

Summary
Recent development of methods to discover and engineer therapeutic TCRs or antibody mimics of TCRs, and to understand their immunology and pharmacology, lag two decades behind therapeutic antibodies. Yet we have every expectation that TCR-based agents will be similarly important contributors to the treatment of a variety of medical conditions, especially cancers. TCR engineered cells, soluble TCRs and their derivatives, TCR mimic antibodies and TCR-based CAR T-cells promise the possibility of highly specific drugs that can expand the scope of immunologic agents to recognize intracellular targets, including mutated proteins and undruggable transcription factors, not accessible by traditional antibodies. Hurdles exist regarding discovery, specificity, pharmacokinetics and best modality of use that will need to be overcome before the full potential of TCR-based agents is achieved. HLA restriction may limit each agent to patient subpopulations and off-target reactivities remain important barriers to widespread development and use of these new agents. In this review we discuss the unique opportunities for these new classes of drugs, describe their unique antigenic targets, compare them to traditional antibody therapeutics and CAR T-cells, and review the various obstacles that must be overcome before full application of these drugs can be realized.
Keywords: TCR gene therapy, Soluble TCR, TCR mimic mAb, HLA restriction, Off-target toxicity, CAR-T-cells
1. Introduction
Methods to produce monoclonal antibodies (mAbs) were described first in the late 1970’s, but the first mAbs for the treatment of cancer were not approved until two decades later. Currently, after 20 more years, mAbs represent an increasingly dominant part of the drug armamentarium for cancers, autoimmune disease, inflammatory processes, infections, and neurologic disorders, among others. Although antibodies and T-cell receptors (TCR) represent the two dominant arms of the adaptive immune response in vertebrates, development of methods to discover and engineer therapeutic TCRs, and to understand their functions and pharmacology, lag two decades behind mAbs. In spite of this, we have every expectation that TCR-based agents will be similarly important future contributors to the treatment of a variety of medical conditions, especially cancers. As with antibodies, there exist now hurdles regarding discovery, specificity, pharmacokinetics and best modality of use that will need to be overcome before the full potential of TCR-based agents is realized.
In addition, the recent success of adoptive cellular immunotherapy with chimeric antigen receptor (CAR)-directed T-cells directed to hematologic malignancies has prompted interest in finding similar approaches for treating solid tumors. CAR molecules, which typically are restricted to cell surface protein targets, have largely been based on the IgG, but TCR-based agents, directed to peptide-MHC targets, have seen increased interest as a strategy to more specifically target solid tumors, in which there is a paucity of tumor-selective cell surface proteins available. Regardless of receptor format, upon binding to cognate tumor antigens, intracellular domains of CAR and TCR can be designed to recruit similar molecules for activating host effector cells for killing.
Why focus on TCR-based agents?
Currently, there are no FDA-approved mAbs that bind to surface antigens exclusive to cancer cells; however, conventional α TCRs can recognize numerous peptide-MHC (pMHC) antigens with exquisite sensitivity and variable specificity, including pMHC on cancer cells in the form of tumor-associated antigens (TAA) and tumor-exclusive neoantigens1. Among the first TAA found to be recognized by TCRs were those derived from MART12, gp1003 MAGE-A4, and Tyrosinase5 all of which were first found to be recognized by either peripheral T-cells or tumor-infiltrating lymphocytes (TIL) from resected melanoma lesions. Similar to TAA, neoantigens produced by somatic mutations exclusive to cancer cells are becoming increasingly appreciated as tumor rejection antigens that can be targeted by TCR therapy. TIL present in several resected solid tumors recognize patient-specific neoantigens6,7. When such TIL are expanded ex vivo and reinfused, they can induce durable regressions in metastatic solid tumors,8,9 thus demonstrating the therapeutic potential of neoantigen targeting. Moreover, various neoantigen qualities, such as clonality, MHC binding properties, and immunogenicity, have been shown to predict response to immune checkpoint blockade10. Because T-cells generated in vivo in patients are endowed with specificity for tumor antigens, there has been significant interest in clinical development of this class of TCR-based agents for cancer immunotherapy.
In this review, we will discuss the principles and uses of TCR and TCR mimic agents, illustrate some of the critical issues that are limiting the development of these agents, provide possible solutions to the problems, and contrast and compare TCRs to monoclonal antibodies (mAbs) and to TCR mimic agents. Although TCR shares structural similarities to mAb, specific features differ markedly between mAb and TCR, rendering TCR more difficult to design as soluble drugs (Table 1). As a consequence, while mAbs have been used in various platforms successfully, ranging from fragments to conjugates to CAR T-cells; TCR’s have had a more limited repertoire of platforms to date. In contrast, when engineered into cells, TCR more easily co-opt T-cell functionality that mAb cannot, requiring the latter to be more radically engineered to be effective drugs. Finally, mAbs are now being discovered and described that share some functions and specificity of the TCR (known as TCR mimic mAbs). Such agents may solve some of the pharmacologic obstacles encountered with TCRs themselves and add considerable scope to mAbs, but may also create unexpected new problems. These issues also will be discussed below.
Table 1.
Hurdles to TCR-based Therapeutics.
| Issues for each class of agent | Possible Solutions Available | Citations |
|---|---|---|
| TCR engineered cells. | ||
| Patient specific cells | Use off the shelf allogenic cells | |
| TCR mispairing | Use cells with CRISPR deleted native TCR; knob and hole paired chains; mouse chains; framework engineering, domain swapped TCRs, single chain TCRs. | 101–106 |
| Immunosuppressive TME | Use of cytokine or chemokine armored cells, deletion of checkpoint molecules, ex vivo selection of optimal subsets or conditioning with cytokines | 241, 254 ,255, 256,280,281. 266, 267,300 |
| Poor Penetration into tumor | Use of cytokine or chemokine armored cells, ex vivo selection of optimal subsets or conditioning with cytokines | 241, 254 ,255, 256,280,281. 266, 267,300 |
| Lack of Persistence | Use of cytokine or chemokine armored cells, overexpression of transcription factors that promote persistence or protect against exhaustion, ex vivo selection of optimal subsets or conditioning with cytokines | 241, 254 ,255, 256,280,281. 266, 267,300,301 |
| Manufacturing logistics | Automated techniques; off the shelf cells including HLA-matching banked cells and differentiation from iPSC | 162,163,168 161 161 165 |
| GVHD if allogeneic cells | Delete native TCR from cells, cell subset selection such as EBV/CMV sensitization, CD137- or CD8-depletion | 176,177. 114.302 |
| Graft rejection, if allogeneic cells | Delete HLA, B2M, & other presentation machinery. Introduce HLA-E or IdeS into cells. | 161–166. |
| Soluble TCR-based agents. | ||
| Low affinity | Affinity maturation | 59. |
| Difficult protein engineering | New technology is advancing | |
| Both soluble and cell-based agents. | ||
| Lack of broad “public” neoantigens | Extensive in silico searches. Empiric MS-based searches. | 52 53. 54, 55,56 41,57 |
| Antigenic heterogeneity | Use of multiple agents; Use of essential (driver oncogene) targets | |
| Escape by HLA loss or downregulation | Pharmacologic interventions | 219–221, 222, 222–224. |
| Escape by antigen presentation loss | Pharmacologic interventions | 219–221, 222, 222–224. |
| Escape by epitope mutation | Use of driver mutations or essential targets | |
| Off-target toxicities (cross-reactivities) | Better in silico screening and empiric screening of TCR | 52 53. 54, 55,56 41,57 |
| On-target, off-tumor toxicities | Better proteomic screening via healthy tissue HLA ligand databases and empiric MS search | 11. 58, |
2. Structural Issues
Traditional antibodies, TCRm antibodies, and TCRs bear structural similarities, belonging to the immunoglobulin superfamily of proteins, but have distinct features that influence their pharmacology and potential applications and platforms. Although TCRm and IgG are largely identical in structure and pharmacologic characteristics, they differ vastly in potential applications and specificity in that TCRm recognize a far larger universe of antigens, including intracellular targets, but the epitopes are HLA restricted. In contrast, TCR-based molecules are similar in recognition properties to TCRm, though of far lower affinity typically, but are much more limited currently in their platform applications because the native TCR structure is usually membrane associated (Table 2). An important difference between the traditional IgG and TCR-like agents is in their specificity. IgG recognize three-dimensional shapes of proteins, carbohydrates, and haptens, among other molecules, which can often confer near-perfect specificity for the target antigen. TCR-based agents recognize a linear peptide sequence buried in the groove of MHC molecules, as well as parts of the MHC sequence adjacent to the peptide. Therefore, the surface area of the recognized epitope bound by the TCR agent is limited, and the possibility of cross-reactive epitopes, both from recognition of the MHC and from sequence similarities to other peptides in the proteome that may be presented, is significant11,12. This distinction makes the discovery and development of specific TCR-based agents more complicated. In contrast, by selecting TCR directly from humans, such as from TILs, many cross-reactivities may be avoided because the thymus filters out most cross-reactive TCR during T-cell development 13.
Table 2.
Typical Features of Immunoglobulin Super Family Therapeutic Agents
| Feature | IgG Antibody | TCR mimic | TCR |
|---|---|---|---|
| Isoforms | Multiple | Multiple | Alpha/beta or gamma/delta |
| Structure | Homodimer | Homodimer | Heterodimer |
| Mass (Daltons) | 150,000 | 150,000 | 40,000/ 80,000 |
| Affinity (typical; native) | High: 0.1–10nM | High: 0.1–10nM | Low: 0.1–10uM |
| Target antigens | All accessible molecules | Peptide/MHC | Peptide/MHC; Lipid, peptide, metabolite/CD1, MR1, HLA-E; Non peptidic-phospho-antigen/BNT3A1 |
| HLA restriction | No | Yes | Yes and No |
| Soluble forms (native) | Yes | Yes | No |
| Membrane bound (native) | No | No | Yes |
| Typical Platforms: | |||
| ● Native | Yes | Yes | Yes |
| ● Fc modified | Yes | Yes | N/A |
| ● Truncated forms | Various | Various | Yes |
| ● Bispecific forms | Yes | Yes | Yes |
| ● ADC* | Yes | Yes | No |
| ● RIC* | Yes | Yes | No |
| ● CAR* or T-cell | Yes | Yes | Yes |
| Half-life (soluble forms) | Long (weeks) | Long (weeks) | Short (hours) |
| Specificity | High | Variable | Variable |
| Marketed | Multiple | No | One (as of 2022) |
| Current clinical Indications | Diverse & many | Cancer | Cancer |
| Discovery/development | Simple | Complex | Complex |
3. Immunologic hurdles in selecting appropriate antigenic targets for TCR based agents
HLA restriction.
CD8+ T-cells detect and eliminate abnormal cells by recognizing peptide fragments of processed proteins that are presented by human leukocyte antigen class I (HLA I). HLA is highly polymorphic, with each variant (allotype) characterized by a different peptide binding groove, resulting in allotype-restricted peptide binding motifs. In humans, three classical HLA class I genes (HLA-A, HLA-B, and HLA-C) are expressed in nucleated cells with up to six different allotypes per individual. The classical antigen presentation pathway for HLA class I presented peptides involves the proteolytic cleavage of proteins in the proteasome followed by the peptide fragment translocation to the ER by TAP; after further trimming, individual peptides get loaded onto HLA-class I molecules and transported to the cell surface for presentation to CD8 T-cells. However, alternate peptide presenting mechanisms exist, as can be seen in humans lacking TAP, that are still able to present peptides on cell surface, though with much lower abundance.
Characteristics of peptide antigens.
The peptides presented on HLA class I can be foreign (e.g virus, bacteria) or self. A recent study found that all proteins can potentially give rise to presented peptides 14. However, presented peptides are often skewed towards proteins with a high abundance and high turnover rates 15.
Cancer-associated aberrant protein expression includes products of mutated oncogenes, passenger mutated genes, tumor suppressor genes, oncofetal genes, aberrantly or overexpressed genes, abnormal glycoproteins, and post-translationally modified proteins. In theory, these aberrant proteins or protein fragments can produce peptide fragments that can be presented on HLA class I where they can be detected by CD8+ T-cells. Hereby, a distinction is made between self-antigens and neo-antigens. Self-antigens derive from proteins that can also be found on other tissues, but are overexpressed or re-expressed in cancerous cells. Prominent examples are lineage-specific tumor-associated antigens (TAA) such as MART-1 and CEA, cancer germline antigens (CGA), including NY-ESO-1, which is usually exclusively expressed in testicular germ cells, but is re-expressed in various cancer cells due to genomic instability (for example, in 40% of epithelial ovarian cancer, 75% of synovial cell sarcoma, 25% of melanoma), MAGE, or PRAME. Neo-antigens are peptides that are exclusively found on cancer cells (tumor-specific antigens) and result from nonsynonymous somatic mutations, frameshift mutations, and sometimes from post-translational modifications such as phosphorylation or glycosylation. Due to the enormous heterogeneity between individuals in their allotypes and the resulting heterogeneity of the immunopeptidome between individuals, most neoantigens identified are patient-specific (that is, “private”). Targeting private neoantigens requires individual customization of TCR posing significant logistical and financial challenges. However, gain of function mutations in a cancer driver gene critical for tumor survival that is shared among patients with particular HLA allotype are called “public” neoantigens. Such targets might be used in broader populations of patients16. Recent studies have shown the successful identification of a public neoantigen derived from a PIK3CA mutation as well as the identification of four different TCRs that are able to detect this neoantigen in an HLA-A03 context which is one of the most prevalent HLA allotypes 17.
Using TCR T-cells for target identification.
T-cell-based immunotherapy is partly based on the assumption that T-cells found endogenously in the host can specifically detect and eliminate cancer cells. While the endogenous cytotoxic T-cell response is often insufficient to protect against tumor development due to the immunosuppressive tumor microenvironment, such TCR may be used to identify the target epitope or create new more potent specific therapeutic agents. Isolated and sequenced TCRs used to produce genetically engineered T-cells infused into the same patient from which they were isolated have shown promising tumor control in clinical trials7,18,19. Rapidly identifying the target peptide as well as the TCR sequence able to react with the target remains one of the major challenges in TCR immunotherapy.
Identifying the TCR alpha and beta chain.
Upon target recognition, T-cells with a TCR able to recognize their target undergo clonal expansion. This expansion can be used to identify clonally expanded TCR sequences that are likely to be specific for antigens presented in a given disease state using single-cell or bulk RNA sequencing. Other high throughput methods for TCR identification include phage, yeast, and T-cell display libraries. A stimulation-induced functional TCR sequencing platform has been described in which naive T-cells from healthy donors are subjected to stimulation with autologous DC electroporated with a mutant or the respective wild-type driver oncogene17. Using qPCR to detect INFgamma, wells that are preferentially reactive towards the mutant antigen are further stimulated and subjected to sequencing in order to identify the TCR alpha and beta chain sequences.
To optimize the activity of genetically engineered T-cells, TCRs are often affinity enhanced by introducing mutations into the CDRs, which make direct contact with the pMHC complex. Since the immune system preferentially deletes high-affinity TCRs (Kd< 6uM) 20 in favor of low-affinity TCRs to prevent autoimmune reactions and to maintain highly promiscuous T-cells that are reactive against a wide range of antigens, affinity enhancement may lead to T-cells with increased off-target reactivity to structurally similar self-peptides, which can lead to severe or lethal toxicity in patients21.
Why do TCRs have off-targets?
The affinity of T-cell receptor (TCR) for its target is determined by its complementarity-determining regions (CDRs) on each TCR alpha and beta chain. This highly variable sequence results from genetic rearrangement and diversification. There are six CDRs per TCR, and they typically recognize a peptide presented in the context of the major histocompatibility complex (MHC), which, in humans, is the human leukocyte antigen (HLA) 22.
There are two classes of canonical MHCs: class I MHC molecules are expressed in almost every nucleated cell in the body and present processed intracellular protein products. In contrast, class II MHCs are restricted to immune cells and present peptides derived typically from phagocytosis. Humans have six HLA class I alleles and six HLA class II alleles. High polymorphism results in the human population having more than 25,000 different HLA class I and 10,000 HLA class II alleles 23. The diversity of these genes is primarily due to variations in the amino acid sequence within the peptide-binding cleft, increasing the variety of peptides displayed. The potential combination of peptide:MHC is predicted to be over 10E15 24 and becomes even larger once all possible post-translational modifications are taken into consideration, such as phosphorylation, oxidation, glycosylation, and citrullination, among others 25.
However, it is estimated that there are only ~10E12 T-cells in the human body collectively representing ~10E8 TCRs 26, millions of times less than needed to recognize every epitope individually. If a TCR were to bind only one cognate peptide:MHC pair, it would fail to mount a protective immune response against the actively evolving microbiome, viruses, and oncogenic mutations. Therefore it is necessary that TCRs have to be cross-reactive, with each TCR capable of recognizing thousands of, and possibly up to a million, different peptide:MHC complexes 27. This hypothesis has been validated while elucidating the mechanism of T-cell development and selection, as well as activation. A single peptide expressed in the thymus may lead to the elimination of polyclonal T-cells, and a monoclonal T-cell may be activated by multiple different foreign peptides. Such binding degeneracy provides the advantage of a single TCR being able to recognize similar pathogenic peptide groups and confer a wider protective effect 28.
However, this raises a concern regarding cross-reactive and auto-reactive therapeutic TCRs. Fortunately, all developing thymocytes undergo positive and negative selection in the thymus. TCRs that can recognize self-MHC molecules expressed by the cortical thymic epithelial cells (cTECs) are positively selected and migrate to the medulla, where they encounter multiple self-peptides presented by medullary thymic epithelial cells (mTECs) and resident dendritic cells (DCs). TCRs that bind too strongly to self-peptides are eventually eliminated by inducing apoptosis (central tolerance), leading to a final pool of T-cells unlikely to be autoreactive24.
Autoreactive T-cells that escape selection and encounter their ligand in the periphery may remain inactive, given that TCR engagement without co-stimulatory signaling leads to T-cell anergy (peripheral tolerance) or the induction of regulatory T-cell differentiation (iTregs)29,30. Another mechanism to keep self-reactive T-cells quiescent is by anatomical exclusion. The brain, central nervous system, eyes, and testes 31 avoid auto-reactivity by actively maintaining an immunosuppressive microenvironment either by secretion of immunosuppressive cytokines, selective homing of tolerogenic immune cells, limited lymphatic drainage 32 or formation of a physical barrier 31.These layers of protection in the periphery against self-reactive T-cells are evidenced by their prevalence during steady state. Prior work has speculated that the total number of potential auto-reactive T-cells is in the range of 1–10% 33,34, and more recent claims have suggested that this number may be even as high as 30% of the total immature effector T-cell population. Therefore, it would not be uncommon to identify TCRs with self-reactivity potential. These cells, if taken out of their quiescent steady-state environment and introduced in the context of TCR-based cell therapies, could cause significant damage to the host, as was lethally evident in certain clinical trials35, further emphasizing the importance of rigorous testing for potential off-targets before the application of a specific TCR in patients.
Consequences of off-targets.
Off-target toxicities may be due to (1) cognate-targeted antigens also being expressed in healthy tissue and (2) cross-reactivity to structurally similar peptides. In patients successfully treated with TCRs targeting MART1 and gp100, some patients developed severe side effects due to the target antigen also being expressed in melanocytes in the skin, ear, and eyes 36,37. Another clinical trial targeting CEA showed severe transient inflammatory colitis in three patients due to its expression on normal intestinal cells38. These studies show the limitation of using tumor-associated antigens as targets in cancer immunotherapy.
To optimize genetically engineered T-cells, TCRs are often affinity enhanced by introducing mutations into the CDRs, that bind to the MHC complex. Affinity enhancement, however, often increases the risk of T-cells’ off-target reactivity because these engineered T-cells bypass the natural negative selection process to self-peptides39. While the threshold for negative selection in the thymus was proposed to be < 6uM, affinity-enhanced TCRs often have logs higher affinity reaching the nanomolar range or even the picomolar range 40,41. Natural T-cell function has been proposed to plateau at an affinity of 5 uM 42 to 10 uM 43, and further increases in affinity may not lead to an increase in function.
In different trials, affinity-enhanced TCRs targeting MAGE A3 cross-reacted with peptides derived from self-proteins, leading to lethal toxicity in four patients. In one clinical trial (NCT01273181), the murine-derived TCR was affinity-enhanced through site directed mutagenesis in the CDR2 region, inducing tumor regression in 5/9 patients; however, also leading to lethal toxicities in two patients due to cross reactivity to a MAGE-A12 peptide expressed in the brain 44. In two separate trials (NCT01350401 and NCT01352286), an affinity-enhanced TCR against MAGE A3 was cross-reactive to a peptide derived from the cardiomyocyte protein Titin, leading to cardiogenic shock and death of two patients 21,35.
Methods of identifying off-targets.
The identification of peptides presented in healthy tissue is crucial for excluding those peptides as targets for immunotherapy. Bioinformatic tools help by analyzing sequencing data from healthy tissue. Tools such as NetMHCpan45 can assess how well peptides from the human proteome bind to different HLA alleles. Other computational methods can identify off-target peptides by considering factors such as charge, hydrophobicity, and structural information like predicted accessible surface area. The BLOSUM algorithm46 47,48 is commonly used, as it allows for peptides of different lengths and can find biologically relevant off-targets by using evolutionary and functional similarities between amino acids. This is achieved by blasting potential sequences to the human reference proteome.
However, these approaches have a high false discovery rate, do not reliably represent what is actually presented by the cell, and do not reliably predict T-cell reactivity. Recent optimizations in mass spectrometry and bioinformatic tools have advanced the field of immuno-peptidomics of healthy tissue49,50. Projects such as the Human HLA Ligand Atlas and the Immune Epitope Database (IEDB) will improve to assess whether the target is also expressed in healthy tissue 51. However, the sensitivity limit of detection for mass spectrometry is currently low, making the detection of infrequently-presented peptides difficult.
An empiric approach is anticipate potential TCR off-targets is to use alanine scans 52 by replacing each amino acid residue in a peptide sequence with an alanine and testing T-cell responses. This approach has the advantage of measuring the actual human T-cell response to an epitope. However, this method may not be effective in identifying significant interactions if the substituted amino acid is structurally similar to alanine and typically relies on single alanine substitutions that does not reflect the diversity of structural modifications. Thus, alanine scans tend to favor identification of TCR interactions with larger and charged amino acids.
The X-scan method is similar to the alanine scan, but instead of substituting with alanine, it individually substitutes one position in the peptide sequence with each of the 19 other amino acids while keeping all other positions unchanged 53. This results in 162 possible substitutions in a 9mer. Another method for screening peptides to identify TCR off-targets is to use combinatorial peptide libraries (CPLs) 54, where one position in the peptide sequence is held constant while the remaining positions are changed to any other amino acid. The peptides resulting from CPL scans are screened in subpools to determine TCR reactivity. Compared to alanine scans, X-scans and CPLs offer a more comprehensive understanding and ranking of potentially cross-reactive peptides by allowing for a wider range of peptides to be screened.
In vitro methods have limitations as they rely on predicted peptides that are based on the known target ligand sequence, and subsequently, cannot evaluate cross-reactivity of highly divergent sequences. Therefore, more empiric methods utilizing large libraries, where the peptide target is genetically encoded into expression systems, have been developed. Yeast-, baculovirus-, and phage-based display libraries of peptides, 55,56, 41,57 have been employed. In these methods, human MHC is expressed with the peptide attached by a linker. However, for these systems to work, the MHC must fold, and the peptide must bind the MHC properly, which may not successfully occur due to species-specific differences.
The PresentER system was developed to enable the upscaled testing of tens of thousands of candidate peptides for their presentation using endogenous human MHC 11. This system involves transducing TAP1- and TAP2-deficient T2 cells with a library of peptides along with an endoplasmic reticulum signaling sequence. Cross-reactive peptides are identified through DNA sequencing of the transduced minigene encoding potential off-target peptide sequences. Another library screening technique, called signaling and antigen-presenting bifunctional receptors (SABR) 58, involves expressing peptides linked to MHC receptors fused to intracellular CD3ζ and CD28 domains. The target cells are identified through fluorescence, and the presented target peptides are subsequently identified through sequencing as well. In contrast to the genetic encoding of short antigenic peptides used in PresentER and other libraries, SABR libraries encode larger numbers of amino acid sequences including all known A2 binding epitopes from IEDB database. However, both methods rely on HLA-binding or peptide cleavage algorithms. Therefore, these screens must be combined with mass spectrometry data or use of T-cells as surrogates for further validation.
4. Soluble TCR-based Therapies
Non-cellular TCR-based therapies bypass many of the limitations of an adoptive T-cell transfer approach. Two main approaches are via a TCR or an antibody that mimics a TCR’s reactivity.
ImmTacs:
Examples of the most clinically advanced soluble TCR therapies are the Immune Mobilizing Monoclonal TCRs Against Cancer (ImmTac) molecules, which comprise a soluble disulfide stabilized, affinity enhanced TCR fused to an anti-CD3 single chain variable fragment (ScFv). One arm of the ImmTac molecule engages pMHC, while the anti-CD3 ScFv arms engage CD3 on T-cells, redirecting powerful polyclonal T-cells to kill the targets. ImmTACs thus overcome the challenges of natural TCRs as soluble drugs (weak affinity towards tumor antigens, difficulties in manufacturing, lack of solubility). An ImmTAC molecule, Tebentafusp, (reactive with a gp100 epitope presented by HLA-A2) was the first approved soluble TCR therapy for the treatment of adult patients with unresectable or metastatic uveal melanoma in the United States and the European Union in 2022.
For TCRs, a relatively small number of mutations is sufficient to improve their affinity to the 100 picomolar range, while still maintaining specificity. In addition, the removal of the transmembrane domain and the addition of a non-native disulphide bond creates a soluble protein with exceptional stability 59. Each of the four described ImmTAC molecules (reactive with gp100/HLA-A*02:01, MAGE-A3/HLA-A*01:01, Melan-A/MART-1/HLA-A*02:0, and NY-ESO-1/ HLA-A*02:01) generated were able to redirect T-cells to tumor cell lines presenting the respective tumor-associated peptide antigens. The affinity of the TCR receptor component correlates closely with the degree of T-cell activation and, importantly, provides greater sensitivity to the expected low numbers of cell surface target antigens. ImmTACs are the first soluble bispecific agents to combine high-affinity recognition of MHC-presented tumor antigens with the simultaneous redirection and activation of bulk T-cells 60,61. Therapeutic ImmTac molecules targeting other tumor antigens PRAME, PIWIL1, or MAGE-A4 in the complexes of HLA-A2 or A24 have been recently developed, and some of these agents have entered clinical trials. Others target viral epitopes such as hepatitis B virus (HVB) and human immunodeficiency virus (HIV) 62,63. As a therapeutic class, ImmTACs offer a tailored, off-the-shelf solution possessing high specificity, in turn mediating efficacious cancer cell cytotoxicity.
TCR mimic monoclonal antibodies (TCRm).
The application of mAb or CAR T-cells in cancer therapy remains limited by the lack of cancer-specific cell surface targets not found on normal cells. Most targets in clinical development are tissue lineage antigens that are shared with normal tissues; therefore, targeting these conventional surface proteins with a high potency of modalities such as CAR-T, bispecific mAbs (bisAbs) or antibody-drug conjugates (ADCs) often causes on-target, off-tumor toxicities. In contrast to hematologic cells, in which deletion of a lineage (for example, B cells) may be tolerated by the patient for moderate time periods, the lack of specific antigens particularly limits the therapeutic applications of these agents among patients with AML or most solid tumors. To target the larger universe of intracellular tumor antigens, a new class of mAbs, TCRm, has been developed. TCRm mAbs are designed to recognize peptide/MHC complexes, similar to TCRs. However, the traditional antibody structure also allows the advantages and versatility of a mAb: easy protein engineering, high affinity and specificity, long half-lives in plasma, solubility, and off-the-shelf dosing flexibility 64. Most importantly, a mAb can be engineered to various formats to improve its therapeutic potency 65. While TCRm can access intracellular peptide/HLAs, the antibody structure offers possible advantages of intrinsic effector functions of mAbs and advanced therapeutic antibody formats. These include antibody-dependent cellular cytotoxicity (ADCC) either as an Ig or as a bispecific format, complement-dependent cytotoxicity (CDC), CAR T-cells, and antibody-dependent cellular phagocytosis (ADCP). In addition, mAbs can serve as antigen-specific vehicles that specifically deliver potent cytotoxic agents such as toxins, drugs, or radionuclides to cancer cells.
Several murine TCRms were developed to monitor antigen processing and presentation in mouse models as experimental tools 66. In the last decade, the use of TCRm mAbs for cancer therapy was greatly advanced. Traditionally, TCRm antibodies have been difficult to generate by conventional hybridoma technology. Advances in antibody display library methodology provided a breakthrough leading to the isolation of many mouse and human TCRm antibody fragments such as Fabs or scFvs, as well as several full-length human TCRm, thus allowing the investigation of these TCRms as potential therapeutic agents. Following the first two therapeutic TCRm mAbs, a murine hybridoma-generated TCRm (8F4) reactive with the myeloid leukemia antigen proteinase 3-derived epitope PR-1 (VLQELNVTV) presented by HLA-A*02:0167 and the first fully human TCRm, ESK1, specific for a Wilm’s tumor protein 1 (WT1)-derived epitope/HLA-A*02:01 complex 68, a growing number of TCRm targeting various tumor or viral antigens have been reported (Table 3). TCRm 8F4 has been humanized and engineered to bispecific antibody (BisAb) and was in clinical trials. ESK1, has been converted to bispecific T-cell engager (BiTE) and CAR T formats, radioconjugates, and also engineered to enhance Fc functions, demonstrating versatile usage of a TCRm mAb in various therapeutic settings as a typical mAb 69,70. A TCRm specific for an epitope derived from alpha fetal protein (AFP) in the context of HLA-A2 has entered clinical trial in a CAR T-cell format for hepatocellular carcinoma 71.
Table 3.
Human TCRm and Their Formats
| Antigen target | HLA restriction | Diseases | Formats | Citations |
|---|---|---|---|---|
| Proteinase 3 | A*02:01 | Myeloid Leukemias | IgG, BisAb, CAR T-cell | 67,309 |
| WT1 | A*02:01 | Leukemias and various solid tumors | IgG, BiTE, full length BisAb, CAR T-cell | 68–70,74 |
| PRAME | A*02:01 | Leukemias and various solid tumors | IgG, BiTE, CAR T-cell | 310 |
| FOXP3 | A*02:01 | Tregs, FOXP3+ T-cell malignancies and other types of cancers | IgG, BiTE | 311 |
| HPV-E7 | A*02:01 | Cervical cancer, many other HPV-associated tumors, head and neck cancers | IgG, BiTE | 312 |
| pIRS2 | A*02:01 | Ovarian, breast, colon, pancreatic, hepatocellular carcinoma, neuroblastoma, glioblastoma, melanoma, prostate, bladder, NSLC, CLL, MCL | IgG, BisAb | 77 |
| p53 mutation (R175H) | A*02:01 | Multiple myeloma, ovarian cancer and many solid tumors | Fab, scDb | 313 |
| Ras G12V | A*03 A*01 |
Wide range of solid tumors: pancreatic, colon, ovarian and more | scDb | 314 |
| Epstein Barr Virus | A*02:01 | B cell lymphoma and carcinoma | IgG | 315 |
| WT1 | A*24:02 | Leukemias and various solid tumors | CAR T-cell | 316 |
| Minor HA-H1 | A*02:01 | Leukemias | CAR T-cell | 317 |
| AFP | A*02:01 | Hepatic carcinoma | CAR T-cell | 71 |
| hCG-beta | A*02:01 | Ovarian, colon, and breast cancer | hIgG1, mIgG2a | 318 |
| NY-ESO-1 | A*02:01 | Melanoma and solid tumors | Fab, CAR T-cell | 79,319 |
| MAGE-A1 | A*01:01 | Melanoma | CAR T-cell | 78 |
| GP100 | A*02:01 | Melanoma | CAR T-cell | 320 |
| MUC-1 | A*02:01 | Breast cancer | Fab | 321 |
| hTERT | A*02:01 | Melanoma and prostate cancer; | Fab | 322 |
| HIV | A*02:01 | HIV | scDb | 323 |
| NDC80 | A*02:01 | Leukemias and various solid tumors | CART | 323,324 |
Only TCRm mAbs against human targets are listed.
TCRm-CAR T-cells.
In comparison to antigen targets of traditional antibodies, which may exist in the tens to hundreds of thousands on the cell surface, peptide/HLA complexes are typically low-density antigens on the cell surface, ranging from less than 10 to hundreds per cell 72. While antibody maturation has often been used to increase the antigen antibody interactions, using CAR T-cells to increase avidity has been shown to be an efficient way to overcome this hurdle. The first TCRm-CAR T, derived from ESK1, showed potent activity against leukemias in vivo70. Recently, more than a dozen more TCRm have been engineered into CAR T-cell formats recognizing NY-ESO-1, gp100, MAGE-A1, minor antigens, among other antigen, in the context of HLA molecules (Table 3).
Bispecific mAbs (BisAb).
Similar to the ImmTACs above, BisAbs are designed to recognize both a cancer antigens and an effector cell antigen and they comprise a large family of molecules, with a wide variety of formats. Such bispecific molecules function by recruiting and activating polyclonal T-cells, NK cells, or other effector cells. The successive conceptual and technical innovations in generating bisAbs have led to the extensive collection of over 100 BisAbs known today 73.
Bispecific T-cell engager molecules (BiTE) are a subtype of BisAb, composed of a scFv specific for tumor antigen on one arm, linked to a scFv for CD3 on the other arm. BiTEs are completely devoid of constant regions of the antibodies, with a small size (55KDa) and are highly flexible, thus enabling close interactions between CD3T-cells and cancer cells, and consequently facilitating potent polyclonal cytotoxicity of CD3 T-cells against cancer cells. Such a BiTE molecule functions by recruiting and activating polyclonal T-cells at tumor sites, thereby bypassing MHC restriction and co-stimulation, while retaining epitope specificity needed for traditional TCRs. Upon crosslinking, T-cells are activated to form an immunologic synapse, which induces apoptosis in tumor cells via the perforin/granzyme B pathways 74. Blinatumomab, an anti-CD19 and anti-CD3 BiTE, is the first BisAb approved by FDA in 201675. Bispecific molecules directed against targets in low abundance like MHC presenting specific epitopes, require an extremely high potency to be effective. ESK1-BiTE was the first TCRm-based BiTE, which showed superior cytotoxicity than an Ig form against a wide range of tumor cells expressing WT1 in vitro and in vivo in mice. Interestingly, The ESK1-BiTE also induced robust secondary CD8 T-cell responses against other epitopes via epitope spreading 69. Such a mechanism may be important for long-lasting anti-tumor immunity by controlling the outgrowth of tumor cells that have lost the target protein or that have downregulated the primary target during tumor evolution. This biological function is possibly analogous to that of the check-point blockade antibodies, which unleashes tumor-specific T-cell responses that had been suppressed or dormant in the tumor microenvironment. In addition, as a small molecule, BiTEs may penetrate more easily than CAR T-cells into the tumor microenvironment (TME) of solid tumors, where it can bridge tumor targets with tumor-infiltrating lymphocytes (TILs). Moreover, BiTE can be delivered by CAR T-cells, achieving dual targeting strategy 76.
Full length BisAbs.
The omission of antibody Fc domains from BiTEs, also has pharmacokinetic implications; BiTEs have a short plasma half-life (4–5 hours), which requires continues infusion and are therefore not ideal as convenient drugs. To overcome this problem, various bisAbs with full length antibody architecture have been developed to engage targets with CD3 T-cells, while silencing the Fc domains of the antibody. For the low-density antigens such as peptide/HLA complexes, bivalent mAb structures would provide more stable binding. Recently, a TCRm 11D06, specific for WT1 RMF epitope presented by HLA-A2, was engineered to a bivalent mAb (in a 2+1 format) IgG with a prolonged half-life. We engineered five different BisAbs derived from a TCRm specific for the phosphopeptide derived from insulin receptor substrate 2 (pIRS2) in the context of HLA-A2 molecule. Among which, we found that mAbs 1+1 and 2+2 format structures, effectively redirected T-cell cytotoxicity against the tumor cells 77. These studies demonstrated that a variety of currently advanced bisAb formats can be applied to TCRm as well.
Challenges for TCR mimics and solutions.
Similar to TCRs, TCRm also recognize a linear peptide sequence bound to HLAs; therefore, cross-reactivity to other similar complexes poses a potentially significant toxicity challenge. One argument against TCRm usage vs TCR, is that TCRm are not naturally selected structures filtered by thymic selection to preferentially recognize foreign, and not self, peptide-HLA complexes. In addition, most selection methods using sequence libraries that may introduce unnatural unstable structures. Therefore, TCRm may never completely mimic natural TCR recognition 72. TCRs generally dock onto peptide-HLA complexes using a conserved canonical binding mode, forming a large binding interface between the TCR and peptide-HLA, enabling broader contacts across both peptide backbones and HLA heavy chain. In earlier studies of TCRm, X-ray crystallography studies have shown that the binding of the TCR-mimic antibody to MAGE-1(161–169)–HLA-A*01:01 was focused on the HLA-α1 helix with no contact between the antibody and N-terminal MAGE-A1 peptide residues 78. A similar phenomenon was reported for ESK1, that the ESK1 Fab primarily interacts with the N-terminal residue of the peptide and HLA-A*02:01 12. However, other binding motifs of TCRms have also been reported. One TCR-mimic antibody engineered to bind to the NY-ESO-1(aa 157–165)/HLA-A*02:01 epitope adopts a TCR-like canonical binding geometry. In this study, crystal structures of 2 Fab antibodies to NY-ESO-1 peptide (SLLMWITQV) presented by HLA-A*0201 were compared to a TCR recognizing the same pMHC,1G4. Binding to the central methionine-tryptophan peptide motif and orientation of binding were almost identical for Fabs and TCR 79.
Alanine substitution assays have shown that various peptide residues could be recognized by TCRm, depending on the individual TCRm mAbs. For example, a TCRm mAb specific for the PRAME peptide/HLA-A*02:01 mainly recognized C-terminal residues of the peptide 80. A recent TCRm mAb to WT1 RMF/HLA-A2 recognized peptide residues 1, 3, 5 and 6 74. A TCRm (6B1) generated for the phosphopeptide pIRS2/HLA-A2 complex had an alanine scan that showed that the mAb primarily recognized the phosphate on the serine of the residue 4, which closely resembled the TCR recognition of the phosphopeptides/HLA-A2 complexes 77. Although a growing number of TCRms have been reported, most lack detailed analyses of recognition mode and specificity data. As a result, the factors that contribute to the recognition modes of TCRms remain complex and unclear. Even the well-established model of TCR-peptide/MHC interactions has also been constantly updated with exceptions, as a recent study revealed a reverse docking topology relative to the established TCR/p/MHC docking paradigm 81. Future work will focus on discovering TCR mimic mAbs that better recognize peptide/MHC complexes with fine specificity and with TCR-like conformations. This may be achieved by more rigorous screening algorithms, better filtering of hits, and structure-based analyses.
Another way to improve the selection process of finding better TCR-likeTCRm, could be the design of improved the phage libraries and protein re-engineering to create molecules that engage peptide/MHC in a manner structurally similar to that of conventional αβ-TCRs 82. Crystallographic analysis of one selected pMHC-restricted Ab revealed highly peptide-specific recognition, validating this engineering strategy.
Improved screening strategies to select TCRms that interact with the amino acids of the peptide/HLA complex that are broader and more central may be preferred as well. Specificity to desired middle amino acids should reduce binding to many potential human proteomic off-target peptides. With this strategy, we were able to select more specific TCRm clones for the WT1 RMF/HLA-A2 complex than we had identified before (unpublished).
Furthermore, the availability of more crystallographic studies would provide direct structural information to improve our current understanding of the interactions between TCRm and the peptide/HLA complexes. Recent studies of TCRs have demonstrated that off-target peptides do not need to share sequence, physiochemical, or backbone geometry with the cognate peptide and that peptides, HLAs, and TCRs all have flexibility and adaptability during the TCR recognition of the peptide/HLAs 83,84. This leads to a question if such conformational plasticity also exists in the TCRm recognition that are not always captured by crystallographic analysis alone. Although conventional mAb binding to protein targets is fundamentally different from TCR recognition, TCRm, which recognize peptide/HLA, may share certain similarity with TCRs. Thus, it is vital to understand the dynamic characteristics of peptide/HLA interactions with TCRm. To better understand the contribution of allostery, protein dynamics, and protein flexibility, during peptide/HLA interactions with TCRm mAbs, dynamic studies using isotope-edited infrared spectroscopy, nuclear magnetic resonance (NMR), Förster resonance Energy transfer (FRET), and molecular dynamics (MD) stimulation, may offer new insights into the recognition of TCRm to peptide/HLA complex. Such methods have shed light on both TCR-p/MHC interactions, antibody orientation and function 85–88.
5. Cancer vaccines
As cancer vaccines are not a direct use of a TCR-based drug, but rather a means to induce a host TCR-based response in which the host provides the cytolytic agent, we will only briefly discuss their uses and issues for comparison here. Cancer vaccines consist of synthetic peptides, mRNAs, DNAs or proteins derived from tumor antigens that are used for active vaccinations to induce or boost naturally occurring tumor-reactive T-cells’ TCR that recognize peptides presented by MHCs. Cell-based vaccines have also been tried using dendritic cells loaded with tumor antigens or modified tumor cells 89. Cancer vaccines have been the subject of intense preclinical and clinical investigation for a variety of malignancies over the past 40 years, however, the successful clinical translation from bench to marketing approval has been elusive. Many clinical trials of cancer vaccines, including our studies 90,91 have shown to be able to induce vaccine-specific immune responses. However, responses alone do not always translate, lacked into immediate clinical benefits especially in the setting of active, bulky cancers or leukemia. Because most cancer vaccines were targeting TAAs, a major obstacle is the induction of potent adaptive immune responses against self-antigens that is limited by the inherent self-tolerance of the host.
The recent success of checkpoint blockade therapy and recent advances in neoantigen identification revived the enthusiasm for current cancer vaccine development 92,93. The adaptive immune system’s ability to discriminate between “non-self” and “self,” coupled with the vast diversity of T-cell repertoire, yields neoantigen-specific T-cells that are present in the blood or TILs of cancer patients. The key role of neoantigens in antitumor immune responses has been demonstrated in patients with solid tumors, whose tumors showed substantial regression after treatment with adoptively transferred neoantigen-specific T-cells 6,8,94. However, neoantigens are generally patient tumor-specific, requiring a patient-specific vaccine to be prepared, making this approach logistically complex and expensive.
Clinical experience suggests that cancer vaccines are safe and can elicit long-term immune memory responses important for durable disease control 89,95,96. This suggests that vaccines may be particularly well-suited in the minimal residual disease setting. In addition, neoantigens are key targets of checkpoint blockade immunotherapy-driven responses; therefore, priming tumor-specific T-cells and mobilizing them to the tumor, vaccine therapies could help checkpoint blockade to unleash T-cell-mediated tumor-specific responses. While several neoantigen vaccines have been tested in human trials, from historical experience, combinations of neoantigen vaccines with checkpoint blockade and other therapies may achieve better therapeutic efficacy 97. While most neoantigen-targeting vaccines are patient-specific, new searches for public neoantigens such as p53 mutations and RAS mutations, could offer a broader application of vaccines 98,99.
6. Cellular TCR-based therapeutic approaches: Choosing the right cell vehicle
Engineering cells with a tumor antigen-specific TCR requires a sufficient quantity of healthy cells for expansion ex-vivo before infusion and an appropriate effector capable of achieving the desired response. If the cell source is the autologous patient, this precondition may limit the types of cells that may be used, especially if the patient has a comorbidity or received prior therapy that reduces cell numbers. The necessity for patient-specific cells and the difficulties of controlling doses, proliferation and persistence of cells once infused, may limit optimal clinical applications at this time. Allogeneic off-the-shelf sources would overcome some of these limitations, but are less well-described and clinically developed. Here we discuss the different types of immune cells that can be engineered with tumor antigen-specific TCR-based agents for adoptive T-cell therapies against cancer.
CD8 T-cells:
Cytotoxic CD8 T-cells, as the most efficient cancer-killing cells that inherently recognize MHC-class I-associated antigens via their TCR 100 have been a top choice of cells to express an exogenous TCR. However, the presence of native TCR within these cells poses challenge. For example, exogenous TCR chains can mis-pair with endogenous TCR αβ chains, which could lead to less specific activity, cross-reactivity towards self-antigens, autoimmunity, and reduced potency. Solutions to this issue include introduction of cysteines into the constant regions or the use of murine constant regions, framework region engineering, domain-swapping, single chain exogenous TCRs, and knocking-out endogenous TCR αβ chains (including knocking in the new TCR into the TCR alpha site)101–106. TCR-engineered CD8 T-cells generally need to be infused together with helper CD4 T-cells for optimal function107. Early TCR-engineered T-cell therapies 108used allogeneic T-cells with exogenous TCR targeting MART-1AFP, CEA, GD2, gp100, MAGE-A3, MAGE-A4, Mesothelin, and NY-ESO-1 among others 60,108–111
CD4 T-cells:
Because CD4+ T-cells make up two-thirds of the total blood T-cell population, CD4 T-cells are being investigated for their cancer-killing efficiency, after engineering them to express tumor pMHC-class I restricted exogenous TCR 112. Challenges to this approach include a reduced potency due to a lack of CD8 co-receptors on CD4 T-cells and, as described above, mispairing of exogenous TCR with endogenous TCR of CD4 T-cells. Strategies to overcome these issues include the transfer of CD8αß co-receptor genes and improving the pairing of exogenous TCR using techniques discussed above 113. For example, one clever and robust approach was to make therapeutic CD4+ T-cells capable of providing MHC Class I-restricted immunity against MHC Class II-negative tumors by use of MHC Class I-restricted CD4+ T-cells specific for Epstein-Barr virus (EBV) and cytomegalovirus (CMV) that recognized HLA-A2/peptide multimers 114. In a xenogeneic mouse model, this work demonstrated that human TCR and CD8 genes engineered into CD4+ T-cells conferred efficient protection against the growth of tumors expressing the EBV or CMV antigens recognized by the TCR.
γδ T -cells:
Gamma-delta (γδ) T-cells are an alternative cytotoxic effector population that can be engineered to express tumor-antigen specific αβ TCR 115,116. Since γδ TCR chains do not pair with αβ TCR chains, γδ TCR are not subject to the problems associated with the use of exogenous αβ TCR chains, such as incorrect mispairing with endogenous TCR leading to alloreactivity and GvHD 117. Many studies have successfully demonstrated engineering of cytotoxic γδ T-cells expressing HLA-class I restricted αβ TCR 118,119. In a similar approach, γδ T-cells could also be equipped with TCR derived from iNKT to target CD1d-restricted tumor antigens 120.
The γδ T-cells have limited expression in the blood, with only 1–10% of total circulating T-cells making manufacturing difficult 121,122. Therefore, in an alternate approach, αβ T-cells can be armed with tumor-specific TCR from γδ T-cells. Hence arming abundantly available αβ T-cells with γδ TCR will make them kill tumor cells in an HLA-independent manner 123. In addition, expression of γδ TCR downregulates the endogenous αβ TCRs, thereby reducing the chance of off-target HLA-antigen recognition and alloreactivity by engineered T-cells.
NK-cells:
Natural killer (NK) cells are innate lymphoid cells with the inherent ability to identify and kill cancer and virus-infected cells 124. They can identify the cancer cells in a TAA and pMHC-independent manner and kill them via several cytotoxic mechanisms such as inducing apoptosis by Fas-FasL interaction, secreting cytolytic molecules such as perforin and granzyme, ADCC, and secreting cytokines that can recruit cells of other innate and adaptive immunity 125–128. Blood-derived primary NK cells and the NK cell line “NK-92” have provided rapid killing of cancer cells in allogeneic settings without causing significant graft-versus-host disease 129–132. CAR -NK cells have also reached human trials and appear to be safe and effective 133. Hence, tumor antigen-directed TCR-engineered NK cells may be alternative off-the-shelf, ready-to-use allogenic cells with enhanced anti-tumor effector functions that combine the effect of TCR mediated tumor cells lysis as well as NK cells’ intrinsic activation mechanisms. However, engineering NK cells with a functional exogenous TCR also requires the expression of exogenous CD3 molecules, as NK cells do not express CD3 components 134,135. For example, enhanced HLA-B*07:02 restricted BOB1-specific TCR-engineered NK cell efficacy against B-cell leukemia compared with TCR-negative NK cells has been shown 105.
NK T-cells:
Natural Killer T-cells (NKT) share the properties of both conventional T-cells and NK cells. They express NK cell’s specific markers and semi-invariant αβ TCR that recognizes lipids and glycolipids antigens presented by CD1d molecules 136,137. There are two types of NKT-cells; Type-1 NKT-cells with limited TCR diversity, also called invariant NKT-cell (iNKT-cells), and other CD1d restricted T-cells called Type-2 NKT-cells. These NKT-cells are naturally potent cytotoxic against cancer cells and also confine immunosuppressive myeloid cells in the tumor microenvironment via CD1d-cognate detection, stimulating anti-tumor responses irrespective of the CD1d expression by cancer cells 138–141. Since CD1d molecules are identical in all individuals, NKT-cells can be adoptively transferred across MHC barriers without the risk of allo-reaction and graft vs. host disease 142. Hence, allogenic NKT-cells also can be exploited as readily available, off-the-shelf donor-unrestricted effector cells for adoptive cell therapies against cancer 143–147. Adoptive cell therapy with tumor antigen-redirected exogenous TCR-engineered NKT-cells could provide combinatorial anti-tumor effects by utilizing both the exogenous tumor-specific TCR to recognize pMHC on tumor and CD1d restricted endogenous TCR against the cancer cells that could boost the overall therapeutic effect. TCR-engineered iNKT-cells demonstrated efficacy against various tumor models 148 in which bispecific effector functions for CD1d- and MHC-restricted antigens were seen.
CIK-cells:
The cytokine-induced killer (CIK) cells are heterogeneous populations of ex-vivo differentiated immune cells with high tumor-killing potency and characteristics of both NK cells and cytotoxic T-cells 149,150. Among them, CD3+CD56+ cells are the most efficient cytotoxic CIK cells, which can kill tumor cells in both MHC-dependent 151 and independent manners by deploying effector molecules such as NKG2D, TRAIL, FasL, DNAM-1, NKp30, LFA-1, perforin and granzyme secretion 152–155. Hence, strategies for engineering CIK cells with tumor antigen-redirected TCR could provide an adequate number of effector cells for adoptive cell therapy with the possibility to target surface and intracellular antigens. CIK cells genetically engineered to express HLA-A2+ restricted anti-Mart-1 and anti-NY-ESO-1 melanoma-antigens specific exogenous TCRs can kill tumor cells in a cognate pMHC specific manner and also maintain their MHC-independent anti-tumor activity 156.
Hematopoietic Stem Cells:
Adoptive cell therapy with tumor antigen-specific TCR-engineered hematopoietic stem cells (HSCs) could provide a continuous supply of effector T-cells against tumors by replacing the exhausted T-cells in the tumor microenvironment. In addition, the expression of exogenous TCR in HSCs will suppress the expression of endogenous TCR via allelic exclusion, which might solve the problem of TCR mismatch and off-target reactivity. However, an exogenous TCR c-terminal linked to CD3z or a co-expressed CD3 may be required to produce fully functional cells. Autologous or donor-matched CD34 positive HSCs can easily be isolated from peripheral blood stem cells, umbilical cord blood, or bone marrow for TCR engineering and transplantation 157. For example, antigen-specific HLA-restricted cytolytic activity by modified T-cells differentiated from NY-ESO-1 and anti-p53-antigens-specific TCR-engineered UCB were demonstrated 158.
7. Protecting cells from host attack
Universal “off-the-shelf” allogeneic don,or cells engineered with tumor antigen-specific TCR are proposed to solve many logistical hurdles of autologous T-cell therapy. However, a mismatch in donor and recipient HLA haplotypes can lead to either host rejection or cell graft-versus-host-disease (GVHD) 159. Ongoing strategies to evade the allo-rejection include HLA-matching to the donor or lymphodepletion of the recipient. However, these strategies are not completely effective and often toxic 160 161Gene editing of the donor cells may provide alternative approaches (Figure 1). For example, deleting endogenous genes of TCRα/β chains, HLA, β2-microglobulin (B2M), and MHC class II transactivator (CIITA) may shield donor cells; alternatively adding genes for HLA-E, alloimmune defense receptor (ADR) and immunoglobulin-degrading enzyme of Streptococcus pyogenes (IdeS) have been attempted to improve the persistence and functionality of the infused allogeneic cells 161–166. Deleting the genes of endogenous TCRα/β chains also can significantly reduce the chances of mispairing with exogenous TCR, potential off-target reactivity, and rejection 105,164. Similarly, deleting genes of B2M and CIITA blocks the expression of HLA class I and II on the cell surface, making these cells not detectable by the recipient T-cells. While deleting the B2M gene leads to the downregulation of all HLA-class I molecules on the cell surface, it also puts these cells at risk of host NK cell killing. Therefore, to escape NK cells’ attack, non-polymorphic exogenous HLA class E and G genes can be inserted in these cells 161–163. Expression of ADR on the cell surface has increased evasion of host T-cell cytotoxicity 165. Expressing IdeS can protect cells from any potential antibody attack on the injected allogeneic cells 166. Similarly, overexpressing CD47 in the donor cells, a “don’t eat me” signal molecule could stop macrophage-mediated phagocytosis of the injected cells 167.
Figure 1. Protecting the Engineered cell.
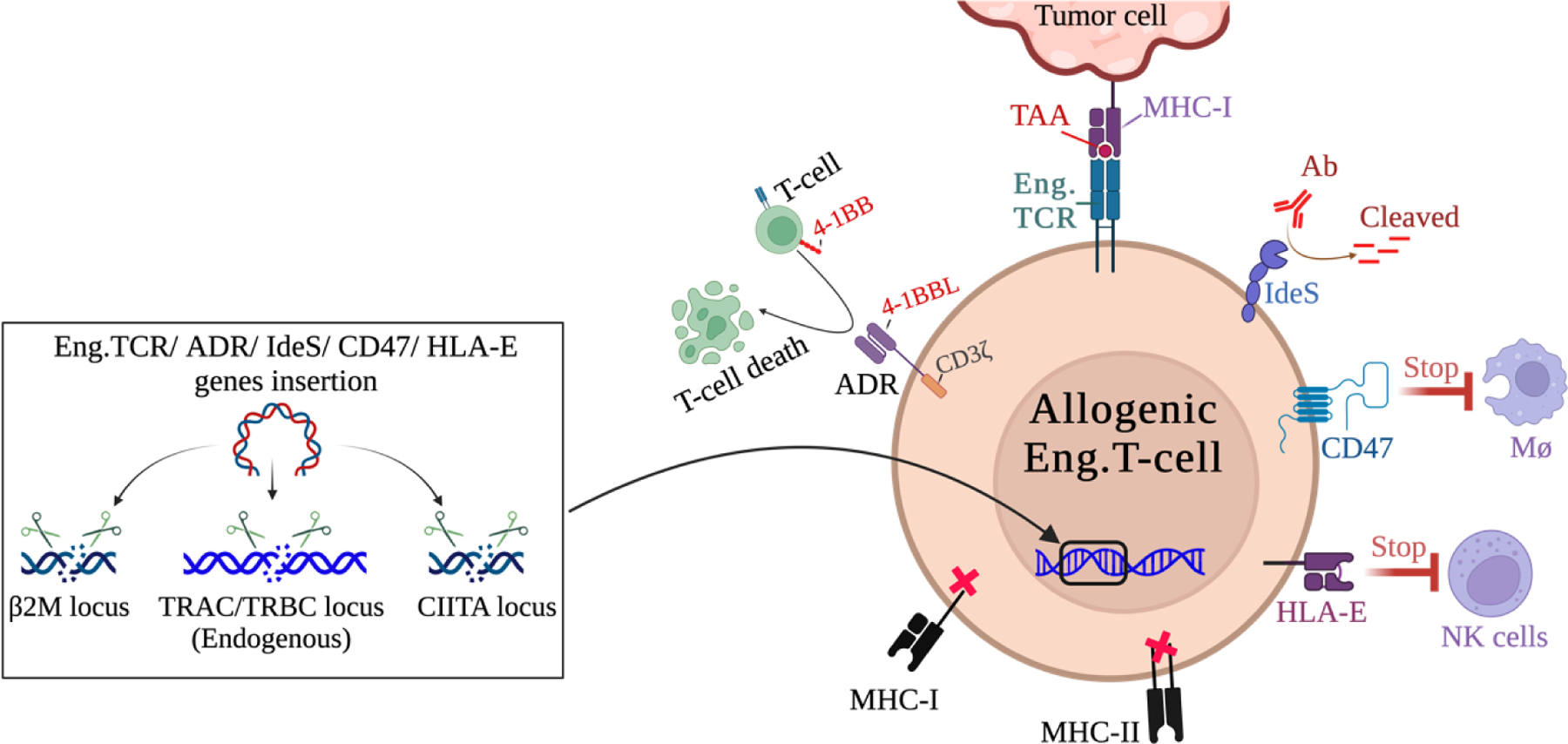
There are two general strategies to generate stealthy, off-the-shelf allogenic donor cells for adoptive cell therapy. Methods include editing endogenous genes of TCRα/β chains, β2-microglobulin (B2M), and MHC class II transactivator (CIITA); by CRISPR mediated gene deletion or disruption of their expression by insertion of new genes such as engineered TCR can reduce recognition by the host. Alternatively, alloimmune defense receptor (ADR), “Do not eat me” CD47, Streptococcus pyogenes (IdeS), and HAL-E proteins can protect allogeneic cells from rejection by the host immune cells. See section 7 for more information. All figures were created by using BioRender.
Unfortunately, with multiple genetic edits, there are risks of structural genomic abnormalities and lack of uniformity in every cell. One possible solution to this problem would be performing serial gene edits in iPSC or HSC to make a single clone-based uniform cell line that can be deposited for future use. These gene-edited stem cells could be differentiated into tumor antigen-specific TCR-engineered T-cells for adoptive cell therapy without the risk of batch-to-batch variability 162,163,168
8. Comparing a TCR versus CAR as the receptor for an effector cell.
CAR T-cell therapy involves genetically combining high-affinity single-chain variable fragments (scFv) of mAb with enhanced intracellular T-cell activating domains and transducing them into T or NK effectors. It has resulted in remarkable clinical results in B cell neoplasms 133,169, but has demonstrated limited benefit in solid tumors. There is still a need for enhanced specificity and potency, as well as mitigation of common side effects, such as cytokine release syndrome (CRS)170, which may be due to abnormally strong signal transduction CD3ζ.
CARs have the advantage of MHC-independent antigen recognition, making this therapy more easily adaptable across different patient populations171. (Figure 2). As a consequence, a major drawback for CAR T-cell therapies, unlike TCR T-cells, is their inability to target intracellular antigens. Cell surface tumor antigens are generally expressed on normal tissues as well172. A prominent example is CAR T-cell therapy targeting CAIX for renal cell carcinoma patients which resulted in off-tumor toxicity at the bile ducts173. TCR T-cell therapies circumvent this roadblock through targeting of intracellular antigens in the context of MHC, and therefore access the enormous immunopeptidome that may be cancer specific174. Additionally, CRS severity is known to correlate with high tumor burden and high T-cell therapy dosing, highlighting T-cell overactivity as a major contributor 170. Because of the low target antigen density, as well as more natural control of T-cell activation and function via the TCR, TCR T-cells may also be less toxic with a decreased incidence of cytokine release syndrome171,175. Recent advances have allowed for endogenous TCR deletion with the incorporation of the transgenic TCR using CRISPR-Cas9 editing to knock out the TRAC and TRBC loci while simultaneously incorporating the new transgenic TCR176,177. This results in increased expression of the transgenic TCR with less mixed dimer formation between the transgenic TCR and endogenous TCR.
Figure 2. Comparison of Characteristics of TCR T-cell and CAR-T-cell formats.
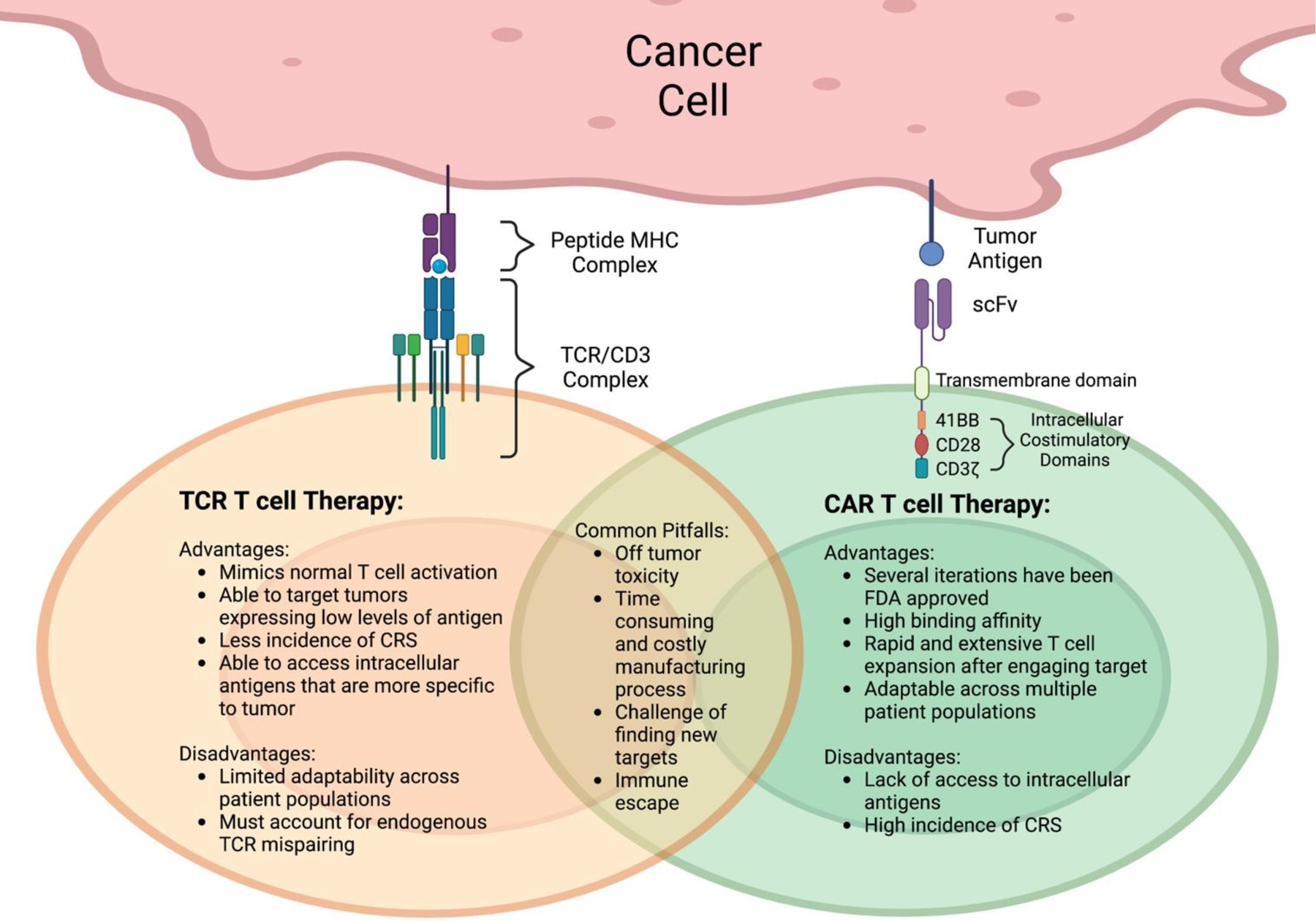
(left) TCR therapy targets a large universe of intracellular tumor antigens that are presented on the cell surface as peptide fragments by MHC molecules. The recognition of the antigen occurs via alpha beta TCR/CD3 complex. (right) Most current CAR-T-cell therapies target cell surface and lineage proteins that are shared between tumor and normal cells. The recognition of the target is through the scFv of an mAb directed to the surface target protein, which was linked to the T-cell activation molecules CD28 or 41BB and CD3 zeta chain. All figures were created by using BioRender.
Though TCR T-cell therapy may be difficult to adapt across multiple patient populations due to its MHC restriction, rapidly expanding identification of epitopes for many of the common HLA types is broadening the scope of accessible targets. This will require advancements in computational and empiric screening strategies. Additionally, TCR T-cell therapies are able to have a cytotoxic effect on cancer cells even at low antigen densities (perhaps 10’s of epitopes) because of the high sensitivity of the TCR to effectively trigger controlled clonal T-cell expansion.
Common pitfalls shared between both CAR T-cell therapies and TCR T-cell therapies include some degree of off-tumor toxicity, lack of rapid and cost-effective product manufacturing, slow identification of truly tumor-specific targets, and immune escape of tumor. Strategies to overcome some of these challenges include soluble mAb and TCR bispecific agents circumventing the need for cell production thus providing efficiency and affordability61. Hybrid approaches, such as TCRm gives CAR cell therapies the ability to access intracellular antigens70. AbTCR is another hybrid approach that gives TCRs typical antibody recognition178,179 and are currently being studied further (Figure 3).
Figure 3. Characteristics of new hybrid T-cell formats.
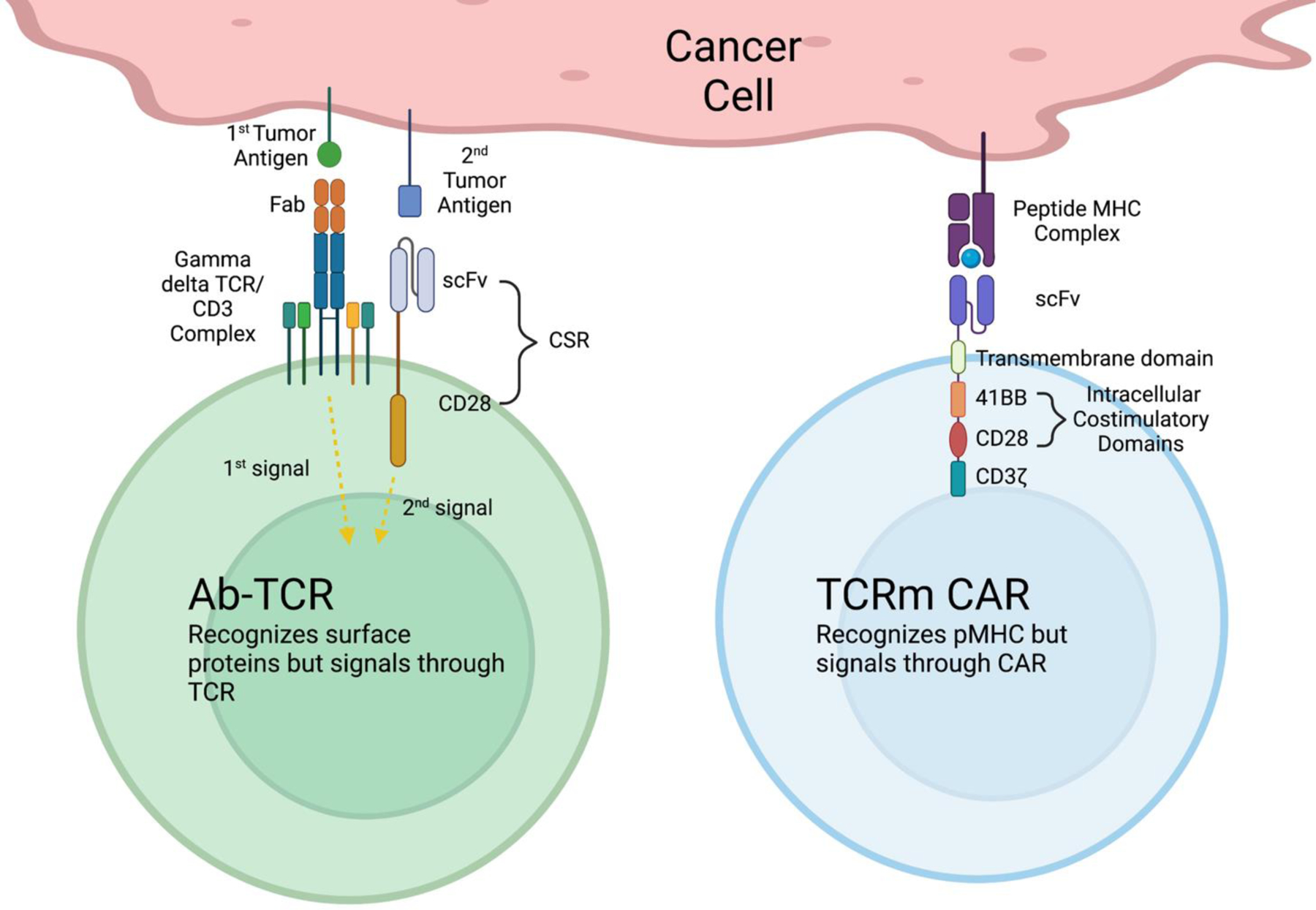
(left). Ab-TCR is a new TCR/CAR-T format, which consists of two separate activation domains: the first domain uses a Fab specific for a tumor antigen, linked to gamma/delta TCR to facilitate a natural T-cell activation. The second domain uses a scFv of an mAb targeting a second tumor antigen, linked to a costimulatory molecule, CD28, down-stream of a signaling molecule needed to for fully activate T-cells and duel targeting tumor antigens This new format of CAR-T-cells could avoid excessive synthetic activation and toxicity caused by traditional CAR-T-cells that assemble T-cell activation molecules in one construct. (right). TCRm CAR-T-cells use traditional CAR-T construct, however, they are able to target intracellular tumor antigen-derived peptide/MHC complexes, by using scFv derived from TCR mimic mAbs. CSR: Costimulatory signaling receptor. All figures were created by using BioRender.
9. Cancer escape mechanisms: Downregulation of epitope presentation
Cancer induced downregulation:
One of many resistance mechanisms in solid tumors to TCR based immunotherapy is the downregulation or loss of cell surface HLA. 180–182. Forty – ninety percent of human tumors are prone to HLA class I loss or downregulation, which is found to correlate with worse clinical responses, shorter overall and progression-free survival, an increase in metastasis183–189 as well as the amount of tumor-infiltrating lymphocytes in the TME190. Thus, dysfunctional HLA antigen presentation may predict resistance to adoptive cell therapy and checkpoint inhibition in a clinical setting190–192.
Genetic mechanisms for HLA loss or downregulation have been grouped into difficult-to-treat, DNA-encoded lesions, and epigenetic, transcriptional, as well as post-transcriptional alterations that are potential therapeutic targets 193. Mutations in structural genes of the pMHC I complex, or the antigen presentation pathway have been shown to abrogate peptide antigen presentation181,193. The genetic HLA locus on chromosome 6p21 is frequently mutated or lost in several cancers, encoding several genes crucial for antigen presentation (i.e. HLA heavy chains, TAP1/2, tapasin)194. Loss of heterozygosity (LOH) associated with chromosome 6p21 is a major mechanism of reduced antigen presentation in several human tumors183,195,196, represented in up to 17% of cancers182. The loss of single HLA class I molecules through somatic mutations in the HLA heavy chain genes have been reported 197,198. Beta-2-microglobulin (B2M), which stabilizes the pMHC complex, is mutated in a variety of cancers, including melanoma, metastatic colon cancer and up to 25% of lymphomas199–202,201,203. Complete loss or a functionally defective allele of TAP1/2 or loss of Tapasin and ERAP has been seen in several solid tumors including, renal cell carcinoma, colorectal-, cervical- and esophageal cancer204–207, 208–210. Because Interferon type I or type II signaling can induce HLA class I expression through Janus kinase and STAT signaling211,212, LOH and mutations in JAK1/2 and STAT, as well as JAK1/2 upstream receptor APLNR also have been found to promote resistance to immune checkpoint blockade199,213–215.
Changes in antigen presentation that are not the result of genetically encoded lesions may allow for therapeutic intervention with small-molecule drugs193. This includes, epigenetic silencing, mainly due to hypermethylation events on key promoters or histone modifications 216–218. Several studies suggest that DNA methyltransferase inhibitors and histone deacetylase (HDACs) inhibitors effectively upregulate HLA class I expression in several cancer types219–221, 222, 222–224. MicroRNA-mediated degradation of mRNA encoding HLA class I heavy chains and transcripts of other crucial members of the peptide presentation pathway (TAP1/2, tapasin, calnexin, etc.)193 may also be a target for intervention. 225,226–228.
Cancers may utilize post-translational mechanisms to degrade HLA proteins, such as endoplasmic reticulum-associated degradation (ERAD)229, autophagy-dependent mechanisms230, and increased lysosomal degradation231. Interestingly, oncogenic signaling mechanisms such as MAPK activation and c-MYC and n-MYC overexpression have been found to reduce HLA class I, TAP, and B2M transcript levels and protein expression232, 233–235. Finally, oxygen tension was found to reduce HLA class I expression in a HIF-1a dependent manner 236.
MHC I downregulation induced by viral infections:
Viruses also use mechanisms to evade immune recognition by downregulation of HLA class I expression when infecting host cells. Due to the focus of this review on TCR-based cancer immunotherapy, this work will not review viral mechanisms for HLA downregulation in detail. For an in-depth review, please refer to 237,238. Of relevance is that by specifically inhibiting steps of the antigen presentation pathway, viral immune-evasins may have the potential to be leveraged pharmacologically in gene therapy, transplantation, and auto-immunity.
10. Cellular micropharmacies
T-cell therapies alone still face many limitations in the treatment of solid tumors 239. One very promising effort to overcome these limitations is the engineering of targeted cellular micropharmacies (TCM), a novel pharmacologic paradigm to genetically engineer or chemically modify immune cells to serve as vectors for drug delivery 240. For example, our Synthetic Enzyme Armed Killer (SEAKER) cells secrete bacterial enzymes that accumulate in the TME. Systemic delivery of a non-toxic prodrug results in localized enzymatic unmasking in the TME, which vastly increases the therapeutic index and potential dose of the unmasked cytotoxic small molecule drug. T-cells are ideal pharmacologic vehicles to deliver payload specifically to tumors since they retain the advantages of adaptive immune cells to allow for a precise localized release of pharmacologic payload that reduces systemic toxicities of highly toxic cancer therapeutics or potent cytokines. There is also the promise of temporal control of payload release and regulation of cellular activity levels by choice of cell type and synthetic gating strategies 240–242, 240,243. In the last 10 years, several TCM constructs have been published, carrying diverse therapeutic payloads, ranging from immune checkpoint- or TAA-targeting antibodies 244–246, scFvs 247–249 and BiTEs 250–253; over proinflammatory cytokines254–265, chemokines266,267 and viral particles268; to ECM degrading269 or pro-drug activating enzymes270, immune modulatory soluble proteins271,272 and small molecule drugs273,274.
mAb blockade of regulatory immune checkpoints like PD-1 or CTLA-4 showed clinical efficacy in several tumors by combating T-cell exhaustion and prolonging tumor-specific immune responses275, but are still limited by low TME penetrance in solid tumors and severe immune-related side effects.275,276 Cellular delivery may solve these problems but have to date been restricted largely to CAR T-cells. Examples include secretion of full-length and scFv mAb to PD-1244, 247–249, and CTLA-4 246, and to CD47 277–279. Cells can also be engineered to secrete specific TAA blockers directly, as BiTEs against EGFR250, CD3, EphA2+251, CD19252, and CD123253.
Cells also are a promising approach to more safely deliver immunomodulatory cytokines (such as IL2, IL7, IL15, IL12, IL18, Flt3 ligand, GMCSF, CCL19, CCL21, ) directly into the TME, initiating or potentiating tumor-specific immune responses, without life-threatening toxicity 241, 254 ,255, 256,280,281. 266, 267 .
11. Clinical Applications of TCR
TCR-based agents currently being studied in the clinic are predominantly in the forms of T-cells genetically modified to express an antitumor TCR and soluble TCR agents. Conventional αβ TCRs can recognize a massive number of peptide-MHC (pMHC) antigens with exquisite sensitivity and variable specificity, including pMHC on cancer cells in the form of tumor-associated antigens (TAA) and tumor-exclusive neoantigens. Among the first TAA found to be recognized by TCRs were those derived from MART1 2, gp100 3 MAGE-A1 4,282, and Tyrosinase 5, all of which were first found to be recognized by either melanoma patient peripheral T-cells or tumor-infiltrating lymphocytes (TIL) from resected melanoma lesions. TIL present in several resected solid tumors recognizes patient-specific neoantigens 6,7,98. When such TIL are expanded ex vivo and reinfused, they can induce durable regressions in solid metastatic tumors 8,283, thus demonstrating the therapeutic potential of neoantigen targeting. Similar to TAA, neoantigens produced by somatic mutations exclusive to cancer cells are becoming increasingly appreciated as tumor rejection antigens that can be targeted by TCR therapy. Moreover, various neoantigen qualities, such as clonality, MHC binding properties, and immunogenicity, have been shown to predict response to immune checkpoint blockade 284,285. Because T-cells generated in vivo in patients are endowed with specificity for tumor antigens, there has been significant interest in the clinical development of a class of TCR-based agents for cancer immunotherapy.
Following the observation that melanoma patient TIL recognizes TAA and can induce cancer regression 286, early clinical studies utilized TAA-specific TCR-transduced T-cells to treat metastatic melanoma 36,287. Though targeting TAA was initially thought to be safe due to their restricted expression, an affinity-enhanced MAGE-A3 TCR was found to exert off-target reactivity to cardiac tissue, causing fatal toxicity when expressed in T-cells adoptively transferred to melanoma and myeloma patients 21,35. Similarly, a MAGE-A3/A12 TCR was found to cause fatal on-target/off-tumor reactivity to neuronal tissue 44. The toxicities observed with affinity-enhanced TCRs targeting conserved TAA have shifted clinical interest towards using patient-derived TCRs to target neoantigens, of which entirely non-self-peptides can be targeted if sufficient somatic mutations are acquired in the tumor. Two allogeneic TCRs targeting the public KRAS G12D/C*0802 neoantigen were used to engineer autologous T-cells, which were reinfused to induce objective regression of metastatic pancreatic cancer 288. In a similar approach, a library of 39 patient-derived TCRs to common TP53 mutations with various HLA restrictions were used to select an allogeneic TCR to redirect patient T-cells to the HLA-A*02-restricted p53 R715H public neoantigen 289. The resulting TCR-engineered T-cells were reinfused and induced objective regression of breast cancer lasting six months. To demonstrate the feasibility of neoantigen calling and TCR identification at a scale to treat a large cohort of patients, a recent effort demonstrated the feasibility of identifying patient-specific neoantigens, their cognate TCRs, and manufacture of neoantigen TCR-engineered T-cells, dosing 16 patients with various solid tumors 290. TCR-engineered T-cells are also being investigated for treating hematologic malignancies, particularly for AML/MDS by targeting the differentially expressed TAA WT1 18,291–293. Interestingly, relapse after WT1 TCR therapy was associated with antigen escape not by WT1 mutation or HLA downregulation but by immunoproteasome regulation 294, a challenge that can be overcome by informed epitope selection.
Given the prominent role of T-cells in clearing viral infections, viral malignancies are expected to be amenable to TCR therapy. To this end, TCRs targeting HPV antigens are under investigation for cell therapy of various HPV+ epithelial malignancies 19. Unlike TAA, HPV targeting can induce objective responses without significant toxicities, a safety feature most likely attributable to the non-self character of viral antigens. HBV antigens are also under clinical investigation for TCR therapy of hepatocellular carcinoma 295–297. In a small cohort, HBV TCR-engineered T-cells could cause stabilization of HBV antigen or DNA levels in most patients and tumor lesion reduction in some patients. The conclusion of future trials will elucidate the potential of TCR-engineered T-cells for treating advanced viral and non-viral cancers. Clinical studies of adoptive cellular therapies utilizing antitumor T-cells from allogeneic sources without genetic modification are reviewed elsewhere 298.
Soluble agents with TCR-like recognition have also generated significant clinical interest. If shown to be efficacious, it may significantly advance cancer immunotherapy by redirecting T-cells to tumor antigens without lengthy and complex ex vivo cell engineering protocols. ImmTACs are the first soluble TCR-based agents to be approved by the FDA 299. Tebentafusp utilizes a TCR domain specific for an HLA-A*02-presented gp100 epitope to redirect T-cell killing to melanoma cells, which manifests in a clinical benefit of a 14% higher 1-year overall survival in uveal melanoma patients. Other ImmTAC molecules currently in clinical trials include IMC-F106C specific for HLA-A*02/PRAME, and IMC-C103C specific for HLA-A*02/MAGE-A4 for the treatment of advanced solid tumors (NCT04262466, NCT03973333). For AML treatment, RO7283420, a T-cell bispecific in IgG format targeting HLA-A*02/WT1, is currently in phase I trials (NCT04580121).
12. Conclusions
Although the TCR-based therapeutic agents are nearly two decades behind mAb-based agents in their scientific and clinical development, TCR-based agents, whether incorporated into cells or as soluble drugs, are poised to be increasingly important therapies for cancer. Recent advances in understanding TCR structure and recognition features has accelerated their transition into both soluble agents, with platforms similar to mAb such as bispecific agents and engineered cells. In principle, the ability of TCRs to recognize truly cancer-specific epitopes, and intracellular targets, unlike traditional antibodies and most small molecules, opens the door to a new class of potentially non-toxic and effective drugs not previously envisioned. The number of potentially useful targets for TCRs will ultimately dwarf that available to traditional mAb. Already, TCR-based tools are available for transcription factors, cancer-germline antigens, oncofetal antigens, neoantigens, post-translationally modified proteins, tumor-associated antigens, and oncogenically mutated proteins. There is every expectation that drugs for each of these classes of targets will become available for use within the decade. Because TCRs are the natural receptor for T-cells, their use may also provide both better potency and control than CAR-engineered cells. However, a number of open areas of study remain, including: 1. Better understanding of the targets and off-targets of the agents, 2. New ways to render the molecules more stable and with longer plasma half-lives when soluble, 3. Controlling TCR protein signaling and protein associations within engineered cells, 4. Improving approaches to affinity enhancement without loss of specificity, 5. Methods of creating drug- or radio-conjugates that may be clinically useful. 6. Automating and expediting the retrieval of patient’s TCRs. Notably, the pace of discovery of tools and prototypes to address these issues has accelerated, and many academic and industrial laboratories are currently tackling these problems. Therefore, the future appears promising.
Funding Information:
NIH RO1 CA55349, PO1 CA23766, NCI P30 008748, R35 CA241894 and NCI 1R50CA265328-01A1.
Conflict of Interests
DAS is on a board of, or has equity in, or income from: Lantheus, Sellas, Iovance, Pfizer, Actinium Pharmaceuticals, OncoPep, Repertoire, Atengen, Sapience, Coimmune, and Eureka Therapeutics. TD is a consultant for Eureka Therapeutics.
References:
- 1.Newell EW, Davis MM. Beyond model antigens: high-dimensional methods for the analysis of antigen-specific T cells. Nat Biotechnol. 2014;32(2):149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawakami Y, Eliyahu S, Delgado CH, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci U S A. 1994;91(9):3515–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawakami Y, Eliyahu S, Delgado CH, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci U S A. 1994;91(14):6458–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Traversari C, van der Bruggen P, Luescher IF, et al. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J Exp Med. 1992;176(5):1453–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brichard V, Van Pel A, Wölfel T, et al. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. 1993;178(2):489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parkhurst MR, Robbins PF, Tran E, et al. Unique Neoantigens Arise from Somatic Mutations in Patients with Gastrointestinal Cancers. Cancer Discov. 2019;9(8):1022–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowery FJ, Krishna S, Yossef R, et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science. 2022;375(6583):877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zacharakis N, Chinnasamy H, Black M, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. 2018;24(6):724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wells DK, van Buuren MM, Dang KK, et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell. 2020;183(3):818–834.e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gejman RS, Jones HF, Klatt MG, et al. Identification of the Targets of T-cell Receptor Therapeutic Agents and Cells by Use of a High-Throughput Genetic Platform. Cancer Immunol Res. 2020;8(5):672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ataie N, Xiang J, Cheng N, et al. Structure of a TCR-Mimic Antibody with Target Predicts Pharmacogenetics. J Mol Biol. 2016;428(1):194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S, Sun J, Chen K, et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021;19(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarkizova S, Klaeger S, Le PM, et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat Biotechnol. 2020;38(2):199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics. 2015;14(3):658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reid Cahn A, Bhardwaj N, Vabret N. Dark genome, bright ideas: Recent approaches to harness transposable elements in immunotherapies. Cancer Cell. 2022;40(8):792–797. [DOI] [PubMed] [Google Scholar]
- 17.Chandran SS, Ma J, Klatt MG, et al. Immunogenicity and therapeutic targeting of a public neoantigen derived from mutated PIK3CA. Nat Med. 2022;28(5):946–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapuis AG, Egan DN, Bar M, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. 2019;25(7):1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagarsheth NB, Norberg SM, Sinkoe AL, et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med. 2021;27(3):419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naeher D, Daniels MA, Hausmann B, Guillaume P, Luescher I, Palmer E. A constant affinity threshold for T cell tolerance. J Exp Med. 2007;204(11):2553–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. [DOI] [PubMed] [Google Scholar]
- 23.Robinson J, Halliwell JA, McWilliam H, Lopez R, Marsh SG. IPD--the Immuno Polymorphism Database. Nucleic Acids Res. 2013;41(Database issue):D1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis MM. T cell receptor gene diversity and selection. Annu Rev Biochem. 1990;59:475–496. [DOI] [PubMed] [Google Scholar]
- 25.Hill JA, Bell DA, Brintnell W, et al. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. J Exp Med. 2008;205(4):967–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286(5441):958–961. [DOI] [PubMed] [Google Scholar]
- 27.Mason D A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today. 1998;19(9):395–404. [DOI] [PubMed] [Google Scholar]
- 28.Kersh GJ, Allen PM. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J Exp Med. 1996;184(4):1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Relland LM, Mishra MK, Haribhai D, Edwards B, Ziegelbauer J, Williams CB. Affinity-based selection of regulatory T cells occurs independent of agonist-mediated induction of Foxp3 expression. J Immunol. 2009;182(3):1341–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhattacharyya ND, Feng CG. Regulation of T Helper Cell Fate by TCR Signal Strength. Front Immunol. 2020;11:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benhar I, London A, Schwartz M. The privileged immunity of immune privileged organs: the case of the eye. Front Immunol. 2012;3:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao S, von der Weid PY. Lymphatic system: an active pathway for immune protection. Semin Cell Dev Biol. 2015;38:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. Autoreactive T cells in healthy individuals. J Immunol. 2004;172(10):5967–5972. [DOI] [PubMed] [Google Scholar]
- 34.Boehncke WH, Brembilla NC. Autoreactive T-Lymphocytes in Inflammatory Skin Diseases. Front Immunol. 2019;10:1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cameron BJ, Gerry AB, Dukes J, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5(197):197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chodon T, Comin-Anduix B, Chmielowski B, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res. 2014;20(9):2457–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Border EC, Sanderson JP, Weissensteiner T, Gerry AB, Pumphrey NJ. Affinity-enhanced T-cell receptors for adoptive T-cell therapy targeting MAGE-A10: strategy for selection of an optimal candidate. Oncoimmunology. 2019;8(2):e1532759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daniels MA, Teixeiro E, Gill J, et al. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature. 2006;444(7120):724–729. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Moysey R, Molloy PE, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005;23(3):349–354. [DOI] [PubMed] [Google Scholar]
- 42.Schmid DA, Irving MB, Posevitz V, et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol. 2010;184(9):4936–4946. [DOI] [PubMed] [Google Scholar]
- 43.Zhong S, Malecek K, Johnson LA, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci U S A. 2013;110(17):6973–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48(W1):W449–w454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Styczynski MP, Jensen KL, Rigoutsos I, Stephanopoulos G. BLOSUM62 miscalculations improve search performance. Nat Biotechnol. 2008;26(3):274–275. [DOI] [PubMed] [Google Scholar]
- 47.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89(22):10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nielsen M, Lundegaard C, Worning P, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12(5):1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klatt MG, Mack KN, Bai Y, et al. Solving an MHC allele-specific bias in the reported immunopeptidome. JCI Insight. 2020;5(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boehm KM, Bhinder B, Raja VJ, Dephoure N, Elemento O. Predicting peptide presentation by major histocompatibility complex class I: an improved machine learning approach to the immunopeptidome. BMC Bioinformatics. 2019;20(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marcu A, Bichmann L, Kuchenbecker L, et al. HLA Ligand Atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J Immunother Cancer. 2021;9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malecek K, Zhong S, McGary K, et al. Engineering improved T cell receptors using an alanine-scan guided T cell display selection system. J Immunol Methods. 2013;392(1–2):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee CH, Salio M, Napolitani G, Ogg G, Simmons A, Koohy H. Predicting Cross-Reactivity and Antigen Specificity of T Cell Receptors. Front Immunol. 2020;11:565096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gray BP, Brown KC. Combinatorial peptide libraries: mining for cell-binding peptides. Chem Rev. 2014;114(2):1020–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holler PD, Holman PO, Shusta EV, O’Herrin S, Wittrup KD, Kranz DM. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci U S A. 2000;97(10):5387–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chlewicki LK, Holler PD, Monti BC, Clutter MR, Kranz DM. High-affinity, peptide-specific T cell receptors can be generated by mutations in CDR1, CDR2 or CDR3. J Mol Biol. 2005;346(1):223–239. [DOI] [PubMed] [Google Scholar]
- 57.Dunn SM, Rizkallah PJ, Baston E, et al. Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci. 2006;15(4):710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joglekar AV, Leonard MT, Jeppson JD, et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat Methods. 2019;16(2):191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liddy N, Bossi G, Adams KJ, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med. 2012;18(6):980–987. [DOI] [PubMed] [Google Scholar]
- 60.Rapoport AP, Stadtmauer EA, Binder-Scholl GK, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21(8):914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dolgin E First soluble TCR therapy opens ‘new universe’ of cancer targets. Nat Biotechnol. 2022;40(4):441–444. [DOI] [PubMed] [Google Scholar]
- 62.Wallace Z, Singh PK, Dorrell L. Combination strategies to durably suppress HIV-1: Soluble T cell receptors. J Virus Erad. 2022;8(3):100082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fergusson JR, Wallace Z, Connolly MM, et al. Immune-Mobilizing Monoclonal T Cell Receptors Mediate Specific and Rapid Elimination of Hepatitis B-Infected Cells. Hepatology. 2020;72(5):1528–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scheinberg DA, Dao T, Liu C. Reaching un-drugable intracellular targets with the long arm of antibodies. Oncotarget. 2013;4(5):647–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dao T, Liu C, Scheinberg DA. Approaching untargetable tumor-associated antigens with antibodies. Oncoimmunology. 2013;2(7):e24678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6(6):715–726. [DOI] [PubMed] [Google Scholar]
- 67.Sergeeva A, Alatrash G, He H, et al. An anti-PR1/HLA-A2 T-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood. 2011;117(16):4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dao T, Yan S, Veomett N, et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med. 2013;5(176):176ra133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dao T, Pankov D, Scott A, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol. 2015;33(10):1079–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rafiq S, Purdon TJ, Daniyan AF, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia. 2017;31(8):1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu H, Xu Y, Xiang J, et al. Targeting Alpha-Fetoprotein (AFP)-MHC Complex with CAR T-Cell Therapy for Liver Cancer. Clin Cancer Res. 2017;23(2):478–488. [DOI] [PubMed] [Google Scholar]
- 72.Lowe KL, Cole D, Kenefeck R, I OK, Lepore M, Jakobsen BK. Novel TCR-based biologics: mobilising T cells to warm ‘cold’ tumours. Cancer Treat Rev. 2019;77:35–43. [DOI] [PubMed] [Google Scholar]
- 73.Segues A, Huang S, Sijts A, Berraondo P, Zaiss DM. Opportunities and challenges of bi-specific antibodies. Int Rev Cell Mol Biol. 2022;369:45–70. [DOI] [PubMed] [Google Scholar]
- 74.Augsberger C, Hanel G, Xu W, et al. Targeting intracellular WT1 in AML with a novel RMF-peptide-MHC-specific T-cell bispecific antibody. Blood. 2021;138(25):2655–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanford M Blinatumomab: first global approval. Drugs. 2015;75(3):321–327. [DOI] [PubMed] [Google Scholar]
- 76.Choi BD, Yu X, Castano AP, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37(9):1049–1058. [DOI] [PubMed] [Google Scholar]
- 77.Dao T, Mun SS, Molvi Z, et al. A TCR mimic monoclonal antibody reactive with the “public” phospho-neoantigen pIRS2/HLA-A*02:01 complex. JCI Insight. 2022;7(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hulsmeyer M, Chames P, Hillig RC, et al. A major histocompatibility complex-peptide-restricted antibody and t cell receptor molecules recognize their target by distinct binding modes: crystal structure of human leukocyte antigen (HLA)-A1-MAGE-A1 in complex with FAB-HYB3. J Biol Chem. 2005;280(4):2972–2980. [DOI] [PubMed] [Google Scholar]
- 79.Stewart-Jones G, Wadle A, Hombach A, et al. Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci U S A. 2009;106(14):5784–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chang AY, Dao T, Gejman RS, et al. A therapeutic T cell receptor mimic antibody targets tumor-associated PRAME peptide/HLA-I antigens. J Clin Invest. 2017;127(7):2705–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gras S, Chadderton J, Del Campo CM, et al. Reversed T Cell Receptor Docking on a Major Histocompatibility Class I Complex Limits Involvement in the Immune Response. Immunity. 2016;45(4):749–760. [DOI] [PubMed] [Google Scholar]
- 82.Yang X, Nishimiya D, Löchte S, et al. Facile repurposing of peptide-MHC-restricted antibodies for cancer immunotherapy. Nat Biotechnol. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yin Y, Mariuzza RA. The multiple mechanisms of T cell receptor cross-reactivity. Immunity. 2009;31(6):849–851. [DOI] [PubMed] [Google Scholar]
- 84.Riley TP, Hellman LM, Gee MH, et al. T cell receptor cross-reactivity expanded by dramatic peptide-MHC adaptability. Nat Chem Biol. 2018;14(10):934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Szabó Á, Szöllősi J, Nagy P. Principles of Resonance Energy Transfer. Curr Protoc. 2022;2(12):e625. [DOI] [PubMed] [Google Scholar]
- 86.Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol. 2011;9:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arkin IT. Isotope-edited IR spectroscopy for the study of membrane proteins. Curr Opin Chem Biol. 2006;10(5):394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tokunaga Y, Takeuchi K. Role of NMR in High Ordered Structure Characterization of Monoclonal Antibodies. Int J Mol Sci. 2020;22(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saxena M, van der Burg SH, Melief CJM, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021;21(6):360–378. [DOI] [PubMed] [Google Scholar]
- 90.Maslak PG, Dao T, Bernal Y, et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. 2018;2(3):224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zauderer MG, Tsao AS, Dao T, et al. A Randomized Phase II Trial of Adjuvant Galinpepimut-S, WT-1 Analogue Peptide Vaccine, After Multimodality Therapy for Patients with Malignant Pleural Mesothelioma. Clin Cancer Res. 2017;23(24):7483–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Z, Lu M, Qin Y, et al. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front Immunol. 2021;12:672356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cercek A, Lumish M, Sinopoli J, et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N Engl J Med. 2022;386(25):2363–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tran E, Robbins PF, Lu YC, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375(23):2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cafri G, Gartner JJ, Zaks T, et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J Clin Invest. 2020;130(11):5976–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carreno BM, Magrini V, Becker-Hapak M, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ott PA, Hu-Lieskovan S, Chmielowski B, et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell. 2020;183(2):347–362.e324. [DOI] [PubMed] [Google Scholar]
- 98.Malekzadeh P, Pasetto A, Robbins PF, et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J Clin Invest. 2019;129(3):1109–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Levin N, Paria BC, Vale NR, et al. Identification and Validation of T-cell Receptors Targeting RAS Hotspot Mutations in Human Cancers for Use in Cell-based Immunotherapy. Clin Cancer Res. 2021;27(18):5084–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zinkernagel RM. Cellular immune recognition and the biological role of major transplantation antigens. Biosci Rep. 1997;17(2):91–111. [DOI] [PubMed] [Google Scholar]
- 101.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67(8):3898–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66(17):8878–8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thomas S, Mohammed F, Reijmers RM, et al. Framework engineering to produce dominant T cell receptors with enhanced antigen-specific function. Nat Commun. 2019;10(1):4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bethune MT, Gee MH, Bunse M, et al. Domain-swapped T cell receptors improve the safety of TCR gene therapy. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Morton LT, Reijmers RM, Wouters AK, et al. Simultaneous Deletion of Endogenous TCRalphabeta for TCR Gene Therapy Creates an Improved and Safe Cellular Therapeutic. Mol Ther. 2020;28(1):64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kamali E, Rahbarizadeh F, Hojati Z, Frodin M. CRISPR/Cas9-mediated knockout of clinically relevant alloantigenes in human primary T cells. BMC Biotechnol. 2021;21(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70(21):8368–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Johnson LA, Heemskerk B, Powell DJ Jr., et al. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. 2006;177(9):6548–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. 2016;22(1):26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhu W, Peng Y, Wang L, et al. Identification of alpha-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology. 2018;68(2):574–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chhabra A MHC class I TCR engineered anti-tumor CD4 T cells: implications for cancer immunotherapy. Endocr Metab Immune Disord Drug Targets. 2009;9(4):344–352. [DOI] [PubMed] [Google Scholar]
- 113.Rath JA, Bajwa G, Carreres B, et al. Single-cell transcriptomics identifies multiple pathways underlying antitumor function of TCR- and CD8alphabeta-engineered human CD4(+) T cells. Sci Adv. 2020;6(27):eaaz7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xue SA, Gao L, Ahmadi M, et al. Human MHC Class I-restricted high avidity CD4(+) T cells generated by co-transfer of TCR and CD8 mediate efficient tumor rejection in vivo. Oncoimmunology. 2013;2(1):e22590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dong R, Zhang Y, Xiao H, Zeng X. Engineering gammadelta T Cells: Recognizing and Activating on Their Own Way. Front Immunol. 2022;13:889051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fisher J, Anderson J. Engineering Approaches in Human Gamma Delta T Cells for Cancer Immunotherapy. Front Immunol. 2018;9:1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Saito T, Hochstenbach F, Marusic-Galesic S, Kruisbeek AM, Brenner M, Germain RN. Surface expression of only gamma delta and/or alpha beta T cell receptor heterodimers by cells with four (alpha, beta, gamma, delta) functional receptor chains. J Exp Med. 1988;168(3):1003–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dorrie J, Krug C, Hofmann C, et al. Human adenovirus-specific gamma/delta and CD8+ T cells generated by T-cell receptor transfection to treat adenovirus infection after allogeneic stem cell transplantation. PLoS One. 2014;9(10):e109944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.van der Veken LT, Hagedoorn RS, van Loenen MM, Willemze R, Falkenburg JH, Heemskerk MH. Alphabeta T-cell receptor engineered gammadelta T cells mediate effective antileukemic reactivity. Cancer Res. 2006;66(6):3331–3337. [DOI] [PubMed] [Google Scholar]
- 120.Shimizu K, Shinga J, Yamasaki S, et al. Transfer of mRNA Encoding Invariant NKT Cell Receptors Imparts Glycolipid Specific Responses to T Cells and gammadeltaT Cells. PLoS One. 2015;10(6):e0131477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Saura-Esteller J, de Jong M, King LA, et al. Gamma Delta T-Cell Based Cancer Immunotherapy: Past-Present-Future. Front Immunol. 2022;13:915837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kabelitz D, Serrano R, Kouakanou L, Peters C, Kalyan S. Correction to: Cancer immunotherapy with gammadelta T cells: many paths ahead of us. Cell Mol Immunol. 2020;17(10):1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marcu-Malina V, Heijhuurs S, van Buuren M, et al. Redirecting alphabeta T cells against cancer cells by transfer of a broadly tumor-reactive gammadeltaT-cell receptor. Blood. 2011;118(1):50–59. [DOI] [PubMed] [Google Scholar]
- 124.Zhang Y, Huang B. The Development and Diversity of ILCs, NK Cells and Their Relevance in Health and Diseases. Adv Exp Med Biol. 2017;1024:225–244. [DOI] [PubMed] [Google Scholar]
- 125.Prager I, Liesche C, van Ooijen H, et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med. 2019;216(9):2113–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Prager I, Watzl C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J Leukoc Biol. 2019;105(6):1319–1329. [DOI] [PubMed] [Google Scholar]
- 127.Alderson KL, Sondel PM. Clinical cancer therapy by NK cells via antibody-dependent cell-mediated cytotoxicity. J Biomed Biotechnol. 2011;2011:379123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roda JM, Parihar R, Magro C, Nuovo GJ, Tridandapani S, Carson WE 3rd. Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res. 2006;66(1):517–526. [DOI] [PubMed] [Google Scholar]
- 129.Miller JS, Soignier Y, Panoskaltsis-Mortari A, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–3057. [DOI] [PubMed] [Google Scholar]
- 130.Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–2100. [DOI] [PubMed] [Google Scholar]
- 131.Tonn T, Schwabe D, Klingemann HG, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15(12):1563–1570. [DOI] [PubMed] [Google Scholar]
- 132.Arai S, Meagher R, Swearingen M, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10(6):625–632. [DOI] [PubMed] [Google Scholar]
- 133.Daher M, Rezvani K. Outlook for New CAR-Based Therapies with a Focus on CAR NK Cells: What Lies Beyond CAR-Engineered T Cells in the Race against Cancer. Cancer Discov. 2021;11(1):45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mensali N, Dillard P, Hebeisen M, et al. NK cells specifically TCR-dressed to kill cancer cells. EBioMedicine. 2019;40:106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Parlar A, Sayitoglu EC, Ozkazanc D, et al. Engineering antigen-specific NK cell lines against the melanoma-associated antigen tyrosinase via TCR gene transfer. Eur J Immunol. 2019;49(8):1278–1290. [DOI] [PubMed] [Google Scholar]
- 136.Salio M, Silk JD, Jones EY, Cerundolo V. Biology of CD1- and MR1-restricted T cells. Annu Rev Immunol. 2014;32:323–366. [DOI] [PubMed] [Google Scholar]
- 137.Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol. 2007;5(6):405–417. [DOI] [PubMed] [Google Scholar]
- 138.Smyth MJ, Thia KY, Street SE, et al. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191(4):661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gorini F, Azzimonti L, Delfanti G, et al. Invariant NKT cells contribute to chronic lymphocytic leukemia surveillance and prognosis. Blood. 2017;129(26):3440–3451. [DOI] [PubMed] [Google Scholar]
- 140.Wu L, Van Kaer L. Natural killer T cells in health and disease. Front Biosci (Schol Ed). 2011;3(1):236–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Exley MA, Friedlander P, Alatrakchi N, et al. Adoptive Transfer of Invariant NKT Cells as Immunotherapy for Advanced Melanoma: A Phase I Clinical Trial. Clin Cancer Res. 2017;23(14):3510–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wolf BJ, Choi JE, Exley MA. Novel Approaches to Exploiting Invariant NKT Cells in Cancer Immunotherapy. Front Immunol. 2018;9:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Simonetta F, Lohmeyer JK, Hirai T, et al. Allogeneic CAR Invariant Natural Killer T Cells Exert Potent Antitumor Effects through Host CD8 T-Cell Cross-Priming. Clin Cancer Res. 2021;27(21):6054–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Delfanti G, Dellabona P, Casorati G, Fedeli M. Adoptive Immunotherapy With Engineered iNKT Cells to Target Cancer Cells and the Suppressive Microenvironment. Front Med (Lausanne). 2022;9:897750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nelson A, Lukacs JD, Johnston B. The Current Landscape of NKT Cell Immunotherapy and the Hills Ahead. Cancers (Basel). 2021;13(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Courtney AN, Tian G, Metelitsa LS. Natural killer T cells and other innate-like T lymphocytes as emerging platforms for allogeneic cancer cell therapy. Blood. 2023;141(8):869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ramos CA, Courtney AN, Robinson SN, et al. Allogeneic NKT Cells Expressing a CD19-Specific CAR in Patients with Relapsed or Refractory B-Cell Malignancies: An Interim Analysis. Blood. 2021;138.33410895 [Google Scholar]
- 148.Delfanti G, Cortesi F, Perini A, et al. TCR-engineered iNKT cells induce robust antitumor response by dual targeting cancer and suppressive myeloid cells. Sci Immunol. 2022;7(74):eabn6563. [DOI] [PubMed] [Google Scholar]
- 149.Garofano F, Gonzalez-Carmona MA, Skowasch D, et al. Clinical Trials with Combination of Cytokine-Induced Killer Cells and Dendritic Cells for Cancer Therapy. Int J Mol Sci. 2019;20(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Guo Y, Han W. Cytokine-induced killer (CIK) cells: from basic research to clinical translation. Chin J Cancer. 2015;34(3):99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pievani A, Borleri G, Pende D, et al. Dual-functional capability of CD3(+)CD56(+) CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. 2011;118(12):3301–3310. [DOI] [PubMed] [Google Scholar]
- 152.Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. 1991;174(1):139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Linn YC, Wang SM, Hui KM. Comparative gene expression profiling of cytokine-induced killer cells in response to acute myloid leukemic and acute lymphoblastic leukemic stimulators using oligonucleotide arrays. Exp Hematol. 2005;33(6):671–681. [DOI] [PubMed] [Google Scholar]
- 154.Kuci S, Rettinger E, Voss B, et al. Efficient lysis of rhabdomyosarcoma cells by cytokine-induced killer cells: implications for adoptive immunotherapy after allogeneic stem cell transplantation. Haematologica. 2010;95(9):1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Durrieu L, Lemieux W, Dieng MM, et al. Implication of different effector mechanisms by cord blood-derived and peripheral blood-derived cytokine-induced killer cells to kill precursor B acute lymphoblastic leukemia cell lines. Cytotherapy. 2014;16(6):845–856. [DOI] [PubMed] [Google Scholar]
- 156.Elia AR, Circosta P, Sangiolo D, et al. Cytokine-induced killer cells engineered with exogenous T-cell receptors directed against melanoma antigens: enhanced efficacy of effector cells endowed with a double mechanism of tumor recognition. Hum Gene Ther. 2015;26(4):220–231. [DOI] [PubMed] [Google Scholar]
- 157.Gschweng E, De Oliveira S, Kohn DB. Hematopoietic stem cells for cancer immunotherapy. Immunol Rev. 2014;257(1):237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhao Y, Parkhurst MR, Zheng Z, et al. Extrathymic generation of tumor-specific T cells from genetically engineered human hematopoietic stem cells via Notch signaling. Cancer Res. 2007;67(6):2425–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Perkey E, Maillard I. New Insights into Graft-Versus-Host Disease and Graft Rejection. Annu Rev Pathol. 2018;13:219–245. [DOI] [PubMed] [Google Scholar]
- 160.Nissani A, Lev-Ari S, Meirson T, et al. Comparison of non-myeloablative lymphodepleting preconditioning regimens in patients undergoing adoptive T cell therapy. J Immunother Cancer. 2021;9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hoerster K, Uhrberg M, Wiek C, Horn PA, Hanenberg H, Heinrichs S. HLA Class I Knockout Converts Allogeneic Primary NK Cells Into Suitable Effectors for “Off-the-Shelf” Immunotherapy. Front Immunol. 2020;11:586168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mattapally S, Pawlik KM, Fast VG, et al. Human Leukocyte Antigen Class I and II Knockout Human Induced Pluripotent Stem Cell-Derived Cells: Universal Donor for Cell Therapy. J Am Heart Assoc. 2018;7(23):e010239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gornalusse GG, Hirata RK, Funk SE, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol. 2017;35(8):765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Torikai H, Reik A, Liu PQ, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119(24):5697–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mo F, Watanabe N, McKenna MK, et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat Biotechnol. 2021;39(1):56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Peraro L, Bourne CM, Dacek MM, et al. Incorporation of bacterial immunoevasins to protect cell therapies from host antibody-mediated immune rejection. Mol Ther. 2021;29(12):3398–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Deuse T, Hu X, Agbor-Enoh S, et al. The SIRPalpha-CD47 immune checkpoint in NK cells. J Exp Med. 2021;218(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhao W, Lei A, Tian L, et al. Strategies for Genetically Engineering Hypoimmunogenic Universal Pluripotent Stem Cells. iScience. 2020;23(6):101162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Finck AV, Blanchard T, Roselle CP, Golinelli G, June CH. Engineered cellular immunotherapies in cancer and beyond. Nat Med. 2022;28(4):678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Teppert K, Wang X, Anders K, Evaristo C, Lock D, Künkele A. Joining Forces for Cancer Treatment: From “TCR versus CAR” to “TCR and CAR”. Int J Mol Sci. 2022;23(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wei F, Cheng XX, Xue JZ, Xue SA. Emerging Strategies in TCR-Engineered T Cells. Front Immunol. 2022;13:850358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lamers CH, Sleijfer S, van Steenbergen S, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21(4):904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Malviya M, Saoudi A, Bauer J, Fillatreau S, Liblau R. Treatment of experimental autoimmune encephalomyelitis with engineered bi-specific Foxp3+ regulatory CD4+ T cells. J Autoimmun. 2020;108:102401. [DOI] [PubMed] [Google Scholar]
- 175.Campana LG, Mansoor W, Hill J, et al. T-Cell Infiltration and Clonality May Identify Distinct Survival Groups in Colorectal Cancer: Development and Validation of a Prognostic Model Based on The Cancer Genome Atlas (TCGA) and Clinical Proteomic Tumor Analysis Consortium (CPTAC). Cancers (Basel). 2022;14(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Crowther MD, Dolton G, Legut M, et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat Immunol. 2020;21(2):178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Oh SA, Seki A, Rutz S. Ribonucleoprotein Transfection for CRISPR/Cas9-Mediated Gene Knockout in Primary T Cells. Curr Protoc Immunol. 2019;124(1):e69. [DOI] [PubMed] [Google Scholar]
- 178.Xu Y, Yang Z, Horan LH, et al. A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discov. 2018;4:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.He P, Liu H, Zimdahl B, et al. A novel antibody-TCR (AbTCR) T-cell therapy is safe and effective against CD19-positive relapsed/refractory B-cell lymphoma. J Cancer Res Clin Oncol.. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Anderson P, Aptsiauri N, Ruiz-Cabello F, Garrido F. HLA class I loss in colorectal cancer: implications for immune escape and immunotherapy. Cell Mol Immunol. 2021;18(3):556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hazini A, Fisher K, Seymour L. Deregulation of HLA-I in cancer and its central importance for immunotherapy. J Immunother Cancer. 2021;9(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Montesion M, Murugesan K, Jin DX, et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discov. 2021;11(2):282–292. [DOI] [PubMed] [Google Scholar]
- 183.Maleno I, Lopez-Nevot MA, Cabrera T, Salinero J, Garrido F. Multiple mechanisms generate HLA class I altered phenotypes in laryngeal carcinomas: high frequency of HLA haplotype loss associated with loss of heterozygosity in chromosome region 6p21. Cancer Immunol Immunother. 2002;51(7):389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Maleno I, Romero JM, Cabrera T, et al. LOH at 6p21.3 region and HLA class I altered phenotypes in bladder carcinomas. Immunogenetics. 2006;58(7):503–510. [DOI] [PubMed] [Google Scholar]
- 185.Seliger B, Ritz U, Abele R, et al. Immune escape of melanoma: first evidence of structural alterations in two distinct components of the MHC class I antigen processing pathway. Cancer Res. 2001;61(24):8647–8650. [PubMed] [Google Scholar]
- 186.Meissner M, Reichert TE, Kunkel M, et al. Defects in the human leukocyte antigen class I antigen processing machinery in head and neck squamous cell carcinoma: association with clinical outcome. Clin Cancer Res. 2005;11(7):2552–2560. [DOI] [PubMed] [Google Scholar]
- 187.Squire R, Fowler CL, Brooks SP, Rich GA, Cooney DR. The relationship of class I MHC antigen expression to stage IV-S disease and survival in neuroblastoma. J Pediatr Surg. 1990;25(4):381–386. [DOI] [PubMed] [Google Scholar]
- 188.Cordon-Cardo C, Fuks Z, Drobnjak M, Moreno C, Eisenbach L, Feldman M. Expression of HLA-A,B,C antigens on primary and metastatic tumor cell populations of human carcinomas. Cancer Res. 1991;51(23 Pt 1):6372–6380. [PubMed] [Google Scholar]
- 189.Park HS, Cho U, Im SY, et al. Loss of Human Leukocyte Antigen Class I Expression Is Associated with Poor Prognosis in Patients with Advanced Breast Cancer. J Pathol Transl Med. 2019;53(2):75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Paulson KG, Voillet V, McAfee MS, et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun. 2018;9(1):3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gurjao C, Liu D, Hofree M, et al. Intrinsic Resistance to Immune Checkpoint Blockade in a Mismatch Repair-Deficient Colorectal Cancer. Cancer Immunol Res. 2019;7(8):1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Luo N, Formisano L, Gonzalez-Ericsson PI, et al. Melanoma response to anti-PD-L1 immunotherapy requires JAK1 signaling, but not JAK2. Oncoimmunology. 2018;7(6):e1438106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Taylor BC, Balko JM. Mechanisms of MHC-I Downregulation and Role in Immunotherapy Response. Frontiers in Immunology. 2022;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Frontiers in Immunology. 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yu S, Zhao Z, Chen L, et al. HLA loss of heterozygosity-mediated discordant responses to immune checkpoint blockade in squamous cell lung cancer with renal metastasis. Immunotherapy. 2021;13(3):195–200. [DOI] [PubMed] [Google Scholar]
- 196.Chowell D, Morris LGT, Grigg CM, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359(6375):582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Chang CC, Pirozzi G, Wen SH, et al. Multiple structural and epigenetic defects in the human leukocyte antigen class I antigen presentation pathway in a recurrent metastatic melanoma following immunotherapy. J Biol Chem. 2015;290(44):26562–26575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Maeurer MJ, Gollin SM, Storkus WJ, et al. Tumor escape from immune recognition: loss of HLA-A2 melanoma cell surface expression is associated with a complex rearrangement of the short arm of chromosome 6. Clin Cancer Res. 1996;2(4):641–652. [PubMed] [Google Scholar]
- 199.Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375(9):819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Toth DF, Raderer M, Wadsak W, Karanikas G. Beta-2 microglobulin as a diagnostic parameter in non-Hodgkin lymphoma: a comparative study with FDG-PET. Anticancer Res. 2013;33(8):3341–3345. [PubMed] [Google Scholar]
- 202.Middha S, Yaeger R, Shia J, et al. Majority of B2M-Mutant and -Deficient Colorectal Carcinomas Achieve Clinical Benefit From Immune Checkpoint Inhibitor Therapy and Are Microsatellite Instability-High. JCO Precis Oncol. 2019;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Castro A, Ozturk K, Pyke RM, Xian S, Zanetti M, Carter H. Elevated neoantigen levels in tumors with somatic mutations in the HLA-A, HLA-B, HLA-C and B2M genes. BMC Med Genomics. 2019;12(Suppl 6):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ling A, Löfgren-Burström A, Larsson P, et al. TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. Oncoimmunology. 2017;6(11):e1356143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kaklamanis L, Townsend A, Doussis-Anagnostopoulou IA, Mortensen N, Harris AL, Gatter KC. Loss of major histocompatibility complex-encoded transporter associated with antigen presentation (TAP) in colorectal cancer. Am J Pathol. 1994;145(3):505–509. [PMC free article] [PubMed] [Google Scholar]
- 206.Cromme FV, Airey J, Heemels MT, et al. Loss of transporter protein, encoded by the TAP-1 gene, is highly correlated with loss of HLA expression in cervical carcinomas. Journal of Experimental Medicine. 1994;179(1):335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tanaka K, Tsuchikawa T, Miyamoto M, et al. Down-regulation of Human Leukocyte Antigen class I heavy chain in tumors is associated with a poor prognosis in advanced esophageal cancer patients. Int J Oncol. 2012;40(4):965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Shionoya Y, Kanaseki T, Miyamoto S, et al. Loss of tapasin in human lung and colon cancer cells and escape from tumor-associated antigen-specific CTL recognition. Oncoimmunology. 2017;6(2):e1274476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mehta AM, Jordanova ES, Kenter GG, Ferrone S, Fleuren GJ. Association of antigen processing machinery and HLA class I defects with clinicopathological outcome in cervical carcinoma. Cancer Immunol Immunother. 2008;57(2):197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Stratikos E, Stamogiannos A, Zervoudi E, Fruci D. A role for naturally occurring alleles of endoplasmic reticulum aminopeptidases in tumor immunity and cancer pre-disposition. Front Oncol. 2014;4:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Chang CH, Hammer J, Loh JE, Fodor WL, Flavell RA. The activation of major histocompatibility complex class I genes by interferon regulatory factor-1 (IRF-1). Immunogenetics. 1992;35(6):378–384. [DOI] [PubMed] [Google Scholar]
- 212.Majoros A, Platanitis E, Kernbauer-Hölzl E, Rosebrock F, Müller M, Decker T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front Immunol. 2017;8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sade-Feldman M, Jiao YJ, Chen JH, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8(1):1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gao J, Shi LZ, Zhao H, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167(2):397–404.e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Patel SJ, Sanjana NE, Kishton RJ, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548(7669):537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yoshihama S, Roszik J, Downs I, et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc Natl Acad Sci U S A. 2016;113(21):5999–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nie Y, Yang G, Song Y, et al. DNA hypermethylation is a mechanism for loss of expression of the HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis. 2001;22(10):1615–1623. [DOI] [PubMed] [Google Scholar]
- 218.Ye Q, Shen Y, Wang X, et al. Hypermethylation of HLA class I gene is associated with HLA class I down-regulation in human gastric cancer. Tissue Antigens. 2010;75(1):30–39. [DOI] [PubMed] [Google Scholar]
- 219.Luo N, Nixon MJ, Gonzalez-Ericsson PI, et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun. 2018;9(1):248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Srivastava P, Paluch BE, Matsuzaki J, et al. Immunomodulatory action of the DNA methyltransferase inhibitor SGI-110 in epithelial ovarian cancer cells and xenografts. Epigenetics. 2015;10(3):237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Saleh MH, Wang L, Goldberg MS. Improving cancer immunotherapy with DNA methyltransferase inhibitors. Cancer Immunol Immunother. 2016;65(7):787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26(37):5420–5432. [DOI] [PubMed] [Google Scholar]
- 223.Moufarrij S, Srivastava A, Gomez S, et al. Combining DNMT and HDAC6 inhibitors increases anti-tumor immune signaling and decreases tumor burden in ovarian cancer. Sci Rep. 2020;10(1):3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mora-García Mde L, Duenas-González A, Hernández-Montes J, et al. Up-regulation of HLA class-I antigen expression and antigen-specific CTL response in cervical cancer cells by the demethylating agent hydralazine and the histone deacetylase inhibitor valproic acid. J Transl Med. 2006;4:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fan F, Lu J, Yu W, et al. MicroRNA-26b-5p regulates cell proliferation, invasion and metastasis in human intrahepatic cholangiocarcinoma by targeting S100A7. Oncol Lett. 2018;15(1):386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lazaridou MF, Gonschorek E, Massa C, et al. Identification of miR-200a-5p targeting the peptide transporter TAP1 and its association with the clinical outcome of melanoma patients. Oncoimmunology. 2020;9(1):1774323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Mari L, Hoefnagel SJM, Zito D, et al. microRNA 125a Regulates MHC-I Expression on Esophageal Adenocarcinoma Cells, Associated With Suppression of Antitumor Immune Response and Poor Outcomes of Patients. Gastroenterology. 2018;155(3):784–798. [DOI] [PubMed] [Google Scholar]
- 228.Colangelo T, Polcaro G, Ziccardi P, et al. Proteomic screening identifies calreticulin as a miR-27a direct target repressing MHC class I cell surface exposure in colorectal cancer. Cell Death Dis. 2016;7(2):e2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wang Y, Wang X, Cui X, et al. Oncoprotein SND1 hijacks nascent MHC-I heavy chain to ER-associated degradation, leading to impaired CD8(+) T cell response in tumor. Sci Adv. 2020;6(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Yamamoto K, Venida A, Yano J, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581(7806):100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Fang Y, Wang L, Wan C, et al. MAL2 drives immune evasion in breast cancer by suppressing tumor antigen presentation. The Journal of Clinical Investigation. 2021;131(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Brea EJ, Oh CY, Manchado E, et al. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunology Research. 2016;4(11):936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.van ‘t Veer LJ, Beijersbergen RL, Bernards R. N-myc suppresses major histocompatibility complex class I gene expression through down-regulation of the p50 subunit of NF-kappa B. Embo j. 1993;12(1):195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Bernards R, Dessain SK, Weinberg RA. N-myc amplification causes down-modulation of MHC class I antigen expression in neuroblastoma. Cell. 1986;47(5):667–674. [DOI] [PubMed] [Google Scholar]
- 235.Versteeg R, Noordermeer IA, Krüse-Wolters M, Ruiter DJ, Schrier PI. c-myc down-regulates class I HLA expression in human melanomas. Embo j. 1988;7(4):1023–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sethumadhavan S, Silva M, Philbrook P, et al. Hypoxia and hypoxia-inducible factor (HIF) downregulate antigen-presenting MHC class I molecules limiting tumor cell recognition by T cells. PLoS One. 2017;12(11):e0187314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Schuren AB, Costa AI, Wiertz EJ. Recent advances in viral evasion of the MHC Class I processing pathway. Curr Opin Immunol. 2016;40:43–50. [DOI] [PubMed] [Google Scholar]
- 238.van de Weijer ML, Luteijn RD, Wiertz EJ. Viral immune evasion: Lessons in MHC class I antigen presentation. Semin Immunol. 2015;27(2):125–137. [DOI] [PubMed] [Google Scholar]
- 239.Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer Journal. 2021;11(4):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gardner TJ, Bourne CM, Dacek MM, et al. Targeted Cellular Micropharmacies: Cells Engineered for Localized Drug Delivery. Cancers (Basel). 2020;12(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Hawkins ER, D’Souza RR, Klampatsa A. Armored CAR T-Cells: The Next Chapter in T-Cell Cancer Immunotherapy. Biologics. 2021;15:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Rebecca CA, Hannah EH- P, Misty RJ. To go or not to go? Biological logic gating engineered T cells. Journal for ImmunoTherapy of Cancer. 2022;10(4):e004185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Yu H, Yang Z, Li F, Xu L, Sun Y. Cell-mediated targeting drugs delivery systems. Drug Deliv. 2020;27(1):1425–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Suarez ER, Chang de K, Sun J, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7(23):34341–34355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Yin Y, Boesteanu AC, Binder ZA, et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Molecular Therapy - Oncolytics. 2018;11:20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Xie YJ, Dougan M, Jailkhani N, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proceedings of the National Academy of Sciences. 2019;116(16):7624–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Rafiq S, Yeku OO, Jackson HJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nature Biotechnology. 2018;36(9):847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Li S, Siriwon N, Zhang X, et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor–Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clinical Cancer Research. 2017;23(22):6982–6992. [DOI] [PubMed] [Google Scholar]
- 249.Harrasser M, Gohil SH, Lau H, et al. Inducible localized delivery of an anti-PD-1 scFv enhances anti-tumor activity of ROR1 CAR-T cells in TNBC. Breast Cancer Research. 2022;24(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Choi BD, Yu X, Castano AP, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nature Biotechnology. 2019;37(9):1049–1058. [DOI] [PubMed] [Google Scholar]
- 251.Iwahori K, Kakarla S, Velasquez MP, et al. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Mol Ther. 2015;23(1):171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Velasquez MP, Torres D, Iwahori K, et al. T cells expressing CD19-specific Engager Molecules for the Immunotherapy of CD19-positive Malignancies. Sci Rep. 2016;6:27130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Bonifant CL, Szoor A, Torres D, et al. CD123-Engager T Cells as a Novel Immunotherapeutic for Acute Myeloid Leukemia. Mol Ther. 2016;24(9):1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Heemskerk B, Liu K, Dudley ME, et al. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum Gene Ther. 2008;19(5):496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Hsu C, Hughes MS, Zheng Z, Bray RB, Rosenberg SA, Morgan RA. Primary Human T Lymphocytes Engineered with a Codon-Optimized IL-15 Gene Resist Cytokine Withdrawal-Induced Apoptosis and Persist Long-Term in the Absence of Exogenous Cytokine1. The Journal of Immunology. 2005;175(11):7226–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hoyos V, Savoldo B, Quintarelli C, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kerkar SP, Muranski P, Kaiser A, et al. Tumor-Specific CD8+ T Cells Expressing Interleukin-12 Eradicate Established Cancers in Lymphodepleted Hosts. Cancer Research. 2010;70(17):6725–6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4(3):e994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Molecular Therapy - Oncolytics. 2018;8:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Pegram HJ, Lee JC, Hayman EG, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Reports. 2017;21(11):3205–3219. [DOI] [PubMed] [Google Scholar]
- 262.Avanzi MP, G. van Leeuwen D, Li X, et al. IL-18 Secreting CAR T Cells Enhance Cell Persistence, Induce Prolonged B Cell Aplasia and Eradicate CD19+ Tumor Cells without Need for Prior Conditioning. Blood. 2016;128(22):816–816.27301861 [Google Scholar]
- 263.Lai J, Mardiana S, House IG, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nature Immunology. 2020;21(8):914–926. [DOI] [PubMed] [Google Scholar]
- 264.Ward JE, McNeel DG. GVAX: an allogeneic, whole-cell, GM-CSF-secreting cellular immunotherapy for the treatment of prostate cancer. Expert Opinion on Biological Therapy. 2007;7(12):1893–1902. [DOI] [PubMed] [Google Scholar]
- 265.Tang L, Zheng Y, Melo MB, et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nature Biotechnology. 2018;36(8):707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Pang N, Shi J, Qin L, et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. Journal of Hematology & Oncology. 2021;14(1):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nature Biotechnology. 2018;36(4):346–351. [DOI] [PubMed] [Google Scholar]
- 268.Cole C, Qiao J, Kottke T, et al. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med. 2005;11(10):1073–1081. [DOI] [PubMed] [Google Scholar]
- 269.Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nature Medicine. 2015;21(5):524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Gardner TJ, Lee JP, Bourne CM, et al. Engineering CAR-T cells to activate small-molecule drugs in situ. Nat Chem Biol. 2022;18(2):216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Dacek MM, Kurtz KG, Wallisch P, et al. Potentiating antibody-dependent killing of cancers with CAR T cells secreting CD47-SIRPα checkpoint blocker. Blood. 2023;141(16):2003–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Roybal KT, Williams JZ, Morsut L, et al. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell. 2016;167(2):419–432.e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Dong X, Chu D, Wang Z. Leukocyte-mediated Delivery of Nanotherapeutics in Inflammatory and Tumor Sites. Theranostics. 2017;7(3):751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Siriwon N, Kim YJ, Siegler E, et al. CAR-T Cells Surface-Engineered with Drug-Encapsulated Nanoparticles Can Ameliorate Intratumoral T-cell Hypofunction. Cancer Immunology Research. 2018;6(7):812–824. [DOI] [PubMed] [Google Scholar]
- 275.Jacob JB, Jacob MK, Parajuli P. Review of immune checkpoint inhibitors in immuno-oncology. Adv Pharmacol. 2021;91:111–139. [DOI] [PubMed] [Google Scholar]
- 276.Dobosz P, Stepien M, Golke A, Dzieciatkowski T. Challenges of the Immunotherapy: Perspectives and Limitations of the Immune Checkpoint Inhibitor Treatment. Int J Mol Sci. 2022;23(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Weiskopf K, Ring AM, Ho CC, et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341(6141):88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Chao MP, Alizadeh AA, Tang C, et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell. 2010;142(5):699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Liu X, Pu Y, Cron K, et al. CD47 blockade triggers T cell–mediated destruction of immunogenic tumors. Nature Medicine. 2015;21(10):1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Głowacki P, Rieske P. Application and Design of Switches Used in CAR. Cells. 2022;11(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Nguyen KG, Vrabel MR, Mantooth SM, et al. Localized Interleukin-12 for Cancer Immunotherapy. Frontiers in Immunology. 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–1647. [DOI] [PubMed] [Google Scholar]
- 283.Rosenberg SA. Immersion in the search for effective cancer immunotherapies. Mol Med. 2021;27(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wells DK, van Buuren MM, Dang KK, et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell. 2020;183(3):818–834 e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Tran E, Urba WJ, Leidner R. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. Reply. N Engl J Med. 2022;387(6):574. [DOI] [PubMed] [Google Scholar]
- 289.Kim SP, Vale NR, Zacharakis N, et al. Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-cell Receptor-Engineered T Cells Targeting Common p53 Neoantigens in Human Solid Tumors. Cancer Immunol Res. 2022;10(8):932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Foy SP, Jacoby K, Bota DA, et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature. 2023;615(7953):687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Tawara I, Kageyama S, Miyahara Y, et al. Safety and persistence of WT1-specific T-cell receptor gene-transduced lymphocytes in patients with AML and MDS. Blood. 2017;130(18):1985–1994. [DOI] [PubMed] [Google Scholar]
- 292.Morris EC, Tendeiro-Rego R, Richardson R, et al. A Phase I Study Evaluating the Safety and Persistence of Allorestricted WT1-TCR Gene Modified Autologous T Cells in Patients with High-Risk Myeloid Malignancies Unsuitable for Allogeneic Stem Cell Transplantation. Blood. 2019;134. [Google Scholar]
- 293.Cossette D, Aiyer S, Kimball C, et al. Clinical-Scale Production and Characterization of Ntla-5001-a Novel Approach to Manufacturing CRISPR/Cas9 Engineered T Cell Therapies. Blood. 2021;138. [Google Scholar]
- 294.Lahman MC, Schmitt TM, Paulson KG, et al. Targeting an alternate Wilms’ tumor antigen 1 peptide bypasses immunoproteasome dependency. Sci Transl Med. 2022;14(631):eabg8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Qasim W, Brunetto M, Gehring AJ, et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J Hepatol. 2015;62(2):486–491. [DOI] [PubMed] [Google Scholar]
- 296.Meng F, Zhao J, Tan AT, et al. Immunotherapy of HBV-related advanced hepatocellular carcinoma with short-term HBV-specific TCR expressed T cells: results of dose escalation, phase I trial. Hepatol Int. 2021;15(6):1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Tan AT, Meng F, Jin J, et al. Immunological alterations after immunotherapy with short lived HBV-TCR T cells associates with long-term treatment response in HBV-HCC. Hepatol Commun. 2022;6(4):841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Molvi Z, O’Reilly RJ. Allogeneic Tumor Antigen-Specific T Cells for Broadly Applicable Adoptive Cell Therapy of Cancer. Cancer Treat Res. 2022;183:131–159. [DOI] [PubMed] [Google Scholar]
- 299.Nathan P, Hassel JC, Rutkowski P, et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. New Engl J Med. 2021;385(13):1196–1206. [DOI] [PubMed] [Google Scholar]
- 300.Zhao Z, Shi L, Zhang W, et al. CRISPR knock out of programmed cell death protein 1 enhances anti-tumor activity of cytotoxic T lymphocytes. Oncotarget. 2018;9(4):5208–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nature Biotechnology. 2018. [DOI] [PubMed] [Google Scholar]
- 302.Kim W, Kim J, Jung D, et al. Induction of lethal graft-versus-host disease by anti-CD137 monoclonal antibody in mice prone to chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2009;15(3):306–314. [DOI] [PubMed] [Google Scholar]
- 303.Schroeder HW Jr., Cavacini L Structure and function of immunoglobulins. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Mariuzza RA, Agnihotri P, Orban J. The structural basis of T-cell receptor (TCR) activation: An enduring enigma. J Biol Chem. 2020;295(4):914–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Morath A, Schamel WW. alphabeta and gammadelta T cell receptors: Similar but different. J Leukoc Biol. 2020;107(6):1045–1055. [DOI] [PubMed] [Google Scholar]
- 306.He Q, Liu Z, Liu Z, Lai Y, Zhou X, Weng J. TCR-like antibodies in cancer immunotherapy. J Hematol Oncol. 2019;12(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Duan Z, Ho M. T-Cell Receptor Mimic Antibodies for Cancer Immunotherapy. Mol Cancer Ther. 2021;20(9):1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Janeway C, Janeway C. Immunobiology : the immune system in health and disease. 5th ed. New York: Garland Pub; 2001. [Google Scholar]
- 309.Herrmann AC, Im JS, Pareek S, et al. A Novel T-Cell Engaging Bi-specific Antibody Targeting the Leukemia Antigen PR1/HLA-A2. Front Immunol. 2018;9:3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Chang AY, Dao T, Gejman RS, et al. A therapeutic T cell receptor mimic antibody targets tumor-associated PRAME peptide/HLA-I antigens. J Clin Invest. 2017;127(9):3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Dao T, Mun SS, Scott AC, et al. Depleting T regulatory cells by targeting intracellular Foxp3 with a TCR mimic antibody. Oncoimmunology. 2019;8(7):1570778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Dao T, Mun S, Korontsvit T, et al. A TCR mimic monoclonal antibody for the HPV-16 E7-epitope p11-19/HLA-A*02:01 complex. PLoS One. 2022;17(3):e0265534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Hsiue EH, Wright KM, Douglass J, et al. Targeting a neoantigen derived from a common TP53 mutation. Science. 2021;371(6533). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Douglass J, Hsiue EH, Mog BJ, et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci Immunol. 2021;6(57). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Ahmed M, Lopez-Albaitero A, Pankov D, et al. TCR-mimic bispecific antibodies targeting LMP2A show potent activity against EBV malignancies. JCI Insight. 2018;3(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Akahori Y, Wang L, Yoneyama M, et al. Antitumor activity of CAR-T cells targeting the intracellular oncoprotein WT1 can be enhanced by vaccination. Blood. 2018;132(11):1134–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Inaguma Y, Akahori Y, Murayama Y, et al. Construction and molecular characterization of a T-cell receptor-like antibody and CAR-T cells specific for minor histocompatibility antigen HA-1H. Gene Ther. 2014;21(6):575–584. [DOI] [PubMed] [Google Scholar]
- 318.Verma B, Neethling FA, Caseltine S, et al. TCR mimic monoclonal antibody targets a specific peptide/HLA class I complex and significantly impedes tumor growth in vivo using breast cancer models. J Immunol. 2010;184(4):2156–2165. [DOI] [PubMed] [Google Scholar]
- 319.Held G, Matsuo M, Epel M, et al. Dissecting cytotoxic T cell responses towards the NY-ESO-1 protein by peptide/MHC-specific antibody fragments. Eur J Immunol. 2004;34(10):2919–2929. [DOI] [PubMed] [Google Scholar]
- 320.Zhang G, Wang L, Cui H, et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep. 2014;4:3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Cohen CJ, Hoffmann N, Farago M, Hoogenboom HR, Eisenbach L, Reiter Y. Direct detection and quantitation of a distinct T-cell epitope derived from tumor-specific epithelial cell-associated mucin using human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of T cells. Cancer Res. 2002;62(20):5835–5844. [PubMed] [Google Scholar]
- 322.Lev A, Denkberg G, Cohen CJ, et al. Isolation and characterization of human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of T cells directed toward the widely expressed tumor T-cell epitopes of the telomerase catalytic subunit. Cancer Res. 2002;62(11):3184–3194. [PubMed] [Google Scholar]
- 323.Sengupta S, Board NL, Wu F, et al. TCR-mimic bispecific antibodies to target the HIV-1 reservoir. Proc Natl Acad Sci U S A. 2022;119(15):e2123406119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Klatt MG, Dao T, Yang Z, et al. A TCR mimic CAR T cell specific for NDC80 is broadly reactive with solid tumors and hematologic malignancies. Blood. 2022;140(8):861–874. [DOI] [PMC free article] [PubMed] [Google Scholar]