

Summary
Hypertension is a major cause of morbidity and mortality in patients with hypertrophic cardiomyopathy (HCM), suggesting a potential role for mechanics in HCM pathogenesis. Here, we developed an in vitro physiological model to investigate how mechanics acts together with HCM-linked myosin binding protein C (MYBPC3) mutations to trigger disease. Micro-heart muscles (μHM) were engineered from induced pluripotent stem cell (iPSC)-derived cardiomyocytes bearing MYBPC3+/− mutations and challenged to contract against substrates of different elasticity. μHMs that worked against substrates with stiffness at or exceeding the stiffness of healthy adult heart muscle exhibited several hallmarks of HCM, including cellular hypertrophy, impaired contractile energetics, and maladaptive calcium handling. Remarkably, we discovered changes in troponin C and T localization in MYBPC3+/− μHM that were entirely absent in 2D culture. Pharmacologic studies suggested that excessive Ca2+ intake through membrane-embedded channels underlie the observed electrophysiological abnormalities. These results illustrate the power of physiologically relevant engineered tissue models to study inherited disease with iPSC technology.
Subject areas: Mechanobiology, Biological sciences, Tissue engineering, Stem cells research
Graphical abstract
Highlights
-
•
Substrate mechanics and MYBPC3+/− mutation trigger early hallmarks of HCM using iPSC
-
•
Differential troponin complex localization was observed in MYBPC3+/− tissues
-
•
MYBPC3+/− tissues exhibit impaired contractile energetics and slower kinetics
-
•
Excessive channel activity underlies abnormal Ca2+ handling in MYBPC3+/− tissues
Mechanobiology; Biological sciences; Tissue engineering; Stem cells research
Introduction
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiomyopathy and the leading cause of sudden cardiac death in the young.1 HCM is known to have incomplete penetrance: for example, HCM patients from the same family, and even twins, harboring identical sarcomere genomic variants can develop vastly different clinical phenotypes.2,3 Thus, non-genetic, environmental factors such as hypertension and associated myocardial stiffening may play a synergistic role in developing an overt HCM phenotype.4,5,6 Hypertension causes increased afterload on the heart, and this increased mechanical loading induces cardiac hypertrophy.7 How HCM genetics and mechanical stress on the heart might synergize to unmask a pathologic phenotype of HCM remains unknown.8 Advances in tissue engineering and stem cell biology have improved structural and functional maturity of induced pluripotent stem cell (iPSC)-derived cardiomyocytes, making these cells valuable tools for studying inherited heart disease.9,10,11,12 However, unlike mouse models where transverse aortic constriction has been used to study pressure overload effects on disease pathophysiology,6 there have been few in vitro engineered heart tissue-based studies that investigate how HCM mutations alter cardiomyocyte hypertrophic remodeling in response to mechanical loading.4,5
HCM is linked to genetic mutations of protein components of the contractile sarcomere apparatus of cardiomyocytes. Mutations in myosin binding protein C (MYBPC3) predominate, and the vast majority of HCM-linked MYBPC3 genomic variants are loss-of-function mutations.13,14,15,16 Hallmarks of the HCM phenotype include myocyte hypertrophy, structural disarray, and the propensity for malignant cardiac arrhythmia. Current medical managements include beta-blockers, calcium channel blockers, and the novel myosin inhibitor, mavacamten.8 Interestingly, valsartan, an afterload reducing agent, has been found to inhibit the progression of the HCM phenotype, but only when the drug is given before the HCM phenotype fully manifests in animal models, suggesting that environmental factors like afterload may play a critical role in disease manifestation.17,18 Nevertheless, the precise molecular mechanisms through which these drugs can reduce and potentially prevent hypertrophic remodeling and/or abnormal calcium handling is not clear.19,20
Clinical and animal studies of HCM suggest that cellular and sarcomeric disarray within cardiomyocytes may be directly related to the sarcomere mutation, with fibrosis and electrophysiological abnormalities being secondary.21,22 In iPSC models, sarcomere disarray was reported with MYH7 mutations. However, no clear sarcomere disarray was seen in MYBPC3−/− cardiomyocytes.5,15,23,24,25 In addition to structural disorganization of the sarcomere, altered Ca2+ intake and increased Ca2+ sensitivity is often described in HCM.26,27,28,29 For example, analysis of cardiomyocytes from HCM patients’ surgical biopsies has shown reduced expression of sarcoplasmic reticulum Ca2+ ATPase (SERCA) and inefficient contractile energetics.30 Studies using iPSC models have identified abnormalities in the Ca2+ transient with contractile deficits.5,28 However, iPSC models also identified sarcomere mutation induced contractile dysfunction, which were independent of Ca2+ signaling.15,23,24 Thus, it remains unclear whether Ca2+ dysfunction in the setting of HCM is triggered by environmental factors.
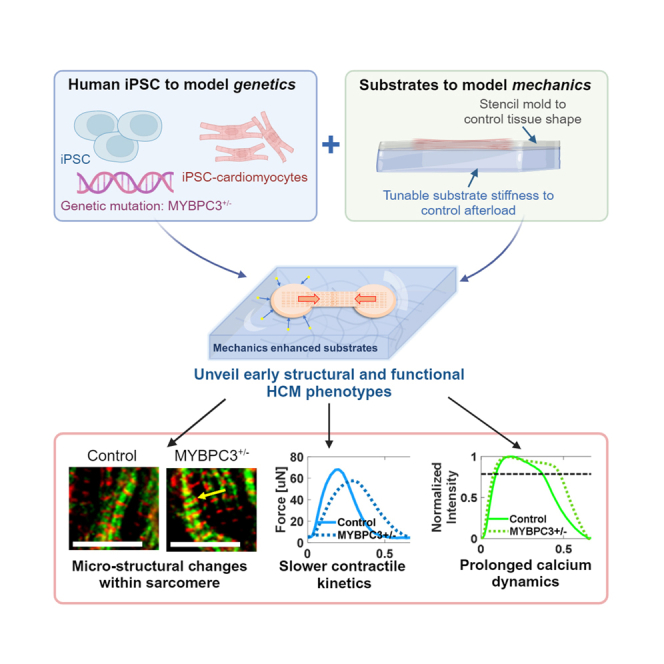
Here, we investigated how substrate mechanics to contractility, together with a physiologically relevant 3D culture environment, impact early development of HCM phenotypes in MYBPC3+/− and isogenic control iPSC-derived micro-heart muscle arrays (μHM). We hypothesized that increased passive stiffness to contractility would mimic the effects of hypertension induced afterload to trigger early pathogenesis of HCM. To provide this substrate mechanics, we formed μHM on poly(dimethyl siloxane) (PDMS) substrates with elasticity (tensile young’s moduli) ranging from 0.4 kPa to 114 kPa to mimic myocardial stiffnesses ranging from the embryonic developmental stage up to a fibrotic adult heart muscle (Figure 1).21,31,32 Within μHMs, MYBPC3+/− cardiomyocytes exhibited cellular hypertrophy, along with changes in sarcomere morphology, contractility, and calcium handling, all of which were absent in 2D monolayers. The changes were exacerbated by increasing the stiffness of the substrate that μHMs contracted against. Pharmacologic studies pointed toward excessive calcium intake through L-type channels, rather than SERCA dysfunction or calcium buffering at the myofilaments, as the most likely explanation for aberrant Ca2+ transients and contractility. Notably, we also identified micro-scale deficits in troponin complex in MYBPC3+/− tissues. These studies suggest that early initiation of the HCM phenotype may involve micro-scale sarcomere reorganization, which depends on mechanical afterload sensitivity, emphasizing the value of engineered tissue models in uncovering pathogenic mechanisms in the early stages of HCM.
Figure 1.

Schematic of mechanical overload enhanced system
(A) Human iPSC differentiation using Wnt signaling pathway, and cardiomyocytes were metabolically purified using lactate. High density of cardiomyocytes mixture was filled into “dog bone” shaped stencil mold to form micro-heart muscle arrays (μHM).
(B) Tissue within “dog bone” shaped mold was formed on PDMS substrates with desired mechanical stiffness mimicking different cardiac resistance during contraction.
(C) iPSC-μHM action potential, calcium and traction forces were measured through fluorescence high speed imaging. Scale bars: 500 μm.
Results
Mechanics enhanced substrates unveil HCM genotype-dependent cellular hypertrophy in μHM
As HCM does not manifest in utero, our expectation was that culture environments that do not induce structural and physiologic maturation would not lead to appreciable differences between MYBPC3+/− and isogenic control iPSC-cardiomyocytes. Conversely, disease phenotypes would develop in physiologically relevant, aligned 3D tissues working against substrates with postnatal levels of stiffness. Consistent with this expectation and prior studies,4,33 there was no evidence for hypertrophy in MYBPC3+/− iPSC-cardiomyocytes in 2D culture in standard tissue culture polystyrene plates (Figure S2). Within μHM, increasing substrate stiffness triggered cellular hypertrophy in both genotypes. For control tissues, cardiomyocytes within μHM formed on 114 kPa substrates had the largest mean projected cellular area (77 ± 24 μm2), which was significantly higher than the projected area of cardiomyocytes in other conditions. Interestingly, MYBPC3+/− cardiomyocytes reached peak cell size when cultured in μHM working against 15 kPa substrates (mean cellular area of 86 ± 26 μm2) and did not exhibit further hypertrophy when substrate stiffness increased beyond 15 kPa. Compared to isogenic controls, MYBPC3+/− tissues revealed a significant increase in cellular cross-sectional area (Figures 2A, 2B, and S4). This difference was exacerbated on substrates mimicking the stiffness of healthy heart muscle (15 kPa) or hearts at intermediate stages of fibrosis (65 kPa),32,34 with MYBPC3+/− cardiomyocytes exhibiting over a 30% increase in projected cell area. In contrast, on very stiff (114 kPa) substrates, cardiomyocytes within control μHM exhibited substantial hypertrophy, blunting genotype-linked differences (Figure 2B). This result mimics prior observations from clinical and mouse studies.25,35 Specifically, MYBPC3 deficient mice had similar heart mass compared to wild-type controls at baseline (no mechanical stress) but exhibited overly exuberant cardiac hypertrophy in response to hypertension triggered by transaortic constriction surgery.6
Figure 2.

Substrate stiffness trigger iPSC-μHM morphological adaptation and hypertrophy
(A) Representative WGA staining for control and MYBPC3+/− iPSC-μHM at 15 kPa, larger cellular cross-sectional area was seen for MYBPC3+/− tissues.
(B) Mechanical stiffnesses trigger cellular hypertrophy. MYBPC3+/− has larger cell area compared to control tissue, especially at 15 and 65 kPa conditions, however, less genotype related hypertrophy was seen at 114 kPa (n = 75–140 per group).
(C) Representative sarcomere α-actinin and DAPI co-stain indicates both control (top) and MYBPC3+/− (bottom) iPSC-μHM has better sarcomere alignment at 15 kPa condition. Less organized sarcomere was observed at ultrasoft 0.4 kPa and stiff 114 kPa conditions.
(D) Average angle between individual sarcomere to tissue shaft. 15 kPa condition has the most organized sarcomere, both genotypes adapt to environmental stiffness similarly (n = 12–25 per group).
(E) Average sarcomere lengths were similar between genotypes, 15 kPa condition has slightly longer sarcomere compared to other conditions (n = 17–30 per group).
(F) Spontaneous beat rate changes for control and MYBPC3+/− 15 kPa tissues in response for isoproterenol.
(G and H) Spontaneous (G) and 1.5 Hz (H) pacing Ca2+ intake for control and MYBPC3+/− 15 kPa tissues in response for isoproterenol (n = 5–12 per group). ∗, ∗∗∗, ∗∗∗∗ indicate p value less than 0.05, 0.001 and 0.0001, respectively. Error bar: SD. Scale bars: 50 μm, insets: 25 μm.
Despite the profound cellular hypertrophy in MYBPC3+/− tissues, overall Z-disk organization, as depicted by staining for sarcomeric α-actinin (ACTN2) was similar between MYBPC3+/− and control iPSC-cardiomyocytes cultured in 2D on tissue culture plastic and in 3D μHM (Figures 2C–2E, 4B, and 4C). In both MYBPC3+/− and isogenic control μHM, increasing substrate stiffness from 0.4 kPa to 15 kPa markedly enhanced cellular alignment, quantified by measuring sarcomere orientation relative to tissue orientation (tissue shaft direction, Figure S5), illustrating mechano-induced structural maturation.36 Notably, μHM formed on stiff 114 kPa substrates showed poor sarcomere organization as indicated by a high divergence in sarcomere alignment to the tissue shaft (Figures 2C and 2D). Sarcomere disarray is strongly related to myocardial disarray (e.g., spatial disorganization of cardiomyocytes), which is one of the first pathological description of HCM in patients and is also a risk factor for sudden death.37,38 Our result concurs with clinical observations about load induced sarcomere disarray.21,25,37
Figure 4.

Mechanical stiffness triggers structural maturation of troponin C
(A) MYBPC3+/− genotype has significantly less MYBPC3, troponin T and C expression compared to control (n = 4–10 per 4A group).
(B and C) Similar MYBPC3, TNNC1, and TNNT2 structural were seen between control and MYBPC3+/− purified cardiomyocytes. Both MYBPC3 and TNNT2 have striated structure, however, troponin C is filamentous with localization at cellular nuclei (middle column). Targeted protein was merged with ACTN2, representative sarcomere traces were shown below each merged image, red indicates ACTN intensity, blue, green, cyan indicate MYBPC3, TNNC1, and TNNT2 respectively.
(D and E) Representative immunostaining for μHM formed on 15 kPa substrates. Compared to 2D sarcomere are widened in 3D, similar MYBPC3 localization was seen between the genotypes. Strikingly, we observed striated TNNC1 protein, yet both TNNC1 and TNNT2 protein have different localization in reference to ACTN2 compared to control tissues. ∗ and ∗∗ indicate p values less than 0.05 and 0.01. Error bars: SD. Scale bars: 10 μm.
To study whether the structural abnormalities were linked to differential adrenergic tone of MYBPC3+/− μHM, as suggested by previous studies of HCM and other cardiomyopathies,39,40 we stressed the μHMs with isoproterenol to determine their capacity for beta-adrenergic stimulation. Acute isoproterenol treatment revealed a significant increase in spontaneous beat rate and calcium intake for both genotypes on 0.4 and 15 kPa conditions (Figures 2F, 2G, and S6). Notably, during spontaneous beating, MYBPC3+/− tissues exhibited a similar, inotropic response to high dose isoproterenol (1 μM) as the isogenic control, suggesting preserved adrenergic reserve in the MYBPC3+/− tissues (Figures 2G and S6). However, MYBPC3+/− tissues exhibited a significantly lower spontaneous beat rate compared to the isogenic control at higher doses (Figures 2F and S6). In addition, when challenged with 1.5 Hz field pacing, the MYBPC3+/− tissues formed on 15 kPa conditions failed to maintain the adrenergic tone at high (1 μM) isoproterenol doses, potentially because of the slower contraction kinetics of MYBPC3+/− tissues (Figure 2H), yet this phenotype was absent at ultrasoft 0.4 kPa conditions (Figure S6). Despite this small change in isoproterenol response at higher dose, the overall adrenergic responsiveness was grossly preserved in MYBPC3+/− μHM, in contrast to the lack of adrenergic response in some iPSC-based models of dilated cardiomyopathy.41 Together with the evidence for cellular hypertrophy in the absence of gross sarcomere disarray, these results suggest that our model reflects an early stage HCM phenotype.39,40
Aberrant contractile kinetics and energetics in MYBPC3+/− μHM
Clinical observations and mouse models of MYBPC3-linked HCM have suggested impairment of both diastolic relaxation and systolic contraction.42,43,44,45 Here, we observed that μHM with MYBPC3+/− deficiency exhibited much slower contraction kinetics compared to isogenic controls (Figures 3A and 3C). During spontaneous contraction, a hypercontractility phenotype was observed in MYBPC3+/− μHM when substrate stiffness approached fibrotic levels (65 and 114 kPa, Figure 3B), consistent with the clinical phenotype of HCM, our own prior studies on μHM, and work by others on MYBPC3 mutant engineered heart muscle beating at low frequencies (≤1 Hz).4,23 However, at 15 kPa, where the spontaneous contractility is similar between the genotypes, analysis of the contractile power curves revealed that the slower contraction kinetics of MYBPC3+/− was associated with significantly higher energy consumption (Figures 3E and 3F). Moreover, MYBPC3+/− μHM exhibited asymmetric power generation due to the slower rising phase of the contraction (Figure 3E). Likely because of their slower contraction kinetics, MYBPC3+/− μHM exhibited significantly reduced contractility when field paced at a moderately high beat rate (1.5 Hz) and were hypocontractile on 15 kPa conditions (Figure 3D). Interestingly, at higher stiffnesses, the energy consumption during spontaneous contraction was diminished (via a marked reduction in contraction velocity) and was not different between genotypes (Figure 3F). These observations are consistent overall with prior animal and human iPSC-cardiomyocyte studies, where slower contraction kinetics in HCM cardiomyocytes, thought to be due to sarcomere abnormalities and disrupted super relaxed state, have been described.24,27,46
Figure 3.

MYBPC3+/− iPSC-μHM is associated with slower contraction kinetics together with impaired energy consumption
(A) Representative spontaneous force curves for both control and MYBPC3+/− iPSC-μHM at 15 kPa, slower force rising phase was seen in MYBPC3+/− iPSC-μHM.
(B) During spontaneous contraction, a hypercontractility phenotype was seen in MYBPC3 tissues, especially at stiffer conditions (n = 5–37 per group).
(C) Representative pacing force curves for both control and MYBPC3+/− iPSC-μHM at 15 kPa, slower force kinetics with minimal resting phase was seen in MYBPC3+/− iPSC-μHM.
(D) At 1.5 Hz pacing, MYBPC3+/− iPSC-μHM is hypocontractile at 15 kPa (n = 6–25 per group).
(E) Representative power curve for control and MYBPC3+/− iPSC-μHM at 15 kPa, a phenotype of non-uniform power distribution was seen in MYBPC3+/− iPSC-μHM.
(F) Integrated energy consumption indicated MYBPC3+/− iPSC-μHM consume more power compared with control at 15 kPa (n = 5–6 per group). ∗ and ∗∗ indicate p value less than 0.05 and 0.01. Error bars: SD.
Differential troponin localization associated with MYBPC3+/− μHM
Despite few differences observed in cardiomyocyte α-actinin organization (Figure 2) or changes in transcription of ACTN2 (Figure S7), we discovered that, along with the expected genetically encoded reduction in transcription of MYBPC3, transcript levels for troponin C (TNNC1), and cardiac troponin T (TNNT2) were also reduced substantially in MYBPC3+/− tissues (Figure 4A). To determine whether these RNA changes might be correlated to sarcomeric organization, we examined protein structure by immunostaining iPSC-cardiomyocytes in monoculture along with longitudinal cryosections of μHM. For purified iPSC-cardiomyocytes (differentiation day 30) cultured in 2D on tissue culture polystyrene substrates, we observed a clear striated structure of ACTN2, TNNT2, and MYBPC3. In contrast, TNNC1 exhibited filamentous and nucleus localization in 2D iPSC-cardiomyocytes of both genotypes. This was reflected strongly in representative line traces of the sarcomere proteins, indicating significantly less periodicity of TNNC1 compared to other sarcomere proteins (Figures 4B and 4C).
In contrast to 2D cardiomyocyte cultures, cardiomyocytes within cryosections of control 3D μHM formed on physiological 15 kPa substrates showed more organized TNNC1 structure, with appropriate sarcomeric localization (Figures 4D and 4E). Moreover, within μHM of both genotypes, individual sarcomeres appeared to be wider than sarcomeres from 2D iPSC-cardiomyocytes (Figures 4B–4E). Interestingly, in MYPBC3+/− μHM, while MYBPC3 itself appeared to have appropriate localization, both TNNC1 and TNNT2 appear to be mis-localized. In isogenic control tissues, we observed physiological alternating localization of troponins with sarcomeric α-actinin containing Z-disks, exemplified most readily in line-scans of the fluorescence micrographs. In contrast, within MYBPC3+/− μHM, both troponin proteins exhibited potentially pathologic overlap with the Z-disk (Figures 4D and 4E).
Mechanics exacerbates calcium handling dysfunction in MYBPC3+/− μHM
Calcium plays a critical role in cardiac contraction and relaxation; dysregulation in calcium can lead to abnormal contraction, arrhythmias, and structural changes in HCM.28,47 Thus, we assessed both action potential waveforms and calcium handling of the μHM (Figures 5A and 5B). In 2D iPSC-cardiomyocytes (differentiation day 30), we observed similar Ca2+ transients and action potential kinetics in cells of both genotypes, although MYBPC3+/− iPSC-cardiomyocytes exhibited higher cytosolic calcium amplitude (Figures 5C and 5D). However, culturing cardiomyocytes in μHM unveiled marked Ca2+ handling abnormalities in MYBPC3+/− cells (Figures 5B–5F). Most striking was a prolonged systolic Ca2+ plateau phase (Figures 5B and 5D) that was comparable to clinical observations of impaired myocardial relaxation, especially under chronic pressure overload conditions.26,48 Abnormalities of Ca2+ dynamics in MYBPC3+/− μHM, especially the prolonged systolic plateau, were exacerbated when μHM substrate stiffness exceeded 0.4 kPa (Figure 5D). Nevertheless, despite significant prolongation in Ca2+ transient, differences in action potential waveform morphology were relatively subtle (Figures 5A–5F). These findings suggest that the primary electrophysiological difference between the HCM-prone MYBPC3+/− tissues and isogenic controls is in Ca2+ handling, as opposed to a global electrophysiology change. Meanwhile, we found that control tissues could adapt to mechanical stiffnesses up to 114 kPa through changes in their physiology, such as increased calcium decay and action potential durations; however, MYBPC3+/− tissues develop disease phenotypes at physiological 15 kPa, but did not exhibit further adaptation to higher mechanical stiffnesses, indicating that calcium dynamics for MYBPC3+/− tissues were less sensitive to environmental stress beyond the stiffness (15 kPa) which induced initial calcium transient prolongation.
Figure 5.

Mechanical stiffness trigger Ca2+ mishandling in MYBPC3+/− iPSC-μHM
(A and B) Representative action potential and calcium traces for both control and MYBPC3+/− iPSC-μHM at 15 kPa condition. Significantly prolonged Ca2+ during the peak was observed for MYBPC3+/− iPSC-μHM.
(C) Compared to 2D monolayer (n = 17–18 for monolayer group), iPSC-μHM has more Ca2+ intake, further, MYBPC3+/− tissue has significantly higher Ca2+ intake compared to control (n = 24–37 per tissue group).
(D) 2D monolayer has similar Ca2+ kinetics compared to control (n = 17–18 per monolayer group). However, when cells formed in 3D tissue, a significantly longer Ca2+ plateau was observed at elevated substrate stiffnesses (n = 24–47 per tissue group).
(E and F) Heatmap of action potential and Ca2+ transient during spontaneous contraction (E) and 1.5 Hz paced contraction. Control tissue has prolonged action potential and Ca2+ transient at stiffness, while 15 kPa has the shortest action potential and Ca2+ transient. MYBPC3+/− tissues always have significantly longer Ca2+ plateau compared to control, while MYBPC3+/− tissues are less responsive to mechanical stiffness. ∗∗∗ and ∗∗∗∗ indicate p values less than 0.001 and 0.0001. Error bars: SD.
Pharmacological studies indicate L-type Ca2+ channel dysfunction in MYBPC3+/− μHM
The prolonged Ca2+ plateau phase in MYBPC3+/− tissues was not associated with transcriptional change of the L-type Ca2+ channel (CACNA1c, Figure 6A). A slight inhibition of SERCA transcript was observed in MYBPC3+/− μHM (Figure 6B). SERCA is responsible for removal of >70% of cytoplasmic Ca2+,49 and SERCA transcript levels were observed to be reduced in cardiac biopsies.26,30 Thus, to test whether Ca2+ mishandling was caused by SERCA inhibition, we administered a SERCA-specific inhibitor, thapsigargin.50 Thapsigargin treatment significantly increased Ca2+ upstroke duration for both genotypes (Figure 6C). However, SERCA inhibition did not cause a prolonged Ca2+ plateau phenotype in the control tissues (Figure S8). In addition, this drug also produced a trend toward hypercontractility in both genotypes, though the magnitude of the change was only significant in control tissues (Figure 6D). While the contractile response was notable, the fact that both genotypes exhibited similar Ca2+ handling changes in response to SERCA inhibition suggests against SERCA dysfunction being the primary cause for observed Ca2+ mishandling (Figures 5D–5F).
Figure 6.

Pharmacological probes target specific Ca2+ mishandling mechanisms associated with MYBPC3
(A and B) Ca2+ regulation gene expression, similar CACNA1c and SERCA expression was seen between genotypes or stiffnesses (n = 4–10 per group).
(C) Thapsigargin (Thap) has similar drug effects for both genotypes, significantly prolonged Ca2+ upstroke duration (n = 10–42 per group).
(D) Thapsigargin resulted in higher contractility, especially for control tissues (n = 6–19 per group).
(E) Mavacamten (MYK) did not change Ca2+ transient significantly, although there is a trend toward shortened Ca2+ plateau for MYBPC3+/− tissues (n = 4–24 per group).
(F) Significantly reduced contractility was seen for both genotypes with Mavacamten (n = 5–8 per 6F group).
(G and H) Control and MYBPC3+/− μHM in response to verapamil. MYBPC3+/− tissues showed more resistance to verapamil blocking. Higher doses of verapamil however reduce the Ca2+ plateau duration in MYBPC3+/− μHM.
(I and J) Ca2+ plateau duration in response to verapamil for both control and MYBPC3+/− μHM, higher IC50 is required for higher mechanical resistance (n = 5–29 per 6I group, n = 5–17 per 6J group).
(K and L) Contractility in response to verapamil for both control and MYBPC3+/− μHM, higher IC50 is required for higher mechanical resistance (n = 4–20 per 6K group, n = 10–20 per 6L group). ∗, ∗∗, ∗∗∗, and ∗∗∗∗ indicate p values less than 0.05, 0.01, 0.001 and 0.0001. Error bars: SD. n > 4, 15 kPa tissues at 1.5 Hz were selected for comparison.
Dysfunction in Ca2+ handling in HCM has been suggested by some to originate directly from excessive Ca2+ buffering at myofilaments.51,52 To test this possibility in our μHM, we acutely inhibited myosin using mavacamten.53 While others have reported that chronic mavacamten exposure can normalize Ca2+ handling in engineered heart muscle with HCM mutations,54 we observed only a very small change in the length of the systolic Ca2+ plateau in MYBPC3+/− tissues with acute mavacamten dosing (Figures 6E and S9). However, this dose did markedly reduce contractility in both genotypes (Figure 6F). These results suggest that changes in the myofilament invoked by mavacamten do not acutely impact the overall Ca2+ transient. Consistent with our results, chronic mavacamten treatment was suggested to impact Ca2+ cycling through indirect means in prior work by others.55
Finally, as drug probe studies suggested against SERCA dysfunction or myofilament calcium buffering as mechanisms for calcium handling dysfunction in MYBPC3+/− tissues, we investigated a potential role for calcium channels using the Ca2+ channel blocker verapamil, which is widely prescribed for HCM patients.56 Strikingly, MYBPC3+/− μHM exhibited markedly higher resistance to verapamil compared to isogenic control tissues: only very subtle changes in calcium handling were seen at 0.1 μM verapamil. In contrast, control tissues showed a significantly negative inotropic effect (Figure 6G). Unlike mavacamten, verapamil treatment significantly reduced the prolonged Ca2+ plateau phase observed in MYBPC3+/− μHM (Figure 6H). Importantly, MYBPC3+/− tissues’ resistance to verapamil increased when the tissues worked against higher stiffnesses 15 kPa (compared to fetal-like stiffness 0.4 kPa; Figures 6I, 6J, and S10). This result suggests that although Ca2+ intake was increased in MYBPC3+/− cardiomyocytes at baseline (Figure 5C), mechanical stress provokes more exaggerated dysfunction of calcium handling. As with calcium handling, the impact of verapamil on force production in MYBPC3+/− μHM was markedly less than the effect on control tissues, with the MYBPC3 deficient tissues exhibiting a much higher IC50: 474 vs. 112 on 0.4 kPa condition and 327 vs. 60 on 15 kPa conditions (Figures 6K and 6L).
Discussion
In this study, we leveraged iPSC technology and a physiologically relevant engineered 3D micro-tissue system, in which tissues performed mechanical work against substrates with tunable stiffness, to simulate the combined effects of mechanical load and genetics in HCM pathogenesis. Our results mirror clinical findings, which indicate that coexistent hypertension worsens the prognosis of HCM patients, but also that HCM can develop in patients with blood pressures that would not be considered abnormal.30,57 By expanding the range of the passive stiffness cardiac tissues contract against from our previous studies,4,33 we were able to simulate the impact of both physiologic increases in tissue stiffness (0.4–15 kPa) and pathologic increases in tissue stiffness (15–65 or 114 kPa (Figure 1). Applying this system, we discovered and quantified how mechanics can trigger several early-stage structural, physiologic, and pharmacologic hallmarks of HCM.58
In the valsartan for attenuating disease evolution in early sarcomeric hypertrophic cardiomyopathy (VANISH) clinical trial, investigators studied the impact of valsartan on early-stage HCM patients, who are less than 30 years old and do not exhibit severe left ventricular hypertrophy (LVH). Valsartan blocks the angiotensin II receptor to inhibit pathological cardiac remodeling.17 Interestingly, a composite clinical score that weighed several early metrics of cardiac function, suggested potential benefits of valsartan in this patient cohort. It is consistent with preclinical studies in HCM mouse models, whereby administration of losartan to HCM mice abrogated the development of LVH and fibrosis, when administered early, but did not appear to be beneficial if administered once hypertrophy was apparent.59 While one of the key proposed mechanisms of action for valsartan was reduction of TGF-β signaling, direct analysis of circulating TGF-β levels in VANISH study patients showed minimal differences between valsartan and placebo treatment groups.60 In contrast, valsartan-treated patients exhibited a significantly reduction of systolic and diastolic blood pressure compared to placebo patients.17 This is consistent with the known ability of valsartan to reduce blood pressure by blocking angiotensin receptors.61 Thus, afterload reduction may be a key means for the efficacy of angiotensin receptor blockers like valsartan in treating early-stage HCM. Conversely, the presence of increased afterload, even subtle increases that would not lead to hypertrophic remodeling in patients who do not harbor sarcomeric genome variants, may be important for early HCM pathogenesis of HCM. In another study of pre-clinical HCM, diltiazem, when administered before the overt HCM phenotype, appeared to improve early LV remodeling in HCM, particularly for MYBPC3 carriers.18 These results suggested that afterload may play a pathogenic role in early-stage HCM patients and aggressive control of either afterload or contractility in the pre-clinical setting may ameliorate predisposition to overt disease. Our in vitro system lends support to this hypothesis, and allowed us to further elucidate how the interaction between mechanics and genetics impacts HCM development.
We observed minimal differences in phenotypes between MYBPC3+/− and control cardiomyocytes cultured on standard 2D tissue culture polystyrene, or in 3D micro-tissues contracting against ultrasoft (0.4 kPa) substrates mimicking fetal heart condition. For example, when contracting against 0.4 kPa substrates, both control and MYBPC3+/− tissues had similar cardiac contractility (Figures 3B–3D and 3F) and calcium dynamics (Figure 5D). Moreover, similar to 2D monolayer iPSC-cardiomyocytes, tissues formed on 0.4 kPa substrates had less aligned sarcomeres regardless of genotype. In contrast, cells of both genotypes exhibited robust sarcomere alignment and organization when cultured on substrates with physiologic stiffness (15 kPa, Figure 2C). Our results are concordant with previous in vitro studies showing that physiologically relevant mechanical stimuli can increase engineered heart tissue maturity,9,36,62 and enhance sarcomere alignment.63,64,65 Likewise, studies using mouse hearts observed that an initial passive stretch can significantly increase the peak contractility of the left ventricular free wall, in both circumferential and longitudinal directions.66 Those results indicate the adaptive behavior of cardiac tissue to external mechanical stresses. In contrast, when μHM of either genotype contracted against very stiff (114 kPa) substrates, we observed marked sarcomere disarray/misalignments (Figure 2C). Similar disarray has been reported in HCM patients, further highlighting the importance of incorporating mechanical cues into iPSC-based in vitro disease models.9,37,67,68
Surprisingly, while the MYPBC3 protein itself exhibited normal localization, we discovered a unique structural defect in troponin complex in MYBPC3+/− tissues. This phenotype was masked in 2D culture and may be linked to structural maturation of the sarcomere triggered by 3D physiological environment (Figures 4B–4E). Within this tissue setting, we observed increased sarcomere width, and physiologic localization of TNNC1 in isogenic control tissues (Figures 2C and 4B–4E). In purified 2D cardiac monolayers, we observed that TNNC1 was primarily localized to the nucleus rather than the sarcomere. Similar mis-localization of this protein has been reported by others in neonatal rat ventricular cardiomyocyte and iPSC-cardiomyocytes.69,70 Importantly, our observation of defective localization of TNNC1 and TNNT2 in μHM mirrors clinical observations of HCM patients, where cardiac cell degeneration is associated with structural defects including an abortive form of sarcomerogenesis.71 The micro-defects of troponin complex may be an early structural deformation associated with MYBPC3 mutations, potentially relating the physiological phenotypes observed. For example, the troponin complex is strongly linked to heart muscle contraction and can cause Ca2+ sensitivity.72,73,74 Further, troponins have important implications in cardiomyopathy, with identified troponin mutations causing hypertrophic, dilated, and restrictive myopathies.47,75,76
Besides the unique troponin structural defects reported here (Figure 4), we also captured marked changes in the Ca2+ transient for the MYBPC3+/− tissues, which exhibited significantly higher Ca2+ intake and a prolonged systolic Ca2+ plateau (Figures 5B and 5D). Ca2+ mishandling has been reported in both human and mice HCM models, with potential mechanisms including dysfunction of Ca2+ channels, disrupted Ca2+ buffering, and stress induced Ca2+ sensitivity.27,28,29,30,47,77 While SERCA inhibition has been reported in obstructive HCM patient samples, we observed similar SERCA function between control and MYBPC3+/− tissues, indicating SERCA may not be the primary cause of the prolonged Ca2+ plateau in our μHM model (Figures 6C and 6D).30,78 Meanwhile, the hypothesis of altered myofilament Ca2+ buffering has been supported by studies with recombinant protein and isolated pig heart samples associated with thin filament proteins like troponin.29,55 However, we and others observed that mavacamten, which shifts myosin into their super-relaxed state,45 has much less effect on intracellular Ca2+ compared to its effect on contraction inhibition (Figures 6E and 6F). This indicates calcium buffering at the myofilaments may not directly cause calcium abnormalities in HCM. Nevertheless, whether chronic depression of HCM-cardiomyocyte contractility with mavacamten can cause adaptive reduction of calcium buffering needs to be further investigated.47,54,79 We did observe that aberrant Ca2+ handling is associated with a much slower contraction kinetics and impaired energy consumption in MYBPC3+/− μHM (Figures 3 and 5).
Our observations of differential verapamil responses between control and MYBPC3+/− μHM suggest an excessive activation of L-type calcium channel in MYBPC3+/− μHM, potentially altering the intercellular Ca2+ homeostasis, and leading to impaired force generation kinetics and hypertrophy (Figures 6G and 6H).27,46 Notably, while MYBPC3+/− μHM on 0.4 kPa substrates had similar verapamil response to controls, the genotype-linked difference was readily observed on substrates with physiologic 15 kPa stiffness (Figures 6I–6L). Consistent with our observations of hypertrophy (Figure 2B) and contractile energetics (Figure 3), we found that substrate mechanics to contractility—linked to substrate stiffness in vitro and hypertension in vivo—plays a critical role in early-stage pathogenesis of MYBPC3-linked HCM.4,80,81
In conclusion, we developed a μHM model that leverages substrates with tunable stiffness to investigate how non-genetic factors (e.g., hypertension) can synergize with genomic variants in triggering pathogenesis of HCM. Our simple PDMS based physiological model provides a valuable tool to understand the pathogenesis of HCM. This model proved useful for capturing early phenotypes of HCM disease, creating a tool for investigating disease prevention, and developing novel pharmacological targets. Importantly, this system can be widely employed to explore other inherited cardiomyopathies and investigate combined effects of mechanics and genetics.
Limitation of the study
There are several limitations to the present μHM model. First, this engineered tissue system does not recapitulate naturally occurring transmural variations in heart muscle alignment and mechanics. Nevertheless, we and others have observed that 3D heart tissue model with enhanced mechanical stimuli do engender robust cardiomyocyte alignment, cell-cell connectivity and cellular maturation (Figure 2C).9,82 Second, our contractility measurements are based on traction force microscopy, where deformation of the underlying substrate was quantified. When modeling stiff fibrotic conditions (elasticity >100 kPa), this led to bead deflection that was very small, increasing the noise in contractility and power measurements, a challenge that exists in many engineered tissue system (e.g., posts) in which contractile motion is small. Finally, while our tissues do have a significant fraction of non-cardiac stromal cells, they lack a true cardiac fibroblast population, which could in the future be added to the tissues to determine how Extracellular Matrix within the tissue and tissue mechanical properties evolve dynamically during early HCM pathogenesis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal Cardiac α-actinin (ACTN2, Clone EA-53, IHC 1:1000) | Sigma | CAT# A7811; RRID: AB_476766 |
| Rabbit monoclonal Cardiac α-actinin (ACTN2, Clone 1E11, IHC 1:100) | Sigma | CAT#ZRB1169; RRID: AB_3094613 |
| Mouse monoclonal Cardiac Troponin C (TNNC1, Clone 12G3, IHC 1:100) | Santa Cruz | CAT# sc-52263; RRID: AB_675914 |
| Mouse monoclonal Cardiac Troponin T (TNNT2, Clone 11-13, IHC 1:100) | Thermo Fisher | Cat#MA5-12960; RRID: AB_11000742 |
| Mouse monoclonal Myosin Binding Protein C (MYBPC3, Clone e-7, IHC 1:100) | Santa Cruz | CAT# sc-137180; RRID: AB_2017317 |
| Mouse monoclonal Human Fibronectin (IHC 1:250) | Developmental Studies Hybridoma Bank-DSHB | CAT# HFN 7.1; RRID: AB_528244 |
| Goat anti-Mouse IgG, Secondary Antibody, Alexa Fluor™ 488 | Thermo Fisher | CAT#A-11001; RRID: AB_2534069 |
| Goat anti-Mouse IgG, Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher | CAT#A-21235; RRID: AB_2535804 |
| Goat anti-Mouse IgG, Secondary Antibody, Alexa Fluor™ 594 | Thermo Fisher | CAT#A-11032; RRID: AB_2534091 |
| Goat anti-Rabbit IgG, Secondary Antibody, Alexa Fluor™ 488 | Thermo Fisher | CAT#A-11008; RRID: AB_143165 |
| Chemicals, peptides, and recombinant proteins | ||
| PDMS-Sylgard 184 | DOW.INC | CAT#1317318 |
| PDMS-Sylgard 527 | DOW.INC | CAT#2270030 |
| Formlabs Clear Resin Cartridge | Formlabs | CAT# RS-F2-GPCL |
| Agar | Fisher Scientific | CAT# BP9744500 |
| 1H,1H,2H,2H-Perfluorooctyltriethoxysilane (fluoro-silane) | Sigma | CAT# 667420 |
| (3-Aminopropyl)triethoxysilane (APTES) | Sigma | CAT# A3648 |
| Glutaraldehyde, 50% aq. soln. | Thermo Fisher | CAT# A10500-22 |
| Fibronectin bovine plasma | Sigma | CAT#F4759 |
| Ethanolamine hydrochloride, 99+% | Thermo Fisher | CAT# 217042500 |
| Poloxamer 188 Non-ionic Surfactant (100X) | Thermo Fisher | CAT# 24040032 |
| Corning Matrigel | Sigma | CAT# CLS356234 |
| Knock-out DMEM | Thermo Fisher | CAT# 10829018 |
| Essential 8™ Medium Kit (E8) | Thermo Fisher | CAT# A1517001 |
| Y-27632 Rock Inhibitor (Ri) | BioGems | CAT# 1293823 |
| CHIR 99021 | BioGems | CAT# 2520691 |
| IWP-2 | BioGems | CAT# 6866167 |
| L-Ascorbic acid (AA) | Fisher Scientific | A61100 |
| Sodium L-lactate, 98+% | Thermo Fisher | CAT# L14500.14 |
| RPMI 1640 | Thermo Fisher | CAT# 11875119 |
| 50X B-27 Complete Serum-Free Supplement (B27c) | Thermo Fisher | CAT# 0080085SA |
| 50X B-27 Minus Insulin Serum-Free Supplement (B27i) | Thermo Fisher | CAT# A1895601 |
| Qualified Fetal Bovine Serum (FBS) | Thermo Fisher | CAT# 26140079 |
| Knockout DMEM | Thermo Fisher | CAT# 10829018 |
| NEAA 100X | Thermo Fisher | CAT#11140050 |
| GlutaMAX 100x | ||
| PowerUp™ SYBR™ green master mix | ThermoFisher | CAT# A25743 |
| Triton-X-100 | Fisher Scientific | CAT# ICN807423 |
| Bovine Serum Albumin | Sigma | CAT# A2153 |
| Goat serum | Sigma | CAT#01-6201 |
| Agarose | Sigma | CAT# BP1356-100 |
| Sucrose | Sigma | CAT#8510-500GM |
| WGA, tetramethylrhodamine conjugate (IHC 1:100, 10μg/mL) | Invitrogen | CAT#W849 |
| WGA 350 (IHC 1:100, 10μg/mL) | Invitrogen | CAT#W11263 |
| Draq5 (IHC 1:1000, 5μM) | Thermo Fisher | CAT# 65-0880-92 |
| Phalloidin 488 (IHC 1:100) | Thermo Fisher | CAT#A12379 |
| Hoescht33342 (IHC 1μg/mL) | Thermo Fisher | CAT# H21492 |
| ProLong Gold | Thermo Fisher | CAT# P36934 |
| Isoproterenol hydrochloride | Sigma | CAT#420355 |
| Verapamil | Thermo Fisher | CAT#J61535 |
| Mavacamten | MedChem Express | CAT# HY-109037 |
| Thapsigargin | Fisher Scientific | CAT#11-381 |
| Dimethyl sulfoxide (DMSO) | Sigma | CAT# 276855 |
| Critical commercial assays | ||
| RNAqueous®-Micro Kit | Invitrogen | CAT#AM1931 |
| Super Script III Reverse Transcriptase Kit | Invitrogen | CAT#18080093 |
| Deposited data | ||
| Mendeley Data | https://doi.org/10.17632/rpbj83wpxw.1 | This paper |
| Custom built action potential and calcium code | https://github.com/huebschlab | Oguntuyo et al. Tissue Eng. C. 2022.83 |
| Custom build sarcomere analysis code | https://github.com/huebschlab | This paper |
| Experimental models: Cell lines | ||
| Isogenic Control iPSC (Wild Type C, GCaMP6f knock-in) | Gladstone Institutes of Cardiovascular Disease | A. J. S. Ribeiro, Circ Res. 2017.84 |
| MYBPC3+/- iPSC (in Wild Type C background with GCaMP6f knock-in) | Gladstone Institutes of Cardiovascular Disease | Ribeiro, Circ Res. 2017.84 |
| Oligonucleotides | ||
| GAPDH: F: CTCTGCTCCTCCTGTTCGAC R: TTAAAAGCAGCCCTGGTGAC |
IDT | N/A |
| ACTN2: F: GCTTCTACCACGCTTTTGCG R: CATTCCAAAAGCTCACTCGCT |
IDT | N/A |
| TNNT2: F: TTCACCAAAGATCTGCTCCTCGCT R: TTATTACTGGTGTGGAGTGGGTGTGG |
IDT | N/A |
| MYBPC3: F: GGCATGCTAAAGAGGCTCAA R: TCTTGTGGCCTTTGCTCAC |
IDT | N/A |
| SERCA: F: ACCCACATTCGAGTTGGAAG R: CCAACGAAGGTCAGATTGGT |
IDT | N/A |
| CACNA1C: F: GGAGAGTTTTCCAAAGAGAG R: TTTGAGATCCTCTTCTAGCTG |
IDT | N/A |
| TNNC1: F: ATGAGCTGAAGATAATGCTG R: AACTCCAGGAACTCATCATAG |
Sigma | N/A |
| Software and algorithms | ||
| ImageJ | National Institute of Health | N/A |
| MATLAB 2023 | MATLAB | Version 2023b |
| GraphPad Prism 10 | GraphPad Software | Version 10 |
| BioRender.com | BioRender.com | N/A |
| Other | ||
| Spin coater | Laurell Technologies | CAT#WS-650MZ- 23NPPB |
| Plasma Chamber | HARRICK Plasma | CAT# PDC-001 |
| Planar Biaxial Test Machine | TestResources | CAT# 574 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Nathaniel Huebsch (nhuebsch@wustl.edu).
Materials availability
This study does not generate new reagents.
Data and code availability
-
•
Data: All data within the main and supplemental figures within this study are published through Mendeley Data, making them publicly accessible. DOIs are listed in the key resources table. Any additional raw data are available upon request from the lead contact.
-
•
Code: All original code has been deposited at the Github repository and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
iPSC culture and differentiation
MYBPC3+/- iPSC and isogenic controls of the Wild Type C background (WTC; Coriell GM2525) harboring the knock-in GCamP6f calcium indicator were cultured in Essential 8 media with Matrigel coated plate (day -3 to day 0). Matrigel was diluted 1:100 with knockout DMEM. When confluent, iPSCs were differentiated to cardiomyocytes using Wnt pathway modulators: from day 0 to day 6, RPMI1640 with R27 minus insulin (R-) was used as the base media. On day 0, R- was supplemented with 6 μM CHIR99021 and 150 μg/mL L-ascorbic acid. Exactly 48 hours after the day 0 media change, media was changed to R- supplemented with 5 μM IWP2 and 150 μg/mL L-ascorbic acid (days 2 to 4). On day 4, media was changed to R- supplemented with 150 μg/mL L-ascorbic acid. From day 6 and onward, RPMI1640 with R27 containing insulin (R+) was supplied.4,33 Media was changed on days 6, 8, 11 and every 3 days thereafter. Beating of the 2D monolayer is normally observed by day 6-10 of differentiation. On differentiation day 14, beating cardiomyocyte monolayers were singularized gently with 0.25% trypsin and replated onto Matrigel coated tissue culture plastic at a density of 250,000 cells/cm2 into Dulbecco’s Modified Eagle Media (DMEM) supplemented with 20% Fetal Bovine Serum (FBS) and 10 μM Y-27632 for 2 days (days 14-16). Media was then changed to R+ for 3 days (days 16-19) followed by cardiac purification. From days 19 to day 21 of differentiation, the media was changed to lactate-based metabolic selection media (RPMI1640 without glucose, supplemented with 4mM lactate and Non-Essential Amino Acids, NEAA). On Days 21 and 24, the monolayer was washed with Dulbecco’s PBS (dPBS) and the media was changed again to fresh metabolic selection media.33 After a total of 5 days culture in metabolic selection media, media for purified iPSC-cardiomyocytes was changed to R+ from day 24 to day 27 for post-selection recovery. Cardiomyocytes purities were quantified using flow cytometry analysis of cardiac troponin T positive cells. At differentiation days 27-30, purified iPSC-cardiomyocytes were used for making micro heart muscle (Figure 1).4
Method details
Material characterization
To address our hypothesis that mechanics can initiate HCM pathogenesis in vitro, we formed μHM on PDMS substrates ranging in elastic modulus from 0.4 kPa up to 114 kPa (Figures 1 and S1) (17, 29, 30). Polydimethylsiloxane (PDMS) elastomer was used for making substrates and stencil molds. Elastic modulus of the material was characterized using a planar biaxial testing machine through tensile test.33 Ultrasoft 0.4 kPa substrates mimicking the embryonic heart microenvironment were made using sylgard 527 (5:4 ratio of component A to component B). Substrates with rigidity ranging from values near that of healthy adult human heart (15 kPa) to fibrosis-like (65kPa and 114kPa) substrates were made by mixing sylgard 184 into sylgard 527 with ratio of 1:40, 1:10 and 1:5.21,31,32
“Dog bone” shaped stencil molds to control the shape of micro-muscles were made of sylgard 184 using 3D printed masters.85 Briefly, “dog bone” positive 3D prints were generated from FormLabs Clear Resin™. To avoid toxicity from direct molding sylgard 184 off the 3D prints, 1.5% agar gels were cast directly off to make “dog bone” negative. Sylgard 184 was then molded off the agar gel to get a positive replica of the original 3D print. Finally, the PDMS replicas of the 3D prints were treated with 1H,1H,2H,2H-Perfluorooctyltriethoxysilane (fluoro-silane, 0.25 mL) in a vacuum chamber through vapor deposition for 72 hours, before sylgard 184 stencils were cast off these replica PDMS molds.33,85 Sylgard 184 was cured against the replica mold under tight compression to form the “dog bone” shaped through-holes within the 1mm-thick PDMS stencils.85
Device fabrication
μHM forming stencils were prepared on soft substrates based on our previously described methods.4,33 Briefly, a thick PDMS base layer (∼ 200 μm thick) and a thinner PDMS/fluorescent bead layer (∼ 5 μm thick) were applied to glass coverslips (20mm x 20mm) by spin coating. Soft PDMS substrates were cured sequentially overnight at 60°C. To prevent floating of the coverslips during culture and imaging, substrates were fixed in 6 well plates using either soft or stiff PDMS and cured overnight. Substrates were then covalently bound to the extracellular matrix protein fibronectin following seven steps:1) plasma oxidation (pressure/flow rate: 580−680 mTorr) for 90 s at high power (30 W). 2) Amino silane (5 v/v % of (3-Aminopropyl)triethoxysilane), APTES, in methanol) was immediately poured onto the plasma treated substrate in 6 well plates. Plates were wrapped with parafilm to reduce the amount of air getting into the reaction, and then shaken gently for 1 hour at room temperature. 3) Unreacted APTES was depleted with 3X 5 minutes washes in methanol, followed by one overnight dPBS wash. The long dPBS was eliminates residual APTES that was not covalently bonded to the substrate.4 4) Amine groups from APTES were next grafted to glutaraldehyde by applying a 2.5 wt/v% solution of glutaraldehyde in dPBS, which reacted for 2 hours at room temperature on an orbital shaker. 5) Covalent bonding of fibronectin to the exposed aldehyde groups from the glutaraldehyde was then achieved by reacting the substrates with 20 μg/mL fibronectin in dPBS for at least 30 minutes at room temperature on the shaker. 6) Unreacted aldehydes and fibronectin were washed off the surface by 3x 5 minutes washes with dPBS followed by 3x5 minutes rinses with deionized water. 7) Unreacted aldehyde groups were further quenched by exposing the substrates to 2.5 w/v% ethanolamine in dPBS (45 minutes at 37°C) followed by 3x 5 minutes dPBS washes.
While preparing soft substrates, rigid sylgard 184 stencils were exposed to 10% Pluronic F68 in dPBS (30 minutes incubation at room temperature with magnetic stirring followed by 3 dPBS washes and 1 methanol wash). Finally, stencils were physiosorbed onto the substrates by applying a small drop of methanol to the substrate and allowing the methanol to evaporate out from under the stencil at 60°C (normally within 2 hours).4 Prior to tissue seeding onto the substrates, the devices were disinfected by applying 70% ethanol overnight.
iPSC-μHM formation and optimization
Purified cardiomyocytes were gently dissociated using 0.25% trypsin EDTA (3-4 minutes, up to 10 minutes) and seeded into each “dog bone” shaped mold with 3 μL of 7 x 107 cell/mL (210,000 cells). The cells were then incubated at 37°C with 5% CO2 for 2 hours before the entire culture well was filled with replating media (DMEM supplemented with 20% FBS, 150 μg/mL ascorbic acid and 10 μM Y27632). Tissue compaction and alignment was typically observed within 24 hours.4 At least 4 high quality tissue batches with cardiomyocytes purity of 85±10% were selected for study. Tissue quality was determined by tissue shape, absence of visible cellular death, and maintenance of adhesion to the substrates (Figure S3). We used tissue gross shape as our first quality control metric because of our recent findings that overall μHM shape impacts physiology.85 Tissues that deviated significantly from the expected “dog bone” shape or were delaminated from the substrate were excluded. We observed that the inclusion rate for tissues was not dependent on cardiomyocyte genotype or substrate stiffnesses. Altogether, these quality control metrics led to a more consistent and narrower range of μHM physiologic behaviors compared to what was previously possible.33
Gene expression analysis
Expression of sarcomere genes and calcium genes were evaluated with quantitative RT-PCR.4 Detailed gene primer sequence in key resources table. Cell or tissue lysis and RNA recovery were performed using RNAqueous®-Micro Kit (Thermo Scientific). At least 320 ng RNA per sample was synthesized for cDNA using Super Script III Reverse Transcriptase Kit (Thermo Scientific). Gene expression was performed using Applied Biosystems Step One Plus Light Cycler instrument using SYBR green master mix (Thermo Scientific). Relative gene expression for each sample was normalized to its housekeeping gene GAPDH.
Immunostaining
μHM were incubated in dPBS until cessation of spontaneous beating, and then step fixed with 1%, 2%, 3% and 4% paraformaldehyde at 4°C, with each fixation step at least 1 hour.9 Next, tissues were embedded into 1% wt/v agar followed by protection in sucrose and embedding into OCT as described previously.33 8 μm cryosections were obtained on a cryotome, and subsequently used for immunostaining for sarcomeric α-actinin, myosin binding protein C, troponin T, troponin C. Wheat germ agglutinin (WGA) was used for staining cell membranes, and cell nuclei were stained with Hoechst 33342. See key resources table for detailed description of antibodies used. Stained tissue sections were mounted in ProLong Gold and scanned using a Fluoview FB1200 confocal microscope (Olympus, Tokyo, Japan). Cell sizes were sampled throughout the entire tissue, which was quantified by drawing individual cross-sectional areas of cell using ImageJ. The regional cellular size quantification within either tissue shaft or knobs sees Figure S4. The sarcomere properties were measured exclusively within the tissue shaft region, where it was possible to observe whether there is sarcomere misalignment relative to the tissue shaft direction. Tissue alignments were measured by using sarcomeric α-actinin staining, the angles of individual sarcomeres in relevant to the tissue shaft were measured through custom built MATLAB code (available at: github.com/huebschlab). For example, 0 degree means perfectly aligned to the tissue shaft direction, 90 degree means perpendicular to the tissue direction, indicating misalignment. More than 4 individual sarcomere angles were averaged for each tissue (Figure S5). Sarcomere profiles (Figure 4) were plotted by selecting representative sarcomere regions in ImageJ and normalized by the mean intensity value of the specific sarcomere trace.
High-speed fluorescent imaging
Physiology of iPSC-μHM at tissue day 10 (corresponding to day 40 of iPSC-cardiomyocytes differentiation) was analyzed using a high-speed imaging system (Eclipse Ts2R, Nikon; Tokyo, Japan) equipped with a digital CMOS camera (Hamamatsu ORCA-Flash4.0 V2; Japan).33 The imaging stage was set to a constant 37°C temperature using a thermal plate (Tokai Hit; Shizuoka, Japan).4 Field pacing of tissue (1.5 Hz, 20msec Bipolar pulses, <20V) during imaging was achieved using graphite electrodes (MyoPacer, Ion Optix, USA). 1.5 Hz field pacing was chosen because it is slightly above the average spontaneous beating rate of the cardiac tissues.
Contractility characterization
Traction force microscopy (TFM) was used to quantify tissue contractility. Tissue peak force was calculated through using substrate elasticity and measuring green fluorescence beads displacement via open-source software described previously490. For TFM, we assumed the Poisson’s ratio of PDMS was 0.49, detailed parameters for TFM analysis are based on prior work.33,84,86 For contractility measurements, we focus on the knob region of the tissue where the tissue adheres and contracts against the substrate (exert forces on substrates). We sampled contractility and calcium data randomly from one randomly selected knob from each tissue.
Mechanical energy consumption calculation
Instantaneous contractile power was calculated at each imaging timepoint by multiplying force and velocity obtained from TFM calculations. These curves were used to calculate energy consumed during contraction and relaxation.84 Briefly, positive (contraction), and negative (relaxation) regions of the power curve from each contraction cycle, were integrated by calculating the area under the curve. Positive energy consumption was used for comparison between μHM of each genotype.
Action potential and calcium measurements
Membrane potential was measured by labeling tissue overnight with 1 μM BeRST-1 voltage sensitive dye in phenol red free RPMI-C.87 Action potential waveforms were captured by imaging in the Cy5 channel. For action potential, we captured the entire tissue. Calcium transients were captured by imaging endogenously expressed GcaMP6f in the GFP channel.4 Calcium and contractility are strongly coupled, therefore, similar to contractility measurements, calcium dynamics measurements were measured at the tissue knobs regions (one randomly selected knob per tissue), where tissues maintain strong mechanical coupling to the substrate. BeRST-1 and GCaMP6f videos were analyzed via custom, open-source MATLAB code (available at: huebschlab.wustl.edu and github.com/huebschlab); intensity profile and kinetics parameters were automatically extracted.83 For calcium transient dynamics, initial baseline to 80% of the intensity was measured as UPD80, plateau region of more than 80% intensity was measured, decay times from end of the plateau to 50 and 75% decay of intensity was characterized. Beats per minute (BPM) were characterized by GCaMP6f and BeRST-1 videos of spontaneously beating tissues. For action potential waveforms, initial baseline to peak intensity was measured as UPD, and the time from action potential initiation to 30% decay from peak intensity (APD30), along with the time from action potential initiation to 80% or 90% decay from peak intensity (APD80 and APD90, respectively), were measured.
Pharmacology studies
The potential mechanism for pathologic calcium handling in HCM-prone MYBPC3+/- μHM was investigated using pharmacological probes. Specifically, isoproterenol (0-1 μM) L-type Ca2+ blocker verapamil (0 up to 5 μM), SERCA inhibitor thapsigargin (0, 5 μM) and myosin inhibitor mavacamten (0, 0.5 μM).50,53,88 Drugs were first diluted in dimethyl sulfoxide (DMSO), then serial diluted in RPMI/C media with supplements. When applying to tissue culture, 10% of the existing media was replaced by 10x target dose of the drug. Acute drug administration was performed by at least 45 minutes incubation at 37°C with 5% CO2 prior imaging. The highest final concentration of DMSO in cell culture media was 0.01% in these imaging studies.
Quantification and statistical analysis
GraphPad 9.3.1 was used for statistics and data visualization. Data are presented with mean ± standard deviation (SD) with n > 3 biological repeats detailed in figure legends. One-way or two-way analysis of variance (ANOVA) was used followed by multiple comparison test between groups was performed using Holm Sidak’s method. For two-way ANOVA, there are two independent variables, genotypes (control and MYBPC3+/-) and substrate stiffnesses (0.4, 15, 65, and 114 kPa) that affect the overall data variation. We investigated each factors’ significance. For example, whether differential cellular size is triggered by either stiffness factor or genotype factor or both. The p value less than 0.05 was considered statistically significant.
Acknowledgments
We thank Drs. Guy M Genin, Sharon Cresci, Michael Vahey, Lori Setton, and Jianjun Guan for helpful discussions and use of the equipment. We thank Dr. Evan Miller for providing BeRST1 dye. This work was supported by the Departments of Biomedical Engineering and Mechanical Engineering and Material Science at Washington University in St. Louis, the American Heart Association (19CDA34730016 to N.H., predoctoral fellowship 828938 to J.G., TPA 970198 to N.H.), the Center for Engineering Mechanobiology—National Science Foundation Science & Technology Center CMMI No. 15-48571 (G.R.), and the National Institutes of Health (R01 HL159094 to N.H.).
Author contributions
Conceptualization: J.G. and N.H.; methodology: J.G. and G.R.; software: D.S. and D.B.; validation: J.G., H.J., and D.S; formal analysis: J.G., H.J., and D.S; investigation: J.G., H.J., D.S., and L.A.; resources: N.H.; data curation: J.G. and D.S.; writing—original draft: J.G., J.D.M., and N.H.; writing—review and editing: J.G., J.D.M., and N.H.; supervision: J.S. and N.H.; funding acquisition: N.H.
Declaration of interests
The authors declare no competing interests.
Published: May 10, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109954.
Supplemental information
References
- 1.Lorenzini M., Norrish G., Field E., Ochoa J.P., Cicerchia M., Akhtar M.M., Syrris P., Lopes L.R., Kaski J.P., Elliott P.M. Penetrance of Hypertrophic Cardiomyopathy in Sarcomere Protein Mutation Carriers. J. Am. Coll. Cardiol. 2020;76:550–559. doi: 10.1016/j.jacc.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rowin E.J., Maron B.J., Maron M.S. The Hypertrophic Cardiomyopathy Phenotype Viewed Through the Prism of Multimodality Imaging: Clinical and Etiologic Implications. JACC. Cardiovasc. Imaging. 2020;13:2002–2016. doi: 10.1016/j.jcmg.2019.09.020. [DOI] [PubMed] [Google Scholar]
- 3.Repetti G.G., Kim Y., Pereira A.C., Ingles J., Russell M.W., Lakdawala N.K., Ho C.Y., Day S., Semsarian C., McDonough B., et al. Discordant clinical features of identical hypertrophic cardiomyopathy twins. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2021717118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo J., Jiang H., Oguntuyo K., Rios B., Boodram Z., Huebsch N. Interplay of Genotype and Substrate Stiffness in Driving the Hypertrophic Cardiomyopathy Phenotype in iPSC-Micro-Heart Muscle Arrays. Cell. Mol. Bioeng. 2021;14:409–425. doi: 10.1007/s12195-021-00684-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma Z., Huebsch N., Koo S., Mandegar M.A., Siemons B., Boggess S., Conklin B.R., Grigoropoulos C.P., Healy K.E. Contractile deficits in engineered cardiac microtissues as a result of MYBPC3 deficiency and mechanical overload. Nat. Biomed. Eng. 2018;2:955–967. doi: 10.1038/s41551-018-0280-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barefield D., Kumar M., Gorham J., Seidman J.G., Seidman C.E., de Tombe P.P., Sadayappan S. Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. J. Mol. Cell. Cardiol. 2015;79:234–243. doi: 10.1016/j.yjmcc.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saucerman J.J., Tan P.M., Buchholz K.S., McCulloch A.D., Omens J.H. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 2019;16:361–378. doi: 10.1038/s41569-019-0155-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ommen S.R., Mital S., Burke M.A., Day S.M., Deswal A., Elliott P., Evanovich L.L., Hung J., Joglar J.A., Kantor P., et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020;142:e558–e631. doi: 10.1161/CIR.0000000000000937. [DOI] [PubMed] [Google Scholar]
- 9.Ronaldson-Bouchard K., Ma S.P., Yeager K., Chen T., Song L., Sirabella D., Morikawa K., Teles D., Yazawa M., Vunjak-Novakovic G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature. 2018;556:239–243. doi: 10.1038/s41586-018-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinberger F., Mannhardt I., Eschenhagen T. Engineering Cardiac Muscle Tissue: A Maturating Field of Research. Circ. Res. 2017;120:1487–1500. doi: 10.1161/CIRCRESAHA.117.310738. [DOI] [PubMed] [Google Scholar]
- 11.Wijnker P.J.M., van der Velden J. Mutation-specific pathology and treatment of hypertrophic cardiomyopathy in patients, mouse models and human engineered heart tissue. Biochim. Biophys. Acta, Mol. Basis Dis. 2020;1866 doi: 10.1016/j.bbadis.2020.165774. [DOI] [PubMed] [Google Scholar]
- 12.Liu C., Oikonomopoulos A., Sayed N., Wu J.C. Modeling human diseases with induced pluripotent stem cells: From 2D to 3D and beyond. Development. 2018;145:dev156166. doi: 10.1242/dev.156166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maron B.J., Maron M.S. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–255. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- 14.Chang H.J., Lynm C., Glass R.M. Hypertrophic cardiomyopathy. JAMA. 2009;302:1720. doi: 10.1001/jama.302.15.1720. [DOI] [PubMed] [Google Scholar]
- 15.Helms A.S., Tang V.T., O'Leary T.S., Helms A.S., Wauchope M., Friedline S., Wasserman A.H., Smith E.D., Lee L.M., Wen X.W., et al. Effects of MYBPC3 loss-of-function mutations preceding hypertrophic cardiomyopathy. JCI Insight. 2020;5 doi: 10.1172/jci.insight.133782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marston S., Copeland O., Gehmlich K., Schlossarek S., Carrier L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J. Muscle Res. Cell Motil. 2012;33:75–80. doi: 10.1007/s10974-011-9268-3. [DOI] [PubMed] [Google Scholar]
- 17.Ho C.Y., Day S.M., Axelsson A., Russell M.W., Zahka K., Lever H.M., Pereira A.C., Colan S.D., Margossian R., Murphy A.M., et al. Valsartan in early-stage hypertrophic cardiomyopathy: a randomized phase 2 trial. Nat. Med. 2021;27:1818–1824. doi: 10.1038/s41591-021-01505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho C.Y., Lakdawala N.K., Cirino A.L., Lipshultz S.E., Sparks E., Abbasi S.A., Kwong R.Y., Antman E.M., Semsarian C., González A., et al. Diltiazem treatment for pre-Clinical hypertrophic cardiomyopathy sarcomereMutation carriers: A pilot randomized trial to modify disease expression. JACC. Heart Fail. 2015;3:180–188. doi: 10.1016/j.jchf.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frey N., Luedde M., Katus H.A. Mechanisms of disease: Hypertrophic cardiomyopathy. Nat Rev Cardiol. 2012;9:91–100. doi: 10.1038/nrcardio.2011. [DOI] [PubMed] [Google Scholar]
- 20.Maron B.A., Wang R.S., Carnethon M.R., Rowin E.J., Loscalzo J., Maron B.J., Maron M.S. What Causes Hypertrophic Cardiomyopathy? Am. J. Cardiol. 2022;179:74–82. doi: 10.1016/j.amjcard.2022.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varnava A.M., Elliott P.M., Sharma S., McKenna W.J., Davies M.J. Hypertrophic cardiomyopathy: The interrelation of disarray, fibrosis and small vessel disease. Heart. 2000;84:476–482. doi: 10.1136/heart.84.5.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tardiff J.C., Hewett T.E., Palmer B.M., Olsson C., Factor S.M., Moore R.L., Robbins J., Leinwand L.A. Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J. Clin. Invest. 1999;104:469–481. doi: 10.1172/JCI6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohn R., Thakar K., Lowe A., Ladha F.A., Pettinato A.M., Romano R., Meredith E., Chen Y.S., Atamanuk K., Huey B.D., Hinson J.T. A Contraction Stress Model of Hypertrophic Cardiomyopathy due to Sarcomere Mutations. Stem Cell Rep. 2019;12:71–83. doi: 10.1016/j.stemcr.2018.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roest A.S.V., Liu C., Morck M.M., Kooiker K.B., Jung G., Song D., Dawood A., Jhingran A., Pardon G., Ranjbarvaziri S., et al. Hypertrophic cardiomyopathy β-cardiac myosin mutation (P710R) leads to hypercontractility by disrupting super relaxed state. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2025030118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trembley M.A., Quijada P., Agullo-Pascual E., Tylock K.M., Colpan M., Dirkx R.A., Myers J.R., Mickelsen D.M., De Mesy Bentley K., Rothenberg E., et al. Mechanosensitive gene regulation by myocardin-related transcription factors is required for cardiomyocyte integrity in load-induced ventricular hypertrophy. Circulation. 2018;138:1864–1878. doi: 10.1161/CIRCULATIONAHA.117.031788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coppini R., Ferrantini C., Yao L., Fan P., Del Lungo M., Stillitano F., Sartiani L., Tosi B., Suffredini S., Tesi C., et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013;127:575–584. doi: 10.1161/CIRCULATIONAHA.112.134932. [DOI] [PubMed] [Google Scholar]
- 27.Coppini R., Ferrantini C., Mugelli A., Poggesi C., Cerbai E. Altered Ca2+ and Na+ homeostasis in human hypertrophic cardiomyopathy: Implications for arrhythmogenesis. Front. Physiol. 2018;9:1391–1416. doi: 10.3389/fphys.2018.01391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lan F., Lee A.S., Liang P., Sanchez-Freire V., Nguyen P.K., Wang L., Han L., Yen M., Wang Y., Sun N., et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–113. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson P., Liu X., Sparrow A., Patel S., Zhang Y.H., Casadei B., Watkins H., Redwood C. Hypertrophic cardiomyopathy mutations increase myofilament Ca2 buffering, alter intracellular Ca2 handling, and stimulate Ca2-dependent signaling. J. Biol. Chem. 2018;293:10487–10499. doi: 10.1074/jbc.RA118.002081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helms A.S., Alvarado F.J., Yob J., Tang V.T., Pagani F., Russell M.W., Valdivia H.H., Day S.M. Genotype-Dependent and -Independent Calcium Signaling Dysregulation in Human Hypertrophic Cardiomyopathy. Circulation. 2016;134:1738–1748. doi: 10.1161/CIRCULATIONAHA.115.020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engler A.J., Carag-Krieger C., Johnson C.P., Raab M., Tang H.Y., Speicher D.W., Sanger J.W., Sanger J.M., Discher D.E. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: Scar-like rigidity inhibits beating. J. Cell Sci. 2008;121:3794–3802. doi: 10.1242/jcs.029678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinz B. Tissue Stiffness, Latent TGF-β1 Activation, and Mechanical Signal Transduction: Implications for the Pathogenesis and Treatment of Fibrosis. Curr. Rheumatol. Rep. 2009;11:120–126. doi: 10.1007/s11926-009-0017-1. [DOI] [PubMed] [Google Scholar]
- 33.Guo J., Simmons D.W., Ramahdita G., Munsell M.K., Oguntuyo K., Kandalaft B., Rios B., Pear M., Schuftan D., Jiang H., et al. Elastomer-Grafted iPSC-Derived Micro Heart Muscles to Investigate Effects of Mechanical Loading on Physiology. ACS Biomater. Sci. Eng. 2021;7:2973–2989. doi: 10.1021/acsbiomaterials.0c00318. [DOI] [PubMed] [Google Scholar]
- 34.Conrad C.H., Brooks W.W., Hayes J.A., Sen S., Robinson K.G., Bing O.H. Myocardial Fibrosis and Stiffness With Hypertrophy and Heart Failure in the Spontaneously Hypertensive Rat. Circulation. 1995;91:161–170. doi: 10.1161/01.CIR.91.1.161. [DOI] [PubMed] [Google Scholar]
- 35.Yildiz M., Oktay A.A., Stewart M.H., Milani R.V., Ventura H.O., Lavie C.J. W.B. Saunders; 2020. Left ventricular hypertrophy and hypertension. Preprint at. [DOI] [PubMed] [Google Scholar]
- 36.Zhang D., Shadrin I.Y., Lam J., Xian H.Q., Snodgrass H.R., Bursac N. Tissue-engineered cardiac patch for advanced functional maturation of human ESC-derived cardiomyocytes. Biomaterials. 2013;34:5813–5820. doi: 10.1016/j.biomaterials.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finocchiaro G., Sheikh N., Leone O., Westaby J., Mazzarotto F., Pantazis A., Ferrantini C., Sacconi L., Papadakis M., Sharma S., et al. Arrhythmogenic potential of myocardial disarray in hypertrophic cardiomyopathy: Genetic basis, functional consequences and relation to sudden cardiac death. Europace. 2021;23:985–995. doi: 10.1093/europace/euaa348. [DOI] [PubMed] [Google Scholar]
- 38.Ma W., Gong H., Jani V., Lee K.H., Landim-Vieira M., Papadaki M., Pinto J.R., Aslam M.I., Cammarato A., Irving T. Myofibril orientation as a metric for characterizing heart disease. Biophys. J. 2022;121:565–574. doi: 10.1016/j.bpj.2022.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doste R., Coppini R., Bueno-Orovio A. Remodelling of potassium currents underlies arrhythmic action potential prolongation under beta-adrenergic stimulation in hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2022;172:120–131. doi: 10.1016/j.yjmcc.2022.08.361. [DOI] [PubMed] [Google Scholar]
- 40.Elesber A., Nishimura R.A., Rihal C.S., Ommen S.R., Schaff H.V., Holmes D.R. Utility of Isoproterenol to Provoke Outflow Tract Gradients in Patients With Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2008;101:516–520. doi: 10.1016/j.amjcard.2007.09.111. [DOI] [PubMed] [Google Scholar]
- 41.Sun N., Yazawa M., Liu J., Han L., Sanchez-Freire V., Abilez O.J., Navarrete E.G., Hu S., Wang L., Lee A., et al. Patient-Specific Induced Pluripotent Stem Cells as a Model for Familial Dilated Cardiomyopathy. Sci. Transl. Med. 2012;4:130ra47. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Dijk S.J., Paalberends E.R., Najafi A., Michels M., Sadayappan S., Carrier L., Boontje N.M., Kuster D.W., van Slegtenhorst M., Dooijes D., et al. Contractile Dysfunction Irrespective of the Mutant Protein in Human Hypertrophic Cardiomyopathy with Normal Systolic Function. Circ. Heart Fail. 2012;5:36–46. doi: 10.1161/CIRCHEARTFAILURE.111.963702. [DOI] [PubMed] [Google Scholar]
- 43.Mearini G., Stimpel D., Geertz B., Weinberger F., Krämer E., Schlossarek S., Mourot-Filiatre J., Stoehr A., Dutsch A., Wijnker P.J.M., et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat. Commun. 2014;5:5515. doi: 10.1038/ncomms6515. [DOI] [PubMed] [Google Scholar]
- 44.Oliva-Sandoval M.J., Ruiz-Espejo F., Monserrat L., Hermida-Prieto M., Sabater M., García-Molina E., Ortiz M., Rodríguez-García M.I., Núñez L., Gimeno J.R., et al. Insights into genotype-phenotype correlation in hypertrophic cardiomyopathy. Findings from 18 Spanish families with a single mutation in MYBPC3. Heart. 2010;96:1980–1984. doi: 10.1136/hrt.2010.200402. [DOI] [PubMed] [Google Scholar]
- 45.Toepfer C.N., Wakimoto H., Garfinkel A.C., McDonough B., Liao D., Jiang J., Tai A.C., Gorham J.M., Lunde I.G., Lun M., et al. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aat1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrantini C., Coppini R., Pioner J.M., Gentile F., Tosi B., Mazzoni L., Scellini B., Piroddi N., Laurino A., Santini L., et al. Pathogenesis of hypertrophic cardiomyopathy is mutation rather than disease specific: A comparison of the cardiac troponin T E163R and R92Q mouse models. J. Am. Heart Assoc. 2017;6 doi: 10.1161/JAHA.116.005407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith G.L., Eisner D.A. Lippincott Williams and Wilkins; 2019. Calcium Buffering in the Heart in Health and Disease. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lorell B.H., Grossman W. Cardiac hypertrophy: The consequences for diastol. J. Am. Coll. Cardiol. 1987;9:1189–1193. doi: 10.1016/S0735-1097(87)80326-1. [DOI] [PubMed] [Google Scholar]
- 49.MacLennan D.H., Kranias E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 50.Lytton J., Westlin M., Hanley M. Thapsigargin Inhibits the Sarcoplasmic or Endoplasmic Reticulum Ca-ATPase Family of Calcium Pumps. J. Biol. Chem. 1991;266:17067–17071. doi: 10.1016/S0021-9258(19)47340-7. [DOI] [PubMed] [Google Scholar]
- 51.Semsarian C., Ahmad I., Giewat M., Georgakopoulos D., Schmitt J.P., McConnell B.K., Reiken S., Mende U., Marks A.R., Kass D.A., et al. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J. Clin. Invest. 2002;109:1013–1020. doi: 10.1172/JCI200214677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sparrow A.J., Sievert K., Patel S., Chang Y.F., Broyles C.N., Brook F.A., Watkins H., Geeves M.A., Redwood C.S., Robinson P., Daniels M.J. Measurement of Myofilament-Localized Calcium Dynamics in Adult Cardiomyocytes and the Effect of Hypertrophic Cardiomyopathy Mutations. Circ. Res. 2019;124:1228–1239. doi: 10.1161/CIRCRESAHA.118.314600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Green E.M., Wakimoto H., Anderson R.L., Evanchik M.J., Gorham J.M., Harrison B.C., Henze M., Kawas R., Oslob J.D., Rodriguez H.M., et al. Heart disease: A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;1979:617–621. doi: 10.1126/science.aad3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sewanan L.R., Park J., Rynkiewicz M.J., Racca A.W., Papoutsidakis N., Schwan J., Jacoby D.L., Moore J.R., Lehman W., Qyang Y., Campbell S.G. Loss of crossbridge inhibition drives pathological cardiac hypertrophy in patients harboring the tpm1 e192k mutation. J. Gen. Physiol. 2021;153 doi: 10.1085/jgp.202012640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sparrow A.J., Watkins H., Daniels M.J., Redwood C., Robinson P. Mavacamten rescues increased myofilament calcium sensitivity and dysregulation of Ca2+ flux caused by thin filament hypertrophic cardiomyopathy mutations. Am. J. Physiol. Heart Circ. Physiol. 2020;318:715–722. doi: 10.1152/ajpheart.00023.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bonow R.O., Rosing D.R., Bacharach S.L., Green M.V., Kent K.M., Lipson L.C., Maron B.J., Leon M.B., Epstein S.E. Effects of Verapamil on Left Ventricular Systolic Function and Diastolic Filling in Patients with Hypertrophic Cardiomyopathy. Circulation. 1981;64:787–796. doi: 10.1161/01.CIR.64.4.787. [DOI] [PubMed] [Google Scholar]
- 57.Luo Q., Chen J., Zhang T., Tang X., Yu B. Retrospective analysis of clinical phenotype and prognosis of hypertrophic cardiomyopathy complicated with hypertension. Sci. Rep. 2020;10 doi: 10.1038/s41598-019-57230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lehman S.J., Crocini C., Leinwand L.A. Targeting the sarcomere in inherited cardiomyopathies. Nat. Rev. Cardiol. 2022;19:353–363. doi: 10.1038/s41569-022-00682-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teekakirikul P., Eminaga S., Toka O., Alcalai R., Wang L., Wakimoto H., Nayor M., Konno T., Gorham J.M., Wolf C.M., et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J. Clin. Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim Y., Mastali M., Van Eyk J.E., Orav E.J., Vissing C.R., Day S.M., Axelsson Raja A., Russell M.W., Zahka K., Lever H.M., et al. Transforming Growth Factor-β Analysis of the VANISH Trial Cohort. Circ. Heart Fail. 2023;16 doi: 10.1161/CIRCHEARTFAILURE.122.010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Siddiqui N., Husain A., Chaudhry L., Alam S., Mitra M., Bhasin P.S. Pharmacological and Pharmaceutical Profile of Valsartan: A Review. J. Appl. Pharmaceut. Sci. 2011;01:12–19. [Google Scholar]
- 62.Leonard A., Bertero A., Powers J.D., Beussman K.M., Bhandari S., Regnier M., Murry C.E., Sniadecki N.J. Afterload promotes maturation of human induced pluripotent stem cell derived cardiomyocytes in engineered heart tissues. J. Mol. Cell. Cardiol. 2018;118:147–158. doi: 10.1016/j.yjmcc.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eschenhagen T., Fink C., Remmers U., Scholz H., Wattchow J., Weil J., Zimmermann W., Dohmen H.H., Schäfer H., Bishopric N., et al. Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: A new heart muscle model system. Faseb. J. 1997;11:683–694. doi: 10.1096/fasebj.11.8.9240969. [DOI] [PubMed] [Google Scholar]
- 64.Nunes S.S., Miklas J.W., Liu J., Aschar-Sobbi R., Xiao Y., Zhang B., Jiang J., Massé S., Gagliardi M., Hsieh A., et al. Biowire: A platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat. Methods. 2013;10:781–787. doi: 10.1038/nmeth.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bian W., Badie N., Himel H.D., Bursac N. Robust T-tubulation and maturation of cardiomyocytes using tissue-engineered epicardial mimetics. Biomaterials. 2014;35:3819–3828. doi: 10.1016/j.biomaterials.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neelakantan S., Kumar M., Mendiola E.A., Phelan H., Serpooshan V., Sadayappan S., Avazmohammadi R. Multiscale characterization of left ventricle active behavior in the mouse. Acta Biomater. 2023;162:240–253. doi: 10.1016/j.actbio.2023.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin T.G., Kirk J.A. Under construction: The dynamic assembly, maintenance, and degradation of the cardiac sarcomere. J. Mol. Cell. Cardiol. 2020;148:89–102. doi: 10.1016/j.yjmcc.2020.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frazier A.H., Ramirez-Correa G.A., Murphy A.M. Molecular mechanisms of sarcomere dysfunction in dilated and hypertrophic cardiomyopathy. Prog. Pediatr. Cardiol. 2011;31:29–33. doi: 10.1016/j.ppedcard.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asumda F.Z., Chase P.B. Nuclear cardiac troponin and tropomyosin are expressed early in cardiac differentiation of rat mesenchymal stem cells. Differentiation. 2012;83:106–115. doi: 10.1016/j.diff.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 70.Wu H., Lee J., Vincent L.G., Wang Q., Gu M., Lan F., Churko J.M., Sallam K.I., Matsa E., Sharma A., et al. Epigenetic Regulation of Phosphodiesterases 2A and 3A Underlies Compromised β-Adrenergic Signaling in an iPSC Model of Dilated Cardiomyopathy. Cell Stem Cell. 2015;17:89–100. doi: 10.1016/j.stem.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maron B.J., Ferrans V.J., Roberts W.C. Ultrastructural Features of Degenerated Cardiac Muscle Cells in Patients With Cardiac Hypertrophy. Am. J. Pathol. 1975;79:387–434. [PMC free article] [PubMed] [Google Scholar]
- 72.Li M.X., Hwang P.M. Elsevier B.V.; 2015. Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Katrukha I.A. Maik Nauka Publishing/Springer SBM; 2013. Human cardiac troponin complex. structure and functions. Preprint at. [DOI] [PubMed] [Google Scholar]
- 74.Siddiqui J.K., Tikunova S.B., Walton S.D., Liu B., Meyer M., de Tombe P.P., Neilson N., Kekenes-Huskey P.M., Salhi H.E., Janssen P.M.L., et al. Myofilament Calcium Sensitivity: Consequences of the effective concentration of troponin I. Front. Physiol. 2016;7 doi: 10.3389/fphys.2016.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dewan S., McCabe K.J., Regnier M., McCulloch A.D., Lindert S. Molecular Effects of cTnC DCM Mutations on Calcium Sensitivity and Myofilament Activation - An Integrated Multiscale Modeling Study. J. Phys. Chem. B. 2016;120:8264–8275. doi: 10.1021/acs.jpcb.6b01950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lin D., Bobkova A., Homsher E., Tobacman L.S. Functional analyses of troponin T mutations that cause hypertrophic cardiomyopathy: Insights into disease pathogenesis and troponin function. Proc. Natl. Acad. Sci. USA. 1998;95:14406–14410. doi: 10.1073/pnas.95.24.14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cingolani H.E., Pérez N.G., Cingolani O.H., Ennis I.L. The Anrep effect: 100 years later. Am. J. Physiol. Heart Circ. Physiol. 2013;304:175–182. doi: 10.1152/ajpheart.00508.2012.-Myocardial. [DOI] [PubMed] [Google Scholar]
- 78.Toischer K., Kögler H., Tenderich G., Grebe C., Seidler T., Van P.N., Jung K., Knöll R., Körfer R., Hasenfuss G. Elevated afterload, neuroendocrine stimulation, and human heart failure increase BNP levels and inhibit preload-dependent SERCA upregulation. Circ. Heart Fail. 2008;1:265–271. doi: 10.1161/CIRCHEARTFAILURE.108.785279. [DOI] [PubMed] [Google Scholar]
- 79.Sorsa T., Pollesello P., Solaro R.J. Kluwer Academic Publishers; 2004. The Contractile Apparatus as a Target for Drugs against Heart Failure: Interaction of Levosimendan, a Calcium Sensitiser, with Cardiac Troponin C. [DOI] [PubMed] [Google Scholar]
- 80.Leite-Moreira A.F., Correia-Pinto J., Gillebert T.C. Afterload induced changes in myocardial relaxation: A mechanism for diastolic dysfunction. Cardiovasc. Res. 1999;43:344–353. doi: 10.1016/S0008-6363(99)00099-1. [DOI] [PubMed] [Google Scholar]
- 81.Truitt R., Mu A., Corbin E.A., Vite A., Brandimarto J., Ky B., Margulies K.B. Increased Afterload Augments Sunitinib-Induced Cardiotoxicity in an Engineered Cardiac Microtissue Model. JACC. Basic Transl. Sci. 2018;3:265–276. doi: 10.1016/j.jacbts.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Simmons D.W., Malayath G., Schuftan D.R., Guo J., Oguntuyo K., Ramahdita G., Sun Y., Jordan S.D., Munsell M.K., Kandalaft B., et al. Engineered tissue geometry and Plakophilin-2 regulate electrophysiology of human iPSC-derived cardiomyocytes. APL Bioeng. 2024;8 doi: 10.1063/5.0160677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oguntuyo K., Schuftan D., Guo J., Simmons D., Bhagavan D., Moreno J.D., Kang P.W., Miller E., Silva J.R., Huebsch N. Robust, Automated Analysis of Electrophysiology in Induced Pluripotent Stem Cell-Derived Micro-Heart Muscle for Drug Toxicity. Tissue Eng. C Methods. 2022;28:457–468. doi: 10.1089/ten.tec.2022.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ribeiro A.J.S., Schwab O., Mandegar M.A., Ang Y.S., Conklin B.R., Srivastava D., Pruitt B.L. Multi-imaging method to assay the contractile mechanical output of micropatterned human iPSC-derived cardiac myocytes. Circ. Res. 2017;120:1572–1583. doi: 10.1161/CIRCRESAHA.116.310363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Simmons D.W., Schuftan D.R., Ramahdita G., Huebsch N. Hydrogel-Assisted Double Molding Enables Rapid Replication of Stereolithographic 3D Prints for Engineered Tissue Design. ACS Appl. Mater. Interfaces. 2023;15:25313–25323. doi: 10.1021/acsami.3c02279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dogru S., Aksoy B., Bayraktar H., Alaca B.E. Poisson’s ratio of PDMS thin films. Polym. Test. 2018;69:375–384. doi: 10.1016/j.polymertesting.2018.05.044. [DOI] [Google Scholar]
- 87.Huang Y.L., Walker A.S., Miller E.W. A Photostable Silicon Rhodamine Platform for Optical Voltage Sensing. J. Am. Chem. Soc. 2015;137:10767–10776. doi: 10.1021/jacs.5b06644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huebsch N., Loskill P., Deveshwar N., Spencer C.I., Judge L.M., Mandegar M.A., Fox C.B., Mohamed T.M.A., Ma Z., Mathur A., et al. Miniaturized iPS-Cell-Derived Cardiac Muscles for Physiologically Relevant Drug Response Analyses. Sci. Rep. 2016;6:24726–24812. doi: 10.1038/srep24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: All data within the main and supplemental figures within this study are published through Mendeley Data, making them publicly accessible. DOIs are listed in the key resources table. Any additional raw data are available upon request from the lead contact.
-
•
Code: All original code has been deposited at the Github repository and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.