

Summary
Hepatocellular carcinoma (HCC) is a major global cause of death, with epithelial-mesenchymal transition (EMT) and cancer stem cell (CSC)-like properties contributing to its metastasis. DEAD box helicase 56 (DDX56) is involved in carcinogenesis, but its role in EMT induction and stem phenotype maintenance is unclear. This study assessed the impact of DDX56 absence on HCC cell stemness and EMT. DDX56 was found to be overexpressed in HCC tissues, correlating with disease stage and prognosis. In vitro, DDX56 stimulated tumor cell proliferation, migration, invasion, EMT, and stemness. It also enhanced maternal embryonic leucine-zipper kinase (MELK)-mediated forkhead box protein M1 (FOXM1) expression, regulating cancer stemness and malignant traits. In vivo, DDX56 knockdown in tumor-bearing mice reduced tumorigenicity and lung metastasis by modulating the MELK-FOXM1 signaling pathway. Collectively, DDX56 initiates stem cell-like traits in HCC and promotes EMT via MELK-FOXM1 activation, shedding light on HCC pathogenesis and suggesting a potential anti-cancer therapeutic target.
Subject areas: Molecular biology, Cell biology, Cancer
Graphical abstract
Highlights
-
•
DDX56 is upregulated in HCC and related to pathological stage and prognosis
-
•
DDX56 triggers EMT process and cancer stemness of HCC cells
-
•
DDX56 promotes MELK-FOXM1, regulating cancer stemness and malignant phenotypes
-
•
DDX56 knockdown reduces the tumorigenicity and lung metastasis via MELK-FOXM1
Molecular biology; Cell biology; Cancer
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common malignant tumor and the third leading cause of cancer-related deaths worldwide.1 At present, due to the lack of effective early detection means, some patients have been diagnosed at advanced stages, thus delaying the best time for treatment and leading to poor prognosis.2 At the same time, tumor metastasis and recurrence are also the main causes of poor prognosis.3 Therefore, it is of great clinical significance to further explore prognostic biomarkers and promising therapeutic targets associated with HCC progression and metastasis.4
Epithelial-mesenchymal transition (EMT) enables epithelial tumor cells to acquire a mesenchymal phenotype, which not only enhances the invasion and metastasis of tumor cells but also endows tumor cells with stem cell-like properties such as self-renewal abilities.5,6 The process of EMT and tumor stemness generation requires the involvement of diversified host factors. Thus, the search for key regulators of this process to provide molecular markers for early diagnosis and therapeutic targets is a hot research topic in the field of HCC therapy.7,8
Maternal embryonic leucine-zipper kinase (MELK) is a member of the Snfl/AMP-activated protein kinase (AMPK) kinase family, which is mainly involved in multifarious physiological and pathological processes such as cell cycle regulation, cell proliferation, tumorigenesis, and apoptosis.9 MELK was found to regulate phosphorylation and activation of oncogenic transcription factor forkhead box protein M1 (FOXM1) through direct interaction,10,11,12 with the latter playing a decisive role in regulating embryonic development and stem cell stemness maintenance.13,14 In glioma12,15,16,17 and HCC,18 the MELK-FOXM1 signaling pathway is widely involved in the maintenance of tumor cell stemness and plays a pivotal role in the tumorigenesis and progression.
DEAD box helicase 56 (DDX56) is an RNA helicase, which is overexpressed in a variety of malignant tumors, such as colorectal cancer,19 osteosarcoma,20 lung cancer,21,22 and gastric cancer,23 and plays a crucial role in tumor cell proliferation, apoptosis, cell cycle, invasion, and metastasis. In this study, we found that DDX56 was significantly elevated in HCC tissues by analyzing The Cancer Genome Atlas-Liver Hepatocellular Carcinoma (TCGA-LIHC) dataset, which may be associated with the pathological stage and prognosis, suggesting that DDX56 may be a potential indicator for clinical prognosis of HCC and a promising target for targeted therapy.
Herein, we further investigated the effects of DDX56 on various biological behaviors of HCC cells using human HCC cell lines, nude mouse tumorigenicity assays, and various experimental techniques. Our project also explored the interaction between DDX56 and the MELK-FOXM1 signaling pathway, thereby regulating the EMT process and stemness of tumor cells, which provided the basis and potential targets for early diagnosis and treatment of HCC.
Results
DDX56 is highly expressed in HCC and is associated with poor prognosis
We first analyzed the differential expression of DDX56 in HCC tissues and normal samples retrieved from the TCGA project. Two public databases, including tumor immune estimation resource (TIMER) (Figure 1A) and The University of Alabama at Birmingham Cancer data analysis portal (UALCAN) (Figure 1B) showed that DDX56 was highly expressed in HCC tumor tissues. The UALCAN online analysis tool also suggested the relationship of DDX56 with tumor stage (Figure 1C). The gene expression profiling interactive analysis (GEPIA) website was used to analyze the effects of DDX56 differential expression on the prognosis of HCC based on TCGA survival data, and the results showed that the overall and disease-free survival of the DDX56 high-expression group was lower than that of the low-expression group (Figure 1D). Subsequently, the mRNA and protein levels of DDX56 in HCC tissues were detected using qRT-PCR and immunohistochemical (IHC), and the results showed that DDX56 expression was upregulated in tumor tissues compared with the normal samples (Figures 1E and 1F). In addition, the same trend of DDX56 expression was observed in HCC cell lines (Figure 1G).
Figure 1.

DDX56 is highly expressed in HCC and is associated with poor prognosis
(A) TIMER web server showed the boxplots presenting DDX56 expression in tumor and normal tissues from the TCGA project.
(B) UALCAN portal showed the boxplots presenting DDX56 expression in tumor and normal tissues from the TCGA-LIHC project.
(C) UALCAN portal showed the expression of DDX56 in LIHC samples based on tumor stage.
(D) GEPIA website performed overall survival or disease free survival analysis based on DDX56 expression.
(E) Relative DDX56 mRNA levels were compared between HCC samples and normal adjacent tissues (n = 50).
(F) IHC staining of DDX56 expression in HCC tissues and normal adjacent tissues (scale bars: 200 μm; 50 μm).
(G) Relative DDX56 mRNA levels were determined in HCC cell lines and hepatic epithelial cell line THLE-2. Data are represented as means ± SD of triplicate experiments. ∗∗p < 0.01, ∗∗∗p < 0.001 indicate statistical significance.
DDX56 promotes HCC cell proliferation in vitro
To further investigate the biological functions of DDX56 in HCC, we overexpressed DDX56 by lentiviral infection or silenced DDX56 by shRNA in HepG2 and Huh7 cell lines. The transfection efficiencies were confirmed by means of western blotting (Figure 2A). The proliferation analysis using cell counting kit-8 (CCK-8) (Figure 2B), colony formation (Figure 2C), and EdU (Figure 2D) assays showed that overexpression of DDX56 in HepG2 and Huh7 cells promoted cell proliferation, whereas knock down inhibited cell proliferation.
Figure 2.

DDX56 promotes HCC cell proliferation in vitro
(A) The protein levels of DDX56 were determined by western blotting in HepG2 and Huh7 cells after transfection.
(B–D) CCK-8 (B), colony formation (C), EdU (D; scale bar: 50 μm) assays for the cell proliferation analysis after DDX56 overexpression or knockdown. Data are represented as means ± SD of triplicate experiments. ##p < 0.01, ∗∗∗;###p < 0.001 indicate statistical significance.
DDX56 promotes the migration, invasion, and EMT process of HCC cells
After performing wound healing (Figure 3A) and transwell (Figures 3B and 3C) assays, we found that DDX56 overexpression enhanced the migratory and invasive abilities, while DDX56 interference alleviated cell mobility. The immunoblotting analysis indicated that E-cadherin was significantly decreased by the enforced expression of DDX56, while N-cadherin and vimentin were both upregulated (Figure 3D). However, DDX56 knockdown led to an opposite effect. Moreover, the overexpression of DDX56 induced EMT in both HepG2 and Huh7 cells as characterized by a morphological change from an epithelial-like phenotype to a spindle-shaped mesenchymal phenotype, while DDX56 knockdown restored epithelial morphology (Figure 3E). Taken together, these findings indicated that DDX56 plays a crucial role in regulating HCC proliferation, migration, invasion and EMT progression.
Figure 3.

DDX56 promotes the migration, invasion and EMT process of HCC cells
(A–C) Wound healing (A) and transwell (B and C) assays for the cell migration, invasion analysis after DDX56 overexpression or knockdown.
(D) The protein levels of E-cadherin, N-cadherin, and vimentin were determined by western blotting in HepG2 and Huh7 cells after DDX56 overexpression or knockdown.
(E) Morphological changes in HepG2 and Huh7 cells after DDX56 overexpression or knockdown. Data are represented as means ± SD of triplicate experiments. #p < 0.05, ∗∗;##p < 0.01, ∗∗∗;###p < 0.001 indicate statistical significance.
DDX56 expedites the stem-like properties of HCC cells
Additionally, DDX56 overexpression enhanced the sphere formation abilities of HepG2 and Huh7 cells, a hallmark of cancer stem cells (CSCs), whereas DDX56 suppression inhibited the sphere formation (Figure 4A). Consistent with this, the expression levels of CSC markers, including SOX2, Oct4, and Nanog, were upregulated by DDX56 overexpression but downregulated in DDX56-silenced cells (Figure 4B). These findings suggested that DDX56 plays a pivotal role in promoting a cancer stem-like cell phenotype in HCC cells.
Figure 4.

DDX56 expedites the stem-like properties of HCC cells
(A) The sphere formation abilities of HepG2 and Huh7 cells after DDX56 overexpression or knockdown.
(B) The protein levels of SOX2, Oct4, and Nanog were determined by western blotting in HepG2 and Huh7 cells after DDX56 overexpression or knockdown. Data are represented as means ± SD of triplicate experiments. #p < 0.05, ∗∗;##p < 0.01, ∗∗∗;###p < 0.001 indicate statistical significance.
DDX56 positively regulates MELK-mediated FOXM1 signaling
According to encyclopedia of RNA interactomes (ENCORI) prediction, DDX56 was positively correlated with MELK and FOXM1 in HCC samples (Figure 5A). As demonstrated by western blotting, we confirmed that DDX56 upregulation led to a significant increase in MELK protein and FOXM1 protein expression and phosphorylation levels in HepG2 and Huh7 cells, whereas, silencing of DDX56 expression led to the opposite trend (Figure 5B). To determine whether DDX56 exerted its tumor-promoting effect in vitro through MELK-mediated FOXM1 expression, we silenced MELK in DDX56-overexpressed HepG2 and Huh7 cells; the knockdown efficiency of MELK shRNA was confirmed via the western blotting analysis (Figure 5C). Subsequently, we found that deletion of MELK reversed the upregulation of MELK and FOXM1 protein expression and phosphorylation levels caused by DDX56 overexpression (Figure 5D). Bioinformatic analysis (ENCORI database) revealed that the MELK and FOXM1 promoters contain candidate DDX56 binding motifs. To further verify whether MELK and FOXM1 were the direct targeting genes of DDX56, we performed luciferase assays and chromatin immunoprecipitation (ChIP)-PCR experiments. Luciferase assay showed that DDX56 overexpression increased the expression of luciferase reporter constructs of MELK-WT or FOXM1-WT promoter regions in HepG2 and Huh7 cells (Figure 5E). Furthermore, the ChIP-PCR assays revealed that DDX56 binds to the MELK or FOXM1 promoter region in HepG2 and Huh7 cells, which verified the combination of DDX56 and MELK or FOXM1 promoter in HCC cells (Figure 5F). In clinical HCC tissues, the mRNA expression levels of MELK and FOXM1 were upregulated compared to normal controls (Figure 5G), and positively correlated with DDX56 expression (Figure 5H). The aforementioned experimental results revealed a role for DDX56 in positively regulating the expression of MELK and FOXM1 in HCC cells.
Figure 5.

DDX56 positively regulates MELK-mediated FOXM1 signaling
(A) The correlation analysis of DDX56 with MELK and FOXM1 in HCC samples according to ENCORI prediction.
(B) The protein levels of MELK, p-FOXM1, and FOXM1 in HepG2 and Huh7 cells after DDX56 overexpression or knockdown.
(C) The protein levels of MELK were determined by western blotting in HepG2 and Huh7 cells after shMELK transfection.
(D) The protein levels of MELK, p-FOXM1, and FOXM1 in HepG2 and Huh7 cells after transfection.
(E) Dual luciferase in HepG2 and Huh7 cells transfected with WT, MUT 3′-UTR of MELK or FOXM1.
(F) ChIP assay showing enrichment of DDX56 binding to the MELK or FOXM1 promoter in HepG2 and Huh7 cells. DDX56 binding at the MELK or FOXM1 promoter region is shown relative to input. IgG was used as a negative control (top). Agarose gel electrophoresis of PCR fragments after ChIP (bottom).
(G and H) The mRNA expression of MELK and FOXM1 (G) and their correlation with DDX56 expression (H) in HCC tissues. Data are represented as means ± SD of triplicate experiments. #p < 0.05, ∗∗;##p < 0.01, ∗∗∗;###p < 0.001 indicate statistical significance.
DDX56 regulates HCC cell proliferation, EMT process, and stemness via MELK-FOXM1 signaling pathway
To further confirm whether MELK-FOXM1 signaling pathway was involved in DDX56-mediated malignant behaviors in cell biology, cell rescue experiment was carried out. FOXM1 interference was achieved by shRNA expressing vector, and confirmed by the inhibition of its protein (Figure 6A). As the corresponding results showed, inhibition of MELK or FOXM1 expression in DDX56 overexpressing HepG2 and Huh7 cells reduced cell proliferation (Figures 6B–6D), migration (Figures 7A and 7B), invasion (Figure 7C) capabilities as well as the sphere formation ability (Figure 7D). These results demonstrated that DDX56 could promote HCC progression, which was mainly achieved by regulating the MELK-FOXM1 signaling pathway.
Figure 6.

DDX56 regulates HCC cell proliferation via MELK-FOXM1 signaling pathway
(A) The protein levels of FOXM1 and p-FOXM1 were determined by western blotting in HepG2 and Huh7 cells after shFOXM1 transfection.
(B–D) CCK-8 (B), colony formation (C), EdU (D; scale bar: 50 μm) assays for the cell proliferation analysis in HepG2 and Huh7 cells after transfection. Data are represented as means ± SD of triplicate experiments. #p < 0.05, ##p < 0.01, ∗∗∗;###p < 0.001 indicate statistical significance.
Figure 7.

DDX56 regulates EMT process and stemness via MELK-FOXM1 signaling pathway in HCC cells
(A–C) Wound healing (A) and transwell (B and C) assays for the cell migration and invasion analysis in HepG2 and Huh7 cells after transfection.
(D) The sphere formation abilities of HepG2 and Huh7 cells after transfection. Data are represented as means ± SD of triplicate experiments. ∗∗;##p < 0.01, ∗∗∗;###p < 0.001 indicate statistical significance.
DDX56 regulates MELK-FOXM1 signaling pathway and promotes tumor growth and lung metastasis in vivo
To provide the in vivo evidence for the role of DDX56 during HCC tumorigenesis, the xenograft tumor model was established. The silencing of DDX56 resulted in a reduction of tumor size (Figure 8A) and weight (Figure 8B), accompanied with decreased expression of DDX56, MELK, FOXM1, and Ki-67 (Figure 8C). We also constructed a pulmonary metastasis model to evaluate the in vivo metastatic capacity of DDX56. H&E staining showed that the number and area of lung metastases in the DDX56-interfering group were significantly lower than those in the control group (Figure 8D). These results were further confirmed in a mouse model of orthotopic HCC. In the DDX56-knockdown group, the volume of HCC tumors (Figure 8E) and the expression of DDX56 (Figure 8F) in the mouse liver were significantly inhibited than that in the control group. Together, these findings confirmed that DDX56 knockdown inhibited HCC tumor growth and metastasis in tumor-bearing mice.
Figure 8.

DDX56 regulates MELK-FOXM1 signaling pathway and promotes tumor growth and lung metastasis in vivo
(A) Representative photographs showing the dissected tumors and growth curves of xenograft-bearing nude mice.
(B) Tumor weights of xenograft-bearing nude mice.
(C) Immunohistochemical staining of DDX56, MELK, FOXM1 and Ki-67 expression in the dissected tumors (scale bar: 50 μm).
(D) Representative images of H&E staining from the lung metastasis model (scale bar: 100 μm).
(E and F) HepG2 cells with DDX56 knockdown were transplanted into the left lobe of the mouse liver; representative images showing livers with tumor lesions (E) and IHC staining showing DDX56 expression in mouse liver with tumor lesions (F; scale bars: 100 μm; 50 μm). Data are represented as means ± SD of triplicate experiments. ∗p < 0.05, ∗∗∗p < 0.001 indicate statistical significance.
Discussion
HCC has rapidly ascended as a primary cause of cancer-related fatalities globally, distinguished by its rapid tumor proliferation, invasion of blood vessels, and pronounced resistance to both chemotherapy and targeted therapies.24,25 Although advancements in the detection and treatment of HCC have expanded the potential for curative outcomes, the prognosis remains grim, largely due to the high incidence of postoperative recurrence and metastasis.26 Consequently, there is an imperative need to delve into the molecular underpinnings of HCC’s onset and progression to uncover more effective treatment modalities.
Increasing evidence has demonstrated that the DDX family regulates cell proliferation, which in turn affects tumor progression. For instance, knock down of DDX46 suppresses the proliferation and invasion of gastric cancer.27 Moreover, increased DDX1 expression predicts a poor prognosis and drives the progression of HCC.28 Additionally, DDX10 promotes the proliferation and metastasis of colorectal cancer cells.29 In this study, we analyzed by TCGA database and identified the differential DDX56 mRNA expression between HCC and matched noncarcinoma tissue samples. To our knowledge, the high expression of DDX56 has been involved in different types of cancers,19,20,21,22,23 indicating that DDX56 may become an oncogene and therapeutic target for cancers. In this study, we found that DDX56 expression in cancer tissues of HCC patients was higher than that in normal tissues, and the high expression of DDX56 was correlated with tumor stage and survival rate. Subsequently, the upregulation of DDX56 in HCC was also confirmed by clinical tissue samples and cell lines. These results suggested that DDX56 may serve as a potential biomarker to predict the prognosis of HCC.
EMT is a basic morphological event for epithelial cells to acquire migration and invasion function, which contributes to tumor cell diffusion and distant colonization.30,31 Zhang et al. identified a double-negative feedback loop between DDX56 and Snail in regulating EMT and metastasis in breast cancer.32 A recent study has shown that DDX56 can be functioned as an oncogene to regulate colorectal cancer metastasis via MCM5-dependent EMT pathway.33 Study has shown that DDX56 can be used as the upstream of PTEN-AKT signaling pathway to promote HCC proliferation.34 Our study consistently suggested that DDX56 could enhance the proliferation, migration, and invasion abilities of HCC cells. Further functional analysis demonstrated that DDX56 overexpression downregulated the expression of epithelial marker E-cadherin,35 while increasing the expression of mesenchymal markers N-cadherin and vimentin,36 leading to trans-differentiation from epithelial to fibroblastic-like phenotypes. Taken together, these results suggest that DDX56 can promote the EMT program of HCC cells.
Tumor stem cells are a small subset of tumor cells with self-renewal ability, which is manifested by the increased expression of potential stem cell markers, such as Sox2, Oct4, Nanog, etc.37 EMT has been reported to be an important process that contributes to the acquisition of stemness in tumor cells.38 In this study, we found that the high expression of DDX56 can upregulate the stem cell markers, Sox2, Oct4, and Nanog and enhance the sphere formation ability of HCC cells. Collectively, these data demonstrate that DDX56 promotes the EMT process in HCC cells, which in turn promotes the proliferation, migration, and invasion of HCC cells and enhances the stemness of HCC cells.
It has been reported that MELK is frequently and highly upregulated in various types of human cancers, and that elevated MELK expression is correlated with poor prognosis of cancer patients.39 FOXM1 is a transcription factor of cell cycle progression and is overexpressed in a number of human cancers including HCC.40,41 Mechanistically, FOXM1 activity is regulated by MELK kinase in various pathologic conditions, such as radioresistance,16 chemoresistance,42 vascular calcification,43 and tumorigenesis.12,15,16,17 The oncogenic role of MELK has been documented in HCC development, involving the regulation of the cell cycle progression and mitosis-related genes through FOXM1 pathway.18 However, the role of MELK-FOXM1 signaling in the process of EMT and the maintenance of cell stemness during hepatocarcinogenesis has not been thoroughly investigated. Our data revealed a significant positive correlation between DDX56, MELK, and FOXM1 expression in HCC tissues. Further investigation further confirmed that DDX56 can directly bind to the MELK and FOXM1 promoter to activate the MELK-FOXM1 signaling axis. In addition, the downregulation of MELK or FOXM1 reversed the tumor-promoting effects of DDX56 in HCC cells. It is concluded that the overexpression of DDX56 in HCC cells can activate the function of MELK-dependent FOXM1 signaling, thus irritate the EMT process and stemness of HCC cells. Corresponding to these results, the anti-cancer effects of DDX56 silencing in vivo on tumor growth and lung metastasis were confirmed via the downregulation of MELK-FOXM1 signaling axis.
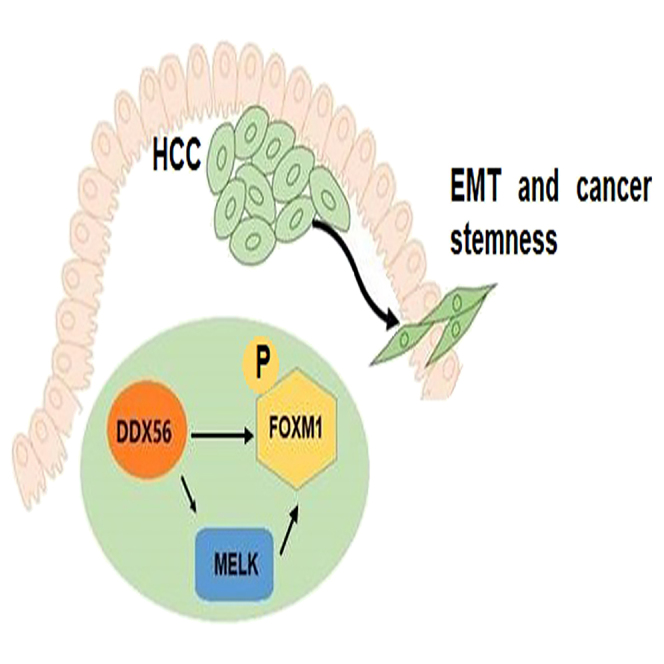
In summary, this study uncovered the relationship between DDX56-mediated MELK-FOXM1 signaling axis and the EMT program, stemness and tumorigenicity of HCC cells, which provides a therapeutic target for HCC (Figure 9).
Figure 9.

Schematic illustrating the carcinogenic mechanism of DDX56 in HCC
We propose that DDX56 maintains cancer cell stemness and EMT in hepatocellular carcinoma via the MELK-FOXM1 signaling.
Limitations of the study
However, some limitations remain to be further addressed. First, the molecular mechanisms through which DDX56 is upregulated in HCC tissues warrant further investigation. Second, the detailed mechanism through which DDX56 promotes EMT process in HCC cells should be examined in future studies. In addition, further studies are required to identify other direct or indirect signaling pathways by which DDX56 regulates EMT and cancer stemness in HCC.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| DDX56 | Abcam | Cat# ab97648; RRID: AB_10679974 |
| E-cadherin | Cell Signaling Technology | Cat# 3195; RRID: AB_2291471 |
| N-cadherin | Cell Signaling Technology | Cat# 13116; RRID: AB_2687616 |
| Vimentin | Cell Signaling Technology | Cat# 5741; RRID: AB_10695459 |
| Sox2 | Cell Signaling Technology | Cat# 3579; RRID: AB_2195767 |
| Oct4 | Cell Signaling Technology | Cat# 2750; RRID:AB_823583 |
| Nanog | Cell Signaling Technology | Cat# 4903; RRID: AB_10559205 |
| MELK | Cell Signaling Technology | Cat# 2274; RRID: AB_2143151 |
| p-FOXM1 | Cell Signaling Technology | Cat# 14170; RRID: AB_2798411 |
| FOXM1 | Cell Signaling Technology | Cat# 20459; RRID: AB_2798842 |
| GAPDH | Cell Signaling Technology | Cat# 5174; RRID: AB_10622025 |
| IgG secondary antibody | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| DDX56 | Abcam | Cat# ab97648; RRID: AB_10679974 |
| MELK | Abcam | Cat# ab129373; RRID: AB_11156762 |
| FOXM1 | Abcam | Cat# ab207298; RRID: AB_3068347 |
| Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| DDX56 | Cell Signaling Technology | Cat# ab115178; RRID: AB_10898837 |
| isotype-matched IgG | Cell Signaling Technology | Cat# 3900; RRID: AB_1550038 |
| Biological samples | ||
| Fifty pairs of HCC and adjacent normal tissues | First Affiliated Hospital of Jinzhou Medical University | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 3000 | Invitrogen | # 2233885 |
| RIPA | Thermo Scientific | 89901 |
| Chemiluminescent Substrates | Invitrogen | WP20005 |
| Critical commercial assays | ||
| CCK-8 | Beyotime Biotechnology | C0038 |
| Cell-Light EdU Apollo488 In Vitro Kit | Guangzhou RiboBio | C10310-3 |
| Dual-Luciferase Reporter Assay System | Promega | E1910 |
| First Strand cDNA Synthesis Kit | Thermo Scientific | K1621 |
| EZ-Magna ChIP A/G Kits | Millipore | 17–408 |
| Deposited data | ||
| TIMER database | TIMER | https://cistrome.shinyapps.io/timer/ |
| UALCAN database | UALCAN | http://ualcan.path.uab.edu/ |
| GEPIA database | GEPIA | http://gepia.cancer-pku.cn |
| ENCORI Pan-Cancer Analysis Platform | ENCORI | https://starbase.sysu.edu.cn/panCancer.php |
| Experimental models: Cell lines | ||
| HepG2 | Cell Bank of Type Culture Collection of the Chinese Academy of Sciences | TCHu 72 |
| Hep3B | Cell Bank of Type Culture Collection of the Chinese Academy of Sciences | SCSP-5045 |
| Huh7 | Cell Bank of Type Culture Collection of the Chinese Academy of Sciences | TCHu182 |
| SNU-387 | Cell Bank of Type Culture Collection of the Chinese Academy of Sciences | SCSP-5046 |
| THLE-2 | the American Type Culture Collection | CRL-2706 |
| Experimental models: Organisms/strains | ||
| BALB/c male nude mice | Experimental Animal Center of Jinzhou Medical University | |
| Oligonucleotides | ||
| shDXX56: 5′-ACTCAAGGAGCTGATATTA-3′ | This paper | N/A |
| shMELK: 5′-CCGGCAGAAACAACAGGCAAACAA TCTCGAGATTGTTTGCCTGTTGTTTCTGTTTTT-3′ |
This paper | N/A |
| shFOXM1: 5′-CCGGGCCAATCGTTCTCTGACAG AACTCGAGTTCTGTCAGAGAACGATTGGCTTTTT-3′ |
This paper | N/A |
| shRNA: 5′-CCGGGCGCGATAGCGCTAATAATTT CTCGAGAAATTATTAGCGCTATCGCGCTTTTT-3′ |
This paper | N/A |
| DDX56-Forward: 5′-CCGCTTATGCTATTCCGATGC-3′ | This paper | N/A |
| DDX56-Reverse: 5′-GCTCCTTGGTAGGAACAAGAACA-3′ | This paper | N/A |
| MELK-Forward: 5′-AACTCCAGCCTTATGCAGAAC-3′ | This paper | N/A |
| MELK-Reverse: 5′-AACGATTTGGCGTAGTGAGTATT-3′ |
This paper | N/A |
| FOXM1-Forward: 5′-ATACGTGGATTGAGGACCACT-3′ |
This paper | N/A |
| FOXM1-Reverse: 5′-TCCAATGTCAAGTAGCGGTTG-3′ |
This paper | N/A |
| GAPDH-Forward: 5′-GGAGCGAGATCCCTCCAAAAT-3′ |
This paper | N/A |
| GAPDH-Reverse: 5′-GGCTGTTGTCATACTTCTCATGG-3′ |
This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 6.0 | GraphPad | https://www.graphpad.com/ |
| PrimerBank database | PrimerBank | http://pga.mgh.harvard.edu/primerbank/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wei Wang (wangwei_lyyy@163.com).
Materials availability
Plasmids and/or cell lines generated in this study are available upon reasonable request. Please contact the lead contact.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Patients and sample collection
The human ethic approval was acquired from First Affiliated Hospital of Jinzhou Medical University (approval No.202254), and our study was also conducted in accordance with the World Medical Association Declaration of Helsinki.44 Written informed consent was obtained from all patients who participated in the study. Fifty pairs of HCC and adjacent normal tissues (46 males and 4 females, Chinese, Han) were collected between January 2019 and December 2019. Patient information is listed in Table S1. All patients who provided samples were confirmed by pathological examination without preoperative chemoradiotherapy. The gender of the patients did not have a significant influence on the study.
Animal experiments
BALB/c male nude mice aged 4 weeks old were used for the in vivo assays. The animal experiment was supported by the Animal Ethics Committee of First Affiliated Hospital of Jinzhou Medical University (approval No.2023037), following the Guide for the Care and Use of Laboratory Animals.45
For tumorigenesis assay, the stably transfected HepG2 cells (1×106 cells dispersed in 150 μL Matrigel) were subcutaneously injected into the flanks of mice. The tumor volume was measured every 7 days. After 28 days, the tumor-bearing mice were euthanized. After weighed, the xenograft tumors were fixed in 10% formalin, dehydrated by gradient ethanol, embedded in paraffin, sliced, processed for IHC staining. The lung metastasis model was established by tail vein injection,46,47 and the lung tissues were taken for conventional hematoxylin and eosin (H&E) staining to observe the formation of lung metastases. To further mimic in situ growth and metastasis, 1×106 HepG2 cells were injected into the left liver lobes of nude mice (5 per group) to construct orthotopic liver xenograft tumor models. Six weeks later, the mice were euthanized, while the tumor sizes were measured based on width and length using vernier calipers and the livers were removed were isolated and subjected to IHC staining.
Method details
Bioinformatics analysis
TIMER (https://cistrome.shinyapps.io/timer/) is an online database used for comprehensive analysis of tumor-infiltrating immune cells and different gene expression levels in different cancer types.48 We used the TIMER database for studying the differential expression of DDX56 between tumor and normal tissues across various cancer types. The UALCAN database (http://ualcan.path.uab.edu/) collects RNA-seq and clinical data of multiple cancer types from TCGA, and it offers a useful platform to analyze gene expression in tumor and normal tissues.49,50 This database was used to analyze the expression of DDX56 in LIHC based on sample types and tumor grade. Differences with a p-value <0.05 were considered to be statistically significant. The online database GEPIA (http://gepia.cancer-pku.cn) is an interactive web that includes 9,736 tumors and 8,587 normal samples from TCGA and the GTEx projects, which analyzes the RNA sequencing expression.51 In this study, GEPIA was used to generate survival curves, including overall survival and disease free survival, based on DDX56 expression in LIHC. The DDX56 co-expression genes were calculated and depicted by Pearson’s correlation test using samples from TCGA samples via ENCORI Pan-Cancer Analysis Platform (https://starbase.sysu.edu.cn/panCancer.php).52
Cell culture and transfection
The human HCC cell lines HepG2 (serial no. TCHu 72), Hep3B (serial no. SCSP-5045), Huh7 (serial no. TCHu182) and SNU-387 (serial no. SCSP-5046) were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Hepatic epithelial cell line THLE-2 (American Type Culture Collection (ATCC) number: CRL-2706) was obtained from the ATCC (Manassas, VA, USA). Cells were cultured in DMEM (Gibco, Carlsbad, CA, USA) containing quantified fetal bovine serum (10%) in an incubator with 95% air, 5% CO2 at 37°C. The pcDNA3.1 plasmid vector used for DDX56 overexpression, DDX56, MELK, or FOXM1 silencing using targeted short hairpin RNA (shRNA), and the corresponding negative controls were designed by GenePharma (Shanghai, China), and prepared for transfection using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). The sequences were shown in Table S2. Cell morphology was observed by an inverted microscopy (Olympus, Tokyo, Japan).
Quantitative real-time PCR (QRT-PCR) assay
Total RNA of tissues and cells was extracted by Trizol (Invitrogen, USA), and cDNA was synthesized using First Strand cDNA Synthesis Kit (Thermo Scientific, Shanghai, China). The procedure of real-time PCR was described as follows: 95°C for 5 min; 45 cycles of 95°C for 15 s and 60°C for 30 s; 95°C for 15 s, 60°C for 1 min and 95°C for 3 min. Primers of DDX56, MELK, FOXM1 and GAPDH as an internal control were retrieved from PrimerBank database (http://pga.mgh.harvard.edu/primerbank/),53 which were listed in Table S3.
Western blot
After exacting from tissue samples and cultured cells using RIPA (Thermo Scientific, China), 30 μg protein were subjected to electrophoresis. The protein was transferred to polyvinylidene fluoride (PVDF) membranes at 250 mA at 4°C for 2 h. After incubation with following indicated antibodies (1:1000 dilution) overnight at 4°C: DDX56 (ab97648; Abcam, Shanghai, China), E-cadherin (#3195), N-cadherin (#13116), Vimentin (#5741), Sox2 (#3579), Oct4 (#2750), Nanog (#4903), MELK (#2274), phospho-FOXM1 (#14170), FOXM1 (#20459) and GAPDH (#5174) (Cell Signaling Technology, Boston, MA, USA), the membranes were washed with tris-buffered saline tween and stained with goat anti-rabbit IgG secondary antibody (#7074; 1:2000 dilution) at room temperature for 2 h. After adding the Chemiluminescent Substrates (Invitrogen, USA), the bands were scanned.
Cell proliferation assays
For CCK-8 assay (Beyotime Biotechnology, Shanghai, China), 100 μL cell suspension was prepared in a 96-well plate at 2 × 103 cells/well, and incubated for 0 h, 24 h, 48 h or 72 h. Then, 10 μL CCK-8 solution was added to each well and incubated for 1 h. The absorbance at 450 nm was measured with a microplate analyzer. For colony formation assay, transfected cells (1 × 103 cells/well) were re-seeded into 6-well plates. Two weeks later, the colony number was counted under microscopy (Olympus, Tokyo, Japan) following specified fixation and staining. For the EdU assay using Cell-Light EdU Apollo488 In Vitro Kit (Guangzhou RiboBio Co., Ltd., Guangzhou, China), 150 μL EdU medium was added to each well, and incubated at 37°C for 2 h. Cells were fixed in 4% paraformaldehyde, washed with 200 μL PBS containing 0.5% Triton X-100 and incubated with 200 μL Apollo staining solution in the dark for 30 min. DAPI (1 mg/L) was added into each well to stain for 20 min in a dark chamber, and then the cells were observed under an inverted fluorescence microscope and photographed.
Cell migration and invasion assay
For wound healing assay, cells (4×106/well) were seeded in a 6-well plate, and the confluent cells were scratched with a 200 μL pipette tip. Images were captured at 0 and 24 h using an inverted microscope (Olympus, Japan), and the wound area was determined. For Transwell assays, a total of 600 μL medium containing 10% FBS was transferred to the lower chamber, while 100 μL serum-free medium containing 4 × 103 cells was added to the upper chamber pre-coated with or without matrigel. Twelve hours later, invasive cells were fixed with paraformaldehyde. After staining with crystal violet, cells were counted under microscopy (Olympus, Japan).
Sphere formation assay
Cells were harvested and prepared into single cell suspension supplemented with 20 ng/mL basic fibroblast growth factor , 10 ng/mL recombinant human epidermal growth factor and 2% B27. After 14 days, the sphere-forming rate was determined when the diameter reached 50 μm.
Dual-luciferase reporter assay
Cells were co-transfected with DDX56 and PGL3-MELK-3′UTR-WT, DDX56 and PGL3-MELK-3′UTR-mutant, DDX56 and PGL3-FOXM1-3′UTR-WT or DDX56 and PGL3-FOXM1-3′UTR-mut using Lipofectamine 3000 (Invitrogen, USA). Twenty-four hours post-transfection, HepG2 and Huh7 cells were seeded into 96-well plates (1×104/well) and luciferase activity was analyzed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA).
Chromatin immunoprecipitation (ChIP) assay
HepG2 and Huh7 cells were cross-linked in 1% formaldehyde; nuclear proteins were extracted using EZ-Magna ChIP A/G Kits (Millipore, Bedford, MA, USA). Immunoprecipitation was performed with anti-DDX56 antibody (ab115178; Abcam, Shanghai, China) or isotype-matched IgG as negative control (#3900; Cell Signaling Technology). The purified DNA fragment was subjected to qRT-PCR analysis. PCR products were analyzed using gel electrophoresis. ChIP data are shown as the percentage of the input normalized to control purifications.
IHC staining
The IHC staining was performed on formalin-fixed, paraffin-embedded sections. Slides (5 μm) were treated with 0.3% hydrogen peroxide for 30 min, followed by incubation overnight with anti-DDX56 (ab97648) using a 1:500 working dilution, anti-MELK (ab129373) using a 1:200 working dilution, FOXM1 (ab207298) using a 1:250 working dilution, anti-Ki67 (ab15580) using a 1:5000 working dilution (all from Abcam, Shanghai, China). The sections were visualized using diaminobenzidine for 10 min, counterstained with hematoxylin, and examined by microscopy.
Quantification and statistical analysis
Values were expressed by the mean ± standard deviation (SD). GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA) was used for data analysis using Student’s t test or ANOVA followed by Tukey’s post hoc test. p < 0.05 was regarded as statistically significance.
Additional resources
Clinical samples of this study were obtained from diagnosed HCC patients who underwent surgical resection, and the study protocol was approved by the ethics committee of First Affiliated Hospital of Jinzhou Medical University (approval No.202254). The animal experiment was supported by the Animal Ethics Committee of First Affiliated Hospital of Jinzhou Medical University (approval No.2023037).
Acknowledgments
This study was supported by Jinzhou Science and Technology Bureau Project (No.204230029), The scientific research team of the First Affiliated Hospital of Jinzhou Medical University (KYTD-2022009)
Author contributions
Conception and design, Q.L., T.W., and W.W.; data analysis and interpretation, X.W., X.Y.G., and T.Y.; manuscript writing, Q.L., X.W., and T.W. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: April 29, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109827.
Supplemental information
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Nhlane R., Kreuels B., Mallewa J., Chetcuti K., Gordon M.A., Stockdale A.J. Late presentation of hepatocellular carcinoma highlights the need for a public health programme to eliminate hepatitis B. Lancet. 2021;398:2288. doi: 10.1016/S0140-6736(21)02138-3. [DOI] [PubMed] [Google Scholar]
- 3.Xia S., Pan Y., Liang Y., Xu J., Cai X. The microenvironmental and metabolic aspects of sorafenib resistance in hepatocellular carcinoma. EBioMedicine. 2020;51 doi: 10.1016/j.ebiom.2019.102610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao M., Yang J.L., Wang D.F., Wang L., Chen Y., Yao D.F. Encouraging specific biomarkers-based therapeutic strategies for hepatocellular carcinoma. World J. Clin. Cases. 2022;10:3321–3333. doi: 10.12998/wjcc.v10.i11.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen T., You Y., Jiang H., Wang Z.Z. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017;232:3261–3272. doi: 10.1002/jcp.25797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babaei G., Aziz S.G.G., Jaghi N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharm. 2021;133 doi: 10.1016/j.biopha.2020.110909. [DOI] [PubMed] [Google Scholar]
- 7.Jayachandran A., Dhungel B., Steel J.C. Epithelial-to-mesenchymal plasticity of cancer stem cells: therapeutic targets in hepatocellular carcinoma. J. Hematol. Oncol. 2016;9:74. doi: 10.1186/s13045-016-0307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flores-Téllez T.N., Villa-Trevino S., Pina-Vazquez C. Road to stemness in hepatocellular carcinoma. World J. Gastroenterol. 2017;23:6750–6776. doi: 10.3748/wjg.v23.i37.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganguly R., Mohyeldin A., Thiel J., Kornblum H.I., Beullens M., Nakano I. MELK-a conserved kinase: functions, signaling, cancer, and controversy. Clin. Transl. Med. 2015;4:11. doi: 10.1186/s40169-014-0045-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maes A., Maes K., Vlummens P., De Raeve H., Devin J., Szablewski V., De Veirman K., Menu E., Moreaux J., Vanderkerken K., De Bruyne E. Maternal embryonic leucine zipper kinase is a novel target for diffuse large B cell lymphoma and mantle cell lymphoma. Blood Cancer J. 2019;9:87. doi: 10.1038/s41408-019-0249-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen L., Wei Q., Bi S., Xie S. Maternal Embryonic Leucine Zipper Kinase Promotes Tumor Growth and Metastasis via Stimulating FOXM1 Signaling in Esophageal Squamous Cell Carcinoma. Front. Oncol. 2020;10:10. doi: 10.3389/fonc.2020.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X., Wang J., Wang Y., Liu G., Li H., Yu J., Wu R., Liang J., Yu R., Liu X. MELK Inhibition Effectively Suppresses Growth of Glioblastoma and Cancer Stem-Like Cells by Blocking AKT and FOXM1 Pathways. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.608082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bella L., Zona S., Nestal de Moraes G., Lam E.W.F. FOXM1: A key oncofoetal transcription factor in health and disease. Semin. Cancer Biol. 2014;29:32–39. doi: 10.1016/j.semcancer.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Yan Q., Fang X., Li C., Lan P., Guan X. Oncofetal proteins and cancer stem cells. Essays Biochem. 2022;66:423–433. doi: 10.1042/EBC20220025. [DOI] [PubMed] [Google Scholar]
- 15.Joshi K., Banasavadi-Siddegowda Y., Mo X., Kim S.H., Mao P., Kig C., Nardini D., Sobol R.W., Chow L.M.L., Kornblum H.I., et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cell. 2013;31:1051–1063. doi: 10.1002/stem.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S.H., Joshi K., Ezhilarasan R., Myers T.R., Siu J., Gu C., Nakano-Okuno M., Taylor D., Minata M., Sulman E.P., et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep. 2015;4:226–238. doi: 10.1016/j.stemcr.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gouazé-Andersson V., Gherardi M.J., Lemarie A., Gilhodes J., Lubrano V., Arnauduc F., Cohen-Jonathan Moyal E., Toulas C. FGFR1/FOXM1 pathway: a key regulator of glioblastoma stem cells radioresistance and a prognosis biomarker. Oncotarget. 2018;9:31637–31649. doi: 10.18632/oncotarget.25827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia H., Kong S.N., Chen J., Shi M., Sekar K., Seshachalam V.P., Rajasekaran M., Goh B.K.P., Ooi L.L., Hui K.M. MELK is an oncogenic kinase essential for early hepatocellular carcinoma recurrence. Cancer Lett. 2016;383:85–93. doi: 10.1016/j.canlet.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 19.Kouyama Y., Masuda T., Fujii A., Ogawa Y., Sato K., Tobo T., Wakiyama H., Yoshikawa Y., Noda M., Tsuruda Y., et al. Oncogenic splicing abnormalities induced by DEAD-Box Helicase 56 amplification in colorectal cancer. Cancer Sci. 2019;110:3132–3144. doi: 10.1111/cas.14163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu C., Zhang X., Kourkoumelis N., Shen Y., Huang W. Integrated Analysis of DEAD-Box Helicase 56: A Potential Oncogene in Osteosarcoma. Front. Bioeng. Biotechnol. 2020;8:588. doi: 10.3389/fbioe.2020.00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui Y., Hunt A., Li Z., Birkin E., Lane J., Ruge F., Jiang W.G. Lead DEAD/H box helicase biomarkers with the therapeutic potential identified by integrated bioinformatic approaches in lung cancer. Comput. Struct. Biotechnol. J. 2021;19:261–278. doi: 10.1016/j.csbj.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Q., Luo X., Terp M.G., Li Q., Li Y., Shen L., Chen Y., Jacobsen K., Bivona T.G., Chen H., et al. DDX56 modulates post-transcriptional Wnt signaling through miRNAs and is associated with early recurrence in squamous cell lung carcinoma. Mol. Cancer. 2021;20:108. doi: 10.1186/s12943-021-01403-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J., Wang Y., Wang J., Zhang S., Yu Z., Zheng K., Fu Z., Wang C., Huang W., Chen J. DEAD-box helicase 56 functions as an oncogene promote cell proliferation and invasion in gastric cancer via the FOXO1/p21 Cip1/c-Myc signaling pathway. Bioengineered. 2022;13:13970–13985. doi: 10.1080/21655979.2022.2084235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Renne S.L., Sarcognato S., Sacchi D., Guido M., Roncalli M., Terracciano L., Di Tommaso L. Hepatocellular carcinoma: a clinical and pathological overview. Pathologica. 2021;113:203–217. doi: 10.32074/1591-951X-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forner A., Reig M., Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–1314. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 26.Pinero F., Dirchwolf M., Pessoa M.G. Biomarkers in Hepatocellular Carcinoma: Diagnosis, Prognosis and Treatment Response Assessment. Cells. 2020;9 doi: 10.3390/cells9061370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L., Xu M., Zhong W., Hu Y., Wang G. Knockdown of DDX46 suppresses the proliferation and invasion of gastric cancer through inactivating Akt/GSK-3β/β-catenin pathway. Exp. Cell Res. 2021;399 doi: 10.1016/j.yexcr.2020.112448. [DOI] [PubMed] [Google Scholar]
- 28.Yuan M., Xu J., Cao S., Sun S. DDX1 is a prognostic biomarker and correlates with immune infiltrations in hepatocellular carcinoma. BMC Immunol. 2022;23:59. doi: 10.1186/s12865-022-00533-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou X., Liu Z., He T., Zhang C., Jiang M., Jin Y., Wu Z., Gu C., Zhang W., Yang X. DDX10 promotes the proliferation and metastasis of colorectal cancer cells via splicing RPL35. Cancer Cell Int. 2022;22:58. doi: 10.1186/s12935-022-02478-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pastushenko I., Blanpain C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019;29:212–226. doi: 10.1016/j.tcb.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Zhang N., Ng A.S., Cai S., Li Q., Yang L., Kerr D. Novel therapeutic strategies: targeting epithelial-mesenchymal transition in colorectal cancer. Lancet Oncol. 2021;22:e358–e368. doi: 10.1016/S1470-2045(21)00343-0. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H., Zhang Y., Chen C., Zhu X., Zhang C., Xia Y., Zhao Y., Andrisani O.M., Kong L. A double-negative feedback loop between DEAD-box protein DDX21 and Snail regulates epithelial-mesenchymal transition and metastasis in breast cancer. Cancer Lett. 2018;437:67–78. doi: 10.1016/j.canlet.2018.08.021. [DOI] [PubMed] [Google Scholar]
- 33.Gao H., Wei H., Yang Y., Li H., Liang J., Ye J., Zhang F., Wang L., Shi H., Wang J., Han A. Phase separation of DDX21 promotes colorectal cancer metastasis via MCM5-dependent EMT pathway. Oncogene. 2023;42:1704–1715. doi: 10.1038/s41388-023-02687-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou H., Du Y., Wei X., Song C., Song J., Xu N., Huang W., Chen L., Yao F., Du D., et al. DDX56 transcriptionally activates MIST1 to facilitate tumorigenesis of HCC through PTEN-AKT signaling. Theranostics. 2022;12:6069–6087. doi: 10.7150/thno.72471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong S.H.M., Fang C.M., Chuah L.H., Leong C.O., Ngai S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018;121:11–22. doi: 10.1016/j.critrevonc.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 36.Yang G., Wang G., Xiong Y., Sun J., Li W., Tang T., Li J. CDC20 promotes the progression of hepatocellular carcinoma by regulating epithelialmesenchymal transition. Mol. Med. Rep. 2021;24 doi: 10.3892/mmr.2021.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.You L., Guo X., Huang Y. Correlation of Cancer Stem-Cell Markers OCT4, SOX2, and NANOG with Clinicopathological Features and Prognosis in Operative Patients with Rectal Cancer. Yonsei Med. J. 2018;59:35–42. doi: 10.3349/ymj.2018.59.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Celia-Terrassa T., Jolly M.K. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition in Cancer Metastasis. Cold Spring Harb. Perspect. Med. 2020;10 doi: 10.1101/cshperspect.a036905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald I.M., Graves L.M. Enigmatic MELK: The controversy surrounding its complex role in cancer. J. Biol. Chem. 2020;295:8195–8203. doi: 10.1074/jbc.REV120.013433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu G., Yan Z., Zhang C., Cheng M., Yan Y., Wang Y., Deng L., Lu Q., Luo S. FOXM1 promotes hepatocellular carcinoma progression by regulating KIF4A expression. J. Exp. Clin. Cancer Res. 2019;38:188. doi: 10.1186/s13046-019-1202-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong J., Xu S., Deng Z., Wang Y., Zhang P. Transcription factor FOXM1 promotes hepatocellular carcinoma malignant progression through activation of the WNT pathway by binding to SETDB1. Tissue Cell. 2023;84 doi: 10.1016/j.tice.2023.102186. [DOI] [PubMed] [Google Scholar]
- 42.Wang K., Zhu X., Yin Y. Maslinic Acid Enhances Docetaxel Response in Human Docetaxel-Resistant Triple Negative Breast Carcinoma MDA-MB-231 Cells via Regulating MELK-FoxM1-ABCB1 Signaling Cascade. Front. Pharmacol. 2020;11:835. doi: 10.3389/fphar.2020.00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei X., Su Y., Li Q., Zheng Z., Hou P. Analysis of crucial genes, pathways and construction of the molecular regulatory networks in vascular smooth muscle cell calcification. Exp. Ther. Med. 2021;21:589. doi: 10.3892/etm.2021.10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.World Medical Association World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 45.Guide for the Care and Use of Laboratory Animals. 2011. [DOI] [Google Scholar]
- 46.Yang N., Chen T., Wang L., Liu R., Niu Y., Sun L., Yao B., Wang Y., Yang W., Liu Q., et al. CXCR4 mediates matrix stiffness-induced downregulation of UBTD1 driving hepatocellular carcinoma progression via YAP signaling pathway. Theranostics. 2020;10:5790–5801. doi: 10.7150/thno.44789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan K., Xie K., Lan T., Xu L., Chen X., Li X., Liao M., Li J., Huang J., Zeng Y., Wu H. TXNDC12 promotes EMT and metastasis of hepatocellular carcinoma cells via activation of beta-catenin. Cell Death Differ. 2020;27:1355–1368. doi: 10.1038/s41418-019-0421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li T., Fan J., Wang B., Traugh N., Chen Q., Liu J.S., Li B., Liu X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017;77:e108–e110. doi: 10.1158/0008-5472.CAN-17-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chandrashekar D.S., Bashel B., Balasubramanya S.A.H., Creighton C.J., Ponce-Rodriguez I., Chakravarthi B.V.S.K., Varambally S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. 2017;19:649–658. doi: 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chandrashekar D.S., Karthikeyan S.K., Korla P.K., Patel H., Shovon A.R., Athar M., Netto G.J., Qin Z.S., Kumar S., Manne U., et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia. 2022;25:18–27. doi: 10.1016/j.neo.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang Z., Li C., Kang B., Gao G., Li C., Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X., Spandidos A., Wang H., Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012;40:D1144–D1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.