

Abstract
The β‐cell relies predominantly on glucose utilization to generate adenosine triphosphate, which is crucial for both cell viability and insulin secretion. The β‐cell has evolved remarkable metabolic flexibility to productively respond to shifts in environmental conditions and changes in glucose availability. Although these adaptive responses are important for maintaining optimal cellular function, there is emerging evidence that the resulting changes in cellular metabolites can impact the epigenome, causing transient and lasting alterations in gene expression. This review explores the intricate interplay between metabolism and the epigenome, providing valuable insights into the molecular mechanisms leading to β‐cell dysfunction in diabetes. Understanding these mechanisms will be critical for developing targeted therapeutic strategies to preserve and enhance β‐cell function, offering potential avenues for interventions to improve glycemic control in individuals with diabetes.
Keywords: β‐Cell dysfunction, Epigenetic change, Glucose metabolism
The β‐cell relies predominantly on glucose utilization to generate adenosine triphosphate, which is crucial for both cell viability and insulin secretion. The β‐cell has evolved remarkable metabolic flexibility to productively respond to shifts in environmental conditions and changes in glucose availability.
INTRODUCTION
The regulation of metabolic homeostasis in mammals is closely tied to complementary actions of insulin, glucagon and somatostatin. These hormones are secreted from pancreatic β‐cells, α‐cells and δ‐cells, respectively, and are controlled by endocrine, paracrine and metabolic regulatory mechanisms 1 . Pancreatic β‐cells are primarily responsible for maintaining glucose homeostasis; loss and/or dysfunction of pancreatic β‐cells results in diabetic phenotypes 1 . Type 1 diabetes is a disease caused by immune‐mediated loss of insulin producing β‐cells, resulting in a life‐long intrinsic inability to maintain glucose homeostasis 2 , 3 . Currently, the prevailing treatment option is insulin replacement therapy. Although there is general acceptance that autoimmunity is the fundamental cause of type 1 diabetes, many important questions remain about the potential contribution of inherent β‐cell dysfunction that might contribute to disease initiation and/or progression 2 . Type 2 diabetes is caused by a complex combination of genetics and environmental factors, and is associated with obesity, insulin resistance and islet dysfunction, including the loss of normal glucose‐stimulated insulin secretion (GSIS) 1 , 4 .
A fundamental feature of pancreatic β‐cells is to maintain blood glucose levels by sensing the glucose as a metabolic fuel. Recent investigations in both type 1 diabetes and type 2 diabetes patients have shown the importance of maintaining mitochondrial function and appropriate metabolic pathways for this process to work optimally 2 , 5 , 6 , 7 , 8 . However, the precise impact of alterations in metabolic flux on the progression of type 1 diabetes and type 2 diabetes remains unclear. Recent studies have suggested that altered intermediates of glucose metabolism can contribute to adverse gene expression changes through altered acetylation or methylation of histone lysine residues 9 , 10 . The purpose of this review article is to evaluate the latest research on the cellular and molecular mechanisms of glucose metabolism that induce epigenetic changes that ultimately alter insulin secretion capacity. Greater insight of the dynamic relationship between glucose metabolism and the epigenome will be necessary to understand some of the underlying mechanisms leading to β‐cell dysfunction in diabetes.
GLUCOSE METABOLISM TRIGGERS ENERGY PRODUCTION AND GSIS
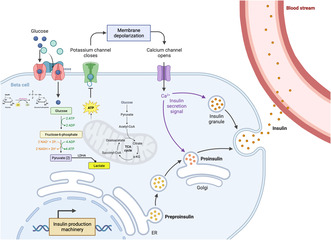
The process of glucose metabolism in islet β‐cells is finely tuned to maintain glucose homeostasis, ensuring that blood glucose levels are kept within a narrow euglycemic range. In response to elevated blood glucose levels, glucose enters the β‐cells through glucose transporter 1 and glucose transporter 3 in humans, and glucose transporter 2 in rodents 2 , 11 , 13 . On entry into the cell, glucose undergoes glycolysis, which involves a series of enzymatic reactions that break glucose down into pyruvate 14 . Unlike many other cell types, pancreatic β‐cells primarily utilize aerobic metabolism (oxidative phosphorylation) for energy production; pyruvate enters the mitochondria where it undergoes further oxidation to produce additional adenosine triphosphate (ATP). The production of ATP leads to KATP‐channel closure, which in turn causes membrane depolarization and the consequent activation of voltage‐gated Ca2+ channels 11 , 12 . The increase in intracellular calcium then triggers insulin granule docking and exocytosis (Figure 1).
Figure 1.
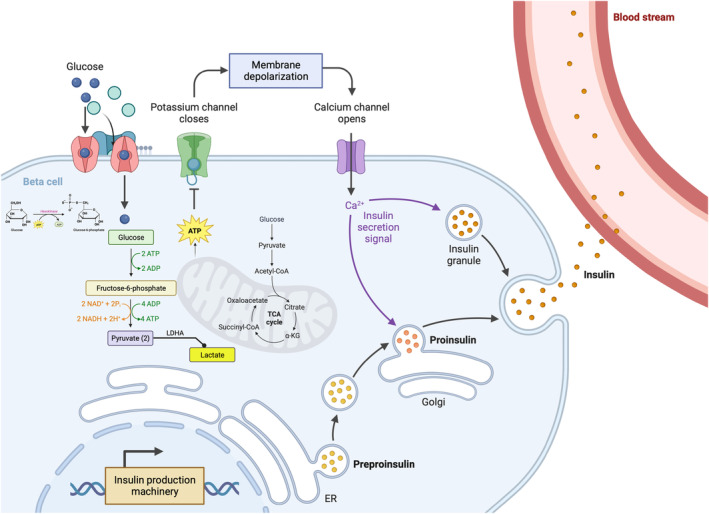
Overview of glucose stimulated insulin secretion in the β‐cell. Glucose enters the cell through glucose transporters (e.g., glucose transporter 2 in mice) and undergoes phosphorylation by hexokinase. Subsequently, glucose enters the glycolytic pathway, leading to the generation of two pyruvates from one glucose molecule. Pyruvate can be converted into acetyl‐coenzyme A or lactate. Adenosine triphosphate (ATP), produced through glycolysis and the mitochondria through the tricarboxylic acid (TCA) cycle, induces membrane depolarization by closing potassium channels. This depolarization event opens calcium channels, initiating the docking and exocytosis of insulin granules. Created with BioRender.com. ER, endoplasmic reticulum; LDHA, lactate dehydrogenase A. ADP, adenosine diphosphate.
ALTERATIONS IN METABOLIC FLUX CONTRIBUTE TO β‐CELL FAILURE
β‐Cells dynamically respond to conditions of increased metabolic demand or stress. For example, high glucose levels can lead to elevated glycolytic flux, and increased production of ATP and other metabolic intermediates. Furthermore, during increased metabolic activity or in situations where oxygen availability is compromised, β‐cells can shift to anaerobic glycolysis, and pyruvate can become converted to lactate in the absence of sufficient oxygen. Although metabolic flexibility is essential for β‐cells' ability to adapt to changing environmental conditions, over time, these changes in metabolic pathways can eventually mediate β‐cell failure. Chronic exposure to high glucose levels and elevated levels of free fatty acids, as seen in type 2 diabetes, can contribute to glucotoxicity and lipotoxicity, resulting in impaired mitochondrial function. Several mitochondrial enzymes have been shown to be impacted in type 2 diabetes patients, including pyruvate dehydrogenase, citrate synthase, isocitrate dehydrogenases (IDH1 and 2) and ATP synthase. Depending on the individual, dysregulation of these enzymes could be a consequence or cause of β‐cell dysfunction. For example, IDH2 messenger ribonucleic acid and protein expression are decreased in two diabetic mouse strains with β‐cell dysfunction, suggesting they are impacted by a diabetic environment 15 . However, inhibition of IDH2 can also result in the suppression of insulin secretion in isolated islets and in mice, suggesting mutations in IDH2 could contribute to an underlying vulnerability of β‐cells to stress conditions 1 , 15 . In addition to mitochondrial dysfunction, lipid toxicity can also induce fatty acid oxidation, which can lead to the impairment of GSIS. The inhibition of fatty acid translocase cluster determinant 36 lowered intracellular peroxide levels in INS‐1 cells, and rescued the decreased GSIS response, 16 , 17 showing that – at least in the short term – β‐cell dysfunction can be reversed. These examples show the importance of preserving normal metabolic pathways to maintain and enhance β‐cell function in diabetes.
ALTERED GLUCOSE METABOLISM CAN LEAD TO EPIGENETIC CHANGES
Altered glucose metabolism can also have profound effects on the epigenome. Metabolism is considered a key regulator of the epigenome, given that nearly all chromatin‐modifying enzymes rely on essential metabolites as cofactors to facilitate their catalytic functions. Consequently, shifts in glucose metabolism dynamically shape the availability of these vital cofactors, influencing the enzymatic processes involved in epigenetic modifications. As described below, these epigenetic modifications, such as deoxyribonucleic acid (DNA) methylation and histone modifications, will influence gene expression in β‐cells to have a significant impact on the development or progression of β‐cell dysfunction in diabetes 10 , 18 , 19 .
DNA AND HISTONE METHYLATION
DNA methylation is a major mechanism for regulating gene expression. There are several DNA methylases that are classified based on their functions and targets. DNMT1 is responsible for maintaining existing DNA methylation patterns during DNA replication, whereas DNMT3a and DNMT3b are involved in the establishment of new DNA methylation patterns in response to environmental cues. DNMT3L modulates the activity of DNMT3a and DNMT3b, and is important for establishing genomic imprinting. S‐adenosyl methionine is a critical cofactor for DNA methyltransferases, and high glucose levels can lead to increased flux through the one‐carbon metabolism pathway, potentially increasing S‐adenosyl methionine levels and impacting DNA methylation patterns (Figure 2). Several studies in rodents and humans have highlighted the importance of DNA methylation in preserving β‐cell identity and function. In rodents, two complementary studies showed the importance of DNMT1 in maintaining β‐cell identity; disruption of DNMT1 expression or targeting in β‐cells resulted in the derepression of α‐cell lineage genes 20 , 21 . In humans, Ling et al. 22 showed that an increase in DNA methylation at the PPARGC1A promoter in type 2 diabetes human islets was associated with reduced insulin gene expression and secretion. This group also identified a cohort of patients with type 2 diabetes that had increased DNA methylation and decreased expression of the essential pancreatic master regulator, PDX1 23 . Additionally, Kameswaran et al. 24 shed light on the importance of appropriate imprinting for maintaining β‐cell function. That study showed that the imprinted DLK1‐MEG3 locus was downregulated in islets from individuals with type 2 diabetes, and the decreased expression correlated with hypermethylation of the MEG3 promoter. Collectively, these findings underscore the potential for altered glucose metabolism to exert profound effects on DNA methylation, thereby influencing gene expression and, consequently, β‐cell function.
Figure 2.
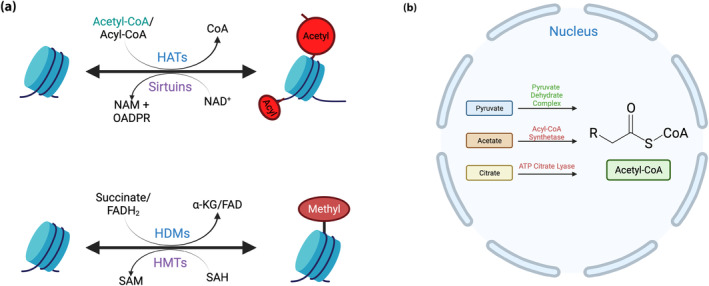
Metabolic intermediates are cofactors for the chromatin‐modifying enzymes. (a) Metabolic intermediates are essential cofactors for chromatin modifying enzymes. (b) Acetyl‐coenzyme A production can be catalyzed from pyruvate, acetate and citrate by different enzymes. CoA, coenzyme A; FADH2, rlavin adenine dinucleotide; HAT, histone acetyltransferases; HDM, histone demethylase; HTM, histone methyltransferase; NAD+, nicotinamide adenine dinucleotide; NAM, nicotinamide; OAADPR, O‐acetyl‐ADP‐ribose; SAH, S‐adenosylhomocysteine; SAM, S‐adenosylmethionine; α‐KG, α‐ketoglutarate. Created with BioRender.com
Consistent with the aforementioned studies cited, lysine‐specific histone demethylase 1A (LSD1/KDM1A) plays a pivotal role in essential cellular processes 25 . The demethylase activity of LSD1 relies on flavin adenine dinucleotide as a cofactor, establishing a strong connection between cellular metabolism and the nutrient environment 26 . LSD1 is a component of transcriptional complexes within the nucleus that regulates nutrient responses in various cell types, including hepatocytes, adipocytes and pancreatic β‐cells 27 . Systemic inhibition of LSD1 results in a reduction of hyperphagia and weight gain, improvement in insulin sensitivity, and prevention of hyperglycemia in obesity models 25 . Furthermore, β‐cell‐specific deletion of LSD1 (Pdx1‐CreERT, MIP‐CreERT) led to insulin hypersecretion, aberrant expression of nutrient‐response genes and histone hyperacetylation 28 . Additionally, genetic variants linked to type 2 diabetes were concentrated at sites bound by LSD1 in human islets. This suggests that LSD1 plays an important role in coupling the nutrient state to the regulation of the islet epigenome and adaptive downstream gene expression 28 .
Histone methylation is another important epigenetic modification that can be influenced by cellular metabolism, as the availability of S‐adenosyl methionine is also crucial for histone methyltransferase activity. Consistently, mice carrying a deletion in the histone arginine methyl transferase, PRMT1, showed impaired expression of genes important for β‐cell function and identity 29 .
HISTONE ACETYLATION
The primary regulation of histone acetylation levels typically involves histone acetyltransferases (HATs) and histone deacetylases (HDACs). Three primary families of HATs have been identified (GNAT, MYST and p300/CBP) that each require acetyl‐coenzyme A (acetyl‐CoA) as the acetyl group donating cofactor 9 . Maintenance of appropriate histone acetylation has been shown to be essential for β‐cell proliferation and function in numerous studies 25 , 28 , 30 , 31 , 32 , 33 , 34 . HDACs are metalloenzymes classified into three main classes based on their protein sequence homologies with yeast deacetylase enzymes 9 , 35 . One group of HDACs, known as class I HDACs, consists of HDAC1, HDAC2, HDAC3 and HDAC8. These are closely associated with the yeast reduced potassium dependency 3 transcriptional regulator. The class II HDACs, is comprised of HDAC4, HDAC5, HDAC7, HDAC9 (class IIa), and HDAC6 and HDAC10 (class IIb), with domains resembling yeast histone deacetylase I. Many different studies have investigated the role of HDACs in β‐cells with mixed results. HDAC inhibition in cell lines and rodent islets has been shown to prevent cytokine‐induced toxicity in β‐cells 36 , 37 , 38 and enhance β‐cell proliferation 39 . Furthermore, mice carrying an inducible β‐cell specific (MIP‐CreERT) deletion of HDAC3 showed increased glucose tolerance and insulin secretion 40 . However, mice carrying a constitutive β‐cell‐specific (Rip‐Cre) deletion of HDAC3 caused an impairment of β‐cell function 41 . This was consistent with a study that showed that the administration of HDAC inhibitors to the rodent β‐cell line β‐TC3 led to a loss of β‐cell identity, with a concomitant elevation in α‐cell marker expression within β‐cells 42 , and a more recent study that reported HDAC inhibition using the pan‐HDAC inhibitor, trichostatin A, appeared to impair pancreatic β‐cell function through an epigenome wide reprogramming 43 . The discrepant results from all these studies suggest that the function of HDACs is more nuanced than appreciated, and a balance of the different HAT and HDAC activities – perhaps in response to external conditions – might function as a rheostat for sensing the metabolic condition of cells and translating it into an appropriate cellular response.
DEPENDENCE OF HISTONE ACETYLATION ON METABOLIC INTERMEDIATES
Although the primary regulation of histone acetylation levels typically involves HATs and HDACs, acetyl‐CoA derived from exported citrate also contributes to the histone acetylation process. Acetyl‐CoA is an essential substrate for HATs that can impact the balance of histone acetylation; during high‐glucose conditions there is an increased flux through glycolysis, leading to higher acetyl‐CoA levels. This implicates acetyl‐CoA as another metabolite that links cellular metabolism to epigenetic regulation (Figure 2a).
Acetyl‐CoA is necessary for both lipid synthesis in the cytosol and histone acetylation in the nucleus 44 , 45 . Acetyl‐CoA functions as a central metabolic intermediate produced during nutrient catabolism, and acts as the substrate for acetylation reactions involving proteins and metabolites, including histone acetylation 46 . The enzyme, ATP citrate lyase (ACLY), converts citrate to acetyl‐CoA for several different cellular pathways, including having a role in augmenting histone acetylation, thereby facilitating chromatin structure and transcriptional activation of cell‐specific genes 45 (Figure 2b).
As ACLY plays a critical role in cellular metabolism, it is tightly regulated to meet the metabolic demands of the cell and regulate appropriate cell‐specific gene expression. The regulation of ACLY has predominantly been investigated in the context of cancer cells, where it is activated by acetylation and AKT‐mediated phosphorylation, and degraded by ubiquitinylation 47 . Recently, in β‐cells, ACLY has been identified as a novel substrate for PTPN2 48 . That study reported that increased tyrosyl phosphorylation of ACLY caused a detrimental effect on metabolic activity and insulin secretion, a result that could be attributed to both its role in the generation of acetyl‐CoA and regulation of gene expression. Furthermore, the observed changes were compounded by a reduction in citrate production 48 (Figure 3). Several inhibitors targeting ACLY have also shown promising outcomes in diverse therapeutic areas, demonstrating that inhibition of ACLY has the potential to modulate inflammation and reduce the progression of atherosclerotic diseases 49 (Figure 3). Further exploration into the mechanisms underlying the translocation of metabolic enzymes into the nucleus, as well as their involvement in epigenetic regulation in the β‐cells, will provide novel information related to the metabolic regulation of cellular functions.
Figure 3.
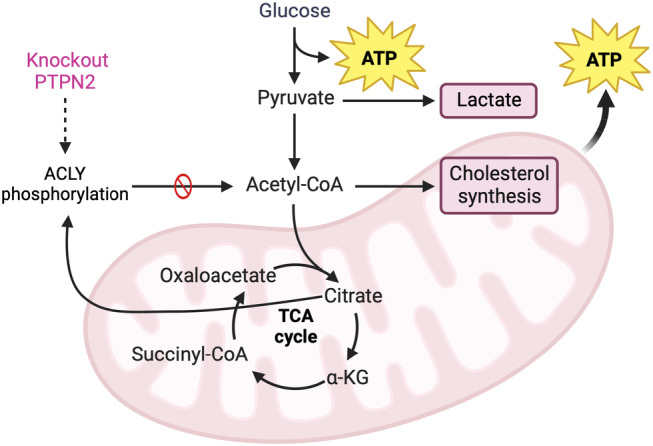
Acetyl‐coenzyme A (acetyl‐CoA) is regulated by adenosine triphosphate (ATP) citrate lyase (ACLY) activity in the cytosol. Schematic of acetyl‐CoA metabolism in the cytosol. Acetyl‐CoA produced by pyruvate dehydrate complex and ACLY. PTPN2, phospho‐tyrosine phosphatase 2; TAC, tricarboxylic acid. Created with BioRender.com.
CONCLUSIONS AND PERSPECTIVES
β‐Cell dysfunction in diabetes involves a complex interplay of molecular changes that affect diverse cellular processes. As discussed in this review, in diabetic conditions, altered glucose metabolism can have profound effects on the epigenome of the β‐cell by altering the availability of metabolites that serve as cofactors for enzymes involved in epigenetic processes. Understanding these molecular changes will be crucial for developing targeted therapeutic strategies to preserve and enhance β‐cell function in diabetes. Research in this area has the potential to identify interventions that can protect β‐cells from stressors, promote their regeneration and ultimately improve glycemic control in individuals with diabetes.
DISCLOSURE
The authors declare no conflict of interest.
Approval of the research protocol: N/A.
Informed consent: N/A.
Registry and the registration no. of the study/trial: N/A.
Animal studies: N/A.
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) RS‐2023‐00208709 (to KCW), National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) R01DK082590 (to LS), R01 DK123560 (to YKK and LS) and Diabetes Research Connection (to YKK).
Contributor Information
Kyu Chang Won, Email: kcwon@med.yu.ac.kr.
Lori Sussel, Email: lori.sussel@cuanschutz.edu.
REFERENCES
- 1. Campbell JE, Newgard CB. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat Rev Mol Cell Biol 2021; 22: 142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim YK, Sussel L, Davidson HW. Inherent Beta cell dysfunction contributes to autoimmune susceptibility. Biomolecules 2021; 11: 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thomas NJ, Jones SE, Weedon MN, et al. Frequency and phenotype of type 1 diabetes in the first six decades of life: A cross‐sectional, genetically stratified survival analysis from UK Biobank. Lancet Diabetes Endocrinol 2018; 6: 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brereton MF, Rohm M, Ashcroft FM. Beta‐cell dysfunction in diabetes: A crisis of identity? Diabetes Obes Metab 2016; 18: 102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fex M, Nicholas LM, Vishnu N, et al. The pathogenetic role of beta‐cell mitochondria in type 2 diabetes. J Endocrinol 2018; 236: R145–R159. [DOI] [PubMed] [Google Scholar]
- 6. Jitrapakdee S, Wutthisathapornchai A, Wallace JC, et al. Regulation of insulin secretion: Role of mitochondrial signalling. Diabetologia 2010; 53: 1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Las G, Oliveira MF, Shirihai OS. Emerging roles of beta‐cell mitochondria in type‐2‐diabetes. Mol Aspects Med 2020; 71: 100843. [DOI] [PubMed] [Google Scholar]
- 8. Vig S, Lambooij JM, Zaldumbide A, et al. Endoplasmic reticulum‐mitochondria crosstalk and Beta‐cell destruction in type 1 diabetes. Front Immunol 2021; 12: 669492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haws SA, Leech CM, Denu JM. Metabolism and the epigenome: A dynamic relationship. Trends Biochem Sci 2020; 45: 731–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lempradl A, Pospisilik JA, Penninger JM. Exploring the emerging complexity in transcriptional regulation of energy homeostasis. Nat Rev Genet 2015; 16: 665–681. [DOI] [PubMed] [Google Scholar]
- 11. Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta‐cells. Nature 1984; 312: 446–448. [DOI] [PubMed] [Google Scholar]
- 12. Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B‐cells. Nature 1984; 311: 271–273. [DOI] [PubMed] [Google Scholar]
- 13. McCulloch LJ, van de Bunt M, Braun M, et al. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol Genet Metab 2011; 104: 648–653. [DOI] [PubMed] [Google Scholar]
- 14. Fridlyand LE, Philipson LH. Glucose sensing in the pancreatic beta cell: A computational systems analysis. Theor Biol Med Model 2010; 7: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang GF, Jensen MV, Gray SM, et al. Reductive TCA cycle metabolism fuels glutamine‐ and glucose‐stimulated insulin secretion. Cell Metab 2021; 33: 804–817.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elumalai S, Karunakaran U, Lee IK, et al. Rac1‐NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells. Redox Biol 2017; 11: 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim YW, Moon JS, Seo YJ, et al. Inhibition of fatty acid translocase cluster determinant 36 (CD36), stimulated by hyperglycemia, prevents glucotoxicity in INS‐1 cells. Biochem Biophys Res Commun 2012; 420: 462–466. [DOI] [PubMed] [Google Scholar]
- 18. Rosen ED. Epigenomic and transcriptional control of insulin resistance. J Intern Med 2016; 280: 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wellen KE, Thompson CB. Cellular metabolic stress: Considering how cells respond to nutrient excess. Mol Cell 2010; 40: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dhawan S, Georgia S, Tschen SI, et al. Pancreatic beta cell identity is maintained by DNA methylation‐mediated repression of Arx. Dev Cell 2011; 20: 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Papizan JB, Singer RA, Tschen SI, et al. Nkx2.2 repressor complex regulates islet beta‐cell specification and prevents beta‐to‐alpha‐cell reprogramming. Genes Dev 2011; 25: 2291–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ling C, Del Guerra S, Lupi R, et al. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 2008; 51: 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang BT, Dayeh TA, Volkov PA, et al. Increased DNA methylation and decreased expression of PDX‐1 in pancreatic islets from patients with type 2 diabetes. Mol Endocrinol 2012; 26: 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kameswaran V, Bramswig NC, McKenna LB, et al. Epigenetic regulation of the DLK1‐MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab 2014; 19: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ramms B, Pollow DP, Zhu H, et al. Systemic LSD1 inhibition prevents aberrant remodeling of metabolism in obesity. Diabetes 2022; 71: 2513–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hino S, Sakamoto A, Nagaoka K, et al. FAD‐dependent lysine‐specific demethylase‐1 regulates cellular energy expenditure. Nat Commun 2012; 3: 758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. The Tabula Muris Consortium , Overall Coordination , Logistical Coordination , et al. Single‐cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018; 562: 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wortham M, Liu F, Harrington AR, et al. Nutrient regulation of the islet epigenome controls adaptive insulin secretion. J Clin Invest 2023; 133: e165208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim H, Yoon BH, Oh CM, et al. PRMT1 is required for the maintenance of mature beta‐cell identity. Diabetes 2020; 69: 355–368. [DOI] [PubMed] [Google Scholar]
- 30. Bompada P, Atac D, Luan C, et al. Histone acetylation of glucose‐induced thioredoxin‐interacting protein gene expression in pancreatic islets. Int J Biochem Cell Biol 2016; 81: 82–91. [DOI] [PubMed] [Google Scholar]
- 31. He F, Li N, Huang HB, et al. LSD1 inhibition yields functional insulin‐producing cells from human embryonic stem cells. Stem Cell Res Ther 2020; 11: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wong CK, Wade‐Vallance AK, Luciani DS, et al. The p300 and CBP transcriptional coactivators are required for beta‐cell and alpha‐cell proliferation. Diabetes 2018; 67: 412–422. [DOI] [PubMed] [Google Scholar]
- 33. Yang XF, Zhou SY, Wang C, et al. Inhibition of LSD1 promotes the differentiation of human induced pluripotent stem cells into insulin‐producing cells. Stem Cell Res Ther 2020; 11: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun X, Wang L, Obayomi SMB, et al. Epigenetic regulation of beta cell identity and dysfunction. Front Endocrinol 2021; 12: 725131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Ruijter AJ, van Gennip AH, Caron HN, et al. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem J 2003; 370: 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chou DH, Holson EB, Wagner FF, et al. Inhibition of histone deacetylase 3 protects beta cells from cytokine‐induced apoptosis. Chem Biol 2012; 19: 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Larsen L, Tonnesen M, Ronn SG, et al. Inhibition of histone deacetylases prevents cytokine‐induced toxicity in beta cells. Diabetologia 2007; 50: 779–789. [DOI] [PubMed] [Google Scholar]
- 38. Lindelov Vestergaard A, Heiner Bang‐Berthelsen C, Floyel T, et al. MicroRNAs and histone deacetylase inhibition‐mediated protection against inflammatory beta‐cell damage. PLoS One 2018; 13: e0203713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Khan S, Jena G. Valproic acid improves glucose homeostasis by increasing Beta‐cell proliferation, function, and reducing its apoptosis through HDAC inhibition in juvenile diabetic rat. J Biochem Mol Toxicol 2016; 30: 438–446. [DOI] [PubMed] [Google Scholar]
- 40. Remsberg JR, Ediger BN, Ho WY, et al. Deletion of histone deacetylase 3 in adult beta cells improves glucose tolerance via increased insulin secretion. Mol Metab 2017; 6: 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen WB, Gao L, Wang J, et al. Conditional ablation of HDAC3 in islet beta cells results in glucose intolerance and enhanced susceptibility to STZ‐induced diabetes. Oncotarget 2016; 7: 57485–57497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kubicek S, Gilbert JC, Fomina‐Yadlin D, et al. Chromatin‐targeting small molecules cause class‐specific transcriptional changes in pancreatic endocrine cells. Proc Natl Acad Sci USA 2012; 109: 5364–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oger F, Moreno M, Derhourhi M, et al. Pharmacological HDAC inhibition impairs pancreatic beta‐cell function through an epigenome‐wide reprogramming. iScience 2023; 26: 107231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wellen KE, Hatzivassiliou G, Sachdeva UM, et al. ATP‐citrate lyase links cellular metabolism to histone acetylation. Science 2009; 324: 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Izzo LT, Trefely S, Demetriadou C, et al. Acetylcarnitine shuttling links mitochondrial metabolism to histone acetylation and lipogenesis. Sci Adv 2023; 9: eadf0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baeza J, Smallegan MJ, Denu JM. Mechanisms and dynamics of protein acetylation in mitochondria. Trends Biochem Sci 2016; 41: 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Icard P, Wu Z, Fournel L, et al. ATP citrate lyase: A central metabolic enzyme in cancer. Cancer Lett 2020; 471: 125–134. [DOI] [PubMed] [Google Scholar]
- 48. Kim YK, Kim YR, Wells KL, et al. PTPN2 regulates metabolic flux to affect beta cell susceptibility to inflammatory stress. Diabetes 2024; 73: 434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Russo M, Pileri F, Ghisletti S. Novel insights into the role of acetyl‐CoA producing enzymes in epigenetic regulation. Front Endocrinol 2023; 14: 1272646. [DOI] [PMC free article] [PubMed] [Google Scholar]