a frameshift and a premature termination codon in exon 2 (Fig. 1b). qRT-PCR analysis confirmed a 3-fold reduction of DPYSL2-B expression in the frameshift iPSC clones relative to controls, consistent with the occurrence of nonsense-mediated decay (Fig. 1c). As expected, CRMP2-B protein was also drastically reduced in frameshift clones (Fig. 1d, e). DPYSL2-A and DPYSL2-C are not expressed in iPSCs [42] and we saw no evidence of CRMP2-A at the expected sizes of 72 kD [12] in our Western blot. Edited iPSC clones were negative for off-target CRISPR editing screened by targeted Sanger sequencing at the single candidate site that passed our prioritization pipeline (Supplementary Fig. 2).
Fig. 1. Generation and characterization of DPYSL2-B frameshift iPSCs.
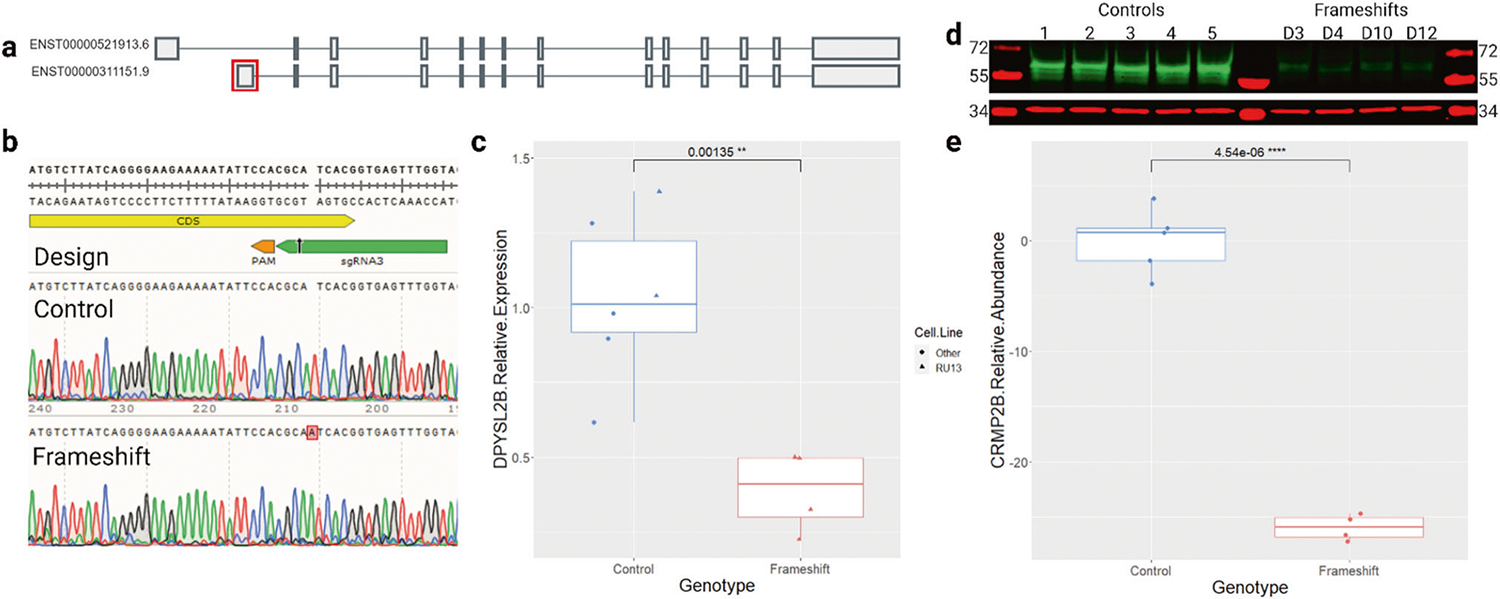
a DPYSL2 isoforms A (top) and B (bottom). The first exon of DPYSL2-B (boxed in red) was targeted to avoid impacting other transcripts. Image adapted from the GTEx portal. b Top panel shows sgRNA3 (green) and PAM (orange) used to create the frameshift mutation in the coding sequence (CDS, yellow) in exon 1. Bottom two panels show Sanger sequencing traces of DNA from control and frameshift iPSCs. c qRT-PCR results showing significantly reduced expression of DPYSL2-B in frameshift iPSC clones (n = 4) compared to control clones (n = 6 including 5 cell lines) (p = 0.001, t-test). Data were normalized to the geometric mean of GAPDH and β-actin loading controls, then to the average of the control clones. d Western blot comparing CRMP2-B (~64 kD, green) abundance in control (n = 5) and frameshift (n = 4) iPSC clones with GAPDH ( ~ 37 kD, red) as a loading control. e Quantification of (d) showing significantly reduced abundance of CRMP2-B in frameshift iPSCs compared to controls (p = 4.5 × 10−6, t-test). Data were normalized to GAPDH and then to the average of the control clones.
Generation and characterization of excitatory glutamatergic neurons
Next, we used an Ngn2-induced differentiation protocol [32, 43, 44] with six frameshift and six unedited control iPSC clones to generate excitatory glutamatergic neurons (Fig. 2a), a cell type consistently implicated in schizophrenia etiology [15, 45, 46]. RNA-sequencing of these twelve clones showed that the neurons expressed appropriate neural and glutamatergic markers, and that the frameshift mutation did not significantly alter expression of these markers, i.e., the identity of the cells (Supplementary Table 4).
Fig. 2. Generation and characterization of DPYSL2-B frameshift glutamatergic neurons.
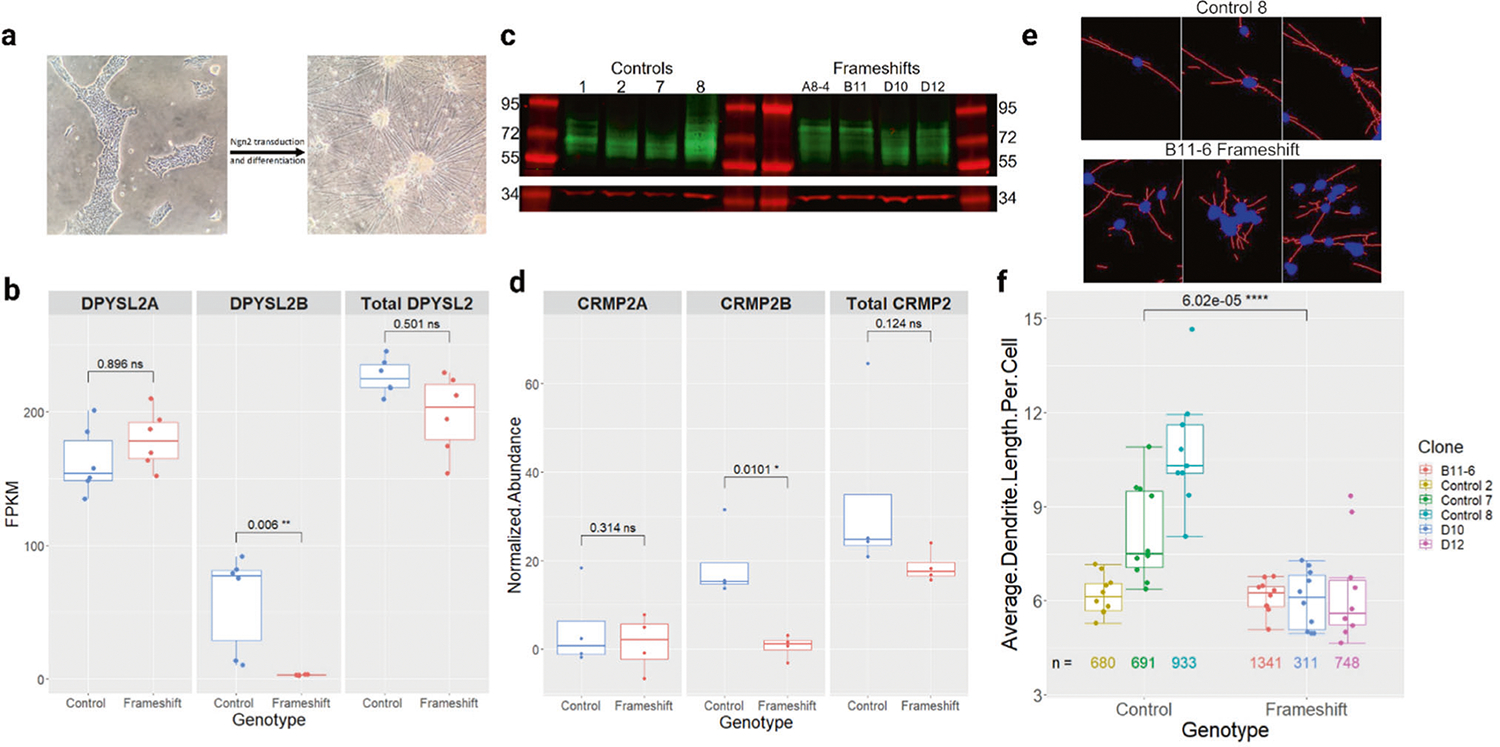
a Representative images of iPSCs before and after Ngn2-induced neuronal differentiation. Scale bars = 1 mm. b Quantification of RNA-seq data showing significantly reduced expression of DPYSL2-B in frameshift (n = 6) neuron clones compared to controls (n = 6) (padj = 0.006, Wald test with Benjamini and Hochberg adjustment). DPYSL2-A expression was not significantly changed, and all other DPYSL2 isoforms were not expressed. c Western blot comparing CRMP2-B (64–72 kD, green) and CRMP2A (72–80 kD, green) abundance in control (n = 5) and frameshift (n = 4) neuron clones with GAPDH ( ~ 37 kD, red) as a loading control. The multiple bands were interpreted to represent post-translational modifications. See the methods section for more detail on isoform distinction. d Quantification of (c). CRMP2-B was significantly reduced in the frameshift clones (p = 0.01, t-test), but CRMP2-A and the total amount of CRMP2 were not changed. Data were normalized to GAPDH. e Representative images of control and frameshift neurons after Neurphology J analysis with soma in blue and dendrites in red. Each image is cropped from the full-size pictures in Supplementary Fig. 1. Scale bars = 1 mm. f Quantification of the average dendrite length per cell in frameshift (n = 3) and control (n = 3) neuron clones. Each dot represents data from one image, with 9–10 images analyzed per clone. The numbers under each boxplot represent the total number of cells that were analyzed for that clone across all the images. The average length of dendrites per cell was significantly reduced in the frameshift clones (p = 6.02 × 10^−5, t-test).
Expression analysis of DPYSL/CRMP family and protein analysis of CRMP2.
Our RNA-seq data showed that as expected, DPYSL2-B expression was significantly reduced in the frameshift neurons relative to control neurons (Fig. 2b). DPYSL2-A expression was higher than DPYSL2-B across both groups and was slightly elevated in the frameshift neurons, but this change was not significant. At the gene level, DPYSL2 was significantly differentially expressed nominally but not after adjustment; we attribute this to the fact that most of the reads came from DPYSL2-A (Fig. 2b). The remaining DPYSL2 isoforms listed in Ensembl were either not expressed (all samples had FPKM < 1; ENST00000523027, ENST00000493789, ENST00000521983, ENST00000474808) or not found in the RNA-seq (ENST00000523690, ENST00000523093). The expression of DPYSL/CRMP –1, −3, −4, and –5 were not significantly changed at the gene level.
At the protein level, as anticipated CRMP2-B was significantly downregulated in the frameshift neurons compared to controls. Interestingly, this reduction was not as profound as it was in the iPSCs. CRMP2-A and the total abundance of CRMP2 did not change meaningfully between the two genotypes (Fig. 2c, d). We noted that the abundance of CRMP2-A was lower than CRMP2-B in all the neuron samples, even though the expression of the DPYSL2-A transcript was higher than the DPYSL2-B transcript. We did not attribute any bands to CRMP2-C because according to the RNA-seq data this isoform was not expressed.
Dendrite morphology analysis.
We next asked if knockout of endogenous DPYSL2-B is sufficient to cause aberrations in dendrite morphology. To investigate this, we differentiated three frameshift and three control clones on rat astrocytes to reduce soma clustering and performed immunocytochemistry with antibodies against the neuronal nucleus marker NEUN and the dendrite marker MAP2 (Fig. 2e, Supplementary Fig. 1). We observed wide variation in the average length of dendrites in the control neurons; but quantitative analysis demonstrated a significant reduction of up to 58% of the average length of dendrites in the frameshift neurons compared to the controls (Fig. 2f). When comparing the images from the control samples (n = 29) and the frameshift samples (n = 30), there was not a statically significant difference between the number of cells per image (cell density) across these two groups (t-test p = 0.95).
Transcriptome analysis.
To investigate how knockout of endogenous DPYSL2-B impacts the neuronal transcriptome, we analyzed our RNA-seq data with two pipelines: Gene set enrichment analysis (GSEA) [47] to identify expression changes of gene sets representing biological pathways; and DESeq2 [40] to identify differential expression of individual genes. Agnostic GSEA using the broad “Hallmarks” and “GO Cellular Component” annotation sets revealed strongly significant (padj ≤ 0.05) downregulation of gene sets involved in mTORC1 signaling, protein secretion, myc targeting, and particularly the immunoglobulin complex; and upregulation of gene sets involved in the epithelial mesenchymal transition. Hypothesis-based GSEA with custom gene sets identified significant downregulation of gene sets related to calcium biology (padj ≤ 0.05). When we used slightly more permissive parameters of padj < 0.1, we also saw significant downregulation of gene sets related to cholesterol biosynthesis. The leading-edge genes contributing to each result are listed in Table 1.
Table 1.
Gene Set Enrichment Analysis Results.
| Annotation Set1 (# gene sets) | Significant Gene Sets | NES | padj | Leading Edge Genes |
|---|---|---|---|---|
| Hallmark (50) | Epithelial Mesenchymal Transition | 1.56 | 0.05 | SERPINE1, JAG1, COL3A, IGFBP3, NOTCH1, SPARC, VEGFA, PLAUR |
| Angiogenesis | 1.47 | 0.08 | ||
| MTORC1 Signaling | −1.54 | 0.05 | MTHFD2, NOLC1, EIF2S1 | |
| Protein Secretion | −1.50 | 0.04 | ||
| MYC Targets | −1.46 | 0.04 | ||
| GO Cellular Component (491) | Immunoglobulin Complex | −2.14 | 0.00 | SYK, JCHAIN, IGLV9-49, IGLV5-52, IGKV1-37, IGKV3D-15, IGKV3-15, IGKV1D-37, IGKV1-12, IGKV1D-8, IGKV3D-7 |
| “Cholesterol” (46) | WP Cholesterol Biosynthesis Pathway | −1.60 | 0.07 | HMGCS1 * , IDI1 * , MVK, HMGCR * , FDFT1, FDPS * , CYP51A1 * |
| Reactome Regulation of Cholesterol Biosynthesis by SREBP_SREBF | −1.56 | 0.07 | ||
| GOBP Regulation of Cholesterol Biosynthetic Process | −1.50 | 0.09 | ||
| “Calcium” (118) | Reactome Role of LAT2 NTAL LAB on Calcium Mobilization | −1.78 | 0.05 | LYN, GAB2, SHC1, PIK3R1, IGKV3-15, MS4A2, IGKV1-12, SYK, SOS1, FCER1G, PIK3CA |
Annotation Sets represent groups of gene sets related to a biological category. Those within quotation marks are custom, hypothesis-driven sets created by searching MSigDB for gene sets with the quoted term. The Normalized Enrichment Score (NES) indicates the degree to which that gene set was enriched for differential expression in the frameshift neurons, with a negative score indicating downregulation and a positive score indicating upregulation. The Leading Edge Genes are those with the largest contribution to the NES.
upregulated genes in Ngn2-induced neurons treated with clozapine [33].
The gene expression (as FPKM) variances of edited and unedited cells were similar (p = 0. 53). DESeq2 analysis with differentiation batch (Supplementary Table 2) as a covariate identified 75 genes with padj < 0.05 (58 upregulated, 17 downregulated) and 138 genes with padj < 0.2, (101 upregulated, 37 downregulated) (Fig. 3a, Supplementary Tables 5 and 6). Of the 75 genes with padj < 0.05, we only found four directly connected in PubMed to DPYSL2/CRMP2: CHN1, IRS1, PLXNA2, and SSTR2. Analysis using the protein interaction database STRING [48] (https://string-db.org/) with the 138 genes at padj < 0.2 at medium confidence and no additional interactors found a significant enrichment for interactions between their protein products, supporting that these proteins are not a random group (Fig. 3c, Supplementary Table 6). To investigate any functional enrichments of these protein products with the whole genome as the statistical background we added a single 1st shell interactor (STRING selected PIK3R2). We observed enrichments in the KEGG diabetes mellitus type II pathway and the Monarch [49] intelligence and cognition phenotypes. Separate analysis of the 101 upregulated genes revealed a significant enrichment of protein products in the GO Cellular Component term Integral component of plasma membrane (GO:0005887, FDR = 0.043) (Supplementary Table 6).
Fig. 3. Transcriptomic analysis of DPYSL2-B frameshift neurons.
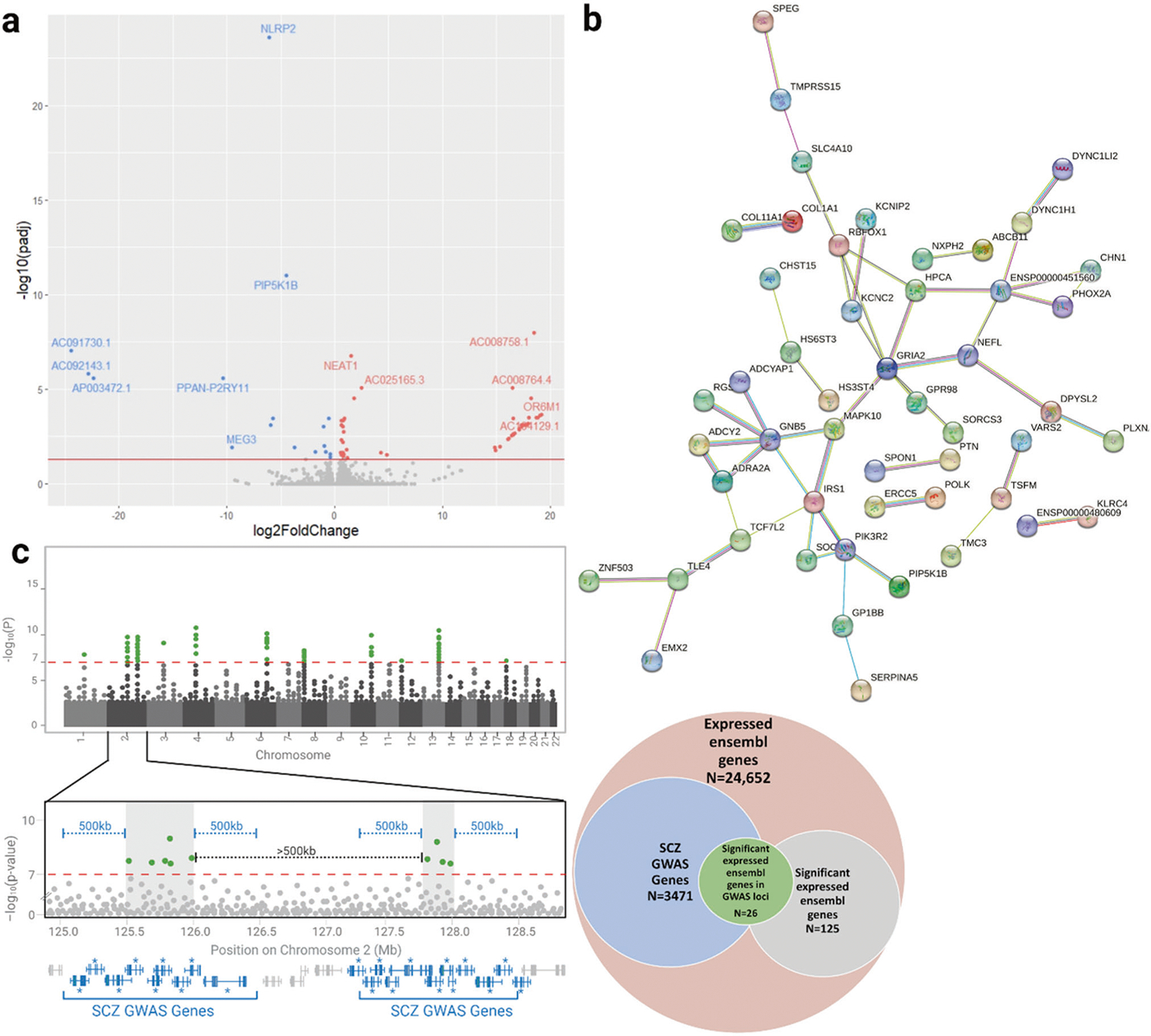
a Volcano plot depicting differential gene expression of the frameshift and control neurons based on DESeq2 analysis of RNA-seq data. The red line indicates genome-wide significance of padj < 0.05. Genes significantly upregulated in the frameshift neurons are in red and downregulated genes are in blue. b Schematic of theoretical GWAS data demonstrating the workflow used to identify genes in schizophrenia risk loci (“SCZ GWAS genes”) from the largest schizophrenia GWAS to date [42]. SNPs reaching significance of 1 × 10−7 (green) were clustered into linkage disequilibrium blocks (gray shaded boxes) at least 500 kb apart. Any genes within 500 kb of these blocks (blue) were considered SCZ GWAS genes. Of the 24,652 genes expressed in the Ngn2-induced neurons, 3471 were GWAS genes. Of the 125 genes that were present in the UCSC gene list and were significant with padj < 0.2 via DESeq2, 26 were GWAS genes. This represents a significant enrichment of our differentially expressed genes in schizophrenia risk loci (p = 0.03, chi squared test). c STRING network demonstrating significant enrichment of protein-protein interactions amongst DPYSL2 and the 132 differentially expressed genes with padj < 0.2. Of the 132 genes, 96 had protein products and 37 of these proteins interacted, representing a 1.77 x enrichment (p = 0.012, see methods for details on statistical analysis).
Review of the 75 differentially expressed genes highlighted DPYSL2’s role in the etiology of mental illness. The functional categories into which many of these genes fall are highly relevant to neuronal function (Table 2). A PubMed search of the 75 genes with the MeSH term “Mental Disorders” found that 42/75 are genes or read-through transcripts of genes already linked to neurological and/or psychiatric disease including NLRP2, MEG3, NEAT1, and TUBB3 (Supplementary Table 6). A parallel analysis demonstrated that the 138 genes at padj <0.2 were enriched for localization within 500 kb of schizophrenia GWAS loci. Specifically, there were ~50% more than expected (chi-square test 1.48x enrichment, p = 0.03; Fig. 3c, Supplementary Table 7). Finally, we used the Enrichr database (https://maayanlab.cloud/Enrichr/) to compare the transcriptome profile of the 75 genes at padj < 0.05 to perturbagen libraries from CMAP, as we had done in our work on HEK293 cells [12], and the Phenotype-Genotype Integrator (PhenGenI). CMAP analysis showed that the 58 upregulated genes were significantly enriched for overlap with genes disrupted by the mTOR inhibitors sirolimus-1001 and LY-294002–6956 (p = 0.003), the atypical antipsychotic sulpiride-1467 (p = 0.034), and the calcium channel blocker tetrandrine-7178 (p = 0.034). Comparison of the 17 downregulated genes with the PhenGenI database revealed overlaps with the atypical antipsychotic olanzapine (p = 0.01) and the mood stabilizer lithium (p = 0.054). These results show that knockout of DPYSL2-B disrupts genes and pathways involved in psychiatric disease.
Table 2.
Functional Categories of DESeq2 significant genes (padj < 0.05).
| Category | Number of Genes | Genes |
|---|---|---|
| Neurotransmission / Neurodevelopment | 14 | GRIA2, PIP5K1B, PHOX2A, EMX2, MEG3, BHCE, CHN1, PRDM8, TMPRSS15, OR6M1, C093525.2 (TBC1D24 and ATP6V0C), AC008758.1 (ZNF709 and ZNF564), Z84488.2 (CALHM6), AC025165 (AVIL) |
| Microtubule/ Cytoskeletal dynamics / Cell Migration | 10 | NEAT1, RBFOX1, PIP5K1B, SPAG6, FLG, SSTR2, PLXNA2, DYNC1H1, TLE4, AC092143.1 (TUBB3) |
| Calcium Signaling/ Cell Adhesion | 8 | ADGRV1, FAT1, SSTR2, PIP5K1B, CLEC4C, RBFOX1, Z84488.2 (CALHM6), AC244517.4 (PCDHB16) |
| Immune response / Inflammation | 9 | NLRP2, HS3ST4, KLRC4, CLEC4C, KLRC4-KLRK1, TXNIP, AL137247.2 (N4BP2L1), Z84488.2 (CALHM6), PPAN-P2RY11 |
| Protein Transport/ Vesicle trafficking | 4 | GOLGA8A, PIP5K1B, ABCB11, AC093525.2 (TBC1D24) |
DISCUSSION
In previous work, we found that the B isoform of DPYSL2 is associated with schizophrenia risk [17] and demonstrated its ability to regulate the cytoskeleton in HEKs [12]. In the present study, we follow up on these results by using CRISPR/Cas9 to create and characterize the first iPSC and neuronal models of endogenous DPYSL2-B knockout.
Knockout of DPYSL2-B confirms link between mTOR and schizophrenia
The mTOR kinase is ubiquitously expressed and regulates proliferation and survival by facilitating protein synthesis, lipid metabolism and mitochondrial biogenesis, and inhibiting autophagy [50–52]. It is a downstream effector of the PI3K/PTEN/Akt signaling cascade which is activated by growth hormones including insulin [52]. In neurons, mTOR influences polarization and axon elongation by regulating the translation of microtubule-associating proteins like Tau and CRMP2 via the mTORC1 complex [13, 53].
We had previously shown in HEK293 cells that DPYSL2-B is regulated by mTOR [12, 17] and reduction of its expression produces complementary signatures to mTOR inhibitors. Here we recapitulate these complementary CMAP signatures in neuronal cells. Additionally, our GSEA showed that DPYSL2-B knockout results in downregulation of genes related to mTORC1 signaling. As a further link to mTOR, we also observed upregulation of the gene IRS1, which encodes an insulin receptor in the PIK3/Akt pathway. These data suggest that DPYSL2-B levels can influence mTOR activity in neurons, and its reduction results in transcriptome-wide downregulation of mTOR signaling genes.
Our findings are supported by numerous other studies which have observed disruption of mTOR signaling in schizophrenia. Our previous analysis of the schizophrenia-associated 13DNR in HEK293 cells found reduced binding of DPYSL2-B RNA to mTOR effector proteins 4EBP, eIF4E, and ELALV4. We also showed that HEK293 cells with the 13DNR were more sensitive to treatment with Rapamycin than 11 DNR controls [12]. Three other recent studies of postmortem schizophrenia brains have observed disruption of various components of the mTOR pathway, including: reduced rpS6 protein abundance and phosphorylation [54, 55]; reduced phosphorylation of Akt and mTOR [56]; increased mTOR abundance [55]; and increased binding of mTOR to mTORC1/mTORC2 complex proteins Rictor and Raptor [56]. Additionally, Akt activation is altered by antipsychotics in the rat brain [54]. Besides schizophrenia, aberrant mTOR signaling has been implicated in various neurodegenerative diseases, likely due to reduced autophagy of toxic protein aggregates [52]. Taken together, these data confirm the role of the DPYSL2-B isoform as a link between mTOR signaling and diseases of the brain including schizophrenia.
Knockout of DPYSL2-B disrupts dendritic morphology and cytoskeletal dynamics
It has long been known that CRMP2 regulates dendrite morphology and axonogensis. Shortening and/or aberrant morphology of neurites has been observed in DPYSL2 knockout mouse models that disrupt all DPYSL2 isoforms [10, 18, 57, 58], as well as in rat hippocampal neurons transfected with DPYSL2-B truncation mutant transgenes [59]. In previous work we demonstrated that a schizophrenia-associated DNR in DPYSL2-B resulted in reduced CRMP2-B protein and marked reduction in the length of cellular projections in HEK293 cells [12]. Here, we further confirm that the DPYSL2-B isoform regulates the cytoskeleton by observing significant reduction of the average length of MAP2+ dendrites in DPYSL2-B knockout neurons (Fig. 2e, f). This morphological difference was supported by our transcriptomic analyses which found differential expression of genes that compose, regulate, and utilize the cytoskeleton (Table 2). AC092143 is a read-through transcript of the neuron-specific tubulin TUBB3, disruptions in which cause “tublinopathies” that manifest with complex neurological and behavioral symptoms [60]. SPAG6 [61] and FLG [62] encode microtubule and actin-associating proteins, respectively, and both have been linked to Alzheimer’s [62, 63]. Several genes regulate the kinase activity of GSK3-β upon CRMP2, including NEAT1 [64–70], TLE4 [2, 71], PLXNA2 [72], PIP5K1B [2, 71], and SSTR2 [73]. RBFOX1 (a neuron-specific splicing factor) regulates many neuronal development genes including those related to the cytoskeleton [74, 75]. As for genes that utilize the cytoskeleton, we observed differential expression of DYNC1H1 (encoding the motor protein dynein, to which CRMP2 is known to directly bind [76]); the Golgi protein GOLGA8A which is suggested to be biomarker for autism spectrum disorder [77]; and the ATP-powered transporter ABCB11. The differential expression of these molecular transport-associated genes was corroborated by GSEA, which found downregulation of genes related to protein secretion. Taken together, these results confirm that DPYSL2-B is necessary for the development of the cytoskeleton, disruption of which leads to aberrations in neuronal structure and function that are relevant to psychiatric disease.
DPYSL2-B regulates cholesterol biosynthesis in neurons
Cholesterol is an important molecule for neuronal function. As a major component of myelin, it is necessary for saltatory conduction of electrical impulses along axons. In the plasma membrane, proper cholesterol content allows for the fluidity necessary for neurotransmission, and cholesterol-enriched lipid rafts facilitate signaling cascades including that of Semaphorin 3A [78]. Cholesterol cannot cross the blood brain barrier and is thus synthesized locally in astrocytes and neurons. Importantly, we have previously shown that application of the antipsychotic clozapine on human iPSC-derived human neurons strongly upregulates cholesterol biosynthesis genes [32].
Our GSEA revealed that knockout of DPYSL2-B downregulated the expression of genes involved in cholesterol biosynthesis. To our knowledge this is the first association of DPYSL2 with cholesterol metabolism in neurons. This finding is supported by a recent study by Chang et al. which demonstrated that CRMP2 can regulate lipid metabolism through multiple mechanisms during adipocyte differentiation in mice [79]. We also observed upregulation of the Semaphorin 3A receptor plexin A2 (PLXNA2), which along with other CRMPs is known to co-localize with lipid rafts [78, 80]. This suggests that downregulation of cholesterol synthesis could disrupt lipid structures that mediate CRMP2-B activity via Semaphorin 3A signaling.
Dysregulation of lipid homeostasis has been implicated in the etiology of numerous brain disorders including schizophrenia, Alzheimer’s, Parkinson’s, and Huntington’s disease [81–83]. Additionally, multiple studies have reported that antipsychotics modulate lipid metabolism and suggested that this contributes to their therapeutic effects [81, 84]. Five out of the seven downregulated leading edge cholesterol biosynthesis genes (starred in Table 1) were among those shown to increase with clozapine treatment in our previous work [32]. The complementary overlap of these genes with clozapine and olanzapine (from our independent CMAP analysis) confirms that knockout of DPYSL2-B captures schizophrenia disease signal, and further cements the link between schizophrenia, lipid homeostasis, and DPYSL2-B/CRMP2-B function.
Clarifying the function of DPYSL2-B vs other DPYSL2 isoforms
Currently there are eight known DPYSL2 transcripts. We observed expression only of DPYSL2-A and DPYSL2-B in our neurons (Fig. 2b–d), which is consistent with other Ngn2 datasets we have generated [32]. Little is known about DPYSL2-A besides that it mainly localizes to the soma and axon, whereas CRMP2-B localizes to soma, axons, and especially dendrites [29, 85]. One study found that these two isoforms have opposing effects on axon elongation and suggested they work in tandem [28]. We observed that knockout of DPYSL2-B did not significantly change expression of DPYSL2-A or CRMP2-A. This suggests that if CRMP2-A does indeed have an opposing effect to CRMP2-B, it does not appear to be mediated by compensatory changes in DPYSL2-A RNA or protein expression. Regarding other functional overlaps, CRMP2-A has been implicated as a molecular brake on carcinogenesis in prostate cancer, lung cancer, and acute myeloid leukemia [86–88] partially due to its suppression of the epithelial mesenchymal transition [86]. Our GSEA revealed that knockout of DPYSL2-B resulted in upregulation of genes related to the epithelial-mesenchymal transition, highlighting a novel functional overlap between these two isoforms. Taken together, our findings confirm that the DPYSL2-B transcript is necessary for proper cytoskeletal and dendritic development and therefore plays an important role in the etiology of mental disorders.
Limitations and future directions
Our experiments were performed on a single iPSC line of an African male. Using only one line reduces noise from genome variation and improves power, however is also raises questions on whether the conclusions are generalizable. While it is possible that the impact of DPYSL2-B disruption could vary between different genetic backgrounds, the concordance of our results with our previous studies in HEK293 cells [12, 17] suggests that at least the results observed in both studies are robust and not genotype-specific.
Although we observed significant reduction of DPYSL2-B RNA and protein in our Ngn2-induced neurons, our Western blot suggested that CRMP2-B was not completely ablated. The presence of residual CRMP2-B protein suggests that there is some bypassing mechanism of the frameshift mutation, perhaps due to alternative start site use, though this has not been confirmed. According to the genome aggregation database (https://gnomad.broadinstitute.org/) human nonsense variants in DPYSL2 are exceedingly rare, are never observed as homozygotes, and never occur in the B isoform prior to the 8th exon. Consequently, DPYSL2 is predicted to have a probability of loss of function intolerance (pLI) score of 0.99 out of 1 suggesting it is an essential gene in vitro. It should be noted that homozygous DPYSL2 knockout mouse models have been successfully generated [10, 18, 57, 58] yet one should consider the different organism and that there may be more buffering systems in vivo that could compensate for disruption of a critical gene.
DPYSL2 is proven to be an important gene for brain development, function and for mental health. As such, more study is warranted to further distinguish the functional properties of its individual isoforms, their exact role in the nervous system and the consequences of their modifications. Our findings are one more small but significant step towards understanding the crucial role of this gene and its individual transcripts in health and disease.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr. Debamitra Das and Dr. Linglei Jiang for their advice on neuronal differentiation and neuronal protein extraction, respectively. Thank you to Dr. Arianna Anzmann, Dr. Sarah Poll and Olivia Sniezek for their guidance on Western blotting, and Cassandra Obie for tissue culture support. Thank you to Lindsay Young for assisting with off-target analysis and Marah Wahbeh for troubleshooting support. We would also like to thank Dr. Vasiliki Machairaki and her lab for their help with immunocytochemistry and fluorescence microscopy. This project was funded by National Institute of Mental Health grants P50 MH094268, R01 MH113215 and RF1 MH122936 to DA. Figures were created with BioRender.com.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41380-023-02186-w.
Reprints and permission information is available at http://www.nature.com/reprints
REFERENCES
- 1.Quach TT, Honnorat J, Kolattukudy PE, Khanna R, Duchemin AM. CRMPs: Critical molecules for neurite morphogenesis and neuropsychiatric diseases. Mol Psychiatry. 2015;20:1037–45. [DOI] [PubMed] [Google Scholar]
- 2.Moutal A, White KA, Chefdeville A, Laufmann RN, Vitiello PF, Feinstein D, et al. Dysregulation of CRMP2 post-translational modifications drive its pathological functions. Mol Neurobiol. 2019;56:6736–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature. 1995;376:509–14. [DOI] [PubMed] [Google Scholar]
- 4.West M, Sutherland DE, Matas AJ. Kidney transplant recipients who die with functioning grafts: serum creatinine level and cause of death. Transplantation. 1996;62:1029–30. [DOI] [PubMed] [Google Scholar]
- 5.Akinaga S, Harada S, Takahashi M, Kaneko A, Kolattukudy P, Goshima Y, et al. Loss of CRMP1 and CRMP2 results in migration defects of Purkinje cells in the X lobule of the mouse cerebellum. Brain Res. 2022;1783:147846. [DOI] [PubMed] [Google Scholar]
- 6.Yamazaki Y, Moizumi M, Nagai J, Hatashita Y, Cai T, Kolattukudy P, et al. Requirement of CRMP2 Phosphorylation in Neuronal Migration of Developing Mouse Cerebral Cortex and Hippocampus and Redundant Roles of CRMP1 and CRMP4. Cereb Cortex. 2022;32:520–7. [DOI] [PubMed] [Google Scholar]
- 7.Chi XX, Schmutzler BS, Brittain JM, Wang Y, Hingtgen CM, Nicol GD, et al. Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J Cell Sci. 2009;122:4351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem. 2009;284:31375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brustovetsky T, Khanna R, Brustovetsky N. CRMP2 is involved in regulation of mitochondrial morphology and motility in neurons. Cells. 2021;10:2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziak J, Weissova R, Jerabkova K, Janikova M, Maimon R, Petrasek T, et al. CRMP2 mediates Sema3F-dependent axon pruning and dendritic spine remodeling. EMBO Rep. 2020;21:e48512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ip JP, Fu AK, Ip NY. CRMP2: functional roles in neural development and therapeutic potential in neurological diseases. Neuroscientist. 2014;20:589–98. [DOI] [PubMed] [Google Scholar]
- 12.Pham X, Song G, Lao S, Goff L, Zhu H, Valle D, et al. The DPYSL2 gene connects mTOR and schizophrenia. Transl Psychiatry. 2016;6:e933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morita T, Sobue K. Specification of neuronal polarity regulated by local translation of CRMP2 and Tau via the mTOR-p70S6K pathway. J Biol Chem. 2009;284:27734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cockman E, Anderson P, Ivanov P. TOP mRNPs: Molecular mechanisms and principles of regulation. Biomolecules. 2020;10:969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, McGrath JA, et al. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am J Hum Genet. 2005;77:918–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey EF, et al. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol Psychiatry. 2000;5:142–9. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Pham X, Zhang L, Chen PL, Burzynski G, McGaughey DM, et al. Functional variants in DPYSL2 sequence increase risk of schizophrenia and suggest a link to mTOR signaling. G3 (Bethesda). 2014;5:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura H, Yamashita N, Kimura A, Kimura Y, Hirano H, Makihara H, et al. Comprehensive behavioral study and proteomic analyses of CRMP2-deficient mice. Genes Cells. 2016;21:1059–79. [DOI] [PubMed] [Google Scholar]
- 19.Nomoto M, Konopaske GT, Yamashita N, Aoki R, Jitsuki-Takahashi A, Nakamura H, et al. Clinical evidence that a dysregulated master neural network modulator may aid in diagnosing schizophrenia. Proc Natl Acad Sci USA. 2021;118:e2100032118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole AR, Noble W, van Aalten L, Plattner F, Meimaridou R, Hogan D, et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J Neurochem. 2007;103:1132–44. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Yang J, Schneider JA, De Jager PL, Bennett DA, Zhang HY. Genome-wide interaction analysis of pathological hallmarks in Alzheimer’s disease. Neurobiol Aging. 2020;93:61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshida H, Watanabe A, Ihara Y. Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer’s disease. J Biol Chem. 1998;273:9761–8. [DOI] [PubMed] [Google Scholar]
- 23.Ozgen HM, Staal WG, Barber JC, de Jonge MV, Eleveld MJ, Beemer FA, et al. A novel 6.14 Mb duplication of chromosome 8p21 in a patient with autism and self mutilation. J Autism Dev Disord. 2009;39:322–9. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki H, Li S, Tokutomi T, Takeuchi C, Takahashi M, Yamada M, et al. De novo non-synonymous DPYSL2 (CRMP2) variants in two patients with intellectual disabilities and documentation of functional relevance through zebrafish rescue and cellular transfection experiments. Hum Mol Genet. 2022;31:4173–82. [DOI] [PubMed] [Google Scholar]
- 25.Weitzdoerfer R, Fountoulakis M, Lubec G. Aberrant expression of dihydropyrimidinase related proteins-2,−3 and −4 in fetal Down syndrome brain. J Neural Transm Suppl. 2001;61:95–107. [DOI] [PubMed] [Google Scholar]
- 26.Khanna R, Moutal A, Perez-Miller S, Chefdeville A, Boinon L, Patek M. Druggability of CRMP2 for Neurodegenerative Diseases. ACS Chem Neurosci. 2020;11:2492–505. [DOI] [PubMed] [Google Scholar]
- 27.Petratos S, Ozturk E, Azari MF, Kenny R, Lee JY, Magee KA, et al. Limiting multiple sclerosis related axonopathy by blocking Nogo receptor and CRMP-2 phosphorylation. Brain. 2012;135:1794–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuasa-Kawada J, Suzuki R, Kano F, Ohkawara T, Murata M, Noda M. Axonal morphogenesis controlled by antagonistic roles of two CRMP subtypes in microtubule organization. Eur J Neurosci. 2003;17:2329–43. [DOI] [PubMed] [Google Scholar]
- 29.Quinn CC, Chen E, Kinjo TG, Kelly G, Bell AW, Elliott RC, et al. TUC-4b, a novel TUC family variant, regulates neurite outgrowth and associates with vesicles in the growth cone. J Neurosci. 2003;23:2815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adikusuma F, Pfitzner C, Thomas PQ. Versatile single-step-assembly CRISPR/Cas9 vectors for dual gRNA expression. PLoS One. 2017;12:e0187236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das D, Peng X, Lam AN, Bader JS, Avramopoulos D. Transcriptome analysis of human induced excitatory neurons supports a strong effect of clozapine on cholesterol biosynthesis. Schizophr Res. 2021;228:324–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Page SC, Sripathy SR, Farinelli F, Ye Z, Wang Y, Hiler DJ, et al. Electrophysiological measures from human iPSC-derived neurons are associated with schizophrenia clinical status and predict individual cognitive performance. Proc Natl Acad Sci USA. 2022;119:e2109395119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho SY, Chao CY, Huang HL, Chiu TW, Charoenkwan P, Hwang E. NeurphologyJ: an automatic neuronal morphology quantification method and its application in pharmacological discovery. BMC Bioinforma. 2011;12:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11:1650–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trubetskoy V, Pardinas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604:502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobrindt K, Zhang H, Das D, Abdollahi S, Prorok T, Ghosh S, et al. Publicly Available hiPSC Lines with Extreme Polygenic Risk Scores for Modeling Schizophrenia. Complex Psychiatry. 2021;6:68–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das D, Sonthalia S, Stein-O’Brien G, Wahbeh M, Feuer K, Colantuoni C, et al. Insights for disease modeling from single cell transcriptomics of iPSC-derived neurons and astrocytes across differentiation time and co-culture. bioRxiv 2022:2022.2006.2015.495952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu W, MacDonald ML, Elswick DE, Sweet RA. The glutamate hypothesis of schizophrenia: evidence from human brain tissue studies. Ann N. Y Acad Sci. 2015;1338:38–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014;511: 421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49:D605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mungall CJ, McMurry JA, Kohler S, Balhoff JP, Borromeo C, Brush M, et al. The Monarch Initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res. 2017;45:D712–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ryskalin L, Limanaqi F, Frati A, Busceti CL, Fornai F. mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders. Int J Mol Sci. 2018;19:2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Na EJ, Nam HY, Park J, Chung MA, Woo HA, Kim HJ. PI3K-mTOR-S6K Signaling Mediates Neuronal Viability via Collapsin Response Mediator Protein-2 Expression. Front Mol Neurosci. 2017;10:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ibarra-Lecue I, Diez-Alarcia R, Morentin B, Meana JJ, Callado LF, Uriguen L. Ribosomal Protein S6 Hypofunction in Postmortem Human Brain Links mTORC1-Dependent Signaling and Schizophrenia. Front Pharm. 2020;11:344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Izumi R, Hino M, Nagaoka A, Shishido R, Kakita A, Hoshino M, et al. Dysregulation of DPYSL2 expression by mTOR signaling in schizophrenia: Multi-level study of postmortem brain. Neurosci Res. 2022;175:73–81. [DOI] [PubMed] [Google Scholar]
- 56.Chadha R, Meador-Woodruff JH. Downregulated AKT-mTOR signaling pathway proteins in dorsolateral prefrontal cortex in Schizophrenia. Neuropsychopharmacology. 2020;45:1059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang H, Kang E, Wang Y, Yang C, Yu H, Wang Q, et al. Brain-specific Crmp2 deletion leads to neuronal development deficits and behavioural impairments in mice. Nat Commun. 2016;7:11773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Makihara H, Nakai S, Ohkubo W, Yamashita N, Nakamura F, Kiyonari H, et al. CRMP1 and CRMP2 have synergistic but distinct roles in dendritic development. Genes Cells. 2016;21:994–1005. [DOI] [PubMed] [Google Scholar]
- 59.Inagaki N, Chihara K, Arimura N, Menager C, Kawano Y, Matsuo N, et al. CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci. 2001;4:781–2. [DOI] [PubMed] [Google Scholar]
- 60.Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140:74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith EF, Lefebvre PA. The role of central apparatus components in flagellar motility and microtubule assembly. Cell Motil Cytoskeleton. 1997;38:1–8. [DOI] [PubMed] [Google Scholar]
- 62.Xiong W, Cai J, Li R, Wen C, Tan H. On Behalf Of The Alzheimer’s Disease Neuroimaging Initiative Adni D. Rare Variant Analysis and Molecular Dynamics Simulation in Alzheimer’s Disease Identifies Exonic Variants in FLG. Genes (Basel). 2022;13:838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Altuna M, Urdanoz-Casado A, Sanchez-Ruiz de Gordoa J, Zelaya MV, Labarga A, Lepesant JMJ, et al. DNA methylation signature of human hippocampus in Alzheimer’s disease is linked to neurogenesis. Clin Epigenetics. 2019;11:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bond CS, Fox AH. Paraspeckles: nuclear bodies built on long noncoding RNA. J Cell Biol. 2009;186:637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sayad A, Omrani MD, Fallah H, Taheri M, Ghafouri-Fard S. Aberrant Expression of Long Non-coding RNAs in Peripheral Blood of Autistic Patients. J Mol Neurosci. 2019;67:276–81. [DOI] [PubMed] [Google Scholar]
- 66.Safari MR, Komaki A, Arsang-Jang S, Taheri M, Ghafouri-Fard S. Expression Pattern of Long Non-coding RNAs in Schizophrenic Patients. Cell Mol Neurobiol. 2019;39:211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sunwoo JS, Lee ST, Im W, Lee M, Byun JI, Jung KH, et al. Altered Expression of the Long Noncoding RNA NEAT1 in Huntington’s Disease. Mol Neurobiol. 2017;54:1577–86. [DOI] [PubMed] [Google Scholar]
- 68.Spreafico M, Grillo B, Rusconi F, Battaglioli E, Venturin M. Multiple Layers of CDK5R1 Regulation in Alzheimer’s Disease Implicate Long Non-Coding RNAs. Int J Mol Sci. 2018;19:2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.He L, Chen Z, Wang J, Feng H. Expression relationship and significance of NEAT1 and miR-27a-3p in serum and cerebrospinal fluid of patients with Alzheimer’s disease. BMC Neurol. 2022;22:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao Y, Wang Z, Mao Y, Li B, Zhu Y, Zhang S, et al. NEAT1 regulates microtubule stabilization via FZD3/GSK3beta/P-tau pathway in SH-SY5Y cells and APP/PS1 mice. Aging (Albany NY). 2020;12:23233–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramakrishnan AB, Sinha A, Fan VB, Cadigan KM. The Wnt Transcriptional Switch: TLE Removal or Inactivation? Bioessays. 2018;40:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmidt EF, Shim SO, Strittmatter SM. Release of MICAL autoinhibition by semaphorin-plexin signaling promotes interaction with collapsin response mediator protein. J Neurosci. 2008;28:2287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang S, Bao Z, Liang QM, Long JW, Xiao ZS, Jiang ZJ, et al. Octreotide stimulates somatostatin receptor-induced apoptosis of SW480 colon cancer cells by activation of glycogen synthase kinase-3beta, A Wnt/beta-catenin pathway modulator. Hepatogastroenterology. 2013;60:1639–46. [PubMed] [Google Scholar]
- 74.Fogel BL, Wexler E, Wahnich A, Friedrich T, Vijayendran C, Gao F, et al. RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum Mol Genet. 2012;21:4171–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee JA, Damianov A, Lin CH, Fontes M, Parikshak NN, Anderson ES, et al. Cytoplasmic Rbfox1 Regulates the Expression of Synaptic and Autism-Related Genes. Neuron. 2016;89:113–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arimura N, Hattori A, Kimura T, Nakamuta S, Funahashi Y, Hirotsune S, et al. CRMP-2 directly binds to cytoplasmic dynein and interferes with its activity. J Neurochem. 2009;111:380–90. [DOI] [PubMed] [Google Scholar]
- 77.Wang Y, Cheng C, Zhang Z, Wang J, Wang Y, Li X, et al. Blood-based dynamic genomic signature for obsessive-compulsive disorder. Am J Med Genet B Neuropsychiatr Genet. 2018;177:709–16. [DOI] [PubMed] [Google Scholar]
- 78.Rosslenbroich V, Dai L, Franken S, Gehrke M, Junghans U, Gieselmann V, et al. Subcellular localization of collapsin response mediator proteins to lipid rafts. Biochem Biophys Res Commun. 2003;305:392–9. [DOI] [PubMed] [Google Scholar]
- 79.Chang YH, Tsai JN, Chang SW, Hsu WT, Yang CP, Hsiao CW, et al. Regulation of Adipogenesis and Lipid Deposits by Collapsin Response Mediator Protein 2. Int J Mol Sci. 2020;21:2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Whitehead SN, Gangaraju S, Slinn J, Hou ST. Transient and bilateral increase in Neuropilin-1, Fer kinase and collapsin response mediator proteins within membrane rafts following unilateral occlusion of the middle cerebral artery in mouse. Brain Res. 2010;1344:209–16. [DOI] [PubMed] [Google Scholar]
- 81.Polymeropoulos MH, Licamele L, Volpi S, Mack K, Mitkus SN, Carstea ED, et al. Common effect of antipsychotics on the biosynthesis and regulation of fatty acids and cholesterol supports a key role of lipid homeostasis in schizophrenia. Schizophr Res. 2009;108:134–42. [DOI] [PubMed] [Google Scholar]
- 82.Zhang J, Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6:254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takahashi N, Sakurai T, Davis KL, Buxbaum JD. Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog Neurobiol. 2011;93:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ferno J, Raeder MB, Vik-Mo AO, Skrede S, Glambek M, Tronstad KJ, et al. Antipsychotic drugs activate SREBP-regulated expression of lipid biosynthetic genes in cultured human glioma cells: a novel mechanism of action? Pharmacogenomics J. 2005;5:298–304. [DOI] [PubMed] [Google Scholar]
- 85.Balastik M, Zhou XZ, Alberich-Jorda M, Weissova R, Ziak J, Pazyra-Murphy MF, et al. Prolyl Isomerase Pin1 Regulates Axon Guidance by Stabilizing CRMP2A Selectively in Distal Axons. Cell Rep. 2015;13:812–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boukouris AE, Zhang Y, Saleme B, Kinnaird A, Zhao YY, Liu Y, et al. A reversible metabolic stress-sensitive regulation of CRMP2A orchestrates EMT/stemness and increases metastatic potential in cancer. Cell Rep. 2022;38:110511. [DOI] [PubMed] [Google Scholar]
- 87.Noura M, Morita K, Kiyose H, Okuno Y, Matsuo H, Koyama A, et al. Albendazole induces the terminal differentiation of acute myeloid leukaemia cells to monocytes by stimulating the Kruppel-like factor 4-dihydropyrimidinase-like 2A (KLF4-DPYSL2A) axis. Br J Haematol. 2021;194:598–603. [DOI] [PubMed] [Google Scholar]
- 88.Noura M, Morita K, Kiyose H, Matsuo H, Nishinaka-Arai Y, Kurokawa M, et al. Pivotal role of DPYSL2A in KLF4-mediated monocytic differentiation of acute myeloid leukemia cells. Sci Rep. 2020;10:20245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.