

Abstract
Cilia play a key role in the regulation of signaling pathways required for embryonic development, including proper formation of the neural tube, the precursor to the brain and spinal cord. Forward genetic screens were used to generate mouse lines that display neural tube defects (NTD) and secondary phenotypes useful in interrogating function. We describe here the L3P mutant line which displays phenotypes of disrupted Sonic Hedgehog signaling and affects the initiation of cilia formation. A point mutation was mapped in the L3P line to the gene Rsg1, which encodes a GTPase-like protein. The mutation lies within the GTP binding pocket and disrupts the highly conserved G1 domain. The mutant protein and other centrosomal and IFT proteins still localize appropriately to the basal body of cilia, suggesting that RSG1 GTPase activity is not required for basal body maturation but is needed for a downstream step in axonemal elongation.
Keywords: Cilia, Neural Tube Defects, RSG1 or CPLANE2, Mouse, Gene Mapping
Results and discussion
Forward genetic screens in mice have utilized ethyl-nitrosourea (ENU) to introduce mutations into the genome as an unbiased means to identify genes necessary for embryonic development (Liu & Eggenschwiler, 2014; Zohn et al., 2005). The ENU-generated mouse line L3P or 3Poly was selected due to disruption of neural tube closure in homozygous mutant embryos (exencephaly at ~30% penetrance) and other embryonic phenotypes of polydactyly (100% penetrant), eye defects (reduced or missing eye, 100%), edema, and embryos typically die by embryonic day E13 (Figure 1a,b). The mutation was generated on a C57Bl/6J background and the line has been out-crossed to C3H/HeJ and to 129/Sv1mJ for >10 generations, with no change in phenotype or penetrance.
Figure 1: L3P mutant mouse embryos display exencephaly, polydactyly, eye defects, and disrupted SHH signaling.

(a) E12.5 Rsg1L3P/L3P mutant embryos show exencephaly and eye defects. (b) E12.5 limbs stained with alcian blue to show cartilaginous skeleton. L3P mutant embryos display polydactyly. Asterisks highlight digit condensations. (c) Shh is expressed relatively normally in L3P limb buds at E10.5 or E11.5 by RNA in situ hybridization but the downstream target Ptch1 is decreased in Rsg1L3P/L3P :Ptch1+/LacZ limb buds as detected by Xgal staining. (d) Shh is present in the notochord but only at low levels in the floor plate of Rsg1L3P/L3P E10.5 embryo neural tubes compared to wildtype. This results in the expression of ISL1/2, LHX3, and PAX6 more ventrally than normal. Yellow bracket indicates domain of expression, blue bracket indicates ventral region where the protein is not present. Stainings for 1c and 1d were performed on a minimum on three embryos of each genotype, representative images are shown.
As polydactyly and neural tube defects (NTDs) are present in other mouse models with defective Sonic hedgehog (SHH) signaling (Harris & Juriloff, 2010; Murdoch & Copp, 2010), we assessed SHH signaling activity in the limb bud and neural tube of homozygous mutant embryos. In the limb, L3P mutants express Shh mRNA at about the same levels as wildtype littermates, although it appears more restricted to the proximal limb in E10.5 and E11.5 embryos (Figure 1c). Patched (Ptch1) is a target of SHH and we analyzed Ptch1 expression by crossing the Ptch1-lacZ transgenic reporter line (Goodrich et al., 1997) into the L3P line for a sensitive readout. L3P;Ptch1-LacZ mutants express lower levels of the LacZ reporter at E10.5 and E11.5.
In the ventral neural tube, graded SHH signals pattern and specify different neuronal progenitor subtypes (Wilson & Maden, 2005). Compared to wildtype littermates, SHH protein is expressed at lower levels in the floorplate in L3P mutants but is unaffected in the notochord (Figure 1d). The reduction in SHH expression in the floorplate predicts a “dorsalized” neural tube, borne out by the ventral expansion of markers of motor neuron progenitors (ISL1/2 and LHX3) and interneuron progenitors marked by PAX6, consistent with decreased SHH protein expression. Together these experiments indicate that SHH signaling is disrupted in L3P homozygous mutants.
SHH requires primary cilia for proper signaling. Moreover, mutations in proteins necessary for ciliogenesis result in polydactyly and NTDs (Huangfu et al., 2003), suggesting that the L3P mutation disrupts the formation or function of primary cilia. In both the neural tube and mouse embryo fibroblasts (MEFs), there is a dramatic decrease in primary cilia (Figure 2a–c). Immunostaining for cilia (ARL13b, which marks the ciliary membrane) in the lumen of E9.5 rostral spinal neural tube showed only trace amounts of cilia in L3P mutants. In MEFs induced to form cilia by a typical 48-hour serum-starvation, staining for cilia and for the basal bodies with γ tubulin (Figure 2b) showed the presence of cilia in approximately 64% of wildtype cells, but in only 0.7% of mutant cells (Figure 2c).
Figure 2: Rsg1L3P/L3P mutant cells lack primary cilia.
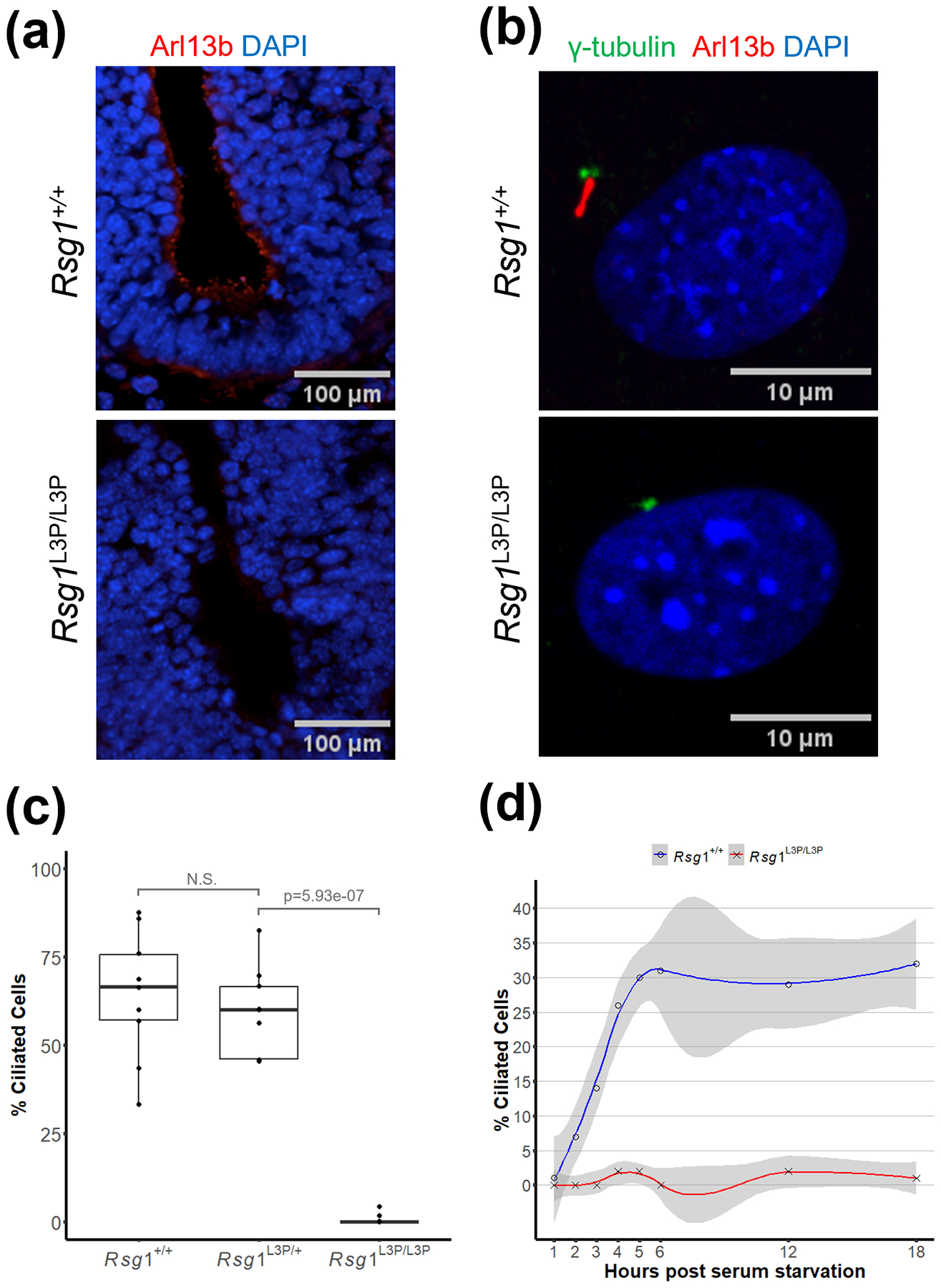
(a) Rsg1L3P/L3P mutant E9.5 embryos lack primary cilia in the lumen of the neural tube, when compared to the wildtype. The presence of primary cilia is visualized by antibody against Arl13b which localizes to the cilia axoneme. Ventral floor plate is at bottom of panel. Immunostaining was performed on a minimum on three embryos of each genotype, representative images are shown. (b) L3P mutant mouse embryonic fibroblasts (MEFs) also lack primary cilia when compared to wildtype. Gamma-tubulin visualizes the location of the basal body and cilia are highlighted by Arl13b. (c) Quantification of MEFs stained as in panel b. MEFs within a 170μm2 field of view were evaluated for the presence of cilia. There were on average 27 cells per image with total cell counts n= 248 for WT; n = 223 for L3P/+; n = 254 for L3P/L3P. (d) Serum starvation fails to induce ciliogenesis in L3P mutant MEFs. Two biological replicates of wildtype and mutant embryos were used for the timed series of ciliogenesis.
To determine if cilia are initiated in the L3P mutant but not maintained, we assessed ciliogenesis in MEFs analyzed at hourly intervals over an 18-hour period of timed serum starvation. For wildtype MEFs, primary cilia formation initiates within two hours of the onset of serum-starvation and by 18 hours, 32% of the cells have formed a primary cilia. However, for L3P mutant MEFs, primary cilia were not observed at any time period, indicating that primary cilia are not initiated (Figure 2d).
It is necessary for the basal body to migrate and associate with the plasma membrane to facilitate nucleation of the ciliary axoneme. Centrosome number and placement near the cell surface appears normal in L3P mutant cells. The mother centriole functions as the microtubule organizing center for the cilia (D’Angelo & Franco, 2009) and can be distinguished by the subdistal and distal appendages. Centrosomal proteins, including Ninein and CEP164, are associated with these appendages and siRNA knock-down of these proteins can result in the loss of primary cilia (Graser et al., 2007; Ishikawa et al., 2005). Immunofluorescence data shows that both proteins are present and localize to the centrioles in wildtype and L3P mutant MEFs upon serum-starvation (Figure 3a,b).
Figure 3: Rsg1L3P/L3P mutant cells retain ability to recruit centrosomal proteins.

Cultured MEFs isolated from wildtype and Rsg1L3PL3P embryos display co-localization of the basal body and (a) Ninein, (b) CEP164, and (c) IFT88. In all images the basal body is marked by γ-tubulin and the primary cilia is visualized by an antibody against Arl13b which localizes to the cilia axoneme. Immunostaining was performed on MEFs from a minimum of three embryos of each genotype and at least 100 cells per embryo visualized, representative images are shown. Scale bars in basal body zoom panels are 1μm. Scale bars in panels that depict the entire cell are 10μm.
A key aspect of ciliogenesis is recruitment of IFT-B proteins to the mother centrioles to promote axoneme assembly. For example, loss of IFT88, a member of the anterograde IFT trafficking group, inhibits production of cilia and is essential in ciliogenesis (Pazour et al., 2000). Immunofluorescence of IFT88 in wildtype and L3P mutant MEFs shows proper localization of IFT88 to the basal body following serum starvation (Figure 3c). Together, these data indicate that centrosome-associated proteins and intraflagellar trafficking proteins are recruited properly to the basal body in the L3P mutant MEFs, despite the lack of cilia production.
To identify the gene disrupted in L3P mutants, we performed classical meiotic mapping using SSLP markers that differentiate between the mutant C57BL/6 DNA and the wildtype 129S1/Sv1mJ DNA. This narrowed the region of the ENU-induced mutation to mouse chromosome 4 between 140.3Mb and 141.8Mb (1.5Mb interval). This region was then subsequently analyzed using mRNA sequencing data.
Sequencing identified the L3P causative ENU-induced mutation within the atypical small GTPase RSG1, which is associated with the CPLANE complex (Toriyama et al., 2016). Knockout or knockdown of individual members of the CPLANE complex, including RSG1, lead to defects in ciliogenesis (Adler & Wallingford, 2017). The L3P mutation is a missense mutation that causes a threonine-to-isoleucine substitution at residue 69 (T69I) within the G1 GTP binding domain (Figure 4a). Although GTPase activity has not been functionally shown for RSG1, the T69I mutation disrupts the conserved G1 GXXXXGK[T/S] motif (Feig, 1999). The threonine in this motif is predicted to play a key role in GTP and Mg2+ binding (Sprang, 1997). Moreover, overexpression of RSG1 T69N construct in Xenopus interferes with ciliogenesis (Gray et al., 2009).
Figure 4: Rsg1L3P mutation affects the GTP binding domain but does not affect RSG1 localization to the basal body.

(a) Protein data bank model of RSG1 (7Q3E) with the GTPase G1 domain shown in blue, bound GTP in red, and the T69I mutation in green. The V169D mutation shown in purple lies outside of the GTP binding domain. (b) Cultured MEFs isolated from wildtype and Rsg1L3P/L3P embryos display co-localization of the basal body and RSG1. (c) There is no statistical difference between colocalization of RSG1 and γ-tubulin in Rsg1L3P/L3P MEFs compared to wildtype. Each datapoint represents quantification from a field of view of cells as shown in 4b with an average of 22 cells per image.
The Anderson lab identified an ENU-induced Rsg1 V169D allele, which lies outside of the G1 GTP binding domain (Figure 4a), and they produced a CRISPR-generated allele that targeted T69 and created a 2 base insertion to create a null allele (Agbu et al., 2018). All three mouse mutant alleles show a similar phenotype of polydactyly, edema and disruption in neural tube patterning. However, they did not report a neural tube defect or eye defects and the loss of cilia appears to be more pronounced in our T69I data of the neural tube and MEFs, relative to their comparable data.
RSG1 localizes to the mother centriole and the transition zone of the primary cilium (Gray et al., 2009). Our T69I mutant protein localizes normally to the basal body based on immunofluorescent images of RSG1 and gamma-tubulin, compared to wildtype MEFs (Figure 4b,c). As the T69I mutation should strongly attenuate predicted GTPase activity, this calls into question a conclusion from the earlier study that GTPase activity is required for RSG1 localization and function (Agbu et al., 2018). Those experiments were performed by transfection of a RSG1 T69N construct into RSG1 V169D mutant MEFs. It is possible that the presence of normal amounts of the hypomorphic V169D protein which localizes to the ciliary base (Agbu et al., 2018) obscured recruitment of the transfected protein. Instead, our data is consistent with that of Gray et al. (Gray et al., 2009) in which a RSG1 mutant construct affecting the same residue showed that RSG1 is still localized, albeit more diffusely to the basal bodies of cilia.
Together our data suggests that RSG1 GTPase activity is not needed for recruitment of RSG1 or other centrosomal or IFT trafficking proteins to the basal body but is needed for a downstream step of axonemal elongation. In summary, we have identified a T69I mutation in the Rsg1/Cplane2 gene that results in a strong loss of function allele and ciliopathy-like defects in mouse embryos, including cranial neural tube defect, polydactyly, eye defects and mid-gestation lethality.
Methods
Mice
Mouse ENU mutagenesis and positional cloning procedures have been previously described (Liu & Eggenschwiler, 2014; Zohn et al., 2005). Prior to gene identification, 3Poly mutants were genotyped using SSLP markers D4dmm35 and D4MIT48. After identification of the A to T mutation at position chr4:141214346 mm10 (creating a threonine-to-isoleucine substitution at residue 69), the presence of the mutation in RSG1 was confirmed with sequencing using primer CAT CTC TGT GTA TCC GGG GC. Ptch1-lacZ transgenic mice were genotyped for lacZ by PCR on genomic DNA using primers CCG AAC CAT CCG CTG TGG TAC and CAT CCA CGC GCG CGT ACA TC. The use of the animals in this research has been approved by the IACUC at University of Colorado.
Whole Mount X-gal Staining
Embryos were dissected and fixed in 4% paraformaldehyde for 1 hr at 4°C. Subsequently, embryos were washed three times for 15 min in Wash Buffer (PBT plus 0.1% Tween-20, 2mM MgCl2, 0.02% NP-40, 0.01% sodium deoxycholate). Embryos were stained overnight in X-gal Staining Solution (Wash Buffer plus 1mg/mL X-gal, 5mM potassium ferrocyanide and 5mM potassium ferricyanide).
Whole Mount RNA In Situ Hybridization
Embryos were fixed in 4% paraformaldehyde then dehydrated in a methanol series. Embryos were rehydrated, then subjected to brief Proteinase K treatment. Embryos were moved into Hybridization Buffer (50% formamide, 1.3X SSC, 5mM EDTA, 50mg/mL yeast tRNA, 0.2% Tween-20, 0.5% CHAPS, 100mg/mL heparin) where they were exposed to DIG-labeled antisense RNAs against Shh (Epstein et al., 1999) at 65°C overnight. Embryos were then treated with RNaseA, and exposed to anti-DIG antibody overnight at 4 °C. Embryos were washed for a full day, then treated with BM-Purple to expose signal. All embryo marker experiments were performed on a minimum on three embryos of each genotype and the results were consistent, so representative images are shown.
Staining of Mouse Skeletons Using Alcian Blue
E12.5 embryos were dissected and fixed in 4% paraformaldehyde overnight. Embryos were then stained overnight in 0.015% alcian blue 8 GX (Sigma) in acid alcohol (75% ethanol/25% glacial acetic acid by volume). Embryos were rinsed one time in acid alcohol for 15 minutes and then placed in fresh acid alcohol for 1–2 hours. Embryos were washed two times in 100% ethanol for two changes of 1 hour each and stored in 100% ethanol prior to imaging.
Tissue culture
Mouse embryonic fibroblasts (MEFs) were isolated from E12.5 embryos. Manually dissociated cells were plated in 6-well tissue culture dishes in DMEM supplemented with 10% fetal bovine serum. After two passages 2.5 ×105 cells were plated onto a coverslip and grown for 24 hours to allow cells to re-adhere. Media was removed and cells were washed briefly with PBS before adding serum-starvation media (0.5% FBS DMEM). Cells were serum-starved for 48 hours to induce ciliogenesis, fixed with cold 100% MeOH, followed by 4%PFA. For the 18-hour timed ciliogenesis experiment, MEFs and coverslips were prepared as described above for two wildtype and two mutant biological replicates. DMEM + 0.5% FBS + 1% pen/strep was added to initiate serum-starvation. After one hour and every hour over an 18-hour period, coverslips were removed and stained as described below for basal bodies/centrosomes (γ tubulin) and cilia (acetylated α-tubulin).
Immunofluorescence
Coverslips containing MEFs were fixed in 100% methanol for 10 minutes at −20°C followed by three washes with 1XPBS. Coverslips were blocked with blocking buffer (0.3% Triton-1XPBS with 1% Normal Goat Serum (NGS)). Blocking buffer was removed and primary antibody added (diluted in blocking buffer) and incubated overnight at 4°C in a dark, humidified chamber. Three washes with 1XPBS were followed by addition of the secondary antibody (Alexa Fluor 488 or 594, Life Technologies) for one hour at room temperature. Coverslips were washed three times with 1XPBS and stained with Hoechst or DAPI (diluted 1:1000 in 1XPBS) for 15 minutes. Coverslips were washed three times with 1XPBS and coverslipped with ProLong Gold Antifade Mountant (ThermoFisher P36930). Primary cilia were visualized using antibodies against ARL13b (diluted 1:500; Dr. Tamara Caspary, Emory University) and acetylated- tubulin (diluted 1:1000; Sigma). Basal bodies/centrosomes were visualized using tubulin (diluted 1:2000, Abcam). Additional antibodies used for staining of ciliary components included Cep164 (1:400 dilution, Dr. Erich Nigg, University of Basel), RSG1 (Novus NBP188322), Ninein (1:400 dilution, Dr. James Sillibourne, Institut Curie), IFT88 (1:400 dilution, Dr. Bradley Yoder, University of Alabama at Birmingham). All embryo immunofluorescence experiments were performed on a minimum on three embryos of each genotype and the results were consistent, so representative images are shown.
Immunohistochemistry
Cilia in the neural tube lumen of the E9.5 wildtype and Rsg1L3P/L3P embryos were stained using an antibody against ARL13b (see above). Neural tube patterning was visualized in frozen transverse sections of the neural tube by immunostaining using antibodies against SSH (Sigma MABD175), ISL1/2 (Sigma AB4326), and PAX6 (Sigma AB2237). Tissue samples were fixed in 4% paraformaldehyde overnight, washed with 1XPBS, sunk in 30% Sucrose/1XPBS solution and transferred to OCT mounting media through a serial gradient (1 hour in each solution: 75% Sucrose solution: 25% OCT, 50% Sucrose solution: 50% OCT, 25% Sucrose solution: 75% OCT, 100% OCT), embedded in OCT and stored at −80oC until sectioning. Samples were cryosectioned to produce 10 micron sections. Sections were incubated for one hour at 37°C and then washed one time with 1XPBS. Addition of primary and secondary antibodies followed by DAPI staining proceeded as described above. Visualization was done at 10x magnification using confocal microscopy (Zeiss). All secondaries were diluted 1:500. Confocal microscopy (Zeiss LSM510 Meta) and magnification of 63X was used to visualize all basal body proteins. Mouse embryonic fibroblasts and cilia were visualized using Nikon Ti Eclipse spinning disk at magnification 20x and 40x.
Bioinformatics
The L3P mutation was identified from mRNA sequencing carried out on RNA collected from WT and L3P/L3P E12.5 MEFs. Library preparation was carried out using the TECAN Universal Plus mRNA-Seq kit. Sequencing was carried out using the Illumina Novaseq platform at a read length of 150bp. Downstream quality control, trimming, sorting, and alignment was carried out using the nf-core RNA-seq v3.1 nextflow pipeline. FASTA files were generated from the alignment for both WT and L3P samples. FASTA files for both WT and L3P samples were cross referenced against assemblies for C57Bl/6J and 129/Sv1mJ backgrounds. Varscan was then used to count and map single nucleotide polymorphism (SNP) locations. Mapped SNPs confirmed the region of chromosome 4 identified by meiotic mapping and which was homozygous for the C57Bl/6J background in L3P mutants. Within the mapped 1.5mB region, there were 20 SNPs that did not match either genetic background, but only one SNP was within a coding region and this resulted in a missense mutation in the Rsg1 gene. This SNP was confirmed by Sanger sequencing and was used for subsequent genotyping.
Statistical analysis
Data points in box plots each represent a field of view containing an average of 22 cells for colocalization studies, and 27 cells for ciliated cell counts. A minimum of 200 cells were visualized in any individual condition. Colocalization of RSG1 and gamma-tubulin was evaluated using the ImageJ plugin JaCoP which generated Pearson’s coefficients between the gamma tubulin and RSG1 channels. Cilia were counted manually using ImageJ by dividing the number cells with centrioles and cilia, by the number of cells with centrioles. For all other experiments where pairwise comparisons were performed, a student’s T-test was used to determine significance.
Acknowledgments
The authors thank Lori Bulwith for assistance with mouse husbandry and Jianfu Chen for Figure 3 microscopy. We thank Dr. Matt Scott for the Ptch1-lacZ mouse. This work was supported by NIH awards to LN (1P01HD104436, 1R01HD081562, U01 HD043478).
Footnotes
Conflict of interest disclosure: The authors declare no competing interests.
Data availability statement:
All data are available in the main text and figures.
References
- Adler PN, & Wallingford JB (2017). From Planar Cell Polarity to Ciliogenesis and Back: The Curious Tale of the PPE and CPLANE proteins. Trends in Cell Biology, 27(5), 379–390. 10.1016/j.tcb.2016.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agbu SO, Liang Y, Liu A, & Anderson KV (2018). The small GTPase RSG1 controls a final step in primary cilia initiation. The Journal of Cell Biology, 217(1), 413–427. 10.1083/jcb.201604048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo A, & Franco B (2009). The dynamic cilium in human diseases. PathoGenetics, 2, 3. 10.1186/1755-8417-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig LA (1999). Tools of the trade: Use of dominant-inhibitory mutants of Ras-family GTPases. Nature Cell Biology, 1(2), E25–E27. 10.1038/10018 [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenković L, Higgins KM, & Scott MP (1997). Altered Neural Cell Fates and Medulloblastoma in Mouse patched Mutants. Science, 277(5329), 1109–1113. 10.1126/science.277.5329.1109 [DOI] [PubMed] [Google Scholar]
- Graser S, Stierhof Y-D, Lavoie SB, Gassner OS, Lamla S, Le Clech M, & Nigg EA (2007). Cep164, a novel centriole appendage protein required for primary cilium formation. The Journal of Cell Biology, 179(2), 321–330. 10.1083/jcb.200707181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray RS, Abitua PB, Wlodarczyk BJ, Szabo-Rogers HL, Blanchard O, Lee I, Weiss GS, Liu KJ, Marcotte EM, Wallingford JB, & Finnell RH (2009). The planar cell polarity effector Fuz is essential for targeted membrane trafficking, ciliogenesis, and mouse embryonic development. Nature Cell Biology, 11(10), 1225–1232. 10.1038/ncb1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MJ, & Juriloff DM (2010). An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Research. Part A, Clinical and Molecular Teratology, 88(8), 653–669. 10.1002/bdra.20676 [DOI] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, & Anderson KV (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature, 426(6962), 83–87. 10.1038/nature02061 [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Kubo A, Tsukita S, & Tsukita S (2005). Odf2-deficient mother centrioles lack distal/subdistal appendages and the ability to generate primary cilia. Nature Cell Biology, 7(5), 517–524. 10.1038/ncb1251 [DOI] [PubMed] [Google Scholar]
- Liu A, & Eggenschwiler J (2014). Identifying Essential Genes in Mouse Development via an ENU-Based Forward Genetic Approach. In Lewandoski M (Ed.), Mouse Molecular Embryology: Methods and Protocols (pp. 95–118). Springer US. 10.1007/978-1-60327-292-6_7 [DOI] [PubMed] [Google Scholar]
- Murdoch JN, & Copp AJ (2010). The relationship between Sonic hedgehog signalling, cilia and neural tube defects. Birth Defects Research. Part A, Clinical and Molecular Teratology, 88(8), 633–652. 10.1002/bdra.20686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, & Cole DG (2000). Chlamydomonas IFT88 and Its Mouse Homologue, Polycystic Kidney Disease Gene Tg737, Are Required for Assembly of Cilia and Flagella. The Journal of Cell Biology, 151(3), 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprang SR (1997). G PROTEIN MECHANISMS: Insights from Structural Analysis. Annual Review of Biochemistry, 66(1), 639–678. 10.1146/annurev.biochem.66.1.639 [DOI] [PubMed] [Google Scholar]
- Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel A-L, Tabler JM, Drew K, Kelley MR, Kim S, Park TJ, Braun D, Pierquin G, Biver A, Wagner K, Malfroot A, Panigrahi I, Franco B, Al-lami HA, … Wallingford JB (2016). The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nature Genetics, 48(6), 648–656. 10.1038/ng.3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson L, & Maden M (2005). The mechanisms of dorsoventral patterning in the vertebrate neural tube. Developmental Biology, 282(1), 1–13. 10.1016/j.ydbio.2005.02.027 [DOI] [PubMed] [Google Scholar]
- Zohn IE, Anderson KV, & Niswander L (2005). Using genomewide mutagenesis screens to identify the genes required for neural tube closure in the mouse. Birth Defects Research Part A: Clinical and Molecular Teratology, 73(9), 583–590. 10.1002/bdra.20164 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available in the main text and figures.