

Abstract
Venom is known as the source of natural antimicrobial products. Previous studies have largely focused on the expression of venom-related genes and the biochemical components of venom. With the advent of metagenomic sequencing, many more microorganisms, especially viruses, have been identified in highly diverse environments. Herein, we investigated the RNA virome in the venom-related microenvironment through analysis of a large volume of venom-related RNA-sequencing data mined from public databases. From this, we identified viral sequences belonging to thirty-six different viruses, of which twenty-two were classified as ‘novel’ as they exhibited less than 90 per cent amino acid identity to known viruses in the RNA-dependent RNA polymerase. Most of these novel viruses possessed genome structures similar to their closest relatives, with specific alterations in some cases. Phylogenetic analyses revealed that these viruses belonged to at least twenty-two viral families or unclassified groups, some of which were highly divergent from known taxa. Although further analysis failed to find venom-specific viruses, some viruses seemingly had much higher abundance in the venom-related microenvironment than in other tissues. In sum, our study provides insights into the RNA virome of the venom-related microenvironment from diverse animal phyla.
Keywords: venom-related microenvironment, RNA virus, virome, phylogenetic analysis
1. Introduction
Venom is a known source of natural antimicrobial products (da Mata et al. 2017). It is generally held that venom and venom-producing tissues are largely sterile environments (Ul-Hasan et al. 2019), and previous studies have largely focused on the biochemical components of venoms and the gene expression of venom-producing tissues (Robinson et al. 2017). However, recent studies have revealed that there may be hidden diverse microbial communities in the microenvironment of venom or venom-producing tissues, such as the venom gland and venom duct (Ul-Hasan et al. 2019; Esmaeilishirazifard et al. 2022). These microorganisms can coexist with the host and even play roles in host reproduction (Zhu et al. 2018). As venom can be used as a defensive weapon in animals, its biochemical components can poison other animals and the microorganisms present in venom may be injected into other animals to cause infection or modulate the host immune reaction (Monteiro et al. 2002; Coffman and Burke 2020).
Metagenomic (DNA) and metatranscriptomic (RNA) sequencing approaches have facilitated the identification of novel and existing microorganisms. In recent years, thousands of novel viruses have been identified in this manner (Shi et al. 2016, 2018; He et al. 2022), with many more to be discovered (Carroll et al. 2018). Viruses are the most abundant biological entities on earth (Paez-Espino et al. 2016), although the vast majority have still to be described (Geoghegan and Holmes 2017; Zhang, Shi, and Holmes 2018).
To date, high-throughput sequencing of venom-producing tissues has resulted in the discovery of many novel venom peptides and accumulation of a large volume of transcriptomic sequencing data (Robinson et al. 2017). However, little is known about the viruses in the venom-related microenvironment (including venom and venom-producing tissues) of animals. Herein, we used these publicly available transcriptome sequencing data to explore the RNA virome in the venom-related microenvironment of diverse animals, representing those in the phyla Chordata, Mollusca, Arthropoda, Annelida, and Cnidaria. In doing so, we identified thirty-six different RNA viruses in thirty-three of the 474 venom-related transcriptome datasets, some of which were phylogenetically distant from known viruses. In addition, we characterized the genomic and phylogenetic features of these viruses and explored their abundance among venom-related tissues and non-venom tissues.
2. Materials and methods
2.1. Dataset retrieving and processing
RNA-sequencing (Seq) data from the venom-related microenvironment of animals was retrieved and downloaded from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database with the keywords ‘venom’ and ‘RNA-seq’. The final data set contained 474 paired-end SRA files from the venom-related microenvironment of 151 species spanning the phyla Chordata, Arthropoda, Mollusca, Annelida, and Cnidaria (Figure S1 and Table S1), with a total of 2,702.87 Gb.
2.2. Sequence assembly and virus discovery
For each RNA-seq dataset, sequencing reads were first adapter trimmed and filtered using Fastp v0.19 (Chen et al. 2018), with the ribosomal RNA reads then removed. The remaining reads were de novo assembled with Trinity v2.5.1 (Grabherr et al. 2011) using default parameters. The assembled contigs were searched against the non-redundant nucleotide (nt) and non-redundant protein (nr) databases from NCBI using Basic Local Alignment Search Tool (BLAST) related toolkits, including BLASTn (Camacho et al. 2009) and Diamond BLASTx (Li et al. 2021), respectively. Viral contigs were identified, and by comparing the virus contigs with the known closest references, viral contigs with near complete genomes were subjected to further analyses.
2.3. Virus genome annotation
Open reading frames (ORFs) within the virus contigs were predicted using the ‘Find ORFs’ function of Geneious v2021.0.1. The closest virus references for each virus contig were downloaded from NCBI. Conserved domains (CD) of the virus contigs and their closest relatives were identified using CD search against the NCBI CD Database.
2.4. Phylogenetic analysis
To infer the evolutionary relationships of the newly identified viruses, the closest relatives and representative viral protein sequences of associated genera were retrieved by BLASTx by comparing the new viral contigs against the nr database (NCBI). Sequences of the relatively well-conserved RNA-dependent RNA polymerase (RdRp) were selected for phylogenetic analysis. Multiple sequence alignment was performed using MAFFT v7.407 (Katoh and Standley 2013) employing the L-INS-I algorithm, with ambiguously aligned regions removed using TrimAl v1.4 (Capella-Gutiérrez, Silla-Martínez, and Gabaldón 2009). Phylogenetic trees were inferred using the maximum likelihood method implemented in IQ-TREE v2.2.0 (Minh et al. 2020) with 1,000 bootstrap replicates, and ModelFinder (Kalyaanamoorthy et al. 2017) was used to find the best-fit substitution model in each dataset.
2.5. Abundance calculation of RNA viruses
Based on the data with ribosomal RNA reads removed, the abundance of the newly found RNA viruses was calculated as Reads Per Million (RPM) using the formula, ‘Viral reads/Total non-rRNA reads × one million’. Bowtie2 v.2.4.2 (Langmead and Salzberg 2012) was used to align reads to the virus genomes. SAMtools v1.10 (H. Li et al. 2009) was used to output the number of matched reads from the Bowtie2 SAM result.
3. Results
3.1. Overview of the venom-related RNA-seq data
The RNA-seq data of the venom and venom-related tissues (including venom gland, venom duct, and venom bulb) were retrieved and downloaded from the NCBI SRA database. Only paired-end sequencing data were analysed. The final dataset finally comprised 474 SRA files from 151 species spanning the phyla Chordata (eighty-four species, 353 SRA files), Arthropoda (thirty-nine species, seventy-three SRA files), Mollusca (twenty-five species, forty-four SRA files), Annelida (one species, two SRA files), and Cnidaria (two species, two SRA files), with total sequencing bases of 2,702.87 Gb (Fig. S1 and Table S1). Further analysis showed that the phylum Chordata mainly included samples from Colubroidea, the phylum Arthropoda mainly included samples from Araneae, Scorpiones, Scolopendromorpha, Diptera, and Hymenoptera, while the phylum Mollusca contained samples from Conidae and Turridae (Fig. S1). A total of 160 SRA files had information on collection locations, involving at least nineteen countries or regions (Fig. S2). The samples were mainly collected from North America (n = 68), Asia (47), and South America (21), and the three countries with the most samples were the USA (n = 60), Brazil (n = 20), and China (n = 16).
3.2. Diverse RNA viruses in the venom-related microenvironment
Sequencing reads were de novo assembled into contigs, which were further analysed by BLAST against the NCBI nr/nt databases. A total of forty-five virus strains with complete or partial genome sequences containing at least the RdRp region were recovered from thirty-three RNA-seq data sets. These forty-five virus strains likely represented thirty-six different viruses (Fig. 1 and Table 1), including twenty-eight negative-sense single-stranded (−ssRNA), thirteen positive-sense single-stranded (+ssRNA), and four double-stranded RNA (dsRNA) viruses (Table S3). BLAST analysis revealed that approximately half of virus contigs were divergent from the known viruses: twenty-two viruses shared less than 90 per cent amino acid identity in RdRp with the most closely related viruses, including sixteen that exhibited <70 per cent amino acid identity to known viruses (Table 1).
Figure 1.
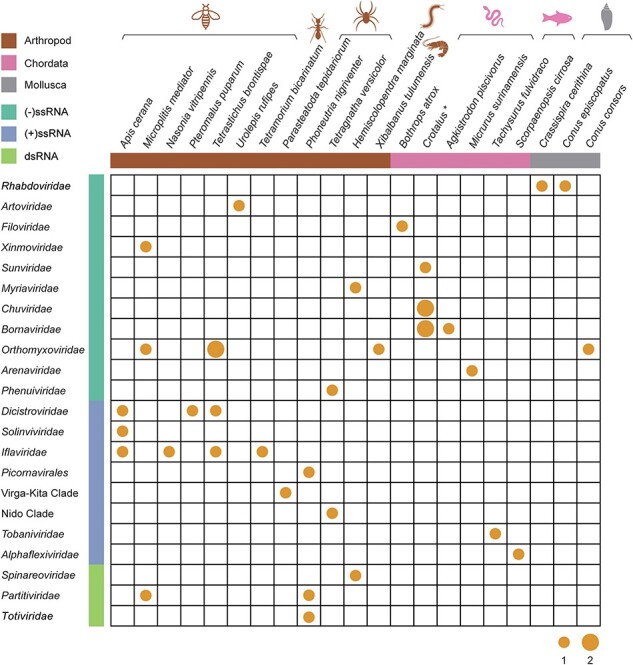
Viral species identified in the venom-related microenvironment from diverse animals. The number of viral species identified in the venom-related microenvironment is summarized according to the respective host organism. The size of the points indicates the number of viral species. The host genus Crotalus marked with a star includes the species Crotalus cerastes, Crotalus durissus terrificus, Crotalus molossus, Crotalus morulus, Crotalus polystictus, and Crotalus totonacus. The colours of the horizontal bars and animal icons represent host groups; brown, pink, and grey indicate Arthropoda, Chordata, and Mollusca, respectively. The aquamarine, royal blue, and light green of the vertical bars represent negative-sense single-stranded, positive-sense single-stranded, and double-stranded RNA viruses, respectively.
Table 1.
Summary of the viruses identified from the venom-related microenvironment in this study.
| No. | Virus name | Host | Baltimore classification | Families or groups | Length (nt) | GenBank accession numbers | Closest relative | RdRp aa identity (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | Crassispira cerithina rhabdovirus b | Crassispira cerithina | (−)ssRNA | Rhabdoviridae | 11,539 | BK064875 | Lyssavirus mokola | 48.09 |
| 2 | Conus episcopatus rhabdovirus b | Conus episcopatus | (−)ssRNA | Rhabdoviridae | 5,738 | BK064874 | Anole lyssa-like virus 1 | 56.79 |
| 3 | Urolepis rufipes peropuvirusa | Urolepis rufipes | (−)ssRNA | Artoviridae | 12,176 | BK064876 | Pteromalus puparum negative-strand RNA virus 1 | 88.35 |
| 4 | Tapajós virus | Bothrops atrox | (−)ssRNA | Filoviridae | 16,181 | FAA04063 | Tapajós virus | 100.00 |
| 5 | Microplitis mediator mononega-like virus strain CHBb | Microplitis mediator | (−)ssRNA | Xinmoviridae | 12,918 | BK064877 | Gudgenby Calliphora mononega-like virus | 53.96 |
| 6 | Crotalus cerastes sunvirus strain Ccera1a | Crotalus cerastes | (−)ssRNA | Sunviridae | 17,220 | BK064879 | Sunshine Coast virus | 86.94 |
| 7 | Hemiscolopendra marginata myriavirusb | Hemiscolopendra marginata | (−)ssRNA | Myriaviridae | 14,106 | BK064881 | Hubei myriapoda virus 8 | 63.97 |
| 8 | Crotalus durissus terrificus chuvirus | Crotalus durissus terrificus | (−)ssRNA | Chuviridae | 10,645 | BK064882 | Herr Frank virus 1 | 91.17 |
| 9 | Crotalus chuvirus strain Ctoto1 a | Crotalus totonacus | (−)ssRNA | Chuviridae | 11,043 | BK064883 | Guangdong red–banded snake chuvirus-like virus | 74.20 |
| 10 | Crotalus totonacus bornavirus strain Ctoto1 | Crotalus totonacus | (−)ssRNA | Bornaviridae | 8,932 | BK064870 | Caribbean watersnake bornavirus | 92.85 |
| 11 | Agkistrodon piscivorus bornavirus | Agkistrodon piscivorus | (−)ssRNA | Bornaviridae | 8,876 | BK064872 | Caribbean watersnake bornavirus | 93.61 |
| 12 | Crotalus molossus bornavirus | Crotalus molossus | (−)ssRNA | Bornaviridae | 8,938 | BK064873 | Mexican black-tailed rattlesnake bornavirus | 100.00 |
| 13 | Conus consors orthomyxo-like virus b | Conus consors | (−)ssRNA | Orthomyxoviridae | 1,431/1,989 | BK064896–BK064897 | Barns Ness dog whelk orthomyxo-like virus 1 | 51.30 |
| 14 | Xibalbanus tulumensis orthomyxo-like virusb | Xibalbanus tulumensis | (−)ssRNA | Orthomyxoviridae | 1,469/1,359/1,328/1,446/1,039 | BK064898–BK064902 | Jos virus | 53.17 |
| 15 | Microplitis mediator orthomyxo-like virus b | Microplitis mediator | (−)ssRNA | Orthomyxoviridae | 2,442/2,484/2,284/1,772/1,711 | BK064903–BK064907 | Hymenopteran orthomyxo-related virus OKIAV173 | 61.07 |
| 16 | Tetrastichus brontispae orthomyxo-like virus 1b | Tetrastichus brontispae | (−)ssRNA | Orthomyxoviridae | 2,434/2,503/2,469/1,786/1,690/1,103 | BK064908–BK064913 | Mason Creek virus | 62.08 |
| 17 | Tetrastichus brontispae orthomyxo-like virus 2 b | Tetrastichus brontispae | (−)ssRNA | Orthomyxoviridae | 2,403/2,548/2,420/1,802/1,707 | BK064914–BK064918 | Longchuan virus | 58.73 |
| 18 | Micrurus arenavirusb | Micrurus surinamensis | (−)ssRNA | Arenaviridae | 6,723/1,890/1,388 | BK064890–BK064892 | Unidentified Reptarenavirus | 40.01 |
| 19 | Tetragnatha versicolor phenuivirus b | Tetragnatha versicolor | (−)ssRNA | Phenuiviridae | 6,145/3,334/853 | BK064893–BK064895 | FinV707 virus | 45.95 |
| 20 | Israeli acute paralysis virus strain SKS | Apis cerana | (+)ssRNA | Dicistroviridae | 9,584 | BK064921 | Israeli acute paralysis virus | 100.00 |
| 21 | Diversinervus elegans virus strain Tbron | Tetrastichus brontispae | (+)ssRNA | Dicistroviridae | 8,832 | BK064919 | Diversinervus elegans virus | 100.00 |
| 22 | Pteromalus puparum dicistrovirus a | Pteromalus puparum | (+)ssRNA | Dicistroviridae | 9,029 | BK064920 | Soybean thrips picorna–like virus 1 | 83.02 |
| 23 | Apis picorna–like virus 4 strain SKS | Apis cerana | (+)ssRNA | Solinviviridae | 10,152 | BK064922 | Apis picorna–like virus 4 | 96.04 |
| 24 | Sacbrood virus strain SKS | Apis cerana | (+)ssRNA | Iflaviridae | 8,792 | BK064925 | Sacbrood virus | 99.68 |
| 25 | Tetrastichus brontispae RNA virus 3 strain CYN | Tetrastichus brontispae | (+)ssRNA | Iflaviridae | 9,971 | BK064923 | Tetrastichus brontispae RNA virus 3 | 100.00 |
| 26 | Nasonia vitripennis virus strain VG | Nasonia vitripennis | (+)ssRNA | Iflaviridae | 10,075 | BK064924 | Nasonia vitripennis virus | 99.69 |
| 27 | Tetramorium bicarinatum iflavirus | Tetramorium bicarinatum | (+)ssRNA | Iflaviridae | 10,231 | BK064926 | Iflaviridae sp. | 99.00 |
| 28 | Phoneutria nigriventer picorna–like virus b | Phoneutria nigriventer | (+)ssRNA | Unclassified Picornavirales | 8,942 | BK064931 | Riboviria sp. | 65.59 |
| 29 | Solenopsis invicta virus 17 strain Ptepi | Parasteatoda tepidariorum | (+)ssRNA | unclassified (Virga-Kita Clade) | 10,267 | BK064929 | Solenopsis invicta virus 17 | 100.00 |
| 30 | Tetragnatha versicolor virus a | Tetragnatha versicolor | (+)ssRNA | unclassified (Nido Clade) | 19,716 | BK064930 | Hubei tetragnatha maxillosa virus 7 | 86.13 |
| 31 | Pelteobagrus fulvidraco oncotshavirus-1 strain VG | Tachysurus fulvidraco | (+)ssRNA | Tobaniviridae | 27,004 | BK064927 | Pelteobagrus fulvidraco oncotshavirus-1 | 100.00 |
| 32 | Scorpaenopsis potexvirusa | Scorpaenopsis cirrosa | (+)ssRNA | Alphaflexiviridae | 6,204 | BK064928 | Hydrangea ringspot virus | 79.87 |
| 33 | Hemiscolopendra marginata reovirus b | Hemiscolopendra marginata | dsRNA | Spinareoviridae | 4,333/4,070/3,147/1,892 | BK064933–BK064936 | Shelly headland virus | 27.66 |
| 34 | Microplitis mediator partiti-like virus b | Microplitis mediator | dsRNA | Partitiviridae | 1,469 | BK064937 | Pennypacker partiti-like virus | 64.11 |
| 35 | Phoneutria nigriventer partiti-like virus b | Phoneutria nigriventer | dsRNA | Partitiviridae | 1,308 | BK064938 | Hubei partiti-like virus 36 | 34.21 |
| 36 | Phoneutria nigriventer toti-like virus b | Phoneutria nigriventer | dsRNA | Totiviridae | 6,790 | BK064932 | Hubei toti-like virus 16 | 53.24 |
Twenty-two viruses shared less than 90 per cent amino acid identity with the most closely related viruses.
Sixteen viruses shared less than 70 per cent amino acid identity with the most closely related viruses.
The virus with the highest abundance (RPM) was Pteromalus puparum dicistrovirus (Dicistroviridae), found in the venom gland of the arthropod Pteromalus puparum (RPM = 59,818.7), followed by Pelteobagrus fulvidraco oncotshavirus-1 strain VG (Tobaniviridae) from the chordate Tachysurus fulvidraco (RPM = 16,523.3), and Tetragnatha versicolor phenuivirus (Phenuiviridae) from the arthropod Tetragnatha versicolor (RPM = 11,612.0) (Table S3). The abundance of different viruses in the venom-related microenvironment also varied sharply across hosts. For example, the abundance of Crotalus cerastes sunvirus and Crotalus chuvirus was different within the same host or among different hosts (Table S3). Although this is likely due to limited data, it may also reflect the infection status and tissue tropism of the viruses.
The viruses identified here came from twenty-six venom-producing animals, involving the phyla Arthropoda (n = 12), Chordata (n = 11), and Mollusca (n = 3). Viruses in the venom-related microenvironment of Arthropoda species were the most common (n = 24), including eleven +ssRNA, nine −ssRNA viruses, and four dsRNA viruses (Fig. 1). These viruses were identified in fourteen Arthropoda RNA-seq datasets, with a detection rate of 19.18 per cent (14/73). Viruses in the venom-related microenvironment of Chordata (n = 18) largely comprised −ssRNA viruses (n = 16), with only two +ssRNA viruses. Viruses were present in sixteen RNA-seq datasets from Chordata species, with a detection rate of 4.53 per cent (16/353). Three −ssRNA viruses were identified in the venom-related microenvironment of Mollusca, with a detection rate of 6.82 per cent (3/44). As the detection rates documented here are based on the available datasets with potential sampling biases, the true prevalence of these viruses in nature is unclear and requires additional investigation.
3.3. Genome organization of the venom-related RNA viruses
Most of the viruses found in the venom-related microenvironment were similar to their closest relatives in terms of genome structure (Figs. 2 and S3). There were, however, some clear structural variations. Seven Crotalus chuvirus sequences were recovered, with RdRp identities to their closest relative (Guangdong red–banded snake chuvirus-like virus) ranging from 71.86 per cent to 74.20 per cent. ORF prediction showed that these chuviruses had similar genome structures to known viruses, although three strains identified in Crotalus polystictus might possess an additional ORF (Fig. S4). The genome sequences of segmented viruses were difficult to recover from sequencing data alone, and the genomes identified were different from those from known viruses. For example, three genome segments were identified for Micrurus arenavirus (Arenaviridae), which were annotated as RdRp, glycoprotein precursor, and nucleoprotein (Fig. 2). However, the closest viruses had only two segments: one with RdRp and a zinc-binding matrix protein and another with a glycoprotein precursor and a nucleoprotein (Fig. S3). Similarly, Tetragnatha versicolor phenuivirus (Phenuiviridae) was identified with three segments (L, M, and S segments). Although the Tetragnatha versicolor phenuivirus S segment contained one predicted ORF (Fig. 2), while the closest viruses have two ORFs, as the S segment obtained here was only partial, it is possible that the additional ORF existed but was not sequenced (Fig. S3).
Figure 2.
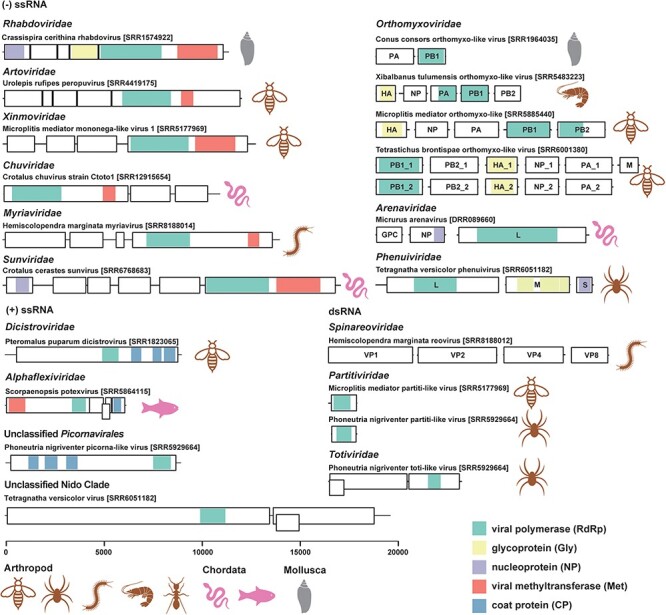
Genome structures of the novel RNA viruses found in the venom-related microenvironment. Genome structures of representative novel RNA viruses (<90 per cent amino acid identity in RdRp with known viruses) are displayed with boxes and coloured blocks. The black boxes represent predicted ORFs, while the coloured blocks indicate predicted functional domains, including viral polymerase (RdRp), glycoprotein (Gly), nucleoprotein (NP), viral methyltransferase (Met), and coat protein (CP), as the legend in bottom right. The host groups of the viruses are also labelled with icons.
3.4. Phylogenetic relationships of the venom-related RNA viruses
Phylogenetic analysis of the forty-five RNA viruses identified from the venom-related microenvironment revealed that they fell within a wide range of taxonomic groups, including at least twenty-two viral families or unclassified groups (Table 1 and Figs 3–5). The −ssRNA viruses were classified into eleven viral families: Rhabdoviridae, Artoviridae, Filoviridae, Xinmoviridae, Sunviridae, Myriaviridae, Chuviridae, Bornaviridae, Orthomyxoviridae, Arenaviridae, and Phenuiviridae (Fig. 3). Thirteen +ssRNA viruses clustered into at least eight viral families and unclassified groups (Fig. 4), while four dsRNA viruses fell into three families: Spinareoviridae, Partitiviridae, and Totiviridae (Fig. 5).
Figure 3.
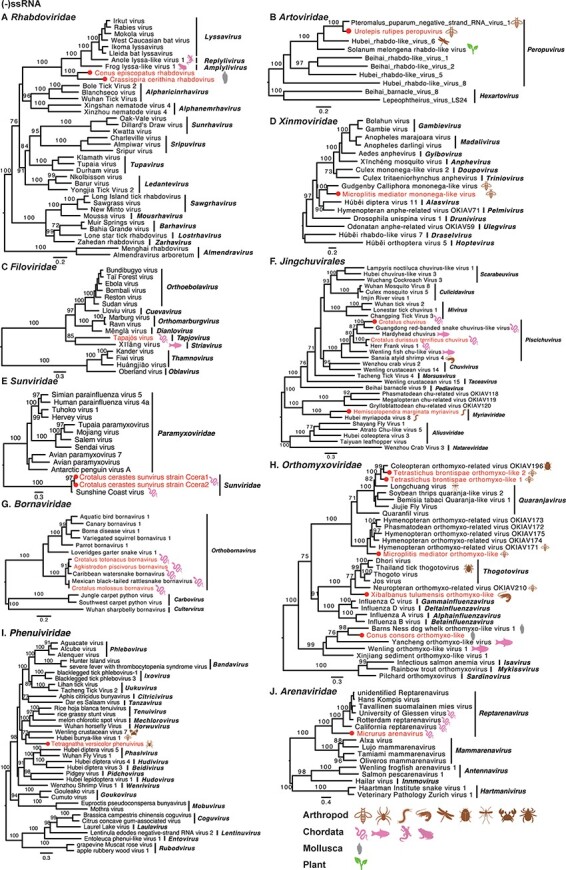
Phylogenetic trees of negative-sense single-stranded RNA viruses. Maximum likelihood phylogenetic trees were estimated using RdRp sequences. The virus names in red were found from venom gland in this study, with their hosts also labelled. The red circles represent viruses sharing <90 per cent amino acid identity with the most closely related viruses.
Figure 4.
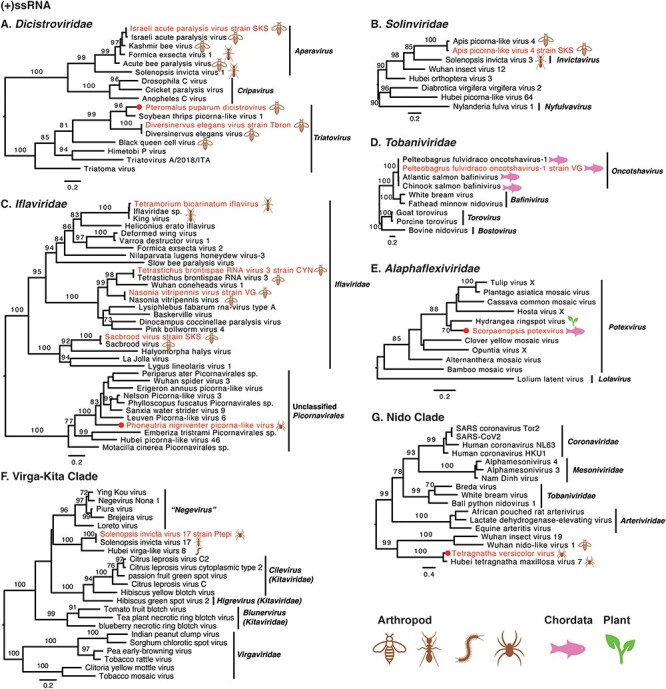
Phylogenetic trees of positive-sense single-stranded RNA viruses. Maximum likelihood phylogenetic trees were estimated using RdRp sequences. The virus names in red were found from venom gland in this study, with their hosts also labelled. The red circles represent viruses sharing <90 per cent amino acid identity with the most closely related viruses.
Figure 5.
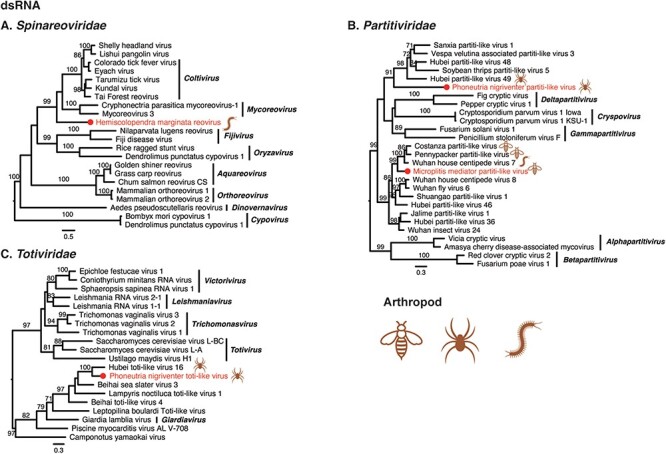
Phylogenetic trees of double-stranded RNA viruses. Maximum likelihood phylogenetic trees were estimated using RdRp sequences. The virus names in red were found from venom gland in this study, with their hosts also labelled. The red circles represent viruses sharing <90 per cent amino acid identity with the most closely related viruses.
3.4.1. Negative-sense single-stranded viruses
Nine −ssRNA viruses were discovered in arthropods, sixteen were discovered in Chordata, and three were found in Mollusca. Phylogenetic analyses revealed that the viruses from the families Artoviridae, Filoviridae, Sunviridae, Chuviridae, and Bornaviridae fell within known genera. The others, however, may represent new taxa (Fig. 3). Two rhabdoviruses (Crassispira cerithina rhabdovirus and Conus episcopatus rhabdovirus) were identified from the venom duct of Mollusca (Gastropoda), which fell basal to lyssaviruses in the Rhabdoviridae phylogeny (Fig. 3A). There are few reports of rhabdoviruses in molluscs, with only two rhabdoviruses recently discovered in freshwater mussels (Mollusca: Bivalvia) (Goldberg et al. 2023). Two strains of Microplitis mediator mononega-like virus were recovered from the venom gland of Microplitis mediator, belonging to the family Xinmoviridae, but were divergent from any established genera (Fig. 3D). Five orthomyxoviruses were found in the venom-related microenvironment, although some were divergent to known genera within the family Orthomyxoviridae (Fig. 3H).
3.4.2. Positive-sense single-stranded viruses
Eleven +ssRNA viruses were discovered in venom glands of arthropod animals, with the other two found in Chordata. Most +ssRNA viruses fell within known genera, although some required further classification (Fig. 4). For instance, Apis picorna–like virus 4 strain SKS was also found in the venom gland of Apis cerana collected from South Korea, exhibiting 96.04 per cent amino acid identity to Apis picorna–like virus 4 in RdRp. Phylogenetic analysis showed that Apis picorna–like virus 4 strain SKS fell into the family Solinviviridae, although none genus has yet been assigned (Fig. 4B). In addition, three viruses had no known assigned taxonomy: Phoneutria nigriventer picorna–like virus (Fig. 4C), Solenopsis invicta virus 17 strain Ptepi (Fig. 4F), and Tetragnatha versicolor virus (Fig. 4G).
3.4.3. Double-stranded viruses
Four dsRNA viruses were identified from the venom-related microenvironment, belonging to the families Spinareoviridae, Partitiviridae, and Totiviridae (Fig. 5). Hemiscolopendra marginata reovirus shared 27.66 per cent amino acid identity with Shelly headland virus in the RdRp. Phylogenetic analysis showed that the virus clustered with the viruses of the genera Mycoreovirus and Coltivirus (Fig. 5A). Microplitis mediator partiti-like virus and Phoneutria nigriventer partiti-like virus shared 64.11 per cent and 34.21 per cent amino acid identity with known viruses (RdRp). Phylogenetic analysis showed that the two viruses were grouped with partiti-like viruses found in arthropods, but were divergent to known genera (Fig. 5B). Phoneutria nigriventer toti-like virus exhibited 53.24 per cent amino acid identity in the RdRp region with known viruses, and phylogenetic analysis revealed that the Phoneutria nigriventer toti-like virus clustered with Hubei toti-like virus 16, both of which may belong to an additional genus in the family Totiviridae (Fig. 5C).
A number of observations can be made for the viruses newly identified here as a whole. First, Tetragnatha versicolor virus, together with Hubei tetragnatha maxillosa virus 7, might represent a previously unclassified virus family (Fig. 4G). Second, it is notable that many newly reported viruses were divergent to classified genera, such as Crassispira cerithina rhabdovirus and Conus episcopatus rhabdovirus (Fig. 3A), Tapajós virus (Horie 2021) (Fig. 3C), Microplitis mediator mononega-like virus (Fig. 3D), Conus consors orthomyxo-like virus (Fig. 3H), Tetragnatha versicolor phenuivirus (Fig. 3I), Solenopsis invicta virus 17 strain Ptepi (Fig. 4F), Hemiscolopendra marginata reovirus (Fig. 5A), Phoneutria nigriventer partiti-like virus and Microplitis mediator partiti-like virus (Fig. 5B), and Phoneutria nigriventer toti-like virus (Fig. 5C), indicative of substantial untapped genetic diversity. Finally, prior to this study, the families Sunviridae and Myriaviridae only had one member each. Herein, we report new members from the two families, thereby enriching their genetic diversity.
3.5. Are the newly identified RNA viruses venom-specific?
To assess whether the newly identified RNA viruses were venom-specific, we retrieved the RNA-seq data of other tissues available under the same SRA BioProjects. This comprised forty-three SRA files from different tissues of eight host species (Table S2). The RNA viruses identified from the venom-related microenvironment were then screened in the other tissues (Fig. 6). Eleven viruses identified from the venom-related microenvironment (Conus consors orthomyxo-like virus, Conus episcopatus rhabdovirus, Pteromalus puparum dicistrovirus, Tetramorium bicarinatum iflavirus, Solenopsis invicta virus 17, Nasonia vitripennis virus strain VG, Israeli acute paralysis virus, Sacbrood virus, Apis picorna–like virus 4, Microplitis mediator mononega-like virus, and Microplitis mediator partiti-like virus) were also detected in other tissues, with abundance ranging from 1.1 to 279,236.7, indicating that these viruses might not be venom-specific (Fig. 6). However, some viruses had clearly higher abundance in the venom-related microenvironment. For example, Conus episcopatus rhabdovirus was enriched in the venom duct (RPM = 19.0), compared with those in radular sac (RPM = 1.2) and salivary gland (RPM = 2.4) (Fig. 6A). Similarly, Tetramorium bicarinatum iflavirus was enriched in the venom gland (RPM = 213.2), while the whole body without the abdomen had a low abundance (RPM = 1.1) of this virus (Fig. 6A).
Figure 6.
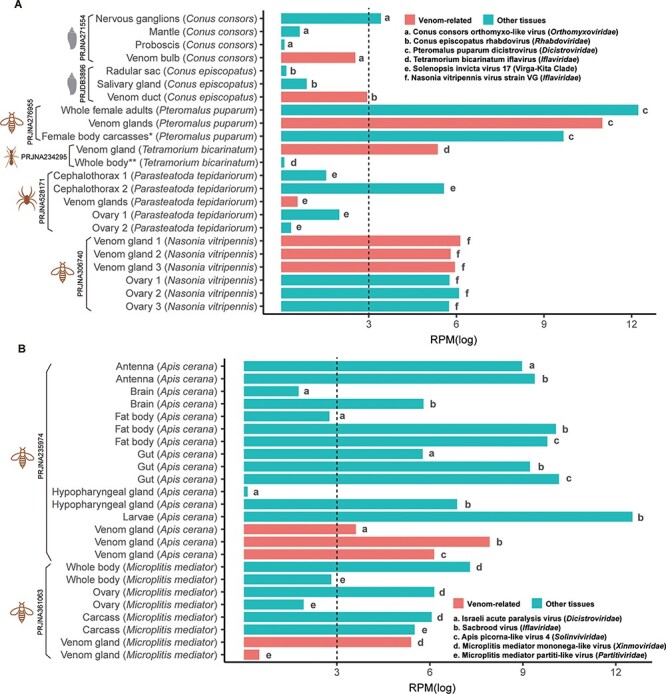
Comparison of the abundance of the RNA viruses from the venom-related microenvironment and other tissues. The SRA files of other tissues were retrieved from the same Bioproject as the venom data. The abundance of the RNA viruses from different tissues was calculated as RPM based on the non-rRNA reads. For visualization and comparison, the RPM values were normalized with log. (A) Only one RNA virus was identified from each host. (B) Multiple RNA viruses were identified from one host. * Female body carcasses without venom apparatus, ** Whole body without abdomen.
In addition, multiple viruses were identified in individual host species, although with variable abundance among tissues (Fig. 6B). Three viruses were identified from the venom glands of Apis cerana—Israeli acute paralysis virus (Dicistroviridae), Sacbrood virus (Iflaviridae), and Apis picorna–like virus 4 (Solinviviridae). Sacbrood virus was also detected in high abundance in larvae, while Israeli acute paralysis virus and Apis picorna–like virus 4 were absent. In addition, Apis picorna–like virus 4 was not detected in the brain and hypopharyngeal gland. Two viruses were found in the venom glands of Microplitis mediator, including Microplitis mediator mononega-like virus (Xinmoviridae) and Microplitis mediator partiti-like virus (Partitiviridae). Besides the venom gland, these two viruses were also found in the carcass, ovary, and whole body, although they were at the lowest abundance in the venom gland. It should be noted, however, that this heterogeneity might also reflect the pooling strategies employed in the sequencing projects, with each pool including multiple samples of the same host.
4. Discussion
There have been few studies on microorganisms in the venom-related microenvironment (Ul-Hasan et al. 2019), with little attention paid to virome diversity. We explored the RNA virome in the venom-related microenvironment using 474 public RNA sequencing datasets. From this, we identified forty-five viruses representing thirty-six viral species belonging to at least twenty-two viral families or groups. These viruses were identified from twenty-six venom-producing animals, representing the phyla Arthropoda, Chordata, and Mollusca, which were collected from at least seven countries. The diverse host and wide geographic range indicate that RNA viruses are commonly present in the venom-related microenvironment. Considering the limited sample size analysed here, we speculate that there are many more viruses to be discovered in the venom-related microenvironment.
Some of the newly described viruses might represent novel viral species and even genera. In particular, twenty-two viruses exhibited <90 per cent RdRp amino acid identity to their most closely related viruses, representing potentially novel viruses. Among these, sixteen viruses had <70 per cent of known viruses in RdRp, indicative of even more divergent taxa. For example, Crassispira cerithina rhabdovirus and Conus episcopatus rhabdovirus shared low identity with known viruses and formed a separated branch, representing a potential new genus.
The number of viral species in the venom-related microenvironment varied sharply across different animals. Arthropods harboured most viruses, involving thirteen viral families or groups, followed by chordates. In addition, viruses described in the families Dicitroviridae, Iflaviridae, and Partitiviridae were only identified from arthropods, while those from Sunviridae, Chuviridae, and Bornaviridae were mostly found in chordates.
It is notable that the abundance of the same virus species in the venom-related microenvironment of different hosts varied sharply and that most of the viruses described here had not previously been described in the venom-related microenvironment. For example, Crotalus cerastes sunvirus was found in venom gland samples of two Crotalus cerastes individuals, but their abundance varied approximately five-fold, from 287.1 to 1431.4. Furthermore, viruses found in the venom-related microenvironment were also detected in other tissues, but showed different abundance levels. For example, Conus episcopatus rhabdovirus was found in high abundance in the venom duct, but it was in much less abundance in the radular sac and salivary gland. Clearly, further investigation is required to understand the potential heterogeneous tissue tropism of these viruses.
In conclusion, we explored the diversity of RNA viruses in the venom-related microenvironment, identifying twenty-two novel viruses and fourteen known viruses. As such, our study sheds light on the hidden diversity of RNA viruses in the venom-related microenvironment, although we may have only documented the tip of the iceberg of virus diversity in this specific microenvironment.
Supplementary Material
Funding
This study was funded by Natural Science Foundation of Shandong Province (ZR2021QH317), Academic Promotion Programme of Shandong First Medical University (2019QL006), and Taishan Scholars Programme of Shandong Province (tsqn202211217).
Contributor Information
Jingkai Ji, School of Life Sciences, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 619 Changcheng Road, Taian 271000, China; Key Laboratory of Emerging Infectious Diseases in Universities of Shandong, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 2 Yingshengdonglu, Taian 271000, China.
Cixiu Li, Key Laboratory of Emerging Infectious Diseases in Universities of Shandong, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 2 Yingshengdonglu, Taian 271000, China; School of Clinical and Basic Medical Sciences, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 6699 Qingdao Road, Ji’nan 250117, China.
Tao Hu, Key Laboratory of Emerging Infectious Diseases in Universities of Shandong, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 2 Yingshengdonglu, Taian 271000, China.
Zhongshuai Tian, Key Laboratory of Emerging Infectious Diseases in Universities of Shandong, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 2 Yingshengdonglu, Taian 271000, China; School of Clinical and Basic Medical Sciences, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 6699 Qingdao Road, Ji’nan 250117, China.
Juan Li, Key Laboratory of Emerging Infectious Diseases in Universities of Shandong, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 2 Yingshengdonglu, Taian 271000, China; School of Clinical and Basic Medical Sciences, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 6699 Qingdao Road, Ji’nan 250117, China.
Lin Xu, Key Laboratory of Emerging Infectious Diseases in Universities of Shandong, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 2 Yingshengdonglu, Taian 271000, China; School of Public Health, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 6699 Qingdao Road, Ji’nan 250117, China.
Hong Zhou, Key Laboratory of Emerging Infectious Diseases in Universities of Shandong, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 2 Yingshengdonglu, Taian 271000, China; School of Clinical and Basic Medical Sciences, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 6699 Qingdao Road, Ji’nan 250117, China.
Edward C Holmes, Sydney Institute for Infectious Diseases, School of Medical Sciences, The University of Sydney, Sydney, New South Wales, Australia; School of Life & Environmental Sciences and School of Medical Sciences, The University of Sydney, Sydney, New South Wales, Australia.
Weifeng Shi, Key Laboratory of Emerging Infectious Diseases in Universities of Shandong, Shandong First Medical University & Shandong Academy of Medical Sciences, No. 2 Yingshengdonglu, Taian 271000, China; Shanghai Institute of Virology, Shanghai Jiao Tong University School of Medicine, No. 227 Chongqingnanlu, Shanghai 200025, China; Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, No. 197 Ruijinerlu, Shanghai 200025, China.
Data availability
The viral sequences obtained here have been submitted to GenBank with accession numbers BK064870–BK064938.
Supplementary data
Supplementary data is available at Virus Evolution Journal online.
Conflict of interest:
The authors declare no competing interests.
References
- Camacho C. et al. (2009) ‘BLAST+: Architecture and Applications’, BMC Bioinformatics, 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J. M., and Gabaldón T. (2009) ‘trimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D. et al. (2018) ‘The Global Virome Project’, Science, 359: 872–4. [DOI] [PubMed] [Google Scholar]
- Chen S. et al. (2018) ‘Fastp: An Ultra-fast All-in-One FASTQ Preprocessor’, Bioinformatics, 34: i884–i90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman K. A., and Burke G. R. (2020) ‘Genomic Analysis Reveals an Exogenous Viral Symbiont with Dual Functionality in Parasitoid Wasps and Their Hosts’, PLoS Pathog, 16: e1009069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Mata E. C. et al. (2017) ‘Antiviral Activity of Animal Venom Peptides and Related Compounds’, Journal of Venomous Animals and Toxins Including Tropical Diseases, 23: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmaeilishirazifard E. et al. (2022) ‘Bacterial Adaptation to Venom in Snakes and Arachnida’, Microbiology Spectrum, 10: e0240821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J. L., and Holmes E. C. (2017) ‘Predicting Virus Emergence amid Evolutionary Noise’, Open Biology, 7: 170189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg T. L. et al. (2023) ‘Plasticity, Paralogy, and Pseudogenization: Rhabdoviruses of Freshwater Mussels Elucidate Mechanisms of Viral Genome Diversification and the Evolution of the Finfish-Infecting Rhabdoviral Genera’, Journal of Virology. 97: e00196–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr M. G. et al. (2011) ‘Full-length Transcriptome Assembly from RNA-Seq Data without a Reference Genome’, Nature Biotechnology, 29: 644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W. T. et al. (2022) ‘Virome Characterization of Game Animals in China Reveals a Spectrum of Emerging Pathogens’, Cell, 185: 1117–29 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie M. (2021) ‘Identification of a Novel Filovirus in a Common Lancehead (Bothrops Atrox (Linnaeus, 1758))’, Journal of Veterinary Medical Science, 83: 1485–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S. et al. (2017) ‘ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates’, Nature Methods, 14: 587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., and Salzberg S. L. (2012) ‘Fast Gapped-Read Alignment with Bowtie 2ʹ’, Nature Methods, 9: 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) ‘The Sequence Alignment/Map Format and SAMtools’, Bioinformatics, 25: 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. et al. (2021) ‘Diamond: A Multi-Modal DIA Mass Spectrometry Data Processing Pipeline’, Bioinformatics, 37: 265–7. [DOI] [PubMed] [Google Scholar]
- Minh B. Q. et al. (2020) ‘IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era’, Molecular Biology and Evolution, 37: 1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro C. L. B. et al. (2002) ‘Isolation and Identification of Clostridium Perfringens in the Venom and Fangs of Loxosceles Intermedia (Brown Spider): Enhancement of the Dermonecrotic Lesion in Loxoscelism’, Toxicon, 40: 409–18. [DOI] [PubMed] [Google Scholar]
- Paez-Espino D. et al. (2016) ‘Uncovering Earth’s Virome’, Nature, 536: 425–30. [DOI] [PubMed] [Google Scholar]
- Robinson S. D. et al. (2017) ‘Venom Peptides as Therapeutics: Advances, Challenges and the Future of Venom-Peptide Discovery’, Expert Review of Proteomics, 14: 931–9. [DOI] [PubMed] [Google Scholar]
- Shi M. et al. (2016) ‘Redefining the Invertebrate RNA Virosphere’, Nature, 540: 539–43. [DOI] [PubMed] [Google Scholar]
- Shi M. et al. (2018) ‘The Evolutionary History of Vertebrate RNA Viruses’, Nature, 556: 197–202. [DOI] [PubMed] [Google Scholar]
- Ul-Hasan S. et al. (2019) ‘The Emerging Field of Venom-Microbiomics for Exploring Venom as a Microenvironment, and the Corresponding Initiative for Venom Associated Microbes and Parasites (Ivamp)’, Toxicon: X, 4: 100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Z., Shi M., and Holmes E. C. (2018) ‘Using Metagenomics to Characterize an Expanding Virosphere’, Cell, 172: 1168–72. [DOI] [PubMed] [Google Scholar]
- Zhu F. et al. (2018) ‘Symbiotic Polydnavirus and Venom Reveal Parasitoid to Its Hyperparasitoids’, Proceedings of the National Academy of Sciences of the United States of America, 115: 5205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The viral sequences obtained here have been submitted to GenBank with accession numbers BK064870–BK064938.