

Abstract
The β-class carbonic anhydrase from the archaeon Methanobacterium thermoautotrophicum (Cab) was structurally and kinetically characterized. Analytical ultracentrifugation experiments show that Cab is a tetramer. Circular dichroism studies of Cab and the Spinacia oleracea (spinach) β-class carbonic anhydrase indicate that the secondary structure of the β-class enzymes is predominantly α-helical, unlike that of the α- or γ-class enzymes. Extended X-ray absorption fine structure results indicate the active zinc site of Cab is coordinated by two sulfur and two O/N ligands, with the possibility that one of the O/N ligands is derived from histidine and the other from water. Both the steady-state parameters kcat and kcat/Km for CO2 hydration are pH dependent. The steady-state parameter kcat is buffer-dependent in a saturable manner at both pH 8.5 and 6.5, and the analysis suggested a ping-pong mechanism in which buffer is the second substrate. At saturating buffer conditions and pH 8.5, kcat is 2.1-fold higher in H2O than in D2O, consistent with an intramolecular proton transfer step being rate contributing. The steady-state parameter kcat/Km is not dependent on buffer, and no solvent hydrogen isotope effect was observed. The results suggest a zinc hydroxide mechanism for Cab. The overall results indicate that prokaryotic β-class carbonic anhydrases have fundamental characteristics similar to the eukaryotic β-class enzymes and firmly establish that the α-, β-, and γ-classes are convergently evolved enzymes that, although structurally distinct, are functionally equivalent.
The thermophilic archaeon Methanobacterium thermoautotrophicum obtains energy for growth by the reduction of CO2 to CH4 and is also an obligate chemolithoautotroph; thus, this organism has a high demand for CO2. Carbonic anhydrase, a zinc-containing enzyme catalyzing the reversible hydration of carbon dioxide (equation 1)
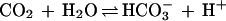 |
1 |
is expected to play an important role in the growth of M. thermoautotrophicum and may have several functions, including transporting HCO3− into the cell and providing CO2 or HCO3− to enzymes that utilize these substrates.
Based on sequence comparisons, carbonic anhydrases belong to three genetically distinct classes (α, β, and γ) which appear to have independent origins (24). The most extensively studied enzymes are those from the α-class, which is composed primarily of mammalian carbonic anhydrases, but also includes enzymes from the green alga Chlamydomonas reinhardtii (19, 20) and the prokaryote Neisseria gonorrhoeae (13). The β-class enzymes are abundant in C3 and C4 monocotyledenous and dicotyledenous plants and green unicellular algae (24, 43), where they are essential for photosynthetic CO2 fixation (6). The most recently identified class of carbonic anhydrase, the γ-class (24), is represented by the prototype Cam from the archaeon Methanosarcina thermophila (2). Even though sequences encoding putative γ-class carbonic anhydrases have been found in prokaryotes from both the Bacteria and Archaea domains (2, 52), Cam is the only γ-class enzyme that has been biochemically characterized (2, 3, 58).
Crystal structures for five α-class mammalian isozymes (CA I to V) (10, 16, 17, 23, 33, 38, 54) and the α-class enzyme from N. gonorrhoeae (26) reveal a monomer in which the dominating secondary feature is an antiparallel β-sheet. The γ-class Cam is remarkably distinct from the α-class carbonic anhydrases in that it is a homotrimer in which each monomer adopts a novel left-handed β-helix fold (28, 36). Even though the α- and γ-classes are notably different in both their tertiary and quaternary structures, both classes contain a catalytically essential zinc ion coordinated by three histidine residues. Recently, the structures of the β-class carbonic anhydrases from both the dicotyledenous plant Pisum sativum (pea) (35) and the red alga Porphyridium purpureum (41) have been solved. Both the P. sativum homo-octamer and the P. purpureum homodimer exhibit a predominantly α-helical secondary structure. Unlike enzymes from the α- and γ-classes, the active site zinc of these β-class enzymes is coordinated by two cysteines and one histidine residue. A conserved aspartate residue appears to serve as a fourth ligand in the P. purpureum enzyme, but not in the P. sativum enzyme.
The kinetic properties of the human α-class isozymes CA I, CA II, and CA III have been extensively investigated and follow a common zinc hydroxide mechanism for catalysis (39, 48). The catalytically active group in this mechanism model is the zinc-bound water, which ionizes to a metal-bound hydroxide ion that attacks CO2. According to the proposed mechanism, the enzyme-catalyzed reaction occurs in two mechanistically distinct steps (where E = enzyme and B = buffer). The first step is the interconversion between carbon dioxide and bicarbonate (equations 2a and 2b), in which the rate is related to the steady-state parameter kcat/Km. The second step is the regeneration of the active form of the enzyme (equations 2c and 2d), involving the rate-determining intramolecular and intermolecular proton transfer events which are reflected in the steady-state parameter kcat:
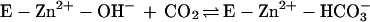 |
2a |
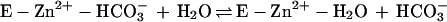 |
2b |
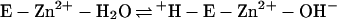 |
2c |
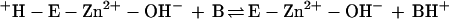 |
2d |
The catalytic mechanism of the M. thermophila γ-class Cam resembles that of human α-class CA II despite significant structural differences in the active sites of these two enzymes (1). The kinetic properties reported for the P. sativum and Spinacia oleracea (spinach) β-class carbonic anhydrases are also consistent with this mechanism (29, 30, 46). Thus, the kinetic analyses of enzymes from all three classes suggest convergent evolution of the catalytic mechanism (1, 36, 39).
The β-class was initially thought to be composed solely of carbonic anhydrases from monocotyledenous and dicotyledenous plants. A mitochondrial β-class carbonic anhydrase was discovered in C. reinhardtii (18), and other enzymes belonging to this class have since been identified in other algae (25, 62). Only two β-class carbonic anhydrases from the Bacteria domain have been purified (22, 53), and the subsequent purification of Cab, a β-class enzyme from the thermophilic archaeon M. thermoautotrophicum, establishes that this class of carbonic anhydrase extends to the Archaea domain (50). Recent work establishes that this class is widely distributed in metabolically diverse prokaryotes representing both the Bacteria and Archaea domains and has ancient origins (50, 52).
Even though the β-class of carbonic anhydrase is the only class with documented enzymes in all three domains, less is known about the biochemistry and overall structural aspects of this class than for either the α-class or the more recently identified γ-class. Herein we report on structural and kinetic studies of Cab, the first for any prokaryotic β-class carbonic anhydrase.
MATERIALS AND METHODS
Analytical ultracentrifugation.
Equilibrium centrifugation was performed with a Beckman model XLI ultracentrifuge. The radial distribution of protein was monitored by A235, A280, or A295, depending upon the concentration loaded. Protein concentrations of Cab were estimated by using A280 and an extinction coefficient (2,740 cm−1 M−1 based on 1 subunit) calculated from the deduced amino acid sequence of the cab gene. Cab was centrifuged at 8,000, 12,000, and 16,000 rpm for 22, 14, and 14 h, respectively. Establishment of equilibrium was verified by the coincidence of the final two scans at each speed. The ultracentrifugation data were fit by using NONLIN (Pharsight Corp., Mountain View, Calif.) and Sedenterp (37).
CD analysis.
Spectra were acquired at 37°C with an Aviv circular dichroism (CD) spectrophotometer, model 62DS. The concentration of the S. oleracea carbonic anhydrase was estimated by using A280 and an extinction coefficient (23,260 cm−1 M−1 based on 1 subunit) calculated from the deduced amino acid sequence of the S. oleracea gene (excluding the 98 N-terminal amino acids encoding the chloroplast transit peptide sequence). Samples (10 μM) of the M. thermoautotrophicum Cab and S. oleracea carbonic anhydrases in 20 mM potassium phosphate (pH 6.8) containing 0.1 M KCl were placed in a cuvette with a 1-mm path length, and the data points obtained were from 320 to 202 nm in 1.0-nm increments. Five spectra were taken for each sample and averaged. The resulting spectra were normalized for direct comparison.
EXAFS.
Zn K-edge X-ray absorption spectroscopy (XAS) data of the as-isolated M. thermoautotrophicum ΔH carbonic anhydrase (Cab) were collected on beam line 7-3 at the Stanford Synchrotron Radiation Laboratory, with the SPEAR ring operating at 3.0 GeV and a 50- to 100-mA current (Table 1) (47). For XAS, 180 μl of Cab (50 mg/ml) in 50 mM potassium phosphate (pH 6.8) containing 35% glycerol was transferred to a Lucite cuvette covered with Mylar adhesive tape as an X-ray transparent window material, capped, and frozen in liquid nitrogen. Extended X-ray absorption fine structure (EXAFS) data analysis and curve fitting were performed by using EXAFSPAK (http://ssrl.slac.stanford.edu/exafspak.html) and Feff v7.0 (5, 44). Multiple-scattering contributions from outer-shell atoms of histidine ligands were quantified as described previously (15), with parameters derived from tetra(imidazole) zinc(II) perchlorate (7).
TABLE 1.
Collection of X-ray absorption spectroscopic data
| Characteristic | Zn EXAFS result |
|---|---|
| Synchrotron radiation facility | Stanford Synchrotron Radiation Laboratory |
| Beamline | 7-3 |
| Current in storage ring (mA) | 50–100 |
| Monochromator crystal | Si[220] |
| Detection method | Fluorescence |
| Detector type | Solid-state arraya |
| Scan length (min) | 25 |
| No. of scans in average | 12 |
| Temp (K) | 10 |
| Energy standard | Zn foil, first inflection |
| Energy calibration (eV) | 9,660.7 |
| E0 (eV) | 9,670 |
| Pre-edge background (eV): | |
| Energy range | 8,657–9625 |
| Gaussian center | 8,638 |
| Width | 750 |
| Spline background energy range; eV | 9,333–9,902 (4) |
| (polynomial order) | |
| 9,902–10,134 (4) | |
| 10,134–10,366 (4) |
The 13-element Ge solid-state X-ray fluorescence detector at the Stanford Synchrotron Radiation Laboratory is provided by the National Institutes of Health Biotechnology Research Resource.
Enzyme purification.
Cab was heterologously produced in Escherichia coli as previously described (50). Thawed cell paste (10 g) was suspended in 20 ml of buffer A (50 mM potassium phosphate [pH 6.8]) and passed twice through a chilled French pressure cell at 138 MPa. The cell lysate was centrifuged at 20,000 × g for 20 min to remove cell debris and then centrifuged at 100,000 × g for 2 h to remove membranes. The supernatant was loaded onto a 50-ml Q Sepharose (Fast Flow) anion-exchange column (Pharmacia) equilibrated with buffer A. After a 100-ml wash, the column was developed with a 400-ml linear gradient from 0 to 0.75 M KCl. The enzyme eluted between 450 and 550 mM KCl, and fractions containing active enzyme were pooled. The fractions containing active enzyme were raised to 1.5 M (NH4)2SO4 and run on a 50-ml Phenyl-Sepharose column (Pharmacia) equilibrated with buffer A plus 1.5 M (NH4)2SO4. After a 100-ml wash, the column was developed with a 400-ml linear gradient from 1.5 to 0 M (NH4)2SO4 with the enzyme eluting at approximately 100 mM (NH4)2SO4. The active fractions were pooled, desalted, and loaded onto a Mono Q 10/10 anion-exchange column (Pharmacia) equilibrated with buffer A. After a 30-ml wash, the column was developed with a 100-ml linear gradient from 0 to 1 M KCl. The enzyme eluted between 460 and 520 mM KCl, and fractions containing active enzyme were pooled, desalted, and stored at −20°C. Carbonic anhydrase activity was measured at room temperature by a modification of the electrometric method of Wilbur and Anderson (60). Protein concentrations were determined by the Bradford method with Bio-Rad dye reagent and bovine serum albumin (Sigma) as the standard (12).
Steady-state kinetics.
Initial rates of CO2 hydration and HCO3− dehydration were determined by stopped-flow spectroscopy (KinTek, State College, Pa.) at 25°C by the changing pH indicator method (34). Saturated solutions of CO2 were prepared by bubbling CO2 into distilled, deionized water (32.9 mM) at 25°C. The CO2 concentration ranged from 6 to 24 mM, and the HCO3− concentration ranged from 5 to 80 mM. The following buffer-pH indicator pairs (and wavelengths) were used: at pH 5.5 to 6.6, MES [2-(N-morpolino)ethanesulfonic acid] (pKa = 6.1)-chlorophenol red (574 nm); at pH 6.8 to 7.2, MOPS [2-(N-morpholino)propanesulfonic acid] (pKa = 7.2)–p-nitrophenol (400 nm); at pH 7.4 to 7.8, HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] (pKa = 7.5)-phenol red (557 nm); at pH 8.0 to 9.0, TAPS [N-tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid] (pKa = 8.4)–m-cresol purple (578 nm). The observed initial rates were corrected for the uncatalyzed rate of the reaction, which was at least five times lower than the catalyzed rate. The steady-state parameters kcat and kcat/Km and their standard errors were then determined by fitting the observed initial rates to the Michaelis-Menten equation.
The buffer dependence was measured at pH 8.5 (TAPS) and 6.5 (MES) by varying the CO2 concentration from 6 to 24 mM and the buffer concentration from 5 to 50 mM, maintaining the ionic strength at 0.1 M by addition of sodium sulfate. The observed initial rates were fit to the Michaelis-Menten equation. To determine the solvent deuterium isotope effect on the kinetic constants, identical buffer solutions were prepared in H2O and D2O. The solvent isotope effect was measured at pH 8.5 (50 mM TAPS). The pH was adjusted until the pH meter readings were identical. This method is appropriate, because the correction of a pH meter reading for pD (pD = meter reading + 0.4) (21) is approximately compensated for by the change in the acid dissociation constant in D2O for weak acids whose pKa values lie between 3 and 10 (pKD − pKH = 0.5 ± 0.1) (8). The CO2 concentrations ranged from 7 to 27 mM based on the saturating solutions of CO2 in D2O at 25°C (38.1 mM). All fits described were performed with KaleidaGraph (Synergy Software, Reading, Pa.).
RESULTS
Analytical ultracentrifugation analysis.
Gel filtration analysis had previously suggested that Cab is a tetramer (50). More precise analytical ultracentrifugation studies were performed to confirm the quaternary structure. A global fit of the data at several speeds and initial concentrations of enzyme could only be satisfactorily modeled as a single species. The molecular mass of 68,080 Da calculated from such a fit is approximately 91% of the tetrameric mass (74,810 Da) calculated from the deduced amino acid sequence. These results establish that Cab is a homotetramer.
CD spectroscopy.
CD spectroscopy was performed on both Cab and the β-class S. oleracea enzyme to characterize and compare the secondary structures. The spectra overlap throughout the far-UV region (Fig. 1). Cab has two intense negative bands centered at 223 nm ([θ] = −7,700.5) and 209 nm ([θ] = −9,970.0). This result is similar to the spectrum obtained for the S. oleracea enzyme (Fig. 1), which has two intense bands centered at 221 nm ([θ] = −11,078) and 209 nm ([θ] = −12,143). These results suggest that the dominant structure for both enzymes is α-helical (31, 32). The CD spectrum of the tetrameric Cab is less intense than that of the octameric S. oleracea enzyme, suggesting that the proportion of α-helical structure in the plant enzyme is greater than that of Cab.
FIG. 1.

Comparison of the CD spectra of the M. thermoautotrophicum and S. oleracea β carbonic anhydrases. Far-UV region spectra of the S. oleracea (solid line) and M. thermoautotrophicum (dotted line) enzymes are shown.
XAS.
The Zn K-edge X-ray absorption spectrum for Cab shows a shift in absorption edge position to lower energy compared to Cam, the γ-class carbonic anhydrase from M. thermophila (1) (Fig. 2A). This shift is indicative of increased electron density at the Zn atom, resulting from an increase in electron-donating ligands such as sulfur. Similarly, the shift of the main peak in the Fourier transform (FT) of β-class Cab (Fig. 2B) to a longer distance, compared with γ-class Cam, is indicative of a larger proportion of sulfur-containing ligands. The decrease in amplitude of the ca. 3- and 4-Å multiple-scattering peaks in the FT indicates that the Zn2+ site of β-class Cab contains fewer histidine ligands than γ-class Cam.
FIG. 2.

XAS analysis of Cab. (A and B) X-ray absorption edge spectra (A) and FTs (B) (k = 2 to 12 Å−1) for Cab, the M. thermoautotrophicum β-class carbonic anhydrase (solid line), and Cam, the γ-class carbonic anhydrase from M. thermophila (dashed line) (1). (C and D) k3-weighted EXAFS (C) and FTs (D) (k = 2 to 12 Å−1) of the β-class Cab (solid line) and calculated results for ZnS2(N/O)2 (dashed line; fit 4, Table 2).
The k3-weighted EXAFS of Cab (Fig. 2C) are best fit by assuming a Zn-S2(N/O)2 coordination environment (cf. fits 3 to 6, Table 2). Bond valence sum (BVS) analysis is an empirical method for correlating the sum of the strength of the metal-ligand bonds, as measured by the bond length, to the oxidation state of the metal (40, 57). BVS confirms that the Zn site has a coordination number of 4 (the BVS value should be approximately equal to the oxidation state of the metal, i.e., 2). The EXAFS can be fit assuming one of the O/N ligands results from histidine imidazole coordination (fit 6, Table 2). This fit results in a slight improvement in goodness-of-fit value (cf. fits 4 and 6, Table 2). However, the Debye-Waller factor (ςas2) for the multiple scattering paths from outer shell atoms indicates that the contribution of this moiety to the overall EXAFS is small. As such, the population of histidine ligands must be disordered to account for the observed ςas2 values. This disorder could result from a heterogeneity among the Zn sites or from a Zn-histidine coordination geometry in which the imidazole ring is tilted such that the Zn-N-C bond angles differ significantly for the two sides of the imidazole ring. Thus, the EXAFS results indicate the zinc site of Cab is coordinated by two sulfur and two O/N ligands, with the possibility that one of the O/N ligands results from histidine (cf. fit 4, Table 2; Fig. 2D).
TABLE 2.
Curve-fitting results for Zn EXAFSa
| Fit | Shell | NS | Ras (Å) | ςas2 (Å2) | ΔE0 (eV) | fb | BVSc |
|---|---|---|---|---|---|---|---|
| 1 | Zn-O | 4 | 2.10 | 0.0054 | 4.38 | 0.131 | 1.37 |
| 2 | Zn-O | 3 | 2.03 | 0.0031 | −1.35 | 0.072 | 1.78 |
| Zn-S | 1 | 2.32 | −0.0011 | ||||
| 3 | Zn-O | 3 | 2.01 | 0.0058 | −3.65 | 0.080 | 2.45 |
| Zn-S | 2 | 2.30 | 0.0035 | ||||
| 4 | Zn-O | 2 | 2.00 | 0.0016 | −4.86 | 0.073 | 2.03 |
| Zn-S | 2 | 2.30 | 0.0022 | ||||
| 5 | Zn-O | 2 | 1.99 | 0.0042 | −5.91 | 0.081 | 2.67 |
| Zn-S | 3 | 2.29 | 0.0057 | ||||
| 6 | Zn-S | 2 | 2.30 | 0.0023 | −4.42 | 0.070 | 2.03 |
| Zn-O,N | 2 | 2.00 | 0.0016 | ||||
| Zn-C | 1 | 2.99 | 0.0092 | ||||
| Zn-C | 1 | [3.06] | [0.0092] | ||||
| Zn-C | 1 | [4.15] | [0.0132] | ||||
| Zn-N | 1 | [4.19] | [0.0132] |
The sample used was M. thermoautotrophicum β-CA. The Stanford Synchrotron Radiation Laboratory file number is ZTC0A (k range = 2 to 12 Å−1). Δk3χ = 12.38. Each group is the chemical unit defined for the multiple scattering calculation. NS is the number of scatterers (or groups) per metal. Ras is the metal-scatterer distance. ςas2 is a mean square deviation in Ras. ΔE0 is the shift in E0 for the theoretical scattering functions. Numbers in square brackets were constrained to be either a multiple of the above value (ςas2) or to maintain a constant difference from the above value (Ras).
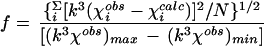 |
Steady-state kinetic measurements.
The pH dependencies of both CO2 hydration and HCO3− dehydration catalyzed by Cab were measured by stopped-flow spectroscopy with the changing pH indicator assay. The progress curves for the hydration of CO2 and dehydration of HCO3− were consistent with Michaelis-Menten kinetics. The efficiency (kcat/Km) for CO2 hydration (Fig. 3A) was several fold greater than that for HCO3− dehydration (Fig. 3B) over the pH range of 6.5 to 7.5, with a 20-fold difference in efficiency at pH 7.0.
FIG. 3.

pH dependence of CO2 hydration and HCO3− dehydration. Activities were measured at 25°C in 50 mM buffer with an ionic strength of 0.1 M. (A) CO2 hydration. Using Cab concentrations of 1.5 to 1.9 μM, the observed steady-state parameters kcat (■) and kcat/Km (□) were determined over a pH range of 6.2 to 9.0. (B) HCO3− dehydration. Using Cab concentrations of 0.8 to 1.8 μM, the kcat/Km (■) was determined over a pH range of 5.5 to 7.5.
Both the steady-state parameters kcat and kcat/Km for CO2 hydration were pH dependent over a range from pH 6.2 to 9.0 and show an increase with increasing pH (Fig. 3A). The CO2 + H2O ⇌ HCO3− + H+ equilibrium becomes increasingly shifted towards HCO3− at increasing pH, requiring the HCO3− dehydration reactions to be performed over the pH range from 5.5 to 7.5. CO2 hydration was not measured below pH 6.2 by this method for the same reasons. The kcat/Km for HCO3− dehydration decreased with increasing pH (Fig. 3B). Neither pH profile could be fitted to a theoretical titration curve with one, two, or three ionizations.
The rate of CO2 hydration determined at both pH 6.5 and 8.5 was found to be strongly dependent on the concentration of buffer (Fig. 4). The steady-state parameter kcat at both pH values was buffer dependent in a saturable manner. Replots of the kcat values yielded an effective Km of 4.8 mM for TAPS at pH 8.5 (Fig. 4A) and 12.7 mM for MES at pH 6.5 (Fig. 4B). These values are typical for the apparent Km of zwitterionic buffers used in this pH range. These results indicate that the buffer behaves kinetically as a second substrate in a ping-pong mechanism, likely accepting a proton from the enzyme during CO2 hydration. The rate constant kcat/Km was not dependent on the concentration of buffer (Fig. 4A and B insets).
FIG. 4.

Buffer dependence of CO2 hydration. CO2 hydration was measured at 25°C at pH 8.5 (A) and pH 6.5 (B) with an ionic strength of 0.1 M and a Cab concentration of 2.0 μM. The CO2 concentration ranged from 6 to 24 mM, and the buffer concentration ranged from 5 to 50 mM. The observed steady-state parameters kcat max and Km, determined by using the Michaelis-Menten equation, were (1.3 ± 0.048) × 104 s−1 and 4.8 ± 0.7 mM, respectively, for TAPS at pH 8.5 and (4.5 ± 0.084) × 103 s−1 and 12.7 ± 0.6 mM, respectively, for MES at pH 6.5. (Insets) Double reciprocal plot of observed initial velocities versus CO2 concentration at different concentrations of TAPS (A) or MES (B) (■, 50 mM; □, 20 mM; ●, 10 mM; ○, 5 mM).
The solvent hydrogen isotope effects on the steady-state parameters for CO2 hydration were measured at pH 8.5 and a concentration of TAPS buffer (50 mM) in which intermolecular proton transfer is not rate limiting. The solvent hydrogen isotope effect on kcat was 2.1 ± 0.1, and no significant effect on kcat/Km (1.2 ± 0.1) was observed.
DISCUSSION
The overall folds of the monomeric mammalian and prokaryotic N. gonorrhoeae α-class carbonic anhydrases are similar, with the antiparallel β-sheet as the dominating secondary structure feature (26). The crystal structure of Cam, the prototypic γ-class carbonic anhydrase, reveals a homotrimer with the monomer adopting a novel left-handed β-helix fold (36). In stark contrast to the mainly β-sheet structures of α- and γ-class carbonic anhydrases, CD analysis of Cab and the S. oleracea enzyme suggests a predominantly α-helical structure. These prokaryotic and eukaryotic β-class carbonic anhydrases are from organisms at the phylogenetic extremes, suggesting this secondary structure feature is common to all β-class enzymes; indeed, the crystal structures of the P. sativum and P. purpureum β-class enzymes reveal a predominance of α-helical structure (35, 41). The content and arrangement of the predicted secondary structure elements for Cab are similar to those of the other β-class carbonic anhydrases (Fig. 5), suggesting a common ancestor, even though the amino acid sequence of Cab is only 23.3% identical to that of the S. oleracea enzyme.
FIG. 5.

Alignment of β-class carbonic anhydrase sequences. An alignment of the amino acid sequences of selected β-class carbonic anhydrases was generated with Clustal X (56). The three conserved zinc ligands are shaded in black, and the two other completely conserved amino acids are shaded in dark gray. The three active site residues not conserved in Cab are shaded in light gray. The open barrels indicate α helices as determined from the P. sativum structure (35), and the arrows represent β strands. The numbering refers to that of the M. thermoautotrophicum Cab amino acid sequence. GenBank accession numbers are as follows: P. sativum (pea), 115471; S. oleracea (spinach), 115472; Oryza sativa (rice), 606817; P. purpureum, 1395172; Coccomyxa sp., 1663720; E. coli CynT, 1657535; and M. thermoautotrophicum ΔH Cab, 1272331.
Nondenaturing polyacrylamide gel electrophoresis and gel filtration chromatography have previously suggested that the β-class carbonic anhydrases from monocotyledenous plants are homodimeric (43) and those from dicotyledenous plants are homooctameric (9); however, a more precise molecular mass has yet to be reported. In contrast to the enzymes isolated from higher plants, gel filtration studies of Cab and the E. coli CynT suggest these prokaryotic enzymes are homotetrameric (22, 50). The more precise analytical ultracentrifugation experiments reported here establish the homotetrameric composition of Cab. Thus, the β-class is distinct from the other two classes in that the β-class carbonic anhydrases are either dimeric, tetrameric, or octameric.
Based on electron microscopy of the chickpea leaf β-class carbonic anhydrase, Aliev et al. (4) presented a model for the quaternary structure of the β-class enzymes from dicotyledenous plants and proposed a 422 (dimer of tetramers) point group symmetry for the eight subunits. Two invariant cysteines (Cys-269 and Cys-272) in the deduced amino acid sequences of β-class carbonic anhydrases from dicotyledenous plants appear necessary for the oligomeric state of the P. sativum chloroplast carbonic anhydrase (9). Studies of these invariant cysteines fit well with the electron microscopy studies indicating a double-layered structure in which each layer is a tetramer. However, the recently solved structure of the P. satvium enzyme indicates the octamer does not have the predicted 422 point of symmetry, but rather has a 222 (dimer of dimers) symmetry (35). The dimeric P. purpureum enzyme, in which each monomer is composed of two internally repeated structures each having an active site (Fig. 5), appears as a tetramer with a pseudo 222 symmetry (41). Why alterations to the invariant Cys-269 and Cys-272 of the dicotyledenous plant enzymes convert the octamer into a tetramer is unclear.
In the P. sativum enzyme, pairs of monomers are joined together through extensive interactions mediated by the α1 and α2 helices and β2 strands (35). The dimer is therefore the basic building block, and nearly all of the protein-protein interactions responsible for forming the loosely packed octamer from dimers are mediated through interactions of the β5 strand. However, the second half of the β5 strand, which mediates most of the oligomerization interactions, is absent in several β-class enzymes, including Cab (Fig. 5). How the dimers are held together in the tetrameric Cab awaits solution of its crystal structure.
XAS analysis clearly indicates that the zinc coordination of β-class Cab is distinct from that of the α- and γ-class carbonic anhydrases (cf. fit 4, Table 2; Fig. 2D). The active site zinc of the α- and γ-class enzymes is coordinated by three histidines and at least one water molecule (1, 28, 61). In the catalytic mechanism of carbonic anhydrase, the zinc-bound water ionizes to a metal-bound hydroxide ion that attacks CO2 (equation 2a). EXAFS results suggest that the active site zinc of β-class Cab is coordinated by two cysteine residues and two oxygen/nitrogen ligands, with the possibility that one of the oxygen/nitrogen ligands derives from histidine (Table 2, fit 4). Our results are nearly identical to EXAFS results previously reported for the S. oleracea enzyme (11, 46). Cys-32, His-87, and Cys-90 of Cab are completely conserved in all known β-class carbonic anhydrase sequences (Fig. 5). The recently solved crystal structures of the P. sativum and P. purpureum β-class carbonic anhydrase confirm that the two conserved cysteines and the conserved histidine are ligands to the active site zinc (35, 41). The second oxygen/nitrogen ligand would be expected to be a water molecule; however, the fourth ligand of the recently solved P. purpureum crystal structure is a conserved aspartate corresponding to Asp-34 of Cab. Nonetheless, several pieces of evidence suggest that the conserved aspartate is unlikely to act as an essential fourth ligand to the active site zinc in β-class carbonic anhydrases. First, this aspartate has previously been shown not to be essential for zinc coordination or catalytic activity. Site-directed mutagenesis studies with the S. oleracea enzyme (11) indicate that alterations to the two conserved cysteine residues and the conserved histidine residue result in inactive variants lacking zinc; however, the variant in which the conserved aspartate was replaced with asparagine retained the active site zinc. Site-directed mutagenesis of the S. oleracea enzyme and Cab indicates that the conserved aspartate is not absolutely required for catalytic activity (41; K. S. Smith, C. J. Ingram-Smith, and J. G. Ferry, unpublished data). Second, the P. purpureum carbonic anhydrase was crystallized at pH 6.75, and previous reports indicate that essentially no activity was detected for this enzyme below pH 7.0 (62), suggesting that the published structure of the P. purpureum enzyme may be of an inactive enzyme. Therefore, Asp-34 is unlikely to be an essential fourth ligand to the active site zinc in Cab. The second oxygen/nitrogen ligand in Cab is most likely a deprotonated water molecule that serves as the zinc hydroxide attacking CO2.
Even though the three zinc ligands (Cys-32, His-87, and Cys-90 of Cab) and two other active site residues (Asp-34 and Arg-36 of Cab) are conserved among all β-class carbonic anhydrases (Fig. 5), key active site residues (Gln-151, Phe-179, and Tyr-205 of the P. sativum enzyme) conserved among the enzymes from dicotyledenous and monocotyledenous plants, algae, and E. coli CynT, are absent in Cab (35). Although the roles of these residues in the P. sativum enzyme have not yet been investigated experimentally, Kimber and Pai (35) propose that Gln-151 may electrophilically activate the CO2 molecule by forming a hydrogen bond with CO2 through its side chain amide. Phe-179 and Tyr-205 form part of an extensive hydrophobic patch whose function may be to ensure that the binding energy of inhibitor molecules is as unfavorable as possible (35). Other members of the same phylogenetic clade as Cab, which consists primarily of sequences from both archaea and gram-positive bacteria species, are also missing these active site residues present in P. sativum (51, 52). In Cab, Gln-151, Phe-179, and Tyr-205 of the P. sativum enzyme are substituted for by histidine (His-23), lysine (Lys-53), and valine (Val-72), respectively (Fig. 5). These substitutions imply that the active site of Cab differs substantially from those of other β-class enzymes. Whether His-23, Lys-53, and Val-72 in Cab play a role similar to that proposed for Gln-151, Phe-179, and Tyr-205 in the plant enzymes remains to be investigated.
Although the active sites of the plant β-class carbonic anhydrases and Cab may be significantly different, the kinetic data presented here suggest that the fundamental catalytic mechanism for Cab is similar to that reported for other β-class enzymes. The steady-state parameter kcat/Km is not dependent on the concentration of buffer, which was shown to act as a second substrate. This result is consistent with the α-class human CA II zinc hydroxide mechanism in which the interconversion of CO2 and HCO3− (equations 2a and 2b), reflected in kcat/Km, is separate from the intramolecular and intermolecular proton transfer steps (equations 2c and 2d) (14, 34, 48). The pH profile of kcat for the hydration of CO2 (Fig. 3A) increases with pH, indicating that an unprotonated form of the enzyme is required for catalytic competence, consistent with nucleophilic attack of a zinc-bound hydroxyl group on CO2. For human CA II, the pH profile of kcat/Km reveals the pKa of the zinc-bound water and the pH profile of kcat reveals the pKa of the proton shuttle residue. The pH profiles of both kcat and kcat/Km in the direction of CO2 hydration for Cab (Fig. 3A) show more complicated behavior and could not be fitted to theoretical curves with one, two, or three ionizations. Nearby ionizable groups may influence these pH profiles to produce multiple pKa values, or the pKa values are lower than 6.0. The steady-state parameter kcat of the β-class S. oleracea carbonic anhydrase was found to be pH dependent, with an apparent pKa of approximately 8.5 in the absence of sulfate (46). Similar to Cab, the pH profiles for both kcat and kcat/Km in the direction of CO2 hydration for the β-class P. sativum carbonic anhydrase are pH dependent, although the profiles were not fitted to theoretical titration curves (29).
No significant hydrogen isotope effect was observed on the steady-state parameter kcat/Km for Cab. This result suggests that D2O imposes no major structural changes in the enzyme and that the catalytic steps up to and including the first committed step of the reaction (equations 2a and 2b) do not contain a rate-contributing proton transfer step, consistent with the zinc-hydroxide mechanism. The solvent hydrogen isotope effect on kcat observed for Cab suggests that an intramolecular proton transfer step is at least partially rate determining (equation 2c). Similar isotope effects on kcat were reported for the bovine CA III α-class carbonic anhydrase (45), the γ-class enzyme Cam (1), and the P. sativum β-class carbonic anhydrase (30). The observed solvent hydrogen isotope effect on kcat of 2.1 for Cab is smaller than the value of 3.8 reported for human α-class CA II (55), but similar to the value reported for human α-class CA IV (27), which follows a mechanism similar to that of CA II.
M. thermoautotrophicum grows optimally at temperatures between 65 and 75°C, and it is expected that the optimal temperature for enzyme activity would fall in this range; however, the decreased solubility of CO2 at these temperatures under atmospheric pressure precludes the determination of accurate kinetic parameters above 25°C. In fact, Cab is the most thermostable carbonic anhydrase yet characterized, retaining greater than 90% activity after incubation at 85°C for 15 min (50). Optimal activity aside, the catalytic efficiency (kcat/Km) for CO2 hydration (Fig. 3A) was several fold greater than that for HCO3− dehydration (Fig. 3B) over the pH range of 6.5 to 7.5, suggesting that the physiological role of Cab is to convert CO2 to HCO3−.
The chemolithoautotrophic M. thermoautotrophicum fixes CO2, and synthesis of oxaloacetate is an important reaction in the CO2-fixation pathways for the methanoarchaea. Oxaloacetate is the starting point of an incomplete reductive citric acid cycle that terminates at α-ketoglutarate and provides precursors for cell material and coenzyme biosynthesis (49). M. thermoautotrophicum possesses two enzymes, pyruvate carboxylase and phosphoenolpyruvate (PEP) carboxylase, for the synthesis of oxaloacetate (49). Bicarbonate has been shown to be the substrate for both of these enzymes; thus, the role of Cab may be to concentrate HCO3− in the vicinity of these enzymes. Similarly, eukaryotic carbonic anhydrase has been shown to provide bicarbonate to both pyruvate carboxylase and PEP carboxylase. The α-class human CA V is a mitochondrial enzyme that provides HCO3− for pyruvate carboxylase in the liver, kidney, and pancreatic islets (42, 59). In the photosynthesis of C4 plants, carbonic anhydrase provides bicarbonate to PEP carboxylase for the initial carboxylation reaction in the fixation of CO2 into C4 acids (6) by rapidly converting CO2 entering the mesophyll cells from the atmosphere to HCO3−.
Conclusions.
Previously, only the plant β-class carbonic anhydrases had been characterized structurally or kinetically. Here we present the first structural and detailed study of a β-class carbonic anhydrase (Cab) from a prokaryote and the first from a chemolithotrophic thermophile. Cab and the enzymes from dicotyledenous plants represent the greatest extremes on the phylogenetic tree of the β-class of carbonic anhydrases (51, 52). The results presented here reveal remarkable similarities between the eukaryotic and prokaryotic enzymes that unite the β-class. Both Cab and the plant enzymes follow a zinc hydroxide mechanism for catalysis. The dominant structure for β-class enzymes is α-helical, and the active site is coordinated by two sulfur and two O/N ligands. These results firmly establish that the α-, β-, and γ-classes are convergently evolved enzymes that, although structurally distinct, are functionally equivalent.
ACKNOWLEDGMENTS
We thank Brandon Doyle for technical assistance with the CD analysis and analytical ultracentrifugation studies and Cheryl Ingram-Smith for assistance with the analytical ultracentrifugation studies. We also thank Matthew Kimber for invaluable discussion of the structure of the P. sativum β-class carbonic anhydrase and David Silverman, Cheryl Ingram-Smith, Brian Tripp, and Christie Brosius for critical reading of the manuscript.
This work was supported by grants from the National Institutes of Health to R.A.S. (GM42025) and J.G.F. (GM44661) and NASA-Ames Cooperative Agreement NCC2-1057 to The Pennsylvania State University Astrobiology Research Center (PSARC). The XAS data were collected at the Stanford Synchrotron Radiation Laboratory (SSRL), which is operated by the Department of Energy, Division of Chemical Sciences. The SSRL Biotechnology program is supported by the National Institutes of Health, Biomedical Resource Technology Program, Division of Research Resources. Support for the X-ray fluorescence detector is from NIH BRS Shared Instrumentation grant RR05648.
REFERENCES
- 1.Alber B E, Colangelo C M, Dong J, Stalhandske C M V, Baird T T, Tu C, Fierke C A, Silverman D N, Scott R A, Ferry J G. Kinetic and spectroscopic characterization of the gamma carbonic anhydrase from the methanoarchaeon Methanosarcina thermophila. Biochemistry. 1999;38:13119–13128. doi: 10.1021/bi9828876. [DOI] [PubMed] [Google Scholar]
- 2.Alber B E, Ferry J G. A carbonic anhydrase from the archaeon Methanosarcina thermophila. Proc Natl Acad Sci USA. 1994;91:6909–6913. doi: 10.1073/pnas.91.15.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alber B E, Ferry J G. Characterization of heterologously produced carbonic anhydrase from Methanosarcina thermophila. J Bacteriol. 1996;178:3270–3274. doi: 10.1128/jb.178.11.3270-3274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aliev D A, Guliev N M, Mamedov T G, Tsuprun V L. Physiochemical properties and quaternary structure of chick pea carboanhydrase. Biokhimiya. 1987;51:1785–1794. [Google Scholar]
- 5.Andkudinov A L, Ravel B, Rehr J J, Conradson S D. Real-space multiple scattering calculation and interpretation of x-ray absorption near-edge structure. Physiol Rev. 1998;58:7565–7576. [Google Scholar]
- 6.Badger M R, Price G D. The role of carbonic anhydrase in photosynthesis. Annu Rev Plant Physiol Plant Mol Biol. 1994;45:369–392. [Google Scholar]
- 7.Bear C A, Duggen K A, Freeman H C. Tetraimidazolezinc(II) perchlorate. Acta Crystallogr Sect B. 1975;31:2713–2715. [Google Scholar]
- 8.Bell R P, editor. The proton in chemistry. Ithaca, N.Y: Cornell University Press; 1959. pp. 183–214. [Google Scholar]
- 9.Bjorkbacka H, Johansson I M, Skarfstad E, Forsman C. The sulfhydryl groups of Cys 269 and Cys 272 are critical for the oligomeric state of chloroplast carbonic anhydrase from Pisum sativum. Biochemistry. 1997;36:4287–4294. doi: 10.1021/bi962825k. [DOI] [PubMed] [Google Scholar]
- 10.Boriack-Sjodin P A, Heck R W, Laipis P J, Silverman D N, Christianson D W. Structure determination of murine mitochondrial carbonic anhydrase V at 2.45-Å resolution: implications for catalytic proton transfer and inhibitor design. Proc Natl Acad Sci USA. 1995;92:10949–10953. doi: 10.1073/pnas.92.24.10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bracey M H, Christiansen J, Tovar P, Cramer S P, Bartlett S G. Spinach carbonic anhydrase: investigation of the zinc-binding ligands by site-directed mutagenesis, elemental analysis, and EXAFS. Biochemistry. 1994;33:13126–13131. doi: 10.1021/bi00248a023. [DOI] [PubMed] [Google Scholar]
- 12.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 13.Chirica L C, Elleby B, Jonsson B H, Lindskog S. The complete sequence, expression in Escherichia coli, purification and some properties of carbonic anhydrase from Neisseria gonorrhoeae. Eur J Biochem. 1997;244:755–760. doi: 10.1111/j.1432-1033.1997.00755.x. [DOI] [PubMed] [Google Scholar]
- 14.Christianson D W, Fierke C A. Carbonic anhydrase: evolution of the zinc binding site by nature and by design. Acc Chem Res. 1996;29:331–339. [Google Scholar]
- 15.Cosper N J, Stalhandske C M V, Iwasaki H, Oshima T, Scott R A, Iwasaki T. Structural conservation of the isolated zinc site in archaeal zinc-containing ferredoxins as revealed by x-ray absorption spectroscopic analysis and its evolutionary implications. J Biol Chem. 1999;274:23160–23168. doi: 10.1074/jbc.274.33.23160. [DOI] [PubMed] [Google Scholar]
- 16.Eriksson A E, Kylsten P M, Jones T A, Liljas A. Crystallographic studies of inhibitor binding sites in human carbonic anhydrase II: a pentacoordinated binding of the SCN− ion to the zinc at high pH. Proteins Struct Funct Genet. 1988;4:283–293. doi: 10.1002/prot.340040407. [DOI] [PubMed] [Google Scholar]
- 17.Eriksson A E, Liljas A. Refined structure of bovine carbonic anhydrase-III at 2.0 angstrom resolution. Proteins Struct Funct Genet. 1993;16:29–42. doi: 10.1002/prot.340160104. [DOI] [PubMed] [Google Scholar]
- 18.Eriksson M, Karlsson J, Ramazanov Z, Gardestrom P, Samuelsson G. Discovery of an algal mitochondrial carbonic anhydrase: molecular cloning and characterization of a low-CO2-induced polypeptide in Chlamydomonas reinhardtii. Proc Natl Acad Sci USA. 1996;93:12031–12034. doi: 10.1073/pnas.93.21.12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukuzawa H, Fujiwara S, Tachiki A, Miyachi S. Nucleotide sequences of two genes CAH1 and CAH2 which encode carbonic anhydrase polypeptides in Chlamydomonas reinhardtii. Nucleic Acids Res. 1990;18:6441–6442. doi: 10.1093/nar/18.21.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukuzawa H, Fujiwara S, Yamamoto Y, Dionisio-Sese M L, Miyachi S. cDNA cloning, sequence, and expression of carbonic anhydrase in Chlamydomonas reinhardtii—regulation by environmental CO2 concentration. Proc Natl Acad Sci USA. 1990;87:4383–4387. doi: 10.1073/pnas.87.11.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glasoe P K, Long F A. Use of glass electrodes to measure acidities in deuterium oxide. J Phys Chem. 1960;64:188–190. [Google Scholar]
- 22.Guilloton M B, Korte J J, Lamblin A F, Fuchs J A, Anderson P M. Carbonic anhydrase in Escherichia coli. A product of the cyn operon. J Biol Chem. 1992;267:3731–3734. [PubMed] [Google Scholar]
- 23.Hakansson K, Wehnert A. Structure of cobalt carbonic anhydrase complexed with bicarbonate. J Mol Biol. 1992;228:1212–1218. doi: 10.1016/0022-2836(92)90327-g. [DOI] [PubMed] [Google Scholar]
- 24.Hewett-Emmett D, Tashian R E. Functional diversity, conservation, and convergence in the evolution of the α-, β-, and γ-carbonic anhydrase gene families. Mol Phylogenet Evol. 1996;5:50–77. doi: 10.1006/mpev.1996.0006. [DOI] [PubMed] [Google Scholar]
- 25.Hiltonen T, Karlsson J, Palmqvist K, Clarke A K, Samuelsson G. Purification and characterisation of an intracellular carbonic anhydrase from the unicellular green alga Coccomyxa. Planta. 1995;195:345–351. doi: 10.1007/BF00202591. [DOI] [PubMed] [Google Scholar]
- 26.Huang S, Xue Y, Sauer-Eriksson E, Chirica L, Lindskog S, Jonsson B H. Crystal structure of carbonic anhydrase from Neisseria gonorrhoeae and its complex with the inhibitor acetazolamide. J Mol Biol. 1998;283:301–310. doi: 10.1006/jmbi.1998.2077. [DOI] [PubMed] [Google Scholar]
- 27.Hurt J D, Tu C, Laipis P J, Silverman D N. Catalytic properties of murine carbonic anhydrase IV. J Biol Chem. 1997;272:13512–13518. doi: 10.1074/jbc.272.21.13512. [DOI] [PubMed] [Google Scholar]
- 28.Iverson T M, Alber B E, Kisker C, Ferry J G, Rees D C. A closer look at the active site of γ-class carbonic anhydrase: high-resolution crystallographic studies of the carbonic anhydrase from Methanosarcina thermophila. Biochemistry. 2000;39:9222–9231. doi: 10.1021/bi000204s. [DOI] [PubMed] [Google Scholar]
- 29.Johansson I M, Forsman C. Kinetic studies of pea carbonic anhydrase. Eur J Biochem. 1993;218:439–446. doi: 10.1111/j.1432-1033.1993.tb18394.x. [DOI] [PubMed] [Google Scholar]
- 30.Johansson I M, Forsman C. Solvent hydrogen isotope effects and anion inhibition of CO2 hydration catalysed by carbonic anhydrase from Pisum sativum. Eur J Biochem. 1994;224:901–907. doi: 10.1111/j.1432-1033.1994.00901.x. [DOI] [PubMed] [Google Scholar]
- 31.Johnson W C J. Analysis of circular dichroism spectra. Methods Enzymol. 1992;210:426–447. doi: 10.1016/0076-6879(92)10022-6. [DOI] [PubMed] [Google Scholar]
- 32.Johnson W C J. Protein secondary structure and circular dichroism: a practical guide. Proteins. 1990;7:205–214. doi: 10.1002/prot.340070302. [DOI] [PubMed] [Google Scholar]
- 33.Kannan K K, Notstrand B, Fridborg K, Lovgren S, Ohlsson A, Petef M. Crystal structure of human erythrocyte carbonic anhydrase B. Three-dimensional structure at a nominal 2.2-Å resolution. Proc Natl Acad Sci USA. 1975;72:51–55. doi: 10.1073/pnas.72.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khalifah R G. The carbon hydroxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isozymes B and C. J Biol Chem. 1971;246:2561–2573. [PubMed] [Google Scholar]
- 35.Kimber M S, Pai E F. The active site architecture of Pisum sativum β-carbonic anhydrase is a mirror image of that of α-carbonic anhydrases. EMBO J. 2000;19:1407–1418. doi: 10.1093/emboj/19.7.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kisker C, Schindelin H, Alber B E, Ferry J G, Rees D C. A left-hand beta-helix revealed by the crystal structure of a carbonic anhydrase from the archaeon Methanosarcina thermophila. EMBO J. 1996;15:2323–2330. [PMC free article] [PubMed] [Google Scholar]
- 37.Lave T M, Shah B D, Ridgeway T M, Pelletier S L. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding S E, Horton J C, Rowe A J, editors. Analytical ultracentrifugation in biochemistry and polymer science. London, United Kingdom: Royal Society of Chemistry; 1992. pp. 90–125. [Google Scholar]
- 38.Liljas A, Kannan K K, Bergsten P C, Waara I, Fridborg K, Strandberg B, Carlbom U, Jarup L, Lovgren S, Petef M. Crystal structure of human carbonic anhydrase C. Nat New Biol. 1972;235:131–137. doi: 10.1038/newbio235131a0. [DOI] [PubMed] [Google Scholar]
- 39.Lindskog S. Structure and mechanism of carbonic anhydrase. Pharmacol Ther. 1997;74:1–20. doi: 10.1016/s0163-7258(96)00198-2. [DOI] [PubMed] [Google Scholar]
- 40.Liu W, Thorp H H. Bond valence sum analysis of metal-ligand bond lengths in metalloenzymes and model complexes. 2. Refined distances and other enzymes. Inorg Chem. 1993;32:4102–4105. [Google Scholar]
- 41.Mitsuhashi S, Mizushima T, Yamashita E, Yamamoto M, Kumasaka T, Moriyama H, Ueki T, Miyachi S, Tsukihara T. X-ray structure of β-carbonic anhydrase from the red alga, Porphyridium purpureum, reveals a novel catalytic site for CO2 hydration. J Biol Chem. 2000;275:5521–5526. doi: 10.1074/jbc.275.8.5521. [DOI] [PubMed] [Google Scholar]
- 42.Parkkila A-K, Scarim A L, Parkkila S, Waheed A, Corbett J A, Sly W S. Expression of carbonic anhydrase V in pancreatic beta cells suggests role for mitochondrial carbonic anhydrase in insulin secretion. J Biol Chem. 1998;273:24620–24623. doi: 10.1074/jbc.273.38.24620. [DOI] [PubMed] [Google Scholar]
- 43.Reed M L, Graham D. Carbonic anhydrase in plants: distribution, properties, and possible physiological roles. Prog Phytochem. 1981;7:47–94. [Google Scholar]
- 44.Rehr J J, Mustre de Leon J, Zabinsky S I, Albers R C. Theoretical x-ray absorption fine structure standards. J Am Chem Soc. 1991;113:5136–5140. [Google Scholar]
- 45.Ren X, Jonsson B H, Millqvist E, Lindskog S. A comparison of the kinetic properties of native bovine muscle carbonic anhydrase and an activated derivative with modified thiol groups. Biochim Biophys Acta. 1988;953:79–85. doi: 10.1016/0167-4838(88)90011-8. [DOI] [PubMed] [Google Scholar]
- 46.Rowlett R S, Chance M R, Wirt M D, Sidelinger D E, Royal J R, Woodroffe M, Wang Y F, Saha R P, Lam M G. Kinetic and structural characterization of spinach carbonic anhydrase. Biochemistry. 1994;33:13967–13976. doi: 10.1021/bi00251a003. [DOI] [PubMed] [Google Scholar]
- 47.Scott R A. Measurement of metal-ligand distances by EXAFS. Methods Enzymol. 1985;117:414–458. [Google Scholar]
- 48.Silverman D N, Lindskog S. The catalytic mechanism of carbonic anhydrase: implications of a rate-limiting proteolysis of water. Acc Chem Res. 1988;21:30–36. [Google Scholar]
- 49.Simpson P G, Whitman W B. Anabolic pathways in methanogens. In: Ferry J G, editor. Methanogenesis: ecology, physiology, biochemistry, and genetics. London, United Kingdom: Chapman & Hall; 1993. pp. 445–472. [Google Scholar]
- 50.Smith K S, Ferry J G. A plant-type (β-class) carbonic anhydrase in the thermophilic methanoarchaeon Methanobacterium thermoautotrophicum. J Bacteriol. 1999;181:6247–6253. doi: 10.1128/jb.181.20.6247-6253.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith K S, Ferry J G. Prokaryotic carbonic anhydrases. FEMS Microbiol Rev. 2000;24:335–366. doi: 10.1111/j.1574-6976.2000.tb00546.x. [DOI] [PubMed] [Google Scholar]
- 52.Smith K S, Jakubziek C, Whittam T S, Ferry J G. Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc Natl Acad Sci USA. 1999;96:15184–15189. doi: 10.1073/pnas.96.26.15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.So A K, Espie G S. Cloning, characterization and expression of carbonic anhydrase from the cyanobacterium Synechocystis PCC6803. Plant Mol Biol. 1998;37:205–215. doi: 10.1023/a:1005959200390. [DOI] [PubMed] [Google Scholar]
- 54.Stams T, Nair S K, Okuyama T, Waheed A, Sly W S, Christianson D W. Crystal structure of the secretory form of membrane-associated human carbonic anhydrase IV at 2.8-Å resolution. Proc Natl Acad Sci USA. 1996;93:13589–13594. doi: 10.1073/pnas.93.24.13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steiner H, Jonsson B H, Lindskog S. The catalytic mechanism of carbonic anhydrase. Hydrogen-isotope effects on the kinetic parameters of the human C isoenzyme. Eur J Biochem. 1975;59:253–259. doi: 10.1111/j.1432-1033.1975.tb02449.x. [DOI] [PubMed] [Google Scholar]
- 56.Thompson J D, Gibson T J, Plewniak F, Jeanmougin F, Higgins D J. The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thorp H H. Bond valence sum analysis of metal-ligand bond lengths in metalloenzymes and model complexes. Inorg Chem. 1992;31:1585–1588. [Google Scholar]
- 58.Tripp B C, Ferry J G. A structure-function study of a proton transport pathway in the γ-class carbonic anhydrase from Methanosarcina thermophila. Biochemistry. 2000;39:9232–9240. doi: 10.1021/bi0001877. [DOI] [PubMed] [Google Scholar]
- 59.Vincent S H, Silverman D N. Carbonic anhydrase activity in mitochondria from rat liver. J Biol Chem. 1982;257:6850–6855. [PubMed] [Google Scholar]
- 60.Wilbur K M, Anderson N G. Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem. 1948;176:147–154. [PubMed] [Google Scholar]
- 61.Yachandra V, Powers L, Spiro T G. X-ray absorption spectra and the coordination number of Zn and Co carbonic anhydrase as a function of pH and inhibitor binding. J Am Chem Soc. 1983;105:6596–6604. [Google Scholar]
- 62.Yagawa Y, Muto S, Miyachi S. Carbonic anhydrase of a unicellular red alga Porphyridium cruentum R-1. I. Purification and properties of the enzyme. Plant Cell Physiol. 1987;28:1253–1262. [Google Scholar]