

Abstract
Palladium-catalyzed C(sp3)–H functionalization presents an efficient strategy to construct a variety of carbon–carbon bonds. However, application of this approach towards the preparation of five-membered benzo-fused carbocycles via the most simplifying C–H activation logic has not been realized. In this Article, we report a palladium-catalyzed annulation reaction between gem-dimethyl containing amides and 1-bromo-2-iodoarenes that effectively constructs two Calkyl–Caryl bonds and provides access to a variety of five-membered benzo-fused compounds. In this transformation, the dihaloarene is stitched to the gem-dimethyl moiety via two sequential β-C(sp3)–H arylations utilizing the differential reactivity of the 1,2-difunctionalized electrophile. This annulation reaction is enabled by a dual ligand system comprising of an N-acyl glycine and a pyridine-3-sulfonic acid that synergistically promotes the palladium stitching and provides the bicyclic products. This method displays a broad substrate scope and shows excellent amide compatibility. We also demonstrate the synthetic potential of this annulation by synthesizing echinolactone D.
Graphical Abstract
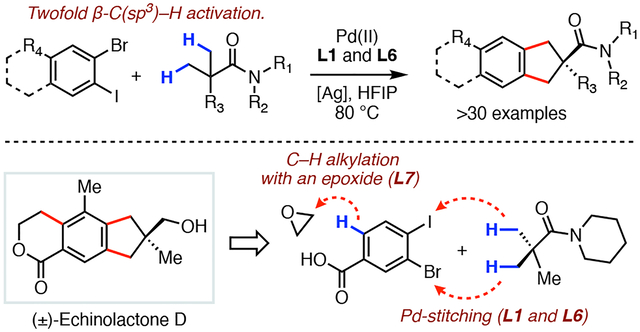
INTRODUCTION.
Organic synthesis relies heavily on transformations that can efficiently construct all-carbon ring structures. A particularly important subset of these scaffolds are benzo-fused carbocycles, which can be routinely found in many biologically active natural products as well as in pharmaceuticals.1 In the context of C–H functionalization, preparation of these scaffolds has extensively relied on intramolecular cyclization strategies (Scheme 1A). For instance, the Baudoin group developed a Pd(0/II)-catalyzed annulative C(sp3)–H arylation of both aryl bromides or aryl chlorides utilizing phosphine-based ligands.2 The use of chiral ligand scaffolds have later rendered this reaction protocol asymmetric.3 In 2010, Chen and co-workers disclosed an intramolecular coupling between aryl iodides and methylene C–H bonds that used the strongly directing 8-aminoquinoline auxiliary.4 The recent development of L,X-transient directing groups has also allowed for this type of reactivity to be extended to intramolecular cyclizations of aldehydes and methyl ketones.5 Furthermore, in 2021 our group reported a Pd(II/IV)-catalyzed cross-dehydrogenative-coupling (CDC) between β-(Csp3)–H bonds of carboxylic acids and pendant aryl C–H bonds by means of six- or seven-membered palladacycles.6
Scheme 1.
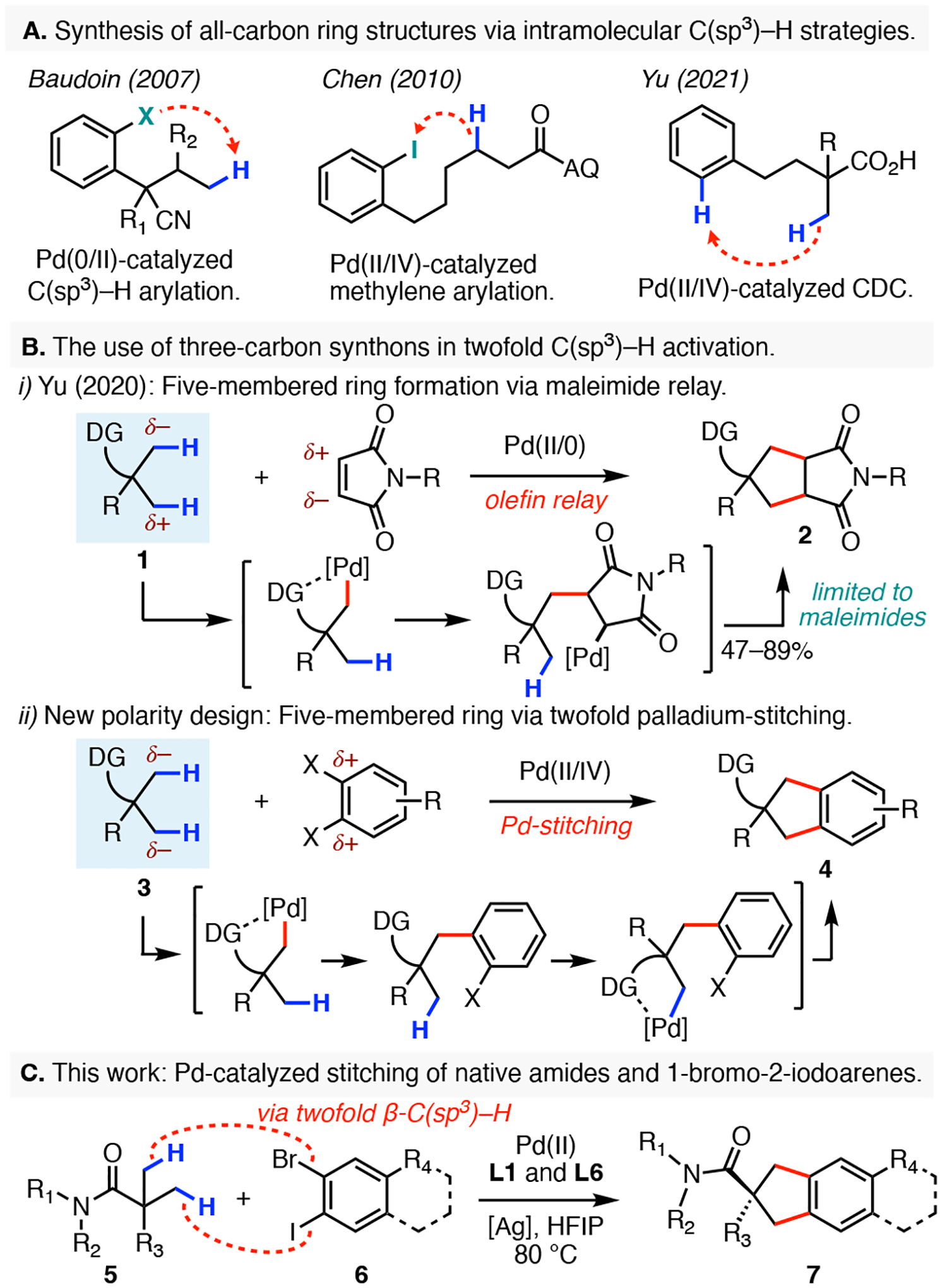
Pd-catalyzed benzannulation via stitching 1,3-C(sp3)–H bonds with dihaloarenes.
Despite the significant advances in C(sp3)–H functionalization reactions,7 the preparation of the five-membered benzo-fused carbocycles via the most straightforward double C–H activation logic has not been realized (Scheme 1B). Specifically, simultaneous dual excision of the carbocycle’s Calkyl–Caryl bonds would reveal two fragments: an alkyl three-carbon synthon and a 1,2-difunctionalized arene, in which their simplest synthetic equivalents, namely a gem-dimethyl fragment and a dihaloarene, could be readily obtained with minimal pre-functionalization effort (Scheme 1B). In the forward sense, these fragments could be stitched together via two independent but sequential β-C(sp3)–H arylation reactions at the gem-dimethyl groups of 3 to provide the bicycle 4. Although a previous study from our group showed that the use of such three-carbon alkyl synthon was possible in case of coupling to maleimides, direct translation of this reactivity towards benzo-fused carbocycle synthesis presents a considerable challenge (Scheme 1B).8 Specifically, the maleimide relay transformation was optimized to exploit the polarity-guided nature of the migratory insertion step, whereby the three-carbon alkyl synthon could be envisioned as a 1,3-dipole 1. However, our proposed strategy to access carbocycles requires the three-carbon gem-dimethyl synthon to exhibit the reactivity of a 1,3-dianion equivalent 3 in order to match the polarity requirements for coupling with dihaloarenes. This type of reactivity modulation of the synthon’s polarity, which would arguably be very challenging with classical organic chemistry, could be realized through the application of a different palladium redox cycle in C–H functionalization chemistry. To turn the 1,3-dipole into a 1,3-dianion synthon, the original Pd(II/0) cycle employed for the maleimide relay is replaced with a dual Pd(II/IV) reaction manifold that can effectively achieve the necessary polarity matching with the dihaloarene electrophile and enable the desired carbocycle synthesis. Evidence for a related reaction protocol was recently reported by Gandon and Sahoo in a tandem C–C and C–O bond forming C–H annulation reaction utilizing a bidentate directing group.9
As part of our program aimed at developing new reactions that rapidly increase molecular complexity, we herein report a palladium-catalyzed annulation reaction between gem-dimethyl containing amides (5) and 1-bromo-2-iodoarenes (6, Scheme 1C). Our transformation features a broad substrate scope, shows excellent amide compatibility, and can also be extended to the preparation of a tricyclic benzo-fused scaffolds (7). The key to success was the discovery of a dual ligand system, comprising of an N-acyl glycine and a pyridine-3-sulfonic acid, that efficiently promotes the sequential palladium catalyzed stitching to arrive at fully annulated benzo-fused products.
RESULTS AND DISCUSSION.
At the onset of our work, we envisioned that the transformation between the 1-bromo-2-iodobenzene (8) and N,N-dimethylpivalamide (9) may proceed through the following steps (Scheme 2A). First, a Pd(II/IV)-catalyzed β-C(sp3)–H arylation of the amide 9 with the more reactive aryl iodide component of the electrophile would form the first Calkyl–Caryl and provide the linear intermediate 10.10 Next, a second β-C(sp3)–H activation directed by the weakly coordinating carbonyl group would generate the alkylpalladium species 11, that can subsequently undergo an intramolecular oxidative addition with the pendant aryl-bromide to furnish the second high-valent palladium(IV) intermediate 12. Finally, a reductive elimination would form the second Calkyl–Caryl bond and stitch the two coupling partner together providing the fully annulated bicycle 13. However, we recognized that several potential pitfalls may interfere with the development of our annulative transformation. First, we identified that the alkylpalladium species 11 may undergo a competitive diarylation reaction with a second equivalent of the dihaloarene due to the increased reactivity of the aryl-iodide component. Next, the second carbon–carbon bond forming step is expected to proceed via oxidation of 11 with the pendant aryl bromide to access the high-valent Pd(IV) intermediate 12. While there is precedence for invoking this type of reactivity with aryl bromides, this high energy intermediate may be difficult to access.11
Scheme 2.
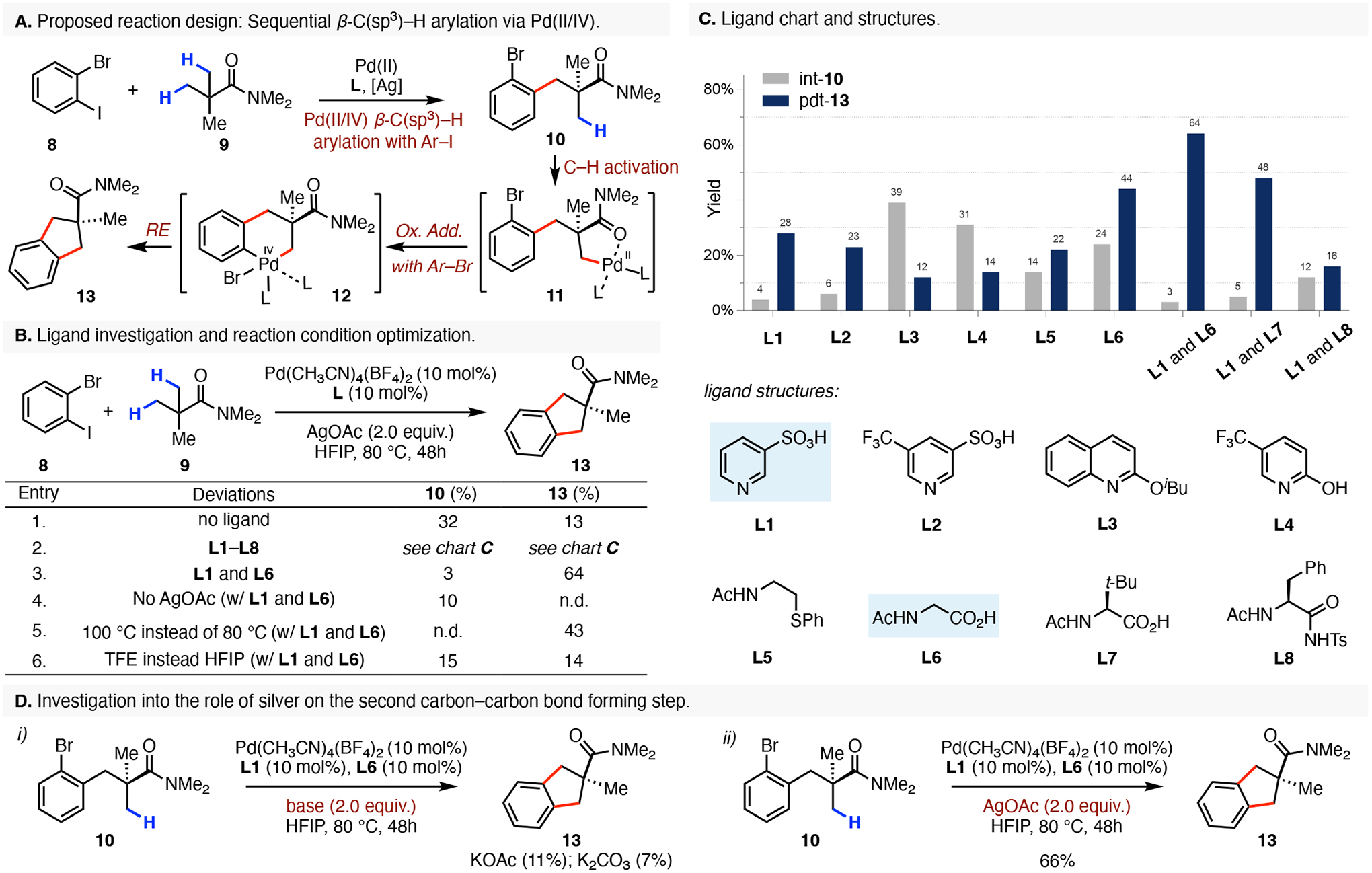
Ligand and reaction condition optimization for the stitching of 1,3-C(sp3)–H bonds and 1-bromo-2-iodoarenes.a,b
aConditions: 8 (0.15 mmol), 9 (0.10 mmol), Pd(CH3CN)4(BF4)2 (10 mol%), Ligand (L) (10 mol%), AgOAc (2.0 equiv), HFIP (0.5 mL), 80 °C, 48h. bYields were determined by 1H NMR analysis of an unpurified product mixture using CH2Br2 as an internal standard.
We started our investigation by first evaluating ligand effects (Scheme 2B). In the absence of any ligand, product formation was very low (13%) and we also observed a significant quantity (32%) of the linear intermediate 10 suggesting that a ligand is needed to promote the second Calkyl–Caryl bond formation (Entry 1). Guided by the findings from our previous study, we first examined the pyridinesulfonic acid ligands L1 and L2.8 We were pleased to find that both of these ligands generated the desired product in modest 28% and 23% yields, respectively (Scheme 2C). However, in both instances we identified large amounts of the unreacted starting amide 9 and only negligible quantities of the linear intermediate needed to arrive at the fully annulated bicycle. Next, we examined several additional ligand scaffolds such as the pyridine-based L3, the monodentate pyridone L4, or the bidentate N-acyl aminoethyl phenyl thioether L5. These ligands did not improve the yield of the product formation (12–22%). We were pleased to find that the use of commercially available N-acyl glycine (L6) resulted in a substantial increase in product formation to 44% and also furnished 24% of the intermediate 10. We reasoned that we may be able to further drive product formation by converting the outstanding linear intermediate with an addition of a second ligand to the reaction mixture.12 Specifically, ligand L1 appeared as a worthy candidate because it displays efficient conversion of the intermediate to the product, as evidenced by only 4% of 10 remaining in the reaction mixture. To our delight, the combination of L1 and L6 ligands displayed synergistic effects in our reaction and provided the fully coupled bicyclic product 13 in 64% yield. Further attempts with L1 and N-acyl L-tert-leucine (L7) possessing a favorable Thorpe−Ingold effect did not further increase the reaction yield.13 Additional optimization of the reaction conditions such as increasing the reaction temperature (80 °C to 100 °C, Entry 5) or switching the reaction solvent from hexafluoroisopropanol (HFIP) to trifluoroethyl alcohol (TFE, Entry 6) were also unproductive.
Next, we sought to investigate the role of silver salts on our annulation reaction. In the absence of silver acetate, we did not detect any of the desired bicyclic product (Scheme 2B, Entry 4). However, we were specifically interested in probing the effects of silver on the second carbon–carbon bond forming step which could provide an insight into whether this step proceeds through a Pd(II/IV) or a Pd(0/II) reaction manifold. It is well known that due to the presence of a highly acidic a-hydrogen, HFIP is capable of generating Pd(0) by means of a Pd(II)-hydride and a base.14 Accordingly, we subjected the linear intermediate 10 to our optimized reaction conditions using two equivalents of potassium acetate or potassium carbonate instead of the silver acetate and observed significantly decreased product formations of 11% and 7%, respectively (Scheme 2D). However, treating 10 with our optimized reaction conditions restored reactivity and delivered the fully annulated bicycle 13 in a yield that was comparable to the intermolecular coupling (64% vs 66%). Taken together, the strong reactivity dependence on silver salts suggests that the second Calkyl–Caryl stitching step proceeds via a Pd(II/IV) cycle and that any potential Pd(0) species are likely rapidly reoxidized with the presence of two equivalents of silver acetate in our reaction mixture.15
With the optimal reaction conditions in hand, we first investigated the substrate scope with respect to the amide residue (Scheme 3). Using a simple amide bond coupling protocol, we were able to prepare a range of amide precursors for our transformation. We found that a variety of symmetrical and unsymmetrical tertiary amides (15a–15c) were well tolerated and all provided the bicyclic products in yields ranging from 56 to 68%. Next, we evaluated several substrates possessing a cyclic dialkyl amine such as the azetidine (15d), pyrrolidine (15e), piperidine (15f), azepane (15g), or morpholine (15h). These substrates all delivered the desired products in moderate yields (48–60%). Secondary amides 15i and 15j with the weakly acidic N–H bond generated the bicyclic products in modest 29% and 33% yield, respectively. Lastly, the Weinreb amide 15k displayed a significantly reduced reactivity and provided the product in a 19% yield. We attribute this result to the potential unproductive bidentate coordination of the catalyst to the carbonyl and the –OMe groups that can impede the desired dual β-(sp3)–H activation.
Scheme 3.
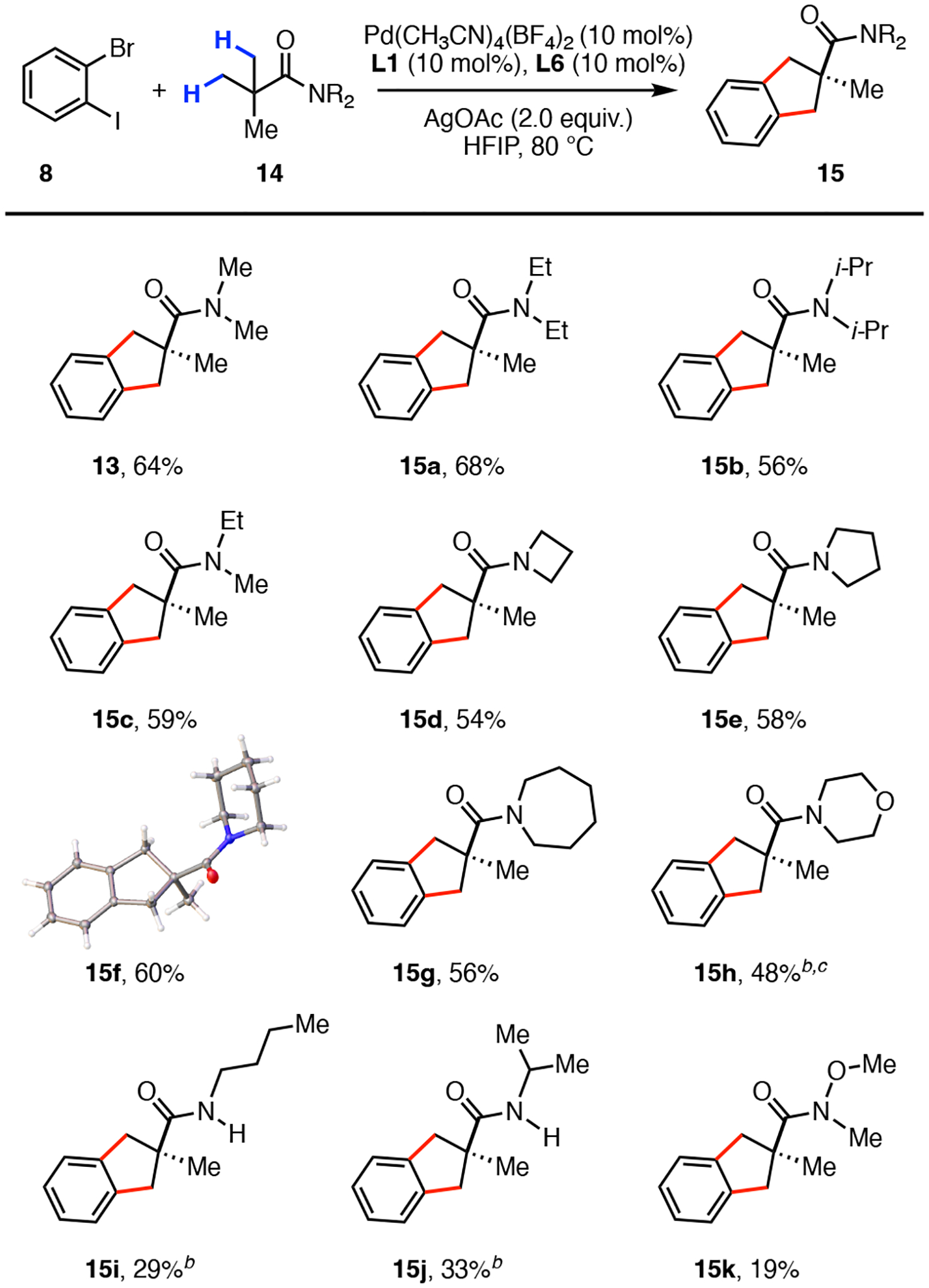
Substrate scope of the amide residue.a,b,c
aConditions: 8 (0.15 mmol), 14 (0.10 mmol), Pd(CH3CN)4(BF4)2 (10 mol%), L1 (10 mol%), L6 (10 mol%), AgOAc (2.0 equiv), HFIP (0.5 mL), 80 °C, 48h. bPd(CH3CN)4(BF4)2 (20 mol%), L1 (20 mol%), L6 (20 mol%). c72h.
Next, we examined the substrate scope with respect to the gem-dimethyl containing amides, and found that a wide variety of substituents at the β-position were compatible with our transformation (Scheme 4A). For example, the alkyl bearing substrates (18a–18c) provided the stitched bicyclic products in 47–61% isolated yields. The cyclic ring containing cyclobutane (18d) and tetrahydropyran (18e) amides furnished the products in 44% and 40% yield, respectively. In the case of 18e, we found that Pd(OAc)2 proved to be the superior palladium source. Next, substrates having chelating methoxy substituents provided the desired products 18g and 18h in 34% and 52% yield. The phthalimide-18f and the methyl ester-18i showed reduced reactivity under our optimized reaction conditions. However, increasing the palladium and ligand loadings by twofold resulted in the formation of 18f and 18i in 47% and 42% yield, respectively. Our protocol was also successfully applied to derivatization of Gemfibrozil,16 the oral therapeutic used to treat high cholesterol. Subjecting a dimethylamide analog to our developed reaction conditions with Pd(OAc)2 resulted in formation of the benzo-fused derivative 18j in 33% yield.
Scheme 4.
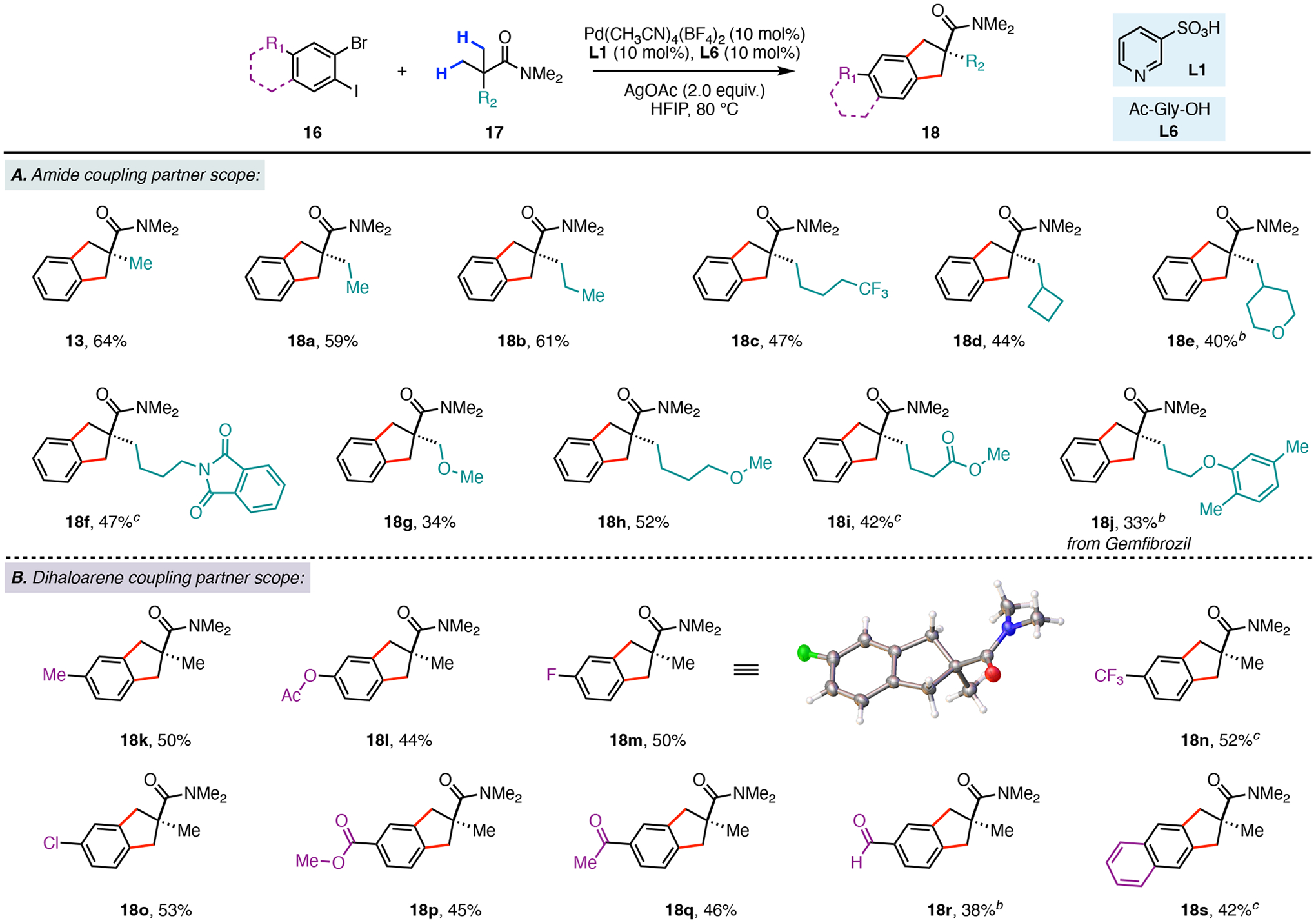
Substrate Scope with Respect to the Dimethyl Amide and 1-Bromo-2-iodoarene Coupling Partners.a,b,c
aConditions: 16 (0.15 mmol), 17 (0.10 mmol), Pd(CH3CN)4(BF4)2 (10 mol%), L1 (10 mol%), L6 (10 mol%), AgOAc (2.0 equiv), HFIP (0.5 mL), 80 °C, 48h. bPd(OAc)2, (20 mol%), L1 (20 mol%), L6 (20 mol%). cPd(CH3CN)4(BF4)2 (20 mol%), L1 (20 mol%), L6 (20 mol%).
Our reaction protocol was also compatible with the incorporation of a variety of functional groups on the 1-bromo-2-iodoarene coupling partner (Scheme 4B). The electron-rich arenes 18k and 18l provided the desired products in 50% and 44% yield, respectively. Halogenated substrates such as the fluorine (18m), trifluoromethyl (18n), and chlorine (18o) showed good reactivity and generated the desired bicyclic products in 46–50% yields. Moreover, the aryl chloride and aryl fluoride products possess useful synthetic handles that could be used for future product elaboration. We also found that the electron-withdrawing methyl ester (18p), methyl ketone (18q), and aldehyde (18r) substituents were tolerated and furnished the annulated products in 45%, 46%, and 38% yield. Our reaction could also be able applied towards the preparation of the tricyclic benzo-fused compound 18s, which was isolated in 42% yield. Lastly, we observed that coupling partners with substitution ortho to the bromide or the iodide positions displayed poor reactivity (see the Supporting Information).
We were also interested in determining if additional vicinal arene electrophiles could be productively annulated to generate our bicyclic products (Scheme 5A). When we subjected the 1,2-dibromobenzene (20) to our reaction conditions, we observed product formation in only 7% yield. In case of the 2-iodophenyl trifluoromethanesulfonate 21, we failed to detect any of the desired product formation in our rection mixture. However, the 1,2-diiodobenzene (22) showed encouraging reactivity and generated 13 in 28% yield. Compared to our standard 1-bromo-2-iodobenzene electrophile, these results indicate that the 1-bromo-2-iodoarenes are ideal coupling partners for our palladium-catalyzed stitching annulation and that the differential reactivity of the of the aryl bromide and the aryl iodide components is needed to achieve high levels of conversion.
Scheme 5.
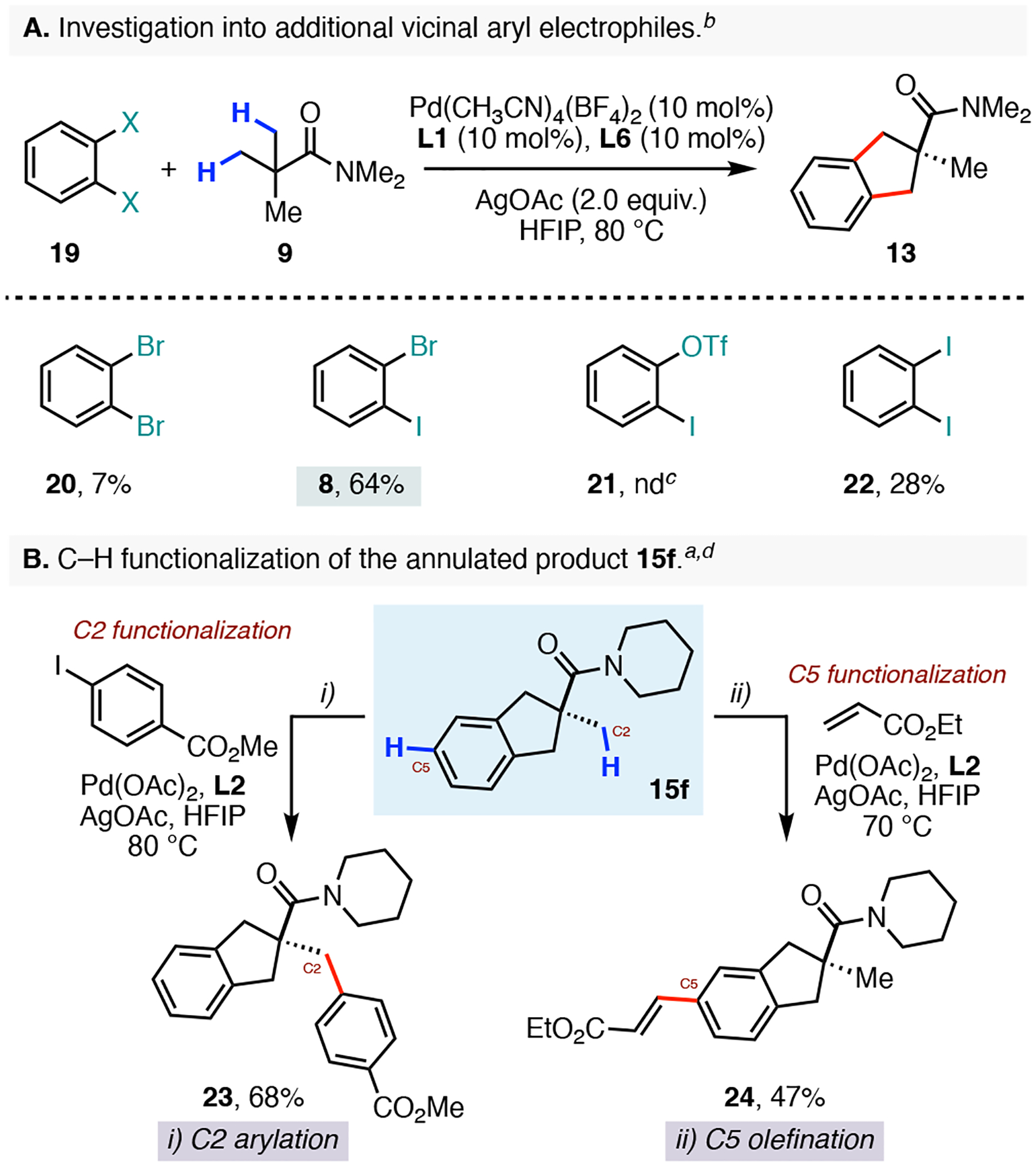
Dihaloarenes and Divergent Derivatization.a,b,c,d
aConditions: 15f (0.10 mmol), methyl 4-iodobenzoate or ethyl acrylate (0.20 mmol), Pd(OAc)2 (10 mol%), L2 (12 mol%), AgOAc (2.0 equiv), HFIP (0.5 mL), 70 or 80 °C, 24h. bYields were determined by 1H NMR analysis. cNone detected. dIsolated Yields.
Next we sought to functionalize the C2 methyl substituent of our product 15f using the previously reported β-C(sp3)–H arylation10 and olefination17 reactions for native amides. To this end, treatment of 15f with methyl 4-iodobenzoate and 5-(trifluoromethyl)-pyridine-3-sulfonic acid (L2) resulted in an efficient arylation reaction of the C2 methyl group to generate 23 in 68% isolated yield (Scheme 5B). However, using the same ligand (L2) and ethyl acrylate as the olefin coupling partner provided the unexpected C5 nondirected olefination product 24 in 47% yield. In this transformation, we did not observe any olefination reactivity at the C2 position. The divergent reactivity of L2 with substrate 15f clearly highlights the subtle differences in electronic and steric demands for the two Pd(II/0) and Pd(II/IV) reaction manifolds. Moreover, this result underscores the ability of C–H functionalization reactions to effectively generate diverse analogues of privileged scaffolds.
To demonstrate the synthetic utility of our annulation protocol, we applied this method to the first synthesis of the bioactive illudane sesquiterpene echinolactone D (29, Scheme 6). Isolated from the Japanese indigenous fungus Echinodontium japonicum in 2006, echinolactone D (29) showed promising ability to stimulate radicle elongation growth in a lettuce seedling bioassay at 100 ppm.1b Our short synthesis of 29 started with the developed palladium-catalyzed annulation reaction between the commercially available methyl 3-bromo-4-iodobenzoate (16p) and the pivalamide 14f, which was followed by a hydrolysis (lithium hydroxide) of the methyl ester to provide the free acid bicycle 25 in 51% yield. Next, a palladium-catalyzed ortho C–H alkylation with ethylene oxide established the tricyclic framework of the natural product and formed the lactone 26 (56%).18 Subsequent electrophilic bromination with tribromoisocyanuric acid (TBCA) in trifluoracetic acid proceeded exclusively at the C3 position and formed the crystalline aryl bromide 27 in 92% yield.19 Installation of the remaining carbon–carbon bond was accomplished via a Suzuki–Miyaura cross-coupling reaction between the aryl bromide and potassium methyltrifluoroborate to provide the methylated compound 28 (80%).20 Lastly, a hydrolysis (sulfuric acid) of the amide moiety was followed by exhaustive reduction of the resulting carboxylic acid with borane dimethylsulfide that completed our synthesis and delivered (±)-echinolactone D (29) in 66% yield. Our short synthesis of 29, featuring the newly developed palladium-catalyzed annulation reaction as the key step, highlights the capability of this method to provide synthetically useful intermediates and suggests that it may find use in syntheses of other benzo-fused natural products.
Scheme 6.
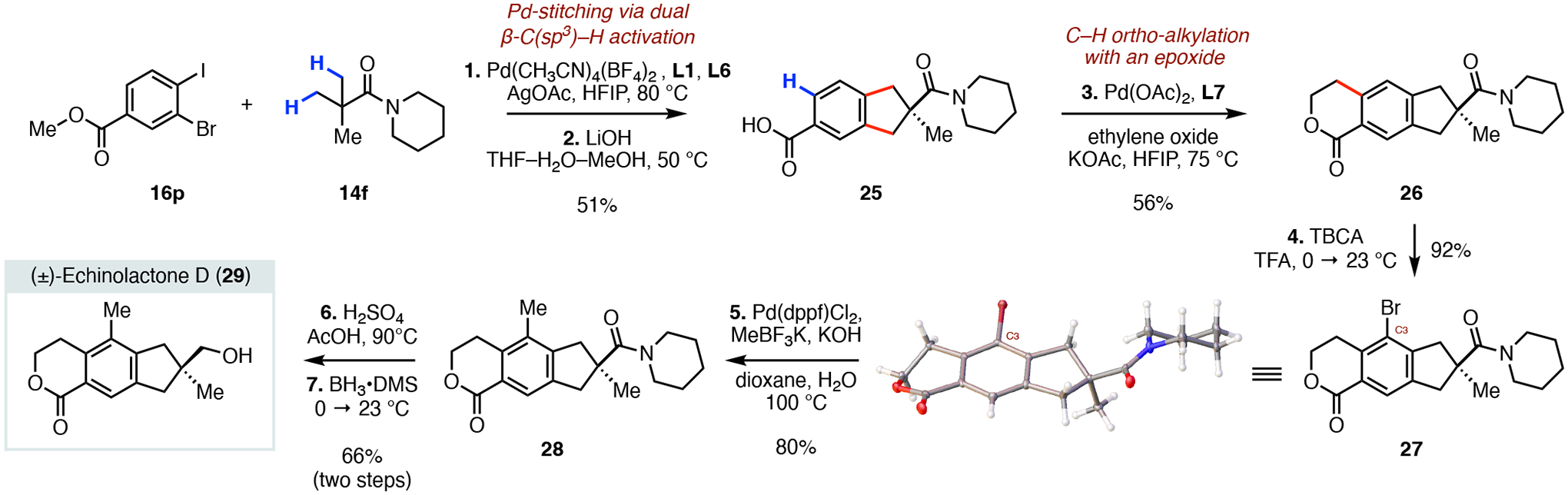
Short synthesis of (±)-echinolactone D (29) enabled by a palladium-catalyzed stitching strategy.a,b
aReagents and conditions: (1) Pd(CH3CN)4(BF4)2 (20 mol%), L1 (20 mol%), L6 (20 mol%), AgOAc (2.0 equiv), HFIP, 80 °C; (2) LiOH (5.0 equiv), THF−H2O−MeOH (1:1:1 v/v), 50 °C; (3) Pd(OAc)2 (2 mol%), L7 (4 mol%), ethylene oxide (2.0 equiv), KOAc (1.0 equiv), HFIP, 75 °C; (4) TBCA (0.35 equiv), TFA, 0→23 °C; (5) Pd(dppf)Cl2 (20 mol%), MeBF3K (1.5 equiv), KOH (3.0 equiv), Dioxane−H2O (5:1 v/v), 100 °C; (6) H2SO4 (20.0 equiv), AcOH, 90 °C; (7) BH3•DMSO (1.5 equiv), THF, 0→23 °C. bTBCA = Tribromoisocyanuric acid.
CONCLUSION.
In summary, we have developed a palladium-catalyzed C–H annulation reaction that effectively unites gemdimethyl fragments and 1-bromo-2-iodoarenes to synthesize a variety of five-membered benzo-fused carbocycles. This transformation proceeds via two tandem β-C(sp3)–H arylation reactions, where the dihaloarene electrophile is stitched to the gemdimethyl groups via two Calkyl–Caryl bond forming events. Moreover, this protocol was enabled by the discovery of a dual ligand system comprising of an N-acyl glycine (L1) and a pyridine-3-sulfonic acid (L6), that synergistically promote the sequential palladium stitching to provide the fully annulated bicyclic products. Our method displayed a broad substrate scope, showed excellent amide compatibility, and could also be used to prepare tricyclic scaffolds. We also demonstrated the synthetic utility of our transformation with a short racemic synthesis of the bioactive isolate echinolactone D (29) that was accomplished in a total of seven steps.
Supplementary Material
ACKNOWLEDGMENT
Financial support from The Scripps Research Institute and the NIH (NIGMS R01 GM084019) is gratefully acknowledged. We thank S. Chan and A. Herron for helpful suggestions in preparing the manuscript. We acknowledge Dr. Jason Chen, Brittany Sanchez, and Quynh Nguyen Wong of the TSRI Automated Synthesis Facility for assistance with HRMS and Prep HPLC. We also acknowledge M. Gembicky (UCSD) for X-ray analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Full experimental details, product characterization, and X-ray crystallographic data (PDF).
The authors declare no competing financial interests.
REFERENCES
- 1.(a) Buysse D; Bate G; Kirkpatrick P, Ramelteon. Nat. Rev. Drug Discov 2005, 4, 881–882. [DOI] [PubMed] [Google Scholar]; (b) Suzuki S; Murayama T; Shiono Y, Echinolactones C and D: Two Illudalane Sesquiterpenoids Isolated from the Cultured Mycelia of the Fungus Echinodontium japonicum. J. Chem. Sci 2006, 61, 1295–1298. [Google Scholar]; (c) Vilums M; Heuberger J; Heitman LH; Ijzerman AP, Indanes–Properties, Preparation, and Presence in Ligands for G Protein Coupled Receptors. Med. Res. Rev 2015, 35, 1097–1126. [DOI] [PubMed] [Google Scholar]; (d) Cheng H-G; Jia S; Zhou Q, Benzo-Fused-Ring Toolbox Based on Palladium/Norbornene Cooperative Catalysis: Methodology Development and Applications in Natural Product Synthesis. Acc. Chem. Res 2023, 56, 573–591. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hitce J; Retailleau P; Baudoin O, Palladium-Catalyzed Intramolecular C(sp3)–H Functionalization: Catalyst Development and Synthetic Applications. Chem. Eur. J 2007, 13, 792–799. [DOI] [PubMed] [Google Scholar]; (b) Rousseaux S; Davi M; Sofack-Kreutzer J; Pierre C; Kefalidis CE; Clot E; Fagnou K; Baudoin O, Intramolecular Palladium-Catalyzed Alkane C−H Arylation from Aryl Chlorides. J. Am. Chem. Soc 2010, 132, 10706–10716. [DOI] [PubMed] [Google Scholar]
- 3.(a) Martin N; Pierre C; Davi M; Jazzar R; Baudoin O, Diastereo- and Enantioselective Intramolecular C(sp3)–H Arylation for the Synthesis of Fused Cyclopentanes. Chem. Eur. J 2012, 18, 4480–4484. [DOI] [PubMed] [Google Scholar]; (b) Holstein PM; Vogler M; Larini P; Pilet G; Clot E; Baudoin O, Efficient Pd0-Catalyzed Asymmetric Activation of Primary and Secondary C–H Bonds Enabled by Modular Binepine Ligands and Carbonate Bases. ACS Catal 2015, 5, 4300–4308. [Google Scholar]
- 4.Feng Y; Wang Y; Landgraf B; Liu S; Chen G, Facile Benzo-Ring Construction via Palladium-Catalyzed Functionalization of Unactivated sp3 C−H Bonds under Mild Reaction Conditions. Org. Lett 2010, 12, 3414–3417. [DOI] [PubMed] [Google Scholar]
- 5.(a) St John-Campbell S; Bull JA, Intramolecular palladium(II)/(IV) catalysed C(sp3)–H arylation of tertiary aldehydes using a transient imine directing group. Chem. Commun 2019, 55, 9172–9175. [DOI] [PubMed] [Google Scholar]; (b) Provencher PA; Bay KL; Hoskin JF; Houk KN; Yu J-Q; Sorensen EJ, Cyclization by C(sp3)–H Arylation with a Transient Directing Group for the Diastereoselective Preparation of Indanes. ACS Catal 2021, 11, 3115–3127. [Google Scholar]
- 6.Zhuang Z; Herron AN; Liu S; Yu J-Q, Rapid Construction of Tetralin, Chromane, and Indane Motifs via Cyclative C–H/C–H Coupling: Four-Step Total Synthesis of (±)-Russujaponol F. J. Am. Chem. Soc 2021, 143, 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Desai LV; Hull KL; Sanford MS, Palladium-Catalyzed Oxygenation of Unactivated sp3 C−H Bonds. J. Am. Chem. Soc 2004, 126, 9542–9543. [DOI] [PubMed] [Google Scholar]; (b) Giri R; Chen X; Yu J-Q, Palladium-Catalyzed Asymmetric Iodination of Unactivated C–H Bonds under Mild Conditions. Angew. Chem. Int. Ed 2005, 44, 2112–2115. [DOI] [PubMed] [Google Scholar]; (c) Zaitsev VG; Shabashov D; Daugulis O, Highly Regioselective Arylation of sp3 C−H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc 2005, 127, 13154–13155. [DOI] [PubMed] [Google Scholar]; (d) Zhu Y; Chen X; Yuan C; Li G; Zhang J; Zhao Y, Pd-catalysed ligand-enabled carboxylate-directed highly regioselective arylation of aliphatic acids. Nat. Commun 2017, 8, 14904. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shen P-X; Hu L; Shao Q; Hong K; Yu J-Q, Pd(II)-Catalyzed Enantioselective C(sp3)–H Arylation of Free Carboxylic Acids. J. Am. Chem. Soc 2018, 140, 6545–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhuang Z; Yu C-B; Chen G; Wu Q-F; Hsiao Y; Joe CL; Qiao JX; Poss MA; Yu J-Q, Ligand-Enabled β-C(sp3)–H Olefination of Free Carboxylic Acids. J. Am. Chem. Soc 2018, 140, 10363–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ghiringhelli F; Uttry A; Ghosh KK; van Gemmeren M, Direct β- and γ-C(sp3)−H Alkynylation of Free Carboxylic Acids**. Angew. Chem. Int. Ed 2020, 59, 23127–23131. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Chan HSS; Yang J-M; Yu J-Q, Catalyst-controlled site-selective methylene C–H lactonization of dicarboxylic acids. Science 2022, 376, 1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Yang J-M; Lin Y-K; Sheng T; Hu L; Cai X-P; Yu J-Q, Regio-controllable [2+2] benzannulation with two adjacent C(sp3)–H bonds. Science 2023, 380, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park H; Yu J-Q, Palladium-Catalyzed [3 + 2] Cycloaddition via Twofold 1,3-C(sp3)–H Activation. J. Am. Chem. Soc 2020, 142, 16552–16556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh A; Grimblat N; Sau S; Saha A; Gandon V; Sahoo AK, Palladium(II)-Catalyzed Annulative Difunctionalization of Two Inert C(sp3)–H Bonds by a Bifunctional Reagent. ACS Catal 2023, 7627–7636. [Google Scholar]
- 10.Park H; Chekshin N; Shen P-X; Yu J-Q, Ligand-Enabled, Palladium-Catalyzed β-C(sp3)–H Arylation of Weinreb Amides. ACS Catal 2018, 8, 9292–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Xu L-M; Li B-J; Yang Z; Shi Z-J, Organopalladium(IV) chemistry. Chem. Soc. Rev 2010, 39, 712–733. [DOI] [PubMed] [Google Scholar]; (b) Sehnal P; Taylor RJK; Fairlamb IJS, Emergence of Palladium(IV) Chemistry in Synthesis and Catalysis. Chem. Rev 2010, 110, 824–889. [DOI] [PubMed] [Google Scholar]; (c) Shi H; Herron AN; Shao Y; Shao Q; Yu J-Q, Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator. Nature 2018, 558, 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shi H; Lu Y; Weng J; Bay KL; Chen X; Tanaka K; Verma P; Houk KN; Yu J-Q, Differentiation and functionalization of remote C–H bonds in adjacent positions. Nat. Chem 2020, 12, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Qi X; Wang J; Dong Z; Dong G; Liu P, Compatibility Score for Rational Electrophile Selection in Pd/NBE Cooperative Catalysis. Chem 2020, 6, 2810–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Chen G; Zhuang Z; Li G-C; Saint-Denis TG; Hsiao Y; Joe CL; Yu J-Q, Ligand-Enabled β-C–H Arylation of α-Amino Acids Without Installing Exogenous Directing Groups. Angew. Chem. Int. Ed 2017, 56, 1506–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen H; Wedi P; Meyer T; Tavakoli G; van Gemmeren M, Dual Ligand-Enabled Nondirected C−H Olefination of Arenes. Angew. Chem. Int. Ed 2018, 57, 2497–2501. [DOI] [PubMed] [Google Scholar]; (c) Chen H; Mondal A; Wedi P; van Gemmeren M, Dual Ligand-Enabled Nondirected C–H Cyanation of Arenes. ACS Catal 2019, 9, 1979–1984. [Google Scholar]; (d) Wedi P; Farizyan M; Bergander K; Mück-Lichtenfeld C; van Gemmeren M, Mechanism of the Arene-Limited Nondirected C−H Activation of Arenes with Palladium**. Angew. Chem. Int. Ed 2021, 60, 15641–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. We examined the enantioselectivity of the chiral ligand L7 alone and in combination with L1 in our transformation but in both instances the ee was <4% (see the Supporting Information).
- 14.(a) Yu C; Sanjosé-Orduna J; Patureau FW; Pérez-Temprano MH, Emerging unconventional organic solvents for C–H bond and related functionalization reactions. Chem. Soc. Rev 2020, 49, 1643–1652. [DOI] [PubMed] [Google Scholar]; (b) Grünwald A; Heinemann FW; Munz D, Oxidative Addition of Water, Alcohols, and Amines in Palladium Catalysis. Angew. Chem. Int. Ed 2020, 59, 21088–21095. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bhattacharya T; Ghosh A; Maiti D, Hexafluoroisopropanol: the magical solvent for Pd-catalyzed C–H activation. Chem. Sci 2021, 12, 3857–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Weibel J-M; Blanc A; Pale P, Ag-Mediated Reactions: Coupling and Heterocyclization Reactions. Chem. Rev 2008, 108, 3149–3173. [DOI] [PubMed] [Google Scholar]; (b) Daugulis O; Do H-Q; Shabashov D, Palladium- and Copper-Catalyzed Arylation of Carbon−Hydrogen Bonds. Acc. Chem. Res 2009, 42, 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen X; Engle KM; Wang D-H; Yu J-Q, Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem. Int. Ed 2009, 48, 5094–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lotz MD; Camasso NM; Canty AJ; Sanford MS, Role of Silver Salts in Palladium-Catalyzed Arene and Heteroarene C–H Functionalization Reactions. Organometallics 2017, 36, 165–171. [Google Scholar]
- 16.(a) Honorato JM, R. M; Purroy A, The use of gemfibrozil in the treatment of primary hyperlipoproteinaemia. Proc. R. Soc. Med 1976, 69, 78–79. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Todd PA; Ward A, Gemfibrozil. Drugs 1988, 36, 314–339. [DOI] [PubMed] [Google Scholar]
- 17.Park H; Li Y; Yu J-Q, Utilizing Carbonyl Coordination of Native Amides for Palladium-Catalyzed C(sp3)−H Olefination. Angew. Chem. Int. Ed 2019, 58, 11424–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng G; Li T-J; Yu J-Q, Practical Pd(II)-Catalyzed C–H Alkylation with Epoxides: One-Step Syntheses of 3,4-Dihydroisocoumarins. J. Am. Chem. Soc 2015, 137, 10950–10953. [DOI] [PubMed] [Google Scholar]
- 19.de Almeida LS; Esteves PM; de Mattos MCS, Superelectrophilic bromination of deactivated aromatic rings with tribromoisocyanuric acid—an experimental and DFT study. Tetrahedron Lett 2009, 50, 3001–3004. [Google Scholar]
- 20.Zeng Z; Zhao Y; Zhang Y, Divergent total syntheses of five illudalane sesquiterpenes and assignment of the absolute configuration. Chem. Commun 2019, 55, 4250–4253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.