

Abstract
Age-related diseases are frequently linked to pathological immune dysfunction, including excessive inflammation, autoreactivity and immunodeficiency. Recent analyses of human genetic data have revealed that somatic mutations and mosaic chromosomal alterations in blood cells — a condition known as clonal haematopoiesis (CH) — are associated with ageing and pathological immune dysfunction. Indeed, large-scale epidemiological studies and experimental mouse models have demonstrated that CH can promote cardiovascular disease, chronic obstructive pulmonary disease, chronic liver disease, osteoporosis and gout. The genes most frequently mutated in CH, epigenetic regulators TET2 and DNMT3A, implicate increased chemokine expression and inflammasome hyperactivation in myeloid cells as a possible mechanistic connection between CH and age-related diseases. In addition, TET2 and DNMT3A mutations in lymphoid cells have been shown to drive methylation-dependent alterations in differentiation and function. Here we review the observational and mechanistic studies describing the connection between CH and pathological immune dysfunction, the effects of CH-associated genetic alterations on the function of myeloid and lymphoid cells, and the clinical and therapeutic implications of CH as a target for immunomodulation.
Introduction
Physiological haematopoiesis leads to the production of blood cells involved in tissue oxygenation, haemostasis and immunity1. Disruption of normal haematopoiesis can cause impaired production or function of red blood cells, platelets or immune cells and occurs in a wide variety of pathological states, including nutrient and growth factor deficiencies, exposure to toxic substances, infection, autoimmunity, neoplasia and inherited bone marrow failure syndromes2–8. In this context, aberrant haematopoiesis is often associated with clinical outcomes related directly to decreased abundance of specific haematopoietic cells (for example, increased bleeding in patients with thrombocytopenia)9. Similarly, clinical indicators of immunodeficiency in patients with aberrant haematopoiesis can also be attributed to cytopenias (for example, increased risk of infection in the setting of lymphopenia or neutropenia)10,11. Interestingly, aberrant haematopoiesis that occurs in patients with certain haematological neoplasms is often accompanied by clinical manifestations of immune dysregulation. For example, autoimmune or autoinflammatory conditions have been reported in 10–25% of patients with myelodysplastic syndromes (MDS) or chronic myelomonocytic leukaemia (CMML)12, while autoantibodies leading to the destruction of red blood cells or platelets develop in approximately 25% of patients with chronic lymphocytic leukaemia (CLL)13.
The mechanistic underpinnings of immune dysfunction in patients with haematological neoplasms have yet to be fully elucidated. However, it has been noted that autoimmune or autoinflammatory conditions can occur several years preceding the diagnosis of a haematological neoplasm, suggesting that pathological changes in immune function occur in a premalignant state. More recently, technological advances in nucleic acid sequencing combined with curation of large clinical datasets have enabled population-wide characterization of somatic genetic alterations in blood cells and the associated clinical phenotypes in hundreds of thousands of individuals. These analyses have revealed that somatic genetic alterations frequently found in patients with haematological malignancies are also common in aged individuals despite lack of evidence of cancer. This phenomenon, referred to as clonal haematopoiesis (CH), has now been linked to a wide range of conditions, including haematological neoplasms and diseases of inflammageing (Box 1). Clonal haematopoiesis of indeterminate potential (CHIP) comprises a subset of CH, defined specifically by the presence of somatic mutations in individual haematological malignancy-associated genes at an allele fraction greater than 2%. Mosaic chromosomal alterations (mCAs), which consist of somatic gains or losses affecting the entirety or parts of a chromosome, represent another type of CH detectable in blood cells. Here, we review the observational studies describing the association between autoimmunity, immunodeficiency and haematological neoplasms as well as the mechanistic connections between the somatic genetic alterations observed in CH and functional dysregulation of myeloid and lymphoid immune cells.
Box 1. Inflammageing.
Ageing is accompanied by gradual changes in the immune landscape. Adaptive immune function declines: B cells display reduced antibody diversity, and T cells develop a more restricted T cell receptor repertoire, impaired effector cell function and skewed memory to effector cell ratio195–197. The result is an overall impaired response to antigenic challenge known as immunosenescence. On the other hand, innate immune responses progress to a chronic baseline activity level, characterized by increased levels of circulating proinflammatory cytokines and low-level infiltration by innate immune cells, such as neutrophils and macrophages. This condition of persistent low-grade sterile inflammation is termed inflammageing198.
Inflammageing is largely mediated by the innate immune system, which mounts immediate, nonspecific immune defenses in response to a wide range of pathogens via the recognition of external pathogen-associated molecular patterns (PAMPs) and intrinsic damage-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs). Stimulation of PRRs leads to the formation of macromolecular platforms, known as inflammasomes, which activate caspase 1-mediated processing of the pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18. During ageing, increasing PAMP and DAMP signals resulting respectively from gut epithelial barrier changes and accumulating genomic damage and oxidative stress induce Toll-like receptor- and inflammasome-mediated inflammatory responses. Senescent cells acquire a hypersecretory phenotype consisting of increased expression of proinflammatory cytokines, chemokines, angiogenic factors, proteases and matrix metalloproteinases, known as the senescence associated secretory phenotype (SASP)199. The expression of proinflammatory cytokines IL-6 and IL-8 is a consistent feature of the SASP and constitutes a significant driver of inflammageing by priming the transcription of the NLRP3 inflammasome in an age-dependent manner.
Several studies have shown that ageing haematopoietic stem cells (HSCs) upregulate NLRP3 inflammasome activity. Aged HSCs demonstrate heightened NLRP3-dependent inflammatory responses to lipopolysaccharide and ATP stimulation compared with young HSCs due to increased mitochondrial oxidative stress200. Similarly, NLRP3 priming and activation are exaggerated in aged human monocytes201. In a timecourse of ageing, mice overexpressing the proinflammatory molecule S100A9 develop increasing NLRP3 activation in bone marrow cells between 2 to 11 months of age202. These studies reveal that inflammageing occurs within both differentiated and undifferentiated haematopoietic compartments.
Broader health implications of inflammageing include an increased susceptibility to latent common viral infections, such as by cytomegalovirus, herpes simplex virus 1 and varicella zoster virus (VZV). Inflammageing also attenuates vaccination responses, for example by promoting skin recruitment of peripheral blood monocytes in response to a VZV challenge, leading to higher levels of T cell-suppressing cyclooxygenases and prostaglandins and reduced VZV-specific T cell responses203. Furthermore, many chronic diseases of ageing, such as metabolic syndrome and atherosclerosis, rheumatic and neurodegenerative diseases, osteoporosis and cancer, are involved in a maladaptive cycle of immune cell infiltration, tissue damage and release of proinflammatory signals204. Many of these diseases are also associated with haematological neoplasia, including clonal haematopoiesis, likely through dysregulated immune signalling by infiltrating haematopoietic cells.
Immune dysregulation in haematological neoplasms
Haematological neoplasms are often associated with concurrent or antecedent indicators of immune dysregulation, including autoimmunity and immunodeficiency. Evidence of immune dysregulation has been observed in patients with myeloid and lymphoid neoplasms, although there are particular diagnoses of autoimmunity and immunodeficiency that are associated with specific cell lineages. In the following sections, we provide a summary of the clinical observational studies describing the manifestations of immune dysregulation in the setting of myeloid and lymphoid neoplasms.
Myeloid neoplasms.
The presence of laboratory and clinical indicators of immune dysregulation in myeloid neoplasms has been recognized since the 1960s14–18. Immune aplastic anaemia shares significant clinicopathological overlap with hypoplastic MDS19, further emphasizing the link between immune state and haematological disease. Several small case series have also suggested that frank rheumatic disease, including vasculitis, relapsing polychondritis, rheumatoid arthritis and polymyalgia rheumatica, are more common in patients with myeloid neoplasms20–29.
Analyses of large, population-based cohorts have revealed associations between myeloid neoplasms and prior autoimmunity or age-related diseases with inflammatory components (i.e., diseases of inflammageing)30–32 (Box 1). Two case-control studies of acute myeloid leukemia (AML) and MDS in the United States and Sweden found that patients were significantly more likely than controls to have antecedent diagnoses of autoimmunity30,31. A recent analysis of approximately 20,000 MDS and CMML cases demonstrated that such patients were more likely to have a history of diseases associated with inflammageing compared with either healthy controls or patients with solid malignancies32. In all three studies, these associations remained statistically significant even when the analyses were restricted to autoimmune or inflammageing-related diagnoses that occurred more than 3–5 years prior to the onset of a myeloid neoplasm. This temporal sequence suggests that autoimmunity may be driven by clonal premalignant cells. Indeed, myeloid clones have been detected within inflammatory skin lesions from patients with and without underlying myeloid neoplasms33–37. In addition, several lines of evidence have suggested that dysregulated inflammation and immune signalling contribute to the development of haematological neoplasms38,39.
Increased secretion of proinflammatory cytokines by monocytes and macrophages also constitutes part of immune dysregulation in myeloid neoplasms. It has been long recognized that MDS, AML and CML show increased serum levels of tumour necrosis factor (TNF), IL-1β and IL-640–44. Increased levels of TNF are associated with bone marrow failure and inferior prognosis in MDS45,46. Lower IL-1β levels predict favourable response to erythropoietin and better survival in MDS47. In CML, higher IL-1β is associated with poor prognosis and blast crisis41. Increased expression of proinflammatory cytokines directly influences the cell fate and function of haematopoietic progenitor cells in myeloid neoplasms. For example, chronic stimulation by IL-1β promotes haematopoietic stem cell (HSC) cell cycle exit and differentiation into myeloid cells in a process dependent on the myeloid transcription factor CEBPA48,49. HSCs lacking Cebpa are resistant to the pro-differentiation effect of IL-1β, thereby gaining a competitive advantage under IL-1 stimulation. IL-1 also selects for Tet2 deficient clonal expansion by promoting HSC cell cycle progression and multilineage differentiation in mice39. Mutations in Tet2, Dnmt3a and Asxl1, which encode the mouse homologues of epigenetic modifiers that are frequently mutated in CH, MDS and myeloid malignancies in humans, further render HSCs more resistant to acute inflammatory stresses from lipopolysaccharide and IL-1β, thereby supporting clonal expansion and malignant evolution via different mechanisms50–52. Finally, increased IL-6 signalling in the setting of myeloid neoplasms has been associated with aggressive disease and poor outcomes in humans and mice43,53–55.
Lymphoid neoplasms.
It is well-established that CLL is associated with increased risk of infectious complications56 due to reduced levels of protective immunoglobulin57,58 and impaired capacity to mount a robust antibody response59. Patients often have a significant clinical history of infection years before the diagnosis of CLL, suggesting that abnormal lymphopoiesis or lymphocyte function creates an immunocompromised state during the premalignant period60. Indeed, the diagnosis of CLL is frequently preceded by a monoclonal B cell lymphocytosis (Box 1), which is also associated with increased risk of infection and abnormalities in serum immunoglobulin levels61,62.
Clinical manifestations of hyperinflammatory immune dysregulation are commonly observed in patients with T and natural killer (NK) cell neoplasms. Angioimmunoblastic T cell lymphoma (AITL) often presents with signs of rheumatic disease, including polyarthritis, leukocytoclastic vasculitis, serositis and autoimmune cytopenias63; NK cell large granular lymphocyte (LGL) leukemia and chronic lymphoproliferative disorder of NK cells (CLPD-NK) are typically associated with autoimmune cytopenias, especially neutropenia; and the macrophage activation syndrome haemophagocytic lymphohistiocytosis (HLH), which is characterized by fever, elevated ferritin, hepatosplenomegaly and pancytopenia, has been especially linked to extranodal NK/T cell lymphoma (ENKTL)64. Altogether, there is a broad spectrum of immune dysregulation and clinical features of inflammation observed in the setting of T and NK cell malignancies.
Clonal haematopoiesis
There is now ample evidence demonstrating that haematological malignancies arise via serial acquisition of mutations by a founding clone65–69, implying the existence of premalignant CH in which genetic lesions are detectable in the absence of disease (Box 2). Additional evidence of CH emerged from studies that reported skewing of X-inactivation and clonal mutations in TET270–72. Subsequent analyses of several genome-wide association studies also observed mCAs in peripheral blood cells from individuals with no evidence of haematological malignancy73,74. In the sections below, we review the genetic, demographic and clinical features of both CH defined by mutations in single genes associated with haematological malignancies and CH defined by mCAs (Fig. 1).
Box 2. Clonal evolution of haematological neoplasms.
In normal haematopoiesis, haematopoietic stem cells differentiate into multipotent progenitors, which eventually give rise to all differentiated cell lineages comprising blood. This cellular hierarchy becomes disrupted in malignant transformation, during which somatic mutations cause an impairment in cell differentiation together with enhanced survival and proliferation. The serial acquisition of driver genetic mutations and chromosomal aberrations in these cancer-initiating clones have been reported in significant detail in several recent studies205–208. Mutation selection in pre-leukemic cells results in an iterative process of clonal expansion and genetic complexity, favouring the accumulation of further mutations and the development of disease. Many of these mutation events are reproducible both in the genes affected and the temporal sequence in which they occur. For example, myeloid diseases are typified by recurrent, early mutations in chromatin modifiers (DNMT3A, TET2, ASXL1, IDH1, IDH2 and EZH2) and RNA splicing machinery (SF3B1, U2AF1, SRSF2 and ZRSR2). Mechanistically, DNMT3A catalyzes de novo DNA methylation whereas TET2 oxidizes methylated CpG to 5-hydroxymethyl cytosine (5-hmC) facilitating further modification to unmodified cysteine; mutations thus cause dysregulated DNA methylation and demethylation, respectively, altering the expression of genes involved in proliferation, differentiation and inflammation via chromatin-level changes209,210. Mutations involving the DNA damage response (TP53 and PPM1D) are strongly associated with therapy-related myeloid neoplasms arising in individuals who had received chemotherapy in the past, indicating a distinct pathogenetic mechanism in this group. With large scale sequencing analyses now being commonplace, multiple studies have demonstrated that these driver mutations, especially those occurring early in disease, are shared with pre-leukemic myeloid conditions such as myelodysplastic syndrome and myelodysplastic or myeloproliferative neoplasms, emphasizing the progressive nature of haematological cancers66,68,194,211. In later stages of acute myeloid leukemia (AML) leukemogenesis, mutations in signal transduction genes (KRAS, NRAS, CBL, JAK2, GNB1, KIT, FLT3 and NPM1) and myeloid transcription factors (EVI1, CEBPA and RUNX1) predominate77,212.
The recognition that clonal haematopoiesis bearing somatic mutations is common among older individuals has established an early signpost in myeloid neoplasia88,213. A formal definition has been established for recognizing clonal haematopoiesis of indeterminate potential (CHIP) as a clinical entity characterized by the presence of somatic mutations in genes associated with haematological malignancies at greater than 2% allele fraction in asymptomatic individuals214–217. With the improvement in sequencing capability, it is now possible to detect clonal haematopoiesis at much greater sensitivity218. However, it is unlikely that these diminutive clones will result in either disease progression or significant morbidity. Early outcome studies based on CHIP ≥2% variant allele frequency (VAF) showed a progression rate of 0.5–1% per year76. In this context, CHIP is analogous to such asymptomatic precursor lesions as high-count monoclonal B cell lymphocytosis and monoclonal gammopathy of undetermined significance, which respectively precede chronic lymphocytic leukemia and small lymphocytic lymphoma and multiple myeloma.
Figure legend: *The rate of progression from CHIP to a myeloid neoplasm varies across different genes. For example, multiple studies have reported that mutations in splicing factor genes or TP53 confer a higher risk of progression83,86–88.
**Among individuals who develop myelodysplastic syndrome (MDS), myeloproliferative neoplasm (MPN), or either disorder after CHIP, the rate of progression to acute myeloid leukemia (AML) varies significantly depending on the identity and characteristics of the pre-AML diagnosis. For example, over a one-year follow-up period, the rate of transformation to AML is 1–2% among patients with the MPN essential thrombocythemia219 but more than 30% among patients with high-risk MDS220.
***The rate of progression from smouldering multiple myeloma (SMM) to multiple myeloma (MM) varies significantly depending on specific SMM disease characteristics, including M protein quantity, percentage of monoclonal plasma cells in the bone marrow and disease cytogenetics221. Over a two-year follow-up period, patients with SMM with low-risk features have a progression rate of less than 10%, whereas those with high-risk features have a progression rate greater than 60%.
Figure 1 |. Features of clonal haematopoiesis.
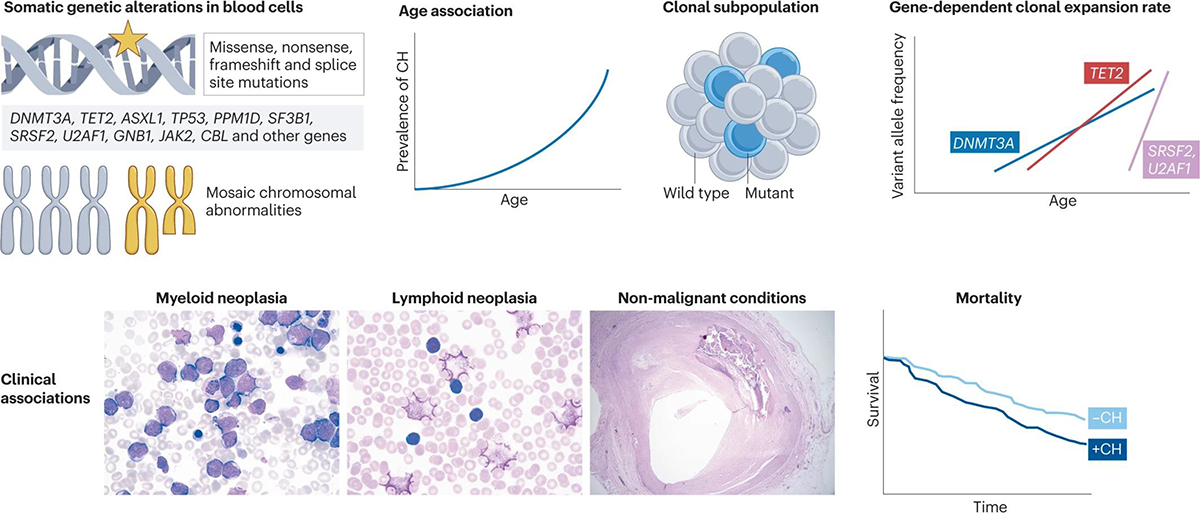
Analysis of human genetic data has led to the discovery of somatic mutations in peripheral blood cells. These mutations comprise non-synonymous variants in genes frequently altered in myeloid malignancies (for example, DNMT3A, TET2 and ASXL1) as well as mosaic chromosomal abnormalities. These genetic alterations occur in a clonal subpopulation of blood cells and are detected more frequently in older individuals. Mutations in different genes tend to follow unique trajectories of clonal expansion over time88–90,194, implying distinct mechanisms of positive selection for different mutations. Clonal haematopoiesis has also been linked to several clinical phenotypes, including incident myeloid and lymphoid neoplasms, non-malignant diagnoses (such as atherosclerosis) and overall mortality. Images courtesy of the Department of Pathology at the Brigham and Women’s Hospital, Boston, USA.
Clonal haematopoiesis associated with individual haematological malignancy genes.
Several independent analyses of more than 30,000 exomes derived from cohort studies of type 2 diabetes and schizophrenia, as well as The Cancer Genome Atlas, demonstrated that ageing individuals without haematological malignancies harbour clonal mutations in their peripheral blood75–77. Mutations were more common in older individuals, occurring in 10–20% of individuals over the age of 70 compared with <5% of individuals under the age of 50. Notably, the majority of CH mutations were in genes associated with MDS and myeloid malignancies. Consistent with the idea of clonal evolution from a premalignant state, the detection of a mutation at the time of DNA collection was associated with a dramatically increased risk of a subsequent myeloid malignancy (Box 2).
Several follow-up analyses with even larger numbers of subjects confirmed that the presence of blood cells harbouring clonal mutations in genes associated with myeloid neoplasia is common in aged individuals and confers an increased risk of mortality, malignancy and non-malignant conditions78–85. The number of mutations, size of the clone(s) and the identity of the gene(s) mutated seem to affect the likelihood of these outcomes, but it is still unclear which individuals with CH are at highest risk. Analysis of peripheral blood DNA collected years before the diagnosis of AML demonstrated that risk of malignant transformation was higher among individuals with a greater number of mutations and among individuals with larger, rapidly expanding clones84,86–90. The risk of AML also varied among different CH-associated genes, likely corresponding to the mechanisms by which mutations promote clonal advantage and/or impact differentiation. For example, mutations in splicing factor genes or TP53 conferred a significantly greater risk of malignant transformation than mutations in DNMT3A or TET2, even though the latter mutations occur more frequently. Future analyses of larger patient cohorts and mechanistic studies will facilitate the discovery of other genetic, demographic, clinical, and laboratory variables that will aid in the risk stratification of individuals with CH.
Another recent study mined exome sequencing data from over 50,000 individuals using a set of target genes somatically mutated in either myeloid or lymphoid malignancies79. Similar to myeloid malignancy-associated mutations, the frequency of somatic mutations in lymphoid malignancy-associated genes increased with age. Lymphoid malignancy-associated mutations were less common than myeloid malignancy-associated mutations, occurring in 1% and 6% of individuals, respectively. Among individuals with lymphoid malignancy-associated mutations, DUSP22, FAT1, KMT2C, KMT2D, SYNE1 and ATM were the genes most frequently affected; consistent with previous studies, DNMT3A, TET2 and ASXL1 were the most frequently mutated myeloid malignancy-associated genes. There was a striking correlation between incident haematological malignancies and the type of premalignant mutation detected. The presence of a lymphoid malignancy-associated mutation conferred an increased risk of developing a lymphoid malignancy (hazard ratio 4.2) but not a myeloid malignancy. Conversely, the presence of a myeloid malignancy-associated mutation conferred an increased risk of developing a myeloid malignancy (hazard ratio 7) but not a lymphoid malignancy, although somatic mutations in DNMT3A and TET2 have also been widely described as oncogenic drivers in several lymphoid neoplasms (Box 3).
Box 3. DNMT3A and TET2 mutations in lymphoid malignancies.
Although typically associated with the development of myeloid neoplasia, recurrent somatic mutations in the epigenetic modifiers DNMT3A and TET2 occur commonly in lymphoid malignancies characterized by autoimmune features, including angioimmunoblastic T cell lymphoma (AITL)222–225, clonal lymphoproliferative disorders of NK cells (CLPD-NK)226, large granular lymphocyte (LGL) leukemias227, T cell acute lymphoblastic leukemia (T-ALL)228–230, and extranodal NK/T cell lymphoma (ENKTL)231 and diffuse large B cell lymphoma (DLBCL)232,233.
In some cases, lymphoid neoplasia appears to develop from an haematopoietic stem cell (HSC) carrying a DNMT3A or TET2 mutation. In AITL, TET2 mutations have been detected in CD34+ progenitor cells with both myeloid and lymphoid differentiation potential225. Similarly, TET2 mutations in patients with CLPD-NK have been detected in both NK and myeloid cells226. Interestingly, families with germline heterozygous TET2 or DNMT3A mutations develop predominantly myeloid neoplasms234–238, whereas rare individuals with germline homozygous TET2 mutations develop either B or T cell lymphomas along with a lymphoproliferative syndrome characterized by autoimmune cytopenias and immunodeficiency239. Finally, several studies have described co-occurrence of lymphoid and myeloid malignancies harbouring DNMT3A or TET2 mutations within the same patient226,240,241. Altogether, these observations strongly suggest that DNMT3A or TET2 mutant HSCs can give rise to both lymphoid and myeloid malignancies; the molecular basis of initiating one or both malignant outcomes has yet to be fully elucidated.
Clonal haematopoiesis associated with mosaic chromosomal abnormalities.
In addition to clonal mutations in individual haematological malignancy genes, CH can be defined by clonal cytogenetic aberrations. In 2012, two independent groups analyzing SNP microarray data from more than 100,000 individuals in total observed that mCAs detected in peripheral blood cells were associated with ageing and conferred an increased risk of developing a haematological malignancy73,74. More recently, analyses of several additional cohorts comprising more than 700,000 individuals in total confirmed these findings and provided novel insights into the laboratory findings and incident clinical diagnoses associated with the presence of specific mCAs91–93. For example, detection of mCAs commonly observed in CLL (for example, events involving chromosomes 3, 12 and 13) were associated with elevated lymphocyte counts and incident diagnosis of CLL; mCAs defined by 9p loss of heterozygosity including the JAK2 locus were associated with elevated levels of red blood cells, platelets and neutrophils and incident diagnosis of a myeloproliferative neoplasm91. By contrast, the most common mCA, loss of chromosome Y, has been associated with increased platelet counts but not consistently with increased risk of neoplasia91,92,94,95.
Multiple studies have described CH with both mCAs and individual gene mutations in the same individual79,81,96. mCAs were more frequent among those with individual CH gene mutations compared to those without individual CH gene mutations. In addition, mCAs defined by loss of heterozygosity tended to affect loci containing individual CH-associated gene mutations. For example, DNMT3A, TET2, TP53 and JAK2 mutations were associated with loss of heterozygosity at chromosomes 2p, 4q, 17p and 9p, respectively.
[H2] Clonal haematopoiesis, mortality and non-malignant disease.
In addition to an increased risk of developing a myeloid malignancy, individuals with mutations in haematological malignancy-associated genes consistently show higher all-cause mortality compared with individuals without mutations75,76,78–84. Surprisingly, the increased risk of death among individuals with mutations is not attributable to haematological malignancy but rather to atherosclerotic cardiovascular disease76,81,82,84,97. Interestingly, all-cause mortality and cardiovascular disease were significantly increased only among individuals with myeloid malignancy-associated mutations; the risk of these adverse outcomes was the same among individuals with lymphoid malignancy-associated mutations and those without any malignancy-associated mutations79.
An increase in mortality unrelated to haematological malignancy was also observed among individuals with mCAs. The cause of this mortality increase has not been determined although it could be linked to an increased risk of infection in individuals with mCAs93, suggesting that mCAs are associated with an immunodeficient phenotype. In this context, higher lymphocyte counts and increased incidence of CLL were observed among individuals with mCAs, indicating that these genetic alterations are markers of the CLL premalignant condition monoclonal B cell lymphocytosis (Box 2). Similar to lymphoid malignancy-associated mutations, mCAs did not confer an increased risk of death due to cardiovascular disease79,81,98, implying that the mechanisms by which mCAs affect the development of non-malignant disease is also distinct from CH with mutations in individual myeloid malignancy-associated genes. It remains unclear if the difference in non-malignant phenotypes between different types of CH is due to differences in the genes mutated or the cell types affected. Interestingly, in the setting of CH with combined mCAs and individual mutations in myeloid malignancy-associated genes, mortality due to incident haematological malignancies or cardiovascular disease was increased compared with either type of CH alone81. This observation was independent of the total number of CH genetic alterations and the presence of individual gene mutations and mCAs affecting the same locus. Further investigation is required to determine the mechanisms by which the co-occurrence of different types of CH alter clonal evolution and non-malignant phenotypes.
Although CH encompasses a wide range of cytogenetic and genetic aberrations, including a substantial fraction of cases with as yet unknown driver mutations75,80, the study of disease associations in CH have focused on CHIP due to the relative consistency and reproducibility of identifying recurrent, high-allele-fraction mutations in genes implicated in haematological malignancies. Since the initial identification of CHIP as a risk factor for cardiovascular disease, CHIP has been found to be significantly associated with other common disorders typified by robust inflammation, ageing or both (Table 1). Using a combination of large scale population-based genetic analysis and mouse models of human disease, CHIP has been associated with heart failure99,100, atrial fibrillation83, chronic kidney disease84,101,102, chronic liver disease103, chronic obstructive pulmonary disease104, gout105, inflammatory bowel disease106, autoimmunity107, osteoporosis108, premature menopause109, venous thromboembolism110, pulmonary hypertension111, post-transplant liver disease112, type II diabetes113, solid cancers83,84,114,115, stroke116 and Alzheimer disease117. Epidemiological tools such as Mendelian randomization have allowed causal inferences to be made regarding the role of CHIP in various diseases83,101,103,118,119.
Table 1 |.
Non-malignant conditions associated with clonal haematopoiesis of indeterminate potential
| Diagnosis | Patient cohorts | Odds ratio | Mouse models | In vivo mouse phenotypes | Refs. |
|---|---|---|---|---|---|
| Alzheimer disease | TOPMed, ADSP | ~0.5–0.8 | NA | NA | 117 |
| Atherosclerotic cardiovascular disease | TOPMed, UKB | ~1.1–1.5 |
Tet2–/– bone marrow → Ldlr
–/–mice fed a high fat diet (myeloid-restricted Tet2–/–sufficient mice) |
↑plaque size; ↑IL-1β, CXCL1, CXCL2, CXCL3 | 76,81,82,84,97,129 |
| Atrial fibrillation | UKB | ~1.1–1.2 | NA | NA | 83 |
| Heart failure | TOPMed, UKB | ~1.1–1.5 |
Tet2–/– bone marrow → wild-type mice with subsequent vascular ligation or constriction Tet2 or Dnmt3a CRISPR-Cas9 edited bone marrow → wild-type mice treated with angiotensin II (myeloid-restricted Tet2–/–sufficient mice) |
↑systolic dysfunction; ↓ejection fraction; ↑fibrosis; ↑IL-1β, CCL2, CCL5 | 99,100 |
| Cerebrovascular accidents | TOPMed, UKB, MGBB | ~1.1–1.2 | NA | NA | 116 |
| Chronic kidney disease | UKB | ~1.1–1.5 | Tet2 or Dnmt3a CRISPR-Cas9 edited bone marrow → wild-type mice treated with angiotensin II | ↑fibrosis | 101,102 |
| Chronic liver disease | FHS, ARIC, UKB, MGBB | ~2–4 |
Tet2–/– or Dnmt3a–/– bone marrow → Ldlr
–/– mice fed a high fat diet (myeloid-restricted Tet2–/– sufficient mice) |
↑steatohepatitis; ↑fibrosis; ↑IL-6, CXCL1, CCL22 | 103 |
| Chronic obstructive pulmonary disease | TOPMed, ICGN, EOCOPD, UKB | ~1.5–2 | Tet2–/– bone marrow → wild-type mice exposed to cigarette smoke and poly(I:C) | ↑emphysema; ↑IFNγ and type I IFNs | 104 |
| Gout | UKB, MGBB | ~1.2–1.7 |
Tet2–/– bone marrow → wild-type mice injected with monosodium urate crystals in the footpad |
↑paw oedema; ↑IL-1β, CCL5, CCL22 | 105 |
| Osteoporosis | UKB | ~1.2–1.5 |
Tet2–/–, Dnmt3a–/– or Dnmt3a R878H bone marrow → Ldlr–/–mice fed a high fat diet or wild-type aged mice fed a regular diet |
↓bone mass; ↑osteoclasts | 108 |
| Premature menopause | UKB, WHI | ~1.1–1.7 | NA | NA | 109 |
ARIC, Atherosclerosis Risk in Communities; ADSP, Alzheimer’s Disease Sequencing Project; CCL, CC-chemokine ligand; CXCL, CXC-chemokine ligand; EOCOPD, Boston Early-Onset COPD Study; FHS, Framingham Heart Study; ICGN, International COPD Genetics Network; IFN, interferon; MGBB, Mass General Brigham Biobank; NA, not available; TOPMed, Trans-Omics for Precision Medicine program; UKB, UK Biobank; WHI, Women’s Health Initiative.
Analyses of epigenetic ageing, which has been associated with a wide range of adverse outcomes in humans120, have refined the relationship between CHIP and non-malignant phenotypes. Individuals with CHIP showed evidence of accelerated epigenetic ageing121,122. Moreover, the increase in all-cause mortality and atherosclerotic cardiovascular disease among individuals with CHIP was observed in those with epigenetic age acceleration but not in those without121. In this context, the likelihood of detecting an association between CHIP and certain phenotypes may depend on factors other than the presence of somatic mutations alone. Indeed, a recent Mendelian randomization analysis to identify diseases and biomarkers linked to CHIP did not find an association with atherosclerotic cardiovascular disease or stroke83. It remains to be determined if additional analyses of CHIP with and without accelerated epigenetic ageing specifically highlight those individuals with significantly increased risk of atherosclerosis or other clinical phenotypes.
Apart from disease associations, population-based studies have shown that individuals with CHIP exhibit overall higher levels of IL-6 and C-reactive protein78,123,124. Specifically, TET2 mutated CHIP is significantly associated with increased serum IL-1β, whereas JAK2 and SF3B1 mutated CHIP correlated with increased serum IL-6 levels123. In a separate study, DNMT3A mutated CHIP was specifically associated with elevated serum interferon-γ (IFNγ) levels in patients with ulcerative colitis106. Although patients with myeloid malignancies have been known to exhibit high levels of pro-inflammatory cytokines43,125,126, these new studies suggest a pattern of gene-specific effects on the inflammatory cytokine milieu in CHIP and related haematological diseases. Another strand of evidence highlighting the influence of CHIP on the inflammatory state lies in the finding that CHIP-mutated haematopoietic cells can be identified at sites of inflammation. TET2, JAK2 and TP53 mutant myeloid cells have been identified in the synovial fluid from individuals with CHIP who have rheumatoid arthritis127. In mice transplanted with Tet2 deficient bone marrow cells, Tet2−/− myeloid cells were enriched in the heart and correlated with increased cardiac hypertrophy and fibrosis128. To summarize, the study of CHIP provides a mechanistic framework for understanding the long-recognized association between myeloid neoplasia and autoimmune and inflammatory conditions as well as molecular insights into diseases of ageing.
Mechanisms of myeloid dysregulation in clonal haematopoiesis
CH has been shown to play a causal role in disease pathogenesis using mouse models transplanted with Tet2−/− bone marrow haematopoietic cells and exposed to specific metabolic and environmental conditions. Tet2-deficient haematopoietic cells exacerbate the development of atherosclerosis in Ldlr−/− mice97,129; interestingly, this effect was observed in the context of myeloid-specific Tet2 deficiency, implying that the presence of a mutation in myeloid cells was sufficient to promote atherosclerosis97,129. Mice transplanted with Tet2-deficient bone marrow cells show increased immune infiltration in models of gout, chronic liver disease and chronic obstructive pulmonary disease103–105. Furthermore, Tet2-deficient macrophages expressed increased levels of several inflammatory chemokines and cytokines compared with wild-type macrophages97,129–131, corroborating human studies in which patients with TET2 mutations had higher circulating levels of the inflammatory chemokine IL-897. Together, these data indicated that increased myeloid-derived inflammation may underlie the association between clonal haematopoiesis and cardiovascular disease.
Mechanistically, the best characterized Tet2-dependent proinflammatory mediator is the NLRP3 inflammasome. The NLRP3 inflammasome is a macromolecular complex that integrates responses to a wide range of damage-associated molecular patterns (DAMPs) that are released into the extracellular space upon cellular damage, including extracellular adenosine triphosphate (ATP), uric acid, reactive oxygen species (ROS), heat shock proteins (HSP), small calcium binding proteins S100A8 and S100A9, nuclear histones and extracellular nucleic acids132. The activation of NLRP3 occurs in two sequential steps. First, priming of the inflammasome occurs via nuclear factor-kB-dependent transcription of NLRP3, caspase 1 and the inactive cytokine forms pro-IL-1β and pro-IL-18. A DAMP or pathogen-associated molecular pattern (PAMP) comprises the second signal, which activates NLRP3 via intracellular potassium and calcium fluxes. Upon activation, NLRP3 rapidly oligomerizes into a multimeric scaffold, recruiting ASC and procaspase 1 into a large perinuclear speck complex. Active caspase 1 cleaves pro-IL-1β and pro-IL-18, causing secretion of biologically active IL-1β and IL-18. NLRP3-mediated inflammatory cytokine release has received intensive study after the discovery that CH is associated with increased IL-1β and IL-678.
Studies implicating NLRP3 inflammasome activity in CH have relied on pharmacological inhibitors of NLRP3, such as the sulfonylurea derivative MCC950, a specific inhibitor of NLRP3 inflammasome that blocks ASC oligomerization133. The pro-atherosclerotic effects of haematopoietic Tet2 deletion in mice can be rescued by MCC950 treatment129. MCC950 treatment also abrogated Tet2-dependent cardiomyocyte hypertrophy and resultant heart failure99. The same group also found that haematopoietic Tet2 deficiency increased the expression of IL-1β in white adipose tissue and led to systemic insulin resistance, and that these effects were suppressed upon NLRP3 inhibition with MCC950113. However, despite its encouraging activity in preclinical models, clinical development of MCC950 was limited by hepatotoxicity in early phase trials. In a study of osteoarthritis, researchers found that genetic knockdown of DNMT3A in chondrocyte and osteoblast cell lines led to promoter hypomethylation of C terminal binding protein genes CtBP1 and CtBP2, resulting in overexpression of CtBP and transcriptional upregulation of NLRP3134. Furthermore, single cell sequencing of peripheral blood cells from patients with heart failure with DNMT3A-mutated CH showed upregulation of NLRP3 and IL-1β expression in monocytes135. These findings suggest that DNMT3A loss-of-function mutations may directly stimulate priming of the NLRP3 inflammasome via transcriptional changes.
In addition to studies of the NLRP3 inflammasome, several reports have implicated a wide range of molecular mechanisms and myeloid cell types involved in CH-related inflammation131,136–146 (Fig. 2). For example, haematopoietic Jak2 V617F mutation also promotes atherosclerosis in hyperlipidemic mice147,148. However, unlike Tet2 and Dnmt3a knockout models of CH, macrophage-specific gain of JAK2 function led to enrichment of mutant macrophages within atherosclerotic plaques. Jak2 gain-of-function macrophages secrete higher levels of IL-1β via activation of the AIM2 inflammasome in response to DNA damage caused by oxidative stress148. Trp53-mutated CH aggravates doxorubicin-related cardiotoxicity via the recruitment of p53-deficient neutrophils, which showed upregulation of genes encoding inflammasome proteins and chemokines149. In a study of osteoporosis, one of the most frequent complications of chronic inflammation, Dnmt3a loss promoted osteoclast differentiation and subsequent bone loss via IRF3–NF-κB-mediated upregulation of IL-20108; interestingly, increased osteoclastogenesis and decreased bone mass were also observed in the setting of Asxl1 deficiency137. Dnmt3a-deficient macrophages showed impaired production of type I IFNs after viral infection or stimulation with various Toll-like receptor agonists; in this context, mice with myeloid cell-specific loss of Dnmt3a were also more susceptible to vesicular stomatitis virus infection145. Altogether, these findings indicate that different genetic drivers in CH promote cardiovascular and other inflammatory phenotypes through various mechanistic pathways to dysregulate the immune response.
Figure 2 |. Functional effects of clonal haematopoiesis-associated mutations in myeloid cells.
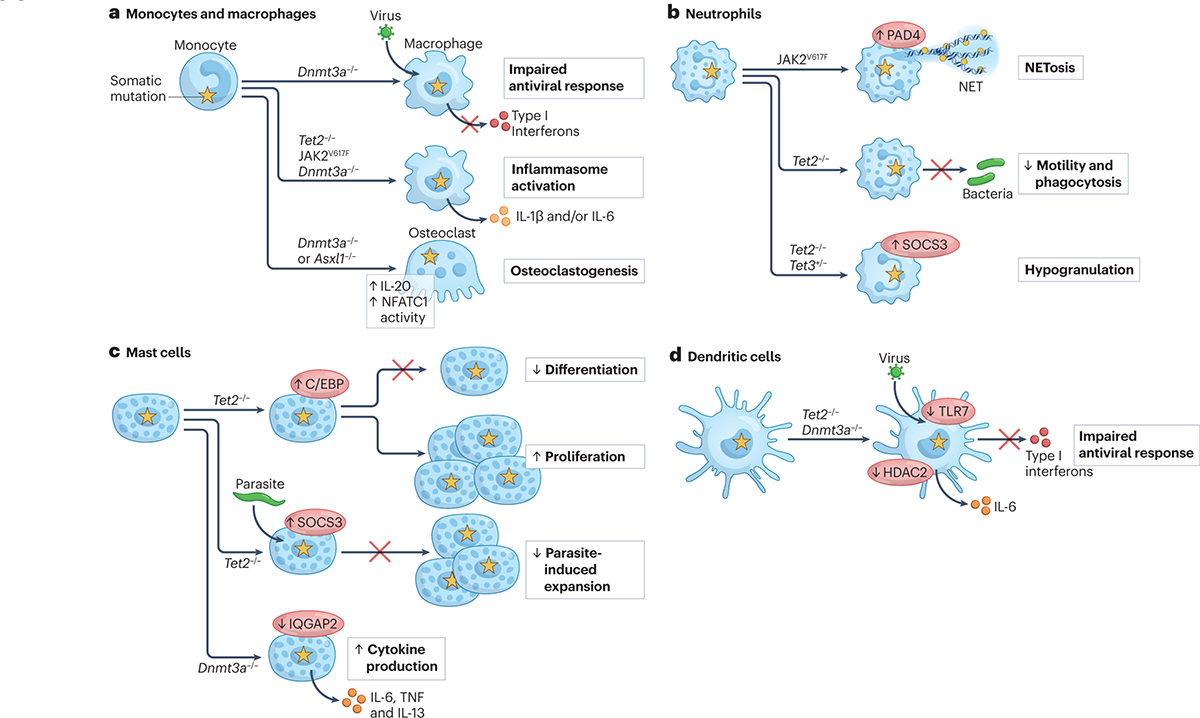
Evidence from mouse models and human observational studies indicate that mutations in genes associated with clonal haematopoiesis can affect the differentiation and function of mature myeloid cells. a | In monocytes and macrophages, the effects of genetic alterations in Tet2, Dnmt3a and Jak2 have been studied most extensively, revealing increased chemokine expression, hyperactivation of the inflammasome and increased IL-1β as common features97,129–131,136,148,188. In response to viral infection and Toll-like receptor (TLR) stimuli, Dnmt3a mutant macrophages produce less type I interferons (IFNs), correlating with reduced survival of virally-infected mice with Dnmt3a-deficient myeloid cells145. Elevated myeloid production of IL-20 in the setting of Dnmt3a deficiency is associated with increased in osteoclastogenesis and osteoporosis108, whereas Asxl1 loss in myeloid cells correlates with increased NFATc1 activity and osteoclast differentiation137. b | In neutrophils, JAK2 V617F is associated with neutrophil extracellular trap (NET) formation and thrombosis in both mouse and human studies138,139. Mouse and zebrafish models have also demonstrated a role for Tet2 and Tet3 in neutrophil mobility, phagocytosis and granule formation140. c | In mast cells, Tet2 loss has contrasting effects – either suppressing mast cell differentiation and stimulating mast cell proliferation via C/EBP transcription factors141, or suppressing mast cell expansion in response to Schistosoma infection142. Mast cells lacking Dnmt3a demonstrated higher cytokine production upon acute stimulation143, whereas Asxl1 truncating mutations promote mast cell differentiation in a multipotent haematopoietic precursor cell line146. d | In dendritic cells, Tet2 loss resulted in higher IL-6 levels and reduced type I IFN production in response to challenge with herpes simplex virus or vesicular stomatitis virus (VSV)131,144. Similarly, Dnmt3a loss reduces type I IFN production in response to VSV145.
Mechanisms of lymphoid dysregulation in clonal haematopoiesis
CH mutations in myeloid malignancy-associated genes can be detected in both myeloid and lymphoid cell subsets150,151, consistent with the occurrence of these mutations in HSCs151–155. Among those with CH mutations in lymphoid cells, DNMT3A and TET2 mutations have been observed most frequently, consistent with their prevalence, and the VAF in lymphoid cells is lower compared with myeloid cells from the same individual. Interestingly, whereas DNMT3A mutations are detectable in myeloid, NK, B and T cells, TET2 mutations appear to be absent from T cells150,151, indicating that different CH mutations may undergo differential selection depending on the haematopoietic lineage. It has long been appreciated that epigenetic modifications play a key role in lymphocyte gene expression156–161. Moreover, studies in both humans and mice have clearly demonstrated that DNMT3A and TET2 regulate lymphocyte differentiation and function, suggesting that lymphoid cells with DNMT3A or TET2 mutations may contribute to CH-associated phenotypes in humans.
[H2] TET2 and DNMT3A in lymphocyte differentiation and function.
Insight into the roles of TET2 and DNMT3A in lymphocytes has come from analysis of mouse models. In mice with T cell-specific Tet2 or Dnmt3a deficiency, T cell subsets develop normally and control of acute viral infection with lymphocytic choriomeningitis virus (LCMV) is unaffected162–164. However, Tet2 or Dnmt3a deficiency is associated with skewing of CD8+ T cells towards a central memory phenotype with relative depletion of short-lived effector cells. This change in differentiation is T cell-intrinsic and leads to increased cytolytic function and control of viral infection upon re-challenge163–166 (Fig. 3). Aberrant cytokine production may also contribute to altered T cell function in the setting of Tet2 or Dnmt3a deficiency. Although Tet2 or Dnmt3a deficiency does not affect the potential of naive CD4+ T cells to differentiate into T helper 1 (TH1), T helper 2 (TH2), T helper 17 (TH17) or regulatory T (Treg) cell subsets in vitro162,167, Tet2-deficient TH1 and TH17 cells produce less IFNγ and IL-17A, respectively; Dnmt3a-deficient CD4+ T cells produce IFNγ aberrantly across all subsets generated in vitro162,167,168 and increased IL-13 upon TH2 cell differentiation in vitro and in vivo169 (Fig. 3).
Figure 3 |. Impact of TET2 and DNMT3A loss in lymphocytes.
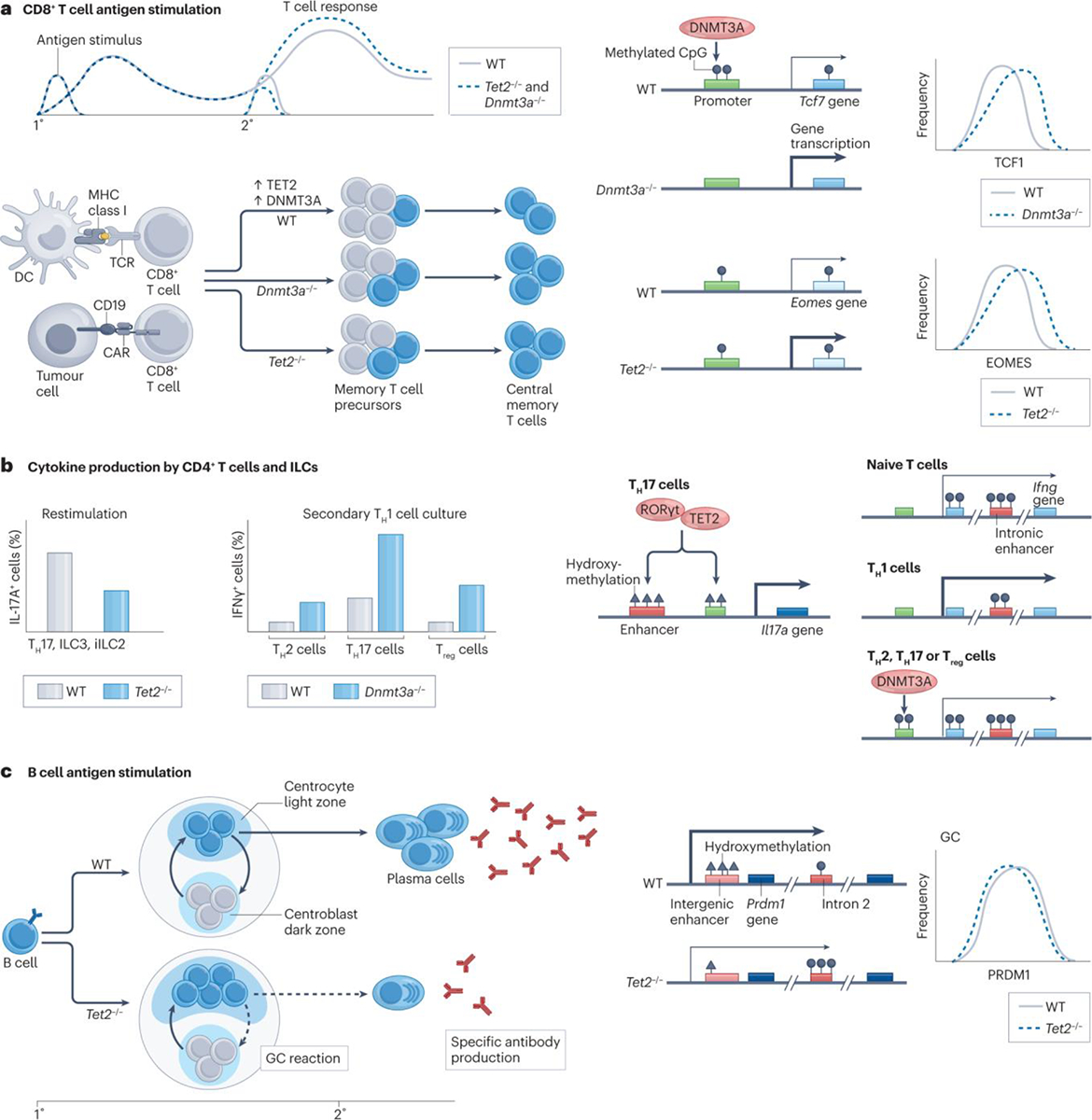
a | In antigen-stimulated mouse CD8+ T cells and human chimeric antigen receptor (CAR) T cells, deficiency in DNMT3A or TET2 is associated with normal primary immune responses but enhanced secondary responses with increased memory precursor and central memory T cell differentiation163–166,183,187. These phenotypic changes correlate with increased expression of the master transcription factors TCF1 (encoded by Tcf7) or EOMES in absence of DNMT3A or TET2, respectively164–166,183,187. While TET2 deficiency does not result in significant methylation changes at the Eomes locus163, DNMT3A deficiency leads to a reduction in Tcf7 promoter methylation, consistent with DNMT3A localization to the Tcf7 promoter in DNMT3A wild-type T cells164. b | In mouse CD4+ T cells and innate lymphoid cell (ILC) subsets, TET2 is recruited to the Il17a enhancer CNS2 via RORγt, leading to increased enhancer and promoter hydroxymethylation and increased Il17a expression162. In this context, IL-17A production by T helper 17 (TH17) cells, group 3 innate lymphoid cells (ILC3s) and inflammatory group 2 innate lymphoid cells (iILC2s) is significantly reduced in the setting of TET2 deficiency162,172. Similarly, DNMT3A localization to the Ifng promoter in T helper 2 (TH2) cells, T helper 17 (TH17) cells and regulatory T (Treg) cells generated in vitro is associated with increased methylation and reduced Ifng expression; reduced Ifng methylation in DNMT3A-deficient TH2, TH17 and Treg cells leads to aberrant IFNγ production by all three subsets upon restimulation with the TH1 cytokine IL-12167. c | In B cells, TET2 deficiency during primary immunization is associated with accumulation of germinal centre B cells, reduced expression of the plasma cell-defining transcription factor PRDM1, impaired plasma cell differentiation and decreased production of antigen-specific antibodies upon secondary immunization173. Reduced PRDM1 expression in the absence of TET2 correlates with hypermethylation within Prdm1 intron 2, which contains a locus that undergoes progressive demethylation during plasma cell differentiation, as well as decreased hydroxymethylation of an intergenic enhancer.
Peripheral B cells and innate lymphoid cell (ILC) subsets also develop normally in unmanipulated mice with cell type-specific deficiency in Tet2 or Dnmt3a162,170–172. Upon model antigen exposure, Tet2-deficient B cells develop a cell-intrinsic increase in germinal centre B cells with impaired capacity to exit the germinal centre reaction, resulting in reduced plasma cell generation and lower circulating antigen-specific antibodies173 (Fig. 3). By contrast, Dnmt3a-deficient B cells respond normally to model antigen exposure but develop an aggressive CLL-like disease with high penetrance170,171. In NK cells, Tet2 is dispensable for activation and maturation during murine cytomegalovirus infection, although cytolytic protein expression and activity against target tumour cells is modestly reduced172. Though the abundance of ILC subsets is unchanged in Tet2-deficient mice, IL-17A production by inflammatory group 2 ILC and group 3 ILC subsets is significantly reduced in the absence of Tet2, similar to TH17 cells. Tet2-deficient group 3 ILCs also produce less IL-22 and are less effective in controlling infection by the enteric bacterial pathogen Citrobacter rodentium172.
Mechanistically, it has become clear that Tet2 and Dnmt3a affect the expression and genomic localization of master transcription factors in lymphocytes after antigen stimulation, leading to alterations in cell fate decisions162,163,171–173. For example, mice with T cell-specific deletion of Dnmt3a show increased CD8+ T cell expression of Tcf7, a transcription factor known to drive memory differentiation174; increased Tcf7 expression is associated with reduced methylation at the Tcf7 promoter, and knockdown of Tcf7 partially rescues the differentiation of effector CD8+ T cells from Dnmt3a knockout164–166,175. In comparison, Tet2-deficient T cells display increased expression of a different transcription factor, Eomes, also involved in the differentiation of memory CD8+ T cells176. Finally, in mice with B cell-specific deletion of Tet2, reduced expression of the plasma cell-defining transcription factor Prdm1 correlated with hypermethylation of the Prdm1 gene locus and impaired plasma cell differentiation173 (Fig. 3).
Epigenetic changes to select cytokine loci in lymphocytes also correlate with altered production of T cell and ILC signature cytokines. In CD4+ T cells differentiated in vitro, TET2 co-localizes on chromatin with the TH1 cell-defining transcription factor T-BET or the TH17 cell-defining transcription factor RORγt to promote targeted DNA hydroxymethylation and expression of IFNγ or IL-17A, respectively162. Conversely, DNMT3A binding and methylation within the Ifng gene locus in TH2-, TH17- and Treg-polarized CD4+ T cells is associated with restrained IFNγ production167 (Fig. 3).
Collectively, these data indicate that Tet2- and Dnmt3a-mediated changes in the epigenetic landscape can promote aberrant lymphocyte differentiation and function (Fig. 3). The extent and role of altered lymphocyte differentiation and function in CH-associated human disease remains largely unclear.
Clonal haematopoiesis and antitumour immunity.
Recent observations indicate that CH can affect outcomes in patients receiving cellular therapies for haematological malignancies. Two independent studies of patients undergoing allogeneic HSC transplant for haematological malignancies reported that the presence of DNMT3A mutant CH in the donor cell product was associated with reduced risk of disease relapse and increased risk of chronic graft-versus-host disease (GVHD)152,155. Interestingly, CH-associated clinical outcomes were dependent on GVHD prophylaxis, as a T cell-depleting post-transplant cyclophosphamide but not calcineurin inhibitor-based regimen, completely abrogated the effect of donor DNMT3A mutant CH on disease relapse and chronic GVHD152. Donor-derived myeloid cells involved in T and NK cell activation might also play a role in the observed effects of donor CH on clinical outcomes in allogeneic transplant. Indeed, recipients of products with DNMT3A mutant donor CH had elevated serum levels of IL-12p70152, which is known to promote the differentiation of IFNγ-producing CD4+ T cells and the cytolytic activity of CD8+ T cells and NK cells177–180.
CH has also been linked to clinical outcomes in patients receiving chimeric antigen receptor (CAR) T cell therapy for haematological malignancies. The presence of CH was associated with increased rates of cytokine release syndrome and neurotoxicity severity in patients receiving CAR T cell therapy for non-Hodgkin lymphoma181,182. Lentiviral integration-mediated disruption of loci encoding genes mutated in CH has also been associated with cell-intrinsic alterations in CAR T cell expansion and differentiation183–185. In a patient receiving anti-CD19 CAR T cell therapy for CLL, lentiviral integration into one TET2 allele, on the background of a heterozygous germline TET2 variant associated with reduced catalytic activity on the other allele, led to dramatic clonal outgrowth with increased central memory CD8+ T cell differentiation, suggesting that the effects TET2 disruption in CAR T cells were the result of biallelic impairment in TET2 function183. Phenotypically, ex vivo CD8+ CAR T cells expressed higher levels of the proliferation marker Ki-67, the cytolytic protein granzyme B and the transcription factor EOMES (Fig. 3); in addition, levels of the senescence marker KLRG1 were reduced186. Notably, knockdown of TET2183 or knockout of DNMT3A187 in experimental models of CAR T cells also led to increased differentiation of central memory CD8+ T cells and antitumour responses dependent on CAR stimulation (Fig. 3), suggesting that inhibition of TET2 or DNMT3A activity could be exploited to enhance the efficacy of CAR T cell therapy.
Future directions
The link between CH and various clinical phenotypes has revealed that immune cells harbouring clonal mutations drive immune dysfunction and age-related diseases in humans. Although the cell lineages and molecular mechanisms involved in aberrant immune function have been partially elucidated in mouse models, there are many outstanding questions related to the cell biology, therapeutic implications and range of immune phenotypes associated with CH.
CH-associated genetic alterations are detectable in both myeloid and lymphoid cells, but it is unclear how the cell-intrinsic effects of a mutation alter immunological interactions with cells that do not have a mutation. In addition, the manner in which cells without clonal mutations participate in driving inflammation and disease in the setting of CH is unknown. Among most individuals with detectable CH, only a small fraction of peripheral blood cells has clonal mutations, and wild-type cells may also contribute to immunopathology. Alternatively, it is possible that enrichment of immune cells with clonal mutations in tissues allows for a relatively small population of cells to drive inflammation and tissue injury independently of wild-type cells.
Several studies have implicated activation of the NLRP3 inflammasome and increased IL-1β production as causal drivers of non-malignant disease observed in human CH with TET2 mutations and mouse models of CH with Tet2 mutations78,97,99,103,105,113,129,188,189. In this context, the existence of pharmacological inhibitors and monoclonal antibodies against multiple components of the inflammasome pathway highlights the possibility of treating CH-associated non-malignant conditions using targeted anti-inflammatory therapies. Indeed, among patients with cardiovascular disease who received the IL-1β neutralizing antibody canakinumab, those with TET2 mutant CH showed a greater reduction in incident major adverse cardiovascular events compared with those with non-TET2 mutant CH and those without any CH190. This finding not only suggests that specific CH-associated mutations may serve as a prognostic biomarker for the efficacy of targeted anti-inflammatory therapies, but also that CH-associated mutations in different genes may require different anti-inflammatory therapies.
Given the distinct cytokine and immune cell profiles observed among individuals with different CH-associated mutations78, it is likely that the inflammatory mechanisms driving non-malignant disease are correlated with the mutated gene. Whether other therapies targeting NLRP3, IL-1β, AIM2 or IL-6 would be effective in treating active CH-associated inflammation has yet to be tested. With the advent of CH-focused clinical programmes191, the role of prophylactic therapy for patients with CH but without evidence of frank disease also remains to be addressed. Importantly, the use of any therapies in the setting of CH alone will require accurate predictive algorithms to identify individuals at highest risk of developing clinically significant malignant and/or inflammatory disease.
Recently, other diseases driven by pathological immune dysfunction have been associated with somatic mutations in haematopoietic cells. Singh et al. reported that autoreactive B cell clones from patients with Sjogren’s syndrome-associated cryoglobulinemic vasculitis harboured mutations in genes recurrently mutated in B cell lymphomas, despite the absence of malignancy at the time of analysis192. Likewise, Beck et al. recently described a severe autoinflammatory condition called VEXAS syndrome, which occurs in late adulthood and is genetically defined by recurrent somatic mutations in UBA1193. Whether there are other autoimmune conditions defined by pathogenic somatic mutations in haematopoietic cells remains to be discovered.
Competing interests
B.L.E. has received research funding from Celgene, Deerfield, Novartis, and Calico and consulting fees from GRAIL. He is a member of the scientific advisory board and shareholder for Neomorph Inc., TenSixteen Bio, Skyhawk Therapeutics, and Exo Therapeutics. The other authors have no competing interests.
References
- 1.Orkin SH & Zon LI Hematopoiesis: An evolving paradigm for stem cell biology. Cell 132, 631–644 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Looker AC, Dallman PR, Carroll MD, Gunter EW & Johnson CL Prevalence of iron deficiency in the United States. Jama-Journal of the American Medical Association 277, 973–976 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Heegaard ED & Brown KE Human parvovirus B19. Clinical Microbiology Reviews 15, 485-+ (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Afdhal N et al. Thrombocytopenia associated with chronic liver disease. Journal of Hepatology 48, 1000–1007 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Nimer SD Myelodysplastic syndromes. Blood 111, 4841–4851 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Babitt JL & Lin HY Mechanisms of Anemia in CKD. Journal of the American Society of Nephrology 23, 1631–1634 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiss G, Ganz T & Goodnough LT Anemia of inflammation. Blood 133, 40–50 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimamura A & Alter BP Pathophysiology and management of inherited bone marrow failure syndromes. Blood Reviews 24, 101–122 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slichter SJ Relationship between platelet count and bleeding risk in thrombocytopenic patients. Transfusion Medicine Reviews 18, 153–167 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Chemaly RF et al. Respiratory viral infections in adults with hematologic malignancies and human stem cell transplantation recipients - A retrospective study at a major cancer center. Medicine 85, 278–287 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Kuderer NM, Dale DC, Crawford J, Cosler LE & Lyman GH Mortality, morbidity, and cost associated with febrile neutropenia in adult cancer patients. Cancer 106, 2258–2266 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Wolach O & Stone R Autoimmunity and inflammation in Myelodysplastic Syndromes. Acta Haematologica 136, 108–117 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Caligaris-Cappio F & Hamblin TJ B-cell chronic lymphocytic leukemia: A bird of a different feather. Journal of Clinical Oncology 17, 399–408 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Finkel HE, Brauer MJ, Taub RN & Dameshek W IMMUNOLOGIC ABERRATIONS IN DI GUGLIELMO SYNDROME. Blood-the Journal of Hematology 28, 634-+ (1966). [PubMed] [Google Scholar]
- 15.Lewis CM & Pegrum GD IMMUNE-COMPLEXES IN MYELOPROLIFERATIVE DISORDERS. Lancet 2, 1151–1153 (1977). [DOI] [PubMed] [Google Scholar]
- 16.Hetzel P & Gee TS NEW OBSERVATION IN CLINICAL SPECTRUMS OF ERYTHROLEUKEMIA - REPORT OF 46 CASES. American Journal of Medicine 64, 765–772 (1978). [DOI] [PubMed] [Google Scholar]
- 17.Enright H et al. PARANEOPLASTIC AUTOIMMUNE PHENOMENA IN PATIENTS WITH MYELODYSPLASTIC SYNDROMES - RESPONSE TO IMMUNOSUPPRESSIVE THERAPY. British Journal of Haematology 91, 403–408 (1995). [DOI] [PubMed] [Google Scholar]
- 18.Colombat PH, Renoux M, Lamagnere JP & Renoux G IMMUNOLOGICAL INDEXES IN MYELODYSPLASTIC SYNDROMES. Cancer 61, 1075–1081 (1988). [DOI] [PubMed] [Google Scholar]
- 19.Young NS, Calado RT & Scheinberg P Current concepts in the pathophysiology and treatment of aplastic anemia. Blood 108, 2509–2519 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green AR, Shuttleworth D, Bowen DT & Bentley DP CUTANEOUS VASCULITIS IN PATIENTS WITH MYELODYSPLASIA. British Journal of Haematology 74, 364–365 (1990). [DOI] [PubMed] [Google Scholar]
- 21.Pagliuca A, Higgins E, Samson D, Humphries S & Mufti GJ PRODROMAL CUTANEOUS VASCULITIS IN MYELODYSPLASTIC SYNDROMES. British Journal of Haematology 75, 444–446 (1990). [DOI] [PubMed] [Google Scholar]
- 22.Hebbar M et al. ASSOCIATION OF MYELODYSPLASTIC SYNDROME AND RELAPSING POLYCHONDRITIS - FURTHER EVIDENCE. Leukemia 9, 731–733 (1995). [PubMed] [Google Scholar]
- 23.Fernandezmiranda C et al. VASCULITIS ASSOCIATED TO MYELODYSPLASTIC SYNDROME - REPORT OF 5 CASES. Medicina Clinica 103, 539–542 (1994). [PubMed] [Google Scholar]
- 24.Savige JA, Chang L, Smith CL & Duggan JC MYELODYSPLASIA, VASCULITIS AND ANTINEUTROPHIL CYTOPLASM ANTIBODIES. Leukemia & Lymphoma 9, 49–54 (1993). [DOI] [PubMed] [Google Scholar]
- 25.Doutre MS et al. CUTANEOUS VASCULITIS AND REFRACTORY-ANEMIA WITH EXCESS OF BLAST CELLS. Annales De Dermatologie Et De Venereologie 114, 97–100 (1987). [PubMed] [Google Scholar]
- 26.Saif MW, Hopkins JL & Gore SD Autoimmune phenomena in patients with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leukemia & Lymphoma 43, 2083–2092 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Giannouli S, Voulgarelis M, Zintzaras E, Tzioufas AG & Moutsopoulos HM Autoimmune phenomena in myelodysplastic syndromes: a 4-yr prospective study. Rheumatology 43, 626–632 (2004). [DOI] [PubMed] [Google Scholar]
- 28.de Hollanda A et al. Systemic and Immune Manifestations in Myelodysplasia: A Multicenter Retrospective Study. Arthritis Care & Research 63, 1188–1194 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Mekinian A et al. Systemic inflammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: a French multicentre retrospective study. Rheumatology 55, 291–300 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Anderson LA et al. Risks of myeloid malignancies in patients with autoimmune conditions. British Journal of Cancer 100, 822–828 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kristinsson SY et al. Chronic Immune Stimulation Might Act As a Trigger for the Development of Acute Myeloid Leukemia or Myelodysplastic Syndromes. Journal of Clinical Oncology 29, 2897–2903 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weeks LD et al. Age-related diseases of inflammation in myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood 139, 1246–1250 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sujobert P et al. Evidence of Differentiation in Myeloid Malignancies Associated Neutrophilic Dermatosis: A Fluorescent In Situ Hybridization Study of 14 Patients. Journal of Investigative Dermatology 133, 1111–1114 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Chavan RN et al. Histiocytoid Sweet syndrome may indicate leukemia cutis: A novel application of fluorescence in situ hybridization. Journal of the American Academy of Dermatology 70, 1021–1027 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Passet M et al. Next-Generation Sequencing in Myeloid Neoplasm-Associated Sweet’s Syndrome Demonstrates Clonal Relation between Malignant Cells and Skin-Infiltrating Neutrophils. Journal of Investigative Dermatology 140, 1873-+ (2020). [DOI] [PubMed] [Google Scholar]
- 36.Magro CM, Kiani B, Li JW & Crowson AN Clonality in the setting of Sweet’s syndrome and pyoderma gangrenosum is not limited to underlying myeloproliferative disease. Journal of Cutaneous Pathology 34, 526–534 (2007). [DOI] [PubMed] [Google Scholar]
- 37.de Fremont GM et al. Myeloid Clonal Infiltrate Identified With Next-Generation Sequencing in Skin Lesions Associated With Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia: A Case Series. Frontiers in Immunology 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trowbridge JJ & Starczynowski DT Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. Journal of Experimental Medicine 2184 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caiado F, Pietras EM & Manz MG Inflammation as a regulator of hematopoietic stem cell function in disease, and clonal selection. Journal of Experimental Medicine 218 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kitagawa M et al. Overexpression of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia 11, 2049–2054 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Wetzler M et al. ALTERED LEVELS OF INTERLEUKIN-1-BETA AND INTERLEUKIN-1 RECEPTOR ANTAGONIST IN CHRONIC MYELOGENOUS LEUKEMIA - CLINICAL AND PROGNOSTIC CORRELATES. Blood 84, 3142–3147 (1994). [PubMed] [Google Scholar]
- 42.Nievergall E et al. TGF-alpha and IL-6 plasma levels selectively identify CML patients who fail to achieve an early molecular response or progress in the first year of therapy. Leukemia 30, 1263–1272 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Pardanani A et al. IPSS-independent prognostic value of plasma CXCL10, IL-7 and IL-6 levels in myelodysplastic syndromes. Leukemia 26, 693–699 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsu HC et al. Circulating levels of thrombopoietic and inflammatory cytokines in patients with acute myeloblastic leukemia and myelodysplastic syndrome. Oncology 63, 64–69 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Gersuk GR et al. A role for tumour necrosis factor-alpha, Fas and Fas-ligand in marrow failure associated with myelodysplastic syndrome. British Journal of Haematology 103, 176–188 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Tsimberidou AM et al. The prognostic significance of cytokine levels in newly diagnosed acute myeloid leukemia and high-risk myelodysplastic syndromes. Cancer 113, 1605–1613 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Musto P et al. LOW SERUM LEVELS OF TUMOR-NECROSIS-FACTOR AND INTERLEUKIN-1-BETA IN MYELODYSPLASTIC SYNDROMES RESPONSIVE TO RECOMBINANT ERYTHROPOIETIN. Haematologica 79, 265–268 (1994). [PubMed] [Google Scholar]
- 48.Pietras EM et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nature Cell Biology 18, 607-+ (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Higa KC et al. Chronic interleukin-1 exposure triggers selection for Cebpa-knockout multipotent hematopoietic progenitors. Journal of Experimental Medicine 218 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai ZG et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 23, 833-+ (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hormaechea-Agulla D et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFN gamma signaling. Cell Stem Cell 28, 1428-+ (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Avagyan S et al. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science 374, 768-+ (2021). [DOI] [PubMed] [Google Scholar]
- 53.Grants JM et al. Altered microRNA expression links IL6 and TNF-induced inflammaging with myeloid malignancy in humans and mice. Blood 135, 2235–2251 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Zhang TY et al. IL-6 blockade reverses bone marrow failure induced by human acute myeloid leukemia. Science Translational Medicine 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reynaud D et al. IL-6 Controls Leukemic Multipotent Progenitor Cell Fate and Contributes to Chronic Myelogenous Leukemia Development. Cancer Cell 20, 661–673 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Twomey JJ INFECTIONS COMPLICATING MULTIPLE-MYELOMA AND CHRONIC LYMPHOCYTIC LEUKEMIA. Archives of Internal Medicine 132, 562–565 (1973). [PubMed] [Google Scholar]
- 57.Itala M, Helenius H, Nikoskelainen J & Remes K INFECTIONS AND SERUM IGG LEVELS IN PATIENTS WITH CHRONIC LYMPHOCYTIC-LEUKEMIA. European Journal of Haematology 48, 266–270 (1992). [DOI] [PubMed] [Google Scholar]
- 58.Brown RK, Read JT, Wiseman BK & France WG THE ELECTROPHORETIC ANALYSIS OF SERUM PROTEINS OF THE BLOOD DYSCRASIAS. Journal of Laboratory and Clinical Medicine 33, 1523–1533 (1948). [PubMed] [Google Scholar]
- 59.Shaw RK et al. INFECTION AND IMMUNITY IN CHRONIC LYMPHOCYTIC LEUKEMIA. Archives of Internal Medicine 106, 467–478 (1960). [Google Scholar]
- 60.Anderson LA, Landgren O & Engels EA Common community acquired infections and subsequent risk of chronic lymphocytic leukaemia. British Journal of Haematology 147, 444–449 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moreira J et al. Infectious complications among individuals with clinical monoclonal B-cell lymphocytosis (MBL): a cohort study of newly diagnosed cases compared to controls. Leukemia 27, 136–141 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Tsai HT et al. Evidence of serum immunoglobulin abnormalities up to 9.8 years before diagnosis of chronic lymphocytic leukemia: a prospective study. Blood 114, 4928–4932 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lunning MA & Vose JM Angioimmunoblastic T-cell lymphoma: the many-faced lymphoma. Blood 129, 1095–1102 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Han LJ et al. Clinical features and treatment of natural killer/T cell lymphoma associated with hemophagocytic syndrome: comparison with other T cell lymphoma associated with hemophagocytic syndrome. Leukemia & Lymphoma 55, 2048–2055 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Kikushige Y et al. Self-Renewing Hematopoietic Stem Cell Is the Primary Target in Pathogenesis of Human Chronic Lymphocytic Leukemia. Cancer Cell 20, 246–259 (2011). [DOI] [PubMed] [Google Scholar]
- 66.Jan M et al. Clonal Evolution of Preleukemic Hematopoietic Stem Cells Precedes Human Acute Myeloid Leukemia. Science Translational Medicine 4, doi: 10.1126/scitranslmed.3004315 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC & Majeti R Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proceedings of the National Academy of Sciences of the United States of America 111, 2548–2553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shlush LI et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506, 328-+ (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chung SS et al. Hematopoietic Stem Cell Origin of BRAFV600E Mutations in Hairy Cell Leukemia. Science Translational Medicine 6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Busque L et al. Nonrandom X-inactivation patterns in normal females: Lyonization ratios vary with age. Blood 88, 59–65 (1996). [PubMed] [Google Scholar]
- 71.Gale RE, Fielding AK, Harrison CN & Linch DC Acquired skewing of X-chromosome inactivation patterns in myeloid cells of the elderly suggests stochastic clonal loss with age. British Journal of Haematology 98, 512–519 (1997). [DOI] [PubMed] [Google Scholar]
- 72.Busque L et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nature Genetics 44, 1179–1181 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laurie CC et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nature Genetics 44, 642–U658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jacobs KB et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nature Genetics 44, 651–U668 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Genovese G et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. New England Journal of Medicine 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jaiswal S et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. New England Journal of Medicine 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xie M et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature Medicine 20, 1472–1478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bick AG et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586, 763-+ (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Niroula A et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nature Medicine 27, 1921-+ (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zink F et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saiki R et al. Combined landscape of single-nucleotide variants and copy number alterations in clonal hematopoiesis. Nature Medicine 27, 1239-+ (2021). [DOI] [PubMed] [Google Scholar]
- 82.Bick AG et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 141, 124–131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kar SP et al. Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nature Genetics 54, 1155-+ (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kessler MD et al. Exome sequencing of 628,388 individuals identifies common and rare variant associations with clonal hematopoiesis phenotypes. medRxiv, 2021.2012.2029.21268342, doi: 10.1101/2021.12.29.21268342 (2022). [DOI] [Google Scholar]
- 85.Genovese G et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. The New England Journal of Medicine 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abelson S et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559, 400-+ (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Desai P et al. Somatic mutations precede myeloid leukemia years before diagnosis. Nature Medicine 24, 1015-+ (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fabre MA et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature 606, 335-+ (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robertson NA et al. Longitudinal dynamics of clonal hematopoiesis identifies gene-specific fitness effects. Nature Medicine, doi: 10.1038/s41591-022-01883-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Watson CJ et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 367, 1449-+ (2020). [DOI] [PubMed] [Google Scholar]
- 91.Loh PR et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559, 350-+ (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Terao C et al. Chromosomal alterations among age-related haematopoietic clones in Japan. Nature 584, 130-+ (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zekavat SM et al. Hematopoietic mosaic chromosomal alterations increase the risk for diverse types of infection. Nature Medicine 27, 1012-+ (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Terao C et al. GWAS of mosaic loss of chromosome Y highlights genetic effects on blood cell differentiation. Nature Communications 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thompson DJ et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 575, 652-+ (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gao T et al. Interplay between chromosomal alterations and gene mutations shapes the evolutionary trajectory of clonal hematopoiesis. Nature Communications 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jaiswal S et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. New England Journal of Medicine 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Loh PR, Genovese G & McCarroll SA Monogenic and polygenic inheritance become instruments for clonal selection. Nature 584, 136-+ (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sano S et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1 beta/NLRP3 Inflammasome. Journal of the American College of Cardiology 71, 875–886 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu B et al. Association of Clonal Hematopoiesis With Incident Heart Failure (vol 78, pg 42, 2021). Journal of the American College of Cardiology 78 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dawoud AAZ, Gilbert RD, Tapper WJ & Cross NCP Clonal myelopoiesis promotes adverse outcomes in chronic kidney disease. Leukemia 36, 507–515 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sano S et al. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circulation Research 123, 335–341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wong WJ et al. Clonal hematopoiesis and risk of chronic liver disease. medRxiv, 2022.2001.2017.22269409, doi: 10.1101/2022.01.17.22269409 (2022). [DOI] [Google Scholar]
- 104.Miller PG et al. Association of clonal hematopoiesis with chronic obstructive pulmonary disease. Blood 139, 357–368 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Agrawal M et al. TET2-mutant clonal hematopoiesis and risk of gout. Blood, doi: 10.1182/blood.2022015384 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang CRC et al. Inflammatory cytokines promote clonal hematopoiesis with specific mutations in ulcerative colitis patients. Experimental Hematology 80, 36–41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hecker JS et al. CHIP and hips: clonal hematopoiesis is common in patients undergoing hip arthroplasty and is associated with autoimmune disease. Blood 138, 1727–1732 (2021). [DOI] [PubMed] [Google Scholar]
- 108.Kim PG et al. Dnmt3a-mutated clonal hematopoiesis promotes osteoporosis. Journal of Experimental Medicine 218 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Honigberg MC et al. Premature Menopause, Clonal Hematopoiesis, and Coronary Artery Disease in Postmenopausal Women. Circulation 143, 410–423 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cordua S et al. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 134, 469–479 (2019). [DOI] [PubMed] [Google Scholar]
- 111.Kimishima Y et al. Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension with ALK1 upregulation in lung neutrophils. Nature Communications 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Newell LF et al. Graft-versus-host disease after liver transplantation is associated with bone marrow failure, hemophagocytosis, and DNMT3A mutations. American Journal of Transplantation 21, 3894–3906 (2021). [DOI] [PubMed] [Google Scholar]
- 113.Fuster JJ et al. TET2-Loss-of-Function-Driven Clonal Hematopoiesis Exacerbates Experimental Insulin Resistance in Aging and Obesity. Cell Reports 33 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Coombs CC et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 21, 374-+ (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ptashkin RN et al. Prevalence of Clonal Hematopoiesis Mutations in Tumor-Only Clinical Genomic Profiling of Solid Tumors. Jama Oncology 4, 1589–1593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bhattacharya R et al. Clonal Hematopoiesis Is Associated With Higher Risk of Stroke. Stroke 53, 788–797 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bouzid H et al. Clonal hematopoiesis is associated with protection from Alzheimer’s disease. medRxiv, 2021.2012.2010.21267552 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nakao T et al. Mendelian randomization supports bidirectional causality between telomere length and clonal hematopoiesis of indeterminate potential. Science Advances 8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Uddin MDM et al. Clonal hematopoiesis of indeterminate potential, DNA methylation, and risk for coronary artery disease. Nature Communications 13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Horvath S & Raj K DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nature Reviews Genetics 19, 371–384 (2018). [DOI] [PubMed] [Google Scholar]
- 121.Nachun D et al. Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell 20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Robertson NA et al. Age-related clonal haemopoiesis is associated with increased epigenetic age. Current Biology 29, R786–R787 (2019). [DOI] [PubMed] [Google Scholar]
- 123.Cook EK et al. Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Advances 3, 2482–2486 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dharan NJ et al. HIV is associated with an increased risk of age-related clonal hematopoiesis among older adults. Nature Medicine 27, 1006-+ (2021). [DOI] [PubMed] [Google Scholar]
- 125.Feng XM et al. Cytokine signature profiles in acquired aplastic anemia and myelodysplastic syndromes. Haematologica-the Hematology Journal 96, 602–606 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tefferi A et al. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. Journal of Clinical Oncology 29, 1356–1363 (2011). [DOI] [PubMed] [Google Scholar]
- 127.De Santis M et al. Mutations Associated with Clonal Hematopoiesis of Indeterminate Potential Are Found in Peripheral Blood and Synovial Fluid Macrophages from Patients with Rheumatoid and Psoriatic Arthritis. Arthritis & Rheumatology 70 (2018). [Google Scholar]
- 128.Wang Y et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. Jci Insight 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fuster JJ et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cull AH, Snetsinger B, Buckstein R, Wells RA & Rauh MJ Tet2 restrains inflammatory gene expression in macrophages. Experimental Hematology 55, 56–70 (2017). [DOI] [PubMed] [Google Scholar]
- 131.Zhang Q et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Swanson KV, Deng M & Ting JPY The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nature Reviews Immunology 19, 477–489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Coll RC et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature Medicine 21, 248-+ (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sun XX et al. DNA methylation is involved in the pathogenesis of osteoarthritis by regulating CtBP expression and CtBP-mediated signaling. International Journal of Biological Sciences 16, 994–1009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Abplanalp WT et al. Clonal Hematopoiesis-Driver DNMT3A Mutations Alter Immune Cells in Heart Failure. Circulation Research 128, 216–228 (2021). [DOI] [PubMed] [Google Scholar]
- 136.Pronier E et al. Macrophage migration inhibitory factor is overproduced through EGR1 in TET2low resting monocytes. Communications Biology 5, 1–15 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rohatgi N et al. ASXL1 impairs osteoclast formation by epigenetic regulation of NFATc1. Blood Advances 2, 2467–2477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wolach O et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Science translational medicine 10, eaan8292 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cook EK et al. Impact of Tet2 Deficiency, and of TET2 Mutations in Clonal Hematopoiesis, on Neutrophil/Granulocyte Immune Function. Blood 138, 2159 (2021).34854882 [Google Scholar]
- 140.Banks KM, Lan Y & Evans T Tet Proteins Regulate Neutrophil Granulation in Zebrafish through Demethylation of socs3b mRNA. Cell Reports 34, 108632 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Montagner S et al. TET2 Regulates Mast Cell Differentiation and Proliferation through Catalytic and Non-catalytic Activities. Cell reports 15, 1566–1579 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shen Q et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature 554, 123–127 (2018). [DOI] [PubMed] [Google Scholar]
- 143.Leoni C et al. Dnmt3a restrains mast cell inflammatory responses. Proceedings of the National Academy of Sciences of the United States of America 114, E1490–E1499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ma S et al. Epigenetic regulator CXXC5 recruits DNA demethylase Tet2 to regulate TLR7/9-elicited IFN response in pDCs. The Journal of Experimental Medicine 214, 1471–1491 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li X et al. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nature Immunology 17, 806-+ (2016). [DOI] [PubMed] [Google Scholar]
- 146.Balasubramani A et al. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nature Communications 6, 7307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang W et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2(V617F) Mice. Circulation Research 123, E35–E47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fidler TP et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 592, 296-+ (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sano S et al. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. Jci Insight 6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Buscarlet M et al. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood 132, 277–280 (2018). [DOI] [PubMed] [Google Scholar]
- 151.Arends CM et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia 32, 1908–1919 (2018). [DOI] [PubMed] [Google Scholar]
- 152.Gibson CJ et al. Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. Journal of Clinical Oncology 40, 189-+ (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gibson CJ et al. Donor-engrafted CHIP is common among stem cell transplant recipients with unexplained cytopenias. Blood 130, 91–94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Boettcher S et al. Clonal hematopoiesis in donors and long-term survivors of related allogeneic hematopoietic stem cell transplantation. Blood 135, 1548–1559 (2020). [DOI] [PubMed] [Google Scholar]
- 155.Frick M et al. Role of Donor Clonal Hematopoiesis in Allogeneic Hematopoietic Stem-Cell Transplantation. Journal of Clinical Oncology 37, 375-+ (2019). [DOI] [PubMed] [Google Scholar]
- 156.Chang SJ & Aune TM Dynamic changes in histone-methylation ‘marks’ across the locus encoding interferon-gamma during the differentiation of T helper type 2 cells. Nature Immunology 8, 723–731 (2007). [DOI] [PubMed] [Google Scholar]
- 157.Chang SJ & Aune TM Histone hyperacetylated domains across the Ifng gene region in natural killer cells and T cells. Proceedings of the National Academy of Sciences of the United States of America 102, 17095–17100 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Schoenborn JR et al. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nature Immunology 8, 732–742 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Winders BR, Schwartz RH & Bruniquel D A distinct region of the murine IFN-gamma promoter is hypomethylated from early T cell development through mature naive and Th1 cell differentiation, but is hypermethylated in Th2 cells. Journal of Immunology 173, 7377–7384 (2004). [DOI] [PubMed] [Google Scholar]
- 160.Bream JH et al. A distal region in the interferon-gamma gene is a site of epigenetic remodeling and transcriptional regulation by interleukin-2. Journal of Biological Chemistry 279, 41249–41257 (2004). [DOI] [PubMed] [Google Scholar]
- 161.Tato C et al. Cutting edge: Innate production of IFN-gamma by NK cells is independent of epigenetic modification of the IFN-gamma promoter. Journal of Immunology 173, 1514–1517 (2004). [DOI] [PubMed] [Google Scholar]
- 162.Ichiyama K et al. The Methylcytosine Dioxygenase Tet2 Promotes DNA Demethylation and Activation of Cytokine Gene Expression in T Cells. Immunity 42, 613–626 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Carty SA et al. The Loss of TET2 Promotes CD8(+) T Cell Memory Differentiation. Journal of Immunology 200, 82–91 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ladle BH et al. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proceedings of the National Academy of Sciences of the United States of America 113, 10631–10636 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ghoneim HE et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Youngblood B et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 552, 404-+ (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Thomas RM, Gamper CJ, Ladle BH, Powell JD & Wells AD De Novo DNA Methylation Is Required to Restrict T Helper Lineage Plasticity. Journal of Biological Chemistry 287, 22900–22909 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gamper CJ, Agoston AT, Nelson WG & Powell JD Identification of DNA Methyltransferase 3a as a T Cell Receptor-Induced Regulator of Th1 and Th2 Differentiation. Journal of Immunology 183, 2267–2276 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yu Q et al. DNA methyltransferase 3a limits the expression of interleukin-13 in T helper 2 cells and allergic airway inflammation. Proceedings of the National Academy of Sciences of the United States of America 109, 541–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Biran A et al. Activation of Notch and Myc Signaling via B-cell-Restricted Depletion of Dnmt3a Generates a Consistent Murine Model of Chronic Lymphocytic Leukemia. Cancer Research 81, 6117–6130 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mahajan VS et al. B1a and B2 cells are characterized by distinct CpG modification states at DNMT3A-maintained enhancers. Nature Communications 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Peng V et al. Whole-genome profiling of DNA methylation and hydroxymethylation identifies distinct regulatory programs among innate lymphocytes. Nature Immunology 23, 619-+ (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dominguez PM et al. TET2 Deficiency Causes Germial Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenes. Cancer Discovery 8, 1632–1653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ferreira DP et al. Central memory CD8(+) T cells derive from stem-like Tcf7(hi) effector cells in the absence of cytotoxic differentiation. Immunity 53, 985-+ (2020). [DOI] [PubMed] [Google Scholar]
- 175.Zhao XD, Shan Q & Xue HH TCF1 in T cell immunity: a broadened frontier. Nature Reviews Immunology 22, 147–157 (2022). [DOI] [PubMed] [Google Scholar]
- 176.Intlekofer AM et al. Effector and memory CD8(+) T cell fate coupled by T-bet and eomesodermin. Nature Immunology 6, 1236–1244 (2005). [DOI] [PubMed] [Google Scholar]
- 177.Perussia B et al. NATURAL-KILLER (NK) CELL STIMULATORY FACTOR OR IL-12 HAS DIFFERENTIAL-EFFECTS ON THE PROLIFERATION OF TCR-ALPHA-BETA+, TCR-GAMMA-DELTA+ LYMPHOCYTES-T, AND NK CELLS. Journal of Immunology 149, 3495–3502 (1992). [PubMed] [Google Scholar]
- 178.Hsieh CS et al. DEVELOPMENT OF TH1 CD4+ T-CELLS THROUGH IL-12 PRODUCED BY LISTERIA-INDUCED MACROPHAGES. Science 260, 547–549 (1993). [DOI] [PubMed] [Google Scholar]
- 179.Salcedo TW, Azzoni L, Wolf SF & Perussia B MODULATION OF PERFORIN AND GRANZYME MESSENGER-RNA EXPRESSION IN HUMAN NATURAL-KILLER-CELLS. Journal of Immunology 151, 2511–2520 (1993). [PubMed] [Google Scholar]
- 180.Asteamezaga M, Dandrea A, Kubin M & Trinchieri G COOPERATION OF NATURAL-KILLER-CELL STIMULATORY FACTOR INTERLEUKIN-12 WITH OTHER STIMULI IN THE INDUCTION OF CYTOKINES AND CYTOTOXIC CELL-ASSOCIATED MOLECULES IN HUMAN T-CELLS AND NK-CELLS. Cellular Immunology 156, 480–492 (1994). [DOI] [PubMed] [Google Scholar]
- 181.Miller PG et al. Clonal hematopoiesis in patients receiving chimeric antigen receptor T-cell therapy. Blood Advances 5, 2982–2986 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Saini NY et al. Clonal hematopoiesis is associated with increased risk of severe neurotoxicity in axicabtagene ciloleucel therapy of large B-cell lymphoma. Blood cancer discovery (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fraietta JA et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558, 307-+ (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Nobles CL et al. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. Journal of Clinical Investigation 130, 673–685 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shah NN et al. Clonal expansion of CAR T cells harboring lentivector integration in the CBL gene following anti-CD22 CAR T-cell therapy. Blood Advances 3, 2317–2322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Voehringer D, Koschella M & Pircher H Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectinlike receptor G1 (KLRG1). Blood 100, 3698–3702 (2002). [DOI] [PubMed] [Google Scholar]
- 187.Prinzing B et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Science Translational Medicine 13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Abplanalp WT et al. Association of Clonal Hematopoiesis of Indeterminate Potential With Inflammatory Gene Expression in Patients With Severe Degenerative Aortic Valve Stenosis or Chronic Postischemic Heart Failure. Jama Cardiology 5, 1170–1175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shin T-H et al. Macaque Clonal Hematopoiesis Model Demonstrates Expansion of TET2-Disrupted Clones and Utility for Testing Interventions. Blood (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Svensson EC et al. TET2-Driven Clonal Hematopoiesis and Response to Canakinumab An Exploratory Analysis of the CANTOS Randomized Clinical Trial. Jama Cardiology 7, 521–528 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Steensma DP & Bolton KL What to tell your patient with clonal hematopoiesis and why: insights from 2 specialized clinics. Blood 136, 1623–1631 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Singh M et al. Lymphoma Driver Mutations in the Pathogenic Evolution of an Iconic Human Autoantibody. Cell 180, 878-+ (2020). [DOI] [PubMed] [Google Scholar]
- 193.Beck DB et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. New England Journal of Medicine 383, 2628–2638 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Williams N et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature 602, 162-+ (2022). [DOI] [PubMed] [Google Scholar]
- 195.Gibson KL et al. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 8, 18–25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fulop T, Larbi A & Pawelec G Human T cell aging and the impact of persistent viral infections. Frontiers in Immunology 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Akbar AN & Fletcher JM Memory T cell homeostasis and senescence during aging. Current Opinion in Immunology 17, 480–485 (2005). [DOI] [PubMed] [Google Scholar]
- 198.Franceschi C et al. Inflamm-aging - An evolutionary perspective on immunosenescence. Molecular and Cellular Gerontology 908, 244–254 (2000). [DOI] [PubMed] [Google Scholar]
- 199.Lasry A & Ben-Neriah Y Senescence-associated inflammatory responses: aging and cancer perspectives. Trends in Immunology 36, 217–228 (2015). [DOI] [PubMed] [Google Scholar]
- 200.Luo HZ et al. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Reports 26, 945-+ (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Andina N et al. Heightened NLRP3 inflammasome activation is associated with aging and CMML diseases severity. bioRxiv, 2022.2001.2027.477992, doi: 10.1101/2022.01.27.477992 (2022). [DOI] [Google Scholar]
- 202.Basiorka AA et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 128, 2960–2975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Picard E et al. The frequency of interleukin-1 beta-producing monocytes is significantly associated with varicella-zoster responses of nursing home residents. Clinical and Experimental Immunology 205, 63–74 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Franceschi C, Garagnani P, Vitale G, Capri M & Salvioli S Inflammaging and ‘Garb-aging’. Trends in Endocrinology and Metabolism 28, 199–212 (2017). [DOI] [PubMed] [Google Scholar]
- 205.Morita K et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nature Communications 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Miles LA et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 587, 477-+ (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.van Galen P et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 176, 1265-+ (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Paguirigan AL et al. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Science Translational Medicine 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Im AP et al. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia 28, 1774–1783 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bowman RL & Levine RL TET2 in Normal and Malignant Hematopoiesis. Cold Spring Harbor Perspectives in Medicine 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Chen JH et al. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nature Medicine 25, 103-+ (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lindsley RC et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 125, 1367–1376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mitchell E et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 606, 343-+ (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Steensma DP et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Khoury JD et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36, 1703–1719 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Arber DA et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemia: Integrating Morphological, Clinical, and Genomic Data. Blood, doi: 10.1182/blood.2022015850 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Dohner H et al. Diagnosis and Management of AML in Adults: 2022 ELN Recommendations from an International Expert Panel. Blood, doi: 10.1182/blood.2022016867 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Young AL, Challen GA, Birmann BM & Druley TE Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nature Communications 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Dunbar AJ, Rampal RK & Levine R Leukemia secondary to myeloproliferative neoplasms. Blood 136, 61–70 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Greenberg PL et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 120, 2454–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mateos MV et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer Journal 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Odejide O et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 123, 1293–1296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Yao WQ et al. Angioimmunoblastic T-cell lymphoma contains multiple clonal T-cell populations derived from a common TET2 mutant progenitor cell. Journal of Pathology 250, 346–357 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Schwartz FH et al. TET2 mutations in B cells of patients affected by angioimmunoblastic T-cell lymphoma. Journal of Pathology 242, 129–133 (2017). [DOI] [PubMed] [Google Scholar]
- 225.Quivoron C et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell 20, 25–38 (2011). [DOI] [PubMed] [Google Scholar]
- 226.Pastoret C et al. Linking the KIR phenotype with STAT3 and TET2 mutations to identify chronic lymphoproliferative disorders of NK cells. Blood 137, 3237–3250 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Olson TL et al. Frequent somatic TET2 mutations in chronic NK-LGL leukemia with distinct patterns of cytopenias. Blood 138, 662–673 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Bensberg M et al. TET2 as a tumor suppressor and therapeutic target in T-cell acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences of the United States of America 118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Aref S, El Menshawy N, El-Ghonemy MS, Abou Zeid T & El-Baiomy MA Clinicopathologic Effect of DNMT3A Mutation in Adult T-Cell Acute Lymphoblastic Leukemia. Clinical Lymphoma Myeloma & Leukemia 16, 43–48 (2016). [DOI] [PubMed] [Google Scholar]
- 230.Bond J et al. DNMT3A mutation is associated with increased age and adverse outcome in adult T-cell acute lymphoblastic leukemia. Haematologica 104, 1617–1625 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Gao LM et al. Somatic mutations in KMT2D and TET2 associated with worse prognosis in Epstein-Barr virus-associated T or natural killer-cell lymphoproliferative disorders. Cancer Biology & Therapy 20, 1319–1327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Reddy A et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171, 481-+ (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lacy SE et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood 135, 1759–1771 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kaasinen E et al. Impact of constitutional TET2 haploinsufficiency on molecular and clinical phenotype in humans. Nature Communications 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Duployez N et al. Familial myeloid malignancies with germline TET2 mutation. Leukemia 34, 1450–1453 (2020). [DOI] [PubMed] [Google Scholar]
- 236.Wu X et al. Pedigree investigation, clinical characteristics, and prognosis analysis of haematological disease patients with germline TET2 mutation. Bmc Cancer 22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Russler-Germain DA et al. The R882H DNMT3A Mutation Associated with AML Dominantly Inhibits Wild-Type DNMT3A by Blocking Its Ability to Form Active Tetramers. Cancer Cell 25, 442–454 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ferris MA et al. DNMT3A overgrowth syndrome is associated with the development of hematopoietic malignancies in children and young adults. Blood 139, 461–464 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Spegarova JS et al. Germline TET2 loss of function causes childhood immunodeficiency and lymphoma. Blood 136, 1055–1066 (2020). [DOI] [PubMed] [Google Scholar]
- 240.Couronne L, Bastard C & Bernard OA TET2 and DNMT3A Mutations in Human T-Cell Lymphoma. New England Journal of Medicine 366, 95–96 (2012). [DOI] [PubMed] [Google Scholar]
- 241.Tiacci E et al. High-Risk Clonal Hematopoiesis as the Origin of AITL and NPM1-Mutated AML. New England Journal of Medicine 379, 981–984 (2018). [DOI] [PubMed] [Google Scholar]